

Abstract
TGF-β has multiple roles and gene products (TGF-β1, -β2, and -β3), which make global targeting of TGF-β undesirable. Expression of TGF-β requires association with milieu molecules, which localize TGF-β to the surface of specific cells or extracellular matrices. Here, we found that LRRC33 was specifically associated with TGF-β1, not -β2 and -β3, and was required for surface display and activation of TGF-β1 on tumor-infiltrating myeloid cells. Loss of LRRC33-dependent TGF-β1 activation slowed tumor growth and metastasis by enhancing innate and adaptive anti-tumor immunity in multiple mouse syngeneic tumor models. LRRC33 loss resulted in a more immunogenic microenvironment, with decreased myeloid-derived suppressor cells, more active CD8+ T and NK cells, and more skewing toward tumor-suppressive M1 macrophages. LRRC33 loss and PD-1 blockade synergized in controlling B16.F10 tumor growth. Our results demonstrate the importance of LRRC33 in tumor biology and highlight the therapeutic potential of dual blockade of the LRRC33/TGF-β1 axis and PD-1/PD-L1 in cancer immunotherapy.
Keywords: Cancer, Myeloid cells, TGF-β1, Immunotherapy, PD-1
INTRODUCTION
The three transforming growth factor-βs (TGF-β) have many important biological roles, and TGF-β1 is especially important in regulating wound-healing, immune responses, and tumor biology. Mice deficient in TGF-β1 die within a few weeks after birth from uncontrolled lymphocyte proliferation and autoimmunity. TGF-β inhibits CD8+ killer T cells and CD4+ Th1 and Th2 cells but stimulates regulatory T cells (Tregs). TGF-β also inhibits killing by natural killer (NK) and NKT cells and inhibits migration and expression of co-receptors and cytokine effectors by dendritic cells (DCs). Thus, TGF-β polarizes immune infiltrating cells in tumor microenvironments (TME) towards non-responsiveness (1,2).
TGF-β expression correlates with poor prognosis for cancer patients and has long been considered an important therapeutic target (3). TGF-β has emerged as an important tumor driver associated with anti-PD-1/PD-L1 resistance in metastatic melanoma (4), and inhibition of TGF-β enhances immune-checkpoint blockade efficacy in multiple preclinical tumor models (5–7). Myeloid cells in the tumor microenvironment (TME) play an important role in tumor progression (8–11). High infiltration of immune-suppressive myeloid cells correlates with poor prognosis and resistance to checkpoint blockade therapy. These cells include Mac-1+F4/80+ tumor-associated macrophages (TAMs), Gr-1+Mac-1+ myeloid-derived suppressor cells (MDSCs), and Mac-1+CD11c+ DCs, which infiltrate the TME in tumor-bearing mice, as well as in cancer patients. Tumor-associated myeloid cells are the predominant sources of TGF-β1 in the TME (2) and are important in tumor immune-evasion (12). Hematopoietic cells preferentially express TGF-β1, and selective TGF-β1 inhibition is sufficient to overcome resistance to checkpoint blockade therapy (5,13–15).
Here, we tested whether TGF-β1 associated with a specific milieu molecule, leucine-rich repeat-containing glycoprotein 33 (LRRC33) (16), was important in tumor biology. Early in intracellular biosynthesis, the large, N-terminal pro-domain of TGF-β co-associates noncovalently and through disulfide bonds with milieu molecules. Besides LRRC33, milieu molecules include GARP (LRRC32) and latent TGF-β binding proteins (LTBP) 1, 3, and 4 (17). Before secretion of milieu molecule-TGF-β complexes, furin cleaves between the TGF-β pro- and growth factor domains. Milieu molecule-TGF-β complexes remain latent in the environment, where the milieu molecule anchors them on the cell surface or in the extracellular matrix until activation releases the growth factor from the complex. TGF-β1 is activated by integrins αVβ6 and αVβ8, which bind to an RGDLXXI/L motif in their prodomains and release the TGF-β growth factor from latency (17,18). LRRC33 is 34% identical in amino acid sequence with its paralogue GARP, and both have transmembrane domains that localize them to cell surfaces. LRRC33 is expressed on myeloid cells, especially on monocytic cells including microglia and macrophages, that also express TGF-β1, and hence make LRRC33-TGF-β1 complexes. This complex appears to be the only type of TGF-β1 complex in macrophages and microglia, as demonstrated in Lrrc33−/− mice, where these myeloid cells are deficient not only in surface expression of LRRC33, but also in cell surface expression and activation of TGF-β1 (16).
In this study, we preferred to use the original gene name Lrrc33, which is functionally agnostic. Several different functions have been ascribed to this gene, including acting as a milieu molecule for TGF-β1 (16), negatively regulating Toll-like receptors (19), and acting as a negative regulator of reactive oxygen species (NRROS)(20). This gene was renamed from LRRC33 after a request to the Human Genome Organization (HUGO) Gene Nomenclature Committee. The name for the protein remains LRRC33, and UniProt changed its longer name to “transforming growth factor beta activator LRRC33” after the demonstration that LRRC33 is a milieu molecule required for expression and activation of TGF-β1 (16). However, the characterization of LRRC33 as a negative regulator of reactive oxygen species has been questioned (16). In brief, the effects on macrophage reactive oxygen species (20) and innate immune function in Lrrc33−/− mice (19) are readily explained by the findings that TGF-β is a potent negative regulator of reactive oxygen species generation by macrophages (21) and suppresses innate immune function (1), respectively. The striking phenotype of ascending paraparesis and death by five months in Lrrc33−/− mice was not revealed in the original report (20); however, a subsequent report by the same group shows that mice deficient in both Lrrc33 and Cybb, an essential component of oxidases, have the same neurological defects as Lrrc33-deficient mice (22). Thus, reactive oxygen species are unrelated to the only overt phenotype of Lrrc33-deficient mice. In contrast, the phenotype of Lrrc33-deficient mice is similar to TGF-β1-deficient mice carrying a TGF-β1 transgene in T lymphocytes to protect against autoimmunity (23) and to mice deficient in the TGF-β1-activating integrin αVβ8 (24,25). Patients deficient in LRRC33 (reported as NRROS following requirements of human genetics journals to use the HUGO gene name) are reported to have severe infantile-onset neurodegeneration, with similarities to human loss of TGF-β1 function and to the phenotype of Lrrc33 deficiency in mice (26,27). Our previous findings that LRRC33 associates with TGF-β1 were independently confirmed. Complexes with wild-type LRRC33 and TGF-β1 are expressed on the cell surface, whereas LRRC33 with C-terminal missense mutations can complex intracellularly with TGF-β1 and cannot be expressed on the cell surface (26). The authors of the reports on human deficiency have joined in requesting that HUGO change the gene name back to LRRC33.
Here, we found that LRRC33 associates with TGF-β1 and not TGF-β2 or TGF-β3. Genetic ablation of Lrrc33 significantly decreased tumor growth and metastasis in several syngeneic tumor models and enhanced anti-PD-1 treatment efficacy. We also defined the cellular mechanisms underlying the role of LRRC33/TGF-β1 complexes in downregulating immunity in the TME. Our studies reveal a more selective, cell type-specific cancer target for blocking TGF-β1 in the TME.
MATERIALS AND METHODS
Study approval and mice
All animal experiments were approved by the Institutional Animal Care and Use Committees (IACUC) at Harvard Medical School and Boston Children’s Hospital. All Lrrc33+/+ (WT), Lrrc33+/− (heterozygous), and Lrrc33−/− (KO) mice were on the C57BL6/NJ (CD45.2+) background, had been further backcrossed onto C57BL6/NJ, and were genotyped and maintained as described (16). Mice with no noticeable neurological symptoms (6–8 weeks old) were used for all experiments. All KO, heterozygous, and WT mice were littermates. B6 CD45.1 (Strain #002014) and C57BL/6J Rag1−/− mice (Strain #002216) were from the Jackson laboratory. Lrrc33+/− mice were bred with Rag1−/− mice to obtain F1 mice, among which Lrrc33 heterozygote-genotyped mice were used for breeding. Lrrc33−/− offspring which lacked CD3+ T cells in tail vein blood, as shown by flow cytometry, were Rag1−/− and were used as Lrrc33−/− Rag1−/− mice; Rag1−/− mice used in experiments were littermates that were Lrrc33+/+ and lacked CD3+ T cells.
