

Abstract
BACKGROUND & AIMS:
In celiac disease (CeD), gluten induces immune activation, leading to enteropathy. TAK-101, gluten protein (gliadin) encapsulated in negatively charged poly(dl-lactide-co-glycolic acid) nanoparticles, is designed to induce gluten-specific tolerance.
METHODS:
TAK-101 was evaluated in phase 1 dose escalation safety and phase 2a double-blind, randomized, placebo-controlled studies. Primary endpoints included pharmacokinetics, safety, and tolerability of TAK-101 (phase 1) and change from baseline in circulating gliadinspecific interferon-γ–producing cells at day 6 of gluten challenge, in patients with CeD (phase 2a). Secondary endpoints in the phase 2a study included changes from baseline in enteropathy (villus height to crypt depth ratio [Vh:Cd]), and frequency of intestinal intraepithelial lymphocytes and peripheral gut-homing T cells.
RESULTS:
In phase 2a, 33 randomized patients completed the 14-day gluten challenge. TAK-101 induced an 88% reduction in change from baseline in interferon-γ spot-forming units vs placebo (2.01 vs 17.58, P = .006). Vh:Cd deteriorated in the placebo group (−0.63, P = .002), but not in the TAK-101 group (−0.18, P = .110), although the intergroup change from baseline was not significant (P = .08). Intraepithelial lymphocyte numbers remained equal. TAK-101 reduced changes in circulating α4β7+CD4+ (0.26 vs 1.05, P = .032), αEβ7+CD8+ (0.69 vs 3.64, P = .003), and γδ (0.15 vs 1.59, P = .010) effector memory T cells. TAK-101 (up to 8 mg/kg) induced no clinically meaningful changes in vital signs or routine clinical laboratory evaluations. No serious adverse events occurred.
CONCLUSIONS:
TAK-101 was well tolerated and prevented gluten-induced immune activation in CeD. The findings from the present clinical trial suggest that antigen-specific tolerance was induced and represent a novel approach translatable to other immune-mediated diseases. ClinicalTrials.gov identifiers: NCT03486990 and NCT03738475.
Keywords: Gliadin, Antigen-specific Immune Tolerance
Graphical Abstract

Celiac disease (CeD) is an immune-mediated disorder triggered by gluten ingestion in individuals expressing human leukocyte antigen (HLA)-DQ2 or HLA-DQ8, resulting in damage to the small intestine.1 CeD is one of the most common autoimmune disorders, affecting more than 1% of the global population.2
Ingestion of prolamin proteins, primarily gliadin, results in an abnormal T-cell response in patients with CeD. After gluten exposure, gliadin is processed and presented to CD4+ T cells, initiating an inflammatory reaction characterized by CD4+ T cell production of interferon (IFN)-γ and the activation of CD8+ T cells and γδ T cells that express the gut-homing or retention integrins α4β7 or αEβ7.3–5 The activated CD8+ T cells increase in the small intestine epithelium with gluten exposure and are thought to mediate intestinal damage.1,6–8
Manifestations of CeD are diverse, and include gastrointestinal symptoms and extraintestinal disorders, including anemia, osteoporosis, neurologic disease, and dermatitis herpetiformis.9 No medications are currently approved for the treatment of CeD, and the only available management strategy is a gluten-free diet (GFD). Diet modification alone is inadequate to achieve clinical and histologic remission in many patients owing to heterogeneous sensitivity to gluten and frequent inadvertent gluten exposure from widespread contamination of food.10–12 In addition, the requirement for lifelong adherence to a strict GFD places a high burden on both patients and caregivers. There is therefore a critical unmet need for effective nondietary therapies for CeD.
Induction of gliadin-specific immune tolerance is a promising therapeutic solution for CeD that targets mechanisms initiating disease pathology instead of mitigating the effects of gluten exposure. Tolerogenic inhibition of specific immune responses is a highly sought-after therapeutic goal for immune-mediated diseases, including autoimmunity, allergy, and transplant rejection. Negatively charged poly(dl-lactide-co-glycolide) (PLGA)-antigen (Ag) nanoparticles have been developed to deliver specific antigens that induce tolerogenic inhibition via a non-inflammatory process. In rodents, PLGA-Ag nanoparticle-induced tolerance to model antigen is dependent on particle uptake via the macrophage receptor with collagenous structure (MARCO) scavenger receptor by tolerogenic antigen-presenting cells (APCs) in the splenic marginal zone and liver.12,13 These APCs lead to anergy within Ag-specific effector T cells and activate populations of Ag-specific regulatory T cells.13–16 This approach has been shown to be effective in mouse experimental autoimmune encephalomyelitis, murine type 1 diabetes induced by adoptive transfer of activated diabetogenic epitope-specific CD4+ or CD8+ transgenic T cells, and a mouse model of CeD induced by transfer of gliadin-specific T cells.15,17–22 TAK-101 (formerly TIMP-GLIA), is composed of gliadin encapsulated in PLGA-Ag nanoparticles. We posited that TAK-101 may induce immune tolerance sufficient for the treatment of CeD.
Here, we present data for the safety, tolerability, pharmacokinetics (PK), and efficacy of TAK-101 from the induction of gliadin-specific T-cell tolerance in patients with biopsy-confirmed CeD in phase 1 and phase 2a studies.
Methods
Trial Design
These studies complied with the ethical principles of Good Clinical Practice in accordance with the World Medical Association Declaration of Helsinki: ethical principle for medical research involving human subjects.23 Written informed consent was obtained from all participants, and all study procedures were performed under institutional review board approval. All authors had access to the study data and reviewed and approved the final manuscript.
Patients were aged 18 to 75 years (phase 1) or 18 to 70 years (phase 2a), were HLA-DQ2 or HLA-DQ8 positive with biopsy-confirmed CeD, and maintained a GFD for ≥6 months before screening, with quiescent CeD symptoms and negative serum anti-tissue transglutaminase 2 immunoglobin A. Patients were required to have no known gluten exposure for at least 10 days before screening and to be willing to maintain a GFD during the study.
Phase 1 study (NCT03486990).
A first-in-human safety study was conducted from January 2018 to May 2019 (see Supplementary Material): TAK-101 was evaluated in patients with CeD in a 2-part trial consisting of a single ascending dose cohort (Part A) and a repeated ascending dose cohort (Part B; 2 doses: day 1 and 8). TAK-101 (0.1–8 mg/kg up to a maximum of 650 mg) was administered by a 30-minute intravenous infusion in accordance with the dose escalation shown in Figure 1A.
Figure 1.

Phase 1 and phase 2a study designs. (A) Phase 1 safety study (n = 23): Part A (single ascending intravenous doses of TAK-101, n = 17) followed by Part B (2 ascending intravenous doses administered on days 1 and 8, n = 6) assessed the safety, tolerability, and pharmacokinetics of TAK-101 and established the dose to be used in the phase 2a study. (B) Phase 2a proof-of-concept study (n = 34) in patients infused with placebo or TAK-101, 8 mg/kg, on days 1 and 8. All patients underwent a 14-day oral GC beginning on day 15 consisting of 12 g/d for the first 3 days and 6 g/d for the following 11 days. BL, baseline; GC, gluten challenge. aDay 29 was equivalent to 1 day after the 14-day gluten challenge.
Phase 2a Study (NCT03738475).
A randomized, double-blind, proof-of-concept study was conducted from January 2019 to July 2019 (see Supplementary Material). Patients with CeD were randomized in a 1:1 ratio (via iMedNet electronic case report forms on day 1) to pretreatment with 2 intravenous doses of placebo (normal saline) or TAK-101 (8 mg/kg, up to a maximum of 650 mg) on days 1 and 8, via a 30-minute intravenous infusion. All patients subsequently underwent a 14-day oral gluten challenge (12 g/d of gluten for 3 days followed by 6 g/d for 11 days beginning 7 days after the second infusion of TAK-101 or placebo) (Figure 1B). Investigators, patients, and all study staff with direct patient contact were blinded to treatment assignment. A designated unblinded pharmacist (or otherwise qualified personnel) at each site prepared each dose and had no contact with the patients and minimal contact with other site study personnel. The study protocol is registered and accessible at ClinicalTrials.gov (NCT03738475).
Study Drug
Details of TAK-101 nanoparticle synthesis and characterization are provided in the Supplementary Materials.
