

Abstract
A pharmacokinetic-pharmacodynamic (PK-PD) modeling approach was developed to investigate the epileptogenic activity of imipenem in rats. Initially, animals received an intravenous infusion of imipenem at a rate of 2.65 mg min−1 for 30 min. Blood samples were collected for drug assay, and an electroencephalogram (EEG) was recorded during infusion and postinfusion. A dramatic delay was observed between concentrations of imipenem in serum and the EEG effect; this effect was accompanied by tremors and partial seizures. Indirect-effect models failed to describe these data, which were successfully fitted using an effect compartment model. The relationship between effect and concentration at the effect site was best described by a spline function. The elimination rate constant from the effect compartment was severalfold lower than that from the central compartment. The robustness of the model was then confirmed after administering the imipenem dose over 60 and 90 min. In conclusion, the successful PK-PD modeling of the imipenem EEG effect in rats constitutes a major improvement for better prediction of the epileptogenic risk associated with this antibiotic.
Antibiotics may occasionally induce central nervous system (CNS) side effects, including seizures, in humans. These are observed with fluoroquinolones (5, 39) and β-lactams, in particular carbapenems (33). In this family the leading compound, imipenem, presents a relatively high risk of convulsions (1, 3), especially in patients with renal impairment or with bacterial meningitis (2, 41).
Various experimental approaches have been developed to investigate the convulsion liabilities of antibiotics in vivo, including direct intracerebroventricular injection (15, 40). However, this approach does not take into consideration the CNS diffusion characteristics of these drugs, which may have major impacts on their convulsant activity (11). The proconvulsive activity of antibiotics on pentylenetetrazol has also been investigated (8, 30), but many parameters may contribute to the observed proconvulsant effects, making data interpretation complex and potentially misleading (27).
An in vivo experimental approach to the study of the pharmacokinetic-pharmacodynamic (PK-PD) contributions to the convulsant activity of fluoroquinolones alone (11, 12) or in association with other drugs (13, 25) has recently been developed. This approach is based upon previous studies which showed that for convulsant compounds, such as pentylenetetrazol (31) or theophylline (32), the cerebrospinal fluid (CSF) was part of the biophase, meaning that the convulsant activities of these substances are directly related to their concentrations in CSF (7). However, unlike fluoroquinolones, the concentration of imipenem in CSF is not predictive of its convulsant activity (20).
Electroencephalogram (EEG) recording was then considered as a suitable alternative. This approach had occasionally been used to assess the epileptogenic effects of drugs, including antibiotics (14, 16, 17), and could be considerably improved by coupling quantitative EEG recording with PK-PD modeling analysis, as previously done to investigate the desirable CNS effects of drugs (6, 26).
MATERIALS AND METHODS
Animals.
This work was done in accordance with the Principles of Laboratory Animal Care. Male Sprague-Dawley rats (Depres Breeding Laboratories, St. Doulchard, France) with a body weight between 320 and 350 g were housed in the animal breeding facilities of the laboratory (authorization no. 0028). The animals were placed in wire cages in a 12-h light-dark cycle for 1 week to adjust to the new environment and to overcome stress possibly incurred during transit. They had free access to food (AO4; U. A. R., Villemoisson, France) and water.
Surgery.
Five days before the experiment, each rat had five cortical EEG electrodes implanted while under anesthesia (1.25 mg of ketamine [Ketalar], at a concentration of 50 mg ml−1; Parke Davis Laboratories, France, and 0.5 mg of xylazine hydrochloride [Rompum]; Bayer Laboratories, France). The electrodes were screwed into little holes drilled into the skull at the following positions, in relation to bregma: 2 mm anterior, 2 mm lateral (F1 and F2); 4 mm posterior, 2 mm lateral (P1 and P2); and 4 mm anterior, 2 mm lateral (reference electrode). The stainless steel electrodes were connected to a miniature plug fixed to the skull with dental cement. One day before the experiment, two permanent polyethylene catheters were implanted while each rat was under anesthesia (thiopental sodium, 60 mg kg of body weight−1; Sanofi Laboratories, France): one in the left femoral vein for drug administration and the other one in the left femoral artery for blood sample collection. Animals were housed individually in plastic cages. Food was withdrawn 12 h before the experiment, but animals had free access to water until drug administration.
Solutions for administration.
Imipenem monohydrate-sodium cilastatin salt (Tienam; Merck, Sharp & Dohme Laboratories, France), was used to prepare a 15.9-mg ml−1 solution of imipenem in 0.9% NaCl.
Drug dosing and blood sampling.