Cell lines and culture
B16.F10 cells and LLC1 cells were purchased from the ATCC in 2017. MC38 cells and CAGA cells (28) were gifts from the Arlene Sharpe lab at Harvard Medical School and Thomas Thompson lab at University of Cincinnati, respectively. All cell lines were frozen in liquid nitrogen. For experiments, cell aliquots were thawed, and used for no more than 2–3 weeks, or about 5–10 passages. B16.F10 cells produced black tumors and metastases and were resistant to PD-1 antibody in vivo as reported, whereas MC38 cells were susceptible to PD-1 antibody in vivo as reported. CAGA cells produced luminescence after TGF-β stimulation as expected. Cells were cultured in DMEM supplemented with 10% FBS (Sigma-Aldrich), 4 mM L-glutamine (Corning), and either 1% penicillin/streptomycin (Corning) or for CAGA cells 100 μg/mL of G418 (KSE Scientific). Expi293F cells were cultured in suspension in Expi293 Expression Medium (Life Technologies) with no antibiotics. Full length αV-GFP, β6, Flag-tagged proTGF-β1, proTGF-β2, proTGF-β3 and HA-tagged LRRC33 or GARP were transiently transfected into Expi293F cells using jetPRIME (Polyplus) according to the manufacturer’s instructions. Flow cytometry was used to detect αVβ6 expression via GFP and tagged proteins by staining with anti-DYKDDDDK Flag (clone L5) and anti-HA (clone 12CA5). All cells were cultured at 37°C in a humidified 5% CO2 atmosphere. All cell lines were tested every three months and found free from mycoplasma contamination. Transiently transfected cells were used immediately 48 hours after.
Immune cell subsets in naïve WT and Lrrc33−/− mice
Spleen, thymus, and lung tissues were taken from 7 week–old WT and Lrrc33−/− mice. The flat end of the syringe plunger was used to mince the spleen or thymus in a 70-μm cell strainer by crushing the tissues in gentle circular motions to release the splenocytes or thymocytes. For lung extravascular DC analysis, mice were injected i.v. with 3 μg of mouse CD45.2 antibody to label intravascular leukocytes, and mice were euthanized 3 minutes post-injection. Lungs were harvested and digested in 1 mg/mL of Liberase TM (Roche) and 20 μg/mL of DNaseI (Roche) for 20 minutes at 37°C; ACK (ammonium-chloride-potassium) Lysing Buffer (Thermo Fisher) was used to lyse red blood cells.
Mouse syngeneic tumor models and pulmonary metastasis
Tumor cells (1×105 B16.F10, 3×105 MC38, or 1×104 LLC1 cells) in 100 μL PBS were subcutaneously (s.c.) injected into the shaved right flank of mice (C57BL6/NJ Lrrc33+/+, Lrrc33+/− Lrrc33−/−, Rag1−/−, and Lrrc33−/−Rag1−/−). Once palpable, tumors were measured every 2–3 days (long diameter and short diameter) with a caliper. KO mice were housed in the same cages as control WT or heterozygote mice, which supported better survival of KO mice and also facilitated blinded tumor size measurements (randomly picking up each mouse and reading/recording its tumor size and animal ID number without knowing its genotype). WT recipient mice that received WT or KO bone marrow were housed in separate cages and therefore were not studied blinded. Tumor volume was determined using the formula for a solid ellipsoid: L × W2/2, where L is the longer diameter and W is the shorter diameter. Mice were sacrificed when tumors were ulcerated, bleeding, or > 2 cm. For antibody treatments, mice were given 10 mg/kg antibody via intraperitoneal injection every 3 days, starting when tumors became palpable, with an average tumor volume of 30 mm3. Dosing schedule is shown in figures. The following antibodies were used: anti-PD-1 (BioXCell clone RMP1–14; cat. BE0146), 21D1 (a gift from Scholar Rock, Inc.) (29), IgG control (BioXCell Rat IgG2a), or human IgG4 (recombinant IgG expressed in Expi293F cells and purified from supernatant by protein G and Sephadex S200 chromatography). For treatment with PD-1 or 21D1 antibodies, we chose mice with similar tumor sizes for antibody and isotype control treatments. A larger number of WT and KO mice were injected with tumors than needed for treatment studies. On the first day of treatment, tumors were measured and ranked by size. A sufficient number of mouse pairs with tumors of similar size were chosen and one from each pair was randomly assigned to the treatment group and the other in the pair to the isotype control group.
For assessment of pulmonary metastasis, 3×105 B16.F10 cells were transferred intravenously via tail vein injection. After 14 days, lungs were perfused using PBS and fixed overnight in 4% paraformaldehyde (PFA) solution. Fixed tissues were sent to the Rodent Pathology Core at Harvard Medical School for paraffin embedding, sectioning, and hematoxylin and eosin histologic staining. The number of metastatic foci was determined using an Olympus BX51 microscope on sections taken every 100 μM throughout the whole lung for at least 5 animals per group.
Whole bone marrow transplantation
Congenic mouse transplant recipients (10-week-old CD45.1+ C57BL6/NJ) were lethally irradiated (950 rad; RS-2000, Rad Source) and tail-vein injected with 8×106 freshly harvested whole bone marrow cells from femurs of WT or Lrrc33−/− donor mice (10-week-old CD45.2+ C57BL6/NJ). Transplant recipients were given water containing 30 mg/L neomycin sulfate, 30 mg/L kanamycin sulfate and 50 mg/L gentamycin sulfate for 4 weeks. MC38 tumor cells were injected 10 weeks post-transplant when CD45.2+ bone marrow chimerism averaged over 90% in peripheral blood, showing successful transplantation.
Cell isolation
B16F10 or MC38 tumors were excised and weighed 14 days after s.c. tumor cell injection, cut into 2 mm sized pieces, and enzymatically dissociated in RPMI-1640 medium with 5% FBS, 0.1% 2-mercaptoethanol, 1 mg/mL collagenase IV (Roche), 20 μg/mL Dnase I (Roche) at 37 °C for 30 minutes during rotation at 180 rpm. Cell mixtures were pipetted up and down to further dissociate cells and then filtered through a 70-μm cell strainer to obtain single-cell suspensions. To isolate tumor-infiltrating leukocytes, cells were centrifuged, and pellets were re-suspended in 40% Percoll (GE Healthcare) and slowly layered on 70% Percoll for centrifugation at 2200 rpm for 30 minutes at room temperature. Leukocytes from the 40% to 70% Percoll interface and other cells from the pellet were washed twice with PBS.
For myeloid cell purification, Petri dishes (100 mm × 15mm, Falcon REF351029, not tissue culture-treated) were pre-coated with 5 μg/mL of specific antibodies in 0.05 M pH 9.0 Tris buffer at 4°C overnight and then washed with PBS before adding 1×107 cells in RPMI1640 medium with 10% FBS. After attachment at 37°C for 2 hours in a 5% CO2 incubator, dishes were washed four times with PBS, adherent cells were detached with 0.25% trypsin/0. 5mM EDTA (Corning) in PBS, diluted in RPMI1640/10% FBS and centrifuged. Cells were either resuspended in RPMI1640/10% FBS for further purification, in FreeStyle medium for TGF-β assays, or in TRIzol (Invitrogen) for RNA isolation.
Using the above protocol, whole bone marrow cells or tumor leukocytes were panned with anti-CD3 (clone 17A2, Biolegend) and anti-CD19 (clone 6D5, Biolegend) to obtain T and B cells for RNA isolation. Non-adherent cells were then added to anti-Mac-1-coated (clone M1/70, Biolegend) plates to enrich myeloid cells for RNA isolation or for further purification. To further purify myeloid populations, cells were resuspended as above and added to dishes pre-coated with antibodies against F4/80 (clone BM8, Biolegend), CD11c (clone N418, Biolegend), and Gr-1 (clone RB6–8C5, Biolegend) to enrich macrophages, DCs, and MDSCs or granulocytes, respectively.
Flow cytometry
Flow cytometry on tumor samples from models was not performed in a blinded manner. For cell surface staining, cells were pre-incubated with 5 μg/mL of anti-mouse CD16/32 Fc blocker (BioLegend, catalog # 101319) in FACS buffer (2% FBS/PBS) for 15 minutes on ice and stained with 2 μg/mL of fluorophore-conjugated antibodies at 4°C for another 30 minutes. Antibodies to surface markers were all from Biolegend: Alexa Fluor 647 anti-mouse CD45.2 (clone 104), Pacific Blue™ anti-mouse CD45 (clone 30-F11), Alexa Fluor 700 anti-mouse CD45.2 (clone 104), FITC-anti-mouse CD3 (clone 17A2), PE-anti-mouse CD4 (clone GK1.5), PE-anti-mouse CD8a (clone 53–6.7), APC-anti-mouse CD8a (clone 53–6.7), APC-anti-mouse NK1.1(clone pk136), PE-Cy7- anti-mouse Mac-1 (clone M1/70), PE-anti-mouse F4/80 (clone BM8), PE-anti-mouse Gr1 (clone RB6–8C5), APC- anti-mouse Ly-6C (clone HK1.4), FITC-anti-mouse Ly6G (clone 1A8), Brilliant Violet 421 anti-mouse CD103 (clone 2E7), APC anti-mouse CD103 (clone 2E7), PE-anti-mouse CD11c (clone HL3), APC-anti-mouse MHC-II (clone AF6–120.1), FITC anti-mouse MHC-II (clone AF6–120.1), PE-anti-mouse CD326 (EpCAM) (clone caa7–9G8), anti-alpha smooth muscle actin (α-SMA, clone 1A4), and Brilliant Violet 421 anti-mouse TGF-β1 prodomain (clone TW7–16B4)(30).
