

Abstract
Adhesion to the surface of moist, dynamic, biological tissues is important in many fields. Currently, tissue adhesives commonly used in clinical practice remain far from ideal, exhibiting either poor tissue compatibility or weak tissue adhesion. Here, we designed biocompatible hydrogels comprising polysaccharides with polyacrylamide and exhibiting promising cytocompatibility, antibacterial activity, and excellent tissue adhesion. Alginate/chitosan-based hydrogels covalently cross-linked to the tissue surface in order to achieve admirable tissue adhesion. Additionally, the mechanical properties of the hydrogels were significantly enhanced with the addition of polyacrylamide, which synergistically promoted their enhanced adhesion. Importantly, the hydrogels exhibited good biocompatibility and reasonable antibacterial activity that promoted wound recovery during use as wound dressings. These results suggested the efficacy of the developed tissue adhesives for applications in biomedical fields, as well as broadening potential hydrogel use in tissue engineering.
By combination of alginate/polyacrylamide/chitosan, tough antibacterial hydrogels are designed for applications as tissues adhesives to promote wound healing.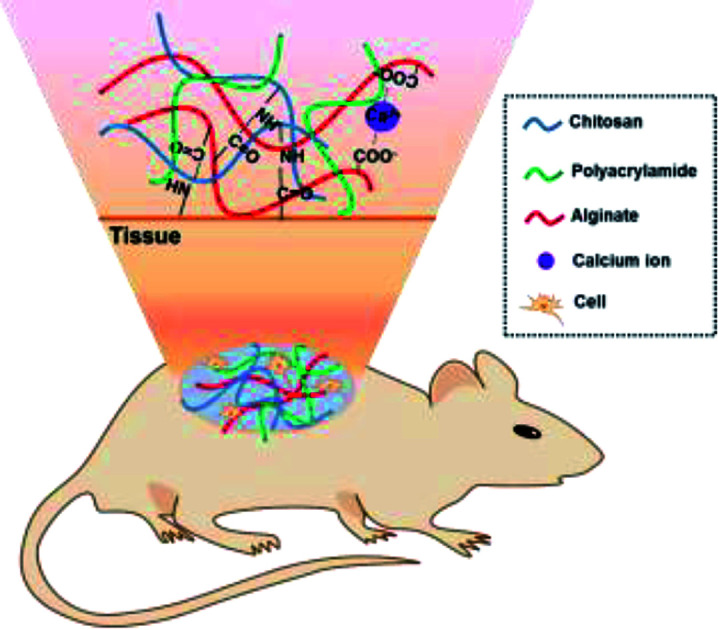
Introduction
Adhesion to wet and dynamic surfaces (including biological tissue) is important in many areas,1 and adhesives that can strongly bind to biological tissue have a wide range of potential applications, including tissue repair,2,3 drug delivery,4,5 and in biomedical devices.6,7 Among these, the repair of tissue, including skin,8 cartilage,9 and internal organs,10 represents an extremely common and urgent problem in the clinic. Although sutures and staples have been widely used to reconnect incisions in order to recover tissue structure and function, piercing tissues to place sutures and staples can further damage the surrounding wound area and increase the risk of infection. Therefore, tissue-adhesive materials have attracted attention as alternatives for sealing and reconnecting tissues or for incorporating implant devices into tissues based on their versatility and ease of application.11 The commonly used super glue cyanoacrylate is the strongest tissue adhesive currently in use;12 however, it is cytotoxic and exhibits poor histocompatibility.13 Additionally, mussel-inspired adhesives14 adhere weakly to tissues, as their adhesion mainly relies on relatively weak physical interactions. Ideal tissue adhesives must be designed to have excellent cell affinity in order to allow adherence of cells and tissues, thereby promoting tissue regeneration after implantation. Moreover, tissue adhesives need to exhibit good mechanical and tissue-adhesion properties in order to facilitate fixation with/to surrounding tissue during surgery.15
Hydrogel is a highly cross-linked, water-swellable, hydrophilic polymer with a structure similar to that of natural extracellular matrix and potentially capable of effectively solving the biocompatibility problem.16,17 Hydrogels have been extensively studied and demonstrated great potential as biocompatible materials in many therapeutic applications.18 These materials can be divided into natural and synthetic materials.19 Naturally occurring polymers, such as chitosan,20 alginate,21 hyaluronic acid,22 collagen,23 and gelatin,24 are biodegradable and typically functionalized in advance using integrins to promote cell adhesion.25 However, due to their potential immunogenicity