

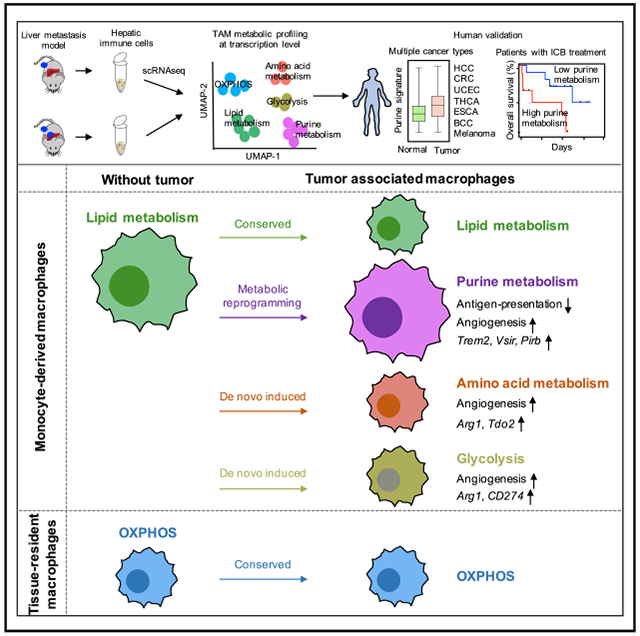
SUMMARY
Tumor-associated macrophages (TAMs) are a major cellular component in the tumor microenvironment (TME). However, the relationship between the phenotype and metabolic pattern of TAMs remains poorly understood. We performed single-cell transcriptome profiling on hepatic TAMs from mice bearing liver metastatic tumors. We find that TAMs manifest high heterogeneity at the levels of transcription, development, metabolism, and function. Integrative analyses and validation experiments indicate that increased purine metabolism is a feature of TAMs with pro-tumor and terminal differentiation phenotypes. Like mouse TAMs, human TAMs are highly heterogeneous. Human TAMs with increased purine metabolism exhibit a pro-tumor phenotype and correlate with poor therapeutic efficacy to immune checkpoint blockade. Altogether, our work demonstrates that TAMs are developmentally, metabolically, and functionally heterogeneous and purine metabolism may be a key metabolic feature of a pro-tumor macrophage population.
Graphical abstract
In brief
Li et al. examine the metabolic heterogeneity of tumor-associated macrophages (TAMs). They demonstrate metabolic patterns of TAMs correlate with their functional characteristics, and increased purine metabolism is a feature of TAMs with pro-tumor and terminal differentiation phenotype. They confirm these observations in patients with multiple types of cancer.
INTRODUCTION
Tumor-associated macrophages (TAMs) are important components of the tumor microenvironment (TME). TAMs are associated with multiple aspects of tumor pathobiology, such as immunosuppression, neovascularization, metastasis, and resistance to therapy (Ding et al., 2009; Forssell et al., 2007; Mantovani et al., 2008; Qian and Pollard, 2010; Ruffell et al., 2012; Sica and Mantovani, 2012; Zou, 2005). However, macrophages also mediate tumor phagocytosis and can uptake, process, and present antigens to T cells and promote anti-tumor immunity. In addition, macrophages may directly mediate tumor killing and are associated with improved patient outcome in some types of cancer (Forssell et al., 2007; Shimura et al., 2000; Welsh et al., 2005). We postulate that the controversies among the previous studies may be attributed to the heterogeneity of TAM ontogeny, phenotype, metabolism, and their functional characteristics in vivo. We reason that a deeper understanding of TAM heterogeneity is critical for eventually developing mechanism-informed, TAM-targeted, effective cancer immunotherapy (Mantovani et al., 2021; Wen et al., 2020; Zhang et al., 2019, 2020).
Transcriptome profiling at the single-cell level yields invaluable information on intracellular signaling responses, cell-type-specific pathways, potential functionalities, and activities of major metabolic pathways. For example, the heterogeneity of human and mouse TAMs in different pathological conditions have been recently analyzed at the single-cell level (Cheng et al., 2021; Zhang et al., 2019). It has been reported that metabolic changes can reshape the states of immune cells in the TME (Bian et al., 2020; Habtetsion et al., 2018; Li et al., 2018). Along these lines, recent reports suggest that metabolic alteration is accompanied by the phenotypic and functional change of TAMs in the TME (Vitale et al., 2019; Xia et al., 2020). However, the interdependent patterns of development, metabolism, and function of TAMs remain largely unraveled in both tumor-bearing mice and patients with cancer. Here, we applied single-cell RNA sequencing (scRNA-seq) to mononuclear cells isolated from livers with or without tumors in subcutaneous-tumor-bearing mice. We revealed the metabolic landscape of hepatic TAMs in the context of liver metastasis and demonstrated the intertwined relationship of development, metabolism, and function in TAMs. We extended our analysis on tumor-bearing mice to 8 cohorts of cancer patients and correlated TAM transcriptomics with patient survival and clinical efficacy to immune checkpoint blockade therapy (ICB). Our work demonstrates that TAMs are developmentally, metabolically, and functionally heterogeneous, and purine metabolism may be a key metabolic feature of pro-tumor TAMs. We suggest that targeting purine metabolic TAMs represents a novel approach for cancer immunotherapy.
RESULTS
Heterogeneity of TAMs within the liver tumor microenvironment
We have previously demonstrated that hepatic macrophages contribute to systemic immunosuppression and resistance to immunotherapy in preclinical murine models and cancer patients with liver metastasis (Yu et al., 2021). To characterize macrophages in the liver TME, we isolated hepatic mononuclear cells from mice bearing MC38 subcutaneous tumor with and without MC38 tumors in the liver (Yu et al., 2021) and performed scRNA-seq on these hepatic mononuclear cells. We computationally analyzed cells expressing typical macrophage markers, including Lyz2, Csf1r, Cd68, and Lgals3, but not dendritic cell (DC) marker Zbtb46 or neutrophil markers Ly6g and Csf3r. We performed graph-based clustering and Uniform Manifold Approximation and Projection (UMAP) analyses on the integrated projector of 8,056 isolated macrophages (Figures 1A and 1B). After clustering, we identified 14 transcriptionally distinct macrophage subsets (Figures 1A and 1B). Next, we split the integrated dataset into 2 projections, with 2,021 hepatic macrophages from mice without liver tumors (normal macrophages) and 6,035 hepatic macrophages from mice with liver tumors (TAMs) (Figure 1C). We found that the relative size of clusters 0, 1, 4, 5, 6, 9, 12, and 13 were increased, and the size of clusters 2, 3, 7, 10, and 11 were decreased in the tumor samples compared with normal samples (Figures 1C and 1D). These data reveal high levels of heterogeneity of intrahepatic TAMs and suggest an important impact of tumors on TAM composition (Figures 1C and 1D).
Figure 1. Heterogeneity of TAMs within the liver TME.
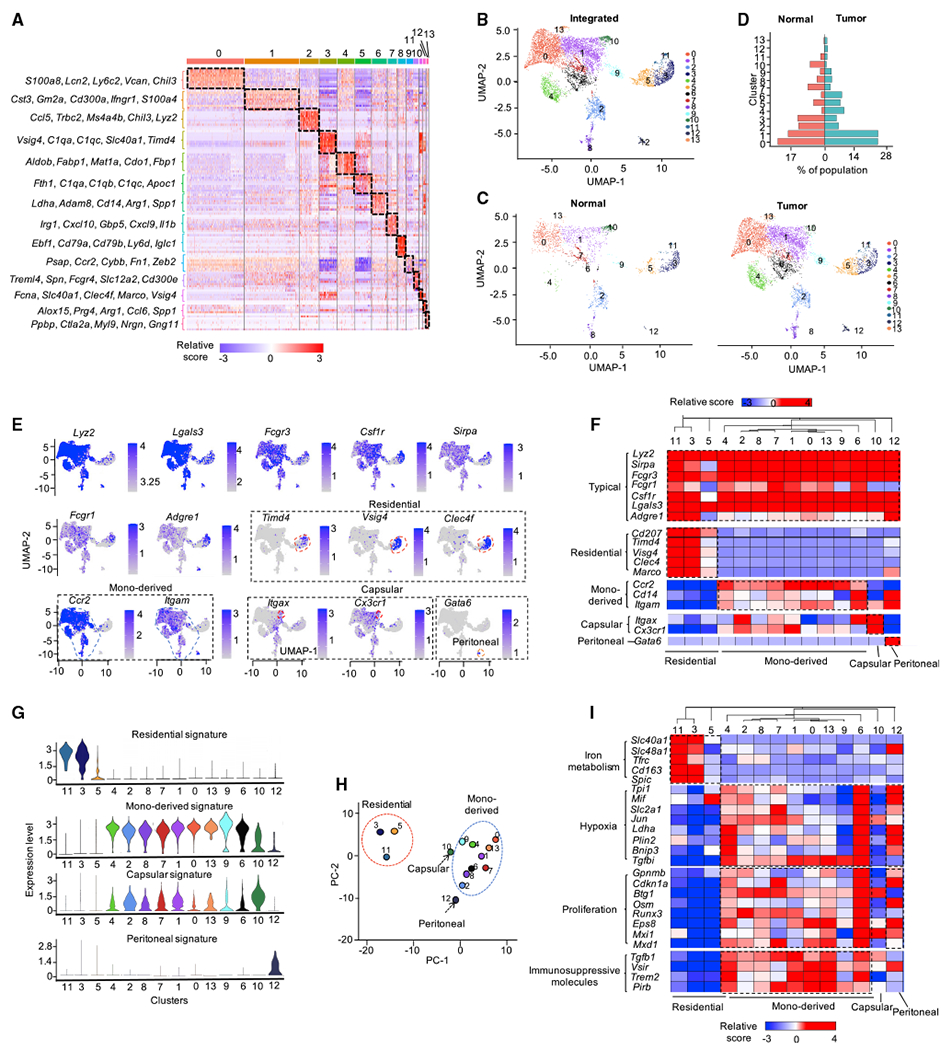
(A) Gene expression heatmap of 14 macrophage clusters.
(B and C) UMAP visualization of integrated (B) and split projections (C) from tumor and normal sample, color-coded based on the clusters; each dot represents a single cell.
(D) Proportions of each macrophage cluster in tumor and normal sample.
(E) UMAP showing the expression levels of 14 selected marker genes for macrophages; expression levels are color-coded as gray: not expressed and blue: expressed.
(F) Heatmap visualization of the expression of markers for typical macrophages, Kupffer cells, MDMs, LCMs, and PMs.
(G) Violin plots showing the enrichment of gene signatures of Kupffer cells, MDMs, LCMs and PMs in macrophages, determined by multiple features analysis of Seurat v. 3.
(H) Principal-component analysis (PCA) for 14 macrophage clusters for all marker genes in each cluster. Averaged position of all cells in each cluster were represented; dots are colored by Seurat cluster, as shown in (B).
(I) Heatmap showing the expression level of iron metabolism-, hypoxia-, proliferation-related genes and T-cell-co-inhibitory molecules in Kupffer cells, MDMs, LCMs, and PMs; color of each heatmap cell represents the relative expression level of each gene (Z score).
Hepatic macrophages exhibit different origins, locations, and times of establishing residency in the liver (Blériot and Ginhoux, 2019; MacParland et al., 2018). To elucidate the potential developmental origin of those hepatic macrophages in mice with and without liver tumors, we reanalyzed these 14 clusters by querying the expression levels of 7 typical macrophage markers and 11 cell-of-origin markers. We identified 4 types of macrophages, including liver resident macrophages (Kupffer cells) (Davies et al., 2013), monocyte-derived macrophages (MDMs) (Serbina and Pamer, 2006), liver capsular macrophages (LCMs) (Blériot and Ginhoux, 2019; Sierro et al., 2017), and peritoneal macrophages (PMs) (Blériot and Ginhoux, 2019; Wang and Kubes, 2016) (Figures 1E–1G). As expected, all 4 macrophage populations expressed typical macrophage markers, including Lyz2, Lgals3, Fcgr3, Csf1r, Fcgr1, and Adgre1 (encoding F4/80) (Figures 1E and 1F). Among 14 clusters, clusters 3, 5, and 11 showed expression of Timd4 (encoding TIM4), and Vsig4, and Clec4f and were determined as Kupffer cells (Figures 1F and 1G); while clusters 0, 1, 2, 4, 6, 7, 8, 9, and 13 expressed Ccr2, Cd14, and Itgam and were determined as MDMs (Figures 1F and 1G). Cluster 10 expressed Itgax and Cx3cr1 and was defined as LCMs (Figures 1F and 1G). Cluster 12 expressed Gata6 and was defined as PMs (Gautier et al., 2014; Rosas et al., 2014; Xia et al., 2020) (Figures 1F and 1G). Principal-component analysis (PCA) on variable genes in each cluster confirmed that clusters in each individual group harbor similar cell identity (Figure 1H).
