

Abstract
Pulmonary arterial hypertension (PAH) is characterized by increased resistance in the pulmonary arterioles as a result of remodeled blood vessels. We sought all available epidemiologic data on population‐based prevalence, incidence, and 1‐year survival of PAH as part of the Global Burden of Disease Study. We performed a systematic review searching Global Index Medicus (GIM) for keywords related to PAH between 1980 and 2021 and identified population‐representative sources of prevalence, incidence, and mortality for clinically diagnosed PAH. Of 6772 articles identified we found 65 with population‐level data: 17 for prevalence, 17 for incidence, and 58 reporting case fatality. Reported prevalence ranged from 0.37 cases/100,000 persons in a referral center of French children to 15 cases/100,000 persons in an Australian study. Reported incidence ranged from 0.008 cases/100,000 person‐years in Finland, to 1.4 cases/100,000 person‐years in a retrospective chart review at a clinic in Utah, United States. Reported 1‐year survival ranged from 67% to 99%. All studies with sex‐specific estimates of prevalence or incidence reported higher levels in females than males. Studies varied in their size, study design, diagnostic criteria, and sampling procedures. Reported PAH prevalence, incidence, and mortality varied by location and study. Prevalence ranged from 0.4 to 1.4 per 100,000 persons. Harmonization of methods for PAH registries would improve efforts at disease surveillance. Results of this search contribute to ongoing efforts to quantify the global burden of PAH.
Keywords: population health, pulmonary arterial hypertension, survival
BACKGROUND
Pulmonary arterial hypertension (PAH) is a progressive and life‐threatening condition characterized by pulmonary vascular remodeling, increased resistance to blood flow through the pulmonary vasculature, and eventually right heart failure. An evaluation of current observed disease rates, with careful attention to the complex case definitions, is needed. The definition of PAH (World Health Organization [WHO] Group 1) has evolved over time and comprises a group of diseases including idiopathic, heritable, or drug‐induced PAH, previously called primary pulmonary hypertension. The updated WHO classification now includes the conditions associated with congenital heart disease, connective tissue disease, HIV, portal hypertension, and schistosomiasis. The diseases included in each subgroup have evolved over time. Systematic reviews of the epidemiologic literature for PAH must carefully account for the manner in which studies reported different definitions as classification systems evolved.
The Global Burden of Disease (GBD) study is a multinational effort to produce comparable, consistent measures of disease burden for national and subnational populations. 1 The study leverages epidemiological relationships between prevalence, incidence, and mortality for hundreds of diseases to produce age‐, sex‐, and location‐specific estimates from 1990 onwards for use by scientists, policymakers, and health departments. While not explicitly estimated in prior iterations, the upcoming GBD 2020 study includes the first‐ever global and national estimates of PAH. To inform epidemiologic modeling efforts of PAH for GBD 2020, we attempted to identify all published population‐level data from 1990 to the present, including prevalence, incidence, and mortality, to produce high‐quality estimates of the burden of this fatal and understudied disease.
Previous systematic reviews of the literature have focused on non‐Group 1 pulmonary hypertension or specific subpopulations afflicted with PAH. 2 , 3 , 4 , 5 , 6 A recent review focused on PAH prevalence and incidence from 2003 to 2020, 7 but did not address case fatality. As the GBD estimates a full‐time series from 1990 to the present and includes all global locations, the inclusion of data before 2003 is essential, along with estimates of mortality with and without therapies. The purpose of this review was to rigorously focus on population representativeness in reviewing reported PAH prevalence, incidence, and mortality rates over the past three decades to identify inputs for future estimation of PAH for the GBD study.
METHODS
Case definition
Our literature review was designed to include all sources since 1990 while maintaining awareness of the evolving classification schema. Primary pulmonary hypertension was commonly reported before the Second World Symposium on Pulmonary Hypertension in 1998, which established five categories of pulmonary hypertension. After 1998, idiopathic, familial, and drug‐induced cases of pulmonary hypertension began to be reported as WHO Group 1 PAH.
Until 2019, hemodynamic criteria for PAH included a mean pulmonary arterial pressure (mPAP) of ≥25 mmHg and a pulmonary arterial capillary wedge pressure of <15 mmHg. In 2019, diagnostic criteria were revised to define PAH as an mPAP >20 mmHg (≥25 mmHg by previous guidelines), a wedge pressure ≤15 mmHg, and a pulmonary vascular resistance ≥3 Wood units. 8 This revision is significant as estimates based on older hemodynamic criteria would represent a potential underestimation compared with the newer criteria.
Because echocardiography can lead to both under‐ and overdiagnosis of PAH in comparison to right heart catheterization (RHC), careful attention was paid to the manner in which echocardiography was used by each study protocol. Only six studies, with five reporting only survival and one reporting survival and prevalence, allowed the use of echocardiography to aid in the diagnosis of PAH for a subset of patients. Even for these studies, RHC was the primary mode of diagnosis and echocardiography was used with adherence to imaging guidelines and explicit exclusion for signs of left‐sided systolic or diastolic dysfunction. Only one study, performed in resource‐limited settings in Africa, was designed prospectively not to require RHC, however, assessments were performed by clinical specialists with a protocol intended to identify pulmonary hypertension and including formal evaluation of left‐heart function. 9
The GBD study selects a case definition for each cause to ensure comparability between data sources and estimates and allows for adjustments to correct for any bias due to alternate definitions. The case definition for this literature review was based on physician diagnosis of WHO Group 1 PAH with supporting diagnostic evidence via RHC or, in only one case, echocardiography alone. PAH identified by the International Classification of Diseases (ICD) codes was included if the study authors had reviewed the patients' medical records and confirmed the diagnosis. We excluded studies restricted to subtypes within WHO Group 1 PAH, such as registries capturing only congenital, drug‐induced, or idiopathic PAH.
WHO Groups 2, 3, 4, and 5 pulmonary hypertension were excluded if recognized as such in the source manuscript.
Data sources
Disease registries are one of the most important sources for understanding the demographics, clinical presentation, and outcomes of PAH. The first national registry of PAH was established in the United States in the early 1980s through the National Institutes of Health (NIH). In the coming decades, registries were established in the United States, France, United Kingdom, Spain, Germany, and Sweden. 10 These registries vary in enrollment criteria, coverage, representativeness, number of centers, and type of participating centers. Other efforts to describe PAH have used health system or hospital‐derived administrative facility data using ICD codes.
Search methodology
We conducted a structured review of the literature to identify all primary data sources reporting population‐representative estimates of prevalence, incidence, and case fatality of PAH. Our Preferred Reporting Items for Systematic Reviews and Meta‐Analyses (PRISMA) checklist is included in the appendix. We searched the Global Index Medicus (GIM), a library that indexes PubMed and international libraries including Latina American and Caribbean Health Sciences Literature and African Index Medicus, between 1980 and 2021 (GIM search string) and included all peer‐reviewed manuscripts. Since the original search in 2018, GIM has removed PubMed from its indexing; to account for this we searched PubMed independently for results from 2018 to 2021 and deduplicated the results in the final count. We reviewed results and added sources based on input from experts in the field of pulmonary vascular disease (S. R., P. L., P. C.).
We screened the titles and abstracts of all results. Full‐text review was performed for all studies with reported prevalence, incidence, or case fatality of PAH. We did not review studies reporting solely on Pulmonary Hypertension Groups 2–5. Studies were included based on the operant definition and classification at the time of the publication. As such, we report on a composite of several PAH definitions. Title/abstract screening and full‐text extraction were performed by two reviewers and arbitrated by a third. All included studies were reviewed by A. B. D., S. E. B, C. J., and G. R. Search strings are included in the appendix.
In full‐text review, we screened for representativeness, diagnostic criteria, and study type. To obtain representative population‐level estimates, we excluded studies that focused on one or only a few subpopulations. For example, we excluded studies in which the authors focused on only one race/ethnicity group among many within a country. Other studies were excluded if they focused on subgroups such as specific social groups or classes, or types of employees, data not representative of the location or geographic locations likely to lead to bias. Studies that sampled larger populations were included if the sampling method was representative of the population. We excluded papers without extractable data, such as descriptive reports of registries or when stratified in such a way as to obscure population‐level results.
