

Abstract
Diabetic patients can develop degenerative corneal changes, termed diabetic keratopathy, during the course of their disease. Topical insulin has been shown to reduce corneal wound area and restore sensitivity in diabetic rats, and both the insulin receptor (IR) and insulin-like growth factor 1 receptor (IGF-1R) stimulate cell signaling of the PI3K-Akt pathway. The purpose of this study was to assess a mechanism by which improved wound healing occurs by characterizing expression within the PI3K-Akt pathway in corneal epithelial and stromal cells. In vitro scratch tests were used to evaluate wound healing outcomes under variable glucose conditions in the presence or absence of insulin. Protein expression of intracellular kinases in the PI3K pathway, stromal cell markers, and GLUT-1 was evaluated by immunoblotting.TGF-β1 expression was evaluated by ELISA. Insulin promoted in vitro wound healing in all cell types. In human corneal epithelial cells, insulin did not induce PI3K-Akt signaling; however, in all other cell types evaluated, insulin increased expression of PI3K-Akt signaling proteins compared to vehicle control. Fibroblasts variably expressed α-SMA under all treatment conditions, with significant increases in α-SMA and TGF-β1 occurring in a dose-dependent manner with glucose concentration. These results indicate that insulin can promote corneal cellular migration and proliferation by inducing Akt signaling. Exogenous insulin therapy may serve as a novel target of therapeutic intervention for diabetic keratopathy.
Keywords: Diabetes mellitus, Insulin, Glucose, Corneal wound healing, Transforming growth factor-beta (TGF-β), Alpha-smooth muscle actin (α-SMA)
1. Introduction
Diabetes mellitus is currently the most prevalent endocrinopathy in the United States and is characterized by persistent hyperglycemia (Schmidt 2018; Kerner and Brückel 2014; Guthrie and Guthrie 2004). Both type 1 and type 2 diabetes continue to affect increasing numbers of human patients, and the CDC projects approximately 500 million people globally will have the disease by 2030 (Schmidt 2018; Centers for Disease Control, 2014; American Diabetes Association 2013). Nearly half of all diabetic patients develop some degree of degenerative corneal disease, termed diabetic keratopathy, due to compromised epithelial barrier function, morphologic changes to the epithelial and endothelial cells, hypertrophy of the epithelial basement membrane, and diminished corneal sensitivity (Schultz et al., 1981; Gekka et al., 2004; Awata et al., 1988; Meyer et al. 1988; Azar et al., 1992; Kaji 2005). Delayed epithelial healing predisposes diabetic patients to sight-threatening complications, such as progressive thinning of the epithelium, resulting in secondary corneal injury and potential scar formation (Gekka et al., 2004; Desai and Lavingia 1987). Therefore, developing novel ways to improve corneal wound healing is of great importance in maintaining the corneal integrity in diabetic patients (Kaji 2005; Abdelkader et al., 2011).
Diabetes is also among the most common chronic endocrine diseases in the dog, and the diabetic canine patient suffers morbidity associated with corneal changes including endothelial loss, reduced corneal sensitivity, and decreased tear production resulting in discomfort and visual impairment (Kador et al., 1990; Yee et al., 1985; Good et al., 2003; Williams et al., 2007; Cullen et al., 2005). While neuropathic dry eye and increased risk of surface infection occur frequently in the diabetic dog, corneal fragility and delayed wound healing are not clinical entities generally reported.
Numerous growth factors have been identified and examined for their potential roles in corneal stability and wound healing including insulin-like growth factor (IGF), epidermal growth factor (EGF), transforming growth factor-β (TGF-β), and IL-6, among others (Abdelkader et al., 2011; Nagano et al., 2003; Maldonado and Furcht 1995; Saika et al., 2004; S. Wilson et al., 1993; Nishida et al., 1992). While treatment of diseased corneas with specific growth factors may have promise, to date, none have proven consistently effective, leaving an unmet need for therapies (Abdelkader et al., 2011). Topical insulin has been shown to enhance healing of cutaneous defects in both diabetic and non-diabetic rats (Apikoglu-Rabus et al., 2010). With respect to the cornea, insulin therapy resulted in reduced wound area and improved corneal sensitivity in diabetic rats when compared to treatment with a vehicle control (Zagon et al., 2007; Zagon et al. 2006). Use of insulin to improve corneal re-epithelialization has been limited to rodents and rabbits, with rare reports in humans (Zagon et al., 2007; Zagon et al. 2006; Yang et al., 2020; Klocek et al., 2009; A. L. Wang et al., 2017; Fai et al., 2017; Cruz-Cazarim et al., 2019; Módulo et al., 2009; Saragas et al., 1985).
Both insulin and IGF-1 have been shown to regulate numerous cellular mechanisms throughout the body (Knudsen et al., 2012; Lawrence et al. 2007; Lizcano and Alessi 2002). Ligand binding of the insulin receptor (IR) and IGF-1 receptor (IGF-1R) induces cell signaling events through activation of a receptor tyrosine kinase (RTK), which phosphorylates and subsequently recruits the Insulin Receptor Substrate (IRS) to activate and convert other kinases, downstream in the PI3K signaling cascade, to their active forms, ultimately promoting cellular proliferation, migration, and glucose metabolism through phosphorylated Akt (pAkt) and mTOR (p-mTOR) (De Meyts et al., 1995; Ersahin et al. 2015; Saltiel and Kahn 2001; Schultze et al., 2012; Morita et al., 2015). Both the IR and IGF-1R are heterotetrameric transmembrane structures with two extracellular alpha subunits, forming the ligand binding domain, and two intracellular beta subunits with tyrosine kinase activity (Lawrence et al. 2007). The IR and IGF-1R have the capacity to heterodimerize to form a hybrid receptor (Hybrid-R), comprising one α and one β subunit from each receptor (De Meyts and Whittaker 2002; Adams et al., 2000). Previous studies have demonstrated that the Hybrid-R exhibits a much lower affinity to insulin than the IR (Lawrence et al. 2007; Adams et al., 2000; Ullrich et al., 1986). Additionally, there is extensive cross-reactivity of both insulin as a ligand and the RTKs with which it is capable of binding, as insulin, IGF-1, and IGF-2 all possess the capacity to act as ligands for the IR, IGF-1R, and Hybrid-R, and both IGF-1 and −2 show high affinity for the IGF-2R (Lawrence et al. 2007; De Meyts et al., 1995; Wu et al. 2012; Siddle 2011; Pandini et al., 2002; Rechler and Nissley 1985).
The current standard of care for diabetes-associated corneal pathology in both physician and veterinary medicine is insufficient to retain comfort and vision in affected patients, and the need exists to explore alternative or adjunctive therapies for better control of ocular surface disease (Abdelkader et al., 2011; Lambiase et al., 1998). While previous studies have evaluated the concentration of glucose in the tear film of diabetic and non-diabetic human and canine patients and the possible effect that elevated tear film glucose concentration has on the development of ocular surface disease in diabetes, no study has yet to evaluate the use of exogenous insulin in an cellular model of diabetic corneal wound healing (Desai and Lavingia 1987; Good et al., 2003; Cullen et al., 2005; Cousen et al., 2007; Alves et al., 2008; Giardini and Roberts 1950; Goebbels 2000; Krumholz et al., 2005; Lane et al., 2006; Sen and Sarin 1980). Additionally, little work has been done to evaluate the mechanism of action for insulin’s capacity to attenuate delayed diabetic corneal wound healing, and no studies have evaluated this mechanism in an in vitro model of diabetic corneal wound healing or characterized the effects of ambient glucose and exogenous insulin on corneal cell phenotype in vitro (Zagon et al., 2007; Zagon et al. 2006; Klocek et al., 2009).
The objectives of the current study were to evaluate the cytotoxicity of insulin on human and canine corneal cells, determine whether insulin promotes corneal wound healing using an in vitro model, and characterize the effects of glucose and insulin on stromal cell phenotype. Additional goals included characterization of GLUT-1 expression in normal corneal cells and IR and IGF-1R expression in normal and diabetic corneas from both human and canine donors.
2. Materials & methods
2.1. Tissues and cell culture
2.1.1. Human corneal epithelial cells
An immortalized human telomerized corneal epithelial (hTCEpi) cell line was generously gifted by Dr. Danielle Robertson at the Department of Ophthalmology, University of Texas Southwestern Medical Center (Robertson et al., 2005). hTCEpi were maintained in KGM-2 medium (Lonza, Walkersville, MD) containing 0.15 mM calcium and supplemented with 0.4% bovine pituitary extract, 0.1% human EGF, 0.1% hydrocortisone, 0.1% transferrin, 0.1% epinephrine, and 0.1% gentamicin sulfate amphotericin B. Cells were incubated at 37 °C and 5% CO2 and were allowed to grow to 90% confluence before passage.
2.1.2. Canine corneal epithelial cells
Primary canine corneal epithelial cell (CEC) cultures were obtained under an approved Institutional Animal Care and Use Committee protocol from canine cadaveric globes (n = 12) whose cause of death was unrelated to this study. Within 2 h of enucleation, primary canine CEC were cultured by placing excised corneas in Dispase II (Gibco, Gaithersburg, MD) and incubating overnight at 4 °C. The following morning, corneas were transferred to 37 °C for 4h. The corneal epithelium was debrided, and debrided cells were suspended in supplemented KGM-2 medium. Cell suspensions were pelleted by centrifuging at 4 °C for 10min (1000×g), and pellets were resuspended in medium before seeding. Cells were incubated at 37 °C and 5% CO2 and were allowed to grow to 90% confluence before passage; cells were passaged no more than five times.
2.1.3. Human and canine stromal fibroblasts
Primary stromal fibroblast cultures were obtained from deidentified cadaveric globes (n = 7) provided by the Department of Anatomy and the Body Donor Service at The Ohio State University or from the National Disease Research Interchange (Philadelphia, PA) or canine cadavers (n = 6 donors; 12 globes) whose cause of death was unrelated to this study. Following receipt of freshly-enucleated globes, corneal epithelium was debrided, and a 50% depth keratectomy was made in the axial corneas to ensure isolation of corneal stroma. The excised stromal tissue was allowed to adhere to culture dishes prior to being covered with low glucose (5.55 mM) Dulbecco’s modified Eagle’s media (DMEM; Gibco) containing 1% antibiotics/antimycotics and 10% fetal bovine serum at 37 °C and 5% CO2. Growth medium was replenished every three days. Cultures of fibroblasts were allowed to grow to 90% confluence before passage; cells were passaged no more than five times. Western blot analysis evaluating vimentin and cytokeratin expression verified the stromal origin of the cell culture (data not shown).