For intracellular staining, cells were first treated and stained for cell surface markers as described above, and then fixed and permeabilized with the BD Cytofix/Cytoperm kit and stained according to the manufacturer’s instructions. For intracellular TNFα and IFNγ, prior to cell surface staining, cells were cultured in RPMI 1640 medium supplemented with 10% FBS and BD leukocyte activation cocktail following manufacturer’s instruction at 37°C for 4 hours. To detect Foxp3, the eBioscience Foxp3/Transcription Factor Staining Buffer Set was used following the manufacturer’s instructions. Antibodies were from Biolegend: PE anti-human/mouse granzyme B (clone QA18A28), APC anti-mouse TNFα (clone MP6-XT22), Brilliant Violet 421 rat anti-mouse IFNγ (clone XMG1.2), Brilliant Violet 421 anti-mouse Foxp3 (clone MF-14), and PE-anti-mouse CD206 (clone C068C2). For phosphorylated (p)SMAD staining, cells were fixed and permeabilized using BD Phosflow Lyse/Fix Buffer and BD Phosflow Perm Buffer III and stained with BD Phosflow Alexa Fluor 647 anti-Smad2 (pS465/pS467)/Smad3 (pS423/pS425) (clone O72–670) (BD Biosciences, San Jose, CA) following the manufacturer’s instructions. Samples were run on a BD Canto II, and data analysis was performed using FlowJo.
TGF-β activation assay
CAGA cells (1.5×104/ well) seeded in 96-well flat plates were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin in a 5% CO2 incubator at 37°C. After 4 hours, plates were inverted, flicked, and wicked to remove excess medium. Purified myeloid, B16.F10, or MC38 cells (1 ×105 unless otherwise indicated) were co-cultured with mock or αVβ6 transiently transfected Expi293F cells (2×104) in a total of 100 μL Freestyle medium; in some cases, medium included 21D1 anti-TGF-β or human IgG4 isotype control. For standard curves, recombinant human TGF-β1 growth factor (#T7039, Sigma) at 20 ng/mL and 11 2-fold dilutions thereof in Freestyle medium was added to CAGA cells. After 24 hours, supernatants were removed, and 40 μl non-diluted cell lysate was assayed for luciferase activity using Luciferase Assay System (Promega cat# E1501) according to the manufacturer’s instructions. Plates were read on a Biotek Synergy H1. TGF-β concentrations were calculated in PRISM9 from the luciferase activity using a standard curve.
Quantitative (q)RT-PCR
RNA from B16F10 and MC38 cell lines, CD45− and CD45+ cells, lymphocytes, myeloid cells, and tumor cells isolated from tumors was extracted using the Direct-zol RNA mini prep kit (Zymo Research) and 1 ug of RNA was reverse-transcribed using SuperScript II (Life Technologies) with oligo(dT18) in 10 μl total volume according to the manufacturer’s instruction. 1 μl of 5 × diluted cDNA product was used for template in qRT-PCR, which was performed using the Power SYBR Green PCR Master Mix (Invitrogen) on a CFX-Connect detection system (Bio-Rad, Hercules, CA) with Bio-Rad CFX Manager 3.1 software. Duplicate wells were performed for each sample and mRNA expression was determined relative to Gapdh (ΔCt = Ct gene of interest - Ct gapdh) with Ct representing cycle threshold. Primers used to murine genes were as follows: Lrrc33 forward ACTGCAGCTTCCCAA GGA, Lrrc33 reverse TGGGTACCGAAGCAAGGT; Tgfb1 forward CTCCCGTGGCTTCTAGTGC, Tgfb1 reverse GCCTTAGTTTGGACAGGATCTG; Tgfb2 forward CTTCGACGTGACAGACGCT, Tgfb2 reverse GCAGGGGCAGTGTAAACTTATT; Tgfb3 forward CCTGGCCCTGCTGAACTTG, Tgfb3 reverse TTGATGTGGCCGAAGTCCAAC; Gapdh forward GTTGTCTCCTGCGACTTCA, Gapdh reverse GGTGGTCCAGGGTTTCTTA.
Flow cytometry of Expi293F transfectants
Flag-tagged TGF-βs and HA-tagged LRRC33 or GARP were transfected into Expi293F cells as described above. After 48 hours, cells were incubated with the primary antibodies in FACS buffer (PBS with 1% BSA) on ice for 30 minutes at 2 μg/mL: APC anti-DYKDDDDK tag (clone L5, Biolegend) and mouse anti-HA which was purified by protein G and Sephadex S200 chromatograpy from supernatant of 12CA5 hybridoma (31). After washing, the cells were incubated with Alexa 647-goat anti-mouse IgG (H+L) (Invitrogen) secondary antibody for 30 minutes, washed, and analyzed on a FACS Canto II (BD Biosciences) and analyzed with FlowJo.
Immunoprecipitation (IP) and Western blot (WB)
Flag-tagged TGF-βs and HA-tagged LRRC33 or GARP-Expi293F transfectants (~1×107/ml) were lysed at 4°C for 30 minutes in 500 μl of 150 mM NaCl, 50 mM Tris-HCL pH8.0, 1% Triton-x100 (Sigma-Aldrich), 0.1% SDS (Sigma-Aldrich), 0.5% sodium deoxycholate, 1mM EDTA, and protease inhibitor cocktail (Roche) and centrifuged at 14,000 rpm at 4°C for 5 minutes. For IP, cell lysates (~400 μL) were incubated with 2 μg/mL of anti-Flag M2 (Sigma, F1804) overnight at 4°C with rotation and then 10 μL of protein G Sepharose beads (50% suspension in PBS) (GE Healthcare) was added for 2 hours at 4°C with rotation. For WBs, 12.5 μl of 5X SDS sample buffer was added to 50 μl of cell lysate or 60 μl of 1X SDS sample buffer was added to pelleted, washed beads from IP. Samples were heated at 95°C in sample buffer containing 5 mM N-ethylmaleimide (Calbiochem) or 5% β-mercaptoethanol and 12 μl per lane was subjected to non-reducing and reducing SDS-polyacrylamide gel electrophoresis (PAGE), respectively. Proteins were transferred to PVDF membrane (Bio-Rad) using Trans-Blot Turbo System (Bio-RAD) and probed with 2 μg/mL of specific antibodies (anti-Flag M2, Sigma F1804; anti-HA, 12CA5), and detected with 1 μg/mL of HRP-conjugated secondary antibodies (Abcam, ab6789). Blots were developed using the SuperSignal West Pico Plus Chemiluminescent Substrate (Thermo Fisher) and were imaged using an iBright FL1000 imager (Invitrogen).
Statistical analyses
For flow cytometry data, statistical analyses were performed using GraphPad Prism 9 software, and statistical significance was determined by unpaired Student’s t-test. *p<0.05, **p<0.01, ***p<0.001.
RESULTS
TGF-β1 specifically associates with LRRC33
LRRC33 can form non-covalent complexes, as well as covalent complexes, through disulfide bonds with TGF-β1 (16), but complex formation with other TGF-βs has not been tested. GARP is a paralog of LRRC33 with 34% sequence identity, associates with TGF-β1, and has a role in tumor immunity (32), but like LRRC33, whether it can associate with TGF-β2 and TGF-β3 is uncharacterized. To better understand the roles of both LRRC33 and GARP in modulating tumor immune responses, we examined their specificity for different TGF-β isoforms. Flag-tagged TGF-βs and HA-tagged LRRC33 or GARP were expressed together or separately in Expi293F cells. Expression of TGF-β1 on the cell surface was increased by co-expression with either LRRC33 or GARP (Fig. 1A). In contrast, LRRC33 did not promote TGF-β2 or TGF-β3 expression on the cell surface, whereas GARP augmented TGF-β2 and TGF-β3 expression on the cell surface (Fig. 1A). Furthermore, LRRC33 expression on the cell surface was increased by co-expression with TGF-β1, but not by TGF-β2 or TGF-β3 (Fig. 1A).
Figure 1. LRRC33 is specific for TGF-β1.

(A) Expi293F cells were transfected with the indicated plasmids. 48 hours later, cell surface expression of HA-tagged LRRC33 or GARP or FLAG-tagged TGF-β1, TGF-β2, or TGF-β3 pro-forms was detected by flow cytometry. Histograms are representative of triplicate transfectants in the same experiment. Inset numbers show mean±SEM of mean fluorescence intensity. (B, C) Immunoprecipitation followed by Western blotting. Expi293F cells were transfected with plasmids encoding LRRC33 (L), TGF-β1 (T1), TGF-β2 (T2), or TGF-β3 (T3) pro-forms as in (A). Cell lysates were subjected to (B) reducing or (C) non-reducing SDS-PAGE either directly or after immunoprecipitation (IP) and then Western blotted as shown under images. (B, C) Blank spaces: separate lanes from the same gel that were imaged together and then moved. Results are representative of two independent experiments.