The rats in group I (n = 6, weight [mean ± standard deviation] = 335 ± 15 g) received an intravenous (i.v.) infusion of imipenem-cilastatin at a rate of 2.65 mg of imipenem per min for 30 min, corresponding to a dose of 80 mg of imipenem. The rats in groups II and III received the same dose of imipenem, either at an infusion rate of 1.325 mg min−1 for 60 min (group II, n = 6, weight = 335 ± 15 g) or at an infusion rate of 0.883 mg min−1 for 90 min (group III, n = 6, weight = 335 ± 15 g). Drug infusions started between 9:00 a.m. and 12:00 p.m.
The femoral vein cannula was connected to a motor-driven syringe pump (Program 2; Vial Inc., France) containing the imipenem solution. Arterial blood samples (300 μl) were collected in dry tubes immediately before and at the following times after infusions had started: 15, 30, 40, 50, 60, 75, 90, 105, and 120 min (group I); 30, 60, 70, 80, 90, 105, 120, 135, and 150 min (group II); or 30, 60, 90, 100, 110, 120, 135, 150, 165, and 180 min (group III). Blood samples were immediately centrifuged to collect serum. Blood was replaced by an equal volume of 0.9% NaCl solution. Before storage, samples were diluted (1:1, vol/vol) with a stabilizer (0.5 M HEPES buffer, pH 6.8; ethylene glycol; h.p.l.c.-grade water [1:0.5:0.5, vol/vol/vol]) and kept frozen at −80°C until analysis.
EEG measurements.
On the day of the experiment each rat was maintained in a plastic bol, and the miniature plug was connected to a moving connector to record the EEG signal. Bipolar EEG leads (F1-P1, F2-P2, F1-F2, and P1-P2) were continuously recorded using a paper polygraph (System 50,000 EEG recorder; Van Gogh, Medelec, France). The signal was band-pass filtered from 0.3 to 75 Hz. The EEG signal from the left hemisphere cortical lead (F2-P2) was simultaneously sampled at 256 Hz and analyzed online by fast Fourier transform (FFT) to determine the EEG total power in the frequency band from 0.5 to 30 Hz (Brain Wave Systems Co., Thornton, Colo.). The FFTs were calculated every 2 s, giving a first EEG power trend which could be visualized before being stored on the hard disk. Subsequently, after artifact removal from this power trend, a data reduction was calculated by averaging this first FFT trend every 1 min, resulting in a secondary trend. Consequently, each data point of the second trend was the mean of 30 consecutive points of the first trend. Corresponding data points of the second trend were used as effect measures for PK-PD modeling. The EEG recordings started 10 min before imipenem infusion and continued until the signals had returned to their baseline values.
Drug analysis.
A previously described high-performance liquid chromatography assay (4) was used with minor modifications for imipenem determination. Proteins from serum samples were precipitated by the addition of methanol (1:2, vol/vol), the mixture was centrifuged, and the supernatant was injected. Separation was performed with a Nucleosil C8 (5 μm, 250 by 0.4 mm [inner diameter]) column. The mobile phase consisted of 0.2 M aqueous borate buffer, pH 7.2, containing 15% (vol/vol) methanol, and the flow rate was 1 ml min−1. The retention time of imipenem was equal to 5.5 min. The chromatographic system consisted of a model L 6000 Merck-Hitachi pump and a Waters 717 plus refrigerated autosampler connected to a Waters 484 UV absorbance detector (λ = 313 nm). Chromatographic data were recorded and processed using a Waters 746 integrator. The limit of quantitation of imipenem was 0.5 μg ml−1 in serum. Intraday coefficients of variation calculated at two concentrations were equal to or less than 10%. Corresponding interday coefficients were equal to or less than 13%.
Modeling procedures.
A one-compartment open model with zero-order input (R0) was used to characterize the serum concentration-versus-time profiles of imipenem during and after infusion:
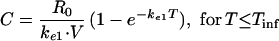 |
1 |
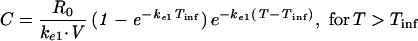 |
2 |
where C is the concentration of imipenem in serum at time T, Tinf is the duration of infusion, ke1 is the elimination rate constant, and V is the apparent volume of distribution.
The pharmacokinetic parameters were then fixed, and the pharmacodynamic models were regressed to the EEG data for each individual rat, using the nonlinear least-squares program WinNonlin (version 1.1; SCI Software, Cary, N.C.). An indirect-response model (9) and an effect compartment model (35) were applied for analysis of the PK-PD relationship, with uniform weights according to the constant variance. The baseline value, P0, was estimated by averaging the 10th min of the EEG record.