To validate the potential developmental origin of macrophage subsets, we queried several conventional functional characteristics of hepatic macrophages. Liver resident macrophages showed activity of iron metabolism (Figure 1I), which is an accessory characteristic of Kupffer cells (Blériot and Ginhoux, 2019). In contrast, most of the MDM clusters showed high hypoxia and proliferation activities (Figure 1I). Moreover, we evaluated expression of several immunosuppressive genes in TAMs, including Tgfb1 (encoding transforming growth factor β [TGFβ]), Vsir (encoding VISTA [V-domain immunoglobulin suppressor of T cell activation]), Pirb (encoding ILT2), and Trem2 (encoding TREM2 [triggering receptor expressed on myeloid cells 2]). Apart from Kupffer cells, MDMs, LCMs, and PMs manifested immunosuppressive potential, as shown by high expression of Tgfb1, Vsir, Pirb, and Trem2 (Figure 1I). Altogether, we transcriptionally identified 14 hepatic macrophage clusters derived from 4 potentially different developmental origins. Our results reveal high levels of heterogeneity of macrophages in the liver TME.
TAMs manifest high metabolic diversification
Metabolism is known to affect the phenotype and function of immune cells, including T cells (Bian et al., 2020; Cascone et al., 2018; Li et al., 2018) and macrophages (Wang et al., 2021; Xia et al., 2020) in the TME. To gain comprehensive and unbiased insights into the metabolic activities of macrophage subsets in the liver TME, we performed gene set enrichment analysis (GSEA) on 14 macrophage clusters against 60 metabolic pathways in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Figure 2A). Interestingly, the 14 macrophage clusters showed distinct metabolic activities (Figures 2B and 2C). For example, clusters 0/1/13 showed higher purine metabolism (Figure 2B); clusters 2/7/9/10/11 had relatively higher activity of lipid metabolism; cluster 3 was featured with higher iron and lipid metabolism (Blériot and Ginhoux, 2019); cluster 4 was characterized by a mixed metabolic profile, as shown by high oxidative phosphorylation (OXPHOS) and high lipid and amino acid metabolism; cluster 5 showed high activity of glycolysis, lipid metabolism, iron metabolism, and OXPHOS (Figure 2B); clusters 6/12 were characterized by high glycolysis activity (Figure 2B); and cluster 8 showed minor metabolic activity. These data suggest that TAMs in the liver microenvironment manifest diverse metabolic activities.
Figure 2. TAMs manifest high metabolic diversification.

(A) Schematic representing the workflow of metabolic pathway activity analysis for each macrophage cluster with scRNA-seq.
(B) Landscape of the activity of different metabolic pathways in different macrophage populations (left). Enrichment analysis by GSEA; color of each heatmap cell represents the value calculated with formula: +/−Log2|NES/FDR|. Red: significantly up-regulated (nominal p < 0.05); blue: significantly down-regulated (nominal p< 0.05); gray: pathways not enriched (nominal p > 0.05). Heatmap on the right shows the ratio between tumor and normal sample and the polarization state as well as the origin, defined in Figure 1G, for each macrophage population; AA: amino acid metabolism; NES: normalized enrichment scores, FDR: false discovery rate.
(C) The activity of selected metabolic pathway in selected macrophage populations. Normalized enrichment scores and nominal p values are calculated by Kolmogorov-Smirnov test.
(D) Workflow of metabolic heterogeneity analysis of macrophages with scRNA-seq; GO: the gene ontology; Lipid: lipid metabolism; Purine: purine metabolism; Gly: glycolysis; AA: amino acid metabolism; OX: oxidative phosphorylation.
(E) UMAP plots showing the metabolic clusters of macrophages. The color of each dot indicates the dominant metabolic cluster of each cell, determined using 1,310 metabolic genes.
(F) GSEA score (+/−Log2|NES/FDR|) of 5 metabolic clusters against 5 metabolic gene signatures. Red: positively enriched; green: negatively enriched; white: not enriched.
(G–I) UMAP plots showing the metabolic clusters of macrophages. The color of each dot indicates the developmental origin (G) and the sample (H) as well as the polarization state of each macrophage (I).
(J) The expression level of genes from SLC family members, ABC transporters, and pump and ion channels. Fold change: each metabolic cluster compared with other clusters.
(K) Enrichment analysis of transcriptional factors (TF) associated with the 5 metabolic clusters using BART (binding analysis for regulation of transcription) (Wang et al., 2018), with top 10 highly expressed genes in each cluster; top 5 significantly enriched TF in each cluster represented; red grid: top 5 or not; white grid: not available.
(L) The metabolic landscape of macrophage populations from normal and tumor in the context of 14 original macrophage populations, as shown in Figure 1C, the dot color was coded based on the dominant metabolism, as shown in Figure 2E.
(M) Relative proportion of cells in each metabolic cluster, as shown in Figure 2L, versus samples from normal and tumor. Chi-squared test was used to calculate the significance between tumor and normal groups; *p < 0.05, **p < 0.01, and ***p < 0.001.
(N) Immunofluorescence staining of CLEC4F and F4/80 in liver tissue with tumor.
(O) Sankey diagram showing the distribution of macrophages in terms of origin and metabolic clusters.
Based on this finding, we decided to explore the fine-grained metabolic heterogeneity of hepatic macrophages at the single-cell level. To this end, we focused our analyses on 1,310 genes with established metabolic function. We performed clustering and gene ontology (GO) analysis as well as assigned dominant metabolic identity (Figure 2D). In line with previous metabolic activity analysis (Figure 2B), the 8,056 macrophages showed heterogeneous metabolic patterns (Figures S1A and S1B). Based on the similarity analysis with PCA (Figure S1C) and Pearson correlation matrix (Figure S1D), we classified those hepatic macrophages into 5 metabolic clusters, featured with lipid metabolism, purine metabolism, amino acid (AA) metabolism, and OXPHOS as well as glycolysis (Figures 2E and S1E). Further GO analysis on the top enriched genes (Figure S1F) and GSEA on metabolic gene signatures (Figure 2F) in each metabolic cluster showed that each defined metabolic type was dominantly enriched in corresponding metabolic clusters (Figures 2F and S1F).
Metabolic clustering could potentially discriminate macrophages with different origins (Figure 2G), including normal macrophages and TAMs (Figure 2H). Interestingly, when macrophages were classified into M1- and M2-like types, they exhibited mixed patterns in terms of metabolic profiles (Figures 2E, 2I, and S1G), developmental origins (Figures 2G, 2I, and S1H), sample origins (Figures 2H, 2I, and S1I), and functional features (Figure S1J). These data suggest that classification of TAMs into M1 and M2 types bear certain limitations, and classification of TAMs at the basis of their metabolic profiles could provide complementary and novel information on TAM heterogeneity.
Nutrients and metabolites can be transported by their solute carriers (SLCs) and are prerequisites for metabolic activities. In line with different metabolic activities in TAM subsets identified in our analysis, several SLC transporters were up-regulated in their corresponding metabolic clusters (Figure 2J). For example, SLC2A1, a glucose transporter, was highly expressed in the glycolysis cluster, while SLC25A15, an ornithine translocase, was highly expressed in the AA cluster (Figure 2J). In addition, distinct transcription factors were predicted to regulate the expression of top 10 highly expressed genes in each metabolic cluster (Figure 2K). To identify the metabolic differences between normal macrophages and TAMs, we split the integrated and metabolic labeled macrophage projector into two projectors (Figure 2L). The presence of tumor cells dramatically changed the metabolic profiles of hepatic macrophages (Figures 2L and 2M). Intriguingly, metabolic changes were largely derived from MDMs because metabolic profiles of residential Kupffer cells in the TME remained stable compared with Kupffer cells in normal liver (Figures 2L, S1K, and S1L). Immunofluorescence staining showed that Kupffer cells largely resided at the peritumor area compared with TAMs in the liver (Figure 2N), suggesting that the localization of Kupffer cells may shape their metabolic profile. MDMs in normal liver were homogenously enriched with lipid metabolism (Figures S1K and S1L), while MDMs from the TME exhibited heterogeneous profiles (Figures S1K–S1M), including purine metabolism, lipid metabolism, AA metabolism, glycolysis, and OXPHOS (Figures 2L and 2M). Notably, all tumor-induced metabolic clusters, including purine (44.4%), AA (11.9%), and glycolysis (11.4%), were predominantly derived from MDMs and contributed to 67.7% of the TAMs (Figures 2M and 2O). Collectively, these results indicate that TAMs, especially MDMs, were heavily metabolically reprogrammed in the liver TME.
Metabolic profiles correlate to distinct functional programs in TAMs
Metabolic profile is coupled with phenotype and functionality of TAMs (Vitale et al., 2019; Xia et al., 2020). To evaluate this notion in hepatic TAMs, we identified differentially expressed genes (DEGs) (Figure S2A) among 5 metabolic clusters (Figure 2E) in macrophages in a supervised manner. Pathway enrichment analysis in these DEGs demonstrated that distinct functional programs were enriched in different metabolic clusters (Figure S2B). In particular, the lipid metabolism cluster was enriched in “regulation of lymphocyte activation” and “cargo recognition for endocytosis.” The purine metabolism cluster was enriched in “angiogenesis” and “myeloid cell differentiation.” The glycolysis metabolism cluster was enriched in “HIF-1-alpha transcription factor pathway” and “phagocytosis.” The AA metabolism cluster was enriched in “cysteine and methionine metabolism” and “arginine metabolic process.” The OXPHOS metabolism cluster was enriched in “scavenging of heme from plasma” (Figure S2B).
To further investigate whether TAM metabolic profiles were related with their functionalities in the context of tumor, we manually queried several well-known macrophage functional programs, including phagocytosis, antigen presentation, angiogenesis, immunosuppression, and M1/M2-like polarization, in different metabolic clusters (Figures 3A–3I). TAMs with glycolysis showed the highest ability of phagocytosis (Figures 3A–3C). TAMs with purine metabolism showed the lowest capability of antigen presentation, compared with TAMs with other metabolic profiles (Figures 3D–3F). Moreover, TAMs with tumor-induced metabolic features, including purine metabolism, AA metabolism, and glycolysis (Figure 2L), expressed high levels of pro-angiogenic genes (Figures 3G–3I). Among the immunosuppressive molecules we examined, Tdo2 was enriched in TAMs with AA metabolism; Cd274, Nt5e, and Entpd1 were enriched in TAMs with glycolysis metabolism; Arg1 was enriched in TAMs with glycolysis as well as AA metabolism; and Trem2 and Vsir were enriched in TAMs with purine metabolism (Figure S2C). As expected, most TAMs showed M2-like phenotype rather than M1-like phenotype (Figures S2D and S2E). As a validation, we observed that macrophages in the same metabolic status tended to show similar functional gene patterns as assessed in PCA, regardless of the sample sources (Figures 3J and S2F). Taken together, these data suggest that different metabolic patterns correlate with distinct functional programs in TAMs.
Figure 3. Metabolic profiles correlate to distinct functional programs in TAMs.
