

Abstract
The enzyme urease is an essential colonizing factor of the notorious carcinogenic pathogen Helicobacter pylori (H. pylori), conferring acid resistance to the bacterium. Recently, antibiotic resistant strains have emerged globally with little to no alternative treatment available. In this study we propose novel urease inhibitors capable of controlling infection by H. pylori and other pathogenic bacteria. We employed hierarchal computational approaches to screen new urease inhibitors from commercial chemical databases followed by in vitro anti-urease assays. Initially ROCS shape-based screening was performed using o-chloro-hippurohydroxamic acid followed by molecular docking studies. Out of 1.83 million compounds, 1700 compounds were retrieved based on having a ROCS Tanimoto combo score in the range of values from 1.216 to 1.679. These compounds were further screened using molecular docking simulations and the 100 top ranked compounds were selected based on their Glide score. After structural classification of the top ranked compounds, eight compounds were selected and purchased for biological assays. The plausible binding modes of the most active compounds were also confirmed using molecular dynamics (MD) simulations. Compounds 1, 2 and 3 demonstrated good urease inhibitory properties (IC50 = 0.32, 0.68 and 0.42 μM) compared to the other compounds. Enzyme kinetic studies revealed that compounds 1 and 3 are competitive inhibitors while 2 is a mixed type inhibitor of the urease enzyme. Cell based urease inhibition and MTT assay showed that these compounds blocked H. pylori urease activity, affecting bacterial growth and acid tolerance.
The enzyme urease is an essential colonizing factor of the notorious carcinogenic pathogen Helicobacter pylori (H. pylori), conferring acid resistance to the bacterium.
1. Introduction
Urease (urea amidohydrolase), is a metal containing enzyme which catalyzes the hydrolysis of urea leading to the formation of ammonia and carbon dioxide. It is present in a wide variety of organisms including plant, fungi and algae.1,2 Urease producing bacteria have a negative effect on human health. Urease plays a major part in the pathologies caused by H. pylori. These bacteria survive at low pH during colonization leading to gastric and peptic ulcers, in some cases leading to cancer.3 Urease activity in H. pylori infection is essential for colonization of the bacterium as urease knockout bacteria lose the ability to establish infection. Since rising antimicrobial resistance in bacterial populations has rendered current therapeutic regimens ineffective in controlling the infection, novel therapeutic targets and compounds should be explored as viable alternatives.4H. pylori relies on urease activity for bacterial survival in low pH environment of the stomach, therefore targeting urease activity can eradicate the bacterium in early stages of the infection. Since the structure, molecular weight, amino acid sequence of urease greatly depends upon its origin. The bacterial ureases are hetero polymeric molecules having three subunits, α, β and γ whereas the urease from jack beans are homo hexameric molecules having six α subunits. Despite the difference in the structure, the active site of the enzyme, largely remains conserved. The active site is always located in α subunits having binuclear nickel center. Due to involvement of urease in bacterial infections, designing of novel urease inhibitors are of great research interest. The current eminence of urease inhibitors is quite restricted. However mostly six different classes of inhibitors including phosphate derivatives (phenylphosphorodiamidates, phosphorotriamides, phosphorylamides),5 barbituric analogues,6 thiourea derivatives,7 five- and six-membered heterocycles, natural products and metal complexes are available in literature,8 the structures of few good urease inhibitors are presented in Fig. 1. To date, hydroxamic acid derivative acetohydroxamic acid (AHA) is known as the best urease inhibitors to treat H. pylori infection. It was approved as a drug by U.S. Food and Drug Administration in May 1983.
Fig. 1. Literature reported scaffolds as urease inhibitor.

Established methods for drug designing involve synthesis of new compounds and then to assess them using in vitro biological assays. However, over the past few decades computer aided drug designing has emerged as a tool to identify new active scaffolds against different biological targets.9–11 Rapid Overlay of Chemical Structure (ROCS), is a 3D shape-based screening method that is found to be very successful in identifying chemically diverse compounds possessing similar or better bioactivity than query compound(s).12 Several published applications of ROCS demonstrated its usefulness in identification of new molecules against different protein targets.13,14 In this study, we report the identification of new urease inhibitors using hierarchal virtual screening approach in which first ligand-based approach followed by molecular docking studies were performed to screen urease inhibitors from commercial chemical database. Then best scored compounds were structurally clustered and out of those nine were selected for their biological assays. In vitro studies revealed that three compounds were inhibiting urease activity at low micro molar concentration in a competitive manner. Similarly, cell based urease inhibition and MTT assay showed that these compounds blocked H. pylori urease activity which retarded bacterial growth and impacted its acid tolerance.
2. Materials and methods
2.1. Database preparation
HTS collection library of enamine database containing 1 834 363 compounds were retrieved and prepared using Openeye software tools. The druglike filter introduced by Openeye software was used to eliminate the unwanted molecules before virtual screening process. OMEGA software15 was used to generate three dimensional multi conformer database after removal of undesired compounds. Maximum 200 conformers were generated by using root-mean-square deviation (RMS) = 0.5 Å, and Ewindow = 10 kcal mol−1 values between them. The Merck Molecular Force Field (MMFF) was used to assign the charges to the conformers.
2.2. Protein and small molecules structure preparation
Jack bean urease protein with PDB code 4H9M was downloaded from the protein databank (http://www.rcsb.org). The protein structure was prepared using the “protein preparation wizard” available in the Schrodinger software. Missing side chains of the amino acid were added using Prime modeling tool. The hydrogen atoms were also added to entire protein and co-crystallized reagents were removed except nickel ions. Since urease enzyme is a metalloenzyme, therefore nickel ion in the active site was assigned a partial positive charge. Then restrained minimization of prepared crystal structure was performed using OPLSA_2005 force field. The structure was allowed to relax up to 0.3 Å from original conformation to remove all steric clashes. Similarly, small molecules exhibiting the anti-urease activities were sketched and prepared using ligprep module implemented in the Schrodinger software package. Different tautomeric and ionization states of compounds were generated along with their different conformations.