2.1.4. Human and canine stromal isolates
Globes from normal and diabetic human and canine donors were obtained from The Ohio State University Department of Anatomy and the Body Donor Service and the Veterinary Medical Center Applied Pathology Service, respectively. The human normal donors (n = 2) were males, 54 and 94 years old, while the human type 2 diabetic donors (n = 2) were males, 60 and 81 years old. The normal canine donors (n = 2) were male, approximately 5 and 10 years old, while the diabetic canine donors (n = 2) were male, 7 and 12 years old. The corneal stroma was isolated as described above and homogenized for immunoblot analysis.
2.2. In vitro cytotoxicity
Confluent cultures of each cell type were treated with varying concentrations of human recombinant insulin (SAFC Biosciences, Lenexa, KS) or vehicle control (0.01M HCl; solvent for lyophilized insulin) for 48h, or 2% Triton X-100 for the last 4h as a positive cytotoxicity control. The insulin concentration used in the present study (2.4 μM or 2U/5 mL media) was extrapolated from a previous in vivo study and was comparable to the concentration used in in vitro evaluations of insulin treatment of human CEC (Zagon et al., 2007; Titone et al. 2018). All treatments were evaluated in triplicate. Cellular monolayers were monitored at 12h intervals to assess for apparent cytopathology and were imaged using a Nikon Eclipse Ts2 inverted phase contrast digital microscope (Tokyo, Japan).
Supernatants were harvested and subject to a commercially-available colorimetric Cytotoxicity (LDH) Detection Kit (Roche, Penzberg, Germany), per the recommended protocol. Briefly, 100 μL of supernatant from each sample was transferred to a 96-well plate, to which 100 μL of reaction mixture was added to each well. Each sample was run in triplicate. The plate was incubated for 30min at 20 °C. Absorbance was measured at 492 nm using a Tecan Infinite M200 ELISA reader (Männedorf, Switzerland). Background activity for each condition was subtracted from experimental values before comparison. A Student’s t-test was performed to assess for statistical significance (α = 0.05) for each cell type using GraphPad Prism (v 9.2.0, San Diego, CA).
2.3. In vitro scratch assay
Confluent cultures of human and canine CEC and stromal cells were trypsinized, resuspended in serum-free glucose-supplemented media (0, 0.92, or 6 mM), and seeded at 1 ×105 cells/well into an Ibidi Culture-Insert (Ibidi, Martinsried, Germany; n = 12 wells/group) and allowed to grow for 24h; 0.92 mM glucose simulated the glucose concentration in the tear film of diabetic humans, and 6 mM glucose simulated the canine diabetic ocular surface (Cullen et al., 2005; Sen and Sarin 1980). Human stromal cells were not viable in 0 mM glucose conditions for greater than 24h, as such, these cells were only incubated in 0.92 and 6 mM concentrations. After the culture inserts had been removed, cells were treated with vehicle or 2.4 μM human recombinant insulin in 0, 0.92, or 6 mM glucose-supplemented media. Mannitol (20 mmol/L) was used as an osmotic control; no differences were noted in any experiments when compared to glucose-treated cells (data not shown). The monolayers were imaged at regular intervals using inverted phase contrast microscopy, and the wound area in the center of the each well was determined using Image J software (National Institutes of Health, Rockville, MD). Epithelial cells were monitored at 12h intervals for 24h, and stromal cells were monitored at 6h intervals for 12h due to the faster rates of monolayer reconstitution observed in fibroblasts. The cellular monolayers and medium were harvested at experimental endpoints and stored at −80 °C until analysis. To account for variability in the size and shape of the initial wound area observed microscopically, all wound area measurements were calculated as mean percent wound area remaining, relative to the size of the original area. A univariate analysis of variance with least significant difference (LSD) post-hoc evaluation was performed to assess for statistical significance (α = 0.05).
2.4. Immunoblotting
Homogenized donor corneal stroma and cellular monolayers were solubilized in a 50 mM Tris lysis buffer containing 150 mM sodium chloride, 1% Triton X-100, and Halt™ protease and phosphatase inhibitors (Thermo, Waltham, MA). Protein concentration was determined using a Pierce 660 Protein Assay with a standard BSA curve. Protein lysates were separated by 10% SDS-polyacrylamide gel electrophoresis and then transferred to a nitrocellulose membrane and blocked in 5% dry milk. Membranes were incubated with anti-GLUT1 (1:200, Santa Cruz Biotechnology, Santa Cruz, CA), anti-keratocan (1:750, Biorbyt, St. Louis, MO), anti-CD34 (1:2000, Abcam, Cambridge, UK), anti-αSMA (1:2000, Abcam), anti-PI3K (1:1000, Abcam), anti-Akt (1:1000, Cell Signaling Technology, Danvers, MA), anti-phospho Akt (1:1000, Cell Signaling Technology), anti-mTOR (1:1000, Cell Signaling Technology), anti-phospho mTOR (1:1000, Cell Signaling Technology), or anti-β-actin (1:1000, Thermo) overnight at 4 °C. Anti-rabbit, -goat, -donkey, or -mouse IgG horseradish peroxidase conjugated secondary antibody (1:5000, Thermo) was applied for 2h at room temperature. Blots were visualized, and protein band densities were evaluated in triplicate by densitometry analysis using the ChemiDoc Imaging Station (Bio-Rad Laboratories, Hercules, CA) and normalized relative to expression of β-actin or the unphosphorylated protein of interest. Positive controls for keratocan and CD34 included normal human corneal stroma and kidney lysate, respectively. A two-way analysis of variance with Tukey’s multiple comparisons test was performed to assess for statistical significance (α = 0.05).
2.5. TGF-β1 ELISA
Culture media were subject to a commercially available, species-specific TGF-β1 Quantikine® ELISA (R&D Systems, Inc., Minneapolis, MN), per the recommended protocol. The absorbance of the samples and standards was measured using a Tecan Infinite M200 ELISA reader at 540 nm with a 450 nm reference wavelength. A two-way analysis of variance with Tukey’s multiple comparisons test was performed to assess for statistical significance (α = 0.05).
2.6. IR and IGF-1R immunohistochemistry
Standard avidin-biotin complex immunohistochemistry was performed, as previously described (Colitz et al., 2000). Briefly, fresh normal human corneas (n = 4) and diabetic human corneas (n = 4), obtained from the Department of Anatomy and the Body Donor Service at The Ohio State University, were fixed in 10% neutral-buffered formalin and were submitted to the Comparative Pathology and Mouse Phenotyping Shared Resource at The Ohio State University College of Veterinary Medicine for paraffin-embedding and sectioning. Formalin-fixed paraffin-embedded sections of normal canine cornea (n = 6) were obtained from previous studies and diabetic canine cornea (n = 3) were obtained from The Ohio State Veterinary Medical Center Applied Pathology Service following a medical records search performed by the Health Information Section at The Ohio State University Veterinary Medical Center. Slides were incubated with primary antibodies (anti-IR or anti-IGF-1R, Abcam) for 36h at 4 °C at a dilution of 1:50 and biotinylated donkey anti-rabbit IgG (Thermo) for 1h at room temperature at a dilution of 1:500. Stained samples were visualized using a Nikon Eclipse 80i digital microscope. Positive controls consisted of human breast carcinoma (n = 3) and murine liver (n = 5), and negative controls were processed without the primary antibody. Controls were performed concurrently with each batch of slides.
3. Results
3.1. In vitro cytotoxicity
There were no significant differences in the amount of LDH released between treatment with vehicle or human recombinant insulin across any cell type, as shown in Fig. 1.
Fig. 1. Insulin is not toxic to human or canine corneal cells.

There were no significant differences in LDH release when comparing insulin and control in treated human CEC or fibroblasts (n = 7), or canine CEC (n = 12) or fibroblasts (n = 12) in vitro (Student’s t-test).
3.2. GLUT-1 expression
All cell types expressed the 55 kDa GLUT-1 protein with significantly greater expression in CEC compared to fibroblasts in both humans (p = 0.006) and dogs (p = 0.0116; Fig. 2).
Fig. 2. GLUT1 is expressed in both human and canine corneal cells.
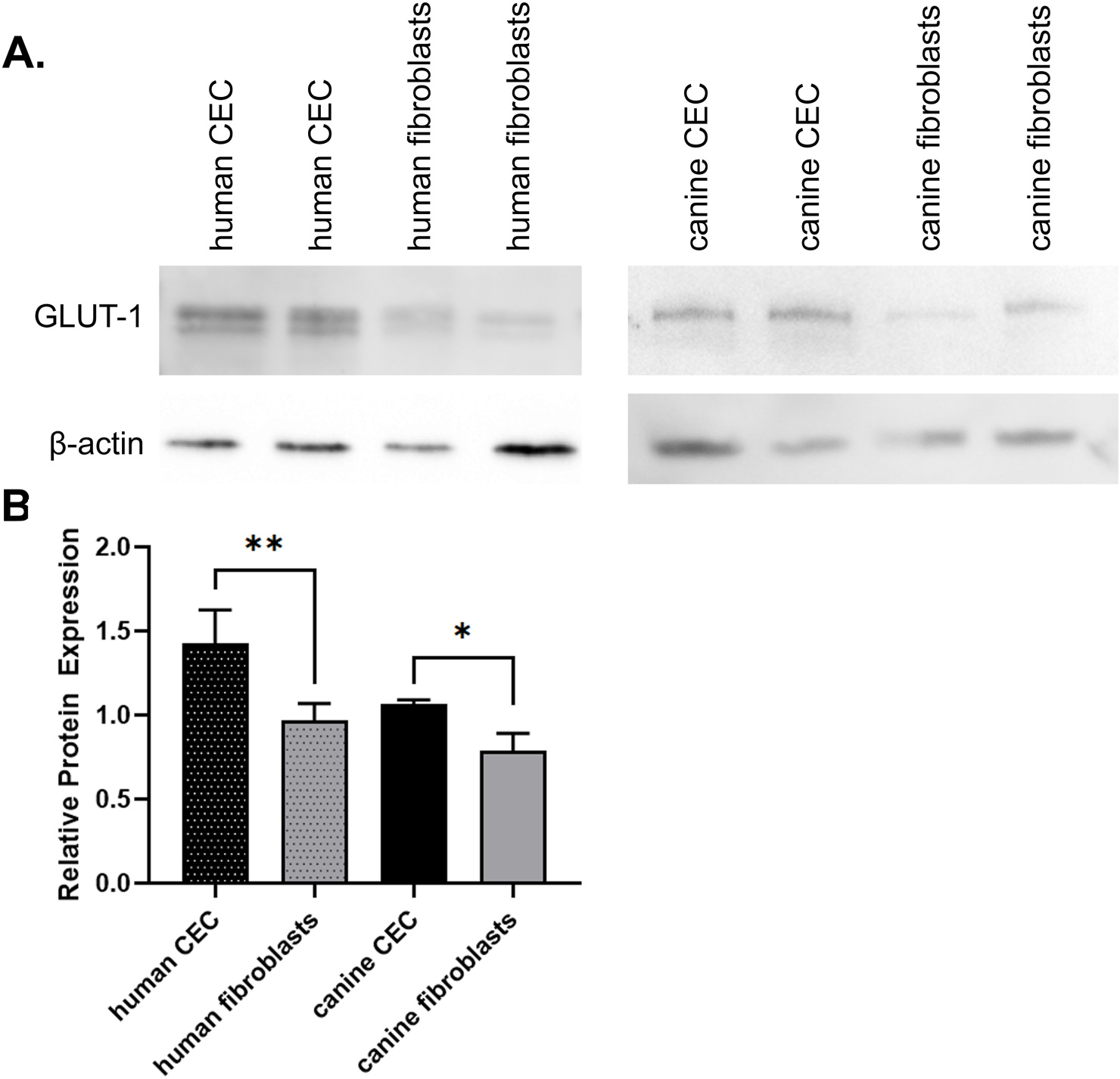
Western blot analysis (A) demonstrates GLUT1 expression in human and canine CEC (n = 12) and stromal cells (n = 7 and 12, respectively). (B) Quantification of GLUT1 expression relative to β actin (ANOVA, *p < 0.05, **p < 0.01).