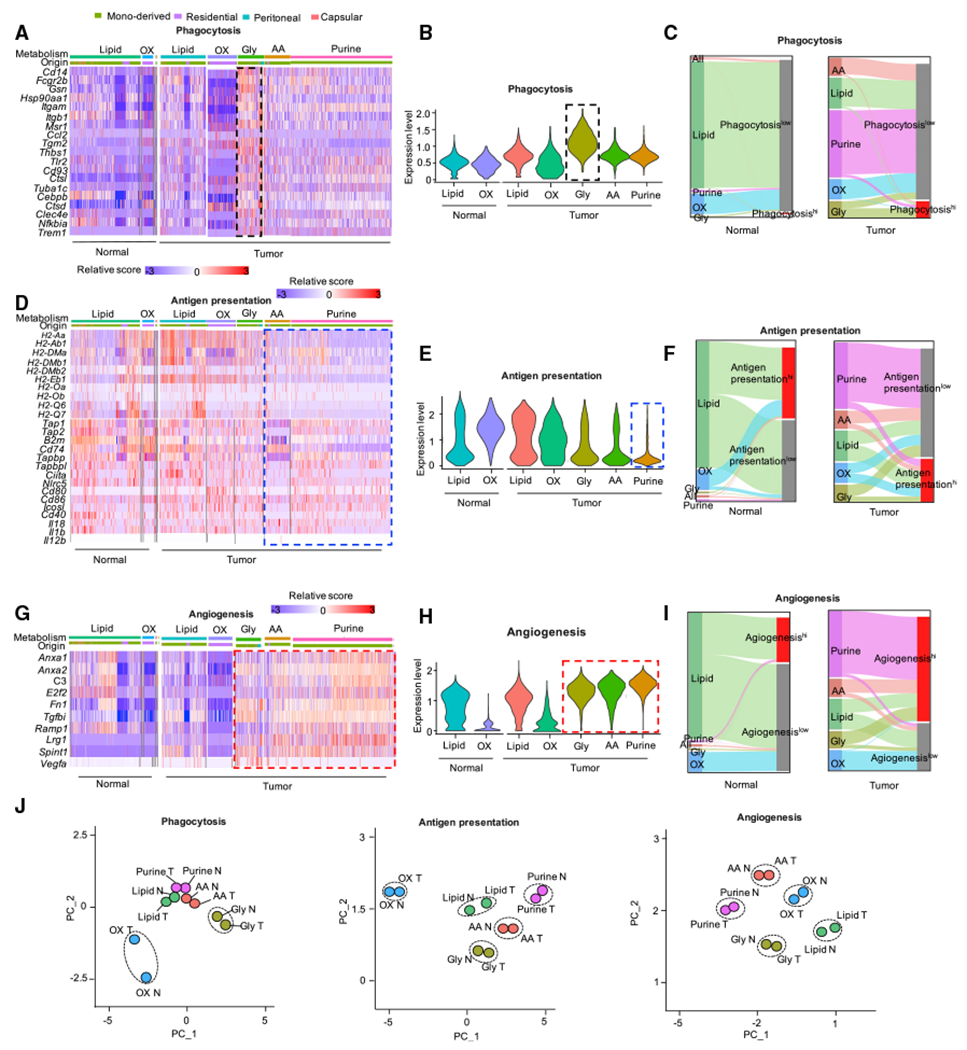
(A–I) Heatmap, violin plots, and Sankey diagrams showing the expression level of genes from phagocytosis (A–C) and antigen presentation (D–F) as well as angiogenesis (G–I) pathways in metabolic cluster of AA, Gly, lipid, OXPHOS, and purine in normal and tumor. Each violin represents the score of each signature (B, E, and H); Sankey diagrams show the proportion of macrophages expressed relative functional gene signature in context of five metabolic clusters; hi: expression score >1 (C, F, and I).
(J) PCA of 5 metabolic clusters with the expression level of phagocytosis-(left) and antigen presentation (middle) as well as angiogenesis (right) gene signature. Cells from normal and tumor sample were analyzed separately; each dot shows the averaged position of cells in each cluster; N: normal, T: tumor.
Purine metabolic TAMs display terminal and immunosuppressive phenotype
Based on the observed associations between metabolic patterns and functional programs in TAMs, we asked if metabolic remodeling of TAMs was coordinated with their cellular differentiation. We used RNA velocity analysis via scVelo (Traag et al., 2019) to infer a pseudo-time axis, representing progression of cellular differentiation in context of metabolic remodeling (Figure 4A). We found that key metabolic-associated genes showed distinct inferred latent time (Figure 4A) and displayed distinct expression level and RNA velocity in corresponding metabolic clusters (Figures 4B and S3A). The data demonstrate pronounced and dynamic changes to metabolic transcriptional programs across macrophage populations (Figures 4A and 4B). Next, we overlaid the RNA velocity stream, which characterizes the direction and rate of changes to cellular transcriptional states based on the relative abundance of spliced and un-spliced transcripts (La Manno et al., 2018), with the metabolic projection as defined above (Figures 2E, 4C and S3B) (Traag et al., 2019). RNA velocity analysis revealed different cell states of the 5 metabolic populations (Figures 4C and S3B); among them, purine metabolic macrophages were at the end stage (Figures 4C and S3B). Additional terminal state analysis based on the inferred transition probabilities confirmed this finding (Figure 4D). Next, by integrating the identified terminal differentiation states with the tissue source of the macrophages, we observed that purine metabolic macrophages served as the terminal stage population in the tumor, whereas some lipid metabolic macrophages served as the terminal stage population in normal liver (Figures 4D and 4E). The endpoint state of purine metabolic macrophages was also confirmed using another pseudo-time algorithm of partition-based graph abstraction (PAGA) (Wolf et al., 2019) (Figure 4F).
Figure 4. RNA velocity analysis identifies terminal-differentiated immunosuppressive macrophage subset.
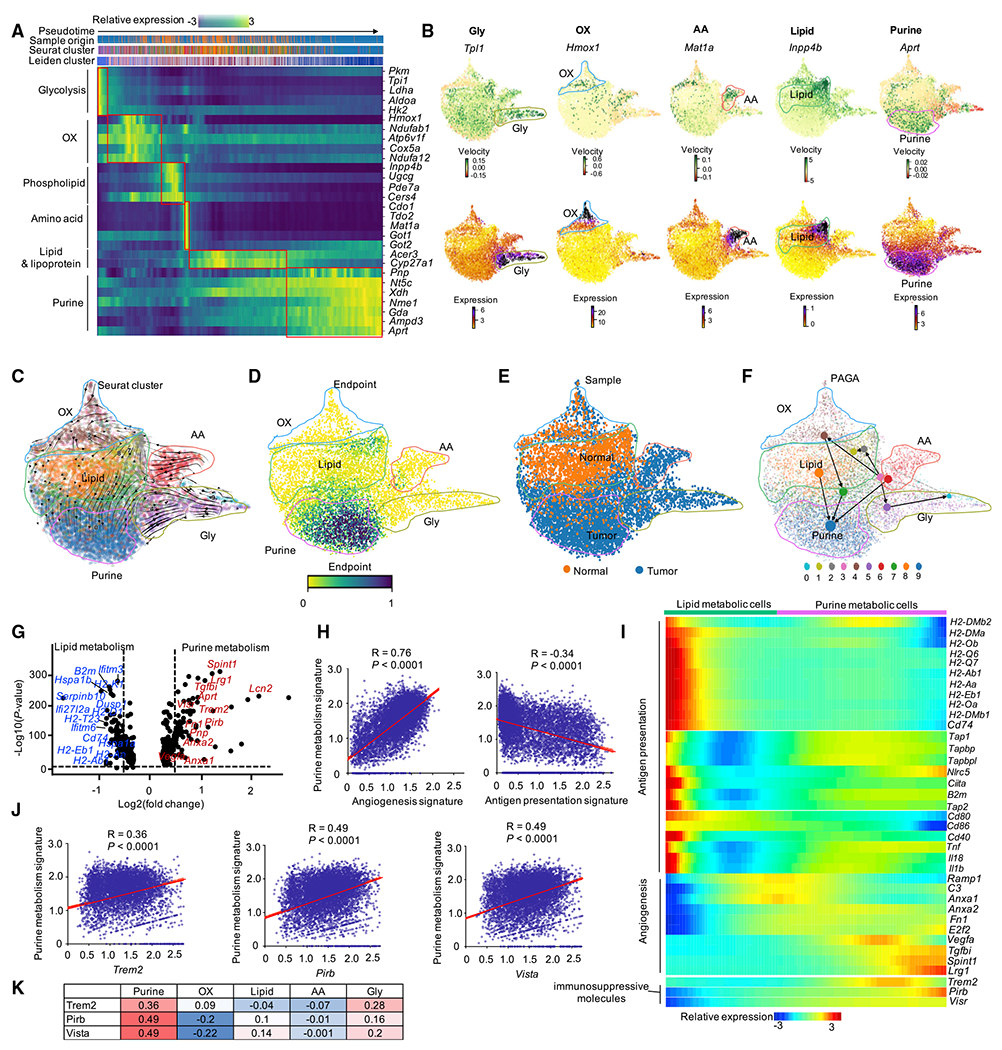
(A) Gene expression dynamics of selected metabolic maker genes in each cluster ordered along latent time inferred by RNA velocity analysis with scVelo. Cells were labeled by pseudo-time (first row), sample (second row), Seurat cluster (third row), and Leiden cluster (fourth row) of each cell, respectively.
(B) The RNA velocity and referred expression level of selected metabolic marker genes in each metabolic cluster on the Louvain projection modeled by scVelo. Positive velocity: up-regulated; negative velocity: down-regulated.
(C) RNA velocity of 5 metabolic clusters overlaid with RNA velocity stream. Cells colored by Seurat cluster, as shown in Figure 2E.
(D) Endpoints analysis of macrophages. The endpoints are obtained as stationary states of the velocity-inferred transition matrix, which is given by left eigenvectors corresponding to an eigenvalue of 1, i.e., μend = μendπ. Color bars show the endpoint score of macrophages; dark blue: end.
(E) UMAP embedding of macrophages modeled by scVelo. Cells were colored by the sample; orange: normal; dark blue: tumor.
(F) PAGA graph showing the inferred developmental trajectories for 5 metabolic clusters. PAGA: partition-based graph abstraction.
(G) Volcano plot showing the fold change versus p value of macrophages of cluster 0 (and cluster 1) from normal and tumor (as shown in Figure 2L), Representative genes were highlighted. Red: up-regulated in purine metabolism; blue: down-regulated in purine metabolism. Dashed lines show p < 0.05 and fold change >1.5.
(H) Correlation analysis between gene signatures of purine metabolism (y axis) and functional programs of angiogenesis and antigen presentation (x axis) on macrophages. Pearson’s correlation coefficient (R) and correlation test (p values) were used to evaluate the association between two signatures.
(I) Expression of genes from antigen presentation, angiogenesis signature, and immunosuppressive molecules along with the pseudo-time inferred by Monocle v. 2.
(J) Correlation analysis between purine metabolic gene signatures (y axis) and 3 immunosuppressive molecules (x axis) on macrophages.
(K) Summary of the correlation between 5 metabolic gene signatures with 3 immunosuppressive molecules; Pearson correlation coefficient was shown in each square.
Given that purine metabolic macrophages were located at the terminal stage of the trajectory and comprised 43.8% of all TAMs, we focused our analysis on purine metabolic macrophages. Normal tissue-derived macrophages in clusters 0/1 showed lipid metabolism (Figure 2L, left), while TAMs in clusters 0/1 displayed purine metabolism (Figure 2L, right). This suggests that macrophages in clusters 0/1 were metabolically reprogrammed from lipid metabolism to purine metabolism in the TME, in line with previous differentiation trajectory analysis (Figure 4C). Tumor reprogrammed macrophages with purine metabolism showed reduced expression of genes related to antigen presentation but conversely increased expression of genes related to immunosuppression and angiogenesis (Figures 4G and S3C). Purine metabolic gene-signature-enriched macrophages (Figure S3D, top) were characterized by increased angiogenesis signature (Figure S3D, middle) and reduced antigen presentation scores (Figure S3D, bottom). In support of this, the purine metabolic gene signature showed a strong positive correlation with angiogenesis signature and a negative correlation with antigen presentation gene signature (Figure 4H). Conversely, other metabolic signatures did not show strong correlation with either angiogenesis or antigen presentation gene signatures (Figure S3E and S3F). Further, pseudo-time analysis suggests that macrophages gradually lost the expression of genes related to antigen presentation while acquiring expression of genes related to angiogenesis when changing from lipid metabolism to purine metabolism (Figures 4I and S3G). In addition, purine metabolic macrophages expressed three potential immunosuppressive molecules, including Trem2, Vista, and Pirb (Figure S4A). The expression of those three genes was gradually gained along the pseudo-time (Figure 4I) and correlated with the expression of purine metabolic signature (Figures 4J and 4K) but not the other three metabolic signatures (Figures S4B and Figure 4K). These data suggest that purine metabolic TAMs may be terminally differentiated, mediate immunosuppression, and have angiogenetic potential.