We extracted all data at the most detailed age, sex, and location. Extractors were fluent in Spanish and English and used the support of colleagues or Google Translate to extract studies in other languages. We extracted the dates of the data, the location, the case definition, the study type, the percent of the study that was female, the mean age, and the mPAP (when given). When provided, we extracted confidence intervals or standard errors. When estimates of uncertainty were not published, we extracted cases and sample size and calculated standard errors using Wilson's score.
The review was not registered and study protocols were not published before this study. Data included in this review can be accessed through the Global Health Data Exchange (GHDX, http://ghdx.healthdata.org/).
RESULTS
The search identified 6772 studies through February 5, 2021, of which 405 were selected for full‐text review and 65 were included in our analysis: 17 reporting prevalence estimates, 17 reporting incidence, and 58 reporting case‐fatality rate. Of these, 19 reported on more than one metric. All included studies were published between 1991 and 2021. See Figure 1 for the detailed flow of identified studies using a PRISMA‐style diagram. Thirty‐seven countries and 22 registries were represented (Table 1). Four studies reported only pediatric cases. All studies, except for one, had >50% female cases. All studies with sex‐specific estimates of prevalence or incidence reported higher levels in females than males. The breakdown of subtypes within WHO group 1 PAH reported by studies is included in the Supporting Information Table.
Figure 1.
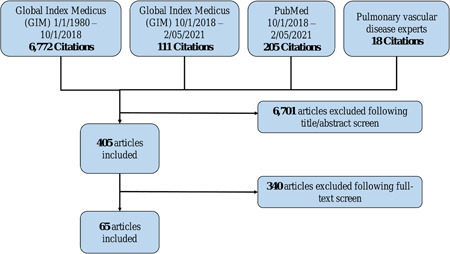
PRISMA diagram.
Results of the systematic review, including number of hits from each data source, number of articles included after title/abstract review, and number of articles included
Table 1.
67 Studies reporting prevalence, incidence, and mortality of PAH identified in systematic review
| First author, publication date | Location | Measure | Diagnosis type | Site type |
|---|---|---|---|---|
| Dubroff, Jason (2020) 11 | Utah | Prevalence | Right heart catheter | Single site |
| Dubroff, Jason (2020) 11 | Utah | Incidence | Right heart catheter | Single site |
| Khou, Victor, (2020) 12 | New Zealand and Australia | Crude mortality rate | Right heart catheter | Registry |
| Kopec, Grzegorz (2020) 13 | Poland | Prevalence | Right heart catheter | Registry |
| Kopec, Grzegorz (2020) 13 | Poland | Incidence | Right heart catheter | Registry |
| Kwiatkowska, Joanna (2020) 14 | Poland | Prevalence | Right heart catheter | Registry |
| Kwiatkowska, Joanna (2020) 14 | Poland | Incidence | Right heart catheter | Registry |
| Kwiatkowska, Joanna (2020) 14 | Poland | 1‐year survival | Right heart catheter | Registry |
| Chazova, I. E. (2019) 15 | Russia | 1‐year survival | Echocardiography + right heart catheter | Registry |
| Gruss, Ana (2019) 16 | Uruguay | Prevalence | Right heart catheter | Single site |
| Gruss, Ana (2019) 16 | Uruguay | Incidence | Right heart catheter | Single site |
| Gruss, Ana (2019) 16 | Uruguay | 1‐year survival | Right heart catheter | Single site |
| Jang, Albert Youngwoo (2019) 17 | South Korea | 1‐year survival | Right heart catheter | Multicenter |
| Gomes, A. (2018) 18 | Portugal | 1‐year survival | Right heart catheter | Single site |
| Strange, Geoff (2018) 19 | Australia | 1‐year survival | Right Heart Catheter | Multicenter |
| Austin, Christopher (2017) 20 | Florida | 1‐year survival | Right heart catheter | Multicenter |
| Gall, Henning (2017) 21 | Germany | 1‐year survival | Right heart catheter | Registry |
| Marques‐Alves, P. (2017) 22 | Portugal | 1‐year survival | Right heart catheter | Registry |
| Quezada Loaiza, Carlos Andres (2017) 23 | Spain | 1‐year survival | Right heart catheter | Single site |
| Tamura, Yuichi (2017) 24 | Japan | 1‐year survival | Right heart catheter | Registry |
| Wang, Le‐Yung (2017) 25 | Taiwan | 1‐year survival | Echocardiography + right heart catheter | Single site |
| Radegran, Goran (2016) 26 | Sweden | 1‐year survival | Right heart catheter | Multicenter |
| Thienemann, Friedrich (2016) 27 | Four African Countries | 6‐month survival | Echocardiography + right heart catheter if available | Registry |
| Vaid, Haris (2016) 28 | Canada | 1‐year survival | electronic data capture | Multicenter |
| Chung, Wook‐Jin (2015) 29 | South Korea | 1‐year survival | Echocardiography + right heart catheter | Multicenter |
| Farber, Harrison (2015) 30 | United States | 1‐year survival | Right heart catheter | Registry |
| Grapsa, Julia (2015) 31 | United Kingdom | 1‐year survival | Right heart catheter | Single site |
| Hoeper, Marius (2015) 32 | Germany | Prevalence | Right heart catheter | Registry |
| Hoeper, Marius (2015) 32 | Germany | Incidence | Right heart catheter | Registry |
| Idrees, Majdy (2015) 33 | Saudi Arabia | 1‐year survival | Right heart catheter | Single site |
| Alves Jr, Jose Leonidas (2015) 34 | Brazil | 1‐year survival | Right heart catheter | Single site |
| Mueller‐Mottet, Severine (2015) 35 | Switzerland | 1‐year survival | Right heart catheter | Registry |
| Adachi, Shiro (2014) 36 | Japan | 1‐year survival | Right heart catheter | Multicenter |
| Del Cerro Marin, M.J. (2014) 37 | Spain | 1‐year survival | Electronic data capture | Multicenter |
| Del Cerro Marin, M. J. (2014) 37 | Spain | Incidence | electronic data capture | Multicenter |
| Del Cerro Marin, M. J. (2014) 37 | Spain | Prevalence | Electronic data capture | Multicenter |
| Jansa, Pavel (2014) 38 | Czech Republic | 1‐year survival | Right heart catheter | Multicenter |
| Jansa, Pavel (2014) 38 | Czech Republic | Incidence | Right heart catheter | Multicenter |
| Jansa, Pavel (2014) 38 | Czech Republic | Prevalence | Right heart catheter | Multicenter |
| Korsholm, Kasper (2014) 39 | Denmark | 1‐year survival | Right heart catheter | Registry |
| Talavera, Maria (2014) 40 | Argentina | 1‐year survival | Right heart catheter | Single site |
| Zijlstra, Willemijn M. H. (2014) 41 | United States and Netherlands | 1‐year survival | Right Heart Catheter | Multicenter |
| Baptista, Rui (2013) 42 | Portugal | 1‐year survival | Right heart catheter | Registry |
| Baptista, Rui (2013) 42 | Portugal | Incidence | Right heart catheter | Registry |
| Ernande, Laura (2013) 43 | France, Belgium | 1‐year survival | Right heart catheter | Multicenter |
| Frost, Adaani (2013) 44 | United States | 1‐year survival | Right heart catheter | Registry |
| Roofthooft, Marcus T. R. (2013) 45 | Denmark | 1‐year survival | Echocardiography + right heart catheter | Multicenter |
| Cogswell, Rebecca (2012) 46 | California | 1‐year survival | Right heart catheter | Single site |
| Cracowski, Jean‐Luc (2012) 47 | France | 1‐year survival | Right heart catheter | Multicenter |