We next tested the association of LRRC33 and TGF-βs by immunoprecipitation (IP) and Western blotting (WB). TGF-β1, TGF-β2, TGF-β3, and LRRC33 were detected in Expi293F transfectant cell lysates when TGF-β and LRRC33 were expressed individually or together (Fig. 1B, upper and middle). HA-tagged LRRC33 interacted with all three FLAG-tagged TGF-βs under reducing conditions when TGF-βs were immunoprecipitated with LRRC33 and blotted with anti-HA antibody (Fig. 1B, lower), although surface expression of LRRC33 was augmented only by TGF-β1, and LRRC33 selectively augmented surface expression of TGF-β1 (Fig. 1A). We, therefore, used a more stringent test and examined ability of LRRC33 to form disulfide bonds with the TGF-βs. Non-reducing SDS-PAGE showed that only TGF-β1 formed a disulfide-linked complex of about 250 kDa with LRRC33 (Fig. 1C). Thus, among the three TGF-βs, only TGF-β1/LRRC33 complexes were capable of forming milieu-molecule/TGF-β disulfide bonds and being transported to the cell surface. Taken together, these results suggest that TGF-β1, and not TGF-β2 and β3, form biologically relevant complexes with LRRC33, whereas all three TGF-βs form complexes with GARP.
Decreased tumor growth and absence of myeloid cell TGF-β1 expression in Lrrc33−/− mice
We tested three different syngeneic tumor lines for growth in Lrrc33−/− mice after s.c. injection into the flank (Fig. 2). B16.F10 melanoma tumors grew more aggressively in WT mice than in Lrrc33−/− mice. Tumor volume was significantly less (p=0.04), and the mass of explanted tumors was 5-fold lower in Lrrc33−/− mice (Fig. 2A–B). MC38 colon carcinoma cells also grew significantly more slowly and had significantly smaller (2.3-fold) masses at explant in Lrrc33−/− mice (Fig. 2C–D). Growth of B16.F10 melanoma and MC38 carcinoma tumors in heterozygous Lrrc33+/− mice was intermediate between growth in WT and homozygous Lrrc33−/− mice at all time points, but was not significantly different than in WT mice (Supplementary Fig. S1). Lewis lung carcinoma (LLC1) tumor cells also grew significantly more slowly and had smaller final masses in Lrrc33−/− mice, although the magnitude of the differences was less (Fig. 2E–F). Thus, all three tested tumor models showed significantly reduced growth in Lrrc33−/− mice.
Figure 2. LRRC33 deficiency decreases syngeneic tumor growth and metastasis.

(A-F) Tumor growth after (A, B) 1×105 B16.F10, (C, D) 3×105 MC38, or (E, F) 1×104 LLC1 cells were subcutaneously injected into the right flank of WT and Lrrc33−/− (knockout, KO) mice. Dashed and solid lines: individual animals and group average, respectively. P-values are for the last time point, as determined by unpaired Student’s t-test. (G-J) B16.F10 melanoma lung metastasis. B16.F10 cells (3×105) were intravenously injected into Lrrc33+/+, Lrrc33+/−, or Lrrc33−/− mice. Metastases were counted 14 days later. (G, H) Lung metastases were counted by gross observation after perfusion. (I, J) H&E staining (10X magnification) to quantitate metastatic lesions as the average number per 6 whole-lung sections (5 mice/group). Data show mean±SEM. *p<0.05, **p<0.01, ***p<0.001, as determined by unpaired Student’s t-test. Results are representative of two or more independent experiments.
Given the importance of TGF-β signaling in myeloid cells for tumor metastasis (33), we also tested a B16.F10 melanoma lung metastasis model. Fourteen days after intravenous injection (i.v.) of tumor cells, Lrrc33−/− mice displayed 56% fewer pulmonary metastatic nodules on the lung surface than WT mice (Fig. 2G–H). More definitive quantitation using hematoxylin and eosin staining of lung tissue sections showed an 82% decrease in metastasis in Lrrc33−/− mice compared to WT mice (Fig. 2I–J). Heterozygous mice also showed significantly decreased lung metastasis (Fig. 2G–J). Overall, our observations showed that loss of LRRC33 slowed tumor growth in three cancer types that vary in immunogenicity, including highly immune-infiltrated MC38 tumors and less-infiltrated B16.F10 and LLC1 tumors, and significantly reduced melanoma lung metastasis.
We next investigated whether loss of LRRC33 abolished pro-TGF-β1 expression on tumor-associated myeloid cells. TGF-β1 prodomain staining and flow cytometry showed that Mac-1+F4/80+ macrophages, Mac-1+CD11c+ DCs, and Mac-1+Gr-1+ MDSCs isolated from Lrrc33−/− B16.F10 tumors lacked cell surface pro-TGFβ1, although staining of MDSCs was weak (Fig. 3A). Similar results were obtained with myeloid cells isolated from MC38 tumors, although staining of MDSCs was weak (Fig. 3B). After permeabilization, pro-TGF-β1 was detected, suggesting that pro-TGF-β1 was still present inside cells with LRRC33 deficiency (Fig. 3B), in agreement with what has been observed in peritoneal macrophages (16) and in transfectants co-expressing human patient LRRC33 mutants and TGF-β1 (26).
Figure 3. Lrrc33−/− tumor-associated myeloid cells lack cell surface pro-TGF-β1 expression and activation.

(A, B) Pro-TGF-β1 expression was measured 14 days after s.c. (A) B16.F10 and (B) MC38 cell injection. Tumors were harvested and dissociated to obtain single-cell suspensions, and cells were analyzed for the indicated markers. Surface and intracellular pro-TGF-β1 was assessed for the indicated immune cell populations by flow cytometry. Gray filled histograms and black curves show control antibody and anti-pro-TGF-β1 staining, respectively. Flow cytometry results were similar for cells from 3 mice/group, and one representative is shown for each. (C) Mac-1+ myeloid cells from (1) whole bone marrow were purified by (2) antibody panning and (3) further panned to isolate DCs, (4) macrophages, and (5) granulocytes. (D, E) Myeloid cell populations were purified [as in (C)] from (D) bone marrow or (E) MC38 tumor and co-cultured with CAGA cells in the presence of mock or αVβ6-transfected Expi293F cells and assayed for TGF-β production. Data show mean±SEM of quadruplicate samples. *p<0.05, **p<0.01, ***p<0.001, as determined by unpaired Student’s t-test. Results in A-D are representative of 2–3 independent experiments.
Loss of cell surface TGF-β1 correlates with loss of activatable TGF-β1 in tumor-associated myeloid cells
Myeloid cell populations were purified from MC38 tumors harvested from WT and Lrrc33−/− mice by negative and positive antibody panning. Cell purity was verified by flow cytometry (Fig. 3C). Cells were co-cultured with CAGA cells, a TGF-β reporter cell line, in the presence or absence of integrin αVβ6-transfected Expi293F cells. Total Mac-1+ myeloid cells, as well as macrophages, DCs, and MDSCs, from tumors in WT mice produced activated TGF-β1, whereas this was not seen with the same cell types isolated from MC38 tumors in Lrrc33−/− mice (Fig. 3D). Activation was dependent on integrin αVβ6. Total Mac-1+ myeloid cells, macrophages, and DCs isolated from Lrrc33−/− bone marrow also lacked activatable TGF-β (Fig. 3E). Comparison of pro-TGF-β1 expression and activatable TGF-β on bone marrow and tumor-infiltrating myeloid cells showed that cells from heterozygous Lrrc33+/− mice were consistently intermediate between cells from WT and KO mice, although not significantly different compared to cells from WT mice (Supplementary Fig. S2).
Loss of LRRC33 biases tumor-infiltrating immune cells toward a more activated phenotype
To investigate the cellular mechanisms connecting loss of LRRC33-dependent TGF-β1 activation to slower tumor growth, we phenotyped MC38 tumor-infiltrating leukocytes 14 days after s.c. tumor cell injection. Although CD45+ immune cell infiltration did not significantly change, CD3+ and CD8+ T cells, as well as NK1.1+ NK cells were significantly increased in Lrrc33−/− compared to WT mice both as frequency among CD45+ cells (Fig. 4A–B,) and per mg of tumor (Supplementary Fig. S3). In contrast, LRRC33 loss did not significantly affect CD4+ T-cell frequency or number. Although Tregs express GARP/TGF-β1 complexes and are regulated by TGF-β1 (13), we did not see a significant difference of tumor Treg infiltration between tumors from WT and Lrrc33−/− mice (Fig. 4C–D). We also evaluated myeloid cell subsets. The proportions of Mac-1+CD11c+ DCs and Mac-1+F4/80+ macrophages were not significantly different between Lrrc33−/− and WT mice (Fig. 4B). Tumor-associated macrophages (TAMs) were not skewed significantly into MHCII+CD206− (M1-like) or MHCII–CD206+ (M2-like) phenotypes (Fig. 4E–F). However, the proportion of CD11b+Gr-1+ MDSCs was significantly decreased in Lrrc33−/− mice (Fig. 4B).