To experimentally validate our observations, we sorted hepatic macrophages (Figure S5A) and examined protein expression of key purine pathway genes, including purine nucleoside phosphorylase (Pnp) and adenine phosphoribosyltransferase (Aprt). Consistent with their transcript levels (Figure 4G), protein levels of PNP and APRT were higher in TAMs compared with macrophages from liver without tumor (Figure 5A). In addition, we found that Trem2 was specifically expressed in macrophages (Figure S5B) and positively correlated with the purine metabolic signature score (Figure 4J). We used TREM2 as a surface marker and sorted TAMs with enriched purine metabolism (Figure S5C). We found that TREM2+ macrophages exhibited higher activity of PNP, and expressed higher levels of AMP and adenosine, two key metabolites at the downstream of APRT (Figures 5B–5E). Moreover, there were more TREM2+ macrophages in liver with tumor compared with liver without tumor (Figures 5F–5H and S5D). TREM2+ macrophages showed lower expression of MHC-II (I-A/I-E) and MHC-I (H-2Db) Figures 5I and 5J), indicating a reduced capacity for antigen-presentation. These data are consistent with our transcript analysis (Figures 4G and 4H). To assess the immunosuppressive potential of this macrophage population, we sorted and co-cultured them with OT-I cells in the presence of cognate peptide SIINFEKL. The proliferation of antigen-specific T cells was significantly decreased in the presence of TREM2+ TAMs (Figures 5K and 5L). The data reveal the immunosuppressive capability of purine-metabolism-enriched, TREM2+ TAMs. Taken together, these results indicate that purine metabolic TAMs are terminally differentiated and immunologically suppressive in the TME.
Figure 5. TREM2+ TAMs display purine metabolic activity and are immunosuppressive.
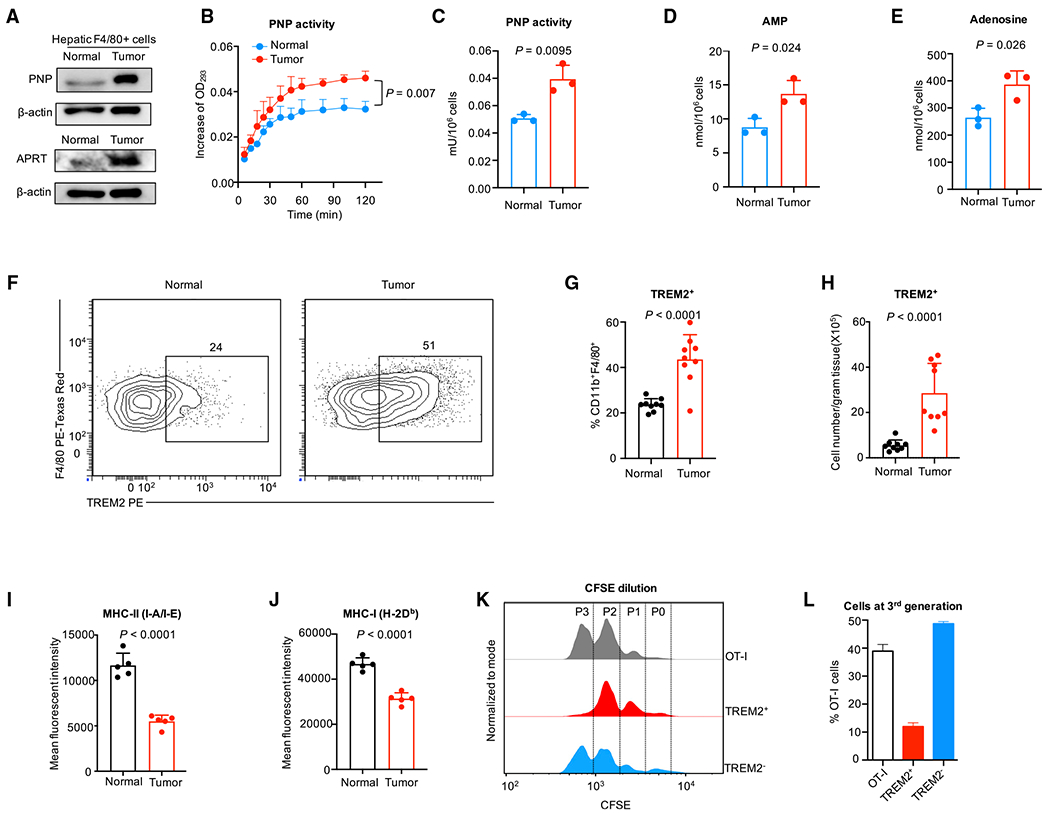
(A) Western blotting of APRT and PNP in F4/80+ cells sorted from liver with and without tumor.
(B and C) Metabolic activity of PNP. PNP was detected in TREM2+ TAMs and hepatic F4/80+ macrophages from normal liver. Student’s t test was used to calculate the significance between two groups.
(D and E) Levels of key purine pathway metabolites. AMP (D) and adenosine (E) were measured in TREM2+ TAMs and hepatic F4/80+ macrophages from normal liver. Student’s t test was used to calculate the significance between two groups.
(F–H) Flow cytometry quantification of TREM2+ hepatic macrophages in liver with and without tumor. Percentage of TREM2+ hepatic macrophages in CD11b+F4/80+ macrophages (G) and cell number of TREM2+ hepatic macrophages (H) are shown. Student’s t test was used to calculate the significance between two groups.
(I and J) Flow cytometry analysis of MHC-II (I-A/I-E) (I) and MHC-I (H-2Db) (J) expression in TREM2+ TAMs and macrophages from liver without tumor. Student t-test was used to calculate the significance between two groups.
(K and L) Flow cytometry analysis of OT-I cell proliferation in the presence or absence of TREM2+ and TREM2− cells purified from liver with tumor. Representative histogram (K) and the percentage of OT-I cells at the third generation (L) are shown. The ratio of TREM2+ or TREM2− cells to OT-I cells is 4:1.
Human TAMs manifest high metabolic heterogeneity
To corroborate our findings in humans, we proceeded with our analysis on human TAMs utilizing two human scRNA-seq datasets, mostly mirroring our experimental model: (1) macrophages in hepatocellular carcinoma (HCC) and adjacent normal tissues (Sharma et al., 2020) and (2) macrophages in colorectal cancer (CRC) and adjacent normal tissues (Zhang et al., 2020). We first analyzed metabolic profiles in human HCC and CRC macrophages (Figures S6A–S6C). Like mouse TAMs, we observed 5 metabolically distinct macrophage clusters in HCC (Figures S6A and S6B) and CRC (Figure S6C). In addition, separated projectors of HCC and CRC and adjacent normal tissues revealed a metabolic homogeneity in normal macrophages and a high metabolic heterogeneity in TAMs (Figures 6A–6D). Moreover, metabolic profiles were gradually shifted from adjacent normal tissues to peripheral tumors and then to core tumors (Figure 6A), suggesting a potent impact of cancer cells on TAM metabolic heterogeneity. By querying the expression of both a combined gene signature and key purine metabolic genes, we could identify a significant number of purine metabolic TAMs in HCC and CRC (Figures 6E–6I). Using pseudo-time analysis (Farrell et al., 2018; Sharma et al., 2020), we confirmed our prior observations that macrophages with purine metabolism are at the terminal differentiation stage (Figures 6J and 6K). Furthermore, the purine metabolic gene signature showed a strong positive correlation with angiogenesis gene signature and immunosuppressive gene TREM2 (Figure 6L). In addition to HCC and CRC, we also observed higher purine gene signature score in TAMs from uterine corpus endometrial carcinoma (UCEC), thyroid carcinoma (THCA), and esophageal carcinoma (ESCA), compared with macrophages from normal adjacent tissues (Cheng et al., 2021)(Figure 6M). Moreover, the purine gene signature score was higher at later stage CRC and ESCA (Figure 6N). These data suggest that purine metabolism is a hallmark of TAMs across multiple cancer types in humans.
Figure 6. Human TAMs manifest high metabolic heterogeneity.
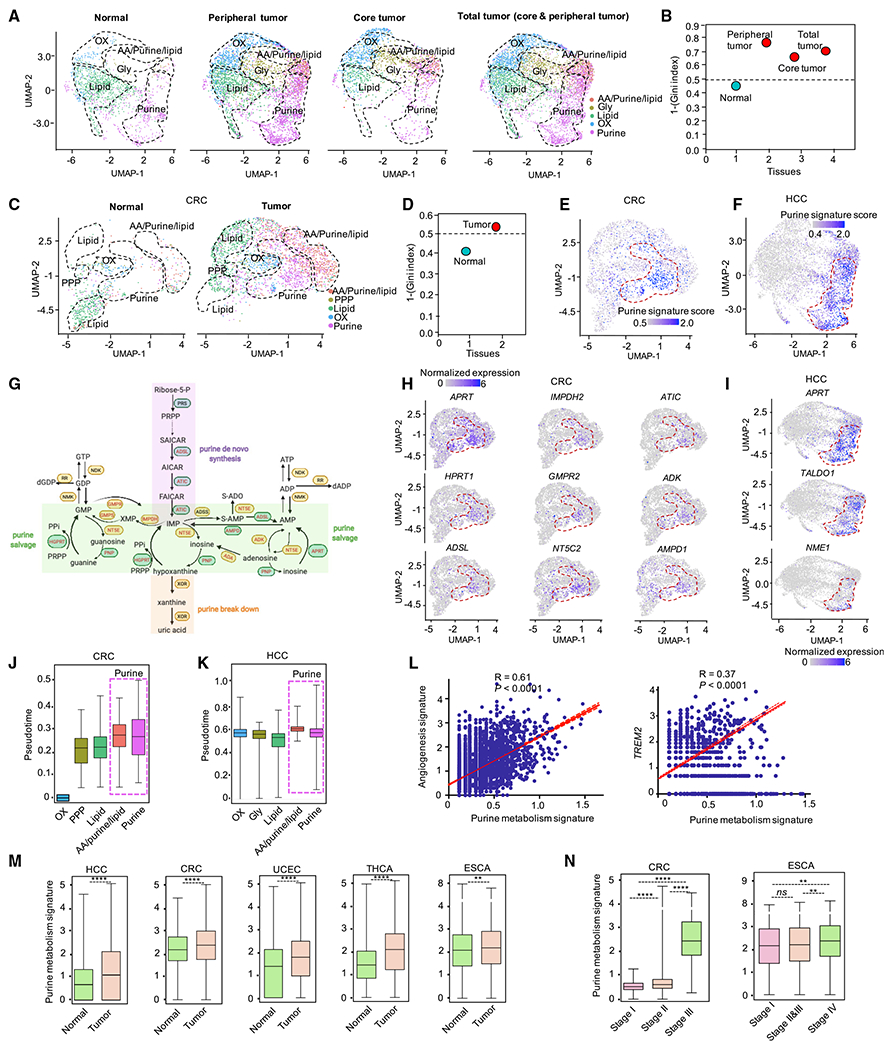
(A) UMAP visualization of split projections of metabolic clusters from macrophages in tissue of adjacent normal, peripheral tumor, core tumor, and total tumor from cohort of HCC. HCC: hepatocellular carcinoma.
(B) Heterogeneity of macrophages in tissue of adjacent normal, peripheral tumor, core tumor, and total tumor from cohort of HCC. Heterogeneity: 1-(Gini index); dashed line: sample with significant diversity.
(C) UMAP visualization of split projections of metabolic clusters from macrophages in tissue of adjacent normal and tumor from cohort of CRC (colorectal cancer).
(D) Heterogeneity of macrophages in tissue of adjacent normal and tumor from cohort of CRC. Heterogeneity: 1-(Gini index). Dashed line: sample with significant diversity.
(E and F) Enrichment of purine metabolic gene signature in each macrophage from cohort of CRC (E) and HCC (F), respectively.
(G) Diagram showing the expression of genes in purine metabolic pathways. Red font: genes enriched, black font: genes not enriched. Data from cohort of CRC were used.
(H and I) UMAP visualization of the expression level of selected purine metabolic genes in macrophages from cohort of CRC (H) and HCC (I), respectively.