| Escribano‐Subias, Pilar (2012) 48 | Spain | 1‐year survival | Right heart catheter | Multicenter |
| Escribano‐Subias, Pilar (2012) 48 | Spain | Prevalence | Right heart catheter | Multicenter |
| Escribano‐Subias, Pilar (2012) 48 | Spain | Incidence | Right heart catheter | Multicenter |
| Ling, Yi (2012) 49 | United Kingdom and Ireland | Prevalence | Right heart catheter | Multicenter |
| Ling, Yi (2012) 49 | United Kingdom and Ireland | 1‐year survival | Right heart catheter | Multicenter |
| Ling, Yi (2012) 49 | United Kingdom and Ireland | Incidence | Right heart catheter | Multicenter |
| Sakao, Seiichiro (2012) 50 | Japan | 5‐year survival | Right heart catheter | Single site |
| Sakao, Seiichiro (2012) 50 | Japan | 5‐year survival | Right heart catheter | Single site |
| Shapiro, Shelley (2012) 51 | United States | 5‐year survival | Right heart catheter | Multicenter |
| Shimony, Avi (2012) 52 | Canada | 1‐year survival | Right heart catheter | Single site |
| Strange, Geoff (2012) 53 | Australia | Prevalence | Right heart catheter | Multicenter |
| Wasywich, C. A. (2012) 54 | New Zealand | 1‐year survival | Electronic data capture | Registry |
| Barst, Robyn (2011) 55 | United States | 1‐year survival | Right Heart Catheter | Registry |
| Frost, Adaani (2011) 44 | United States | Incidence | Right heart catheter | Registry |
| Frost, Adaani (2011) 44 | United States | Prevalence | Right heart catheter | Registry |
| Hurdman, J. (2011) 56 | United Kingdom | 1‐year survival | Electronic data capture | Single site |
| Kirson, Noam Y. (2011) 57 | United States | Prevalence | Electronic data capture | Multicenter |
| Kirson, Noam Y. (2011) 57 | United States | Prevalence | Electronic data capture | Multicenter |
| Low, A. J. (2011) 58 | Australia | 1‐year survival | Right heart catheter | Multicenter |
| Sachdev, A. (2011) 59 | Minnesota | 1‐year survival | Right heart catheter | Single site |
| Van Loon, Rosa Laura E. (2011) 60 | Netherlands | 1‐year Survival | Electronic data capture | Registry |
| Fraisse, Alain (2010) 61 | France | 1‐year survival | Echocardiography + right heart catheter | Multicenter |
| Fraisse, Alain (2010) 61 | France | Prevalence | Echocardiography + right heart catheter | Multicenter |
| Humbert, Marc (2010) 62 | France | 1‐year survival | Right heart catheter | Registry |
| Carrington, Melinda (2008) 63 | Scotland | 1‐year survival | Electronic data capture | Multicenter |
| Kim, Hyung Woo (2008) 64 | South Korea | 5‐year survival | Right heart catheter | Single site |
| Tueller, Claudia (2008) 65 | Switzerland | Prevalence | Right heart catheter | Multicenter |
| Tueller, Claudia (2008) 65 | Switzerland | Incidence | Right heart catheter | Multicenter |
| Tueller, Claudia (2008) 65 | Switzerland | Incidence | Right heart catheter | Multicenter |
| Peacock, A. J. (2007) 66 | Scotland | Prevalence | Electronic data capture | Multicenter |
| Peacock, A. J. (2007) 66 | Scotland | Incidence | Electronic data capture | Multicenter |
| Thenappan, T. (2007) 67 | United States | 1‐year survival | Right heart catheter | Multicenter |
| Humbert, Marc (2006) 68 | France | Prevalence | Right heart catheter | Registry |
| Humbert, Marc (2006) 68 | France | Incidence | Right heart catheter | Registry |
| Sankelo, Marja (2005) 69 | Finland | Prevalence | Electronic data capture | Multicenter |
| Sankelo, Marja (2005) 69 | Finland | Incidence | Electronic data capture | Multicenter |
| Appelbaum, Liat (2001) 70 | Israel | 1‐year survival | Right heart catheter | Multicenter |
| Appelbaum, Liat (2001) 70 | Israel | Prevalence | Right heart catheter | Multicenter |
| Appelbaum, Liat (2001) 70 | Israel | Incidence | Right heart catheter | Multicenter |
| Okada, Osamu (1998) 71 | Japan | 1‐year survival | Right heart catheter | Multicenter |
| Dantzker, David (1994) 72 | United States | 1‐year survival | Right heart catheter | Registry |
| Rajasekhar, D. (1994) 73 | India | 1‐year survival | Right heart catheter | Single site |
| Sandoval, Julio (1994) 74 | Mexico | 1‐year survival | Right heart catheter | Single site |
| D'Alonzo, Gilbert (1991) 75 | United States | 1‐year survival | Right heart catheter | Registry |
Note: First author and publication date of 67 studies reporting prevalence, incidence, or survival of pulmonary arterial hypertension. Location refers to the location of patients or data collection; Measure refers to epidemiologic measures reported in the study; diagnosis type refers to diagnostic criteria used to identify pulmonary arterial hypertension; and site type refers to the type of study or site (e.g., registry, single site) used to identify patients.
Reported prevalence ranged from 0.37 cases/100,000, in a referral center of French children, to 15 cases/100,000, in an Australian study of a large database of echocardiograms (Figure 2). The simple mean of reported prevalence was 3.0 cases/100,000. Twelve studies diagnosed PAH through RHC, one through echocardiography with optional RHC, one through echocardiography and RHC, and three with ICD codes. Two studies reported on primary pulmonary hypertension, while the rest reported on PAH. Restricting to studies diagnosing PAH with RHC, the simple mean of reported prevalence was 3.7 cases/100,000. Restricting to studies diagnosing PAH with ICD codes, the mean of reported prevalence was 1.6 cases/100,000, and reported prevalence ranged from 0.66 cases/100,000 to 3 cases/100,000.
Figure 2.
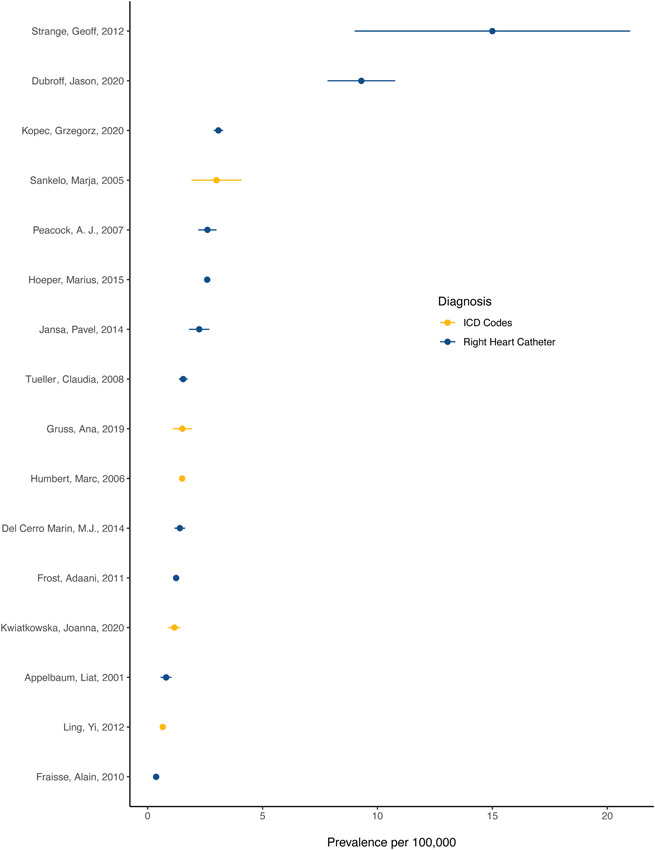
Reported prevalence of pulmonary arterial hypertension in 17 studies identified in systematic review x‐axis text: Prevalence per 100,000. Reported prevalence of pulmonary arterial hypertension per 100,000 as identified by echocardiography, right heart catheterization, echocardiography + right heart catheterization, or electronic data capture. Studies identified by first author and date of publication. Data represent reported prevalence and 95% confidence interval. When a confidence interval was not reported, we used cases and sample size to estimate a standard error using Wilson's score
Reported incidence ranged from 0.008 cases/100,000 person‐years, in a study of discharge records in Finland, to 1.4 cases/100,000 person‐years, in a retrospective chart review at a clinic in Utah, United States (Figure 3). The simple mean of reported incidence was 0.40 cases/100,000 person‐years. Fourteen studies diagnosed PAH through RHC, and three using electronic data capture. No papers reported on the incidence of primary pulmonary hypertension from the older classification schema. Restricting to studies diagnosing PAH with RHC, the simple mean of reported incidence was 0.43 cases/100,000 person‐years. Restricting to studies diagnosing PAH with ICD codes, the mean of reported incidence was 0.21 cases/100,000 person‐years, and reported incidence ranged from 0.11 cases/100,000 person‐years to 0.37 cases/100,000 person‐years.