2.3. Grid generation and molecular docking simulation
The prepared crystal structure of urease enzyme was used as receptor and grid box around the active site containing nickel ions was generated. The center of the grid was defined at X = 17.5, Y = 36.56 and Z = 20.48 with 20 Å length in each dimension. Hydrogen bond or metal–ligand interaction constraint was also defined for the nickel ion present in the active site of enzyme during docking simulations. The hydrogen atoms of thiol and hydroxyl groups of active site amino acids residues were allowed to rotate during molecular docking simulations. The van der Waals radii of receptor atom was set to a scaling factor of 1.0 with partial charge cutoff value set to 0.25 in order to soften the potential of non-polar parts of receptor. During docking simulations top five poses of the compounds were subjected to minimization and best poses were selected based on the highest Glide score.16 For docking simulations, standard precision (sp) mode of Glide was used.
2.4. ROCS query generation and screening
The vROCS graphical interface was used for generating shape and color (hydrogen bond acceptor, hydrogen bond donor, ring) based query of o-chloro-hippurohydroxamic acid. Then ROCS (Rapid Overlay of Chemical Structures) code was run to carry out the screening of prepared chemical database using the generated query. The screened compounds were ranked based on the Tanimoto combo score. When query molecule is aligned with the database molecule it yields the score based on the shape and colors matching. The higher the score the better will be the match. Thus, this match is represented by a combo score which ranges from 0 to 2.
2.5. Molecular dynamics simulation
First, the bonding parameters (bond length, bond angle, torsion angle, and improper torsion angle) and nonbonding van der Waals parameters (radius and well-depths) of three compounds and a lysine Nz-carboxylic acid (KCX) were assigned general amber force field (GAFF). Atomic charges were calculated using Gaussian16 software at the B3LYP/6-31G(d) level. The polarizable continuum model (PCM) was used to consider the solvent effect on the calculation of atomic charges. Antechamber software was used for restrained electrostatic potential (RESP) charge-fitting from the results of quantum chemical calculation. We constructed the trimer model structures by superimposing urease/compound complex structures to three chains of the urease from jack bean (PDB-id: 3la4). All the trimer structures of urease/compound were hydrated using a TIP3P water box, consequently, ∼68 000 water molecules in total were added. The urease/compound complex structures were optimized in 10 000 steps for hydrogen atoms after 10 000 steps for all atoms. For the optimized structure, a 50 ps MD simulation under constant volume (NVT) was performed with constraint for the urease structure. Furthermore, to equilibrate the system, a 1 ns MD simulation was performed under constant pressure (1 atm) and temperature (T = 300 K) (NPT) with a Langevin thermostat with the collision frequency = 1. We used the particle mesh Ewald (PME) method with a direct-space non-bonded cutoff of 12 Å. Bond lengths with hydrogen atoms were constrained with SHAKE algorithm. Using the structure after 1 ns MD simulation, MD simulation was performed for 50 ns. The snapshots were stored every 100 ps, with 500 snapshots in total.
2.6. Pharmacological activities
2.6.1. % urease inhibition assay, IC50 and kinetics study protocol
In vitro studies of the synthesized compounds were performed as reported in our earlier studies.17 Initially activity assay buffer (10 μL, K2HPO4, pH = 6.8–7.0, 50 mmol), H2O (10 μL) and urease enzyme solution (20 μL, jack bean urease UNI-CHEM, U30550-2E) were prepared and poured in each well of 96 well plate. Then each compound (10 μL in vol. and 1000 to 0.4882 μM conc.) was incubated with urease enzyme solution (20 μL) and assay buffer at 37 °C for 10 min. After the incubation, 40 μL of urea substrate with 20 mM conc. was added and incubated again at 37 °C for 10 min. Then 40 μL of phenol reagent (phenol 1.0% w/v, sodium nitroprusside 0.005% w/v) and 75 μL of alkali reagent (sodium hydroxide 0.5%, 0.1% active chlorine from sodium hypochlorite) were added to each well and placed at room temperature for 50 min. For positive control thiourea was used as reference in 1000 to 0.4882 μM concentration. Absorbance of each well was recorded at 625 nm by micro plate reader LT-4500 (Labtech International Ltd, UK) and urease inhibition (%) was calculated by following formula.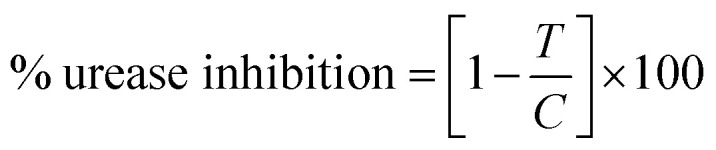 where, T is absorbance of inhibitory well containing synthesized compound and C is absorbance of buffer solutions without compounds. All the in vitro assays were performed in triplicate and results are presented as mean ± SEM. IC50 values of each synthesized compound and thiourea was calculated at 50% inhibition using regression equation. For kinetics studies, binding mechanism of each compound (inhibitor) was determined using Lineweaver Burk plot. Three best active compounds showing good IC50 values were used with different concentration of substrate (urea, 0.5–4.0 mM) to evaluate whether the inhibitor is competitive, non-competitive (mixed) and uncompetitive after determining the Km (app), Vmax (app) from Lineweaver Burk plot. The inhibition constant (Ki) value was also calculated for best active compound by secondary Lineweaver Burk plot using PRISM 7.0.
where, T is absorbance of inhibitory well containing synthesized compound and C is absorbance of buffer solutions without compounds. All the in vitro assays were performed in triplicate and results are presented as mean ± SEM. IC50 values of each synthesized compound and thiourea was calculated at 50% inhibition using regression equation. For kinetics studies, binding mechanism of each compound (inhibitor) was determined using Lineweaver Burk plot. Three best active compounds showing good IC50 values were used with different concentration of substrate (urea, 0.5–4.0 mM) to evaluate whether the inhibitor is competitive, non-competitive (mixed) and uncompetitive after determining the Km (app), Vmax (app) from Lineweaver Burk plot. The inhibition constant (Ki) value was also calculated for best active compound by secondary Lineweaver Burk plot using PRISM 7.0.