Figure 4. LRRC33 deficiency in hematopoietic cells augments both innate and adaptive host immune responses to MC38 tumors.

(A-J) Phenotype of tumor-filtrating immune cells 14 days after s.c. MC38 injection in WT or Lrrc33 knockout (KO) mice. Cells from tumors were stained for the indicated markers and subjected to flow cytometry from five mice/group (one sample of IFNγ-stained cells was lost in (H)). Mean±SEM is shown. P values were determined by unpaired Student’s t-test. Representative flow cytometry profiles for one sample from WT and KO mice are shown in (D, G, I, and J). Overall results are representative of two independent experiments. GzmB: granzyme B. (K) Effect of Rag1 and Lrrc33 deficiency on MC38 tumor growth. Data show mean±SEM for n=6–7 mice. (L, M) Tumor growth in WT mice transplanted with WT or Lrrc33−/− bone marrow cells. (L) Whole bone marrow transplantation chimerism in individual mice. (M) Growth of MC38 tumors in transplant recipients. Data show mean±SEM for n=8 mice/group. In (K, M), P-values are for the last time point, unpaired Student’s t-test: *p<0.05, **p<0.01, ***p<0.001.
We next asked whether the increase in CD8+ T cells and NK cells in Lrrc33−/− mice was accompanied by enhancement of functional activity. CD8+ T cells showed significant increases in the proportion of TNFα+, IFNγ+, and granzyme B+ cells in tumors from Lrrc33−/− mice, including a ~2-fold increase in TNFα-secreting cells (Fig. 4G–I). Tumor-infiltrating granzyme B+ NK cells were also significantly increased in Lrrc33−/− mice (Fig. 4G), along with an increase in the proportion of tumor-infiltrating IFNγ+ NK cells, which increased from 26.7±14.0% in WT to 62.6±3.1% in Lrrc33−/− mice (Fig. 4J).
Experiments in a Rag1−/− background showed that tumors grew significantly faster in Lrrc33−/−Rag1−/− mice than in Lrrc33−/− mice, demonstrating that deficiency of LRRC33 augmented specific immune responses (Fig. 4K). This increase in growth rate was comparable to the faster growth in Rag1−/− mice compared to WT mice. MC38 tumors were also significantly smaller in Lrrc33−/−Rag1−/− mice than in Rag1−/− mice, indicating that deficiency of LRRC33 also augmented innate immunity. Immunophenotyping showing increased TNFα+, IFNγ+, and granzyme B+ CD8+ T cells is consistent with increased adaptive immunity, whereas higher infiltration of IFNγ+ and granzyme B+ NK cells are likely to contribute to the higher innate immunity in Lrrc33−/− mice (Fig. 4G–J). To test the contribution of hematopoietic cells to the effects on tumor growth, we transferred CD45.2+ WT or Lrrc33−/− whole bone marrow cells to lethally irradiated CD45.1+ WT mice. Chimerism was greater than 95% at 10 weeks (Fig. 4L). Chimeric mice that received Lrrc33−/− bone marrow exhibited significantly slower MC38 tumor growth than mice that received WT bone marrow (Fig. 4M).
Because immune responses vary among tumors, we extended examination of tumor-infiltrating leukocytes to the s.c. B16.F10 melanoma model (Fig. 5A–B, Supplementary Fig. S4). CD8+ T cells were significantly increased in tumor infiltrates in Lrrc33−/− compared to WT mice. CD8+ T cells also showed significant increases in the proportion of cells expressing TNFα, IFNγ, and granzyme B, ranging from 1.7 to 2.7-fold (Fig. 5C–D). In contrast to results with CD8+ T cells and results with NK cells in MC38 tumors, NK1.1+ NK cells infiltrating B16.F10 tumors showed no increase in expression of IFNγ and granzyme B (Fig. 5E). Numbers of Mac-1+CD11c+ DCs and Mac-1+F4/80+ macrophages infiltrating B16.F10 tumors showed no differences between WT and Lrrc33−/− mice (Fig. 5F). However, macrophages were skewed in phenotype. MHCII+CD206− (M1-like) TAMs were significantly increased and MHCII−CD206+ (M2-like) were significantly decreased in Lrrc33−/− mice (Fig. 5G–H). In summary, in B16.F10 melanoma, LRRC33 deficiency polarized immune cells toward a more immune reactive phenotype, as in MC38 carcinomas, yet there were also differences from MC38 among T cell, NK cell, and macrophage subpopulations in the way they were skewed.
Figure 5. Slower growth of B16.F10 tumors in Lrrc33 KO mice associates with increased infiltration of effector cells and altered macrophage phenotype.

(A-H) Phenotype of tumor-filtrating immune cells 14 days after s.c. B16.F10 injection. The indicated immune populations were measured by flow cytometry in tumors from five mice/group; mean±SEM. Representative flow cytometry profiles for one sample from WT and KO mice are shown in (D, E, H). P values were determined by unpaired Student’s t-test. Data are representative of two independent experiments.
Although immune cell populations are reported to be normal in Lrrc33−/− mice (20), we evaluated immune cell subsets to test if any of the skewing seen in tumor-infiltrating cells was pre-existing in tumor-free mice (Supplementary Fig. S5). Compared to WT mice, Lrrc33−/− mice had comparable percentages of splenic CD3+, CD4+, and CD8+ T cells, B cells, NK cells, Mac-1+F4/80+ macrophages, Mac-1+CD11c+ DCs, and Mac-1+Gr-1+ granulocytes (Supplementary Fig. S5A). Intracellular functional markers (granzyme B and IFNγ) in splenic CD8+ T cells and NK cells also showed no significant differences (Supplementary Fig. S5B–D). Lrrc33 loss did not significantly affect neither Ly6C+ monocytes nor Ly6G+ polymorphonuclear (PMN) neutrophils among splenic Mac-1+ cells (Supplementary Fig. S5E–F). Thymus CD4+CD8−, CD4−CD8−, CD4−CD8+, and CD4+CD8+ T-cell also showed no difference between WT and KO mice (Supplementary Fig. S5G–H). However, in lungs, although extravascular innate and adaptive immune cells in Lrrc33−/− mice were otherwise normal, there was a significant decrease in CD103highCD11c+CD11b−MHCII+ DCs (Supplementary Fig. S5I–J). This finding may relate to the unusual susceptibility of Lrrc33−/− mice to pneumonia and otitis media (16). Lrrc33 mRNA is selectively expressed on immune cells and tumors, and LRRC33 has only been found on immune cells (16,20,34). We therefore assessed expression in B16.F10 tumor cells. Lrrc33 mRNA was not expressed on B16.F10 tumor cells grown in vivo (Supplementary Fig. S6).
LRRC33 deficiency, TGF-β antibody treatment, and inhibition of SMAD phosphorylation
We next compared deficiency of LRRC33 to inhibition of TGF-β signaling using the 21D1 antibody, which reacts with the growth factor moiety of TGF-β1 with a Kd of <0.5 pM and with that of TGF-β3 with a Kd of 4.9 nM (29). Blocking TGF-β signaling led to significantly stronger inhibition of MC38 tumor growth in WT mice than deficiency of LRRC33 (Fig. 6A). Moreover, 21D1 antibody inhibited MC38 tumor growth to similar extents in Lrrc33−/− and WT mice. Similar results were obtained with B16.F10 tumors, with tumor growth being inhibited similarly in WT and Lrrc33−/− mice (Fig. 6B). These results suggest that TGF-β from sources other than LRRC33/TGF-β1 complexes, including complexes of LTBP and GARP with TGF-β1 or TGF-β3 from host or tumor cells, also contribute to tumor growth.
Figure 6. Blocking TGF-β1 with an antibody inhibits tumor growth and pSMAD2/3 signaling more effectively than LRRC33 deficiency.