and differences between batches, use of these materials is limited.26 By contrast, synthetic polymers, such as poly(ethylene glycol), polyacrylamide, poly(vinyl alcohol), and poly(methyl methacrylate), generally exhibit better mechanical properties and lower immunogenicity relative to natural materials but lack biological function and require biochemical processing prior to in vivo application.27 Therefore, hybrid hydrogels comprising the advantages of both natural and synthetic polymers offer the greatest opportunity for repairing tissue defects and accelerating wound healing.28–30
In this study, we designed a series of hydrogel-based wound dressings comprising sodium alginate, chitosan, and polyacrylamide and possessing good mechanical properties capable of supporting the dynamic motion of tissue, strong tissue adhesion to wet tissue or scaffolds for dressing tissue defects, and promising cytocompatibility and antibacterial activity to promote wound healing. We suggest that these adhesives can be widely used in many biomedical applications, including as tissue adhesives and wound dressings and for tissue repair.
Experimental
Synthesis of hydrogels
To create alginate/chitosan (SC) hydrogels, 2% sodium alginate (Aladdin, Shanghai, China) and 2% chitosan (Nantong Xingcheng Biological Industrial Co. Ltd., Nantong, China) (w/v) were first dissolved in MES (Sigma-Aldrich, St. Louis, MO, USA) buffer (pH 5.0) and stirred overnight until clean solutions were obtained. Precursor solution (10 mL) were mixed with 0.12 g 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), 0.12 g N-hydroxysuccinimide (NHS) and allowed to gel inside of a closed mold at room temperature overnight. To create alginate/chitosan/polyacrylamide (SCM) hydrogel (SCM4, SCM8, and SCM12), 2% sodium alginate and 2% chitosan were dissolved with 4%, 8%, or 12% of acrylamide (w/v), respectively. Additional 2% (w/v) N,N′-methylenebis(arylamide) (MBAA; Sigma-Aldrich) (12 μL), N,N,N′,N′-tetramethylethylenediamine (TEMED; Sigma-Aldrich) (2.7 μL), 0.27 M ammonium persulphate (APS; Sigma-Aldrich) (75 μL), and 0.75 M CaSO4 (Sigma-Aldrich) (64 μL) were syringe mixed with precursor solution containing sodium alginate/chitosan/4% acrylamide, along with EDC and NHS, to give SCM4 hydrogel for instance. Sodium alginate (SA) hydrogels were prepared by mixing 2% sodium alginate solution (w/v) with 0.75 M CaSO4.
Characterization of the physical and chemical performance of the hydrogels
Fourier transform infrared spectroscopy (FTIR)
We performed FTIR analysis of the hydrogels using a Nicolet iS50 FTIR spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) within the range 4500 cm−1 to 400 cm−1. Samples were tested in the form of KBr disks at room temperature.
Scanning electron microscopy (SEM)
Hydrogel morphology was characterized by SEM using an S-3400N II (Hitachi, Tokyo, Japan) at an accelerating voltage of 30 kV. Prior to analysis, the hydrogels were freeze dried, and the surfaces were coated with a thin layer of gold. Pore diameter was calculated using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Evaluation of hydrogel porosity
The different composite hydrogels were lyophilized, and immersed in anhydrous ethanol with a known volume (V1). Then, the mixture were evacuated at 0.08 kPa and the volume of ethanol-perfused hydrogels and ethanol was recorded as V2, and after removal of the hydrogels, the final volume was recorded as V3. Porosity was calculated, as follows: P = (V1 − V3)/(V2 − V3) × 100%.