2.6.2. Bacterial strains and culture
Campylobacter jejuni strain 255 was maintained on modified CCDA agar at 42 °C under microaerophilic conditions. H. pylori strain J166 was kindly provided by Prof. Dr Jay Solnick. H. pylori was cultured on BHI agar/broth with DENT supplement (Oxoid, UK) and 5% calf serum and maintained under microaerophilic conditions at 37 °C. Previously characterized clinical waterborne and foodborne bacterial isolates enteropathogenic Escherichia coli, Pseudomonas aeruginosa, Methicillin resistant staphylococcus aureus (MRSA) and Campylobacter jejuni (Cj 255) were obtained from Microbiology and Public health lab, COMSATS Institute of Information Technology Islamabad, Pakistan used for antibacterial assay.18
2.6.3. Antimicrobial susceptibility test
Antimicrobial susceptibility testing was performed using the broth dilution method. Briefly, 180 μL bacterial culture (CFU ∼ 107) were added into each well of the microtiter along with different concentration of test material (10–100 μg mL−1) and were incubated for incubated for 24 h at 37 °C for enteropathogenic E. coli, P. aeruginosa and methicillin resistant S. aureus and for 48 h under microaerophilic conditions for C. jejuni (at 42 °C) and H. pylori (at 37 °C). Growth inhibition in each well was visuality by the presence/absence of turbidity whereas cellular viability was accessed using, 20 μL of TTC 2,3,5-triphenyltetrazolium chloride (TTC) as indicator in each well. Appearance of pink to red coloration after addition of TTC was indicative of viable bacterial cell whereas absence of color change was indicative of dead bacterial cells. Un-inoculated media was used as a negative control and untreated bacterial cell were used as a positive control.
2.6.4. Cell based urease activity
Effect of the test compounds on urease activity of H. pylori cells was determined using Mc Laren method.19H. pylori (CFU mL−1 ∼ 106) cultures were treated with the test compounds (100 μg mL−1) and incubated at 37 °C for 48 h. The cells were harvested, washed and re-suspended in 100 μL of PBS. This suspension was then mixed with 100 μL of urea broth and change in color of the broth was recorded for 1 h. Development of pink coloration indicated urease activity, whereas no color change indicated absence of urease activity. Untreated H. pylori cells were used as positive control.
2.6.5. Acid stress tolerance assay
Growth inhibition at acidic pH by the addition of urease inhibitors was used to determine acid tolerance of H. pylori as described earlier with minor modifications.20 Bacteria were grown in BHI broth with 5% calf serum and 10 mM urea with or without addition of 100 μg mL−1 of test compound. The pH of cell suspension was adjusted to either 7.0, 5.5 or 3.5 with 1 M HCl. Growth was monitored by recording absorbance at 600 nm after 24 h of incubation. The experiments were evaluated under three different pH conditions in triplicates and repeated twice. To determine bacterial cell viability the same setup was tested by microtitre plate assay using 2,3,5-triphenyltetrazolium chloride (TTC) as indicator. Briefly, 200 μL of bacterial suspension (107 CFU mL−1), treated as described earlier (with or without inhibitor at specific pH) was added in each well of a 96-well plate and incubated for 24 h under microaerophilic conditions. After the specified time, 20 μL of TTC indicator solution (5 mg mL−1 dH2O) was added to each well and incubated for another 15–20 min. Un-inoculated media was used as a negative control and untreated H. pylori used as a positive control. Appearance of pink to light red coloration indicated the presence of viable bacteria whereas absence of color change was indicative of dead bacterial cells.
3. Results and discussion
Urease enzyme plays an essential role in colonization of notorious carcinogenic H. pylori pathogen in human stomach. H. pylori has widespread prevalence and is associated with gastritis, peptic ulcers and gastric cancer.21 The pathogen constitutively expresses an acid neutral urease and its activity is essential in establishing infection.22 It has been observed that loss of urease activity leads to loss of colonization by the bacterium.23–25 Control of the gastritis and other severe complications associated with H. pylori infection focus on eradication of the bacterium which relies on administration of proton pump inhibitors and a combination of antibiotics clarithromycin and amoxicillin or metronidazole. However recent evidences suggest a rise in metronidazole and clarithromycin resistant strains.26,27 Perhaps due to this reason standard triple therapy failed to eradicate infection in a fraction of infected individuals.28 Alternative therapeutic strategies are required to treat such individuals. Due to essential role of H. pylori urease in infection establishment, this bacterial enzyme presents a promising therapeutic target.
3.1. Molecular docking studies of literature retrieved compounds
Different classes of urease enzyme inhibitors have been reported in the literature. Some have shown competitive and other uncompetitive inhibition. Therefore, the best active competitive inhibitors belonging to different structural classes were docked in jack bean urease enzyme.29–32 Docking simulations studies demonstrated that among these classes, o-chloro-hippurohydroxamic acid, a derivative of hydroxamic acid yielded best glide score (−9.229 kcal mol−1) with reasonably good binding mode shown in Fig. 2A. The hydroxamic acid, head group of the compound interacts with the bimetallic center containing nickel atoms whereas the tail part is exposed to the solvent.