Figure 3.
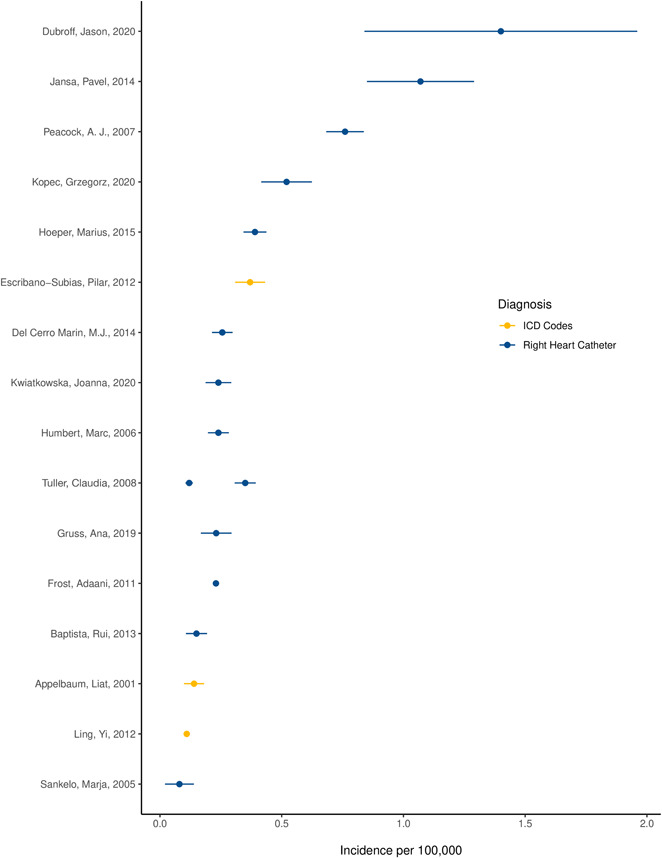
Reported incidence of pulmonary arterial hypertension in 17 studies identified in systemic review x‐axis text: Incidence per 100,000. Reported incidence of pulmonary arterial hypertension per 100,000 as identified by echocardiography or right heart catheterization. Studies identified by first author and date of publication. Data represent reported incidence and 95% confidence interval. When a confidence interval was not reported, we used cases and sample size to estimate a standard error using Wilson's score
Reported 1‐year survival ranged from 67%, in a single‐center observational study in India in 1994, to 99%, in a 2019 study from the national PAH registry in Russia (Figure 4). The average reported 1‐year survival was 86%. Five studies reported 6‐month or 5‐year survival instead of 1‐year survival. Of the sources that reported on mortality, 44 used RHC to diagnose PAH, seven used echocardiography or a combination of echocardiography and RHC, and six with ICD codes. Five studies reported survival for primary pulmonary hypertension. Restricting to studies diagnosing PAH with RHC, the average reported 1‐year survival was 87%. Restricting to studies diagnosing PAH with ICD codes, the average reported 1‐year survival was 84% and reported 1‐year survival ranged from 72% to 91%.
Figure 4.
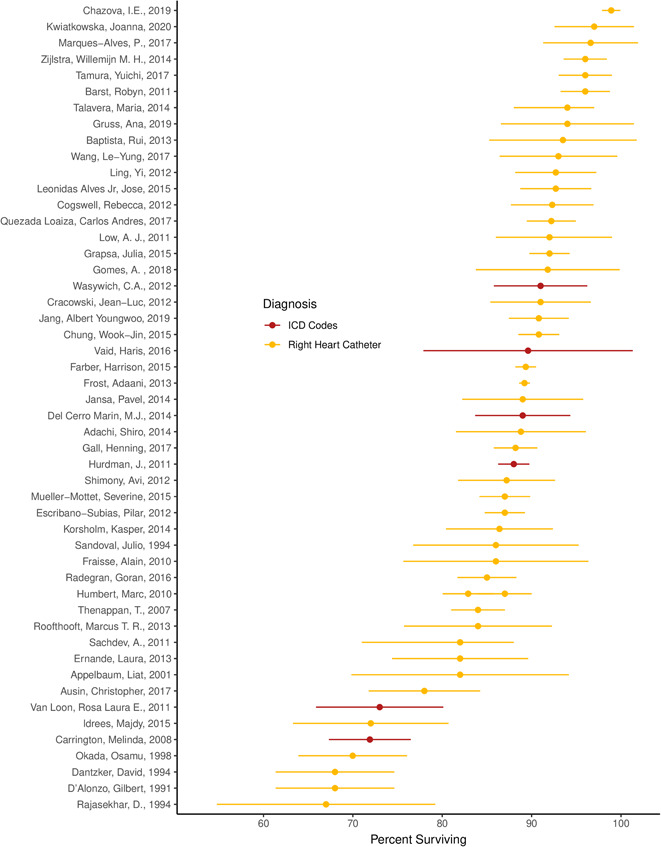
Reported 1‐year survival of pulmonary arterial hypertension in 58 studies identified in systematic review x‐axis text: Percent surviving 1 year. Reported 1‐year survival of pulmonary arterial hypertension per 100,000 as identified by echocardiography, right heart catheterization, echocardiography + right heart catheterization, or electronic data capture. Studies identified by first author and date of publication. Data represent reported incidence and 95% confidence interval. When a confidence interval was not reported, we used cases and sample size to estimate a standard error using Wilson's score
Studies varied in size, case ascertainment strategy, diagnostic criteria, and design. Fifteen papers reported data from a single hospital or referral center, while others reported data from dozens of hospitals, multicenter registries, or national electronic medical systems. Single‐centers include specialty referral centers and nonreferral hospitals. Most studies estimated the catchment area of their center(s) to calculate the prevalence or incidence of PAH; some, like the Australian echocardiography study, performed community‐based sampling or used national insurance databases. Largely, studies with data before 1998 examined primary pulmonary hypertension, and studies with data after 1998 examined PAH. Many studies did not report the diagnostic criteria physicians used to diagnose PAH, and mPAP and echocardiography cutoffs varied slightly between studies. ICD codes used to diagnose PPH and PAH varied over time, due to the transition from ICD‐9 to ICD‐10.
DISCUSSION
Estimates of prevalence, incidence, and mortality varied widely between studies with a 175‐fold difference in incidence between studies and a 40‐fold difference in prevalence. Although there may well be differences in local risk factors for PAH, such as methamphetamine use or congenital heart disease, there are no known risk factors of sufficient strength as to be likely to explain this level of variation. 1 , 6 As such, it is quite likely that a substantial amount of the variability in studies was related to the methodology used by individual studies including the approach to classifying PAH, centralized versus decentralized systems for tabulating PAH cases, and the generalized approach or resource limitations of the unique healthcare system in its ability and rigor in searching out PAH diagnoses. Harmonization of methods for PAH registries, especially diagnostic criteria, would improve efforts at comprehensive disease surveillance.
This search identified 65 studies reporting the prevalence, incidence, and 1‐year survival of PAH between 1991 and 2021 in 37 countries. These data will be used in the upcoming GBD study to estimate the global burden of PAH. The geographies represented in this review are largely in the high‐income world. There were very few studies in Africa, Eastern Europe, South America, and Asia, and papers from low‐ and middle‐income countries often reported survival, not prevalence or incidence. Challenges for the care of pulmonary hypertension in middle‐ and low‐income regions have been well‐described, 76 but further surveillance is required to help understand the geographic variation in PAH incidence, prevalence, and survival.
These estimates are valuable both for resource allocation, but also as a benchmark for individual healthcare systems to understand if they are systematically missing the diagnosis of PAH or applying a label of PAH too liberally. Underdiagnosis has serious implications for individuals who are not recognized to have PAH. Conversely, over‐diagnosis has implications for society who may bear the significant costs for PAH treatment that typically runs to the hundreds of thousands of dollars per patient in highly resourced countries. 77
The current effort found similar estimates of mean PAH prevalence and incidence as previous systematic reviews (incidence 0.4/100,000 person‐years vs. 0.58/100,000 person‐years, prevalence 3/100,000 vs. 5/100,000). 7 The main contribution to differences between estimates was the inclusion of studies dating back to 1990, as well as those where PAH was diagnosed by echocardiography or electronic data capture. We include them to draw attention to the variation in criteria, case ascertainment, and case definition between studies.