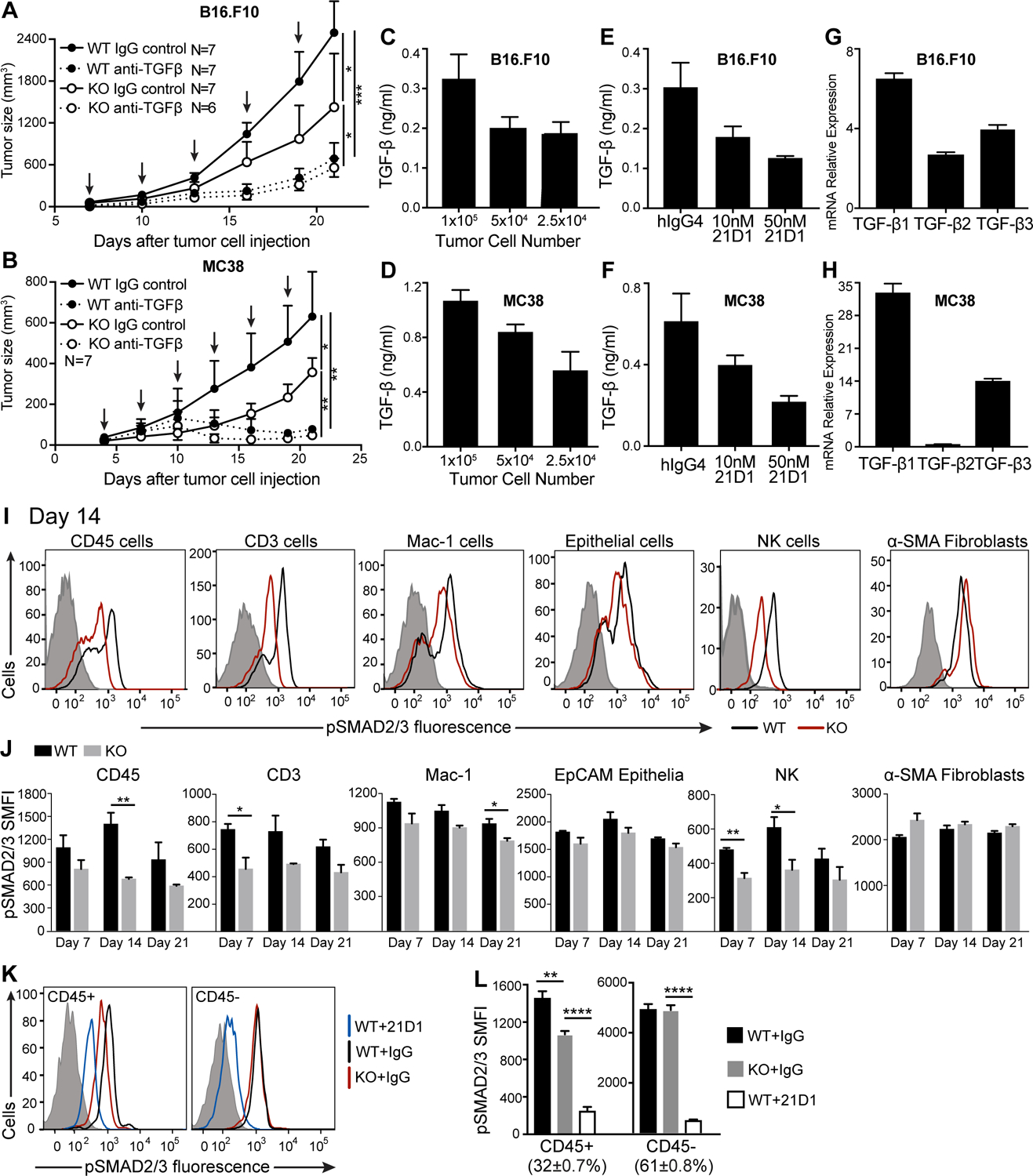
(A, B) Effect of TGF-β antibody 21D1 (specific for the TGF-β1 and TGF-β3 growth factors) on growth of (A) B16.F10 and (B) MC38 tumors in WT or Lrrc33−/− mice. Mice were treated with 10 mg/kg 21D1 antibody or isotype control on indicated days (arrows) after tumor cell s.c. injection. Data show mean±SEM for n=6–7 mice. P-values are shown for last time point, unpaired Student’s t-test. *p<0.05, **p<0.01, ***p<0.001. (C-H) TGF-β release and mRNA expression in (C, E, G) B16.F10 and (D, F, H) MC38 tumor cells. (C, D) TGF-β release from the indicated number of tumor cells measured by co-culture with CAGA cells in the presence of mock-transfected Expi293F cells for 24 hours. Data are mean±SEM of quadruplicate samples. (E, F) Effect of 21D1 or IgG4 control antibody (50 nM) on TGF-β production by tumor cells (1×105) co-cultured with CAGA cells for 24 hours. Data are mean±SEM of quadruplicate samples. (G, H) Tgfb1, Tgfb2, and Tgfb3 mRNA expression relative to Gapdh in B16.F10 and MC38 cell lines determined by qRT-PCR. Expression determined using the 2−ΔCt method is presented as mean±difference of duplicates. Data are representative of two independent experiments. (I, J) Phosphorylated (p)SMAD2/3 in immune cells and non-immune cells in MC38 tumors from WT and Lrrc33−/− mice measured by flow cytometry. (I) pSMAD2/3 expression in the indicated cell types in one representative tumor from WT and Lrrc33−/− mice on day 14. Control antibody: gray histogram, WT: black curve, KO: red curve. (J) pSMAD2/3 expression in tumors from n=3 mice/group on day 7, 14, and 21 after tumor injection (mean±SEM). SMFI: specific mean fluorescence intensity after isotype control subtraction. (K, L) pSMAD2/3 expression measured by flow cytometry in MC38 tumors seven days after treatment of WT mice with 21D1 antibody or treatment of WT or KO mice with IgG4 control antibody. (K) Representative histograms of staining with anti-pSMAD2/3 in the indicated groups. WT+21D1: blue curve; KO+IgG: red curve; WT+IgG: black curve; isotype control staining: grey shaded histogram. (L) pSMAD2/3 SMFI of CD45+ and CD45− cells in 4 mice/group, mean±SEM. The % of CD45+ and CD45- cells in tumors is also shown. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, as determined by unpaired Student’s t-test.
To test for TGF-β production by B16.F10 and MC38 cells, we co-cultured them in vitro with CAGA indicator cells. Both cell lines produced active TGF-β (Fig. 6C, D), and the majority of TGF-β was neutralized by 50 nM 21D1 antibody (Fig. 6E, F). Essentially identical results were obtained with mock and integrin αVβ6-transfected Expi293F cells. Because we used purified TGF-β1 to standardize our assays, we calculated that in our assays, B16.F10 and MC38 cells secreted about 3×10−4 and 11×10−4 pg/cell of TGF-β1 equivalents, respectively, whereas the myeloid cells in MC38 tumors secreted on average 6×10−4 pg/cell. The caveat to these calculations is that although there was a dose-response relationship between the number of tumor cells and TGF-β production, the relationship was not linear (the intermediate cell number in Fig. 6C–D was used for calculations). Quantitative real-time PCR showed high expression of TGFβ1 and less expression of TGFβ3 mRNA in B16.F10 and MC38 cells, less TGF-β2 mRNA in B16.F10 cells, and almost no TGF-β2 mRNA in MC38 cells (Fig. 6G–H).
Although TGF- β is known to directly act on adaptive and innate immune cells in the TME (1), we verified this in the MC38 tumor model using Lrrc33−/− mice by measuring intracellular phosphorylated SMAD2 and SMAD3 (pSMAD2/3). SMAD2/3 phosphorylation was decreased in MC38 tumors in Lrrc33−/− mice compared to WT in immune cells, particularly in CD3+ and NK cells (Fig. 6I–J). Decreased pSMAD2/3 was seen in tumor-infiltrating CD45+, CD3+, Mac-1+, and NK populations in Lrrc33−/− mice at all timepoints measured (days 7, 14, and 21) and reached significance in at least one timepoint for each population (Fig. 6J). There was no significant change in pSMAD2/3 in either tumor epithelial cells or fibroblasts (Fig. 6J). We also compared the effect of the 21D1 antibody to TGF-β1 and β3 on SMAD2/3 phosphorylation 7 days after beginning treatment. Treatment reduced TGF-β signaling in CD45+ immune cells and CD45− non-immune cells alike in MC38 tumors (Fig. 6K–L). In contrast, Lrrc33 deficiency was completely selective, whereby it reduced TGF-β signaling only in immune cells.
LRRC33-deficiency enhances anti-PD-1 efficacy in the B16.F10 model
B16F10 tumors are immune checkpoint therapy-resistant (35,36), in agreement with lack of a significant effect of PD-1 antibody in WT mice (Fig. 7A). However, treating Lrrc33−/− mice with PD-1 antibody significantly reduced B16.F10 tumor growth (Fig. 7A). Tumor size reduction persisted throughout the experiment and was synergistic (combined treatments reduced tumor size more than the sum of the two treatments). Contrasting results were obtained in the PD-1–sensitive MC38 colon carcinoma model (37), in which PD-1 blockade more strongly inhibited MC38 tumor growth than Lrrc33 deficiency (Fig. 7B).
Figure 7. LRRC33 loss and PD-1 blockade synergize in controlling B16.F10 tumor growth by altering the tumor immune landscape.