Fig. 2. (A) Best binding mode of o-chloro-hippurohydroxamic acid (green sticks) in urease active site (cyan sticks). Nickel metal ions are shown in brown color. (B) ROCS shape and colour based query of hydroxamic acid, green colour sphere is hydrophobic, red and blue colours are hydrogen bond acceptor and donor respectively.

3.2. Shape and colour based query generation and virtual screening
Based on highest biological activity, best docking score and reasonably good binding mode among the selected competitive urease inhibitors, the o-chloro-hippurohydroxamic acid best docked pose was used to generate ROCS shape and color based query using vROCS program. The green color shown in Fig. 2B represents hydrophobic regions such as aromatic ring, the blue and red regions show hydrogen bond donors and acceptors respectively. In ROCS query generation only heavy atoms and polar hydrogens were included while nonpolar hydrogen atoms were ignored. Then generated query was subjected to screen Enamine database. This color and shape matching of database compounds give the idea that screened compounds will likely bind at the same binding site where active molecules bind. Another advantage of using shape-based query is to retrieve compounds which possess similar shape with different central scaffold. While running ROCS based screening, different scoring schemes like Shape Tanimoto, Color Tanimoto, ScaledColor, RefTverskyCombo, RefColorTversky and FitColorTversky can be used to rank the molecules. We ranked and selected the compounds based on Tanimoto combo scoring scheme. Tanimoto combo score quantifies both shape and color features overlap between query and target molecule. Maximum shape volume and color overlap of query and template molecule yields Tanimoto combo score of 2.0. Using this scoring scheme 1700 compounds were selected from the database in score range between 1.216 to 1.679. Then to further reduce the size of ROCS based screened compounds, they were docked in urease bimetallic site containing Ni ions. The glide score range of docked compounds were observed from −0.829 to −10.954 kcal mol−1. After docking, top 100 compounds were selected for their structural clustering. The clustering analysis aims at sorting different objects into groups in a way that the degree of association between two objects is maximal if they belong to the same group and minimal otherwise.33 Finally, ten best compounds were selected based on different structures, docking scores and visual inspection. The workflow followed during virtual screening is shown in Fig. 3.
Fig. 3. Hierarchal virtual screening workflow employed in the study.

3.3. In vitro urease inhibition assays of hit compounds
Out of ten, eight compounds were provided by the Enamine vendor. Table 1 shows the results of in vitro anti-urease activities of compounds in which some compounds inhibit enzyme at low micromolar concentrations while others at a higher concentration. Comp. 1 showed the lowest IC50 value (0.32 ± 0.06 μM) and highest percentage inhibition (96.2%). Similarly compound-2 and 3 exhibited IC50 values 0.68 ± 0.03 and 0.42 ± 0.04 respectively with 84.8 and 90.7 percent inhibition, while 4, 5, 6, 7 and 8 exhibited biological activity in the range of 7.83 to 12.53 ± 0.03 μM. The reference thiourea exhibited 22.61 ± 0.23 μM inhibitory activity with 92.3% inhibition. Due to different scaffolds of reported compounds their structure activity relationship cannot be explained at this stage. Inhibition mechanism of three most potent compounds was also investigated using different concentration of compounds (0–20 μM) and substrate (0.5–4.0 mM) by performing various kinetic studies. Lineweaver–Burk plots were used to assess the mode of inhibition by determining the effect of inhibitors (compounds) on Vmax and Km. Vmax of jack bean urease enzyme was not effected in the presence of inhibitors (compound 1 and 2) while Km values of enzyme increase (Fig. 4 and 6) which indicate the competitive inhibition. The decrease in Vmax value and increase in Km value of jack bean urease enzyme (shown in Fig. 5) in the presence of compound-2 indicates a mixed type inhibition of this compound which might interact at the allosteric site or active site of the enzyme.
Virtual screen hits structures, inhibitory activities and kinetics parameters.