Endpoint Measures
Phase 1 study.
The primary endpoint of the phase 1 study was an evaluation of the safety and tolerability of TAK-101 administered intravenously in patients with CeD. Secondary endpoints were the PK characteristics of TAK-101 (measured as plasma nanoparticle-free gliadin concentrations) and establishment of the dose with the best safety and tolerability profile for the phase 2a study.
Phase 2a study.
The primary endpoint of the phase 2a study was change from baseline in T-cell–mediated, gluten-stimulated IFN-γ production in peripheral blood mononuclear cells (PBMCs) on day 6 of gluten challenge in patients treated with TAK-101 compared with placebo, determined using an enzyme-linked immunospot (ELISpot) assay to measure the number of gliadin-specific IFN-γ spot-forming units (SFUs).
Secondary endpoints included the following: reduction in damage to the small intestinal mucosa, measured as the change from baseline in villus height to crypt depth ratio (Vh:Cd) and change in intraepithelial lymphocyte (IEL) density using quantitative histologic assessment of duodenal biopsies and the proportion of participants who have a ≥0.4 decrease in Vh:Cd; the change in percentage of activated CD4+, CD8+, and γδ+ effector memory T cells in the blood expressing either the α4β7 gut-homing or αEβ7 gut-retention integrins, measured using time-of-flight mass cytometry (CyTOF); and gliadin-specific ex vivo T-cell proliferation and cytokine secretion after oral gluten challenge. Measurement of the proportion of other immune cell phenotypes was also conducted using CyTOF, after gluten challenge in patients treated with TAK-101 or placebo. Further secondary endpoints included PK (serum concentration of gliadin) and safety (adverse events [AEs], serious adverse events [SAEs], vital signs, changes in serum deamidated gliadin peptide immunoglobulin G levels, serum complement and cytokine levels, and hematology and serum chemistry).
ELISpot Assay
ELISpot assays for gliadin-specific T-cell–mediated IFN-γ production in PBMCs were performed to measure the increase in the amount of IFN-γ producing T cells after 6 days of gluten challenge.24 PBMCs were collected before gluten challenge and 6 days after the start of gluten challenge. Briefly, PBMCs were isolated from heparinized whole blood using Ficoll-Paque density gradient separation (Lymphoprep; Stemcell Technologies Inc., Vancouver, Canada) and cryopreserved. Before analysis, cells were rested overnight then resuspended in complete RPMI 1640 medium containing 10% heat-inactivated human AB serum and plated at 500,000 cells per well. Cells were unstimulated (negative control), stimulated with an anti-CD3 monoclonal antibody (100,000 cells per well, positive control) or with gliadin epitope mix (12.5 μg/mL of each peptide [deamidated α-gliadin (QLQPFPQPELPYPQPQS) and deamidated ω-gliadin (PFPQPEQPFPW) peptides]) using the IFN-γ ELISpotpro (Mabtech, Naka Strand, Sweden; Cat# 3420-2APW) and performed according to the manufacturer’s protocol.25–28 Peptides were acquired from JPT Peptide Technologies (Acton, Massachusetts, >95% purity). A total of 6 replicates were performed for negative controls and peptide-stimulated cells, and triplicates were completed for positive controls. SFUs were counted using an automated ELISpot reader (AID Multispot; Autoimmun Diagnostika GmbH, Strassberg, Germany). Normalized SFU values were calculated as the average SFU per million cells from unstimulated wells subtracted from the SFU per million cells for each stimulation condition and then averaged.
Vh:Cd and IEL Analysis (Histology)
Quantitative histologic methods were used to determine changes from baseline in Vh, Cd, and IEL density in duodenal biopsies taken during screening and at the end of the 14-day oral gluten challenge in patients receiving TAK-101 or placebo in the phase 2a study.29 Histology samples were assessed centrally (4–6 biopsies per patient per endoscopy). Biopsies were reviewed by the central pathologist.
Biopsies obtained by endoscopy were taken from the distal-most part of the second part of the duodenum, or the third part of the duodenum, before and at the end of gluten challenge. Each biopsy was taken from a fold if possible. One biopsy was taken per pass and immediately placed into 10% neutral buffered formalin; it was then embedded in paraffin and oriented for Vh:Cd evaluation.29 Biopsies were stained with hematoxylin and eosin, and evaluated independently by a senior, experienced, gastrointestinal pathologist blinded to patient identity and study visit. Recuts were performed as necessary to secure optimal orientation (defined as sections of mucosa where the entire villus and adjacent full depth of the crypt ending on the muscularis mucosa could be seen). On the optimally oriented sections, Vh:Cd was determined by measuring the mean height and mean depth of adjacent villi/proliferative crypt zones at ×100 magnification. Vh:Cds derived from at least 3 optimally oriented individual villus crypt units, derived from 4 to 6 biopsies from a single endoscopy, were averaged to produce a representative Vh:Cd for each endoscopy time point.30 Villous lymphocyte infiltration was determined as the average number of IELs per 100 enterocytes. The IEL count was performed at ×400 magnification on the anti-CD3 immunostained slides and 100 enterocytes were counted twice.
Statistical Analysis
Descriptive statistics were used in the phase 1 study. For the phase 2a study, mean changes from baseline in the number of IFN-γ SFUs, Vh:Cd, and IEL density within and between treatment groups were compared using a Wilcoxon signed rank test and a Wilcoxon rank sum test, respectively.
Methodologic details for the measurement of cell numbers by CyTOF, gliadin-specific ex vivo T-cell proliferation and cytokine secretion, PK, safety and tolerability, and full details of all statistical analyses are provided in the Supplementary Materials.
Results
Patient Disposition
Phase 1 study.
Twenty-three adults with CeD were enrolled (Supplementary Figure 1). In the single ascending dose cohort (Part A, n = 17), 2 patients were enrolled for each of the first 2 TAK-101 dose levels (0.1 and 0.5 mg/kg) followed by 3 to 4 patients for each of the subsequent TAK-101 dose levels (1, 2, 4, and 8 mg/kg). In the repeated ascending dose cohort (Part B, n = 6), 2 patients were enrolled for each of the repeat doses (2, 4, or 8 mg/kg) administered 1 week apart. All patients completed the study.
Phase 2a study.
Thirty-four patients with CeD were enrolled (TAK-101, n = 16; placebo, n = 18), and 33 patients (97.1%) completed the study (Supplementary Figure 2). One patient in the TAK-101 group discontinued participation owing to noncompliance with gluten consumption. Five patients (2 receiving TAK-101 and 3 receiving placebo) discontinued gluten challenge before 14 days (because of inability to tolerate the gluten) but completed all other study procedures as scheduled.
Demographic and baseline characteristics of patients in both studies are presented in Supplementary Table 1.
Endpoints
Phase 1 study: TAK-101 safety outcomes.
TAK-101 was well tolerated at doses up to 8 mg/kg after single and repeated intravenous administrations 7 days apart. Mild-to-moderate flushing, headache, back pain, and fatigue were the most commonly reported AEs. One patient who received a single dose of TAK-101 1 mg/kg was reported to have an AE of nonserious “colitis.” There was no documentation of any testing carried out for this AE and the patient was treated with antidiarrheals only. Symptoms resolved within 8 days and the AE was considered unrelated to the study drug. Two patients in the 8 mg/kg single-dose group experienced moderate infusion-related reactions, one of whom discontinued treatment. No patients discontinued treatment owing to AEs in the repeated dose group. No deaths or other SAEs were reported during the study. A summary of treatment-emergent AEs is provided in Table 1. No clinically meaningful changes from baseline were observed in hematology, coagulation, or serum chemistry parameters, vital signs, or results of physical examinations.
Table 1.