The first approach was to correlate the EEG effect with imipenem concentration using an indirect-response model based on the mechanism of action of the drug. Because it has been proposed that the convulsant activity of imipenem was mainly related to inhibition of the GABA system (8), an indirect-response model with inhibition of the loss of the response, frequently referred to as model II by its authors (23), was selected. The general form of the corresponding inhibition function is as follows:
 |
3 |
where Imax represents the maximum inhibition capacity, IC50 represents the imipenem concentration producing 50% of the maximum drug-induced inhibition, and γ is the sigmoidicity factor. However, when the PD effect tends toward infinity without reaching a maximum, as was the case with the total power of the EEG signal of imipenem, Imax must be given a value of one (34). Therefore, a simplified form of equation 3 was used as the inhibition function:
 |
4 |
The rate of change of the EEG effect over time could then be described by
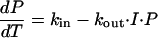 |
5 |
where P is the total power of the EEG signal (response variable representing EEG effect), kin is the zero-order constant for production of the EEG effect, and kout is the first-order rate constant for the loss of the EEG effect.
The second approach used to describe the time course of the effect was to include a hypothetical effect compartment in the PK-PD model, allowing minimization of the hysteresis observed in the serum concentration-EEG effect relationship, according to the following equations:
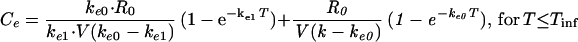 |
6 |
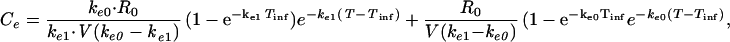 |
7 |
 |
In these equations, Ceis the drug concentration in the effect compartment at time T, and ke0 is the rate constant for elimination of the drug from the effect compartment. The other parameters are the PK parameters previously defined and estimated for being used as constants for the PK-PD modeling (35).
The profile of the EEG effect was described using a spline function derived from the Hill equation (10):
 |
8 |
In this equation, P is the total power (EEG effect) corresponding to Ce, P0 is the baseline effect value, Bn is the combined parameter Emax/EC50n, and n is a factor determining the steepness of the curve.
The goodness-of-fit of each model was assessed by visual inspection and analysis of the residuals, and coefficient of variation percentage associated with parameter estimates (22).
The robustness of the compartment effect model was assessed by comparing the parameters characteristic of the PK-PD model (B, n, and ke0) between groups. The derived parameters, such as the maximum intensity of the EEG signal (Pmax), the corresponding time of occurrence (TPmax), and the period of time elapsed between the end of infusion and the effect peak (ΔT) observed after infusing 80 mg of imipenem at a rate of 1.325 mg min−1 (group II) or 0.833 mg min−1 (group III), were also compared with the corresponding values obtained by simulations using individual parameters characteristic of the PK-PD model estimated in the first part of this study (group I).
The unpaired Student t test was used to compare the simulated and fitted values of the derived parameters (Pmax, TPmax, and ΔT) for each group. Parametric one-way analysis of variance (ANOVA) was used to compare the model parameter (B, n, and ke0) values between groups after applying Bartlett's test to check for homogeneity of variances. A significance level of 5% was selected. Values are reported as the means ± standard deviations.
RESULTS
Following a 30-min infusion of imipenem at a rate of 2.65 mg min−1, corresponding to a dose of 80 mg (group I), the decay of imipenem concentration in serum with time was monoexponential and rapid after infusion was stopped. Limited interindividual variability was observed. PK parameters are presented in Table 1. A dramatic temporal delay between concentration in serum and EEG effect was observed in every individual rat. Only isolated spikes appeared during imipenem infusion, with limited effects on the total power of the EEG signal. Then the frequency and amplitude of the spikes increased dramatically, leading to a relatively sudden increase of the total power, occurring in most animals between one and two half-lives postinfusion. This was accompanied by behavioral troubles, including tremors and partial seizures. The EEG signal reached a maximum at three elimination half-lives postinfusion on average and came back progressively to the baseline. Such a typical behavior is presented in Fig. 1.
TABLE 1.