Greater availability of PAH therapies has led to an improved prognosis as supported by a number of randomized controlled trials showing efficacy and improvement on current regimens including an impact on mortality. 78 We observed higher mortality in studies reporting on data before 1998 than after 1998, although there were only four studies published before 1998. However, differences in case ascertainment, geographic representativeness, and diagnostic criteria make between‐study comparisons difficult. As such, we would recommend caution about over‐interpreting overall estimates of mortality trends.
Several registries were identified that did not meet our strict inclusion criteria due to exclusion of PH subtypes, however, provide important information on the epidemiology of some types of PAH. 79 , 80 , 81 For example, 1‐year survival in idiopathic and familial PAH was found to be 68% in the Chinese Registry for PAH. 82
Additionally, geographic comparison could provide insights into possible risk factors for PAH, as well as health system effectiveness, and location and comorbidity impacts on treatment. The inclusion of PAH in future iterations of the GBD study may provide the impetus for broader data collection efforts. More sophisticated proteomic and metabolomic approaches to classification may help guide future efforts to assess its true burden. 83 The results of this search will be used to inform GBD estimation of PAH and help to quantify its global burden, thereby guiding global efforts to prioritize and treat this often‐overlooked condition.
CONFLICT OF INTERESTS
Paul A. Corris serves on clinical trial committees for Acceleron, Bayer, and Johnson and Johnson.
ETHICS STATEMENT
This study is part of the Global Burden of Diseases, Injuries, and Risk Factors study. The University of Washington IRB Committee approved the Global Burden of Diseases, Injuries, and Risk Factors study, STUDY00009060.
AUTHOR CONTRIBUTIONS
Ms. Emmons‐Bell, Ms. Boon‐Dooley, Dr. Johnson, and Dr. Roth performed the systematic review. Ms. Emmons‐Bell performed the statistical analysis and prepared tables and figures. Ms. Emmons‐Bell, Dr. Johnson, and Dr. Roth drafted the manuscript. Dr. Corris, Dr. Leary, Dr. Rich, and Dr. Yacoub contributed critical analysis and revision of the manuscript.
Supporting information
Supporting information.
ACKNOWLEDGMENT
We acknowledge collaboration with the Pulmonary Vascular Research Institute in the preparation of this manuscript. This study was supported by the Cardiovascular Medical Research and Education Fund and the Bill and Melinda Gates Foundation. The funders had no role in study design.
Emmons‐Bell S, Johnson C, Boon‐Dooley A, Corris PA, Leary PJ, Rich S, Yacoub M, Roth GA. Prevalence, incidence, and survival of pulmonary arterial hypertension: a systematic review for the global burden of disease 2020 study. Pulm Circ. 2022;12:e12020. 10.1002/pul2.12020
REFERENCES
- 1. GBD 2019 Diseases and Injuries Collaborators . Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. The Lancet. 2020;396:1204–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bigna JJ, Noubiap JJ, Nansseu JR, Aminde LN. Prevalence and etiologies of pulmonary hypertension in Africa: a systematic review and meta‐analysis. BMC Pulm Med. 2017;17:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mocumbi AO, Thienemann F, Sliwa K. A Global perspective on the epidemiology of pulmonary hypertension. Can J Cardiol. 2015;31:375–81. [DOI] [PubMed] [Google Scholar]
- 4. Fernandes CJC, Jardim C, Souza R. The global view. Curr Opin Pulm Med. 2019;25:391–7. [DOI] [PubMed] [Google Scholar]
- 5. Awdish R, Cajigas H. Definition, epidemiology and registries of pulmonary hypertension. Heart Fail Rev. 2016;21:223–8. [DOI] [PubMed] [Google Scholar]
- 6. Hoeper MM, Humbert M, Souza R, Idrees M, Kawut SM, Sliwa‐Hahnle K, Jing ZC, Gibbs JS. A global view of pulmonary hypertension. Lancet Respir Med. 2016;4:306–22. [DOI] [PubMed] [Google Scholar]
- 7. Leber L, Beaudet A, Muller A. Epidemiology of pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension: identification of the most accurate estimates from a systematic literature review. Pulm Circ. 2021;11:2045894020977300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG, Souza R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53:1801913. 10.1183/13993003.01913-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bossone E, D'Andrea A, D'Alto M, Citro R, Argiento P, Ferrara F, Cittadini A, Rubenfire M, Naeije R. Echocardiography in pulmonary arterial hypertension: from diagnosis to prognosis. J Am Soc Echocardiogr. 2013;26:1–14. [DOI] [PubMed] [Google Scholar]
- 10. Swinnen K, Quarck R, Godinas L, Belge C, Delcroix M. Learning from registries in pulmonary arterial hypertension: pitfalls and recommendations. Eur Respir Rev. 2019;28:190050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dubroff J, Melendres L, Lin Y, Beene DR, Ketai L. High geographic prevalence of pulmonary artery hypertension: associations with ethnicity, drug use, and altitude. Pulm Circ. 2020;10:2045894019894534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Khou V, Anderson JJ, Strange G, Corrigan C, Collins N, Celermajer DS, Dwyer N, Feenstra J, Horrigan M, Keating D, Kotlyar E, Lavender M, McWilliams TJ, Steele P, Weintraub R, Whitford H, Whyte K, Williams TJ, Wrobel JP, Keogh A, Lau EM. Diagnostic delay in pulmonary arterial hypertension: insights from the Australian and New Zealand pulmonary hypertension registry. Respirology. 2020;25:863–71. [DOI] [PubMed] [Google Scholar]
- 13. Kopeć G, Kurzyna M, Mroczek E, Chrzanowski Ł, Mularek‐Kubzdela T, Skoczylas I, Kuśmierczyk B, Pruszczyk P, Błaszczak P, Lewicka E, Karasek D, Mizia‐Stec K, Tomaszewski M, Jacheć W, Ptaszyńska‐Kopczyńska K, Peregud‐Pogorzelska M, Doboszyńska A, Pawlak A, Gąsior Z, Zabłocka W, Ryczek R, Widejko‐Pietkiewicz K, Waligóra M, Darocha S, Furdal M, Ciurzyński M, Kasprzak JD, Grabka M, Kamiński K, Hoffman P, Podolec P, Torbicki A. Characterization of patients with pulmonary arterial hypertension: data from the Polish Registry of pulmonary hypertension (BNP‐PL). J Clin Med. 2020;9:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kwiatkowska J, Zuk M, Migdal A, Kusa J, Skiba E, Zygielo K, Przetocka K, Werynski P, Banaszak P, Rzeznik‐Bieniaszewska A, Surmacz R, Bobkowski W, Wojcicka‐Urbanska B, Werner B, Pluzanska J, Ostrowska K, Waldoch A, Kopec G. Children and adolescents with pulmonary arterial hypertension: baseline and follow‐up data from the polish registry of pulmonary hypertension (BNP‐PL). J Clin Med. 2020;9:1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chazova IE, Arkhipova OA, Martynyuk TV. Pulmonary arterial hypertension in Russia: six‐year observation analysis of the National Registry. Ter Arkh. 2019;91:25–31. [DOI] [PubMed] [Google Scholar]