3.3. In vitro scratch assay
In the absence of additional glucose, treatment with insulin resulted in significantly improved (p = 0.0004) wound healing in human CEC compared to vehicle control, as shown in Fig. 3A and B. High glucose concentration (6 mM) significantly slowed wound healing compared to 0 mM (p ≤ 0.0001), but there was no significant interaction between glucose and treatment (p = 0.7224).
Fig. 3. Effect of glucose and insulin on cultured human corneal cells.

Mean wound area remaining at 12 or 24 h for human CEC (A, B) and fibroblasts (n = 7; C, D) incubated in 0, 0.92, or 6 mM glucose and treated with vehicle or insulin. (ANOVA, **p < 0.01, ***p < 0.001, ****p ≤ 0.0001). Scale bar = 100 μm.
Insulin treatment resulted in significantly improved (p ≤ 0.001) wound healing in human fibroblasts compared to vehicle control, as shown in Fig. 3C and D. Glucose concentration did not significantly affect wound healing (p = 0.1248) but there was a significant interaction between glucose and treatment (p = 0.0038).
Treatment with insulin resulted in improved wound healing in canine CEC compared to vehicle control; this was a significant change in the 0 mM glucose concentration (p = 0.0082; Fig. 4A and B). Neither glucose concentration (p = 0.4226) nor the interaction between glucose and treatment (p = 0.3724) significantly affected wound healing.
Fig. 4. Effect of glucose and insulin on cultured canine corneal cells.

Mean wound area remaining at 12 or 24 h for canine CEC (n = 12; A, B) and fibroblasts (n = 12; C, D) incubated in 0, 0.92, or 6 mM glucose and treated with vehicle or insulin. (ANOVA, **p < 0.01, ***p < 0.001, ****p ≤ 0.0001). Scale bar = 100 μm.
In canine fibroblasts, treatment with insulin did not result in significant differences in wound healing (p = 0.1624) compared to vehicle control, as shown in Fig. 4C, D. Higher glucose concentration significantly improved wound healing (p ≤ 0.001), and there was a significant interaction between glucose and treatment (p = 0.0322).
3.4. Stromal cell characterization
Human stromal cells variably expressed keratocan, CD34, and α-SMA under all treatment conditions, as shown in Fig. 5A, C. Expression of the keratocyte markers keratocan and CD34 was significantly lower in 6 mM glucose conditions (p = 0.002, p < 0.001, respectively), while α-SMA expression was significantly higher in the 6 mM glucose conditions (p < 0.001). Additionally, expression of keratocan (p = 0.005) and CD34 (p = 0.001) significantly decreased with insulin treatment, while α-SMA expression significantly increased with insulin treatment (p < 0.001). Canine stromal cells also variably expressed keratocan, CD34, and α-SMA protein under all treatment conditions, as shown in Fig. 5B, D. Similar to the findings in human stromal cells, canine stromal cell expression of keratocan (p < 0.001) and CD34 (p < 0.001) significantly decreased with glucose concentration, and α-SMA significantly increased (p < 0.001), in a dose-dependent manner with glucose concentration. Treatment of canine cells with insulin did not result in significant differences in keratocan (p = 0.581) or CD34 expression (p = 0.341); however, α-SMA expression was significantly decreased with insulin treatment compared to vehicle (p = 0.004). Further, corneal stroma from both human and canine diabetic globes showed increased α-SMA protein expression compared to non-diabetic controls (Fig. 5E).
Fig. 5. Glucose concentration alters the human and canine stromal cell phenotype in vitro and in vivo.
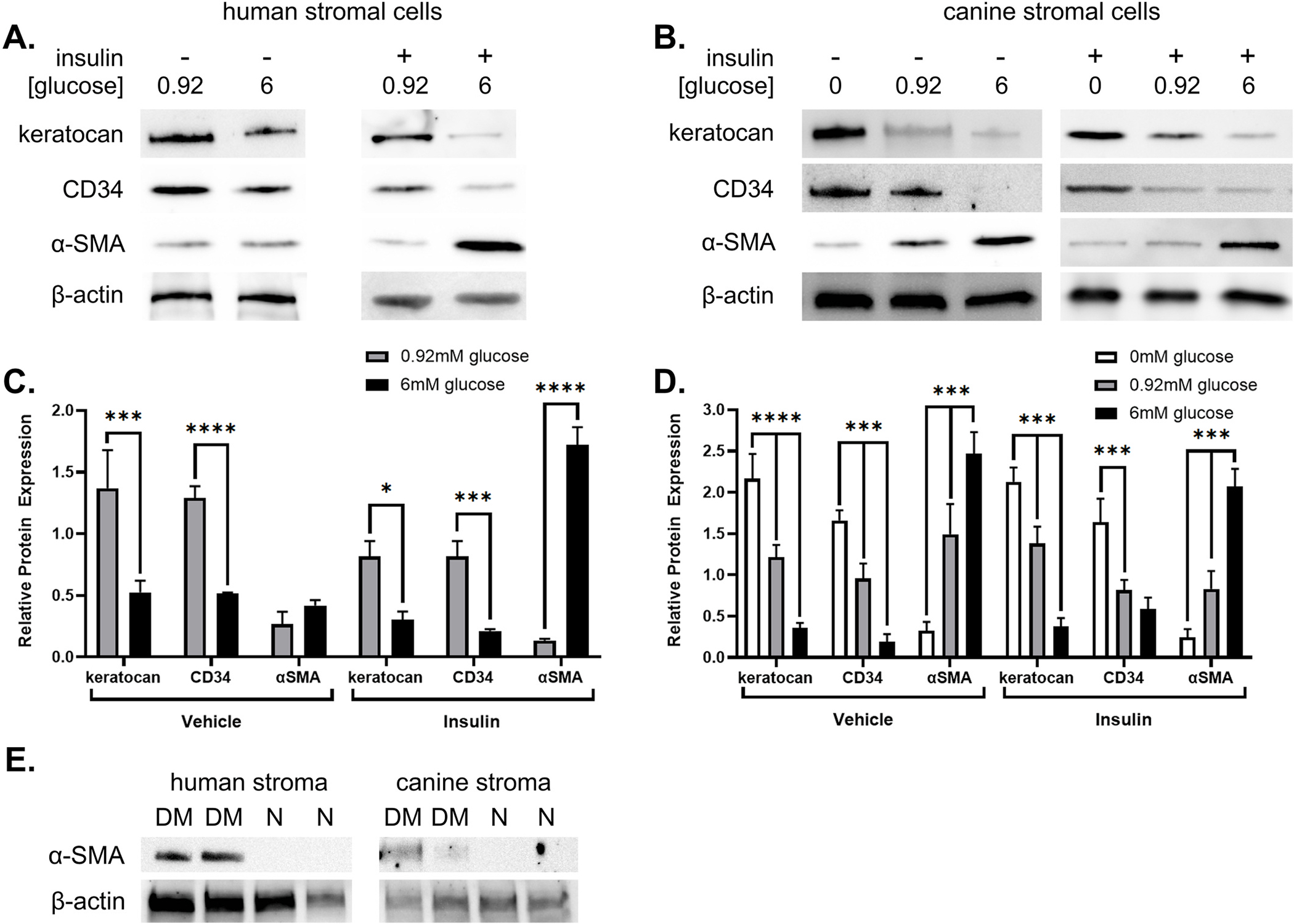
Expression of keratocyte markers in human (n = 7; A, C) and canine (n = 12; B, D) stromal cells (ANOVA, *p < 0.05, ***p < 0.001, ****p ≤ 0.0001). Expression of α-SMA expression in human (n = 2) and canine (n = 2) donor corneal stroma (E).
3.5. TGF-β1 expression
TGF-β1 concentration was significantly increased in culture media from human stromal cells incubated in 6 mM glucose (p < 0.001), as shown in Fig. 6A. Insulin treatment also induced significantly greater (p = 0.004) TGF-β1 concentration compared to vehicle treatment in human stromal cells. TGF-β1 concentration in canine stromal cells significantly increased with glucose concentration (p = 0.001; Fig. 6B); however, insulin treatment did not result in significant differences in TGF-β1 concentration (p = 0.122). Interestingly, TGF-β1 concentration measured in canine stromal cells was approximately 10-fold lower than concentrations measured in human stromal cells.
Fig. 6. Glucose increases stromal cell expression of TGF-β1.

Expression of TGF-β1 in human (n = 7; A) and canine (n = 12; B) stromal cells. (ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001).
3.6. RTK immunohistochemistry
Normal human corneas demonstrated expression of IR and IGF-1R in the perinuclear and cytoplasmic regions of the basal epithelial cells as well as the stromal cells, as shown in Fig. 7A and B. Diabetic human corneas had a similar, although less intense, staining pattern as the normal eyes (Fig. 7C and D). Normal canine corneas also demonstrated expression of the IR and the IGF-1R in the perinuclear and cytoplasmic regions of the basal epithelial cells, as well as the stromal cells (Fig. 7E and F). By comparison, diabetic canine corneas did not express either RTK in epithelial or stromal cells, as shown in Fig. 7G and H.
Fig. 7. IR and IGF-1R are expressed in diabetic human corneas but not in diabetic canine corneas.