(J and K) Pseudo-time analysis of macrophages in each metabolic cluster from cohort of CRC (J) and HCC (K), respectively. Pseudo-time for each macrophage in cohort of CRC was inferred by R package of URD. Pseudo-time for each macrophage in cohort of HCC was extracted directly from the original data (Sharma et al., 2020).
(L) Correlation analysis between gene signature of purine metabolism (x axis) with angiogenesis and TREM2 (y axis) on macrophages. Data from cohort of HCC (Sharma et al., 2020) were used. Pearson’s correlation coefficient (R) and correlation test (p values) were used to evaluate the correlation between the purine metabolic signature and angiogenesis signatures (and TREM2).
(M and N) Pan-cancer analysis of purine metabolic gene expression in macrophages from different tissues (M), and from patients at different grade malignancy (Cheng et al., 2021) (N) Mann-Whitney test was used to calculate the significance between two groups; only CRC and ESCA have enough patients for stage analysis (at least 2 patients at each stage are needed); *p < 0.05, **p < 0.01, and ***p < 0.001. HCC: hepatocellular carcinoma; CRC: colorectal cancer; UCEC: uterine corpus endometrial carcinoma; THCA: thyroid cancer; ESCA: esophageal carcinoma.
Purine metabolic TAMs were associated with clinical outcome
Because purine-metabolism-enriched TAMs harbor a pro-tumor phenotype, we wondered whether purine metabolism in TAMs correlated with clinical response to ICB and cancer patient outcome. We studied 3 patient cohorts that received ICB therapy, including (1) cohort 1, metastatic melanoma patients (Sade-Feldman et al., 2018); (2) cohort 2, metastatic melanoma patients (Jerby-Arnon et al., 2018); and (3) cohort 3, advanced basal cell carcinoma (BCC) (Yost et al., 2019). We found that the levels of purine metabolic gene expression were consistently higher in TAMs from non-responders than responders (Figure 7A; left two panels of Figures 7B and 7C; and 7D–7F). In addition, TAMs from non-responders showed higher expression of purine metabolic genes in post-treatment samples compared with baseline samples (Figures 7B and 7C, right; and 7D–7F). Thus, purine metabolism in TAMs may contribute to both primary and acquired therapeutic resistance. Next, we examined the relationship between the expression level of purine metabolic genes in TAMs and patient survival. We used the coefficient of individual purine metabolic genes in cohort 1 (Sade-Feldman et al., 2018) (Figures 7G–7I) to score the risk of individual patients. We then stratified patients into low- or high-risk groups based on their risk scores (Figure 7G, top). Compared with the low-risk group, purine metabolic genes, such as APRT, NT5C2, and PAICS, were expressed at higher levels in the high-risk group (Figure 7G, bottom), in which most patients were dead (Figure 7G, middle) and had shorter overall survival (Figure 7H). Cox proportional hazards model (Cox-PH) analysis showed that patients with higher purine metabolic gene expression in their TAMs had poor prognosis compared with those with lower purine metabolic gene expression (Figure 7I). Taken together, these results strongly suggest that the expression levels of purine metabolic genes correlate to ICB response and are associated with patient outcomes.
Figure 7. Purine metabolic TAMs were associated with clinical outcome.
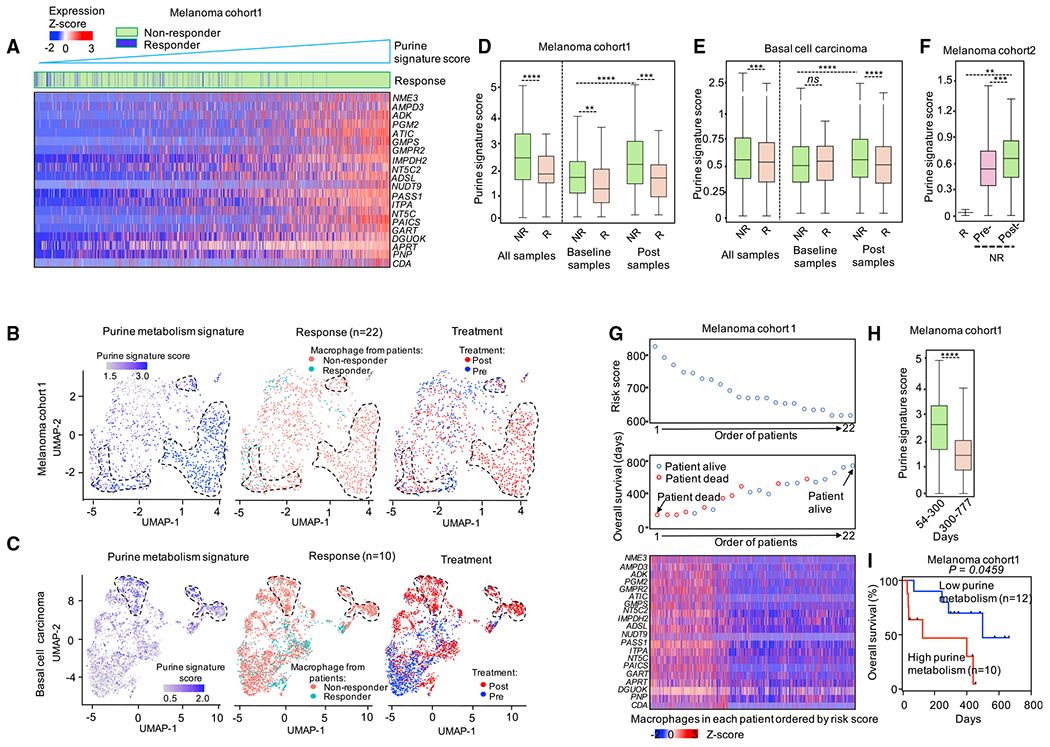
(A) Heatmap showing the expression level of purine metabolic genes in macrophages from patients with melanoma (Sade-Feldman et al., 2018). Macrophages were ordered by the score of purine metabolic gene signature. Macrophages were labeled (the “Response” row) by their origins; green: macrophage from ICB non-responders; blue: macrophage from ICB responders.
(B and C) UMAP plots showing the clusters of macrophages from melanoma cohort 1 (Sade-Feldman et al., 2018) (B) and basal cell carcinoma cohort (C). Cells were colored by the score of purine metabolic gene signature, ICB response of patients, and samples the macrophages were isolated from, respectively.
(D–F) The expression level of purine metabolic genes in macrophages from ICB responders and non-responders over three cohorts (Jerby-Arnon et al., 2018; Sade-Feldman et al., 2018; Yost et al., 2019). The thick line represents the median value, the bottom and top of the boxes are the 25th and 75th percentiles (interquartile range). Whiskers encompass 1.5 times the interquartile range. Mann-Whitney test was used to calculate the significance between two groups; *p < 0.05, **p < 0.01, and ***p < 0.001.
(G) The impact of expression of purine metabolic genes in macrophages on overall survival (OS) in melanoma patients. Upper: the ranked risk score of 22 melanoma patients evaluated by the coefficient of purine metabolic genes. Middle: the OS distribution of the 22 patients ranked according to the risk score (from upper). Bottom: heatmap of the expression pattern of purine metabolic genes in the macrophages from the 22 melanoma patients.
(H) The expression level of purine metabolic genes in macrophages from patients with melanoma. Macrophages were grouped by the OS time of corresponding patients. Mann-Whitney test was used to calculate the significance between two groups; *p < 0.05, **p < 0.01, and ***p < 0.001.
(I) Kaplan–Meier plots of OS for 22 melanoma patients accept ICB therapy (Sade-Feldman et al., 2018). Patients were stratified into low and high purine metabolism groups with the median of the purine metabolism score in macrophages from corresponding melanoma patients. Cox proportional hazards model was used to test the OS difference between low and high group.
DISCUSSION
In this work, we have conducted an scRNA-seq study on hepatic macrophages in mice bearing tumor liver metastases and extended our findings to several cancer patient cohorts. We have found that TAMs are developmentally, metabolically, and functionally heterogeneous and that purine metabolism delineates macrophage population with a consistent pro-tumor phenotype.
According to the classical dualistic theory, macrophages, including TAMs, are classified into M1 and M2 types. M1 macrophages are viewed as pro-inflammatory and cytotoxic while M2 macrophages are anti-inflammatory and promote wound healing. Indeed, in the normal human liver tissues, there are both pro-inflammatory and immune regulatory macrophage subsets (MacParland et al., 2018). However, while we have found that most macrophages showed an M2-like phenotype in the hepatic TME, TAMs manifested high heterogeneity at developmental, metabolic, and functional levels. Ontogenetically, macrophages, including TAMs, could be classified into two populations: circulating MDMs and embryonically originated residential macrophages. In line with this notion, our transcriptome analysis demonstrated the existence of both MDMs and residential macrophages in the liver microenvironment (Mould et al., 2019; Sharma et al., 2020). Previous data suggest that embryonic-derived macrophages and peripheral MDMs may play different and often conflicting roles in different types of tumor models (Franklin et al., 2014; Loyher et al., 2018; Zhu et al., 2017). For example, TIM-4+ peritoneal residential macrophages appear to mediate major immune suppression in ovarian cancer (Xia et al., 2020). Interestingly, our analysis revealed that MDMs are reprogrammed in the context of tumor liver metastasis and exhibit an immunosuppressive and pro-tumor phenotype. Our results suggest that macrophage subsets can play different roles in different disease models or sites. Nonetheless, ontogeny of TAM subsets and potential mechanisms controlling TAM metabolic heterogeneity need additional studies. It is suggested that the metabolic profiles of TAMs may be shaped by both their developmental origin and specific histological TME (Argüello et al., 2020; Ben-Moshe and Itzkovitz, 2019; Ben-Moshe et al., 2019; Kumar et al., 2019). In line with this, we found that Kupffer cells largely reside at the peritumor area. This localization may potentially explain their relatively stable metabolic status in the liver with tumor.
Recent studies have demonstrated that nutrients and metabolites can phenotypically and functionally reshape the states of immune cells, including myeloid cells and T cells, in the TME (Bian et al., 2020; Li et al., 2018; Wang et al., 2021; Wu et al., 2019; Zhao et al., 2016). Accordingly, our metabolic pathway analysis revealed several features of TAMs. First, there was a wide heterogeneity of TAMs in the context of ontogeny (monocyte-derived and residential), tissue origin (normal tissue and tumor), and macrophage polarization status. Second, when we compared TAMs with normal macrophages, we discovered that tumors endow TAMs with specific metabolic profiles, as demonstrated by clusters with high glycolysis and AA metabolism. Additionally, tumors may also reprogram existing metabolic cluster(s), as shown by the metabolic shift from lipid metabolism to purine metabolism. Interestingly, TAM subsets enriched with purine metabolism were terminally differentiated. This was in stark contrast to other TAMs with high activity of other metabolic pathways, such as glycolysis, OXPHOS, lipid metabolism, and AA metabolism. Furthermore, the purine-metabolism-enriched TAMs possessed limited ability of phagocytosis and antigen presentation but expressed high levels of genes related to angiogenesis and immunosuppression. Moreover, high purine metabolism signature score in TAMs correlated with poor therapeutic outcome to immunotherapy in several cancer patient cohorts and was associated with shorter survival. Tumor cell purine metabolism may promote ICB resistance (Keshet et al., 2020; Zhou et al., 2020). Previous studies have demonstrated that targeting CSF-1/CSF-1R signaling pathway can deplete MDMs, resulting in tumor regression in several murine models (Pyonteck et al., 2013; Ries et al., 2014; Yu et al., 2021). However, given the multifactorial roles of TAM subsets, systemic macrophage depletion has not been translated into therapeutic efficacy in patients with cancer (Cassetta and Pollard, 2018). Our work indicates that TAMs are highly heterogeneous, and purine-metabolism-enriched TAMs are detrimental for cancer patients. Given that purine metabolism plays a key role in tumor transformation and progression (Keshet et al., 2020; Zhou et al., 2020), targeting purine metabolism may selectively disable harmful TAM subset and simultaneously impair tumor cells, thereby being a “killing two birds with one stone” approach.
Limitations of the study
A limitation of our analysis is its relative dependence on transcriptomic profiling, while multiple factors, including metabolic protein expression levels, metabolic enzymatic activities, and environmental regulatory mechanisms, may coordinately determine the functional relevance of specific metabolic pathway in tumor immunity. Although we have validated our major conclusions with selective experiments, additional complementary and confirmatory strategies, such as high-throughput metabolic proteomic profiling, and gain- and loss-of-function studies, should be employed to fully characterize TAMs in the future.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact: Weiping Zou (wzou@med.umich.edu). This study did not generate new unique reagents.