Summary of AEs and Treatment-Emergent AEs in the Phase 1 Study
| Part A: single ascending dose | |||||||
|---|---|---|---|---|---|---|---|
| TAK-101 dose | |||||||
| 0.1 mg/kg (n = 2) | 0.5 mg/kg (n = 2) | 1 mg/kg (n = 3) | 2 mg/kg (n = 3) | 4 mg/kg (n = 3) | 8 mg/kg (n = 4) | All doses (n = 17) | |
| Patients with at least 1 treatment-emergent AE, n (%) | |||||||
| AE | 1 (50.0) | 2 (100.0) | 3 (100.0) | 3 (100.0) | 2 (66.7) | 3 (75.0) | 14 (82.4) |
| Grade 3 (severe) AE | 0 (0.0) | 0 (0.0) | 1 (33.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.9) |
| Drug-related AEa | 1 (50.0) | 0 (0.0) | 2 (66.7) | 2 (66.7) | 2 (66.7) | 3 (75.0) | 10 (58.8) |
| AE leading to withdrawal | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (25.0) | 1 (5.9) |
| Treatment-emergent AEs, n | |||||||
| All AEs | 4 | 3 | 10 | 15 | 7 | 16 | 55 |
| Drug-related AEsb | 1 | 0 | 4 | 4 | 6 | 13 | 28 |
| Part B: repeated ascending doses | ||||
|---|---|---|---|---|
| TAK-101 doseb | ||||
| 2 mg/kg (n = 2) | 4 mg/kg (n = 2) | 8 mg/kg (n = 2) | All doses (n = 6) | |
| Patients with at least 1 treatment-emergent AE, n (%) | ||||
| AE | 1 (50.0) | 1 (50.0) | 2 (100.0) | 4 (66.7) |
| Grade 3 (severe) AE | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Drug-related AEa | 0 (0.0) | 1 (50.0) | 1 (50.0) | 2 (33.3) |
| AE leading to withdrawal | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Treatment-emergent AEs, n | ||||
| All AEs | 3 | 5 | 5 | 13 |
| Drug-related AEsa | 0 | 3 | 3 | 6 |
AE, adverse event.
Drug-related AEs are those that the investigator assessed as being possibly or probably related to the study treatment.
Patients received a single intravenous administration of TAK-101 on days 1 and 8.
Phase 1 study: TAK-101 PK outcomes.
Gliadin exposure peaked at the end of each TAK-101 infusion (highest mean ± SD maximum drug serum concentration was 938 ± 26.9 ng/mL, observed in the 8 mg/kg repeat dose cohort) then rapidly declined over the next 4 hours (mean terminal elimination half-life 2.00–4.85 hours across all dose levels in the single and repeated dose cohorts) (Supplementary Table 2). Plasma gliadin concentrations increased with rising doses of TAK-101, and similar PK parameters were observed after single and repeated TAK-101 doses. No accumulation of TAK-101 was observed from day 1 to day 8 in the repeated dose cohort. A maximum feasible dose of 8 mg/kg was determined for TAK-101 administration in the phase 2a study.
Phase 2a study: TAK-101 efficacy outcomes.
In the placebo group, ex vivo gliadin peptide stimulation of PBMCs after 6 days of oral gluten challenge resulted in an approximately 10-fold increase over baseline in mean IFN-γ+ SFUs/106 PBMCs (baseline mean IFN-γ+ SFUs, 1.98; day 20 mean IFN-γ+ SFUs, 19.56; P = < .001, Figure 2A). In the TAK-101 group, the gluten challenge–induced gliadin-dependent T-cell response was reduced by nearly 90% (baseline mean IFN-γ+ SFUs, 3.08; day 20 mean IFN-γ+ SFUs, 5.09; P = .735; Figure 2A). The mean change from baseline was 17.58 SFUs for placebo vs 2.01 SFUs for TAK-101 (P = .006; Figure 2B). In contrast, the number of IFN-γ+ SFUs was equivalent in the 2 treatment groups on stimulation of PBMCs with anti-CD3 (Figure 2C), indicating that TAK-101 acts in an antigen-specific manner.
Figure 2.
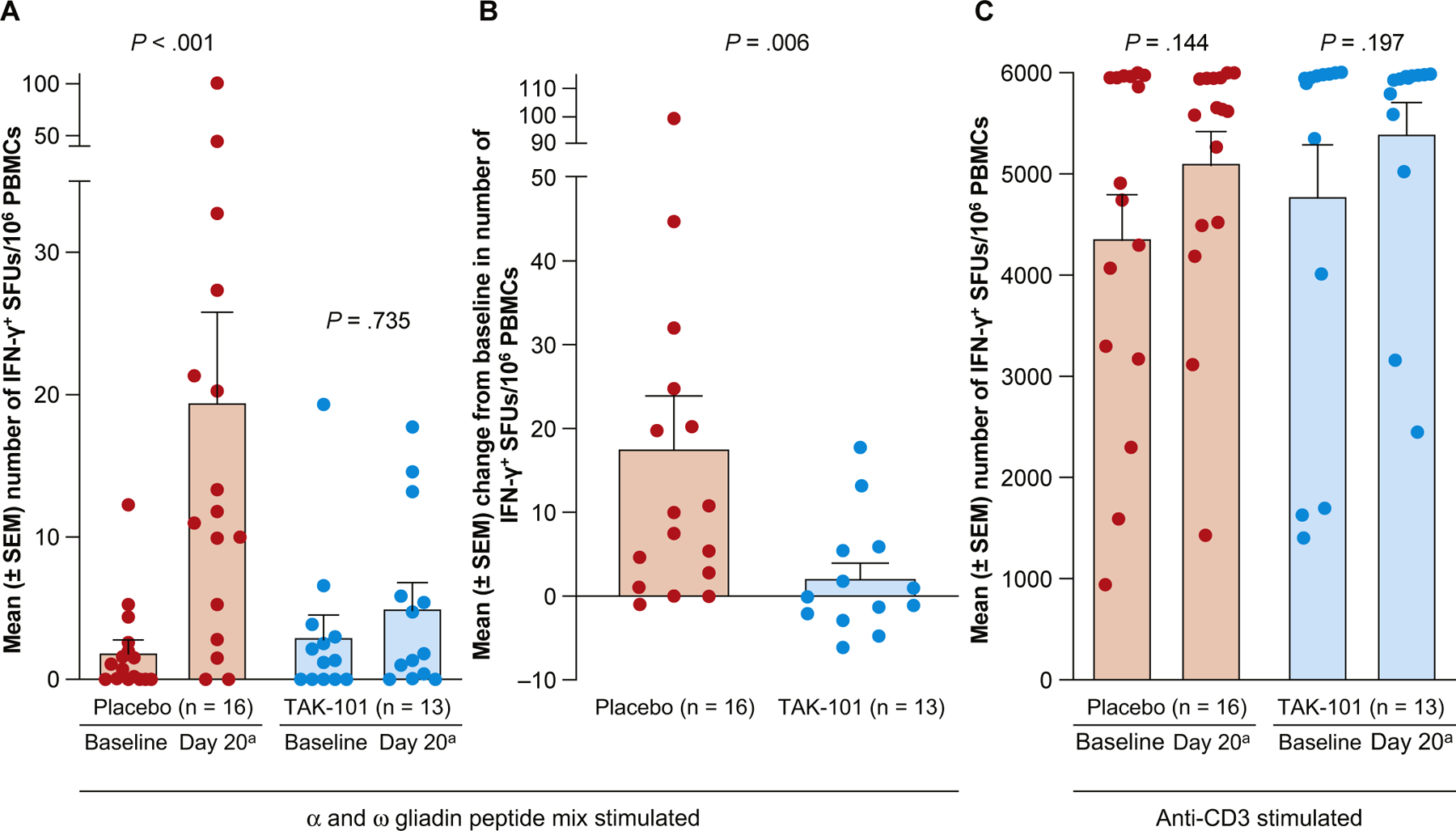
TAK-101 reduces gluten-specific activated T cells in peripheral blood in response to oral gluten challenge. (A) Number of IFN-γ+ SFUs after ex vivo stimulation of PBMCs with an HLA-DQ2-restricted deamidated α- and ω-gliadin peptide mix. (B) Change from baseline in the number of IFN-γ+ SFUs after ex vivo stimulation of PBMCs with a gliadin peptide mix. (C) Number of IFN-γ+ SFUs after ex vivo stimulation of PBMCs with anti-CD3. Induction of IFN-γ+ T cells in the peripheral blood of patients receiving placebo or TAK-101 after oral gluten challenge was examined by ELISpot assay. Patients received placebo (n = 16) or TAK-101 (n = 13) at days 1 and 8, followed by a 14-day oral gluten challenge beginning on day 15. Values for individual patients are shown as circles, and bars represent mean ± SEM. P values for (A) and (C) were calculated using the Wilcoxon signed rank test for the mean change from baseline within each treatment group. The P values for (B) were calculated using the Wilcoxon rank sum test for the mean change from baseline between treatment groups. aDay 20 was equivalent to day 6 of gluten challenge.