- 16. Gruss AI, Pascal G, Chao C, Janssen B, Bedó C, Salisbury JP, Trujillo P, Curbelo P, Grignola JC. Diez años de experiencia de un centro de referencia en hipertensión arterial pulmonar en Uruguay. Rev Médica Urug. 2019;35:193–202. https://revista.rmu.org.uy/ojsrmu311/index.php/rmu/article/view/117 [Google Scholar]
- 17. Jang AY, Chung W‐J. Current status of pulmonary arterial hypertension in Korea. Korean J Intern Med. 2019;34:696–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gomes A, Cruz C, Rocha J, Ricardo M, Vicente M, Melo A, Santos M, Carvalho L, Gonçalves F, Reis A. Pulmonary hypertension: real‐world data from a Portuguese expert referral centre. Pulmonology. 2018;24:231–40. [DOI] [PubMed] [Google Scholar]
- 19. Strange G, Lau EM, Giannoulatou E, Corrigan C, Kotlyar E, Kermeen F, Williams T, Celermajer DS, Dwyer N, Whitford H, Wrobel JP, Feenstra J, Lavender M, Whyte K, Collins N, Steele P, Proudman S, Thakkar V, Keating D, Keogh A, PHSANZ R. Survival of idiopathic pulmonary arterial hypertension patients in the modern era in Australia and New Zealand. Heart Lung Circ. 2018;27:1368–75. [DOI] [PubMed] [Google Scholar]
- 20. Austin C, Burger C, Kane G, Safford R, Blackshear J, Ung R, Ray J, Alsaad A, Alassas K, Shapiro B. High‐risk echocardiographic features predict mortality in pulmonary arterial hypertension. Am Heart J. 2017;189:167–76. [DOI] [PubMed] [Google Scholar]
- 21. Gall H, Felix JF, Schneck FK, Milger K, Sommer N, Voswinckel R, Franco OH, Hofman A, Schermuly RT, Weissmann N, Grimminger F, Seeger W, Ghofrani HA. The Giessen Pulmonary Hypertension Registry: survival in pulmonary hypertension subgroups. J Heart Lung Transplant. 2017;36:957–67. [DOI] [PubMed] [Google Scholar]
- 22. Marques‐Alves P, Baptista R, Marinho da Silva A, Pêgo M, Castro G. Real‐world, long‐term survival of incident patients with pulmonary arterial hypertension. Rev Port Pneumol Engl Ed. 2017;23:124–31. [DOI] [PubMed] [Google Scholar]
- 23. Quezada Loaiza CA, Velázquez Martín MT, Jiménez López‐Guarch, Ruiz Cano MJ, Navas Tejedor P, Carreira PE, Flox Camacho Á, de Pablo Gafas A, Delgado Jiménez JF, Gómez Sánchez MÁ, Escribano Subías P. Trends in pulmonary hypertension over a period of 30 years: experience from a single referral centre. Rev Esp Cardiol Engl Ed. 2017;70:915–23. [DOI] [PubMed] [Google Scholar]
- 24. Tamura Y, Kumamaru H, Satoh T, Miyata H, Ogawa A, Tanabe N, Hatano M, Yao A, Abe K, Tsujino I, Fukuda K, Kimura H, Kuwana M, Matsubara H, Tatsumi K, Japan PH Registry (JAPHR) Network . Effectiveness and outcome of pulmonary arterial hypertension‐specific therapy in Japanese patients with pulmonary arterial hypertension. Circ J. 2017;82:275–82. [DOI] [PubMed] [Google Scholar]
- 25. Wang L‐Y, Lee K‐T, Lin C‐P, Hsu L‐A, Wang C‐L, Hsu T‐S, Ho W‐J. Long‐term survival of patients with pulmonary arterial hypertension at a single center in Taiwan. Acta Cardiol Sin. 2017;33:498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rådegran G, Kjellström B, Ekmehag B, Larsen F, Rundqvist B, Blomquist SB, Gustafsson C, Hesselstrand R, Karlsson M, Kornhall B, Nisell M, Persson L, Ryftenius H, Selin M, Ullman B, Wall K, Wikström G, Willehadson M, Jansson K, Stefan Söderberg j, on behalf of SveFPH and S . Characteristics and survival of adult Swedish PAH and CTEPH patients 2000‐2014. Scand Cardiovasc J SCJ. 2016;50:243–50. [DOI] [PubMed] [Google Scholar]
- 27. Thienemann F, Dzudie A, Mocumbi AO, Blauwet L, Sani MU, Karaye KM, Ogah OS, Mbanze I, Mbakwem A, Udo P, Tibazarwa K, Damasceno A, Keates AK, Stewart S, Sliwa K. The causes, treatment, and outcome of pulmonary hypertension in Africa: insights from the Pan African Pulmonary Hypertension Cohort (PAPUCO) Registry. Int J Cardiol. 2016;221:205–11. [DOI] [PubMed] [Google Scholar]
- 28. Vaid HM, Camacho X, Granton JT, Mamdani MM, Yao Z, Singh S, Juurlink DN, Gomes T. The Characteristics of treated pulmonary arterial hypertension patients in Ontario. Can Respir J. 2016;2016:6279250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chung W‐J, Park YB, Jeon CH, Jung JW, Ko KP, Choi SJ, Seo HS, Lee JS, Jung HO, KORPAH I. Baseline characteristics of the Korean Registry of pulmonary arterial hypertension. J Korean Med Sci. 2015;30:1429–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Farber HW, Miller DP, Poms AD, Badesch DB, Frost AE, Muros‐Le Rouzic E, Romero AJ, Benton WW, Elliott CG, McGoon MD, Benza RL. Five‐year outcomes of patients enrolled in the REVEAL Registry. Chest. 2015;148:1043–54. [DOI] [PubMed] [Google Scholar]
- 31. Grapsa J, Pereira Nunes MC, Tan TC, Cabrita IZ, Coulter T, Smith BC, Dawson D, Gibbs JS, Nihoyannopoulos P. Echocardiographic and hemodynamic predictors of survival in precapillary pulmonary hypertension: seven‐year follow‐up. Circ Cardiovasc Imaging. 2015;8:e002107. [DOI] [PubMed] [Google Scholar]
- 32. Hoeper MM, Huscher D, Pittrow D. Incidence and prevalence of pulmonary arterial hypertension in Germany. Int J Cardiol. 2016;203:612–3. [DOI] [PubMed] [Google Scholar]
- 33. Idrees M, Alnajashi K, Abdulhameed J, Khan A, Batubara E, Alotay A, Fayed A, Aldammas S, Alseif M, Alawwad H, Abusabaa Y, Almobrad M, Kashour T, Registry Taskforce S. Saudi experience in the management of pulmonary arterial hypertension; the outcome of PAH therapy with the exclusion of chronic parenteral prostacyclin. Ann Thorac Med. 2015;10:204–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Alves JL Jr, Gavilanes F, Jardim C, Fernandes C, Morinaga L, Dias B, Hoette S, Humbert M, Souza R. Pulmonary arterial hypertension in the southern hemisphere: results from a registry of incident Brazilian cases. Chest. 2015;147:495–501. [DOI] [PubMed] [Google Scholar]
- 35. Mueller‐Mottet S, Stricker H, Domenighetti G, Azzola A, Geiser T, Schwerzmann M, Weilenmann D, Schoch O, Fellrath JM, Rochat T, Lador F, Beghetti M, Nicod L, Aubert JD, Popov V, Speich R, Keusch S, Hasler E, Huber LC, Grendelmeier P, Tamm M, Ulrich S. Long‐term data from the Swiss pulmonary hypertension registry. Respir Int Rev Thorac Dis. 2015;89:127–40. [DOI] [PubMed] [Google Scholar]
- 36. Adachi S, Hirashiki A, Nakano Y, Shimazu S, Murohara T, Kondo T. Prognostic factors in pulmonary arterial hypertension with Dana Point group 1. Life Sci. 2014;118:404–9. [DOI] [PubMed] [Google Scholar]
- 37. Marín MJC, Rotés AS, Ogando AR, Soto AM, Jiménez MQ, Camacho JLG, Sonnenfeld IR, Bonora AM, Brotons DCA, Galdó AM. Assessing pulmonary hypertensive vascular disease in childhood. Data from the Spanish Registry. Am J Respir Crit Care Med. 2014;190:1421–29. [DOI] [PubMed] [Google Scholar]