| Compound | Enamine ID | Structure | IC50 (μM); mean ± SEM (% inhibition) | a V max (app) (μM min−1) | b K m (app) (mM) | c K i (μM) | Mode of inhibition |
|---|---|---|---|---|---|---|---|
| 1 | Z28824346 |
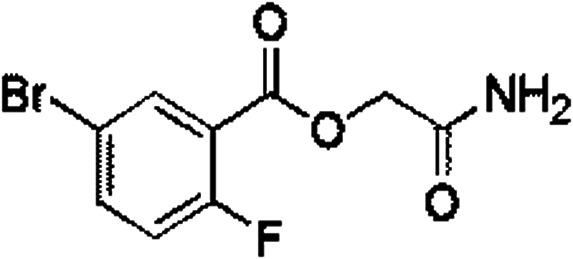
|
0.32 ± 0.06 (96.2) | 3.33 | 3.07 | 9.24 | Competitive |
| 2 | Z422952944 |
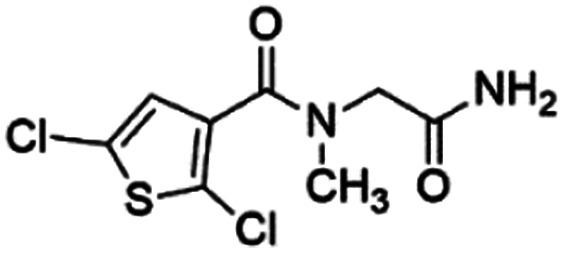
|
0.68 ± 0.03 (84.8) | 5.36 | 4.27 | 7.14 | Mixed |
| 3 | Z826553418 |
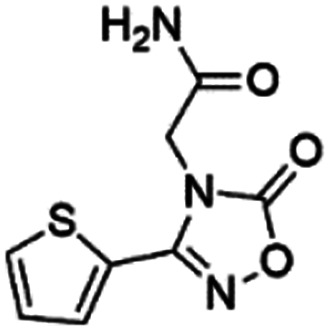
|
0.42 ± 0.04 (90.7) | 9.75 | 6.30 | 9.69 | Competitive |
| 4 | Z31483741 |
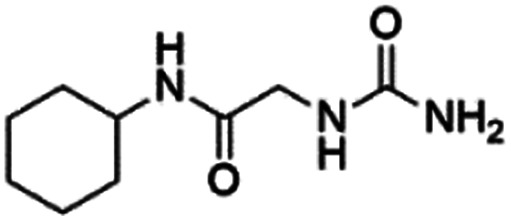
|
12.89 ± 0.11 (86.6) | — | — | — | — |
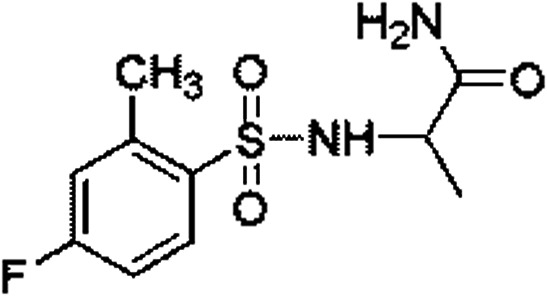
| 5 | Z279766864 |
|
7.83 ± 0.13 (92.4) | — | — | — | — |
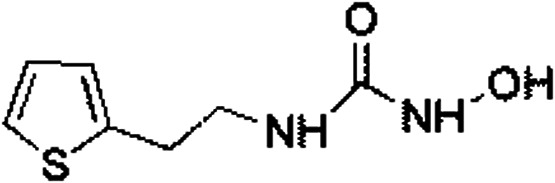
| 6 | Z335452622 |
|
12.24 ± 0.09 (81.1) | — | — | — | — |
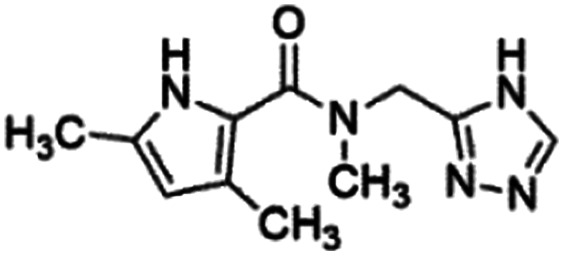
| 7 | Z1420422040 |
|
14.32 ± 0.06 (80.2) | — | — | — | — |
| 8 | Z1748646799 |
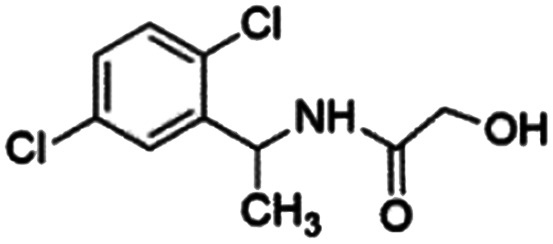
|
12.53 ± 0.03 (87.3) | — | — | — | — |
| dThiourea | 22.61 ± 0.23 (92.3) | 18.61 | 2.18 | 18.18 | Competitive |
V max (app) = maximum velocity of enzymatic activity at 20 μM inhibitor concentration.
K m (app) = Michaelis–Menten constant at 20 μM inhibitor concentration.
K i (μM) = calculated from secondary replot (Lineweaver–Burk).
Standard inhibitor of urease.
Fig. 4. Lineweaver–Burk plots (a and b) for enzymatic kinetics of compound 1.

Fig. 6. Lineweaver–Burk plots (a and b) for enzymatic kinetics of compound 3.

Fig. 5. Lineweaver–Burk plots (a and b) for enzymatic kinetics of compound 2.

Similarly predicted physiochemical/pharmacokinetic properties presented in Table 2 shows that all compounds are following the Lipinski's drug like properties.34 No compound was found to violate acceptable ranges of other properties like water solubility, herg channel blockage, cell permeability, blood/brain barrier co-efficient and binding to human serum albumin.
Calculated physiochemical/pharmacokinetic properties of screened compoundsa.