Vh:Cd decreased from baseline in the placebo group after oral gluten challenge (mean change from baseline to day 29, −0.63; P = .002), consistent with gluten-induced mucosal inflammation and villous atrophy. Vh:Cd was not significantly decreased in the TAK-101 treatment group (mean change from baseline to day 29, −0.18; P = .110; Figure 3A). However, comparison of the mean change between the 2 groups (Figure 3B) did not reach statistical significance (P = .080) (see Supplementary Figure 3 for change in Vh:Cd in individual patients). A decrease in Vh:Cd of at least 0.4 was measured in 8 (53.3%) of 15 patients in the placebo group and 3 (23.1%) of 13 patients treated with TAK-101, but this difference between the groups was not significant (P = .1367).
Figure 3.
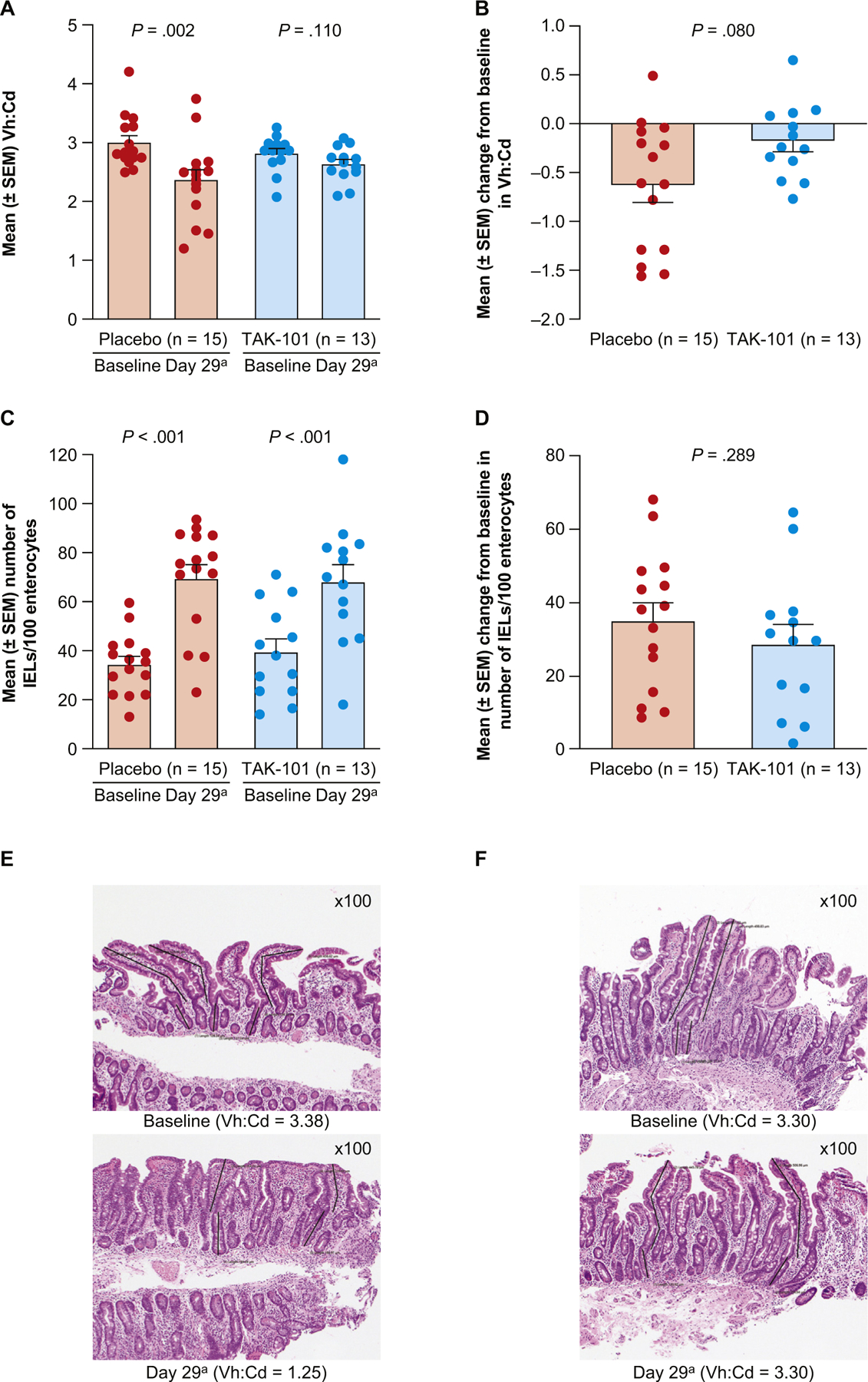
TAK-101 pre-treatment is associated with reduced gluten challenge–induced Vh:Cd deterioration. (A) Vh:Cd. (B) Change from baseline in Vh:Cd at day 29. (C) Number of IELs/100 enterocytes. (D) Change from baseline in number of IELs/100 enterocytes at day 29. (E) Representative baseline and post gluten challenge biopsies from a single patient treated with placebo, showing partial villous atrophy on day 29 compared with baseline or (F) treated with TAK-101, showing normal villous architecture without villous atrophy on day 29. Values for individual patients are shown as circles, and bars represent mean ± SEM. The P values for (A) and (C) were calculated using the Wilcoxon signed rank test for the mean change from baseline within each treatment group. The P values for (B) and (D) were calculated using the Wilcoxon rank sum test for the mean change from baseline between treatment groups (F). aDay 29 was equivalent to 1 day after the 14-day gluten challenge.
The density of IELs increased in response to gluten challenge in patients given either placebo or TAK-101 (P < .001, Figure 3C), with no differences in change from baseline found between the 2 treatment groups (P = .289; Figure 3D).
Representative histopathologic sections from placebo- and TAK-101–treated patients are shown in Figure 3E and Figure 3F, respectively. These photomicrographs show flattening of the villi and reduction in crypt depth in a patient with CeD from the placebo treatment group, in response to the 14-day oral gluten challenge (Figure 3E), compared with a patient in the TAK-101 treatment group (Figure 3F).
The proportion of activated (CD38+) CD4+, CD8+, and γδ+ effector memory T cells was increased 6 days after oral gluten challenge in the peripheral blood of patients who received placebo, but not in patients treated with TAK-101 (P = .002, P < .001, and P < 0.001, respectively) (Figure 4A–C). Gluten-induced increases from baseline in the proportion of CD4+CD38+α4β7+ (Figure 4D), CD8+CD38+αEβ7+ (Figure 4E), and γδ+CD38+αEβ7+ (Figure 4F) T cells were also diminished in patients given TAK-101 compared with those given placebo (P = .013, P = .004, and P = .010, respectively). The relative fold-change in percentages of other immune cell phenotypes, after gluten challenge in individual patients, in the placebo and TAK-101 treatment cohorts, is shown as a heat map in Figure 5. In addition to the cell types that have been previously described as sensitive to gluten challenge,5 we also noted that gut-homing T cells with a reported regulatory phenotype (CD4+, CD25+, CD127−)31 were also increased with gluten challenge and suppressed by TAK-101. Dendritic cells, B lineage cells, natural killer cells, monocytes, and most T-cell subtypes, other than those T cells that expressed α4b7 or αEb7, did not change with gluten challenge.
Figure 4.

TAK-101 prevents the induction of activated T cells bearing gut-homing/retention integrins in response to oral gluten challenge. Percentage of activated (A) CD4+CD38+α4β7+ T cells, (B) CD8+CD38+αEβ7+ T cells, and (C) γδ+CD38+αEβ7+ T cells. Change from baseline in percentage of activated (D) CD4+CD38+α4β7+ T cells, (E) CD8+CD38+αEβ7+ T cells, and (F) γδ+CD38+αEβ7+ T cells. Percentages of activated CD4+, CD8+, and γδ+ T cells bearing gut-homing/retention integrins (α4β7 or αEβ7) in the peripheral blood of patients administered placebo or TAK-101, after 6 days of gluten challenge, were determined by CyTOF. Values for individual patients are shown as circles, and bars represent mean ± SEM. The P values for (A–C) were calculated using the Wilcoxon signed rank test for the mean change from baseline within each treatment group. The P values for (D–F) were calculated using the Wilcoxon rank sum test for the mean change from baseline between treatment groups. aDay 20 was equivalent to day 6 of gluten challenge.