(A, B) Growth of (A) B16.F10 and (B) MC38 tumors injected s.c in WT and KO mice treated i.p. with 10 mg/kg of anti-PD-1 or IgG control antibody on indicated days (arrows). Data are mean±SEM for n=6 mice. P-values are for the last time point, unpaired Student’s t-test; *p<0.05, **p<0.01, ***p<0.001. Data are representative of two independent experiments. (C-G) Mice (4/group) were injected s.c. with B16.F10 cells and treated with antibodies as in (A) on days 7, 10, and 13 and sacrificed on day 15 for flow cytometric analysis of immune cell populations in tumors. Mean± SEM is shown. P-values were determined by unpaired Student’s t-test.
To provide mechanistic insights into how loss of LRRC33 sensitized B16.F10 melanoma to anti-PD-1 treatment, we examined tumor-infiltrating leukocytes by flow cytometry. PD-1 blockade in WT mice did not affect infiltration by CD45+, CD3+, CD4+, CD8+, and NK cells in tumors (Fig. 7C–E). However, the combination of PD-1 blockade with LRRC33 deficiency led to an increase in tumor-infiltrating CD3+ and CD8+ T cells, as well as NK cells, and the increase with the combination was often significant compared to either alone (Fig. 7D). Furthermore, the combination significantly increased granzyme B CD8+ T cells and NK cells relative to deficiency or treatment in WT mice (Fig. 7E–F). TNFα was similarly increased in CD8+ T cells by anti-PD-1 plus LRRC33 deficiency and was significantly increased in the combination relative to either deficiency or treatment alone (Fig. 7F). Moreover, IFNγ in both tumor-infiltrating NK cells and CD8+ cells were increased with deficiency and treatment and were significantly augmented by the combination of LRRC33 deficiency and PD-1 treatment relative to either alone (Fig. 7E–F). These data suggest that loss of LRRC33-dependent TGF-β1 activity potentiates the ability of anti-PD-1 to enhance antitumor immunity by increasing immigration of CD8 and NK cells and enhancing their effector activity.
Myeloid cell infiltration was also affected by treatment. Blockade of PD-1 in Lrrc33−/− mice significantly increased infiltration of DCs (Fig. 7D). Furthermore, MDSCs were decreased by LRRC33 deficiency and PD-1 treatment and further decreased by their combination (Fig. 7D). Phenotyping macrophages for CD206 and MHCII expression showed that LRRC33 deficiency had a more significant effect than anti-PD-1 in both increasing the MHCII+CD206− M1 population and decreasing the MHCII–CD206+ M2 population (Fig. 7G). However, skewing towards M1 was significantly enhanced by addition of anti-PD-1 to LRRC33 deficiency (Fig. 7G). Overall, the results suggest that LRRC33 deficiency leads to a more immunogenic B16.F10 TME and improved anti-PD-1 efficacy. These data emphasize the therapeutic potential of dual blockade of LRRC33/TGFβ1 and PD-1 in cancer immunotherapy.
DISCUSSION
Here, we focused on a selective approach to tumor therapy: inhibiting only one (of many combinations) of the three TGFβ isoforms associated with their five well-characterized milieu molecules, LTBP1, 3, and 4, GARP, and LRRC33 (16,18). We used this approach because in contrast to Tgfb1−/− mice, Lrrc33−/− mice show no autoimmune or inflammatory disease (16,20). We confirmed here, and extended to further myeloid cell types, that LRRC33 associates with TGF-β1 and is required for TGF-β1 expression and activation (16,32). GARP has been known only to associate with TGF-β1 (38–40). Here, we showed that GARP associated with and presented all three TGF-β isoforms on cell surface, whereas LRRC33 only productively interacted with TGF-β1. This is the first time that a milieu molecule has been shown to associate with a unique TGF-β1 isoform.
Tumors grew more slowly in Lrrc33−/− than in WT mice, which we traced to a lack of TGF-β1/LRRC33 complexes and activatable TGF-β1. B16.F10 lung metastasis was also decreased. Whole bone marrow transplantation confirmed that hematopoietic Lrrc33−/− cells were sufficient to slow tumor growth in WT mice, in agreement with the finding that TGF-β1/LRRC33 complexes were primarily expressed on myeloid cell types. Tumors in Lrrc33−/− mice showed increased infiltration of both adaptive immune CD3+ and CD8+ T cells and innate immune NK cells, TAMs were skewed toward an M1-like phenotype, and MDSC infiltration was decreased. Additionally, CD8+ T-cell and NK cell effector phenotype was enhanced. These results are consistent with results from Lrrc33−/− and- Rag1−/− mouse crosses, which showed that Lrrc33−/− lowered tumor growth by increasing both adaptive and innate immune cell responses. Phosphorylation of SMAD2/3 was decreased in infiltrating adaptive and innate immune cells, demonstrating a direct effect of Lrrc33 deficiency on TGF-β signaling in both immune arms (1). We confirmed that Lrrc33−/− deficiency did not affect immune cell subset distributions in spleen or thymus (20); however, we also found a reduction in the CD103highCD11c+CD11b−MHCII+ DC subset in lungs, which might be relevant to the predisposition of Lrrc33−/− mice to pneumonia (16). We observed synergy between Lrrc33 deficiency and PD-1 blockade in slowing growth of checkpoint blockade-resistant B16.F10 tumors. These synergistic effects also contributed to increased infiltration of CD8+ T cells and NK cells and their effector functions, and in skewing the M1/M2 macrophage subset ratio toward a more tumoricidal M1-like phenotype.
Our study suggests that TGF-β release from TGF-β1/LRRC33 complexes is localized at the cellular level within tumors. TGF-β1/β3 antibody, 21D1, more effectively inhibited tumor growth than Lrrc33 deficiency. However, the inhibition of SMAD2/3 phosphorylation in tumor-infiltrating leukocytes by 21D1 was more dramatic compared to Lrrc33 deficiency. Furthermore, SMAD2/3 phosphorylation in non-immune cells in tumors was almost completely inhibited by 21D1 antibody and unaffected by Lrrc33 deficiency. These results demonstrate the high selectivity of the TGFβ1 growth factor that is released from proTGF-β1/LRRC33 complexes within tumors for action on immune cells. As emphasized previously and demonstrated in the central nervous system, activation (release) of TGF-β1 takes place between an αVβ6 or αVβ8 integrin-bearing cell and a cell bearing proTGF-β1/LRRC33, and the released TGF-β acts on cells in the immediate proximity of its site of release (16). We did not identify the relevant integrin-expressing cells in our study; however, DCs are known to express integrin αVβ8 and antibodies to αVβ8 are effective at suppressing tumor growth (41). The selective effect of Lrrc33 deficiency on non-LRRC33 bearing cells, including CD3+, CD8+, and NK cells, suggests that these cells are in close contact with TGF-β1/LRRC33-expressing myeloid cells in tumors, and that an αVβ6 or αVβ8 integrin-expressing cells may also be nearby. Notably, tumor-associated myeloid cells are one of the most important factors in suppressing immune responses to tumors and in inducing resistance to immune checkpoint blockade (42). Furthermore, DCs, macrophages, and MDSCs, all of which have activatable LRRC33/TGF-β1 complexes, accounted for 60–70% of the infiltrating leukocytes in this study.
Previous studies that combined inhibition of TGF-β1 or the TGF-β1/GARP complex with inhibition of PD-1 or PD-L1 showed substantial inhibition of tumor growth in mice and effects on tumor-infiltrating immune cells. Increases in CD8+ T cells, decreases in M2-like macrophages, decreases in MDSCs, and a surprising increase in Foxp3+ Tregs was found in one study (14), and increases in CD8+ T cells were observed in other studies (5,15). In contrast, combination TGF-β1/GARP and PD-1 blockade had no effect on tumor-infiltrating populations (13). However, this and two other studies report enhanced CD8+ T-cell effectors (5,13,14). Our results were most different from TGF-β1/GARP blockade, suggesting that combination of TGF-β1/GARP and TGF-β1/LRRC33 could be additive or synergistic. We found here that CD3− NK cells, which were not examined in these previous studies, were increased in MC38 tumors in Lrrc33−/− mice and in B16.F10 tumors in Lrrc33−/− mice treated with PD-1 antibody.
Our results suggest that targeting the LRRC33 axis of TGF-β1 activation has promise as an immuno-oncology treatment strategy. It will be important to produce antibodies that specifically bind to the TGF-β1/LRRC33 complex and inhibit release of the active TGF-β1 growth factor, just as has been previously achieved for the TGF-β1/GARP complex (30,41). Such antibodies would also allow overcoming two limitations of the current work: (i) the restriction of tumors to the C57BL/6 mouse strain carrying the Lrrc33−/− mutation and (ii) the inability to grow tumors prior to inhibiting the LRRC33 axis of TGF-β1 activation. Antibodies would also enable future translation to patients of the highly selective mechanism of inhibiting the LRRC33 axis of TGF-β1 activation in myeloid cells in the TME.