PK parameters characteristic of imipenem infused i.v. to rats at a dose of 80 mg at various ratesa
| Group | Infusion rate (mg min−1) | Infusion duration (min) | Mean ± SD
|
||||
|---|---|---|---|---|---|---|---|
| Clearance∗ (ml min−1 kg−1) | V∗ (ml kg−1) | ke1 (min−1) | Half-life∗ (min) | Cmax (μg ml−1) | |||
| I (n = 6) | 2.65 | 30 | 14.5 ± 1.7 | 244.1 ± 43.4 | 0.060 ± 0.007 | 11.7 ± 1.7 | 457.4 ± 46.5 |
| II (n = 6) | 1.325 | 60 | 14.4 ± 3.2 | 265.5 ± 40.9 | 0.054 ± 0.010 | 13.1 ± 2.2 | 272.9 ± 47.0 |
| III (n = 6) | 0.883 | 90 | 15.5 ± 4.4 | 277.4 ± 20.3 | 0.055 ± 0.012 | 13.1 ± 2.7 | 177.8 ± 37.0 |
∗, non-statistically significant differences between groups based on one-way ANOVA.
FIG. 1.

Characteristic EEG changes induced by imipenem infusion (80 mg over 30 min) in a typical rat (group I) before (a) and at the end of (b) infusion, at the maximum effect (c), and after return to the baseline (d).
The indirect-effect model was tentatively fitted to these experimental data. However, it failed to described the lag time before the total power sharply increased, and it was only capable of predicting a progressive increase in the EEG signal, as illustrated in Fig. 2.
FIG. 2.

Observed total power of the EEG signal versus time and predicted values (solid line) according to the indirect-effect model with inhibition of the loss of effect, following imipenem infusion (80 mg over 30 min) in the same rat as in Fig. 1.
Much better results were obtained with the effect compartment model. Individual plots of EEG effect versus imipenem concentration in serum showed a spectacular counterclockwise hysteresis, indicating a profound disequilibrium between the concentrations in serum and at the effect site (Fig. 3A). This hysteresis collapsed when the effect was plotted versus concentration in the effect compartment, which could be adequately fitted with a spline function (Fig. 3B). Overall, the effect compartment model provided a good fitting of the relationship between imipenem concentration in serum and EEG effect, as illustrated in Fig. 4, with corresponding parameters accurately estimated (Table 2). The non-statistically significant differences between the parameter values estimated for the three groups (Table 2) and between the observed and predicted values of the derived parameters (Table 3) attest to the robustness of the compartment effect model in various experimental settings. Note that the pharmacokinetic parameters of imipenem were also unaffected by the change in infusion rate (Table 1).
FIG. 3.

Measured total power of the EEG signal versus imipenem concentrations in serum (A) and at the effect site (B), for the same rat as in Fig. 1. The solid line represents the best fit of the data according to the spline function model (equation 8).
FIG. 4.

Concentrations of imipenem in serum and EEG effect versus time for the same rat as in Fig. 1. The broken line represents the best PK fit to the measured concentrations of imipenem in serum, with the following values for PK parameters: V = 254 ml kg−1 and clearance = 14.0 ml min−1 kg−1. The solid line represents the best fit to the measured total power of the EEG signal effect, according to the effect compartment model, with the following values for PD parameters: P0 = 0.17 mV2, B = 0.0151, n = 8.1, and ke0 = 0.0070 min−1.
TABLE 2.
PD parameters characteristic of imipenem infused i.v. to rats at a dose of 80 mg at various rates, derived from the compartment effect modela
| Group | Infusion rate (mg min−1) | Infusion duration (min) | Mean ± SD
|
|||
|---|---|---|---|---|---|---|
| P0∗ (mV2) | B∗ | n∗ | ke0∗ (min−1) | |||
| I (n = 6) | 2.65 | 30 | 0.21 ± 0.07 | 0.0264 ± 0.0147 | 7.1 ± 6.6 | 0.0054 ± 0.0031 |
| II (n = 6) | 1.325 | 60 | 0.16 ± 0.07 | 0.0236 ± 0.0199 | 4.5 ± 4.4 | 0.0099 ± 0.0075 |
| III (n = 6) | 0.883 | 90 | 0.18 ± 0.07 | 0.0155 ± 0.0091 | 5.1 ± 4.1 | 0.0128 ± 0.0083 |
∗, non-statistically significant differences between groups based on one-way ANOVA.
TABLE 3.
Comparisons between simulated parameters characteristic of the EEG effect derived from PK-PD effect compartment modeling conducted in rats from group I and fitted parameters in rats from groups II and IIIa
| Group |
Pmax∗ (mV2)
|
TPmax∗ (min)
|
ΔT∗ (min)
|
|||
|---|---|---|---|---|---|---|
| Simulated | Fitted | Simulated | Fitted | Simulated | Fitted | |
| I (n = 6) | 4.2 ± 1.4 | 64.5 ± 7.6 | 34.5 ± 7.6 | |||
| II (n = 6) | 3.6 ± 1.2 | 3.8 ± 1.7 | 85.5 ± 7.6 | 82.2 ± 10.5 | 25.5 ± 7.6 | 22.2 ± 10.5 |
| III (n = 6) | 2.8 ± 0.9 | 2.0 ± 1.4 | 110.0 ± 7.3 | 104.0 ± 9.5 | 20.0 ± 7.3 | 14.0 ± 9.5 |
Values are presented as means ± standard deviations. ∗, non-statistically significant differences between simulated and fitted values within groups based on the unpaired Student t test.