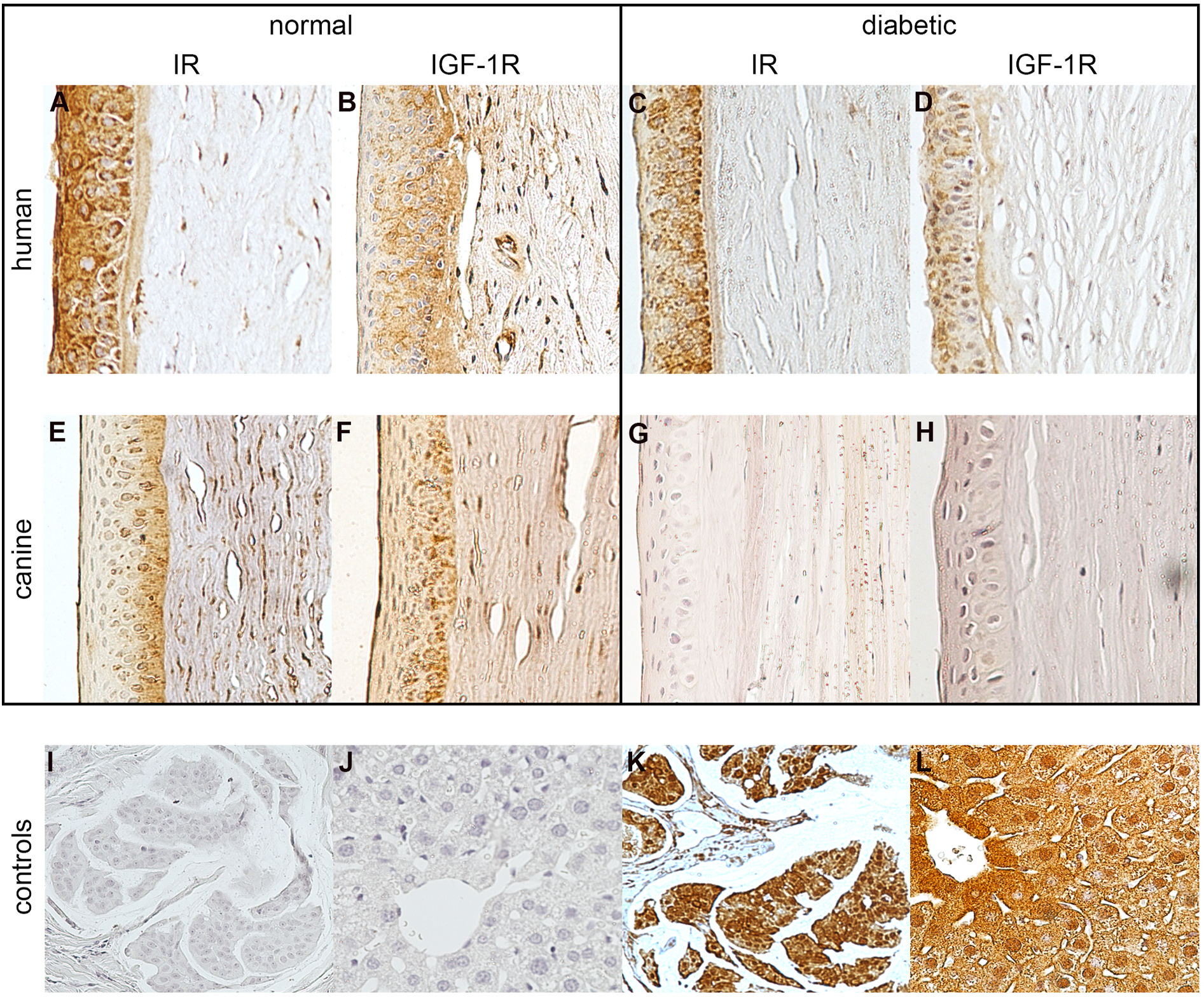
Immunolabeling of the normal human cornea (n = 4) with anti-IR and anti-IGF-1R (A, B), and of the diabetic human cornea (n = 4) with anti-IR and anti-IGF-1R (C, D). Immunolabeling with anti-IR and anti-IGF-1R in normal (n = 6; E, F) and diabetic (n = 3; G, H) canine cornea. Negative controls (I, J) and positive controls including human breast carcinoma (n = 3; K) and murine liver (n = 5; L).
3.7. Kinase expression
As glucose concentration increased, PI3K expression significantly decreased in both vehicle- and insulin-treated human CEC (p ≤ 0.0001, Fig. 8A and B). When evaluating pAkt, vehicle-treated human CEC had significantly decreased expression in the 6 mM glucose concentration (p ≤ 0.0001) when compared to both 0 and 0.92 mM glucose, while insulin-treated human CEC had significantly lower pAkt expression in both 0.92 and 6 mM glucose conditions, compared to 0 mM glucose. Similar to the observed changes in PI3K expression, p-mTOR was significantly decreased (p ≤ 0.001) in both vehicle- and insulin-treated human CEC, as glucose concentration increased.
Fig. 8. Effect of glucose and insulin on PI3K signaling in cultured human corneal cells.

Expression of PI3K, p-Akt, and p-mTOR in wounded human CEC (A, B) and fibroblasts (n = 7; C, D) incubated in 0, 0.92, or 6 mM glucose and treated with vehicle or insulin (ANOVA, *p < 0.05, ***p < 0.001, ****p ≤ 0.0001). Protein expression is relative to β-actin or the unphosphorylated protein of interest.
Unlike human CEC, wounded human stromal cells did not demonstrate significant changes in PI3K or pAkt expression when comparing glucose concentration in either vehicle- or insulin-treated conditions (Fig. 8C and D). It should be noted that as glucose concentration increased, vehicle-treated cells had decreased PI3K and pAkt expression, while insulin-treated cells had increased expression of the kinases. Phosphorylated-mTOR expression was found to be significantly (p < 0.05) decreased in vehicle-treated human stromal cells when comparing glucose concentrations. When insulin treatment was applied, there was a significant increase in expression of the kinases pAkt and p-mTOR in the 6 mM glucose condition (p = 0.01). Although not significant, insulin treatment also increased expression of PI3K within the 6 mM glucose condition.
As shown in Fig. 9A and B, wounded canine CEC had significantly increased expression of PI3K, pAkt, and p-mTOR in insulin-treated cells compared to the vehicle control (p ≤ 0.001). In vehicle-treated CEC, PI3K expression significantly (p < 0.01) decreased when comparing the 0 and 6 mM glucose conditions, and pAkt expression significantly decreased when comparing both 0.92 and 6 mM glucose to 0 mM glucose (p < 0.05, p ≤ 0.0001, respectively); p-mTOR expression did not significantly change with glucose concentrations. In insulin-treated canine CEC, PI3K was significantly (p < 0.05) elevated in the 6 mM glucose condition relative to the 0.92 mM glucose treatment, while pAkt expression significantly (p < 0.01) increased in both 0.92 and 6 mM glucose relative to 0 mM glucose. Phosphorylated-mTOR significantly increased in insulin-treated canine CEC when comparing 6 mM glucose to 0 mM glucose (p < 0.05).
Fig. 9. Effect of glucose and insulin on PI3K signaling in cultured canine corneal cells.

Expression of PI3K, p-Akt, and p-mTOR in wounded canine CEC (n = 12; A, B) and fibroblasts (n = 12; C, D) incubated in 0, 0.92, or 6 mM glucose and treated with vehicle or insulin (ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001, ****p ≤ 0.0001). Protein expression is relative to β-actin or the unphosphorylated protein of interest.
In wounded vehicle-treated canine stromal cells, significant decreases in the expression of PI3K and pAkt were observed as glucose concentration increased (Fig. 9C and D); p-mTOR expression also decreased with increasing glucose concentrations but this was not found to be significant. By comparison, insulin treatment resulted in increased kinase expression, even as glucose concentrations increased.
4. Discussion
Initial determination of in vitro cytotoxicity of insulin in primary canine corneal CEC and fibroblasts was performed, as no previous study has utilized insulin on the canine cornea. Further, determining whether the insulin concentration utilized in this study, extrapolated from the topical dose of 2U administered to the rat ocular surface, induced cytopathic changes to human CEC or stromal cells was critical before proceeding with additional experiments (Zagon et al., 2007). As expected, at the concentration and time points evaluated in the present experiments, insulin did not induce cytopathic changes or increase cytotoxicity. Pre- and post-prandial plasma insulin concentrations in both humans and dogs have been established, and the tear film insulin concentration in humans has been evaluated, all measuring orders of magnitude lower than the concentrations utilized here (Melmed et al., 2011; Gayet et al., 2004; Rocha et al., 2002; Nomura et al., 1990; Pillion et al., 1991). The insulin concentration in human tears, as measured by a radioimmunoassay, has been shown to be 0.404 ng/mL (10−5 μM) (Rocha et al., 2002). Normal canine fasted and post-prandial plasma insulin concentrations range between 18 and 40μU/mL (10−4 μM), and no study has yet to evaluate the insulin concentration of the normal or diabetic canine tear film (Gayet et al., 2004).
Insulin treatment resulted in significantly improved wound healing in human and canine CEC at the 0 mM glucose concentration, suggesting that enhanced epithelial migration or proliferation occurs with exogenous insulin treatment in conditions simulating the normal ocular surface. Interestingly, in the current study the concentration of glucose, alone, significantly affected epithelial wound healing in vitro. High glucose environments have been shown to decrease adhesion and induce down regulation of the integrin α3β1 and laminin-5 compared to normal glucose conditions in human CEC (Fujita et al., 2003; Lu et al., 2006). One study demonstrated attenuated ex vivo corneal epithelial wound healing in high glucose environments and further showed that high glucose induced ROS-mediated suppression of the EGF receptor pathway (Xu et al., 2009). Additional work has documented cross-talk between IGF and EGF in tumor progression, suggesting that alterations in extracellular glucose concentrations may affect cellular proliferation and migration via multiple intracellular pathways (Adams et al. 2004).
There is also substantial data to support glucose-mediated modulation of insulin signaling. Insulin has two high affinity and four low affinity glucose binding sites, which readily become saturated in states of hyperglycemia and alter the affinity of the receptor for the glycated insulin (Zoete et al. 2004). Further, the binding of insulin to the IR has been shown to be glucose-sensitive, and glucose facilitates the upregulation of the IR by insulin (Yu and Eriksson 2000; Root-Bernstein and Vonck 2009). One study demonstrated insulin-like IR peptides are capable of binding glucose with low millimolar Kd (3 mM), supporting the notion that these corresponding IR regions would be sensitive to alterations in serum glucose concentrations within physiologic ranges (Root-Bernstein and Vonck 2009; Güemes et al. 2016). Further, this work clarified previous investigations reporting two-step insulin binding with high affinity glucose Kd of 250 μM and low affinity Kd of 15 mM (Root-Bernstein and Vonck 2009; Anzenbacher and Kalous 1975; Kalous and Anzenbacher 1979). Insulin binding to the adipocyte IR was shown to be three times greater in 5.6 mM glucose conditions compared to 0 mM in another study (Yu and Eriksson 2000). This glycation of insulin has been shown to be both time and concentration-dependent (O’Harte et al., 1996; Abdel-Wahab et al., 1997). Finally, there is evidence to support that the glucose transporters (GLUTs) allosterically complex with the IR once translocated to the plasma membrane, further supporting the feedback of glucose and insulin in physiologic and pathologic cellular metabolism (H. F. Li et al., 1997).