Mechanical evaluation
Tensile/compressive-strength measurements were performed using an electronic universal testing machine (UTM; TFW-58; Shanghai Tuofeng Instrument Technology Co. Ltd., Shanghai, China). Hydrogel samples were made into shape of solid cylinder with 6 mm diameter and 5 cm length and tested on a QMESYS system with tensile jigs at a stretch velocity of 5 mm min−1. For compression testing, samples with 22 mm diameter and 5 mm depth were tested at a displacement rate of 5 mm min−1. All tests of physical properties were performed in a room under strictly controlled conditions (80% humidity and 27 °C) to prevent drying.
Adhesive strength to tissue and chitosan nerve grafting
Lap-shear tensile-stress measurements of the hydrogels were performed using a UTM (TFW-58; Shanghai Tuofeng Instrument Technology Co. Ltd.). Porcine skin was cut into rectangular sections (5.0 × 2.0 cm), and excess fat was removed before transfer to PBS for immediate testing while the tissues remained moist. A pre-gel solution was applied between two pieces of porcine skin, and the samples were allowed to cure for 10 min at 37 °C under humid conditions. We used a tensile tester with a 10 kN load cell, and samples were fixed between the two film clamps at a tensile rate of 0.5 mm min−1. For chitosan nerve grafting, a similar process was performed to link the chitosan nerve grafts (1.0 mm inner diameter and 2 mm outer diameter) with a rat sciatic nerve. Similarly, the samples were maintained in a humid environment before use, with the overlapping area at π × 1 mm × 50 mm. The samples were incubated under the same conditions as the skin and tested using a speed of 0.5 mm min−1.
In vitro/in vivo biological evaluation
Cytotoxicity
The cytotoxicity of the hydrogels was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Beyotime Biotechnology, Beijing, China). Mouse fibroblasts (L929; Cell Bank of the Chinese Academy of Sciences, Beijing, China) were seeded in a 96-well plate at 8000 cells per well and incubated for 4 h to 6 h at 37 °C under 5% CO2. Complete medium comprising Dulbecco's modified Eagle medium (Gibco, Gaithersburg, MD, USA) containing 10% fetal bovine serum (Gibco) was removed, and 100 μL hydrogel extract was added to each well. After incubation for 1, 2, or 3 days with the extraction medium refreshed daily, the medium was replaced with 100 μL of fresh complete medium and assessed by MTT. For each hydrogel formulation, five independent cultures were prepared, and samples with relative cell viability <70% were considered cytotoxic.
Cell attachment test
L929 cells (1 × 105 cells per hydrogel) were seeded onto hydrogels coated onto coverslips and stained with rhodamine phalloidin reagent (Abcam, Cambridge, UK) and Hoechst 33342 (Thermo Fisher Scientific) at 24 h after seeding. Before staining, cells were fixed in 4% (v/v) paraformaldehyde for 20 min, permeabilized in 0.1% (w/v) Triton X-100 (Sigma-Aldrich) for 5 min, and then blocked with 1% bovine serum albumin (Sigma-Aldrich) for 60 min. Actin filaments were stained with rhodamine phalloidin reagent (1:1000) for 60 min, and nuclei were stained with Hoechst 33342 (1:20) for 5 min. The images were examined using a fluorescence microscope (ZEISS-AX10; Carl Zeiss AG, Oberkochen, Germany).
Cell encapsulation
L929 cells were wrapped in hydrogel at a density of 1 × 106 cells per mL, and a confocal laser scanning microscope (Leica, Wetzlar, Germany) was used to observe live/dead staining (LIVE/DEAD viability/cytotoxicity kit; Molecular Probes, Eugene, OR, USA) of the cells on days 1, 2, and 3.
Antibacterial assay for hydrogels
The hydrogels were soaked and washed with sterilized ddH2O for 24 h to remove bacteria. Bacterial suspension (10 μL) in sterilized PBS (106 CFU mL−1) was spread onto each hydrogel surface in the 48-well culture plate and incubated for 4 h at 37 °C. Following incubation, all surviving bacteria in each well was resuspended with 1 mL sterilized PBS and incubated for 16 h at 37 °C. The CFU was determined in the Petri dish. Tests were repeated three times for each group.
