

Abstract
Pulmonary tumor thrombotic microangiopathy (PTTM) is a rapidly progressive subtype of pulmonary hypertension (PH) associated with impaired right ventricular adaptation and very poor prognosis in cancer, and its rapid progression makes antemortem diagnosis and treatment extremely difficult. We describe the case of a 35‐year‐old woman who developed severe PH with subsequent circulatory collapse. The patient was clinically diagnosed with PTTM induced by lung adenocarcinoma harboring the c‐ros oncogene 1 (ROS1) rearrangement within 1–2 weeks, while hemodynamics were stabilized by rescue venoarterial extracorporeal membrane oxygenation support. Crizotinib, an oral tyrosine kinase inhibitor targeting anaplastic lymphoma kinase, MET, and ROS1 kinase domains dramatically resolved PH, resulting in more than 3 years of survival. Targeted gene‐tailored therapy with mechanical support can improve survival in PTTM.
Keywords: crizotinib, lung cancer, pulmonary hypertension, pulmonary tumor thrombotic microangiopathy, venoarterial extracorporeal membrane oxygenation
INTRODUCTION
Pulmonary tumor thrombotic microangiopathy (PTTM) is a rare, lethal clinicopathological entity wherein tumor microemboli in the pulmonary microcirculation induce thrombotic microangiopathy. 1 PTTM is characterized by rapidly progressive respiratory and right‐sided heart failure. The average time from onset of PTTM to death is 9.5 weeks. 1 Therefore, very few cases have been diagnosed antemortem. Some reports have demonstrated the efficacy of imatinib, a tyrosine kinase inhibitor (TKI) against platelet‐derived growth factor (PDGF), in PTTM. 2 , 3 Recent advances in tailoring cancer treatment to the genetic profile could prolong survival in cancer. However, there have been no reports of PTTM with an antemortem diagnosis of genetically analyzed cancer and personalized treatment. Additionally, the effects of targeted molecular therapy on PTTM remain unknown. We describe a patient who recovered from PTTM and has survived for >3 years on crizotinib, a c‐ros oncogene 1 (ROS1) TKI.
CASE PRESENTATION
A 35‐year‐old woman, a nonsmoker without a notable medical history, with an uneventful delivery 8 months ago, was admitted to the referring hospital because of progressive severe dyspnea. Transthoracic echocardiography (TTE) indicated pulmonary hypertension (PH). Computed tomography (CT) revealed lymphadenopathy with an infiltrative shadow in the right middle lobe. An initial diagnosis of pulmonary sarcoidosis was considered. However, the administered steroid pulse therapy did not yield the expected results. Since distal‐type chronic thromboembolic pulmonary hypertension (CTEPH) was suspected based on lung perfusion scintigraphy, the patient was referred to our hospital for further management.
The patient presented with World Health Organization functional class (WHO‐FC) IV symptoms with elevated brain natriuretic peptide (387 pg/ml), a pulse rate of 118 beats/min, and a blood pressure of 121/84 mmHg with noninvasive continuous positive airway pressure ventilation. TTE demonstrated an estimated systolic pulmonary arterial pressure (eSPAP) of 51 mmHg and severe right ventricular systolic dysfunction, with a compressed left ventricle and slight pericardial effusion. Contrast‐enhanced CT demonstrated mediastinal and bilateral hilar lymphadenopathy with an infiltrative shadow in the right middle lobe, without any obvious findings of pulmonary thromboembolism up to distal subsegmental arteries as far as we can see or a mass in any organ indicating malignancy (Figure 1a–c). Lung perfusion scintigraphy demonstrated multiple subsegmental perfusion defects in the subpleural portion of both lungs, which were relatively small compared with the typical characteristics of CTEPH (Figure 1d–g). Therefore, we suspected severe PTTM. Right heart catheterization indicated severe precapillary PH with low output, as evidenced by a mean pulmonary artery wedge pressure of 5 mmHg, mean pulmonary arterial pressure (mPAP) of 45 mmHg, cardiac index of 1.68 L/min/m2, and mixed venous oxygen saturation of 40.3% under arterial oxygen saturation of 90% with noninvasive continuous positive airway pressure ventilation. Pulmonary wedge aspiration cytology showed one atypical cell. Her carcinoembryonic antigen levels were markedly elevated (31.6 ng/ml; normal range, 0.1–5.0 ng/ml).