Previous studies have found that rabbit and human CEC, human stromal fibroblasts, and human limbal stem cells express GLUT-1, a classically insulin-insensitive GLUT; in these cells, the binding of insulin to its receptor did not effectively result in a change in glucose uptake by the cell (Kumagai et al. 1994; Takahashi et al., 2000; Kuipers et al., 2013; Zurawski et al. 1989; Takahashi et al. 1996). Cells that preferentially express GLUT-1 over GLUT-4 transport glucose by facilitated diffusion down the concentration gradient (Leney and Tavaré 2009). These tissues tend to equilibrate the intracellular glucose with the serum glucose concentration and are more susceptible to damage with chronic hyperglycemia, as is the case with diabetes (Leney and Tavaré 2009). Two additional studies demonstrated similar GLUT-1 expression in both human and rat diabetic and non-diabetic CEC (Kumagai et al. 1994; Takahashi et al., 2000). Demonstrating GLUT-1 expression in canine CEC and stromal fibroblasts is a novel finding, as no previous evaluation of GLUT expression in the canine eye was identified in the literature. Collectively, these results support that insulin improves corneal wound healing in vitro independent of cellular glucose uptake.
Insulin promoted wound healing in human stromal cells at both glucose concentrations evaluated, suggesting exogenous insulin facilitates proliferation or migration of stromal fibroblasts in conditions simulating the normal and diabetic ocular surface. One study has previously reported insulin stimulation of cellular proliferation by corneal stromal cells in vitro (Musselmann et al., 2005). Using a univariate analysis of variance, glucose had a significant effect on stromal wound healing, in addition to insulin treatment in these cells. Interestingly, although insulin treatment improved healing in canine stromal cells, the difference was not significant. Additional studies would need to be performed to further evaluate such species differences, but the current study suggests that canine stromal cells are somewhat less responsive to insulin than their human counterparts.
We next evaluated protein expression of α-SMA and TGF-β1 in vehicle and insulin-treated human and canine stromal fibroblasts to determine whether insulin could induce cellular differentiation towards a more myofibroblastic phenotype. TGF-β is present in the tear film and aqueous humor, and TGF-β mRNA and/or protein has been detected in CEC, keratocytes, and stromal fibroblasts (Zheng et al., 2010; Klenkler et al. 2007; Tandon et al., 2010; Utsunomiya et al., 2016; Y. C. Wang et al., 2019; Funderburgh et al., 2001). TGF-β is known to induce α-SMA expression and fibroblast differentiation into the contractile, myofibroblast phenotype (Saika et al., 2004; B. Li and Wang 2011; Tomasek et al., 2002). While the tissue contracture resulting from myofibroblast activity is essential for wound closure, excessive extracellular matrix and collagen secretion can lead to corneal opacity, observed clinically as stromal haze or fibrosis (S. E. Wilson 2012).
To more completely characterize the stromal cell phenotype, expression of the keratocyte markers keratocan and CD34 were also evaluated. In both species, glucose significantly increased expression of myofibroblast markers, with dose-dependent increases in α-SMA and TGF-β1. Treatment with insulin also significantly affected expression of all three proteins and TGF-β1 concentration in human fibroblasts, favoring a more fibrotic phenotype. Interestingly, in canine cells, insulin treatment did not significantly affect TGF-β1 concentration but did result in significantly lower α-SMA expression, suggesting a protective effect against a fibrotic phenotype. Similar to our results in canine stromal cells, bovine stromal cells incubated in insulin-supplemented culture medium retained the dendritic morphology and expression of the keratocyte markers aldehyde dehydrogenase and keratocan (Musselmann et al., 2005). Alpha-smooth muscle actin was also evaluated in corneal stroma obtained from diabetic human and canine donors. Although the sample size was limited, the data confirm that naturally occurring diabetic corneas are potentially more fibrotic than non-diabetic controls. These in vitro findings may account for the increased predisposition to clinical stromal haze or fibrosis following wounding in the diabetic eye, as both epithelial injury and subsequent exposure of the anterior stroma and resident keratocytes to higher glucose concentrations in the diabetic tear film, induce myofibroblast differentiation (Sánchez-Thorin 1998; Hager et al. 2009; Hasan 2010; Espaillat 2012; Bikbov and Surkova 2016; Thoft et al. 1971).
Low glucose conditions have been shown to induce a stromal cell reversion to the keratocyte phenotype in vitro, with diminished expression of α-SMA and other stress proteins and enhanced expression of mesenchymal progenitor cell markers such as CD34 (Foster et al. 2015; Shah et al., 2012). This less differentiated phenotype is more sessile with decreased migratory and contractile potential (Foster et al. 2015; Pinnamaneni and Funderburgh 2012). Demonstrating high expression of the keratocyte markers keratocan and CD34 in human stromal cells incubated in 0 mM glucose conditions would confirm this cellular phenotype; however, human stromal cells were not viable in the absence of glucose. Interestingly, canine stromal cells remained viable in 0 mM glucose conditions. It is unclear why this species-specific difference was noted. Of note, the canine stromal cells with highest expression of keratocyte markers (0 mM glucose) corresponded to the stromal cell treatment group with the slowest wound healing. Slower migration rates would be more expected in a keratocyte population compared to either a fibroblast or myofibroblast population. It should be recognized that all cultured cells evaluated for changes in protein expression were also subject to an in vitro wounding assay, which may be independently capable of stimulating differentiation. Phenotypic reversion is likely to be both passage and density-dependent, with CD34 positive cells being demonstrated in passages four and earlier in a previous study (Foster et al. 2015). The differences in response to insulin treatment and expression of fibrotic markers by human and canine fibroblasts may support an increased glucose sensitivity by human cells and account for the clinical predisposition to corneal fragility and scarring not reported in diabetic dogs. This notion is further supported by less drastic changes in TGF-β1 concentrations and α-SMA expression in diabetic donors and fibroblasts treated in high glucose conditions observed in canine corneas in the current study.
Immunohistochemical evaluation of the normal and diabetic human cornea was consistent with previous studies that have demonstrated expression of the IR and IGF-1R in the corneal epithelium and stroma of normal human cadaveric globes and unaltered IR expression in diabetic eyes (Naeser 1997; Rocha et al., 2002; Wu et al. 2012; Robertson et al. 2012). As no study has previously evaluated insulin signaling receptors in the canine cornea, this is a novel demonstration of perinuclear and cytoplasmic expression of IR and IGF-1R in the normal corneal epithelium and intracellular expression of both by stromal cells. Absent expression of both RTK’s in the diabetic canine cornea and the subsequent divergent cell signaling may account for the differences in clinical manifestations of ocular surface disease between human and canine diabetic eyes; however, the significance of this finding remains to be elucidated. Insulin-induced IR downregulation and post-receptor defects in the glucose transport system have been demonstrated in rat adipocytes and pancreatic acinar cells and adipocytes isolated from insulin-resistant human patients in vitro; accelerated RTK internalization and degradation may contribute to the expression patterns observed in the diabetic canine cornea in this study (Garvey et al. 1985; Okabayashi et al., 1989; Kolterman et al., 1981; Fink et al., 1984). Future studies to further characterize IR, IGF-1R, and Hybrid-R expression in both human and canine corneal cells in response to ambient glucose concentrations and exogenous insulin stimulation may provide additional information regarding the RTK activation kinetics in determining downstream kinase cascade signaling.
Glucose concentration significantly affected expression of PI3K kinases in wounded human CEC. Expression of all kinases, regardless of treatment, was inversely proportional to glucose concentration, and treatment with insulin resulted in significantly lower expression of p-Akt compared to vehicle control. Diminished p-Akt protein expression at 6 mM glucose conditions suggests an attenuated capacity to promote Akt activation in the face of hyperglycemia, which has been previously demonstrated in diabetic skeletal muscle (Krook et al., 1998; Zdychová and Komers 2005). Alternatively, p-Akt expression may have be reduced in 6 mM conditions due to the negative feedback of Akt resulting from high glucose in an attempt to limit further GLUT translocation (Hernandez et al. 2001; Varma et al., 2005). One study demonstrated a 30% reduction in acute activation of Akt by insulin following chronic exposure to high glucose (Nelson et al. 2002). The authors hypothesized that dephosphorylation of Akt may be accelerated by increased expression or activation of phosphoprotein phosphatases, such as PTEN; such a mechanism could explain how p-Akt expression could be significantly lower with insulin treatment at higher glucose concentrations (Nelson et al. 2002).
Glucose concentration did not significantly affect expression of PI3K kinases in wounded human stromal fibroblasts; however, in vehicle controls, p-mTOR expression was significantly lower in 6 mM glucose conditions compared to 0.92 mM. With glucose deprivation, compensatory induction of mTOR has been demonstrated in rat myocytes, suggesting that mTOR may be upregulated in the face of glucose deprivation as a bystander to PI3K, Akt, and GLUT activation (Miniaci et al., 2015). Contrary to these results, however, are studies documenting activation of mTOR with high glucose conditions in rat mesangial cells resulting in protein synthesis through activation of Akt (Das et al., 2014). Additionally, mTOR activity is further induced by high glucose through increased expression of the transcription factor FoxO1, which represses transcription of target genes including antioxidants, catalases, and proline-rich Akt substrate 40 (PRAS40), leading to ROS production and positive feedback for mTOR expression (Das et al., 2014). This is supported by data from the current study in which expression of both p-Akt and p-mTOR was significantly higher in insulin-treated cells compared to vehicle control at the 6 mM glucose concentration. The differential responses of mTOR activation in response to glucose are likely to be species, tissue, and time-dependent. As an example of this, for both canine CEC and fibroblasts, kinase expression was inversely proportional to glucose concentration with vehicle treatment but directly proportional to glucose with insulin treatment.
Collectively, these experiments indicate that PI3K pathway kinase expression significantly increases with insulin treatment in canine corneal cells but not in human corneal cells in vitro. Kinase expression in CEC was evaluated at 24h post-injury, while expression in stromal cells was evaluated at 12h, when monolayers were nearly restored, indicating that stromal cells respond more quickly than CEC in vitro. The kinetics of kinase induction in CEC and stromal cells in vivo are more likely to follow the temporal patterns of wound healing responses, with Akt induction and migratory changes by CEC occurring first, after the initial lag in the healing response, and the insulin-stimulated Akt activity in stromal cells requiring longer to respond. Additionally, signal transduction in vivo is likely to involve significant feedback between the CEC and stromal cells. The basis for the species-specific differential responses in kinase protein expression is unclear but may be contributory to the differences in the clinical course of diabetes between human and canine ophthalmology patients.