In vivo degradation and histologic analysis of hydrogels
In vivo degradation and histology of the hydrogels were evaluated subcutaneously according to international (ISO/10993) and national (GB/T1688) standards. Adult female New Zealand white rabbits (2.5–3.0 kg) were used for in vivo studies. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Nantong University and approved by the Animal Ethics Committee of Nantong University. Hydrogel samples were cut into 10 × 3 mm cylinders and implanted under the mediodorsal skin of rabbits. At designated time intervals (weeks 1, 2, 4, 8, and 12), the rabbits were sacrificed and the samples were processed for further histologic analyses and degradation studies.
Immunofluorescence staining was performed to evaluate the inflammatory reactions by examining expression of CD11b and CD68. Briefly, the sections were incubated with the anti-CD11b rabbit monoclonal antibody (primary antibodies, 1:400, Abcam, 21865-1-AP, Proteintech, USA) and anti-CD68 mouse monoclonal antibody (primary antibodies, 1:400, Abcam, 21865-1-AP, Proteintech, USA) at a volumetric ratio of 1 : 1 overnight at 4 °C. The samples were then treated with Cy3-conjugated affinipure goat anti-rabbit IgG (H + L) (secondary antibodies, 1:400, Abcam, 21865-1-AP, Proteintech, USA) and AlexaFluor 488-conjugated affinipure goat anti-mouse IgG (H + L) (secondary antibodies, 1:400, Abcam, 21865-1-AP, Proteintech, USA) secondary antibodies. Hoechst 33342 (1:20; Thermo Fisher Scientific, USA) was used to stain nuclei. Images were examined using a fluorescence microscope (DM2500; Leica, Germany).
In vivo wound healing
Construction of the injury model
Male Sprague-Dawley rats (∼200 g) were anesthetized and full-thickness round wound (∼2 cm diameter) was created on the back of each rat. Then, the wounds were treated with in situ formed SCM4 hydrogel, SA based on commercial wound dressings (positive control) and saline (negative control). Following treatment, the rats were individually housed in cages for 7 days. The wounds were examined and photographed on days 0, 1, 4, and 7.
Histologic analysis
On days 1, 4, and 7, the rats were perfused with a 4% paraformaldehyde solution, and samples of the wound site were collected. The tissue gradient was dehydrated and subjected to frozen sectioning, followed by hematoxylin and eosin (H&E) staining.
Data analysis
All experimental results were analyzed using Origin 8.0 (OriginLab, Northampton, MA, USA), Image Tool (https://en.bio-soft.net/) software, and Graphpad Prism 5.0 (GraphPad Software, San Diego, CA, USA). Results are expressed as the mean ± standard deviation (SD), and differences between two groups were analyzed by a two-tailed, unpaired Student's t test. A p < 0.05 was considered significant.
Results and discussion
Fabrication of hydrogels and characterization
We designed the hydrogel dressings by optimizing the composition of chitosan, alginate, and polyacrylamide and synthesized the SCM hydrogels by mixing chitosan, alginate and acrylamide solution with cross-linking agents (Fig. 1a). Briefly, divalent calcium ions form bonds with side chain alginic acids and acrylamide cross-links in the presence of MBAA, TEMED, and APS. The tissue adhesion of the hydrogels is attributed to the formation of amide bonds, facilitated by EDC and NHS, between tissue and chitosan/alginate within the matrix. Fig. 1b shows evidences indicating hydrogel uniformity after curing, revealing its fit-to-shape properties.
Fig. 1. Characterization of hydrogels. (a) Schematics illustration of SCM hydrogels, which consist of chitosan, alginate and polyacrylamide, forming strong adhesion with wounded skin. (b) Visual image of hydrogels with various 3D structures. (c) SEM images of hydrogels. (d) FTIR spectra of hydrogels. (e) Compressive and (f) tensile strengths of hydrogels. (g) Photographs of the original, bending, compressed, stretched, twisted, and knotted shapes of hydrogels. Data represent the mean ± SD of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001 vs. the SA hydrogels.