Figure 1.
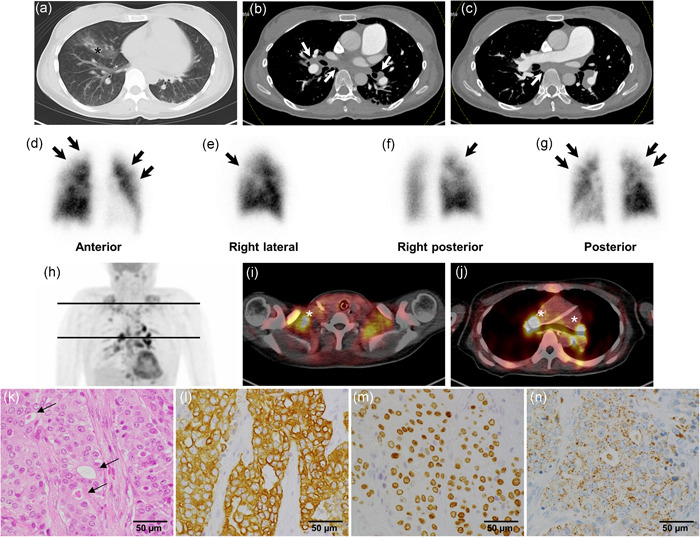
Contrast‐enhanced computed tomography (CT), lung perfusion scintigraphy, 18F‐fluorodeoxyglucose positron emission tomography (FDG‐PET)/CT, and histology of the right subclavian lymph node biopsy specimens during diagnostic evaluation. (a) Chest CT shows an infiltrative shadow in the right middle lobe (black asterisk). (b and c) Contrast‐enhanced CT shows mediastinal and bilateral hilar lymphadenopathy (white arrows) without obvious findings of pulmonary thromboembolism or mass in any organs indicating malignancy. (d–g) Lung perfusion scintigraphy shows multiple subsegmental perfusion defects (black arrows) in the subpleural portion of both lungs. Note: They are relatively small and light compared with the typical characteristics of chronic thromboembolic pulmonary hypertension. (h–j) FDG‐PET/CT shows multiple abnormal uptakes (white asterisks) in the mediastinal, bilateral hilar, bilateral supraclavicular, and subclavian lymph nodes. (k) Lymph nodes are replaced by predominantly solid sheets and nests of tumor cells with very focal gland formation with mucin (arrows), suggesting metastatic adenocarcinoma (hematoxylin and eosin staining). Immunohistochemistry panels show that (l) cytokeratin (CK) 7 (brown), (m) thyroid transcription factor‐1 (brown), and (n) napsin A (brown) are all positive. Considering that p40, CK5/6, CK20, and thyroglobulin were negative markers (data not shown), these results indicate that lung adenocarcinoma is the most probable primary lesion
Despite continuous intravenous anticoagulation therapy, increased inotropic administration, and introduction of sildenafil, a phosphodiesterase type‐5 inhibitor, respiratory and right‐sided heart failure rapidly progressed with circulatory collapse 4 days after her transfer. Following the initiation of venoarterial extracorporeal membrane oxygenation (VA‐ECMO), 18F‐fluorodeoxyglucose positron emission tomography‐CT showed abnormal uptake in the mediastinal, bilateral hilar, bilateral supraclavicular, and subclavian lymph nodes (Figure 1h–j). Surgical biopsy of the right subclavian lymph nodes revealed metastatic adenocarcinoma (Figure 1k). Immunohistochemistry panels indicated lung adenocarcinoma as the most probable primary lesion, as evidenced by positive markers of cytokeratin (CK) 7, thyroid transcription factor‐1, and napsin A (Figure 1l–n), and with negative markers of p40, CK5/6, CK20, and thyroglobulin. We clinically diagnosed PTTM due to advanced lung adenocarcinoma after excluding other causes of PH; associated with some underlying diseases such as connective tissue disease, human immunodeficiency virus infection, or portal hypertension. Notably, oncogenic driver mutation analysis found this adenocarcinoma to harbor the ROS1 rearrangement.