- 38. Jansa P, Jarkovsky J, Al‐Hiti H, Popelova J, Ambroz D, Zatocil T, Votavova R, Polacek P, Maresova J, Aschermann M, Brabec P, Dusek L, Linhart A. Epidemiology and long‐term survival of pulmonary arterial hypertension in the Czech Republic: a retrospective analysis of a nationwide registry. BMC Pulm Med. 2014;14:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Korsholm K, Andersen A, Kirkfeldt RE, Hansen KN, Mellemkjær S, Nielsen‐Kudsk JE. Survival in an incident cohort of patients with pulmonary arterial hypertension in Denmark. Pulm Circ. 2015;5:364–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Talavera ML, Cáneva JO, Favaloro LE, Klein F, Boughen RP, Bozovich GE, Ossés JM, Favaloro RR, Bertolotti AM. Hipertensión arterial pulmonar. Registro de un centro de referencia en Argentina. Revista Americana de Medicina Respiratoria. 2014;14:9. http://www.ramr.org/articulos/volumen_14_numero_2/articulos_originales/articulos_originales_hipertension_arterial_pulmonar_registro_centro_referencia_argentina.pdf [Google Scholar]
- 41. Zijlstra WMH, Douwes JM, Rosenzweig EB, Schokker S, Krishnan U, Roofthooft M, Miller‐Reed K, Hillege HL, Ivy DD, Berger R. Survival differences in pediatric pulmonary arterial hypertension: clues to a better understanding of outcome and optimal treatment strategies. J Am Coll Cardiol. 2014;63:2159–69. [DOI] [PubMed] [Google Scholar]
- 42. Baptista R, Meireles J, Agapito A, Castro G, da Silva AM, Shiang T, Gonçalves F, Robalo‐Martins S, Nunes‐Diogo A, Reis A. Pulmonary hypertension in Portugal: first data from a nationwide registry. BioMed Res Int. 2013;2013:489574. https://pubmed.ncbi.nlm.nih.gov/24228252/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ernande L, Cottin V, Leroux P‐Y, Girerd N, Huez S, Mulliez A, Bergerot C, Ovize M, Mornex JF, Cordier JF, Naeije R, Derumeaux G. Right isovolumic contraction velocity predicts survival in pulmonary hypertension. J Am Soc Echocardiogr. 2013;26:297–306. [DOI] [PubMed] [Google Scholar]
- 44. Frost AE, Badesch DB, Miller DP, Benza RL, Meltzer LA, McGoon MD. Evaluation of the predictive value of a clinical worsening definition using 2‐year outcomes in patients with pulmonary arterial hypertension: a REVEAL Registry analysis. Chest. 2013;144:1521–9. [DOI] [PubMed] [Google Scholar]
- 45. Roofthooft MT, Douwes JM, Vrijlandt EJ, Berger RM. Frequency and prognostic significance of hemoptysis in pediatric pulmonary arterial hypertension. Am J Cardiol. 2013;112:1505–9. [DOI] [PubMed] [Google Scholar]
- 46. Cogswell R, Kobashigawa E, McGlothlin D, Shaw R, De Marco T. Validation of the Registry to Evaluate Early and Long‐Term Pulmonary Arterial Hypertension Disease Management (REVEAL) pulmonary hypertension prediction model in a unique population and utility in the prediction of long‐term survival. J Heart Lung Transplant. 2012;31:1165–70. [DOI] [PubMed] [Google Scholar]
- 47. Cracowski J‐L, Degano B, Chabot F, Labarère J, Schwedhelm E, Monneret D, Iuliano L, Schwebel C, Chaouat A, Reynaud‐Gaubert M, Faure P, Maas R, Renversez JC, Cracowski C, Sitbon O, Yaïci A, Simonneau G, Humbert M. Independent association of urinary F2‐isoprostanes with survival in pulmonary arterial hypertension. Chest. 2012;142:869–76. [DOI] [PubMed] [Google Scholar]
- 48. Escribano‐Subias P, Blanco I, López‐Meseguer M, Lopez‐Guarch CJ, Roman A, Morales P, Castillo‐Palma MJ, Segovia J, Gómez‐Sanchez MA, Barberà JA, REHAP i. Survival in pulmonary hypertension in Spain: insights from the Spanish registry. Eur Respir J. 2012;40:596–603. [DOI] [PubMed] [Google Scholar]
- 49. Ling Y, Johnson MK, Kiely DG, Condliffe R, Elliot CA, Gibbs JS, Howard LS, Pepke‐Zaba J, Sheares KK, Corris PA, Fisher AJ, Lordan JL, Gaine S, Coghlan JG, Wort SJ, Gatzoulis MA, Peacock AJ. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med. 2012;186:790–6. [DOI] [PubMed] [Google Scholar]
- 50. Sakao S, Tanabe N, Kasahara Y, Tatsumi K. Survival of Japanese patients with pulmonary arterial hypertension after the introduction of endothelin receptor antagonists and/or phosphodiesterase type‐5 inhibitors. Intern Med Tokyo Jpn. 2012;51:2721–6. [DOI] [PubMed] [Google Scholar]
- 51. Shapiro S, Traiger GL, Turner M, McGoon MD, Wason P, Barst RJ. Sex differences in the diagnosis, treatment, and outcome of patients with pulmonary arterial hypertension enrolled in the registry to evaluate early and long‐term pulmonary arterial hypertension disease management. Chest. 2012;141:363–73. https://pubmed.ncbi.nlm.nih.gov/21757572/ [DOI] [PubMed] [Google Scholar]
- 52. Shimony A, Fox BD, Afilalo J, Rudski LG, Hirsch A, Langleben D. Pulmonary arterial hypertension in the elderly‐clinical characteristics and long‐term survival. Lung. 2012;190:645–9. https://pubmed.ncbi.nlm.nih.gov/23064491/ [DOI] [PubMed] [Google Scholar]
- 53. Strange G, Playford D, Stewart S, Deague JA, Nelson H, Kent A, Gabbay E. Pulmonary hypertension: prevalence and mortality in the Armadale echocardiography cohort. Heart Br Card Soc. 2012;98:1805–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wasywich CA, Cicovic A, McWilliams T, Coverdale HA, Stewart C, Whyte K. Building a pulmonary vascular service: the 12‐year experience and outcomes of the Auckland pulmonary arterial hypertension clinic. Intern Med J. 2013;43:635–42. [DOI] [PubMed] [Google Scholar]
- 55. Barst RJ, Ivy DD, Foreman AJ, McGoon MD, Rosenzweig EB. Four‐ and seven‐year outcomes of patients with congenital heart disease‐associated pulmonary arterial hypertension (from the REVEAL Registry). Am J Cardiol. 2014;113:147–55. [DOI] [PubMed] [Google Scholar]
- 56. Hurdman J, Condliffe R, Elliot CA, Davies C, Hill C, Wild JM, Capener D, Sephton P, Hamilton N, Armstrong IJ, Billings C, Lawrie A, Sabroe I, Akil M, O'Toole L, Kiely DG. ASPIRE registry: Assessing the Spectrum of Pulmonary hypertension Identified at a REferral centre. Eur Respir J. 2012;39:945–55. [DOI] [PubMed] [Google Scholar]
- 57. Kirson NY, Birnbaum HG, Ivanova JI, Waldman T, Joish V, Williamson T. Prevalence of pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension in the United States. Curr Med Res Opin. 2011;27:1763–8. [DOI] [PubMed] [Google Scholar]
- 58. Low AJ, Fowler D, Manghani MK, Young I, Garsia R, Torzillo P, Youssef P, Celermajer DS. Screening and treating pulmonary arterial hypertension in a tertiary hospital‐based multidisciplinary clinic: the first 200 patients. Intern Med J. 2013;43:32–7. [DOI] [PubMed] [Google Scholar]
- 59. Sachdev A, Villarraga HR, Frantz RP, McGoon MD, Hsiao JF, Maalouf JF, Ammash NM, McCully RB, Miller FA, Pellikka PA, Oh JK, Kane GC. Right ventricular strain for prediction of survival in patients with pulmonary arterial hypertension. Chest. 2011;139:1299–1309. [DOI] [PubMed] [Google Scholar]
- 60. van Loon RL, Roofthooft MT, Hillege HL, ten Harkel AD, van Osch‐Gevers M, Delhaas T, Kapusta L, Strengers JL, Rammeloo L, Clur SA, Mulder BJ, Berger RM. Pediatric pulmonary hypertension in the Netherlands: epidemiology and characterization during the period 1991 to 2005. Circulation. 2011;124:1755–64. [DOI] [PubMed] [Google Scholar]