Figure 5.

Fold-change from baseline in number of immune cell types in peripheral blood in individual patients treated with TAK-101 (n = 13) or placebo (n = 16) post gluten challenge, determined via CyTOF analysis. CM, central memory (T cells); CSMB, class-switched memory B cells; EM, effector memory (T cells); mDC, myeloid dendritic cells; NCSMB, non–class-switched memory B cells; NK, natural killer cells; NKT, natural killer T cells; pDC, plasmacytoid dendritic cells; TCR, T-cell receptor; TEMRA, terminally differentiated effector memory cells reexpressing CD45RA.
Phase 2a study: TAK-101 safety outcomes.
The most common AEs reported during dosing in the phase 2a study were gluten-related gastrointestinal disorders, and included abdominal distention or pain, diarrhea, flatulence, nausea, vomiting, and abnormal gastrointestinal sounds. All AEs were mild or moderate in intensity except gastrointestinal disorders in 1 patient in the TAK-101 group and 2 patients in the placebo group, which were severe (grade 3). No patients experienced an AE with severity of grade 4 or above and no deaths or other serious AEs were reported (Supplementary Table 3).
No effect of TAK-101 was observed on ex vivo T-cell proliferation and no clinically meaningful changes in vital signs, routine clinical laboratory test results, or serum cytokine/chemokine levels occurred.
Further safety and PK results are reported in the Supplementary Materials.
Discussion
Immunologic tolerance is a state of antigen-specific nonresponsiveness to foreign or self-antigens mediated by clonal deletion, clonal anergy, and/or the activity of regulatory T-cell (Treg) subsets.32 In conventional autoimmune diseases, self-tolerance is broken by a variety of mechanisms, including molecular mimicry and bystander activation, leading to self-tissue damage.33 In CeD, oral tolerance to gluten is broken by unknown mechanisms, resulting in activation of gliadin-specific IFN-γ- and interleukin (IL)21-producing CD4+ T cells, and ultimately activated cytotoxic CD8+ T cells.3–5 These cells trigger inflammation in the small bowel, leading to downstream activation of harmful (auto)immune responses, causing further mononuclear cell activation, villous atrophy, and crypt hyperplasia.34–37
Reestablishment of immunologic tolerance is a therapeutic aim for T-helper (Th) cell 1/Th17-mediated autoimmune diseases, Th2-directed antibody-mediated allergic diseases, and CD8-mediated transplant rejection. Currently, none of the attempts to induce tolerance via parenteral administration of soluble antigens, peptides, or altered peptide ligands have led to the development of an approved therapy for reestablishing immunologic tolerance in autoimmune diseases.32,38,39 It has been demonstrated that intravenous administration of protein/peptide antigens delivered by carboxylated PLGA (PLGA-Ag) nanoparticles is an effective method for inducing antigen-specific tolerance in mouse models of autoimmune13–17,20–22 and allergic diseases.40 We chose to initiate clinical testing of the PLGA-Ag tolerance-inducing platform in CeD for multiple reasons, including (1) gliadin-specific T-cell responses to specific HLA-defined peptide epitopes are the known upstream driver of the CeD disease process1,41; (2) Good Manufacturing Practice-grade gliadin can be produced for clinical testing; (3) well-characterized assays are available to measure gliadin-specific T-cell and antibody responses27,28; (4) performance of gluten challenges in a CeD clinical trial is feasible and results in reliable immune activation42; and (5) intestinal biopsies have been developed as the standard methodology used to evaluate intestinal damage and inflammation.43 Direct support for the current clinical study came from findings in a mouse model of CeD induced by the transfer of activated gliadin-specific T cells to C57BL/6-Rag1 recipient mice.18 In this study, intravenous infusion of gliadin-encapsulating PLGA-Ag nanoparticles inhibited the proliferation of gliadin-stimulated T cells and their secretion of IFN-γ and IL17, increased FOXP3 (forkhead box P3) expression by regulatory T cells, decreased antigliadin antibody production, and prevented weight loss and gliadin-induced gut histopathology.18
We evaluated the potential of TAK-101, gliadin encapsulated in PLGA-Ag nanoparticles, to induce immune tolerance in patients with biopsy-confirmed CeD. We chose to encapsulate cGMP-grade gliadin protein extract within PLGA nanoparticles instead of specific gliadin peptides that comprise known immunodominant epitopes. Using the intact gliadin protein extract ensured that all immunodominant gliadin epitopes were encapsulated at equimolar concentrations. Furthermore, although there are immunodominant peptides, especially in HLA-DQ2.5 individuals, there is known to be a range of immunoreactive peptides that differ between individuals. Our goal is to develop a therapy that has the potential to work for all patients with CeD, regardless of HLA type. For this reason, we chose to induce tolerance to gliadin protein extract containing a broad range of epitopes, as prior animal model data suggest that this would be effective at the protein loads achievable.20,21 The presence of deamidated gliadin was confirmed by mass spectrometry to ensure sufficient activity of the gliadin extract.
In our phase 1 and 2a studies, intravenous administration of up to 2 doses of TAK-101 8 mg/kg in patients with CeD on a GFD was well tolerated with an acceptable safety profile. An AE of nonserious “colitis” (with no confirmation by colonoscopy recorded) was reported for 1 patient who received a single dose of TAK-101 1 mg/kg, which began on day 20 post therapy and resolved on day 28. This AE was not observed with higher doses of TAK-101 and it seems unlikely that the event was due to study drug administration or immunosuppression (no antibiotics were administered). No accumulation of TAK-101 was observed from day 1 to day 8 in the phase 1 repeated dose cohort.
In the phase 2a study, the primary efficacy endpoint of reduction in the number of circulating gliadin-specific IFN-γ spot-forming T cells in response to oral gluten challenge, after treatment with TAK-101, was met. It is notable that the degree of downregulation of the gluten challenge–induced gliadin-specific T-cell response was antigen specific with no apparent effect on the overall T-cell responses to mitogenic anti-CD3 T-cell stimulation in patients treated with TAK-101. Furthermore, pretreatment with TAK-101 led to a reduction in the proportion of circulating activated (CD38+) CD4+, CD8+, and γδ+ T cells bearing gut-homing/retention integrins (α4β7 or αEβ7) characteristic of CeD-induced intestinal inflammation, while not affecting other PBMC populations within the blood (Figure 5). The reliability of reduced IFN-γ–producing gliadin-specific cells as a marker for protection has not been confirmed, and further studies are required to fully evaluate the effect of TAK-101 pre-treatment on symptoms in patients with CeD.44 Future studies will also include IL2 measurements after gluten exposure, because secretion of this cytokine in the hours after single-dose gluten challenge has been shown to be a marker of immune response to gluten in humans.44
TAK-101 pretreatment was also associated with a reduction in gluten challenge–induced small intestinal mucosa deterioration (as measured by Vh:Cd). Although there was a significant deterioration in Vh:Cd in the placebo group, a significant change in Vh:Cd was not observed with TAK-101 pretreatment. However, the difference in change of Vh:Cd from baseline between the 2 groups did not reach statistical significance, likely owing to the reduced power of this comparison, which is a ratio of a ratio, in comparison with the analysis of change from baseline within each group. Despite the observed reduction in enteropathy with TAK-101 pretreatment, an equivalent increase in the density of IELs after gluten challenge was observed in both placebo- and TAK-101–treated patients, which is interesting considering the reduced number of gut-homing activated CD8+ T cells seen in the peripheral blood. One hypothesis is that the increase in IEL density in the TAK-101 group may reflect an increased ratio of functional regulatory to effector T-cell subsets in patients receiving TAK-101 vs placebo, in light of the reduced enteropathy in those treated with TAK-101 (regulatory to effector T-cell ratios will be determined in both blood and biopsy samples in future studies). This phenomenon has been observed in animal models including an increased Treg to effector T-cell ratio in the pancreas of nonobese diabetic mice protected from type 1 diabetes by tolerization with PLGA-Ag nanoparticles encapsulating a diabetogenic pancreatic cell autoepitope.17
Contrary to the current results demonstrating the successful induction of gluten-specific tolerance in patients with celiac disease (using the intravenous infusion of PLGA nanoparticles encapsulating intact gliadin), a recently terminated trial showed that the intradermal injection of soluble gliadin peptides (Nexvax44) did not appear to successfully induce a clinically relevant degree of tolerance. There are 2 major differences between the approach to tolerance induction using Nexvax and the approach used in our study: (1) the use of soluble gliadin peptides vs the delivery of full-length gliadin encapsulated in protolerogenic PLGA nanoparticles, and (2) intradermal vs intravenous routes of antigen delivery. Our approach allows for the presentation of all possible gliadin CD4 and CD8 epitopes after uptake and processing of the nanoparticles by tolerogenic APCs. Furthermore, peripheral vaccination is expected to induce a Th1-cell response, whereas the environment of the spleen and liver is known to be immunosuppressive when antigens or even plain nanoparticles are engulfed by splenic/liver (myeloid) APCs or liver sinusoidal endothelial cells. Hence, intradermal or subcutaneous delivery of gliadin peptides is expected to target immunogenic APCs in the skin and draining lymph nodes, whereas intravenous delivery is expected to target tolerogenic APCs in the spleen and liver. We speculate that if intradermal or subcutaneous injected peptides are found in the blood, the presentation by immunogenic APCs tips the balance toward activation rather than regulation.