To determine hydrogel composition, we characterized their chemical structures by FTIR analysis. Fig. 1d shows 1040 cm−1 and 1420 cm−1 as characteristic peaks of –OH and –COOH (carboxylate ions) for sodium alginate,31 respectively. Additionally, we observed a peak at ∼3201 cm−1 characteristic of polyacrylamide32 hydride-NH2 and absence of a peak at 890 cm−1 for the acrylamide monomer CH2 in the spectrum of the composite hydrogel, confirming polymerization of the acrylamide monomer. Moreover, we observed a peak at 2881 cm−1 representing the contraction vibration-absorption peak of chitosan–CH2 and a peak ranging from 1700 cm−1 to 1600 cm−1 representing amide I (C O-shrinkage vibration) and II (N–H-bending vibration) peaks for chitosan, polyacrylamide, and amide formed by condensation of amino and carboxyl groups. These results indicated successful polymerization of each hydrogel containing its corresponding components.
To monitor hydrogel gelation, we performed dynamic time-sweep rheological experiments. Within 100 s, the G′ of the hydrogels all exceeded the G′′, resulting in rapid formation of the hydrogel (Fig. S1†), which meets the time requirements for the process of mixing and injection for clinical administration.33 The G′ of SCM hydrogels significantly increased along with increasing macromolecule content, including chitosan and polyacrylamide, signifying a structurally robust network that maintains its three-dimensional (3D) shape.
We then used SEM to evaluate the microscopic structures of the hydrogels. Fig. 1c shows that all hydrogels displayed porous structures, which are critical for hydrogel use as biomaterials, especially for sealants, because it facilitates absorption of exudates from wounds and increases the concentration of the red blood cells and plates to enhance clotting activity.34 However, as tissue fluids can easily penetrated into and swell the hydrogel, excessive porosity of hydrogel can cause the excessive volume expansion of the hydrogel to squeeze surrounding normal tissues or organs, which induces local injure. We found that pore number increased and pore size decreased along with increases in the solid concentration (chitosan and polyacrylamide) of hydrogels (Fig. S2†), due to higher polymer concentrations result in an increased cross-link density with denser internal structure. Importantly, the pore diameter of the SC/SCM4/SCM8 hydrogels ranged from 100 μm to 200 μm, which is suitable for cell attachment, migration, proliferation, and extracellular matrix production.35 The porosity and swelling properties of a hydrogel play major roles in tissue regeneration.36 As shown in Fig. S3 and S4,† increases in macromolecular content reduced hydrogel porosity and swelling rate. Because surface wettability significantly influences the biocompatibility of biomaterials exposed to tissue, we determined the contact angle (θ) of the hydrogels, finding that all were <90° and decreased slightly along with increased acrylamide content, indicating that the hydrogels exhibited good hydrophilicity (Fig. S5†).37
Mechanical performance of the hydrogels
Application of adhesives to tissue requires their ability to adapt to dynamic movement; therefore, designed hydrogels require specific mechanical properties, including stretchability, compression, and recovery, as well as mechanical stability. Fig. 1g demonstrates flexible mechanical properties of hydrogels in various shapes.
We performed compression and tensile tests to assess the mechanical properties of the hydrogels (Fig. 1e and f). Statistics for compressive strength showed that addition of polyacrylamide significantly increased the compressive strength of the hydrogel (SCM12 was >300-fold higher than that of SA). Additionally, tensile statistical showed increased polyacrylamide content resulted in elevated tensile strength, with those for SA, SC, SCM4, SCM8, and SCM12 gels determined at ∼17.3 kPa, ∼17.6 kPa, ∼26.0 kPa, ∼30.0 kPa, and 79.7 kPa, respectively. Notably, the tensile strengths of the SCM hydrogels were all higher than those of hydrogels constructed based on hydrogen bonds (1.66 ± 0.47 kPa),38 and those of the SCM8 and SCM12 hydrogels exceeded those of hybrid hydrogels containing both covalent and ionic cross-linking (∼29 kPa).30,39 Ideally, the tensile strength of hydrogel dressings should match the strength of the underlying and neighboring tissues in order to assure their integrity and secure wound safety until healed.40,41 These results illustrated that the SCM hydrogels exhibited desirable mechanical properties within the range of Young's modulus of human tissues (1–100 kPa).40
Tissue adhesion of the hydrogels
Strong tissue adhesion is essential for in situ hydrogels during tissue bonding in order to resist mechanical forces during dynamic movement.42 We then investigated hydrogel adhesion to biological tissues. As shown in Fig. 2a, the hydrogels possesses strong tissue adhesion to tissues (including skin, liver and nerve) or chitosan scaffolds. Fig. S6† and 2b shows sustained hydrogel binding to a piece of wet pigskin, even after bending, distortion, water soaking, and stretching, suggesting considerable potential for in vivo applications.