Treatment with crizotinib, an oral TKI targeting anaplastic lymphoma kinase (ALK), MET, and ROS1 kinase domains, was started in combination with sildenafil (30 mg/day) and intravenous epoprostenol (5 ng/kg/min) under sustained VA‐ECMO support. Five days after the initiation of crizotinib (500 mg/day), mPAP significantly decreased from 45 to 28 mmHg, allowing for the discontinuation of VA‐ECMO support. Even after weaning off inotropic agents, the patient had no signs of PH, as evidenced by an eSPAP of <21 mmHg. She was discharged on dual oral PH‐specific therapies 42 days after her transfer. Nine months after crizotinib initiation, she developed progressive disease and received second‐line chemotherapy with cisplatin, pemetrexed sodium hydrate, and bevacizumab, followed by maintenance chemotherapy with pemetrexed sodium and bevacizumab hydrate. Three years later, she was managed by a pulmonologist in an outpatient clinic with WHO‐FC II symptoms.
CLINICAL DISCUSSION
The use of VA‐ECMO for PTTM has been controversial because PTTM characterizes rapid progression associated with malignancy, where it is almost impossible to wean a patient from support. There have been only two reports demonstrating the use of VA‐ECMO for PTTM. 2 , 3 Ogawa et al. 2 reported about a 47‐year‐old female with gastric adenocarcinoma who was supported with VA‐ECMO, which bridged imatinib, a TKI against PDGF, followed by chemotherapy. The patient died 9 months later. Iwashita et al. 3 reported about a 46‐year‐old female supported with VA‐ECMO who died 2 days after weaning. No other reports have described long‐term survival. Recent advances in genetic testing for cancer allow the diagnosis of specific mutations, and molecular targeting therapies are known to improve prognosis. 4 , 5 We considered the possibility of lung cancer associated with some specific alteration including ROS1, because our patient is a young, nonsmoking female. 6 VA‐ECMO allowed complete multimodality imaging, pathologic, and genetic evaluation of PTTM. It has been hypothesized in PTTM that tumor cell embolization into pulmonary arterioles induces the activation of coagulation, platelet aggregation, and release of various cytokines derived from platelets, tumor cells, and endothelial cells including PDGF and vascular endothelial growth factor (VEGF). 1 , 5 Targeted drugs including imatinib and bevacizumab, a VEGF receptor inhibitor, may be effective in some selected cases because PDGF, VEGF, and tissue factor were found to be important molecules in the pathogenesis of PTTM. 1 , 2 , 7 There have been no reports involving a specific mutation for causative malignancy itself. Approximately, 1% of lung adenocarcinomas are driven by a ROS1 rearrangement. 4 , 6 Crizotinib is highly effective in patients with Stage IV ROS1‐rearranged lung adenocarcinoma. 4 Shah et al. 8 reported two cases of PTTM associated with ROS1‐rearranged lung cancer, both of which had been treated with crizotinib before the onset of PTTM. This case builds on previous findings, demonstrating the efficacy of crizotinib on untreated lung cancer and PTTM despite even being initiated under VA‐ECMO. Conversely, Awada et al. reported that crizotinib may exacerbate existing pulmonary arterial hypertension (PAH) in animal models, perhaps via endothelial cell injury. 9 However, Khouri et al. 10 described the association between ALK receptor TKIs (ALK‐TKI) and PAH from the WHO pharmacovigilance database that there was only one case of PAH onset during crizotinib therapy among 18,945 adverse drug reactions with ALK‐TKI, which is very rare compared to lorlatinib, third‐generation ALK‐TKI. Further studies are needed to determine this heterogeneity of ALK‐TKI and whether the resolution of PH by crizotinib is attributed to suppression of the tumor itself, suppression of cytokine release from tumor cells, or both.
CONCLUSION
Genetic diagnosis and targeted molecular therapy for PTTM under VA‐ECMO support have the potential to improve poor prognosis.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interest.
ETHICS STATEMENT
Authors have written informed consent from the patient.
AUTHOR CONTRIBUTIONS
All authors have reviewed, contributed, and approved the manuscript.