A priori, there is no reason why an appropriate mixture of immunodominant gliadin peptides should not induce tolerance, as we have shown that PLGA-Ag nanoparticles encapsulating a cocktail of 4 encephalitogenic myelin peptide epitopes could induce tolerance for the prevention and treatment of disease in separate groups of mice in which experimental allergic encephalomyelitis was induced by each of the individual myelin peptides or by the peptide mixture (manuscript in preparation). In addition, our previous trial in patients with early relapsing–remitting multiple sclerosis, infused intravenously with autologous apoptotic PBMCs coupled with a cocktail of 7 myelin peptides, showed successful induction of tolerance in T cells specific for 4 of the 7 autoepitopes.45 Given these data, it is likely that the major reason for our ability to successfully induce gliadin-specific tolerance is that intravenous infusion of gliadin-encapsulating carboxylated PLGA nanoparticles effectively delivers the antigen to MARCO-expressing “protolerogenic” APCs in the splenic marginal zone and spleen. These same APCs have evolved to clear apoptotic debris from the hematopoietic system to aid in maintaining self-tolerance.13 In contrast, intradermal injection of soluble peptides leads to antigen uptake and presentation by “proimmunogenic” APCs in the dermis and draining lymph nodes that have evolved to activate T cells for protection against infectious agents.
We hypothesize that the apparent efficacy of TAK-101 in patients with CeD can be explained by preclinical studies elucidating the mechanisms by which PLGA-Ag nanoparticles induce tolerance.13,15,16,46 Although we cannot directly assess the tissue immune effects of therapy in humans, results from previous animal model studies suggest that after intravenous infusion, TAK-101 may be taken up predominantly by pro-tolerogenic APCs in the splenic marginal zone and liver expressing the MARCO scavenger receptor that binds polyanionic surfaces. This may lead to processing and HLA-DQ2–restricted presentation of dominant gliadin epitopes by host tolerogenic APCs, upregulation of programmed death-ligand 1 co-inhibitory molecules, and release of IL10 and transforming growth factor β. This could provoke anergy induction in gliadin-specific T cells and activation of gliadin-specific Tregs, which are critical for PLGA-Ag–tolerance induction and maintenance by inhibiting T-cell activation and controlling T-cell trafficking (Figure 6). In contrast, it is believed that intradermal injection of soluble peptides leads to presentation by APCs in the draining lymph nodes, which express high levels of costimulation molecules and are pro-immunogenic and inefficient at inducing anergy and Treg activation.17
Figure 6.

Proposed mechanism of action of TAK-101 based on preclinical animal model and clinical studies. (A) Intravenously administered gliadin-encapsulating PLGA nanoparticles are taken up by tolerogenic APCs in the liver and splenic marginal zone expressing the MARCO scavenger receptor. (B) PLGA particle uptake by APCs induces the upregulation of PD-L1, the release of TGF-β and IL-10, and the processing and presentation of gliadin T-cell epitopes to gliadin epitope-specific T cells. (C) Tolerance is induced and maintained by multiple mechanisms, including T-cell anergy, and the activation of both induced FOXP3+ Tregs (iT-regs) and IL10-producing Tr1 cells. (D) Effective tolerance induction results in the inhibition of activation of and trafficking of gliadin-specific IFN-γ–producing T helper 1 effector cells to the small bowel, protecting the gut from immune-mediated damage. FOXP3, forkhead box P3; IL, interleukin; MHC, major histocompatibility complex; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; PLGA, poly(dl-lactide-co-glycolic acid); TCR, T-cell receptor; TGF, transforming growth factor; Tr1, type 1 regulatory T cell.
PLGA-Ag nanoparticles are believed to target APCs responsible for the daily scavenger receptor-dependent uptake and disposal of vast numbers of hematopoietic cells while maintaining self-tolerance.47 Thus, we propose that carboxylated PLGA-Ag nanoparticles serve as surrogates for apoptotic debris, triggering synergistic tolerance mechanisms that evolved to deal with disposal after normal apoptotic cell death, while avoiding immune activation. The major advantage of this tolerance system is that by varying the antigen(s) encapsulated within the “universal” carboxylated PLGA nanoparticle, it is theoretically possible to treat any immune-mediated disease when the targeted (auto) antigens are known.
This phase 2a trial had limitations, including the small number of patients tested. Although the inclusion criteria covered both HLA-DQ2 and HLA-DQ2/DQ8 individuals (which allowed for measurement of IFN-γ production via ELISpot assay after stimulation with the 5 HLA-DQ2 restricted epitopes), there were no HLA-DQ8+ individuals in the treatment group, owing to the low frequency of this genotype. Larger studies are therefore required to confirm our results in a wider spectrum of celiac-permissible HLA types. Some individuals discontinued gluten consumption earlier than planned. However, even with these limitations, the preliminary evidence indicates TAK-101 may reduce small bowel enteropathy. We were unable to assess whether the protective effects of TAK-101 translated into a reduction in symptoms, as this study design, with weekly patient-reported outcomes, did not allow adequate granularity to assess symptom differences between groups. Furthermore, as gluten challenge is associated with a strong nocebo effect and study enrollment may select for individuals with less severe symptoms on gluten exposure, gluten challenge studies are felt to be suboptimal for assessment of symptoms. The effect of treatment on symptoms in CeD will be rigorously assessed in future studies. The gluten challenge dose used in future studies will likely also be reduced to be in line with real-world gluten consumption levels. The high gluten doses used in this current study were necessary for reliable ELISpot responses (our chosen primary endpoint based on the mechanism of action of TAK-101). In addition, the durability of TAK-101–induced tolerance in humans is unknown at this time. This raises important questions about TAK-101 therapy, such as whether there is a requirement for repeated doses to maintain tolerance, the number and frequency of doses required, and the ability of patients with CeD to resume ingestion of gluten-containing foods, which may reinforce TAK-101–induced activation of gliadin-specific regulatory T cells. Finally, TAK-101–induced tolerance targets T cells, and it is not known how long it takes for preexisting antigliadin and antitissue transglutaminase antibodies to abate in the absence of T-cell help, which may be required for optimal clinical effect. These questions will be explored in larger subsequent clinical trials.
In conclusion, TAK-101 demonstrated a favorable safety profile and efficacy in patients with CeD through inhibition of T-cell activation and possible reduction in the deterioration of Vh:Cd following gluten challenge. These findings support further clinical development of this novel immunotherapy for CeD and other antigen-specific immune diseases.
Supplementary Material
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT
Celiac disease is a well-characterized immune-mediated disorder incited by gliadin, a component of gluten. TAK-101, gliadin encapsulated in nanoparticles, is under development as an antigen-specific immune tolerance therapy for celiac disease.
NEW FINDINGS
TAK-101 was well tolerated in patients with celiac disease undergoing oral gluten challenge, and prevented immune activation and small intestinal mucosal injury, with no evidence of systemic immune suppression.
LIMITATIONS
These small phase 1 and 2 studies were of limited duration. Longer-term effects of TAK-101 on celiac disease symptoms were not fully evaluated.