Fig. 2. Evaluation of tissue adhesion of hydrogels. (a) Photographs of hydrogel adherence to liver, nerve or chitosan scaffolds. (b) Photographs of hydrogel adherence to porcine skin extended immersion underwater. (c) Photographs of hydrogels for nerve grafting. (d) Bond strengths of hydrogels bound to porcine skin. (e) Bond strengths of hydrogels used for nerve grafting. Data represent the mean ± SD of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001 vs. the SA hydrogel.

We evaluated the adhesive strength of the hydrogels using a UTM and according to binding to porcine skin (Fig. 2d) and nerve adhesion using a chitosan conduit (Fig. 2c and e). The statistical results showed that the adhesive strength of the SC hydrogel was twice as strong as the SA hydrogel, suggesting that addition of chitosan enhanced the adhesive strength of the hydrogels due to the formation of covalent bonds between hydrogels and tissue or chitosan conduits. Additionally, we found that adhesive strength was influenced by hydrogel breakage during the test in addition to detachment of hydrogels from the tissue or chitosan conduits. Therefore, increased polyacrylamide content improved the tensile strength of the hydrogels, leading to enhanced adhesive strength. Specifically, the adhesive strength of the SCM12 hydrogel to porcine skin reached 20 kPa and nerve grafting with chitosan was 56 kPa, which was 5- and 6-fold higher than that of the SA hydrogel, respectively. These results demonstrated that the SCM hydrogels displayed adequate adhesion to tissue or chitosan-based scaffolds as hydrogel dressings.
In vitro cytotoxicity and antibacterial evaluation of hydrogels
The ability of biomaterials to allow cells to survive and grow is fundamental to tissue repair and regeneration.43 To evaluate the biocompatibility of hydrogels with L929 fibroblasts, we performed MTT assays to evaluate cell viability and proliferation according to fluorescence staining. The results showed that none of the hydrogels were cytotoxic (cell viability: >95%), and that the cells exhibited normal proliferation on all hydrogels to levels comparable with controls on blank plates (Fig. 3a and S7†). Fig. 3b shows the diffusion pattern of L929 cells seeded on different hydrogel substrates. Cells on all the hydrogels showed uniform spreading and displayed a spindle-like morphology after incubation for 1 day. Based on its pore structure, suitable swelling ratio and advantageous mechanical properties, we chose the SCM4 hydrogel for subsequent experiments involving embedding of L929 fibroblasts and evaluation (Fig. 3c). The experimental results showed adequate cell survival, growth, and proliferation in both SCM4 and control hydrogels over 3 days, indicating that the SCM4 hydrogels held good structural integrity and effectively allowed cell embedding and supported normal cell growth, which are necessary for use as a 3D culture matrix. These above indicated that the SCM hydrogels displayed good cytocompatibility and represent promising candidates for use as wound dressings.
Fig. 3. Evaluation of in vitro cytotoxicity and antibacterial activity. (a) Viability of L929 fibroblasts cultured on hydrogels for 3 days according to MTT assay. (b) F-actin and nuclear staining of L929 cells seeded on hydrogels after 24 h. Scale bar: 20 μm. (c) LIVE/DEAD staining of L929 cells after encapsulation in hydrogels for 1, 2, and 3 days. Scale bar: 200 μm. (d) Evaluation of CFUs obtained from bacterial suspensions of E. coli- and S. aureus-encapsulated hydrogels. (e) Percentage decreases in E. coli- and S. aureus-encapsulated bacteria. Data represent the mean ± SD of three independent experiments. ***p < 0.001 vs. SA.