Aoyama D, Fukui S, Hirata H, Ohta‐Ogo K, Matama H, Tateishi E, Nishii T, Asaumi Y, Toyofuku M, Ikeue T, Ogo T, Ishibashi‐Ueda H, Yasuda S. Crizotinib for ROS1‐rearranged lung cancer and pulmonary tumor thrombotic microangiopathy under venoarterial extracorporeal membrane oxygenation. Pulmonary Circulation. 2022;12:e12047. 10.1002/pul2.12047
REFERENCES
- 1. Godbole RH, Saggar R, Kamangar N. Pulmonary tumor thrombotic microangiopathy: a systematic review. Pulm Circ. 2019;9(2):2045894019851000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ogawa A, Yamadori I, Matsubara O, Matsubara H. Pulmonary tumor thrombotic microangiopathy with circulatory failure treated with imatinib. Intern Med. 2013;52(17):1927–30. [DOI] [PubMed] [Google Scholar]
- 3. Iwashita Y, Hiramoto T, Suzuki K, Hashizume R, Maruyama K, Imai H. Possibility of venoarterial extracorporeal membranous oxygenator being a bridging therapy for hemodynamic deterioration of pulmonary tumor thrombotic microangiopathy prior to initiating chemotherapy: a case report. Medicine. 2018;97(37):e12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mazières J, Zalcman G, Crinò L, Biondani P, Barlesi F, Filleron T, Dingemans AM, Léna H, Monnet I, Rothschild SI, Cappuzzo F, Besse B, Thiberville L, Rouvière D, Dziadziuszko R, Smit EF, Wolf J, Spirig C, Pecuchet N, Leenders F, Heuckmann JM, Diebold J, Milia JD, Thomas RK, Gautschi O. Crizotinib therapy for advanced lung adenocarcinoma and a ROS1 rearrangement: results from the EUROS1 cohort. J Clin Oncol. 2015;33(9):992–999. [DOI] [PubMed] [Google Scholar]
- 5. Abe H, Hino R, Fukayama M. Platelet‐derived growth factor‐A and vascular endothelial growth factor‐C contribute to the development of pulmonary tumor thrombotic microangiopathy in gastric cancer. Virchows Arch. 2013;462(5):523–31. [DOI] [PubMed] [Google Scholar]
- 6. Bergethon K, Shaw AT, Ignatius Ou SH, Katayama R, Lovly CM, McDonald NT, Massion PP, Siwak‐Tapp C, Gonzalez A, Fang R, Mark EJ, Batten JM, Chen H, Wilner KD, Kwak EL, Clark JW, Carbone DP, Ji H, Engelman JA, Mino‐Kenudson M, Pao W, Iafrate AJ. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30(8):863–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Higo K, Kubota K, Takeda A, Higashi M, Ohishi M. Successful antemortem diagnosis and treatment of pulmonary tumor thrombotic microangiopathy. Intern Med. 2014;53(22):2595–99. [DOI] [PubMed] [Google Scholar]
- 8. Shah AT, Bernardo RJ, Berry GJ, Kudelko K, Wakelee HA. Two cases of pulmonary tumor thrombotic microangiopathy associated with ROS1‐rearranged non‐small‐cell lung cancer. Clin Lung Cancer. 2021;22(2):e153–56. [DOI] [PubMed] [Google Scholar]
- 9. Awada C, Grobs Y, Wu WH, Habbout K, Romanet C, Breuils‐Bonnet S, Provencher S, Potus F, Boucherat O, Tremblay E, Martineau S, Paulin R. R‐crizotinib predisposes to and exacerbates pulmonary arterial hypertension in animal models. Eur Respir J. 2021;57(5):2003271. [DOI] [PubMed] [Google Scholar]
- 10. Khouri C, Hlavaty A, Roustit M, Cracowski JL, Chaumais MC, Humbert M, Montani D. Investigating the association between ALK receptor tyrosine kinase inhibitors and pulmonary arterial hypertension: a disproportionality analysis from the WHO pharmacovigilance database. Eur Respir J. 2021;58:2101576. [DOI] [PubMed] [Google Scholar]