Materials availability
This study did not generate new unique reagents.
Data and code availability
The in-house dataset in this study is available in GEO under the accession number of GSE157600 (Yu et al., 2021). Other datasets could be obtained from the original papers listed above and the Supplementary table S1 (Table S1). This paper does not report original code. All software is freely or commercially available and is listed in the STAR Methods description. Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Eight- to ten-week-old female C57BL/6 were ordered from Jackson Laboratory. OT-I mice were ordered from Jackson Laboratory and bred in house. All mice were maintained under SPF housing. Animal studies were conducted under the approval of the Institutional Animal Care & Use Committee (IACUC) at the University of Michigan (PRO00010169, PRO00009373).
Cell lines
Luciferase-expressing MC38 cells (MC38-luc) were established by transfection with lenti-GF1-CMV-VSVG virus expressing GFP and luciferase as previously described (Yu et al., 2021).
Tumor model
We studied MC38 colon cancer metastasis in the livers in murine model. To this end, we established subcutaneous MC38 tumors and inoculated 5 × 105 MC38 tumor cells into liver via intrasplenic injection, followed by immediate splenectomy, as we reported recently (Yu et al., 2021). Control mice have undergone a sham surgery. 14 days after tumor establishment, we isolated hepatic mononuclear cells via density centrifugation. Normal livers and livers bearing MC38 tumors were resected, flushed out blood through the portal vein with PBS, minced, and passed through a 100-uM cell strainer (BD Biosciences). Hepatocytes were depleted through a series of 100 x g centrifugation steps. The supernatant was layered onto 15 mL Ficoll-Paque media and centrifuged at 1000 x g for 20 minutes and stopped without a brake applied. The buffy layer was isolated, washed, and filtered. Three biological replicates were pooled.
METHOD DETAILS
Immunofluorescence
Liver tissues with metastatic tumors were collected and fixed in 4% paraformaldehyde at 4 °C overnight. Tissues were dehydrated in 30% sucrose solution at 4 °C overnight. After dehydration, tissues were frozen in optimal cutting temperature compound. Cryosections at 10 μm were made for immunofluorescent staining. Sections were blocked with 1% BSA (37 °C, 1 hour), stained with PE-F4/80 and anti-CLEC4F overnight at 4 °C, washed and stained with secondary antibody (37 °C, 20 minutes), and counter-stained with DAPI. Tissue sections were analyzed with confocal microscope (Nikon, A1) and imaged with ImageJ.
Magnetic-activated cell sorting (MACS) of hepatic macrophages
Liver mononuclear cells were isolated as previously described. Cell suspensions were blocked with rat serum at 4°C for 30 minutes, labeled with PE-F4/80 (BD Biosciences) or PE-TREM2 (R&D systems) antibodies at 4°C for 30 minutes, washed with PBS twice, labeled with anti-PE microbeads (Miltenyi) at 4°C for 30 minutes, and washed with PBS twice. Then, PE positive cells were sorted by passing through LS columns (Miltenyi) according to manufacturer’s instructions. Purity of the sorted cells (> 90%) were assessed by flow cytometry.
Immunoblots
Sorted macrophages were lysed in RIPA buffer (Thermo Fisher Scientific) supplemented with Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). Protein concentrations of cell lysates were determined using BCA protein assay kit (Thermo Fisher Scientific). Equivalent amounts of total cellular protein were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were then transferred to polyvinylidene fluoride membranes and immunoblotted with the indicated antibodies.
Purine nucleoside phosphorylase (PNP) activity assay
PNP activity was determined by using PNP activity assay kit (Abcam). One million sorted macrophages were lysed in 200 μL PNP activity assay buffer. PNP activity was determined according to manufacturer’s instruction.
OT-I cell proliferation assay
Splenocytes from OT-I transgenic mice were isolated and labeled with carboxyfluorescein succinimidyl ester (CFSE, Thermo Fisher Scientific). CFSE-labeled OT-I splenocytes were stimulated with OVA-SIINFEKL peptides (100 nM) in the presence of IL-2 (5 ng/mL), 2-Mercaptoethanol (55 μM) and seeded at a concentration of 5 ×104 cells per well of flat 96-well plates. Sorted TREM2+ cells or TREM2− cells (2 × 105 cells) were added at the time when the activation started. Two days after coculture, OT-I cell proliferation was determined by CFSE dilution. OT-I cells were gated as CD45+CD3+CD8+ cells.
Flow cytometry
Mononuclear cells were isolated from the livers, blocked with serum, and stained with fluorescently conjugated antibodies. Quantification of cell number was performed using CountBright Absolute Counting Beads (Thermo Fisher). Data analysis was performed on a BD LSRFortessa (BD Bioscience) using BD FACS Diva software. The following antibodies were used: CD45 (Clone 30-F11, BD Biosciences), CD11b (Clone M1/70, BD Biosciences), F4/80 (Clone T45-2342, BD Biosciences), I-A/I-E (Clone M5/114.15.2, Thermo Fisher Scientific), H-2Db (Clone AF6-88.5, BD Biosciences), and TREM2 (FAB17291P, R&D systems).
Single cell sequencing
Single cell library preparation was carried out by using the 10X Genomics Chromium Single Cell 5′ Library and Gel Bead Kit v2 #1000014 (10X Genomics, Pleasanton, CA, USA). Single cell sequencing was performed on the single cells as we recently reported (Yu et al., 2021). Libraries were sequenced on an Illumina HiSeq 2500.
QUANTIFICATION AND STATISTICAL ANALYSIS
Single cell sequencing data analysis
The data processing method for the in-house scRNA-seq dataset was similar to the method described previously (Yu et al., 2021). In brief, basecalls were first converted into FASTQs with bcl2fastq. FASTQ files were aligned to the mice genome (GRCm38.p6) using the STAR aligner as implemented in the pipeline of Cell Ranger (version 2.2). UMI counts, and gene information as well as barcode matrix output from the Cell Ranger workflow were used for downstream analyses. Next, several data quality checks and filtering steps were performed: 1) cells with fewer than 200 genes detected, 2) cells with greater than 30% mitochondrial RNA content, and 3) cells not expressing macrophage markers (Lyz2, Csf1r, Cd68, and Lgal3) and expressing dendritic cell marker (Zbtb46) or neutrophil marker (Ly6g and Csf3r), 4) cells identified as doublet by using DoubletDecon (DePasquale et al., 2019) and Scrublet (Wolock et al., 2019) were excluded from all downstream analyses. Subsequently, the majority of downstream analyses were performed using the Seurat (version 2.3.4) workflow (Satija et al., 2015) as described below: counts on the filtered matrix for each gene were normalized to the total library size with the Seurat ‘NormalizeData’ function (verbose = FALSE). 2000 most highly variable genes identified by the function ‘FindVariableGenes’ from Seurat were used to perform unsupervised clustering. ‘FindIntegrationAnchors’ and ‘IntegrateData’ functions were used to integrate samples with and without liver tumors. Then each integrated feature was centered to a mean of zero and was scaled by the standard deviation with the function ‘ScaleData’ in Seurat. PCA was performed using the function ‘RunPCA’. The ‘FindClusters’ function was used to calculate the clusters of the cells, with the resolution set to 0.8. Next, UMAP v0.3.9 (McInnes et al., 2018) plots were used to visualize clusters of cells localized in the graph-based clusters by using ‘RunUMAP’ function (reduction: pca, dims: 1:20). Markers for each cluster were identified by finding differentially expressed genes between cells from the individual cluster versus cells from all other clusters using the ‘FindAllMarkers’ function (only.pos: TRUE, min.pct: 0.25, logfc.threshold: 0.25). Clusters were further annotated by ‘Enrichr’ software (Kuleshov et al., 2016) with the markers identified above. The scPred packegae was applied to validate the definition of macrophage clusters (Alquicira-Hernandez et al., 2019). Metabolic heterogeneity was evaluated with 1- (Gini index), which was calculated by R package of “ineq”.
Gene signature enrichment
In order to create heatmaps, feature plots, and violin plots for gene set with multiple genes, 10 curated gene expression signatures were interrogated: 1) residential macrophages (RTMs): Timd4, Vsig4, Clec4 (Davies et al., 2013); 2) Monocyte-derived macrophages (MDMs): Ccr2 (Serbina and Pamer, 2006); 3) Liver capsular macrophages (LCMs): Itgax, Cx3cr1 (Blériot and Ginhoux, 2019; Sierro et al., 2017); 4) Peritoneal macrophages (PMs): Gata6 (Blériot and Ginhoux, 2019; Wang and Kubes, 2016); 5) Antigen presentation and processing, the complete signature was collected from databases of MSigDB ((Subramanian et al., 2005) and Mouse Genome Informatics (http://www.informatics.jax.org/go/term/GO:0019882) with number of hsa04612 and GO:0019882, respectively, key genes selected to represent on related heatmaps including: H2-Aa, H2-Ab1, H2-DMa, H2-DMb1, H2-Dmb2, H2-Eb1, H2-Oa, H2-Ob, H2-Q6, H2-Q7, Tap1, Tap2, B2m, Cd74, Tapbp, Tapbpl, Ciita, Nlrc5, Cd80, Cd86, Icosl, Cd40, Il18, Il1b, and Il12b; 6) Phagocytosis: Cd74, Fcgr2b, Gsn, Hsp90aa1, Itgam, Itgb1, Msr1, Ccl2, Tgm2, Thbs1, Tlr2, Cd93, Ctsl, Tuba1c, Cebpb, Ctsd, Clec4w, Nfkbia, and Trem1 (collected from KEGG (Kanehisa et al., 2016)); 7) Angiogenesis: Anxa1, Anxa2, C3, E2f2, Fn1, Tgfbi, Ramp1, Lrg1, Spint1, Vegfa (collected from KEGG (Kanehisa et al., 2016)); 8) M2-like polarization: Alox5, Arg1, Chil3, Cd163, Il10, Il10ra, Il10rb, Irf4, Kif4, Mrc1, Myc, Socs2, and Tgm2 (Mould et al., 2019); 9) M1-like polarization: Azin1, Cd38, Cxcl10, Cxcl9, Fpr2, Il18, Il1b, Irf5, Nifkbiz, Tlr4, Tnf, and Cd80 (Mould et al., 2019); and 10) purine metabolic genes: Nme3, Ampd3, Adk, Pgm2, Atic, Gmps, Gmpr2, Impdh2, Nt5c2, Adsl, Nudt9, Pass1, Itpa, Nt5c, Paics, Gart, Dguock, Aprt, and Pnp (collected from MSigDB) (Subramanian et al., 2005). Individual cells were scored for enrichment of gene signatures using the function of AddModuleScore by setting the ‘ctrl’ (number of control features selected from the same bin per analyzed feature) as 5. Next, standard FeaturePlot and VlnPlot function of Seurat were used to generate the feature plots or violin. Transcriptional factors (TF) that regulated the 5 metabolic clusters were predicted by using BART database (Wang et al., 2018) with top 10 highly expressed genes in each cluster, top 5 significantly enriched TF in each cluster were represented.
M1-like and M2-like macrophage definition
The phenotype of M1-like and M2-like macrophages was defined as the mean expression score calculated by using the function of AddModuleScore of relative gene signatures (Azizi et al., 2018; Zhang et al., 2020). Genes associated with M1-like macrophages included Azin1, Cd38, Cxcl10, Cxcl9, Fpr2, Il18, Il1b, Irf5, Nifkbiz, Tlr4, Tnf, and Cd80 (Mould et al., 2019). Alox5, Arg1, Chil3, Cd163, Il10, Il10ra, Il10rb, Irf4, Kif4, Mrc1, Myc, Socs2, and Tgm2 were used to define the signature of M2-like macrophages (Mould et al., 2019).
Correlation analysis between gene signatures
For correlation analysis between gene signatures of antigen presentation (or angiogenesis) and purine metabolic genes, score of each signature was defined as described above. Then, Pearson correlation analyses and scatter plots were performed and prepared using GraphPad version 6.0.