| ID | MW | HBD | HBA | QP log Po/w | QP log HERG | QPPCaco | QP log BB | QP log Khsa |
|---|---|---|---|---|---|---|---|---|
| Z28824346 | 276.062 | 2 | 4.5 | 0.552 | −2.908 | 119.143 | −0.802 | −0.734 |
| Z422952944 | 267.129 | 2 | 5.5 | 0.741 | −2.344 | 201.733 | −0.391 | −0.782 |
| Z826553418 | 225.222 | 2 | 6.5 | −0.944 | −2.395 | 57.863 | −1.196 | −0.992 |
| Z31483741 | 199.252 | 3 | 3.75 | −0.584 | −1.316 | 37.134 | −1.404 | −0.973 |
| Z279766864 | 260.283 | 2 | 6.25 | 0.278 | −2.734 | 189.587 | −0.86 | −0.803 |
| Z335452622 | 186.228 | 3 | 3.7 | 0.044 | −2.773 | 203.189 | −0.883 | −0.904 |
| Z1420422040 | 233.272 | 2 | 5.5 | 1.34 | −4.332 | 737.139 | −0.693 | −0.297 |
| Z1748646799 | 248.108 | 2 | 4.2 | 1.456 | −2.723 | 679.558 | −0.235 | −0.611 |
MW = molecular weight, HBD = hydrogen bond donor, HBA = hydrogen bond acceptor, QP log Po/w = octanol/water partition coefficient (recommended rage −2.0 to 6.5), QP log HERG = blockage of HERG K+ channels (recommended range < −5), QPPCaco2 = Caco2 cell permeability (recommended range <25 poor, >500 great), QP log BB = brain/blood partition coefficient (recommended range −3.0 to 1.2), and QP log Khsa = binding to human serum albumin (recommended range −1.5 to 1.5).35
3.4. In vitro studies of screened compounds
Antibiotic resistant bacteria have emerged globally with little to no alternative treatment available. In order to control associated diseases, it is important to look for alternative therapeutic targets. Urease being an essential colonization factor for H. pylori holds promising potential as drug target especially for the current emerging multiple drug resistant strains. We therefore also analyzed the potential of our test compounds to inhibit the potent urease activity in H. pylori. Various inhibitor compounds were tested for direct growth inhibition on selected bacterial strains including H. pylori (ESI Fig. 1†). There was no direct effect on bacterial growth inhibition of either tested inhibitors against H. pylori, enteropathogenic E. coli, C. jejuni, P. aeruginosa and methicillin resistant S. aureus (MRSA). Next, screened compounds were tested for their ability to block urease activity in H. pylori liquid culture supplemented with urea broth. All tested compounds possessed urease inhibitory capacity. While none of the compounds showed any direct growth inhibition against H. pylori and other tested bacterial strains, all test compounds exhibited strong urease inhibition in intact bacteria (Fig. 7A). Since urease activity is dispensable in a laboratory setup and urease negative mutants can grow normally on neutral culture media, urease inhibitory activity may not contribute to bacterial cell survival under normal culture conditions.
Fig. 7. Antimicrobial efficacy of hit compounds (A) anti-urease activity of test compounds was determined after 48 hours of pre-treatment with inhibitor compounds at 100 μg ml−1 concentration in presence of urea broth was tested. Experiment was performed in triplicates and results are expressed as mean of two independent experiments. (B) Bacterial growth in presence of acid stress was monitored by measuring suspension absorbance at 600 nm after 24 hours of incubation in presence of test compounds at (100 μg ml−1). Bacterial growth was monitored at 5 and 3.5 pH. Statistical analysis was performed using unpaired t-test. Results are expressed as mean of two independent experiments. * = p value of < 0.05, ** = p value < 0.005, *** = p value < 0.0005. (C) Bacterial viability was determined using MTT assay after 24 hours after addition of test compounds (100 μg ml−1). Viable cells were qualitatively assessed by development of red coloration after addition of 2,3,5-triphenyltetrazolium chloride (TTC). Deep red color was assigned a score of (3) followed by red indicating less viable cells (2), light pink coloration indicating few alive cells (1) and no viability with color development similar to blank (0). Viability was monitored at 5 and 3.5 pH.

It is known that urease of H. pylori confers acid tolerance enabling the bacterium to sustain activity in a highly acidic gastric milieu, prior to contact with the gastric mucosa.36 Surface urease (present in the cytoplasm or surface of the bacterium) increases the bacterium's microenvironmental pH to nontoxic levels promoting bacterial survival in the stomach.37 On the other hand, free urease is inactive.22,38 Urease activity of intact bacteria increases markedly as the pH of media falls below 4.0, providing H. pylori with a means to survive extended exposure to acid in the presence of urea. Therefore internal urease of H. pylori, activated by external acidic pH determines acid resistance.36 In order to test whether these compounds could interfere with the ability of the bacterium to sustain acid stress we monitored H. pylori growth in the presence of test compounds at neutral and acidic pH. The bacteria were exposed to low pH and inhibitor compounds were added to the broth. As shown in Fig. 7B, all tested compounds exhibited growth reduction at pH 5 and 3.5 compared to the untreated control. Meanwhile, inhibitors 1 and 2 significantly retarded bacterial growth at pH 3.5.
Since some rise in turbidity was observed for these compounds, in order to ascertain loss of bacterial viability, a replica plate for each experimental setup was tested using MTT assay in order to determine whether exposure to low pH after addition of compounds is merely delaying bacterial growth or is contributing to cell death. As compared to the untreated control, all tested compounds showed a decrease in cell viability at pH 5 (Fig. 7C). This decrease in cell viability was more pronounced at pH 3.5, with no viability observed for bacteria treated with compounds 1, 2 and 3. Based on our data we propose compound 2 as a suitable candidate for further optimization for H. pylori control. However additional studies for its cytotoxicity must also be undertaken in order to validate its use in humans.
3.5. Molecular dynamics simulations of best identified hits complexes
In order to confirm the binding modes and to evaluate the stability of three most potent compounds with urease enzyme, molecular dynamics simulations were performed using AMBER software. Since the crystal structure of different urease revealed that active site lies on alpha subunit and are independent from other subunits. Root mean square deviation (RMSD) for single monomer backbone and biological assembly in trimeric form was calculated over 50 ns long simulation. The RMSD value for monomer was approximately 3 Å from the complex structure as shown in Fig. 9A, however trimeric complex did not undergo any obvious fluctuation after reaching the equilibrium state. Recently similar observation were observed, when MD studies were performed to understand the urease enzyme structural features and its urea hydrolysis mechanism.39,40 Best docked pose of compound 1, 2 and 3 shown in Fig. 8 were used to calculate their RMSD values in urease trimeric structure and their trajectories are presented in Fig. 9B. MD trajectories showed that ligand receptor complexes of compound 1 and 3 remained stable throughout the 50 ns time. Similarly, the RMSD values of compounds from their docking pose shows very little deviation from their initial complex after getting equilibrium, indicating that the selected poses are reliable, and their complexes are stable. However, compound-2 showed large deviation throughout its simulation time which might be due to its mix type inhibition mechanism. Binding free energies of these compounds with urease was calculated using MM-PBSA method. The total binding free energy calculated for compound 1, 2 and 3 is −28.96 kcal mol−1, −22.96 kcal mol−1 and −25.9885 kcal mol−1 respectively. For these compounds, van der Waals energy (VDWAALS) and electrostatic energy (EEL) contributed more than non-polar solvation energy (ESURF) and other energy functions.