IMPACT
TAK-101 prevented gluten-induced immune activation in CeD. This is the first trial demonstrating antigen-specific immune tolerance in autoimmune disease, which represents a novel approach translatable to other immune-mediated diseases.
Acknowledgments
The authors thank the TAK-101 Clinical Trial Group co-investigators (TAK-101 Study Group): Dr Robert Fogel (Clinical Research Institute of Michigan, LLC, MI, USA); Dr Tobias L. Freitag (Translational Immunology Research Program, University of Helsinki, Finland); Dr Michele Gerber (at the time of this study, Takeda Pharmaceuticals International Inc, USA; now Myeloid Therapeutics, Cambridge, MA, USA); Dr Paul K. Haynes (Indianapolis Gastroenterology Research Foundation, IN, USA); Dr Michael Koren (Jacksonville Center for Clinical Research, Jacksonville, FL, USA); Dr Mark Matson (Prism Clinical Research Saint Paul, MN, USA); Dr Seppo Meri (Translational Immunology Research Program, University of Helsinki, Finland); Dr Thomas H. Oliphant (Innovative Analytics Inc, Kalamazoo, MI, USA); Dr Barbara E. Rizzardi (Advanced Clinical Research, West Jordan, UT, USA); Dr Jocelyn Silvester (Beth Israel Deaconess Medical Center, Boston, MA, USA); Dr Mark Turner (Advanced Clinical Research, West Jordan, UT, USA). A list of study investigators and sites is included in the Supplementary Materials. Medical writing support was provided by Sally McTaggart, PhD, of Oxford PharmaGenesis, Oxford, UK and was funded by Takeda Pharmaceuticals International Co.
The data sets, including the redacted study protocol, redacted statistical analysis plan, and individual participants’ data supporting the results reported in this article, will be made available within 3 months from initial request, to researchers who provide a methodologically sound proposal. The data will be provided after de-identification, in compliance with applicable privacy laws, data protection, and requirements for consent and anonymization.
COUR Pharmaceuticals Development Co, Inc, Northbrook, Illinois was Amy Morris’ affiliation at the time of this study.
Funding
This study was sponsored by COUR Pharmaceuticals Development Co. and supported by Takeda Pharmaceuticals International Co.
Conflict of interest
These authors disclose the following: Ciarán P. Kelly reports receiving advisory fees from Anokion, COUR Pharmaceuticals Development Co, Glutenostics, ImmunogenX, Innovate, Janssen, Merck & Co, Milky Way, and Takeda Pharmaceuticals International Co; holding stock options in COUR Pharmaceuticals Development Co. and Glutenostics; and being a principal investigator on research grants from Aptalis, Merck & Co, and the National Institutes of Health (NIH). Joseph A. Murray reports receiving scientific advisory fees from Chugai Pharma, Janssen, Amgen, Bioniz, Intrexon, Dr Schar, and Inova Diagnostics; being an investigator on research grants from the NIH, ImmunogeniX, Allakos, Takeda Pharmaceuticals International Co, Kanyos, COUR Pharmaceuticals Development Co, Innovate, Provention Bio, and ImmunosanT; and receiving royalties from Torax Medical and Evelo. Daniel A. Leffler reports being a full-time employee of and owning stock options in Takeda Pharmaceuticals International Co. Daniel R. Getts reports being a co-founder and owning stock in COUR Pharmaceuticals Development Co, being a co-founder and Chief Executive Officer of Myeloid Therapeutics, and receiving advisory fees from Takeda Pharmaceuticals International Co. Glennda Smithson reports being a full-time employee of and owning stock in Takeda Pharmaceuticals International Co. M. Roy First reports receiving consulting fees from COUR Pharmaceuticals Development Co. Amy Morris was a full-time employee of COUR Pharmaceuticals Development Co. at the time of this research. Michael Boyne, Adam Elhofy, and Joseph R. Podojil report being full-time employees of and holding stock options in COUR Pharmaceuticals Development Co. Stephen D. Miller reports being a co-founder, a member of the Scientific Advisory Board, and a grantee of and holds stock options in COUR Pharmaceuticals Development Co.; being a paid consultant for COUR Pharmaceuticals Development Co. and Takeda Pharmaceuticals International Co; being a paid consultant and member of the Scientific Advisory Board of NextCure Inc; being a paid consultant for Kite Pharmaceuticals; and being a paid consultant for and member of the Scientific Advisory Board of Myeloid Therapeutics. The remaining authors disclose no conflicts.
Abbreviations used in this paper:
- AE
adverse event
- Ag
antigen
- APC
antigen-presenting cell
- CeD
celiac disease
- CyTOF
time-of-flight mass cytometry
- ELISpot
enzyme-linked immunospot
- GFD
gluten-free diet
- HLA
human leukocyte antigen
- IEL
intraepithelial lymphocyte
- IFN
interferon
- IL
interleukin
- MARCO
macrophage receptor with collagenous structure
- PBMC
peripheral blood mononuclear cell
- PK
pharmacokinetics
- PLGA
poly(dl-lactide-co-glycolide)
- SFU
spot-forming unit
- Th
T helper cell
- Treg
regulatory T cell
- Vh:Cd
villus height to crypt depth ratio
Footnotes
Supplementary Material
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at https://doi.org/10.1053/j.gastro.2021.03.014.
CRediT Authorship Contributions
Ciarán P. Kelly, MD (Conceptualization: Equal; Data curation: Equal; Formal analysis: Equal; Writing – review & editing: Equal).
Joseph A. Murray, MD (Conceptualization: Equal; Data curation: Equal; Formal analysis: Equal; Writing – review & editing: Equal).
Daniel A. Leffler, MD, MS (Conceptualization: Equal; Formal analysis: Equal; Writing – original draft: Lead; Writing – review & editing: Equal).
Daniel R. Getts, MD (Conceptualization: Equal; Formal analysis: Equal; Methodology: Equal; Writing – review & editing: Equal).
Adam C. Bledsoe, MD (Investigation: Equal; Writing – review & editing: Equal).
Glennda Smithson, PhD (Conceptualization: Equal; Formal analysis: Equal; Writing – review & editing: Equal).
M. Roy First, MD (Conceptualization: Equal; Formal analysis: Equal; Investigation: Equal; Writing – review & editing: Equal).
Amy Morris, MBA (Investigation: Equal; Writing – review & editing: Equal).
Michael Boyne, PhD (Formal analysis: Equal; Methodology: Equal; Writing – review & editing: Equal).
Adam Elhofy, PhD (Conceptualization: Equal; Formal analysis: Equal; Methodology: Equal; Writing – review & editing: Equal).
Tsung-Teh Wu, MD (Conceptualization: Equal; Data curation: Equal; Writing – review & editing: Equal).
Joseph R. Podojil, PhD (Conceptualization: Equal; Formal analysis: Equal; Methodology: Equal; Writing – review & editing: Equal).
Stephen D. Miller, PhD (Conceptualization: Equal; Formal analysis: Equal; Writing – review & editing: Equal).