Metabolic activity analysis with GSEA
Ranked fold-change list of differentially expressed genes for each cluster was obtained by comparing cells from the individual cluster (defined above) with cells from all other clusters. To obtain the differentially expressed genes of each cluster, the “FindAllMarkers” function of the Seurat pipeline was used. Next, GSEA was performed with 60 KEGG (Kanehisa et al., 2016) metabolic pathways and the ranked lists by using the “PreRanked” function of GSEA v3.0 (Subramanian et al., 2005). Gene set enrichment nominal p values, NES (Normalized Enrichment Score) values, and adjusted p values (FDRs) were then calculated with 1,000 permutations in the GSEA software, run in Signal2Noise mode. Finally, the NES and nominal p-value were applied to sort each cluster of the enrichment degree of metabolic pathways. Heatmap visualization was performed by using GENE-E (https://software.broadinstitute.org/GENE-E/) with the value obtained (+/−Log2|NES/FDR|).
Metabolic clustering for hepatic macrophages
To obtain metabolic clusters of the 8056 macrophages, 1310 metabolic genes extracted from 60 metabolic pathways were obtained from the KEGG database. A new 1310 genes by 8056 cells matrix was subset from the total 20000 genes by 8056 cells data matrix (tumor and normal samples were integrated as showed above), followed by the creation of Seurat objects. The following steps of clustering cells by metabolic genes were the same as the standard Seurat pipeline described above. With the ‘dims’ parameter set to 20, ten metabolic clusters were obtained with those 1310 input genes and 350 highly variable features.
To identify differentially expressed genes in each of the ten metabolic clusters, the “FindAllMarkers” function of the Seurat pipeline was used to compare cells from an individual cluster with cells from all other clusters. Next, each of the ten metabolic clusters were annotated with the top 10 differentially expressed genes (P < 0.05 and log2(fold change) >=0.25) by using the Enrichr program (Kuleshov et al., 2016). The top one (with fisher exact test p-value ranked as the most significant) enriched metabolic pathway was considered as the dominant metabolic pathway of each cluster. PCA and consensus clustering from scater (McCarthy et al., 2017) package was used to validate the clusters defined above. We found that the cells clustered by Seurat as a cell type could also be grouped together by PCA. Then clusters with the same annotated dominant metabolic pathway were merged and labeled with the same color in corresponding plots. Also, each cell was labeled by other features: 1) Seurat cluster defined by all marker genes, 2) predicted developmental origin as defined above (RTMs, MDMs, LCMs and PMs), 3) tissue source (tumor versus normal), and 4) the polarization states defined above. Finally, the UMAP projection of 20000 genes by 8056 cells was colored with the same color of the 5 merged metabolic clusters and split into two projections of tumor and normal samples. The proportion of each cluster was calculated with the function of “prop.table” in Seurat pipeline.
Trajectory analysis
BAM files aligned using the Cell Ranger pipelines were first sorted by using SAMtools v1.12 (Li et al., 2009). Next, the Velocyto (La Manno et al., 2018) pipeline was used to count spliced and un-spliced reads, and generate two loom files of both tumor and normal samples. Gene-specific velocities were then computed following the scVelo python package (Bergen et al., 2020). Meanwhile, the projection clustered with metabolic genes (as defined in Figure S1A) was embedded with the velocity streams predict by scVelo with the loom files. Finally, plots for the ratio of spliced and un-spliced, for the velocity and the expression of various individual gene were generated based on the velocity calculated by scVelo. Additional developmental trajectory inference algorithms, URD v1.1.0 (Farrell et al., 2018) (Figure 6J) and partition-based graph abstraction (PAGA (Wolf et al., 2019), Figure 4F), were used to confirm the robustness of our results. For URD analysis, cells from the macrophage metabolic cluster characterized by OXPHOS were set as root. The analyses were performed by setting the parameters of knn to 100 and sigma to 16. For PAGA, the pseudo-time was calculated by scanpy v1.4.3 (Wolf et al., 2018). In brief, we followed the pipeline plugged in scVelo and used the same projection generated by scVelo and performed the prediction with the function of scv.pl.paga in scVelo by setting the parameter of basis as umap, the size as 50 and the alpha as 0.3, the min_edge_width as 2 and the node_size_scale as 1.5.
Clinical datasets
We extended our analysis to 8 human scRNA-seq datasets (Table S1), including: (I) Hepatocellular Carcinoma (HCC) and adjacent normal samples from 14 HCC patients (Sharma et al., 2020) (5353 macrophages, Figure 6); (II) Colorectal Cancer (CRC) and adjacent normal samples from 18 CRC patients (Zhang et al., 2020) (3123 macrophages, Figure 6); (III) melanoma samples from 22 metastatic melanoma patients treated with ICB (Sade-Feldman et al., 2018), named as melanoma cohort 1 (1254 macrophages, Figure 7); (IV) melanoma samples from 31 melanoma patients grouped by the response rate and the treatment types (15 untreated, 15 with ICB resistance, and 1 with ICB response) (Jerby-Arnon et al., 2018), named as melanoma cohort 2 (420 macrophages, Figure 7); (V) advanced Basal Cell Carcinoma (BCC) samples from 11 BCC patients with before and after anti-PD-1 treatment in site-matched primary tumors (Yost et al., 2019) (3093 macrophages, Figure 7); (VI) Uterine Corpus Endometrial Carcinoma (UCEC) and adjacent normal samples from 9 UCEC patients (Cheng et al., 2021) (4194 macrophages, Figure 6); (VII) Thyroid Cancer (THCA) and adjacent normal samples from 10 THCA patients (Cheng et al., 2021) (5312 macrophages, Figure 6); (VIII) Esophageal Carcinoma (ESCA) and adjacent normal samples from 7 ESCA patients (Cheng et al., 2021) (4825 macrophages, Figure 6). As there were at least 2 patients in each stage in the CRC and ESCA data sets, we used these two data sets for clinical stage analysis.
Patient outcomes analysis
To evaluate the performance of the expression of purine metabolic genes in TAMs in ICB response, we analyzed the scRNA-seq datasets in 3 cohorts (Jerby-Arnon et al., 2018; Sade-Feldman et al., 2018; Yost et al., 2019). For each dataset, in matrix representation, the normalized expression level of each purine metabolic gene across macrophages (already defined by each original paper) was used. Next, the Z-scores were calculated by subtracting the average among all macrophages of each gene. Then, the Z-score was used as the input for downstream heatmap visualization by GENE-E. GraphPad version 6.0 was used to make boxplots and perform Mann-Whitney test on different groups (such as ICB response and tumor stages).
For patient overall survival (OS) analysis, based on the availability of clinical follow-up information, we used the melanoma cohort 1 (Sade-Feldman et al., 2018). We constructed a Cox proportional hazard model with OS regressed on the expression of purine metabolic genes. First, the value of coefficient for each gene was computed based on the expression levels and the living days as well as the living status (dead or not) of each patient with “coxph test” from the “survival” R package. Second, for each individual patient, a risk score was obtained by taking sum of the value of coefficient*expression level of each gene. The patients were then ranked by their corresponding risk score (Han et al., 2019; Wang et al., 2019) and colored with living status (middle panel of Figure 7G). Finally, the expression level of each purine metabolic gene in each macrophage was represented by a heatmap with patients ordered according to their risk scores (bottom panel of Figure 7G). In addition, to explore the relationship between purine metabolic gene expression in TAMs and with OS, a Kaplan-Meier survival curve was generated. Patients were initially divided into two groups based on the expression levels of purine metabolic genes. Kaplan-Meier survival curves were subsequently generated, and log-rank test p values were calculated by using the “survival” program of GraphPad version 6.0 with the living days and the living status (dead or not) of patients.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Anti-APRT antibody | Abcam | ab196558 |
| Anti-PNP Antibody (D-11) | Santa Cruz | RRID: AB_10709574 |
| PE Rat Anti-Mouse F4/80 | BD Biosciences | RRID: AB_2872009 |
| Human/Mouse TREM2 PE-conjugate Antibody | R&D systems | RRID: AB_884528 |
| Anti-CD45 antibody | BD Biosciences | RRID: AB_1107002 |
| Anti-CD11b antibody | BD Biosciences | RRID: AB_396772 |
| Anti-I-A/I-E | Thermo Fisher Scientific | RRID: AB_2534339 |
| Anti-H-2Db | BD Biosciences | RRID: AB_313512 |
|
| ||
| Critical commercial assays | ||
|
| ||
| CountBright Absolute Counting Beads | Thermo Fisher Scientific | C36950 |
| RIPA buffer | Thermo Fisher Scientific | #89900 |
| Protease and Phosphatase Inhibitor Cocktail | Thermo Fisher Scientific | #78430 |
| BCA protein assay kit | Thermo Fisher Scientific | 23227 |
| AMP Colorimetric Assay Kit | Thermo Fisher Scientific | K229 |
| Adenosine Assay Kit (Fluorometric) | Thermo Fisher Scientific | K327-100 |
| Purine Nucleoside Phosphorylase Activity Assay Kit (Colorimetric) | Abcam | ab204707 |
| Anti-PE Microbeads | Miltenyi | 130-048-801 |
|
| ||
| Experimental models: Cell Lines | ||
|
| ||
| MC38 cells | Walter Storkus | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6J | The Jackson Laboratory | Stock No: 000664 |
| Mouse: OT-I | The Jackson Laboratory | Stock No: 003831 |
|
| ||
| Software and algorithms | ||
|
| ||
| Prism 6.0 | http://www.graphpad.com/faq/viewfaq.cfm?faq=1362 | Commercial |
| Rstudio 3.6.0 | https://rstudio.com | RStudio Team (2020). RStudio: Integrated Development for R. RStudio, PBC, Boston, MA |
| Cellranger 2.2 | https://10xgenomics.com | 10x Genomics |
| Seurat 2.3.4 | https://satijalab.org/seurat/ | Satija et al., 2015 |
| Monocle | http://cole-trapnell-lab.github.io/projects/monocle/ | Version 2.8.0 |
| Scanpy 1.4.3 | https://github.com/theislab/scanpy | Wolf et al., 2018 |
| UMAP 0.3.9 | https://github.com/lmcinnes/umap | McInnes et al., 2018 |
| Enrichr | https://maayanlab.cloud/Enrichr/ | Kuleshov et al., 2016 |
| BART | http://bartweb.org/ | Wang et al., 2018 |
| GSEA 3.0 | https://www.gsea-msigdb.org/gsea/index.jsp | Subramanian et al., 2005 |
| GENE-E | https://software.broadinstitute.org/morpheus/ | N/A |
| scater | https://bioconductor.org/packages/release/bioc/html/scater.html | McCarthy et al., 2017 |
| SAMtools 1.12 | http://www.htslib.org/download/ | Li et al., 2009 |
| Velocyto 1.0 | http://velocyto.org/ | La Manno et al., 2018 |
| scVelo 1.0 | https://scvelo.readthedocs.io/installation.html | Bergen et al., 2020 |
| URD 1.1.0 | https://github.com/farrellja/URD/releases | Farrell et al., 2018 |
| PAGA | https://github.com/theislab/paga | Wolf et al., 2019 |
| DoubletDecon | https://github.com/EDePasquale/DoubletDecon | DePasquale et al., 2019 |
| Scrublet | https://github.com/swolock/scrublet | Wolock et al., 2019 |
| scPred | https://github.com/powellgenomicslab/scPred | Alquicira-Hernandez et al., 2019 |
Highlights.
Single-cell RNA-seq reveals metabolic heterogeneity of TAMs
TAM metabolic patterns correlate with their functional features
Purine metabolism marks TAMs with pro-tumor and terminal phenotype
Purine metabolism signature correlates with patient outcome and response to ICB
ACKNOWLEDGMENTS
This work was supported in part by research grants from the NIH/NCI for W.Z. (CA248430, CA123088, CA099985, CA193136, and CA152470) and A.M.C. (1UM1HG006508), grants for M.C. (V Foundation: T2019-006, Cancer Research Institute, and Fund for Innovation in Cancer Informatics), and the NIH through a University of Michigan Rogel Cancer Center support grant (P30CA46592). We thank all members of the Zou, Cieslik, Green, and Chinnaiyan laboratories for their insightful communication, library preparation, and sequencing data pre-processing.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110609.
DECLARATION OF INTERESTS
We declare that we have no competing interests.