Fig. 9. Calculated RMSD distance. (A) RMSD of urease enzyme trimeric complex and its monomer structure. (B) RMSD distances of docked poses in all simulation trajectories.

Fig. 8. Best docking poses of highly active compounds in urease enzyme active site (green sticks). (A) Compound-1 (yellow sticks) is shown in urease enzyme active site, (B) compound-2 (white sticks) is shown in urease enzyme active site, (C) compound-3 (cyan sticks) is shown in urease enzyme active site. Nickel ions are shown with orange spheres and hydrogen bonding with yellow dotted lines.

4. Conclusion
In this study, we have identified new chemical scaffolds that inhibit urease enzyme activity. Initially shape and color based virtual screening of chemical database was performed and then highly ranked hits were subjected for molecular docking simulations to screen them further on their binding modes basis. The selected compounds inhibited the urease activity in cell based and in vitro experiments. Best active compounds kinetic studies revealed that compound-1 and 3 were competitive inhibitor while 2 was mixed type inhibitor. Further large RMSD fluctuation of compound 2 binding mode also highlighted that it might be binding with urease protein in different manners as revealed by kinetic studies. The study thus shows that by employing different computational approaches with experimental validation can lead to identify new chemical compounds against urease protein.
Compliance with ethical standards
Informed consent
Not applicable.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflicts of interest
We wish to confirm that there are no known conflicts of interest associated with this publication.
Supplementary Material
Acknowledgments
The authors are thankful to HEC-Pakistan for providing financial support to purchase software and hardware for computational studies vide Projects No. 6804/Federal/NRPU/R&D/HEC/2016 & 8094/Balochistan/NRPU/R&D/HEC/2017.
Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02363a
References
- Hameed A. Khan K. M. Zehra S. T. Ahmed R. Shafiq Z. Bakht S. M. Yaqub M. Hussain M. de León A. d. l. V. Furtmann N. Bioorg. Chem. 2015;61:51–57. doi: 10.1016/j.bioorg.2015.06.004. [DOI] [PubMed] [Google Scholar]
- Shi W.-K. Deng R.-C. Wang P.-F. Yue Q.-Q. Liu Q. Ding K.-L. Yang M.-H. Zhang H.-Y. Gong S.-H. Deng M. Bioorg. Med. Chem. 2016;24:4519–4527. doi: 10.1016/j.bmc.2016.07.052. [DOI] [PubMed] [Google Scholar]
- Mobley H. Island M. D. Hausinger R. P. Microbiol. Mol. Biol. Rev. 1995;59:451–480. doi: 10.1128/MMBR.59.3.451-480.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham D. Y. Gastroenterology. 2015;148:719–731. doi: 10.1053/j.gastro.2015.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosikowska P. Berlicki Ł. Expert Opin. Ther. Pat. 2011;21:945–957. doi: 10.1517/13543776.2011.574615. [DOI] [PubMed] [Google Scholar]
- Rauf A. Shahzad S. Bajda M. Yar M. Ahmed F. Hussain N. Akhtar M. N. Khan A. Jończyk J. Bioorg. Med. Chem. 2015;23:6049–6058. doi: 10.1016/j.bmc.2015.05.038. [DOI] [PubMed] [Google Scholar]
- Gull Y. Rasool N. Noreen M. Altaf A. Musharraf S. Zubair M. Nasim F.-U.-H. Yaqoob A. DeFeo V. Zia-Ul-Haq M. Molecules. 2016;21:266. doi: 10.3390/molecules21030266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modolo L. V. de Souza A. X. Horta L. P. Araujo D. P. de Fatima A. J. Adv. Res. 2015;6:35–44. doi: 10.1016/j.jare.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muddassar M. Jang J. W. Gon H. S. Cho Y. S. Kim E. E. Keum K. C. Oh T. Cho S.-N. Pae A. N. Bioorg. Med. Chem. 2010;18:6914–6921. doi: 10.1016/j.bmc.2010.07.010. [DOI] [PubMed] [Google Scholar]
- Bhattarai D. Muddassar M. Jang J. W. Hong S. K. Kim E. E. Oh T. Cho S. N. Pae A. N. Keum G. Curr. Comput.-Aided Drug Des. 2014;10:383–392. doi: 10.2174/1573409911666150414150300. [DOI] [PubMed] [Google Scholar]
- Matsuoka M. Kumar A. Muddassar M. Matsuyama A. Yoshida M. Zhang K. Y. J. J. Chem. Inf. Model. 2017;57:203–213. doi: 10.1021/acs.jcim.6b00649. [DOI] [PubMed] [Google Scholar]
- Rush T. S. Grant J. A. Mosyak L. Nicholls A. J. Med. Chem. 2005;48:1489–1495. doi: 10.1021/jm040163o. [DOI] [PubMed] [Google Scholar]