References
- 1.Green PH, Lebwohl B, Greywoode R. Celiac disease. J Allergy Clin Immunol 2015;135:1099–1106. [DOI] [PubMed] [Google Scholar]
- 2.Singh P, Arora A, Strand TA, et al. Global prevalence of celiac disease: systematic review and meta-analysis. Clin Gastroenterol Hepatol 2018;16:823–836.e2. [DOI] [PubMed] [Google Scholar]
- 3.Malamut G, El Machhour R, Montcuquet N, et al. IL-15 triggers an antiapoptotic pathway in human intraepithelial lymphocytes that is a potential new target in celiac disease-associated inflammation and lymphoma-genesis. J Clin Invest 2010;120:2131–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mazzarella G, Stefanile R, Camarca A, et al. Gliadin activates HLA class I-restricted CD8+ T cells in celiac disease intestinal mucosa and induces the enterocyte apoptosis. Gastroenterology 2008;134:1017–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Han A, Newell EW, Glanville J, et al. Dietary gluten triggers concomitant activation of CD4+ and CD8+ αβ T cells and γδ T cells in celiac disease. Proc Natl Acad Sci U S A 2013;110:13073–13078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fasano A, Catassi C. Clinical practice. Celiac disease. N Engl J Med 2012;367:2419–2426. [DOI] [PubMed] [Google Scholar]
- 7.Green PH, Cellier C. Celiac disease. N Engl J Med 2007; 357:1731–1743. [DOI] [PubMed] [Google Scholar]
- 8.Schuppan D, Junker Y, Barisani D. Celiac disease: from pathogenesis to novel therapies. Gastroenterology 2009; 137:1912–1933. [DOI] [PubMed] [Google Scholar]
- 9.Ludvigsson JF, Aro P, Walker MM, et al. Celiac disease, eosinophilic esophagitis and gastroesophageal reflux disease, an adult population-based study. Scand J Gastroenterol 2013;48:808–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laurin P, Wolving M, Falth-Magnusson K. Even small amounts of gluten cause relapse in children with celiac disease. J Pediatr Gastroenterol Nutr 2002;34:26–30. [DOI] [PubMed] [Google Scholar]
- 11.Leffler DA, Acaster S, Gallop K, et al. A novel patient-derived conceptual model of the impact of celiac disease in adults: implications for patient-reported outcome and health-related quality-of-life instrument development. Value Health 2017;20:637–643. [DOI] [PubMed] [Google Scholar]
- 12.Rubio-Tapia A, Hill ID, Kelly CP, et al. ACG clinical guidelines: diagnosis and management of celiac disease. Am J Gastroenterol 2013;108:656–676; quiz 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Getts DR, Shea LD, Miller SD, et al. Harnessing nanoparticles for immune modulation. Trends Immunol 2015; 36:419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Getts DR, Terry RL, Getts MT, et al. Therapeutic inflammatory monocyte modulation using immune-modifying microparticles. Sci Transl Med 2014; 6:219ra7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Getts DR, Martin AJ, McCarthy DP, et al. Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nat Biotechnol 2012;30:1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCarthy DP, Hunter ZN, Chackerian B, et al. Targeted immunomodulation using protein coated nanoparticles. WIRES Nanomed Nanobiotechnol 2014;8:2148–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prasad S, Neef T, Xu D, et al. Tolerogenic Ag-PLG nanoparticles induce Tregs to suppress activated diabetogenic CD4 and CD8 T cells. J Autoimmun 2018; 89:112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freitag TL, Podojil JR, Pearson RM, et al. Gliadin nanoparticles induce immune tolerance to gliadin in mouse models of celiac disease. Gastroenterology 2020; 158:1667–1681.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freitag TL, Rietdijk S, Junker Y, et al. Gliadin-primed CD4+CD45RBlowCD25− T cells drive gluten-dependent small intestinal damage after adoptive transfer into lymphopenic mice. Gut 2009;58:1597–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hunter Z, McCarthy DP, Yap WT, et al. A biodegradable nanoparticle platform for the induction of antigen-specific immune tolerance for treatment of autoimmune disease. ACS Nano 2014;8:2148–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCarthy DP, Yap JW, Harp CT, et al. An antigen-encapsulating nanoparticle platform for TH1/17 immune tolerance therapy. Nanomedicine 2017;13:191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jamison BL, Neef T, Goodspeed A, et al. Nanoparticles containing an insulin-ChgA hybrid peptide protect from transfer of autoimmune diabetes by shifting the balance between effector T cells and regulatory T cells. J Immunol 2019;203:48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA 2013;310:2191–2194. [DOI] [PubMed] [Google Scholar]
- 24.Tye-Din JA, Stewart JA, Dromey JA, et al. Comprehensive, quantitative mapping of T cell epitopes in gluten in celiac disease. Sci Transl Med 2010;2:41ra51. [DOI] [PubMed] [Google Scholar]
- 25.Beissbarth T, Tye-Din JA, Smyth GK, et al. A systematic approach for comprehensive T-cell epitope discovery using peptide libraries. Bioinformatics 2005;21(Suppl 1):i29–i37. [DOI] [PubMed] [Google Scholar]
- 26.Goel G, King T, Daveson AJ, et al. Epitope-specific immunotherapy targeting CD4-positive T cells in coeliac disease: two randomised, double-blind, placebo-controlled phase 1 studies. Lancet Gastroenterol Hepatol 2017;2:479–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson RP, Degano P, Godkin AJ, et al. In vivo antigen challenge in celiac disease identifies a single transglutaminase-modified peptide as the dominant A-gliadin T-cell epitope. Nat Med 2000;6:337–342. [DOI] [PubMed] [Google Scholar]
- 28.Anderson RP, van Heel DA, Tye-Din JA, et al. T cells in peripheral blood after gluten challenge in coeliac disease. Gut 2005;54:1217–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taavela J, Koskinen O, Huhtala H, et al. Validation of morphometric analyses of small-intestinal biopsy readouts in celiac disease. PLoS One 2013;8:e76163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leffler D, Vanga R, Mukherjee R. Mild enteropathy celiac disease: a wolf in sheep’s clothing? Clin Gastroenterol Hepatol 2013;11:259–261. [DOI] [PubMed] [Google Scholar]
- 31.Liu W, Putnam AL, Xu-Yu Z, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med 2006;203:1701–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller SD, Turley DM, Podojil JR. Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease. Nat Rev Immunol 2007;7:665–677. [DOI] [PubMed] [Google Scholar]
- 33.Munz C, Lunemann JD, Getts MT, et al. Antiviral immune responses: triggers of or triggered by autoimmunity? Nat Rev Immunol 2009;9:246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lebwohl B, Sanders DS, Green PHR. Coeliac disease. Lancet 2018;391:70–81. [DOI] [PubMed] [Google Scholar]
- 35.Nilsen EM, Jahnsen FL, Lundin KE, et al. Gluten induces an intestinal cytokine response strongly dominated by interferon gamma in patients with celiac disease. Gastroenterology 1998;115:551–563. [DOI] [PubMed] [Google Scholar]
- 36.Lahdenpera AI, Holtta V, Ruohtula T, et al. Up-regulation of small intestinal interleukin-17 immunity in untreated coeliac disease but not in potential coeliac disease or in type 1 diabetes. Clin Exp Immunol 2012; 167:226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Monteleone I, Sarra M, Del Vecchio Blanco G, et al. Characterization of IL-17A-producing cells in celiac disease mucosa. J Immunol 2010;184:2211–2218. [DOI] [PubMed] [Google Scholar]
- 38.Turley DM, Miller SD. Prospects for antigen-specific tolerance based therapies for the treatment of multiple sclerosis. Results and problems in cell differentiation 2010;51:217–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Serra P, Santamaria P. Antigen-specific therapeutic approaches for autoimmunity. Nature Biotechnol 2019; 37:238–251. [DOI] [PubMed] [Google Scholar]
- 40.Smarr CB, Yap WT, Neef TP, et al. Biodegradable antigen-associated PLG nanoparticles tolerize Th2-mediated allergic airway inflammation pre- and postsensitization. Proc Natl Acad Sci U S A 2016; 113:5059–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jabri B, Sollid LM. T cells in celiac disease. J Immunol 2017;198:3005–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leonard MM, Silvester JA, Leffler D, et al. Evaluating responses to gluten challenge: a randomized, double-blind, 2-dose gluten challenge trial. Gastroenterology 2021;160:720–733.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adelman DC, Murray J, Wu TT, et al. Measuring change in small intestinal histology in patients with celiac disease. Am J Gastroenterol 2018;113:339–347. [DOI] [PubMed] [Google Scholar]
- 44.Truitt KE, Daveson AJM, Ee HC, et al. Randomised clinical trial: a placebo-controlled study of subcutaneous or intradermal NEXVAX2, an investigational immunomodulatory peptide therapy for coeliac disease. Aliment Pharmacol Ther 2019;50:547–555. [DOI] [PubMed] [Google Scholar]
- 45.Lutterotti A, Yousef S, Sputtek A, et al. Antigen-specific tolerance by autologous myelin peptide-coupled cells: a phase 1 trial in multiple sclerosis. Sci Transl Med 2013; 5:188ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuo R, Saito E, Miller SD, et al. Peptide-conjugated nanoparticles reduce positive co-stimulatory expression and T cell activity to induce tolerance. Mol Ther 2017; 25:1676–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Getts DR, Turley DM, Smith CE, et al. Tolerance induced by apoptotic antigen-coupled leukocytes is induced by PD-L1+ and IL-10-producing splenic macrophages and maintained by T regulatory cells. J Immunol 2011; 187:2405–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.