To protect wounds from external bacterial infection, a biomaterial should also harbor antimicrobial properties.44 Therefore, we evaluated the surface antibacterial activities of all hydrogels in the presence of Escherichia coli (Gram-negative bacterium) and Staphylococcus aureus (Gram-positive bacterium). As shown in Fig. 3d and e, the SA hydrogels showed weak antibacterial effects (20–25%) on both species, whereas hydrogel groups containing chitosan (SC, SCM4, SCM8, and SCM12) killed ∼80% of E. coli after incubation, with bacteriostatic effects significantly better than SA hydrogels (p < 0.001). Additionally, hydrogels containing chitosan exhibited a kill ratio of >90% for S. aureus, demonstrating excellent inherent antibacterial properties. These results suggested that addition of chitosan into hydrogels significantly enhance their antibacterial effects toward E. coli and S. aureus, possibly because the positive-charged amino groups of chitosan damage the bacterial wall via electrostatic interaction with the cytoderm, resulting in release of intracellular fluids.45 Moreover, it is possible that the complicated cytoderm of E. coli results in slightly lower antibacterial activity on the part of chitosan-containing hydrogels.46 These results indicated that the chitosan-containing hydrogels demonstrated good antibacterial activity and would be potentially efficacious for in vivo applications.
In vivo degradation and biocompatibility
To efficiently regenerate injured tissue, the tissue adhesive needs to effectively simulate the native characteristics of the particular tissue.9 To evaluate the in vivo biocompatibility and biodegradability of the SCM hydrogels, SCM4 was implanted under the mediodorsal skin of rabbits, with the non-immunogenic SA hydrogel used as a control.21 As shown in Fig. 4a, hydrogel volume gradually reduced along with prolonged implantation time, and we observed no adverse reactions, such as redness and pustule formation, around the implant sites. Previously, we have demonstrated that chitosan scaffolds can be degraded by the action of enzymes in vivo (e.g. lysozyme), and the degradation rate can be accelerated using chitosan with lower molecular weight.47 Ionically crosslinked alginate hydrogels can undergo slow dissolution through ion exchange of calcium and cause loss of mass.48 Herein, we used chitosan with low molecular weights (∼90 kDa) to give the hybrid hydrogels, which leads faster biodegradation of hydrogels in vivo. We found that the hydrogel was degraded by nearly 50% after 12 weeks, due to the degradation of chitosan and dissociation of individual chains within hydrogels, indicating positive biodegradability of the SCM4 hydrogel and suggesting its suitability as a tissue dressing, given that slow degradation of biomaterials can promote cellular ingrowth and implant replacement with autologous neo-tissue (Fig. 4b).49 Additionally, we evaluated in vivo biocompatibility by fluorescence immunohistochemical staining using macrophages (CD68) and neutrophils (CD11b) to characterize local immune responses. Fig. 4c shows that early acute inflammation was present at only 1- to 2-weeks post-implantation of both the SCM4 and SA hydrogels, respectively; however, no further inflammatory cells were observed after 4 weeks in either group. These results suggested the SCM4 hydrogel as exhibiting good in vivo degradability and biocompatibility, thereby indicating its potential suitability for in vivo applications.
Fig. 4. Evaluation of in vivo degradation and biocompatibility. (a) Photographs of SA and SCM4 hydrogels post-transplant into the subcutaneous space of rabbits at various time points up to 12 weeks. (b) Degradation rate of hydrogels in vivo. (c) Immunohistochemical staining for CD11b and CD68 at 1-, 2-, 4-, 8-, and 12-weeks post-surgery (red: CD11b; green: CD68; blue: Hoechst 33342). Scale bar: 200 μm. Data represent the mean ± SD of three independent experiments.