- da Costa G. V. Ferreira E. F. B. Ramos R. D. da Silva L. B. de Sa E. M. F. da Silva A. K. P. Lobato C. M. Souto R. N. P. da Silva C. H. T. D. Federico L. B. Rosa J. M. C. dos Santos C. B. R. Pharmaceuticals. 2019;12:61. doi: 10.3390/ph12020061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S. U. Ahemad N. Chuah L. H. Naidu R. Htar T. T. RSC Adv. 2019;9:2525–2538. doi: 10.1039/C8RA09318K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins P. C. D. Skillman A. G. Warren G. L. Ellingson B. A. Stahl M. T. J. Chem. Inf. Model. 2010;50:572–584. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren T. A. Murphy R. B. Friesner R. A. Beard H. S. Frye L. L. Pollard W. T. Banks J. L. J. Med. Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Ahmed M. Qadir M. A. Hameed A. Arshad M. N. Asiri A. M. Muddassar M. Biochem. Biophys. Res. Commun. 2017;490:434–440. doi: 10.1016/j.bbrc.2017.06.059. [DOI] [PubMed] [Google Scholar]
- Noreen Z. Khalid N. Abbasi R. Javed S. Ahmad I. Bokhari H. Mater. Sci. Eng. C. 2019;98:125–133. doi: 10.1016/j.msec.2018.12.087. [DOI] [PubMed] [Google Scholar]
- McLaren A. Reshetko L. Huber W. Soil Sci. 1957;83:497–502. doi: 10.1097/00010694-195706000-00011. [DOI] [Google Scholar]
- Toledo H. Valenzuela M. Rivas A. Jerez C. A. FEMS Microbiol. Lett. 2002;213:67–72. doi: 10.1111/j.1574-6968.2002.tb11287.x. [DOI] [PubMed] [Google Scholar]
- Bray F. Ferlay J. Soerjomataram I. Siegel R. L. Torre L. A. Jemal A. Ca-Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- Mobley H. L. Island M. D. Hausinger R. P. Microbiol. Rev. 1995;59:451–480. doi: 10.1128/MMBR.59.3.451-480.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrutis K. A. Fox J. G. Schauer D. B. Marini R. P. Murphy J. C. Yan L. Solnick J. V. Infect. Immun. 1995;63:3722–3725. doi: 10.1128/IAI.63.9.3722-3725.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton K. A. Krakowka S. Infect. Immun. 1994;62:3604–3607. doi: 10.1128/IAI.62.9.3604-3607.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M. Karita M. Morshed M. G. Okita K. Nakazawa T. Infect. Immun. 1994;62:3586–3589. doi: 10.1128/IAI.62.8.3586-3589.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. H. Wong B. C. Mukhopadhyay A. K. Berg D. E. Cho C. H. Lai K. C. Hu W. H. Fung F. M. Hui W. M. Lam S. K. Aliment. Pharmacol. Ther. 2000;14:901–910. doi: 10.1046/j.1365-2036.2000.00795.x. [DOI] [PubMed] [Google Scholar]
- Savoldi A. Carrara E. Graham D. Y. Conti M. Tacconelli E. Gastroenterology. 2018;155:1372–1382.e17. doi: 10.1053/j.gastro.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakoob J. Abid S. Abbas Z. Jafri S. N. Br. J. Biomed. Sci. 2010;67:197–201. doi: 10.1080/09674845.2010.11730319. [DOI] [PubMed] [Google Scholar]
- Amtul Z. Siddiqui R. Choudhary M. Curr. Med. Chem. 2002;9:1323–1348. doi: 10.2174/0929867023369853. [DOI] [PubMed] [Google Scholar]
- Amtul Z. Rasheed M. Choudhary M. I. Rosanna S. Khan K. M. Biochem. Biophys. Res. Commun. 2004;319:1053–1063. doi: 10.1016/j.bbrc.2004.05.036. [DOI] [PubMed] [Google Scholar]
- Jiang X.-Y. Sheng L.-Q. Song C.-F. Du N.-N. Xu H.-J. Liu Z.-D. Chen S.-S. New J. Chem. 2016;40:3520–3527. doi: 10.1039/C5NJ01601K. [DOI] [Google Scholar]
- Juszkiewicz A. Kot M. Zaborska W. Thermochim. Acta. 1998;320:45–52. doi: 10.1016/S0040-6031(98)00397-9. [DOI] [Google Scholar]
- Sastry M. Lowrie J. F. Dixon S. L. Sherman W. J. Chem. Inf. Model. 2010;50:771–784. doi: 10.1021/ci100062n. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A. Drug Metab. Rev. 2004;36:21. [Google Scholar]
- Tahir A. Alharthy R. D. Naseem S. Mahmood N. Ahmed M. Shahzad K. Akhtar M. N. Hameed A. Sadiq I. Nawaz H. Muddassar M. Molecules. 2018;23:1527. doi: 10.3390/molecules23071527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rektorschek M. Buhmann A. Weeks D. Schwan D. Bensch K. W. Eskandari S. Scott D. Sachs G. Melchers K. Mol. Microbiol. 2000;36:141–152. doi: 10.1046/j.1365-2958.2000.01835.x. [DOI] [PubMed] [Google Scholar]
- Sidebotham R. L. Worku M. L. Karim Q. N. Dhir N. K. Baron J. H. Eur. J. Gastroenterol. Hepatol. 2003;15:395–401. doi: 10.1097/00042737-200304000-00010. [DOI] [PubMed] [Google Scholar]
- Scott D. R. Weeks D. Hong C. Postius S. Melchers K. Sachs G. Gastroenterology. 1998;114:58–70. doi: 10.1016/S0016-5085(98)70633-X. [DOI] [PubMed] [Google Scholar]
- Minkara M. S. Ucisik M. N. Weaver M. N. Merz Jr K. M. J. Chem. Theory Comput. 2014;10:1852–1862. doi: 10.1021/ct5000023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estiu G. Merz K. M. J. Am. Chem. Soc. 2004;126:6932–6944. doi: 10.1021/ja049327g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.