DISCUSSION
Quantitative EEG recording constitutes an interesting experimental approach offering a sensitive, objective, and continuous measure of the neurotoxic effects of imipenem in rats. Furthermore, since variations in the total power of the EEG signal were related to behavioral modifications and occurrence of tremor and partial seizures, changes in EEG can be considered as an appropriate surrogate pharmacodynamic endpoint for the investigation of the epileptogenic activity of imipenem in rats. Compared to EEG recording used in previously published studies (14, 16), the integrated PK-PD modeling approach used here constituted a major improvement. Because imipenem is supposed to interact with GABAA receptors (8, 16), an indirect-response model with inhibition of the factors controlling drug response (that is, inhibition of GABAA binding to its receptor sites) was initially selected but was unable to capture the lag time followed by a sudden increase in the EEG effect (Fig. 2).
The ability of the effect compartment model to describe imipenem data suggests that this delay is due to the limited and slow CNS diffusion of imipenem (19, 22, 28), which is also responsible for the important differences observed between the elimination rate constants from the central and effect compartments (Tables 1 and 2). Under similar experimental conditions, no hysteresis was found for midazolam, a drug with extensive and rapid CNS diffusion (26), and only a limited, if any, temporal delay between concentrations and effects with values of kel and ke0 close to each other was observed with synthetic opioids (6).
Although the effect compartment provided satisfactory data fitting in one particular situation (80 mg infused over 30 min), it was important to assess its robustness under various experimental conditions. It was initially decided to change the dose as previously done to discriminate between the effect compartment and indirect-effect models (9, 38). However, preliminary experiments showed that no EEG effect was observed when the dose was reduced by 50% and animals died before the end of the experiment when it was increased by 50%. These results are consistent with the sudden and rapid increase of the EEG effect illustrated in Fig. 3A and with the high value of the sigmoidicity factor n in equation 8 (Table 2). However, this precluded dose modification as a way to assess the robustness of the effect compartment model. Keeping the dose constant but changing the duration of infusion by adjusting the infusion rate was therefore considered. Simulations showed that the effect compartment model predicts that the peak of effect should decrease as the duration of infusion increases.
Results obtained following 60- and 90-min infusions, in particular the decrease of Pmax as infusion duration increases (Table 3 and Fig. 5A) as well as the lack of statistically significant differences between parameter estimates in the various experimental settings (Table 2), attest to the robustness of the compartment effect model. However, a close look at the data suggests that, although not statistically different, the ke0 value had a tendency to increase together with the duration of infusion, indicating that the effect compartment model may not capture all of the complexities of the system. Among these, an active metabolite accumulating with time could contribute to the observed EEG effect. Several authors have suggested that the convulsant activity of imipenem was related to the accumulation of an open lactam metabolite (18, 37), but other results suggest that the parent compound itself would be responsible for the neurotoxicity (29, 36). However, this metabolite is not detected in serum and cannot be included in a PK-PD model. Therefore, the effect compartment model may not be perfect, but at least it was satisfactory in predicting the EEG effect of imipenem in a relatively wide range of experimental settings (Table 3). It provides a new and unique way to approach this complex nonlinear concentration-effect relationship. As an example, it was confirmed experimentally (Table 3) that keeping the dose constant but increasing the duration of infusion (and therefore reducing the input rate) allowed a reduction of the epileptogenic effect. As another consequence of this change in the dosing regimen, Cmax decreases (Table 1) but the area under the concentration-time curve (AUC) does not, since dose and clearance are kept constant. One could argue that the CNS toxicity of imipenem is essentially related to high concentrations in serum and that apart from the temporal delay, peak concentrations in serum would be predictive of the epileptogenic risk. Simulations conducted with the compartment effect model show that this would not be the case, as infusing imipenem at various combined input rates and durations in order to keep the Cmax constant (but not the AUC) would lead to a completely different EEG effect as well. This is illustrated in Fig. 5B, which shows that a slight change in the dosing regimen (for example, infusing imipenem at a rate of 2.50 mg min−1 for 35 min instead of 2.65 mg min−1 for 30 min), which has no effect on Cmax, should result in a dramatic increase of the EEG effect. This confirms that the Cmax and AUC alone are not good predictors of the epileptogenic risk associated with imipenem. Interestingly, the model also predicts that a reduction in imipenem clearance, as encountered in patients with renal insufficiency, should lead to a much more than proportional increase in the epileptogenic effect. This is consistent with the current knowledge gained from clinical practice, that imipenem CNS toxicity is essentially observed in patients with renal impairment (2) and dosage adjustment based upon renal function may not be adequate to prevent seizures (24).