In summary, we have demonstrated that exogenous insulin induces the PI3K/Akt/mTOR pathway through binding of a RTK to promote in vitro wound healing and is safe to use in human and canine corneal cells. Differential responses to variable extracellular glucose conditions and exogenous insulin may contribute to the differences in the clinical presentation of diabetic corneal disease between human and canine patients.
Acknowledgments
This work was supported by grants from Prevent Blindness Ohio, The Ohio Lions Eye Research Foundation, the Cincinnati Eye Institute Foundation, and National Institutes of Health (EY030621, CA016058).
Footnotes
Declaration of competing interest
Neither author has any conflict of interest to declare.
CRediT author contribution statement
C. Peterson: contributed equally to experimental conception and design, Both authors have read and approved the final manuscript. H.L. Chandler: contributed equally to experimental conception and design, Both authors have read and approved the final manuscript.
References
- Abdelkader H, Patel DV, McGhee CN, Alany RG, 2011. New therapeutic approaches in the treatment of diabetic keratopathy: a review. Clin. Exp. Ophthalmol 39 (3), 259–270. 10.1111/j.1442-9071.2010.02435.x. [DOI] [PubMed] [Google Scholar]
- Abdel-Wahab YH, O’Harte FP, Barnett CR, Flatt PR, 1997. Characterization of insulin glycation in insulin-secreting cells maintained in tissue culture. J. Endocrinol 152 (1), 59–67. 10.1677/joe.0.1520059. [DOI] [PubMed] [Google Scholar]
- Adams TE, Epa VC, Garrett TP, Ward CW, 2000. Structure and function of the type 1 insulin-like growth factor receptor. Cell. Mol. Life Sci.: CMLS 57 (7), 1050–1093. 10.1007/PL00000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams TE, McKern NM, Ward CW, 2004. Signalling by the type 1 insulin-like growth factor receptor: interplay with the epidermal growth factor receptor. Growth Factors 22 (2), 89–95. 10.1080/08977190410001700998. [DOI] [PubMed] [Google Scholar]
- Alves MDC, Carvalheira JB, Módulo CM, Rocha EM, 2008. Tear film and ocular surface changes in diabetes mellitus. Arq. Bras. Oftalmol 71 (6 Suppl. l), 96–103. 10.1590/s0004-27492008000700018. [DOI] [PubMed] [Google Scholar]
- American Diabetes Association, 2013. Diagnosis and classification of diabetes mellitus. January Diabetes Care 36 (Suppl. 1), S67–S74. 10.2337/dc13-S067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzenbacher P, Kalous V, 1975. Binding of D-glucose to insulin. Biochim. Biophys. Acta 386 (2), 603–607. 10.1016/0005-2795(75)90303-7. [DOI] [PubMed] [Google Scholar]
- Apikoglu-Rabus S, Izzettin FV, Turan P, Ercan F, 2010. Effect of topical insulin on cutaneous wound healing in rats with or without acute diabetes. Clin. Exp. Dermatol 35 (2), 180–185. 10.1111/j.1365-2230.2009.03419.x. [DOI] [PubMed] [Google Scholar]
- Awata T, Sogo S, Yamagami Y, Yamamoto Y, 1988. Effect of an aldose reductase inhibitor, CT-112, on healing of the corneal epithelium in galactose-fed rats. J. Ocul. Pharmacol 4 (3), 195–201. 10.1089/jop.1988.4.195. [DOI] [PubMed] [Google Scholar]
- Azar D, Spurr-Michaud S, Tisdale A, Gipson I, 1992. Altered epithelial-basement membrane interactions in diabetic corneas. Arch. Ophthalmol 110, 537–540. [DOI] [PubMed] [Google Scholar]
- Bikbov MM, Surkova VK, 2016. Cornea and its changes in diabetes mellitus: the review. Diabetes Mellitus 19 (6), 479–485. 10.14341/dm7972. [DOI] [Google Scholar]
- Centers for Disease Control, 2014. CDC-national diabetes statistics report, 2014 Med. Benefit 31 (14), 3. [Google Scholar]
- Colitz CM, Malarkey D, Dykstra MJ, McGahan MC, Davidson MG, 2000. Histologic and immunohistochemical characterization of lens capsular plaques in dogs with cataracts. Am. J. Veteri. Res 61 (2), 139–143. 10.2460/ajvr.2000.61.139. [DOI] [PubMed] [Google Scholar]
- Cousen P, Cackett P, Bennett H, Swa K, Dhillon B, 2007. Tear production and corneal sensitivity in diabetes. J. Diab. Complicat 21 (6), 371–373. 10.1016/j.jdiacomp.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Cruz-Cazarim ELC, Cazarim MS, Ogunjimi AT, Petrilli R, Rocha EM, Lopez RFV, 2019. Prospective insulin-based ophthalmic delivery systems for the treatment of dry eye syndrome and corneal injuries. Eur. J. Pharm. Biopharm 140 (July), 1–10. 10.1016/j.ejpb.2019.04.014. [DOI] [PubMed] [Google Scholar]
- Cullen CL, Ihle SL, Webb AA, McCarville C, 2005. Keratoconjunctival effects of diabetes mellitus in dogs. Vet. Ophthalmol 8 (4), 215–224. 10.1111/j.1463-5224.2005.00389.x. [DOI] [PubMed] [Google Scholar]
- Das F, Ghosh-Choudhury N, Dey N, Bera A, Mariappan M, Kasinath BS, Ghosh Choudhury G, 2014. High glucose forces a positive feedback loop connecting Akt kinase and FoxO1 transcription factor to activate MTORC1 kinase for mesangial cell hypertrophy and matrix protein expression. J. Biol. Chem 289 (47), 32703–32716. 10.1074/jbc.M114.605196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Meyts P, Ursø B, Christoffersen CT, Shymko RM, 1995. Mechanism of insulin and IGF-I receptor activation and signal transduction specificity. Receptor dimer cross-linking, bell-shaped curves, and sustained versus transient signaling. September Ann. N. Y. Acad. Sci 766, 388–401. 10.1111/j.1749-6632.1995.tb26688.x. [DOI] [PubMed] [Google Scholar]
- De Meyts P, Whittaker J, 2002. Structural biology of insulin and IGF1 receptors: implications for drug design.” nature reviews. Drug Discov. 1 (10), 769–783. 10.1038/nrd917. [DOI] [PubMed] [Google Scholar]
- Desai BM, Lavingia BC, 1987. Cornea thickness and tear glucose levels in diabetes mellitus and normal persons. Indian J. Ophthalmol 35 (5–6), 130–132. [PubMed] [Google Scholar]
- Ersahin T, Tuncbag N, Cetin-Atalay R, 2015. The PI3K/AKT/MTOR interactive pathway. Mol. Biosyst 11 (7), 1946–1954. 10.1039/c5mb00101c. [DOI] [PubMed] [Google Scholar]
- Espaillat A, 2012. Diabetic Eye Diseases: A Comprehensive Review. SLACK Incorporated, Thorofare, NJ. [Google Scholar]
- Fai S, Ahem A, Mustapha M, Mohd Noh UK, Bastion MLC, 2017. Randomized controlled trial of topical insulin for healing corneal epithelial defects induced during vitreoretinal surgery in diabetics. Asia Pac. J. Ophthalmol 6 (5), 418–424. 10.22608/APO.201780. [DOI] [PubMed] [Google Scholar]
- Fink RI, Kolterman OG, Kao M, Olefsky JM, 1984. The role of the glucose transport system in the postreceptor defect in insulin action associated with human aging. J. Clin. Endocrinol. Metabol 58 (4), 721–725. 10.1210/jcem-58-4-721. [DOI] [PubMed] [Google Scholar]
- Foster JW, Gouveia RM, Connon CJ, 2015. Low-glucose enhances keratocyte-characteristic phenotype from corneal stromal cells in serum-free conditions. June Sci. Rep 5. 10.1038/srep10839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita H, Morita I, Takase H, Ohno-Matsui K, Mochizuki M, 2003. Prolonged exposure to high glucose impaired cellular behavior of normal human corneal epithelial cells. Curr. Eye Res 27 (4), 197–203. 10.1076/ceyr.27.4.197.16598. [DOI] [PubMed] [Google Scholar]
- Funderburgh JL, Funderburgh ML, Mann MM, Corpuz L, Roth MR, 2001. Proteoglycan expression during transforming growth factor β-induced keratocyte-myofibroblast transdifferentiation. J. Biol. Chem 276 (47), 44173–44178. 10.1074/jbc.m107596200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvey WT, Olefsky JM, Marshall S, 1985. Insulin receptor down-regulation is linked to an insulin-induced postreceptor defect in the glucose transport system in rat adipocytes. J. Clini. Investi 76 (1), 22–30. 10.1172/JCI111950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayet C, Bailhache E, Dumon H, Martin L, Siliart B, Nguyen P, 2004. Insulin resistance and changes in plasma concentration of TNFalpha, IGF1, and NEFA in dogs during weight gain and obesity. J. Anim. Physiol. Anim. Nutr 88 (3–4), 157–165. 10.1111/j.1439-0396.2003.00473.x. [DOI] [PubMed] [Google Scholar]
- Gekka M, Miyata K, Nagai Y, Nemoto S, Sameshima T, Tanabe T, Maruoka S, Nakahara M, Kato S, Amano S, 2004. Corneal epithelial barrier function in diabetic patients. Cornea 23 (1), 35–37. [DOI] [PubMed] [Google Scholar]
- Giardini A, Roberts JRE, 1950. Concentration of glucose and total chloride in tears. The Br. J. Ophthalmol 34 (12), 737–743. 10.1136/bjo.34.12.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels M, 2000. Tear secretion and tear film function in insulin dependent diabetics. The Br. J. Ophthalmol 84 (1), 19–21. 10.1136/bjo.84.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good KL, Maggs DJ, Hollingsworth SR, Scagliotti RH, Nelson RW, 2003. Corneal sensitivity in dogs with diabetes mellitus. Am. J. Veteri. Res 64 (1), 7–11. 10.2460/ajvr.2003.64.7. [DOI] [PubMed] [Google Scholar]
- Güemes Maria, Rahman Sofia A., Hussain Khalid, 2016. What is a normal blood glucose? Arch. Dis. Child 101 (6), 569–574. 10.1136/archdischild-2015-308336. [DOI] [PubMed] [Google Scholar]
- Guthrie RA, Guthrie DW, 2004. Pathophysiology of diabetes mellitus. Crit. Care Nurs. Q 27 (2), 113–125. 10.1097/00002727-200404000-00003. [DOI] [PubMed] [Google Scholar]