- 61. Fraisse A, Jais X, Schleich J‐M, di Filippo S, Maragnès P, Beghetti M, Gressin V, Voisin M, Dauphin C, Clerson P, Godart F, Bonnet D. Characteristics and prospective 2‐year follow‐up of children with pulmonary arterial hypertension in France. Arch Cardiovasc Dis. 2010;103:66–74. [DOI] [PubMed] [Google Scholar]
- 62. Humbert M, Sitbon O, Yaïci A, Montani D, O'Callaghan DS, Jaïs X, Parent F, Savale L, Natali D, Günther S, Chaouat A, Chabot F, Cordier JF, Habib G, Gressin V, Jing ZC, Souza R, Simonneau G, French Pulmonary Arterial Hypertension Network . Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J. 2010;36:549–55. [DOI] [PubMed] [Google Scholar]
- 63. Carrington M, Murphy NF, Strange G, Peacock A, McMurray JJ, Stewart S. Prognostic impact of pulmonary arterial hypertension: a population‐based analysis. Int J Cardiol. 2008;124:183–7. [DOI] [PubMed] [Google Scholar]
- 64. Kim HW, Kim GB, Je HG, Beak JS, Bae EJ, Noh CI, Choi JY, Yun YS. Pulmonary arterial hypertension in children: a single center experience. Korean Circ J. 2008;38:644–50. https://e-kcj.org/DOIx.php?id=10.4070/kcj.2008.38.12.644 [Google Scholar]
- 65. Tueller C, Stricker H, Soccal P, Tamm M, Aubert JD, Maggiorini M, Zwahlen M, Nicod L, Swiss Society for Pulmonary H. Epidemiology of pulmonary hypertension: new data from the Swiss registry. Swiss Med Wkly. 2008;138:379–84. [DOI] [PubMed] [Google Scholar]
- 66. Peacock AJ, Murphy NF, McMurray JJ, Caballero L, Stewart S. An epidemiological study of pulmonary arterial hypertension. Eur Respir J. 2007;30:104–9. [DOI] [PubMed] [Google Scholar]
- 67. Thenappan T, Shah SJ, Rich S, Gomberg‐Maitland M. A USA‐based registry for pulmonary arterial hypertension: 1982‐2006. Eur Respir J. 2007;30:1103–10. [DOI] [PubMed] [Google Scholar]
- 68. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaici A, Weitzenblum E, Cordier JF, Chabot F, Dromer C, Pison C, Reynaud‐Gaubert M, Haloun A, Laurent M, Hachulla E, Simonneau G. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med. 2006;173:1023–30. [DOI] [PubMed] [Google Scholar]
- 69. Sankelo M, Flanagan JA, Machado R, Harrison R, Rudarakanchana N, Morrell N, Dixon M, Halme M, Puolijoki H, Kere J, Elomaa O, Kupari M, Räisänen‐Sokolowski A, Trembath RC, Laitinen T. BMPR2 mutations have short lifetime expectancy in primary pulmonary hypertension. Hum Mutat. 2005;26:119–24. [DOI] [PubMed] [Google Scholar]
- 70. Appelbaum L, Yigla M, Bendayan D, Reichart N, Fink G, Priel I, Schwartz Y, Richman P, Picard E, Goldman S, Kramer MR. Primary pulmonary hypertension in Israel: a national survey. Chest. 2001;119:1801–6. [DOI] [PubMed] [Google Scholar]
- 71. Okada O, Tanabe N, Yasuda J, Yoshida Y, Katoh K, Yamamoto T, Kuriyama T. Prediction of life expectancy in patients with primary pulmonary hypertension. A retrospective nationwide survey from 1980‐1990. Intern Med Tokyo Jpn. 1999;38:12–6. [DOI] [PubMed] [Google Scholar]
- 72. Dantzker DR. Primary pulmonary hypertension: the American experience. Chest. 1994;105:26S–8S. https://pubmed.ncbi.nlm.nih.gov/8306804/ [DOI] [PubMed] [Google Scholar]
- 73. Rajasekhar D, Balakrishnan KG, Venkitachalam CG, Tharakan JA, Titus T, Subramanian R, Kumar VK, Bhat A, Pillai VR. Primary pulmonary hypertension: natural history and prognostic factors. Indian Heart J. 1994;46:165–70. [PubMed] [Google Scholar]
- 74. Sandoval J, Bauerle O, Palomar A, Gómez A, Martínez‐Guerra ML, Beltrán M, Guerrero ML. Survival in primary pulmonary hypertension. Validation of a prognostic equation. Circulation. 1994;89:1733–44. [DOI] [PubMed] [Google Scholar]
- 75. D'alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, Fishman AP, Goldring RM, Groves BM, Kernis JT. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115:343–9. [DOI] [PubMed] [Google Scholar]
- 76. Hasan B, Hansmann G, Budts W, Heath A, Hoodbhoy Z, Jing ZC, Koestenberger M, Meinel K, Mocumbi AO, Radchenko GD, Sallmon H, Sliwa K, Kumar RK, European Pediatric Pulmonary Vascular Disease Network (EPPVDN), endorsed by AEPC, CSC, PASCAR, PCS, and PCSI . Challenges and special aspects of pulmonary hypertension in middle‐ to low‐income regions: JACC state‐of‐the‐art review. J Am Coll Cardiol. 2020;75:2463–77. [DOI] [PubMed] [Google Scholar]
- 77. Sikirica M, Iorga SR, Bancroft T, Potash J. The economic burden of pulmonary arterial hypertension (PAH) in the US on payers and patients. BMC Health Serv Res. 2014;14:676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Coeytaux RR, Schmit KM, Kraft BD, Kosinski AS, Mingo AM, Vann LM, Gilstrap DL, Hargett CW, Heidenfelder B, Dolor RJ, McCrory DC. Comparative effectiveness and safety of drug therapy for pulmonary arterial hypertension: a systematic review and meta‐analysis. Chest. 2014;145:P1055–63. [DOI] [PubMed] [Google Scholar]
- 79. He J. National, Prospective, Multicenter, Observational Registry Study on pulmonary arterial hypertension or chronic thromboembolic pulmonary hypertension in China. 2017. [cited 2021 July 6]. Clinical Trial Registration NCT01417338. Available from: https://clinicaltrials.gov/ct2/show/NCT01417338
- 80. Hemnes AR, Beck GJ, Newman JH, Abidov A, Aldred MA, Barnard J, Berman Rosenzweig E, Borlaug BA, Chung WK, Comhair S, Erzurum SC, Frantz RP, Gray MP, Grunig G, Hassoun PM, Hill NS, Horn EM, Hu B, Lempel JK, Maron BA, Mathai SC, Olman MA, Rischard FP, Systrom DM, Tang W, Waxman AB, Xiao L, Yuan JX, Leopold JA, PVDOMICS Study G. PVDOMICS: a multi‐center study to improve understanding of pulmonary vascular disease through phenomics. Circ Res. 2017;121:1136–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pulmonary Hypertension Association. 2021. Pulmonary Hypertension Association Registry (PHAR). [cited 2021 July 7]. Available from: https://phassociation.org/phar/
- 82. Jing Z‐C, Xu X‐Q, Han Z‐Y, Wu Y, Deng KW, Wang H, Wang ZW, Cheng XS, Xu B, Hu SS, Hui RT, Yang YJ. Registry and survival study in Chinese patients with idiopathic and familial pulmonary arterial hypertension. Chest. 2007;132:373–9. https://journal.chestnet.org/article/S0012-3692(15)37426-2/fulltext [DOI] [PubMed] [Google Scholar]
- 83. Oldham WM, Hemnes AR, Aldred MA, Barnard J, Brittain EL, Chan SY, Cheng F, Cho MH, Desai AA, Garcia J, Geraci MW, Ghiassian SD, Hall KT, Horn EM, Jain M, Kelly RS, Leopold JA, Lindstrom S, Modena BD, Nichols WC, Rhodes CJ, Sun W, Sweatt AJ, Vanderpool RR, Wilkins MR, Wilmot B, Zamanian RT, Fessel JP, Aggarwal NR, Loscalzo J, Xiao L. NHLBI‐CMREF workshop report on pulmonary vascular disease classification: JACC state‐of‐the‐art review. J Am Coll Cardiol. 2021;77:2040–52. https://pubmed.ncbi.nlm.nih.gov/33888254/ [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.