Supplementary Material
Synopsis.
LRRC33 is required for myeloid cell TGF-β1 activation. LRRC33 deficiency reduces tumor growth in multiple models, boosts antitumor responses by effector cells, and enhances therapeutic efficacy of immune checkpoint blockade, highlighting the LRRC33/TGF-β1 axis as a therapeutic target.
Acknowledgements
We thank Patricia Sousa for help with animal irradiation and whole bone marrow transplantation, the Arlene Sharpe lab for providing the MC38 cell line and consultation on tumor cell culture and injection, Colette Matysiak in the von Andrian Lab for assistance with lung immune cell staining and analysis, Chafen Lu for early work and advice, Vijay Kuchroo for advice, Roderick T. Bronson of the Rodent Pathology Core at Harvard Medical School, Wei Yang and Viet Le for critical comments on the manuscript, and Margaret Nielsen for illustrations. Supported by NIH grant R01-HL159714.
Financial Support:
National Institutes of Health Grant R01-HL159714 (TAS).
Footnotes
Conflict of Interest Statement: The authors declare no potential conflicts of interest.
References
- 1.Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P. The polarization of immune cells in the tumour environment by TGFβ. Nat Rev Immunol 2010;10:554–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Derynck R, Turley SJ, Akhurst RJ. TGFβ biology in cancer progression and immunotherapy. Nature Reviews Clinical Oncology 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Colak S, Ten Dijke P. Targeting TGF-β signaling in cancer. Trends Cancer 2017;3:56–71 [DOI] [PubMed] [Google Scholar]
- 4.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 2017;168:542. [DOI] [PubMed] [Google Scholar]
- 5.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554:544–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018;554:538–43 [DOI] [PubMed] [Google Scholar]
- 7.Lan Y, Zhang D, Xu C, Hance KW, Marelli B, Qi J, et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Sci Transl Med 2018;10. [DOI] [PubMed] [Google Scholar]
- 8.Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGFβ signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 2008;13:23–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol 2009;9:259–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 2004;6:409–21 [DOI] [PubMed] [Google Scholar]
- 11.Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest 2015;125:3356–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 2012;12:253–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Streel G, Bertrand C, Chalon N, Lienart S, Bricard O, Lecomte S, et al. Selective inhibition of TGF-β1 produced by GARP-expressing Tregs overcomes resistance to PD-1/PD-L1 blockade in cancer. Nat Commun 2020;11:4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin CJ, Datta A, Littlefield C, Kalra A, Chapron C, Wawersik S, et al. Selective inhibition of TGFβ1 activation overcomes primary resistance to checkpoint blockade therapy by altering tumor immune landscape. Sci Transl Med 2020;12 [DOI] [PubMed] [Google Scholar]
- 15.Dodagatta-Marri E, Meyer DS, Reeves MQ, Paniagua R, To MD, Binnewies M, et al. α-PD-1 therapy elevates Treg/Th balance and increases tumor cell pSmad3 that are both targeted by α-TGFβ antibody to promote durable rejection and immunity in squamous cell carcinomas. J Immunother Cancer 2019;7:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qin Y, Garrison BS, Ma W, Wang R, Jiang A, Li J, et al. A milieu molecule for TGF-β required for microglia function in the nervous system. Cell 2018;174:156–71 e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hinck AP, Mueller TD, Springer TA. Structural biology and evolution of the TGF-β family. Cold Spring Harb Perspect Biol 2016;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robertson IB, Rifkin DB. Regulation of the bioavailability of TGF-β and TGF-β-related proteins. Cold Spring Harb Perspect Biol 2016;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su X, Mei S, Liang X, Wang S, Liu J, Zhang Y, et al. Epigenetically modulated LRRC33 acts as a negative physiological regulator for multiple Toll-like receptors. Journal of leukocyte biology 2014;95:1–10 [DOI] [PubMed] [Google Scholar]
- 20.Noubade R, Wong K, Ota N, Rutz S, Eidenschenk C, Valdez Pa, et al. NRROS negatively regulates reactive oxygen species during host defence and autoimmunity. Nature 2014;509:235–9 [DOI] [PubMed] [Google Scholar]
- 21.Tsunawaki S, Sporn m, Ding a, Nathan C. Deactivation of macrophages by transforming growth factor-β. Nature 1988;334:260–4 [DOI] [PubMed] [Google Scholar]
- 22.Wong K, Noubade R, Manzanillo P, Ota N, Foreman O, Hackney JA, et al. Mice deficient in NRROS show abnormal microglial development and neurological disorders. Nat Immunol 2017;18:633–41 [DOI] [PubMed] [Google Scholar]
- 23.Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nature neuroscience 2014;17:131–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aluwihare P, Mu Z, Zhao Z, Yu D, Weinreb PH, Horan GS, et al. Mice that lack activity of αVβ6- and αVβ8-integrins reproduce the abnormalities of TGFβ1- and TGFβ3-null mice. J Cell Sci 2009;122:227–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mobley AK, Tchaicha JH, Shin J, Hossain MG, McCarty JH. β8 integrin regulates neurogenesis and neurovascular homeostasis in the adult brain. J Cell Sci 2009;122:1842–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong X, Tan NB, Howell KB, Barresi S, Freeman JL, Vecchio D, et al. Bi-allelic LoF NRROS variants impairing Active TGF-β1 delivery cause a severe infantile-onset neurodegenerative condition with intracranial calcification. Am J Hum Genet 2020;106:559–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith C, McColl BW, Patir A, Barrington J, Armishaw J, Clarke A, et al. Biallelic mutations in NRROS cause an early onset lethal microgliopathy. Acta Neuropathol 2020;139:947–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cash JA EB; Kattamuri C; Nolan K; Zhao H; Sidis Y; Keutmann HT; Thompson TB. Structure of myostatin·follistatin-like 3: N-terminal domains of follistatin-type molecules exhibit alternate modes of binding. J Biol Chem 2012;6:1043–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davies J, Dickinson CD, Marquis DM, Tang Y, Vaillancort PE; Eli Lilley and Company, Indianapolis, IN (US), assignee. Antibodies to TGF-β1. USA patent US 7,619,069 B2. 2009.
- 30.Oida T, Weiner HL. TGF-β induces surface LAP expression on murine CD4 T cells independent of Foxp3 induction. PLoS One 2010;5:e15523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kolodziej PA, Young RA. Epitope tagging and protein surveillance. Methods Enzymol 1991;194:508–19 [DOI] [PubMed] [Google Scholar]
- 32.Cuende J, Lienart S, Dedobbeleer O, van der Woning B, De Boeck G, Stockis J, et al. Monoclonal antibodies against GARP/TGF-β1 complexes inhibit the immunosuppressive activity of human regulatory T cells in vivo. Sci Transl Med 2015;7:284ra56. [DOI] [PubMed] [Google Scholar]
- 33.Novitskiy SV, Pickup MW, Chytil A, Polosukhina D, Owens P, Moses HL. Deletion of TGF-β signaling in myeloid cells enhances their anti-tumorigenic properties. J Leukoc Biol 2012;92:641–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma W, Qin Y, Chapuy B, Lu C. LRRC33 is a novel binding and potential regulating protein of TGF-β1 function in human acute myeloid leukemia cells. PLoS One 2019;14:e0213482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen S, Lee LF, Fisher TS, Jessen B, Elliott M, Evering W, et al. Combination of 4–1BB agonist and PD-1 antagonist promotes antitumor effector/memory CD8 T cells in a poorly immunogenic tumor model. Cancer Immunol Res 2015;3:149–60 [DOI] [PubMed] [Google Scholar]
- 36.Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, et al. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell 2015;162:1242–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, et al. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med 2017;214:895–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stockis J, Colau D, Coulie PG, Lucas S. Membrane protein GARP is a receptor for latent TGF-β on the surface of activated human Treg. Eur J Immunol 2009;39:3315–22 [DOI] [PubMed] [Google Scholar]
- 39.Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, Shevach EM. GARP (LRRC32) is essential for the surface expression of latent TGF-β on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A 2009;106:13445–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang R, Kozhaya L, Mercer F, Khaitan A, Fujii H, Unutmaz D. Expression of GARP selectively identifies activated human FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A 2009;106:13439–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dodagatta-Marri E, Ma H-Y, Liang B, Li J, Meyer DS, Sun K-H, et al. Integrin αvβ8 on T cells is responsible for suppression of anti-tumor immunity in multiple syngeneic models and is a promising target for tumor immunotherapy. bioRxiv 2020:2020.05.14.084913 [Google Scholar]
- 42.De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature 2016;539:443–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lienart S, Merceron R, Vanderaa C, Lambert F, Colau D, Stockis J, et al. Structural basis of latent TGF-β1 presentation and activation by GARP on human regulatory T cells. Science 2018;362:952–6 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.