- Hager A, Wegscheider K, Wiegand W, 2009. Changes of extracellular matrix of the cornea in diabetes mellitus. Graefe’s Arch. Clin. Exp. Ophthalmol 247 (10), 1369–1374. 10.1007/s00417-009-1088-4. [DOI] [PubMed] [Google Scholar]
- Hasan SA, 2010. The cornea in diabetes mellitus. In: Diabetic Retinopathy, 347–55. Springer, New York. 10.1007/978-0-387-85900-2_12. [DOI] [Google Scholar]
- Hernandez R, Teruel T, Lorenzo M, 2001. Akt mediates insulin induction of glucose uptake and up-regulation of GLUT4 gene expression in Brown adipocytes. FEBS (Fed. Eur. Biochem. Soc.) Lett 494 (3), 225–231. 10.1016/s0014-5793(01)02353-5. [DOI] [PubMed] [Google Scholar]
- Kador PF, Akagi Y, Takahashi Y, Ikebe H, Wyman M, Kinoshita JH, 1990. Prevention of retinal vessel changes associated with diabetic retinopathy in galactose-fed dogs by aldose reductase inhibitors. Arch. Ophthalmol 108 (9), 1301–1309. 10.1001/archopht.1990.01070110117035. [DOI] [PubMed] [Google Scholar]
- Kaji Y, 2005. Prevention of diabetic keratopathy. The Br. J. Ophthalmol 89 (3), 254–255. 10.1136/bjo.2004.055541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalous V, Anzenbacher P, 1979. On the mechanism of the insulin-glucose interactions. Acta Diabetol. Lat 16 (2), 169–174. 10.1007/BF02581096. [DOI] [PubMed] [Google Scholar]
- Kerner W, Brückel J, 2014. Definition, classification and diagnosis of diabetes mellitus. Exp. Clin. Endocrinol. Diabetes 122 (7), 384–386. 10.1055/s-0034-1366278. [DOI] [PubMed] [Google Scholar]
- Klenkler B, Sheardown H, Jones L, 2007. Growth factors in the tear film: role in tissue maintenance, wound healing, and ocular pathology. Ocul. Surf 5 (3), 228–239. 10.1016/S1542-0124(12)70613-4. [DOI] [PubMed] [Google Scholar]
- Klocek MS, Sassani JW, McLaughlin PJ, Zagon IS, 2009. Naltrexone and insulin are independently effective but not additive in accelerating corneal epithelial healing in type I diabetic rats. Exp. Eye Res 89 (5), 686–692. 10.1016/j.exer.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen L, Hansen BF, Jensen P, Pedersen TA, Vestergaard Kirsten, Schäffer L, Blagoev B, Oleksiewicz MB, Kiselyov VV, De Meyts P, 2012. Agonism and antagonism at the insulin receptor. PLoS One 7 (12), e51972. 10.1371/journal.pone.0051972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolterman OG, Gray RS, Griffin J, Burstein P, Insel J, Scarlett JA, Olefsky JM, 1981. Receptor and postreceptor defects contribute to the insulin resistance in noninsulin-dependent diabetes mellitus. J. Clini. Investi 68 (4), 957–969. 10.1172/jci110350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook A, Roth RA, Jiang XJ, Zierath JR, Wallberg-Henriksson H, 1998. Insulin-stimulated Akt kinase activity is reduced in skeletal muscle from NIDDM subjects. Diabetes 47 (8), 1281–1286. 10.2337/diab.47.8.1281. [DOI] [PubMed] [Google Scholar]
- Krumholz DM, Lane J, Sack R, Morris C, 2005. Correlation of glucose levels in the tear film and blood in normals and diabetics. Ocul. Surf 3, S84. [Google Scholar]
- Kuipers DP, Scripture JP, Gunnink SM, Salie MJ, Schotanus MP, Ubels JL, Louters LL, 2013. Differential regulation of GLUT1 activity in human corneal limbal epithelial cells and fibroblasts. Biochimie 95 (2), 258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai AK, Glasgow BJ, Pardridge WM, 1994. GLUT1 glucose transporter expression in the diabetic and nondiabetic human eye. Investi. Ophthal. Visual Sci 35 (6), 2887–2894. [PubMed] [Google Scholar]
- Lambiase A, Rama P, Bonini S, Caprioglio G, Aloe L, 1998. Topical treatment with nerve growth factor for corneal neurotrophic ulcers. N. Engl. J. Med 338 (17), 1174–1180. 10.1056/NEJM199804233381702. [DOI] [PubMed] [Google Scholar]
- Lane JD, Krumholz DM, Sack RA, Morris C, 2006. Tear glucose dynamics in diabetes mellitus. Curr. Eye Res 31 (11), 895–901. 10.1080/02713680600976552. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, McKern NM, Ward CW, 2007. Insulin receptor structure and its implications for the IGF-1 receptor. Curr. Opin. Struct. Biol 17 (6), 699–705. 10.1016/j.sbi.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Leney SE, Tavaré JM, 2009. The molecular basis of insulin-stimulated glucose uptake: signalling, trafficking and potential drug targets. J. Endocrinol 203 (1), 1–18. 10.1677/JOE-09-0037. [DOI] [PubMed] [Google Scholar]
- Li B, Wang JHC, 2011. Fibroblasts and myofibroblasts in wound healing: force generation and measurement. J. Tissue Viability 20 (4), 108–120. 10.1016/j.jtv.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HF, Petroll WM, Moller-Pedersen T, Maurer JK, Cavanagh HD, Jester JV, 1997. Epithelial and corneal thickness measurements by in vivo confocal microscopy through focusing (CMTF). Curr. Eye Res 16 (3), 214–221. [DOI] [PubMed] [Google Scholar]
- Lizcano JM, Alessi DR, 2002. The insulin signalling pathway. Curr. Biol.: CB (Curr. Biol.) 12 (7), R236–R238. 10.1016/s0960-9822(02)00777-7. [DOI] [PubMed] [Google Scholar]
- Lu W, Ebihara N, Miyazaki K, Murakami A, 2006. Reduced expression of laminin-5 in corneal epithelial cells under high glucose condition. Cornea 25 (1), 61–67. 10.1097/01.ico.0000179932.21104.3c. [DOI] [PubMed] [Google Scholar]
- Maldonado B, Furcht L, 1995. Epidermal growth factor stimulates integrin-mediated cell migration of cultured human corneal epithelial cells on fibronectin and arginine-glycine-aspartic acid peptide. Investi. Ophthal. Visual Sci 36 (10), 2120–2126. [PubMed] [Google Scholar]
- Melmed, Polonsky SK, Larsen PR, Kronenberg HM, 2011. Textbook of Endocrinology, twelfth ed. Saunders, Philadelphia, PA. [Google Scholar]
- Meyer LA, Ubels JL, Edelhauser HF, 1988. Corneal endothelial morphology in the rat. Effects of aging, diabetes, and topical aldose reductase inhibitor treatment. Investi. Ophthal. Visual Sci 29 (6), 940–948. [PubMed] [Google Scholar]
- Miniaci MC, Dattolo MG, Irace C, Capuozzo A, Santamaria R, Scotto P, 2015. Glucose deprivation promotes activation of MTOR signaling pathway and protein synthesis in rat skeletal muscle cells. Pflueg. Arch. Eur. J. Physiol 467 (6), 1357–1366. 10.1007/s00424-014-1583-2. [DOI] [PubMed] [Google Scholar]
- Módulo CM, Jorge AG, Dias AC, Braz AM, Bertazolli-Filho R, Jordã0 Alceu A., Marchini Sérgio J., Eduardo Rocha M., 2009. Influence of insulin treatment on the lacrimal gland and ocular surface of diabetic rats. Endocrine 36 (1), 161–168. 10.1007/s12020-009-9208-9. [DOI] [PubMed] [Google Scholar]
- Morita M, Gravel SP, Hulea L, Larsson O, Pollak M, St-Pierre J, Topisirovic I, 2015. MTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle 14 (4), 473–480. 10.4161/15384101.2014.991572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselmann K, Alexandrou B, Kane B, Hassell JR, 2005. Maintenance of the keratocyte phenotype during cell proliferation stimulated by insulin. J. Biol. Chem 280 (38), 32634–32639. 10.1074/jbc.M504724200. [DOI] [PubMed] [Google Scholar]
- Naeser P, 1997. Insulin receptors in human ocular tissues. Immunohistochemical demonstration in normal and diabetic eyes. Ups. J. Med. Sci 102 (1), 35–40. 10.3109/03009739709178930. [DOI] [PubMed] [Google Scholar]
- Nagano T, Nakamura M, Nakata K, Yamaguchi T, Takase K, Okahara A, Ikuse T, Nishida T, 2003. Effects of substance P and IGF-1 in corneal epithelial barrier function and wound healing in a rat model of neurotrophic keratopathy. Investi. Ophthal. Visual Sci 44 (9), 3810. 10.1167/iovs.03-0189. [DOI] [PubMed] [Google Scholar]
- Nelson BA, Robinson KA, Buse MG, 2002. Defective Akt activation is associated with glucose- but not glucosamine-induced insulin resistance.” American journal of physiology. Endocrinol. Metabol 282 (3), E497–E506. 10.1152/ajpendo.00438.2001. [DOI] [PubMed] [Google Scholar]
- Nishida T, Nakamura M, Mishima H, Otori T, 1992. Interleukin 6 promotes epithelial migration by a fibronectin-dependent mechanism. J. Cell. Physiol 153 (1), 1–5. [DOI] [PubMed] [Google Scholar]
- Nomura M, Kubota MA, Sekiya M, Hoshiyama S, Imano E, Matushima Y, Ishimoto I, Kawamori R, Kamada T, 1990. Insulin absorption from conjunctiva studied in normal and diabetic dogs. J. Pharm. Pharmacol 42 (4), 292–294. [DOI] [PubMed] [Google Scholar]
- O’Harte FP, Højrup P, Barnett CR, Flatt PR, 1996. Identification of the site of glycation of human insulin. Peptides 17 (8), 1323–1330. 10.1016/s0196-9781(96)00231-8. [DOI] [PubMed] [Google Scholar]
- Okabayashi Y, Maddux BA, McDonald AR, Logsdon CD, Williams JA, Goldfine ID, 1989. Mechanisms of insulin-induced insulin-receptor downregulation. Decrease of receptor biosynthesis and MRNA levels. Diabetes 38 (2), 182–187. 10.2337/diab.38.2.182. [DOI] [PubMed] [Google Scholar]
- Pandini G, Frasca F, Mineo R, Sciacca L, Vigneri R, Belfiore A, 2002. Insulin/ Insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J. Biol. Chem 277 (42), 39684–39695. 10.1074/jbc.M202766200. [DOI] [PubMed] [Google Scholar]
- Pillion DJ, Bartlett JD, Meezan E, Yang M, Crain RJ, Grizzle WE, 1991. Systemic absorption of insulin delivered topically to the rat eye. Investi. Ophthal. Visual Sci 32 (12), 3021–3027. [PubMed] [Google Scholar]