FIG. 5.

Simulated EEG effect for the same rat as in Fig. 1 according to the effect compartment model following imipenem infusions. (A) The same dose (80 mg) administered for various durations, leads to changes in Cmax but not in AUC. Lines correspond to infusions at 0 (bolus), 15, 30, 60, 90, 120, 150, and 180 min; solid lines represent experimental conditions used during this study. (B) Various rates (3.20, 2.85, 2.65, and 2.50 mg min−1 for 20, 25, 30, and 35 min, respectively) lead to changes in doses (64, 71, 80, and 88 mg, respectively) and, therefore, AUC, but peak concentrations in serum (Cmax = 437 μg ml−1) do not change. The solid line corresponds to an experimental condition used in this study (30-min infusion).
In conclusion, the PK-PD model validated in this study represents a major improvement in the comprehension of the epileptogenic activity of imipenem in rats.
ACKNOWLEDGMENT
We are grateful for the excellent technical assistance of Isabelle Martineau.
REFERENCES
- 1.Calandra G B, Brown K R, Grad L C, Ahonkkai V I, Wang C, Aziz M A. Review of adverse experiences and tolerability in the first 2,516 patients treated with imipenem/cilastatin. Am J Med. 1985;78(Suppl. 6A):73–78. doi: 10.1016/0002-9343(85)90104-4. [DOI] [PubMed] [Google Scholar]
- 2.Calandra G B, Lydick E, Carrigan J, Weiss L, Guess H. Factors predisposing to seizures in seriously ill infected patients receiving antibiotics: experience with imipenem/cilastatin. Am J Med. 1988;84:911–918. doi: 10.1016/0002-9343(88)90071-x. [DOI] [PubMed] [Google Scholar]
- 3.Calandra G B, Wang C, Aziz M A, Brown K R. The safety profile of imipenem/cilasatin: worldwide clinical experience based on 3470 patients. J Antimicrob Chemother. 1986;18(Suppl. E.):193–202. doi: 10.1093/jac/18.supplement_e.193. [DOI] [PubMed] [Google Scholar]
- 4.Carlucci G, Biordi L, Vicentini C, Bologna M. Determination of imipenem in human plasma, urine and tissue by high-performance liquid chromatography. J Pharm Biomed Anal. 1990;8:283–286. doi: 10.1016/0731-7085(90)80038-q. [DOI] [PubMed] [Google Scholar]
- 5.Christ W. Central nervous system toxicity of quinolones: human and animal findings. J Antimicrob Chemother. 1990;26(Suppl. B):219–225. doi: 10.1093/jac/26.suppl_b.219. [DOI] [PubMed] [Google Scholar]
- 6.Cox E H, Kerbusch T, Van Der Graaf P H, Danhof M. Pharmacokinetic-pharmacodynamic modeling of the electroencephalogram effect of synthetic opioids in the rat: correlation with the interaction at the Mu-opioid receptor. J Pharmacol Exp Ther. 1998;284:1095–1103. [PubMed] [Google Scholar]
- 7.Danhof M, Levy G. Kinetics of drug action in disease states. I. Effect of infusion rate on phenobarbital concentrations in serum, brain and cerebrospinal fluid of normal rats at onset of loss of righting reflex. J Pharmacol Exp Ther. 1984;229:44–50. [PubMed] [Google Scholar]
- 8.Day I P, Goudie J, Nishiki K, Williams P D. Correlation between in vitro and in vivo models of proconvulsive activity with the carbapenem antibiotics, biapenem, imipenem/cilastatin and meropenem. Toxicol Lett. 1994;76:239–243. doi: 10.1016/0378-4274(95)80008-2. [DOI] [PubMed] [Google Scholar]
- 9.Dayneka N L, Garg V, Jusko W J. Comparison of four basic models of indirect pharmacodynamic responses. J Pharmacokinet Biopharm. 1993;21:457–478. doi: 10.1007/BF01061691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Della Paschoa O E, Mandema J W, Voskuyl R A, Danhof M. Pharmacokinetic-pharmacodynamic modeling of the anticonvulsant and electroencephalogram effects of phenytoin in rats. J Pharmacol Exp Ther. 1998;284:460–466. [PubMed] [Google Scholar]