REFERENCES
- Alquicira-Hernandez J, Sathe A, Ji HP, Nguyen Q, and Powell JE (2019). scPred: accurate supervised method for cell-type classification from single-cell RNA-seq data. Genome Biol. 20, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argüello RJ, Combes AJ, Char R, Gigan J-P, Baaziz AI, Bousiquot E, Camosseto V, Samad B, Tsui J, Yan P, et al. (2020). SCENITH: a flow cytometry-based method to functionally profile energy metabolism with single-cell resolution. Cell Metab. 32, 1063–1075.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, Nainys J, Wu K, Kiseliovas V, Setty M, et al. (2018). Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 174, 1293–1308.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Moshe S, and Itzkovitz S (2019). Spatial heterogeneity in the mammalian liver. Nat. Rev. Gastroenterol. Hepatol 16, 395–410. [DOI] [PubMed] [Google Scholar]
- Ben-Moshe S, Shapira Y, Moor AE, Manco R, Veg T, Halpern KB, and Itzkovitz S (2019). Spatial sorting enables comprehensive characterization of liver zonation. Nat. Metab 1, 899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergen V, Lange M, Peidli S, Wolf FA, and Theis FJ (2020). Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol 38, 1408–1414. [DOI] [PubMed] [Google Scholar]
- Bian Y, Li W, Kremer DM, Sajjakulnukit P, Li S, Crespo J, Nwosu ZC, Zhang L, Czerwonka A, Pawłowska A, et al. (2020). Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 585, 277–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blériot C, and Ginhoux F (2019). Understanding the heterogeneity of resident liver macrophages. Front. Immunol 10, 2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C, Williams LJ, Wang Z, Bristow CA, Carugo A, et al. (2018). Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab. 27, 977–987.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassetta L, and Pollard JW (2018). Targeting macrophages: therapeutic approaches in cancer. Nat. Rev. Drug Discov 17, 887–904. [DOI] [PubMed] [Google Scholar]
- Cheng S, Li Z, Gao R, Xing B, Gao Y, Yang Y, Qin S, and Zhang L (2021). A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 184, 792–809.e723. [DOI] [PubMed] [Google Scholar]
- Davies LC, Jenkins SJ, Allen JE, and Taylor PR (2013). Tissue-resident macrophages. Nat. Immunol 14, 986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePasquale EA, Schnell DJ, Van Camp P-J, Valiente-Alandí Í, Blaxall BC, Grimes HL, Singh H, and Salomonis N (2019). DoubletDecon: deconvolving doublets from single-cell RNA-sequencing data. Cell Rep. 29, 1718–1727.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding T, Xu J, Wang F, Shi M, Zhang Y, Li S-P, and Zheng L (2009). High tumor-infiltrating macrophage density predicts poor prognosis in patients with primary hepatocellular carcinoma after resection. Hum. Pathol 40, 381–389. [DOI] [PubMed] [Google Scholar]
- Farrell JA, Wang Y, Riesenfeld SJ, Shekhar K, Regev A, and Schier AF (2018). Single-cell reconstruction of developmental trajectories during ze-brafish embryogenesis. Science 360, eaar3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forssell J, Öberg Å, Henriksson ML, Stenling R, Jung A, and Palmqvist R (2007). High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin. Cancer Res 13, 1472–1479. [DOI] [PubMed] [Google Scholar]
- Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, and Li MO (2014). The cellular and molecular origin of tumor-associated macrophages. Science 344, 921–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier EL, Ivanov S, Williams JW, Huang SC-C, Marcelin G, Fairfax K, Wang PL, Francis JS, Leone P, Wilson DB, et al. (2014). Gata6 regulates aspartoacylase expression in resident peritoneal macrophages and controls their survival. J. Exp. Med 211, 1525–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habtetsion T, Ding Z-C, Pi W, Li T, Lu C, Chen T, Xi C, Spartz H, Liu K , Hao Z, et al. (2018). Alteration of tumor metabolism by CD4+ T cells leads to TNF-α-dependent intensification of oxidative stress and tumor cell death. Cell Metab. 28, 228–242.e226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Chen M, Fang Q, Zhang Y, Wang Y, Esma J, and Qiao H (2019). Prediction of the prognosis based on chromosomal instability-related DNA methylation patterns of ELOVL2 and UBAC2 in PTCs. Mol. Therapy-Nucleic Acids 18, 650–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, and Leeson R (2018). A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell 175, 984–997.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Sato Y, Kawashima M, Furumichi M, and Tanabe M (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshet R, Lee JS, Adler L, Iraqi M, Ariav Y, Lim LQJ, and Lerner S (2020). Targeting purine synthesis in ASS1-expressing tumors enhances the response to immune checkpoint inhibitors. Nat. Cancer 1, 894–908. [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, and Jagodnik KM (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Sharife H, Kreisel T, Mogilevsky M, Bar-Lev L, Grunewald M, Aizenshtein E, Karni R, Paldor I, Shlomi T, et al. (2019). Intra-tumoral metabolic zonation and resultant phenotypic diversification are dictated by blood vessel proximity. Cell Metab. 30, 201–211.e6. [DOI] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, and Kastriti ME (2018). RNA velocity of single cells. Nature 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, and Marth G (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25,2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, Wei S, Zhao L, Vatan L , Wen B, et al. (2018). Aerobic glycolysis controls myeloid-derived suppressor cells and tumor immunity via a specific CEBPB isoform in triple-negative breast cancer. Cell Metab. 28, 87–103.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loyher P-L, Hamon P, Laviron M, Meghraoui-Kheddar A, Goncalves E, Deng Z, Torstensson S, Bercovici N, Baudesson de Chanville C, Combadière B, et al. (2018). Macrophages of distinct origins contribute to tumor development in the lung. J. Exp. Med 215, 2536–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacParland SA, Liu JC, Ma X-Z, Innes BT, Bartczak AM, Gage BK, Manuel J, Khuu N, Echeverri J, Linares I, et al. (2018). Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun 9, 4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A, and Balkwill F (2008). Cancer-related inflammation. nature 454, 436–444. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Marchesi F, Jaillon S, Garlanda C, and Allavena P (2021). Tumor-associated myeloid cells: diversity and therapeutic targeting. Cell Mol. Immunol 18, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Campbell KR, Lun AT, and Wills QF (2017). Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 33, 1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes L, Healy J, and Melville J (2018). Umap: uniform manifold approximation and projection for dimension reduction. Preprint at arXiv, 1802.03426. [Google Scholar]
- Mould KJ, Jackson ND, Henson PM, Seibold M, and Janssen WJ (2019). Single cell RNA sequencing identifies unique inflammatory airspace macrophage subsets. JCI insight 4, e126556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, and Olson OC (2013). CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med 19, 1264–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian B-Z, and Pollard JW (2010). Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries CH, Cannarile MA, Hoves S, Benz J,Wartha K, Runza V, and Rey-Giraud F (2014). Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 25, 846–859. [DOI] [PubMed] [Google Scholar]
- Rosas M, Davies LC, Giles PJ, Liao C-T, Kharfan B, Stone TC, O’Donnell VB, Fraser DJ, Jones SA, and Taylor PR (2014). The transcription factor Gata6 links tissue macrophage phenotype and proliferative renewal. Science 344, 645–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffell B, Affara NI, and Coussens LM (2012). Differential macrophage programming in the tumor microenvironment. Trends Immunology 33, 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M,Yizhak K, Bjorgaard SL, Ray JP,deBoer CG,Jenkins RW, and Lieb DJ (2018). Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175, 998–1013.e1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serbina NV, and Pamer EG (2006). Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol 7, 311–317. [DOI] [PubMed] [Google Scholar]
- Sharma A, Seow JJW, Dutertre CA, Pai R, Blériot C, Mishra A, Wong RMM, and Singh GSN (2020). Onco-fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell 183, 377–394.e21. [DOI] [PubMed] [Google Scholar]
- Shimura S, Yang G, Ebara S, Wheeler TM, Frolov A, and Thompson TC (2000). Reduced infiltration of tumor-associated macrophages in human prostate cancer: association with cancer progression. Cancer Res. 60, 5857–5861. [PubMed] [Google Scholar]
- Sica A, and Mantovani A (2012). Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest 122, 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierro F, Evrard M, Rizzetto S, Melino M, Mitchell AJ, Florido M, Beattie L, Walters SB, Tay SS, Lu B, et al. (2017). A liver capsular network of monocyte-derived macrophages restricts hepatic dissemination of intraperitoneal bacteria by neutrophil recruitment. Immunity 47, 374–388.e6. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traag VA, Waltman L, and Van Eck NJ (2019). From Louvain to Leiden: guaranteeing well-connected communities. Scientific Rep. 9, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale I, Manic G, Coussens LM, Kroemer G, and Galluzzi L (2019). Macrophages and metabolism in the tumor microenvironment. Cell Metab. 30, 36–50. [DOI] [PubMed] [Google Scholar]
- Wang J, and Kubes P (2016). A reservoir of mature cavity macrophages that can rapidly invade visceral organs to affect tissue repair. Cell 165, 668–678. [DOI] [PubMed] [Google Scholar]
- Wang J, Wang Y, Chu Y, Li Z, Yu X, Huang Z, Xu J, and Zheng L (2021). Tumor-derived adenosine promotes macrophage proliferation in human hepatocellular carcinoma. J. Hepatol 74, 627–637. [DOI] [PubMed] [Google Scholar]
- Wang X, Liao X, Yang C, Huang K, Yu T, Yu L, Han C, Zhu G, Zeng X , Liu Z, et al. (2019). Identification of prognostic biomarkers for patients with hepatocellular carcinoma after hepatectomy. Oncol. Rep 41, 1586–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Civelek M, Miller CL, Sheffield NC, Guertin MJ, and Zang C (2018). BART: a transcription factor prediction tool with query gene sets or epigenomic profiles. Bioinformatics 34, 2867–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh TJ, Green RH, Richardson D, Waller DA, O’Byrne KJ, and Bradding P (2005). Macrophage and mast-cell invasion of tumor cell islets confers a marked survival advantage in non-small-cell lung cancer. J. Clin. Oncol 23, 8959–8967. [DOI] [PubMed] [Google Scholar]
- Wen Y, Lambrecht J, Ju C, and Tacke F (2020). Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol. Immunol 18, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf FA, Hamey FK, Plass M, Solana J, Dahlin JS, Göttgens B, and Rajewsky N (2019). PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 20, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolock SL, Lopez R, and Klein AM (2019). Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 8, 281–291.e289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W-C, Sun H-W, Chen J, OuYang H-Y, Yu X-J, Chen H-T, Shuang Z-Y, Shi M, Wang Z, and Zheng L (2019). Immunosuppressive immature myeloid cell generation is controlled by glutamine metabolism in human cancer. Cancer Immunol. Res 7, 1605–1618. [DOI] [PubMed] [Google Scholar]
- Xia H, Li S, Li X, Wang W, Bian Y, Wei S, Grove S, Wang W, Vatan L, Liu JR, et al. (2020). Autophagic adaptation to oxidative stress alters peritoneal residential macrophage survival and ovarian cancer metastasis. JCI insight 5, e141115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, and McNamara KL (2019). Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med 25, 1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Green MD, Li S, Sun Y, Journey SN, Choi JE, Rizvi SM, Qin A, Waninger JJ, Lang X, et al. (2021). Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat. Med 27, 152–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O’Brien SA, He Y , and Wang L (2020). Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell 181, 442–459.e29. [DOI] [PubMed] [Google Scholar]
- Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, Modak M, Carotta S, Haslinger C, Kind D, et al. (2019). Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell 179, 829–845.e20. [DOI] [PubMed] [Google Scholar]
- Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, Wei S, Crespo J, Wan S, Vatan L, et al. (2016). Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat. Immunol 17, 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Yao Y, Scott AJ, Wilder-Romans K, Dresser JJ, Werner CK, and Sun H (2020). Purine metabolism regulates DNA repair and therapy resistance in glioblastoma. Nat. Commun 11, 3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Herndon JM, Sojka DK, Kim K-W, Knolhoff BL, Zuo C, Cullinan DR, Luo J, Bearden AR, Lavine KJ, et al. (2017). Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity 47, 323–338.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W (2005). Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 5, 263–274. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The in-house dataset in this study is available in GEO under the accession number of GSE157600 (Yu et al., 2021). Other datasets could be obtained from the original papers listed above and the Supplementary table S1 (Table S1). This paper does not report original code. All software is freely or commercially available and is listed in the STAR Methods description. Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.