- Pinnamaneni N, Funderburgh JL, 2012. Concise review: stem cells in the corneal stroma. Stem Cell. 30 (6), 1059–1063. 10.1002/stem.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechler MM, Nissley SP, 1985. The nature and regulation of the receptors for insulin-like growth factors. Annu. Rev. Physiol 47, 425–442. 10.1146/annurev.ph.47.030185.002233. [DOI] [PubMed] [Google Scholar]
- Robertson DM, Li L, Fisher S, Pearce VP, Shay JW, Wright WE, Cavanagh HD, Jester JV, 2005. Characterization of growth and differentiation in a telomerase-immortalized human corneal epithelial cell line. Investi. Ophthal. Visual Sci 46 (2), 470–478. 10.1167/iovs.04-0528. [DOI] [PubMed] [Google Scholar]
- Robertson DM, Zhu M, Wu YC, 2012. Cellular distribution of the IGF-1R in corneal epithelial cells. Exp. Eye Res 94 (1), 179–186. 10.1016/j.exer.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EM, Cunha DA, Carneiro EM, Boschero AC, Saad MJA, Velloso LA, 2002. Identification of insulin in the tear film and insulin receptor and IGF-1 receptor on the human ocular surface. Investi. Ophthal. Visual Sci 43 (4), 963–967. [PubMed] [Google Scholar]
- Root-Bernstein R, Vonck J, 2009. Glucose binds to the insulin receptor affecting the mutual affinity of insulin and its receptor. Cell. Mol. Life Sci.: CMLS 66 (16), 2721–2732. 10.1007/s00018-009-0065-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saika S, Okada Y, Miyamoto T, Yamanaka O, Ohnishi Y, Ooshima A, Liu CY, Weng D, Kao WWY, 2004. Role of P38 MAP kinase in regulation of cell migration and proliferation in healing corneal epithelium. Investi. Ophthal. Visual Sci 45 (1), 100. 10.1167/iovs.03-0700. [DOI] [PubMed] [Google Scholar]
- Saltiel AR, Kahn CR, 2001. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414 (6865), 799–806. 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- Sánchez-Thorin JC, 1998. The cornea in diabetes mellitus. Int. Ophthalmol. Clin 38 (2), 19–36. [PubMed] [Google Scholar]
- Saragas S, Arffa R, Rabin B, Kronish J, Miller D, Mayman C, 1985. Reversal of wound strength retardation by addition of insulin to corticosteroid therapy. Ann. Ophthalmol 17 (7), 428–430. [PubMed] [Google Scholar]
- Schmidt AM, 2018. Highlighting diabetes mellitus. Arterioscler. Thromb. Vasc. Biol 38 (1), e1–8. 10.1161/atvbaha.117.310221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz RO, Van Horn DL, Peters MA, Klewin KM, Schutten WH, 1981. Diabetic keratopathy. Trans. Am. Ophthalmol. Soc 79, 180–199. [PMC free article] [PubMed] [Google Scholar]
- Schultze SM, Hemmings BA, Niessen M, Tschopp O, 2012. PI3K/AKT, MAPK and AMPK signalling: protein kinases in glucose homeostasis. January Expet Rev. Mol. Med 14, e1. 10.1017/S1462399411002109. [DOI] [PubMed] [Google Scholar]
- Sen DK, Sarin GS, 1980. Tear glucose levels in normal people and in diabetic patients. The Br. J. Ophthalmol 64 (9), 693–695. 10.1136/bjo.64.9.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah VS, Harvey SA, Funderburgh ML, Funderburgh JL, 2012. Identifying markers of keratocyte lineage determination. Investi. Ophthal. Visual Sci 53 (14), 4742. [Google Scholar]
- Siddle K, 2011. Signalling by insulin and IGF receptors: supporting acts and new players. J. Mol. Endocrinol 47 (1), R1–R10. 10.1530/JME-11-0022. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Kaminski AE, Zieske JD, 1996. Glucose transporter 1 expression is enhanced during corneal epithelial wound repair. Exp. Eye Res 63 (6), 649–659. 10.1006/exer.1996.0159. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Ohara K, Ohmura T, Takahashi R, Zieske JD, 2000. Glucose transporter 1 expression in corneal wound repair under high serum glucose level. Jpn. J. Ophthalmol 44 (5). [DOI] [PubMed] [Google Scholar]
- Tandon A, Tovey JCK, Sharma A, Gupta R, Mohan RR, 2010. Role of transforming growth factor beta in corneal function, biology and pathology. Curr. Mol. Med 10 (6), 565–578. 10.2174/1566524011009060565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoft RA, Friend J, Dohlman CH, 1971. Corneal glucose concentration. Flux in the presence and absence of epithelium. Arch. Ophthalmol 85 (4), 467–472. 10.1001/archopht.1971.00990050469013. [DOI] [PubMed] [Google Scholar]
- Titone R, Zhu M, Robertson DM, 2018. Insulin mediates de Novo nuclear accumulation of the IGF-1/insulin hybrid receptor in corneal epithelial cells. Sci. Rep 8 (1) 10.1038/s41598-018-21031-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasek James J., Gabbiani Giulio, Hinz Boris, Chaponnier Christine, Robert A. Brown, 2002. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol 3 (5), 349–363. 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- Ullrich A, Gray A, Tam AW, Yang-Feng T, Tsubokawa M, Collins C, Henzel W, Le Bon T, Kathuria S, Chen E, 1986. Insulin-like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 5 (10), 2503–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utsunomiya T, Ishibazawa A, Nagaoka T, Hanada K, Yokota H, Ishii N, Yoshida A, 2016. Transforming growth factor-β signaling cascade induced by mechanical stimulation of fluid shear stress in cultured corneal epithelial cells. Investi. Ophthal. Visual Sci 57 (14), 6382. 10.1167/iovs.16-20638. [DOI] [PubMed] [Google Scholar]
- Varma S, Lal BK, Zheng R, Breslin JW, Saito S, Pappas PJ, Hobson RW, Durán WN, 2005. Hyperglycemia alters PI3k and Akt signaling and leads to endothelial cell proliferative dysfunction. Am. J. Physiol. Heart Circ. Physiol 289 (4), H1744–H1751. 10.1152/ajpheart.01088.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AL, Weinlander E, Metcalf BM, Barney NP, Gamm DM, Nehls SM, Struck MC, 2017. Use of topical insulin to treat refractory neurotrophic corneal ulcers. Cornea 36 (11), 1426–1428. 10.1097/ICO.0000000000001297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YC, Zhang Y, Yeh LK, Liu CY, 2019. TGF beta signaling in stroma is essential for corneal development. Investi. Ophthal. Visual Sci 60 (9), 4155. [Google Scholar]
- Williams DL, Pierce V, Mellor P, Heath MF, 2007. Reduced tear production in three canine endocrinopathies. J. Small Anim. Pract 48 (5), 252–256. 10.1111/j.1748-5827.2007.00349.x. [DOI] [PubMed] [Google Scholar]
- Wilson S, Walker J, Chwang E, He Y, 1993. Hepatocyte growth factor, keratinocyte growth factor, their receptors, fibroblast growth factor receptor-2, and the cells of the cornea. Investi. Ophthal. Visual Sci 34 (8), 2544–2561. [PubMed] [Google Scholar]
- Wilson SE, 2012. Corneal myofibroblast biology and pathobiology: generation, persistence, and transparency. Exp. Eye Res 99 (June), 78–88. 10.1016/j.exer.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YC, Zhu M, Robertson DM, 2012. Novel nuclear localization and potential function of insulin-like growth factor-1 receptor/insulin receptor hybrid in corneal epithelial cells. PLoS One 7 (8), e42483. 10.1371/journal.pone.0042483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu KP, Li Y, Ljubimov AV, Yu FS, 2009. High glucose suppresses epidermal growth factor receptor/phosphatidylinositol 3-kinase/akt signaling pathway and attenuates corneal epithelial wound healing. Diabetes 58 (5), 1077–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Zhang Y, Zhang Z, Dan J, Zhou Q, Wang X, Li W, Zhou L, Yang L, Xie L, 2020. Insulin promotes corneal nerve repair and wound healing in type 1 diabetic mice by enhancing wnt/β-catenin signaling. Am. J. Pathol 190 (11), 2237–2250. 10.1016/j.ajpath.2020.08.006. [DOI] [PubMed] [Google Scholar]
- Yee R, Matsuda M, Kern T, Engerman R, Edelhauser H, 1985. Corneal endothelial changes in diabetic dogs. Curr. Eye Res 4 (7), 759–766. [DOI] [PubMed] [Google Scholar]
- Yu ZW, Eriksson JW, 2000. The upregulating effect of insulin and vanadate on cell surface insulin receptors in rat adipocytes is modulated by glucose and energy availability. Horm. Metab. Res 32 (8), 310–315. 10.1055/s-2007-978642. [DOI] [PubMed] [Google Scholar]
- Zagon I, Klocek M, Sassani J, McLaughlin PJ, 2007. Use of topical insulin to normalize corneal epithelial healing in diabetes mellitus. Arch. Ophthalmol 125 (8), 1082–1088. [DOI] [PubMed] [Google Scholar]
- Zagon I, Sassani J, McLaughlin PJ, 2006. Insulin treatment ameliorates impaired corneal reepithelialization in diabetic rats. Diabetes 55 (4), 1141–1147. [DOI] [PubMed] [Google Scholar]
- Zdychovα J, Komers R, 2005. Emerging role of Akt kinase/protein kinase B signaling in pathophysiology of diabetes and its complications. Physiol. Res 54 (1), 1–16. [DOI] [PubMed] [Google Scholar]
- Zheng X, De Paiva CS, Rao K, Li DQ, Farley WJ, Stern M, Pflugfelder SC, 2010. Evaluation of the transforming growth factor-β activity in normal and dry eye human tears by CCL-185 cell bioassay. Cornea 29 (9), 1048–1054. 10.1097/ico.0b013e3181cf98ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoete V, Meuwly M, Karplus M, 2004. Investigation of glucose binding sites on insulin. Proteins 55 (3), 568–581. 10.1002/prot.20071. [DOI] [PubMed] [Google Scholar]
- Zurawski CA, McCarey BE, Schmidt FH, 1989. Glucose consumption in cultured corneal cells. Curr. Eye Res 8 (4), 349–355. 10.3109/02713688908996382. [DOI] [PubMed] [Google Scholar]