- 11.Delon A, Bouquet S, Huguet F, Brunet V, Courtois P, Couet W. Pharmacokinetic-pharmacodynamic contributions to the convulsant activity of fluoroquinolones in rats. Antimicrob Agents Chemother. 1999;43:1511–1515. doi: 10.1128/aac.43.6.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delon A, Huguet F, Courtois P, Vierfond J M, Bouquet S, Couet W. Pharmacokinetic-pharmacodynamic contributions to the convulsant activity of pefloxacin and norfloxacin in rats. J Pharmacol Exp Ther. 1997;280:983–987. [PubMed] [Google Scholar]
- 13.Delon A, Levasseur L M, Giraudon M, Bouquet S, Couet W. Antagonistic interaction between the convulsant activities of pefloxacin and its main metabolite norfloxacin in rats. Pharm Res. 1999;16:1894–1897. doi: 10.1023/a:1011967813140. [DOI] [PubMed] [Google Scholar]
- 14.De Sarro A, Ammendola D, De Sarro G. Effects of some quinolones on imipenem induced-seizures in DBA/2 mice. Gen Pharmacol. 1994;25:369–379. doi: 10.1016/0306-3623(94)90068-x. [DOI] [PubMed] [Google Scholar]
- 15.De Sarro A, Ammendola D, Zappala M, Grasso S, De Sarro G B. Relationship between structure and convulsant properties of some β-lactam antibiotics following intracerebroventricular microinjection in rats. Antimicrob Agents Chemother. 1995;39:232–237. doi: 10.1128/aac.39.1.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Sarro A, Imperatore C, Mastroeni P, De Sarro G. Comparative convulsant potencies of two carbapenem derivatives in C57 and DBA/2 mice. J Pharm Pharmacol. 1995;47:292–296. doi: 10.1111/j.2042-7158.1995.tb05798.x. [DOI] [PubMed] [Google Scholar]
- 17.De Sarro G, Nava B F, Calapai G, De Sarro A. Effects of some excitatory amino acid antagonists and drugs enhancing γ-aminobutyric acid neurotransmission on pefloxacin-induced seizures in DBA/2 mice. Antimicrob Agents Chemother. 1997;41:427–434. doi: 10.1128/aac.41.2.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dudley M N. Imipenem-cilastatin. The promise of single agent antimicrobial therapy fulfilled? Clin Pharm. 1989;5:760. [PubMed] [Google Scholar]
- 19.Dupuis A, Caillaud A, Pariat C, Courtois P, Couet W, Bouquet S. Comparative cerebrospinal fluid diffusion of imipenem and meropenem in rats. J Pharm Pharmacol. 2000;52:1143–1149. doi: 10.1211/0022357001774912. [DOI] [PubMed] [Google Scholar]
- 20.Dupuis A, Pariat C, Courtois P, Couet W, Bouquet S. Imipenem but not meropenem induces convulsions in DBA/2 mice, unrelated to cerebrospinal fluid concentrations. Fundam Clin Pharmacol. 2000;14:163–165. doi: 10.1111/j.1472-8206.2000.tb00406.x. [DOI] [PubMed] [Google Scholar]
- 21.Gabrielsson J, Weiner D. Pharmacokinetic and pharmacodynamic data analysis: concepts and application. 2nd ed. Stockholm, Sweden: Swedish Pharmaceutical Press; 1997. [Google Scholar]
- 22.Jacobs R F, Kearns G L, Brown A L, Longee D C. Cerebrospinal fluid penetration of imipenem and cilastatin (Primaxin) in children with central nervous system infections. Antimicrob Agents Chemother. 1986;29:670–674. doi: 10.1128/aac.29.4.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jusko W J, Ko H C. Physiologic indirect response models characterize diverse types of pharmacodynamic effects. Clin Pharmacol Ther. 1994;56:406–419. doi: 10.1038/clpt.1994.155. [DOI] [PubMed] [Google Scholar]
- 24.Leo R J, Ballow C H. Seizure activity associated with imipenem use: clinical case reports and review of the literature. DICP Ann. Pharmacother. 1991;25:351–354. doi: 10.1177/106002809102500403. [DOI] [PubMed] [Google Scholar]
- 25.Levasseur L M, Delon A, Greco W R, Faury P, Bouquet S, Couet W. Development of a new quantitative approach for the isobolographic assessment of the convulsant interaction between pefloxacin and theophylline in rats. Pharm Res. 1998;15:1069–1076. doi: 10.1023/a:1011938429379. [DOI] [PubMed] [Google Scholar]
- 26.Mandema J W, Tukker E, Danhof M. Pharmacokinetic-pharmacodynamic modelling of the EEG effects of midazolam in individual rats: influence of rate and route of administration. Br J Pharmacol. 1991;102:663–668. doi: 10.1111/j.1476-5381.1991.tb12230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marchand S, Pariat C, Bouquet S, Courtois P, Couet W. Pharmacokinetic-pharmacodynamic modelling of the convulsant interaction between norfloxacin and biphenyl acetic acid in rats. Brit J Pharmacol. 2000;129:1609–1616. doi: 10.1038/sj.bjp.0703260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Modai J, Vittecoq D, Decazes J M, Meulemans A. Penetration of imipenem and cilastatin into cerebrospinal fluid of patients with bacterial meningitis. J Antimicrob Chemother. 1985;16:751–755. doi: 10.1093/jac/16.6.751. [DOI] [PubMed] [Google Scholar]
- 29.Norrby R S. Neurotoxicity of carbapenem antibacterials. Drug Saf. 1996;15:87–90. doi: 10.2165/00002018-199615020-00001. [DOI] [PubMed] [Google Scholar]
- 30.Patel J B, Giles R E. Meropenem: evidence of lack of proconvulsive tendency in mice. J Antimicrob Chemother. 1989;24(Suppl. A):307–309. doi: 10.1093/jac/24.suppl_a.307. [DOI] [PubMed] [Google Scholar]
- 31.Ramzan I M, Levy G. Kinetics of drug action in disease states. XIV. Effect of infusion rate on pentylenetetrazol concentrations in serum, brain and cerebrospinal fluid of rats at onset of convulsions. J Pharmacol Exp Ther. 1985;234:624–628. [PubMed] [Google Scholar]
- 32.Ramzan I M, Levy G. Kinetics of drug action in disease states. XVI. Pharmacodynamics of theophylline induced seizures in rats. J Pharmacol Exp Ther. 1986;236:708–713. [PubMed] [Google Scholar]
- 33.Schliamser S E, Cars O, Norrby S R. Neurotoxicity of β-lactam: predisposing factors and pathogenesis. J Antimicrob Chemother. 1991;27:405–425. doi: 10.1093/jac/27.4.405. [DOI] [PubMed] [Google Scholar]
- 34.Sharma A, Jusko W J. Characterization of four basic models of indirect pharmacodynamic responses. J Pharmacokinet Biopharm. 1996;24:611–635. doi: 10.1007/BF02353483. [DOI] [PubMed] [Google Scholar]
- 35.Sheiner L B, Stanski D R, Vozeh S, Miller R D, Ham J. Simultaneous modelling of pharmacokinetics and pharmacodynamics: application to d-tubocurarine. Clin Pharmacol Ther. 1979;25:358–371. doi: 10.1002/cpt1979253358. [DOI] [PubMed] [Google Scholar]
- 36.Sunagawa M, Matsumura H, Sumita Y, Nouda H. Structural features resulting in convulsive activity of carbapenem compounds: effect of C-2 side chain. J Antibiot. 1995;48:408–416. doi: 10.7164/antibiotics.48.408. [DOI] [PubMed] [Google Scholar]
- 37.Tse C S T, Vera F H, Desai D V. Seizure-like activity associated with imipenem-cilastatin. Drug Intell Clin Pharm. 1987;21:659–660. doi: 10.1177/1060028087021007-820. [DOI] [PubMed] [Google Scholar]
- 38.Wakelkamp M, Alvan G, Paintaud G. The time of maximum effect for model selection in pharmacokinetic-pharmacodynamic analysis applied to frusemide. Br J Clin Pharmacol. 1998;45:63–70. doi: 10.1046/j.1365-2125.1998.00637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wallace K L. Antibiotic-induced convulsions. Crit Care Clin. 1997;13:741–762. doi: 10.1016/s0749-0704(05)70367-5. [DOI] [PubMed] [Google Scholar]
- 40.Williams P D, Bennett D B, Comereski C R. Animal model for evaluating the convulsive liability of β-lactam antibiotics. Antimicrob Agents Chemother. 1988;32:758–760. doi: 10.1128/aac.32.5.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wong V K, Wright H T, Ross L A, Mason W H, Inderlied C B, Kim K S. Imipenem/cilastatin treatment of bacterial meningitis in children. Pediatr Infect Dis J. 1991;10:122–125. doi: 10.1097/00006454-199102000-00009. [DOI] [PubMed] [Google Scholar]