In vivo wound healing
We then evaluated the hydrogels using a full-thickness skin-defect model in order to demonstrate the potential wound-healing efficacy of the SCM4 hydrogel. As shown in Fig. S8,† SCM4 hydrogel could strongly adhere to the skin-defect sites on the mouse wound closure model. Fig. 5a shows the wound contraction of the SCM4, SA, and saline groups on days 0, 1, 4, and 7, respectively. As expected, the wound recovery of the group using the SCM4 hydrogel was improved relative to that of the control group (Fig. 5a and b), with the SCM4 hydrogel exhibiting excellent wound-repair and skin-regeneration activities at days 1 and 4 relative to the control and SA groups. This activity was attributed to the antibacterial activity and hemostatic performance of the chitosan,50 as well as the moist wound environment provided by the hydrogel dressing. Furthermore, as shown in Fig. 5c, H&E staining images of investigating the interface indicate that hydrogel maintained structural integrity and remained adherent to the defect site and the surrounding tissue during in vivo wound healing test. The tight and seamless interface between the SCM4 hydrogel and surrounding tissue, due to its strong adhesion with skin tissue, promoted faster cell migration into the SCM4 hydrogel from tissue as compared with that observed in the SA group (Fig. 5c). After 4 days, we observed cell migration into the hydrogel in the SA group, with complete tissue infiltration and fusion within the hydrogel at day 7. These results suggested that the in situ SCM4 hydrogel possessed excellent shape-matching and tissue-integration capabilities, allowing it to mimic host-tissue properties. Moreover, no additional fixation was necessary, and the hydrogel exhibited good tolerance for post-operative treatment, thereby demonstrating efficacy for wound repair.
Fig. 5. Evaluation of the therapeutic effects of the SCM4 hydrogel. (a) Wounds at days 0, 1, 4, and 7. (b) Wound-healing rate according to various treatments. (c) H&E staining (days 1, 4, and 7) of tissue section harboring hydrogels in the rat skin-wound model (black dotted line indicates the interface of the skin and the hydrogel; yellow arrow indicates cells growing into the hydrogel). Scale bar: 200 μm. Data represent the mean ± SD of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001 vs. control.

Conclusions
In summary, we developed a new type of tissue-bonded hydrogel based on the incorporation of chitosan, alginate, and polyacrylamide. These hydrogels achieved rapid gelation and excellent tissue adhesion to wet tissue to promote tissue repair, as well as good mechanical, biocompatibility, and antibacterial properties, thereby meeting the key requirements for modern tissue adhesives. We believe that these tissue adhesives will broaden the impact of hydrogel use in tissue engineering and have great potential for use in wound dressings, implantation surgery, and tissue repair.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
Xiaoxuan Tang and Xinyi Gu contributed equally to this work. This research was financially supported by the National Key Research and Development Program of China (Project No: 2018YFC1105603, 2017YFA0701304 and 2016YFC1101603), the National Science Foundation of China (Project No: 31830028, 81671823, 21702112) and Jiangsu Provincial Key Medical Center.
Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02017f
Notes and references
- Li J. Celiz A. D. Yang J. Yang Q. Wamala I. Whyte W. Seo B. R. Vasilyev N. V. Vlassak J. J. Suo Z. Mooney D. J. Science. 2017;357:378–381. doi: 10.1126/science.aah6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duflo S. Thibeault S. L. Li W. H. Shu X. Z. Prestwich G. D. Tissue Eng. 2006;12:2171–2180. doi: 10.1089/ten.2006.12.2171. [DOI] [PubMed] [Google Scholar]
- Sharma B. Fermanian S. Gibson M. Unterman S. Herzka D. A. Cascio B. Coburn J. Hui A. Y. Marcus N. Gold G. E. Elisseeff J. H. Sci. Transl. Med. 2013;5:167ra6. doi: 10.1126/scitranslmed.3004838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prausnitz M. R. Langer R. Nat. Biotechnol. 2008;26:1261–1268. doi: 10.1038/nbt.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. Y. Mooney D. J. Nat. Rev. Mater. 2016;1:16071. doi: 10.1038/natrevmats.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche E. T. Wohlfarth R. Overvelde J. T. B. Vasilyev N. V. Pigula F. A. Mooney D. J. Bertoldi K. Walsh C. J. Adv. Mater. 2014;26:1200–1206. doi: 10.1002/adma.201304018. [DOI] [PubMed] [Google Scholar]
- Feiner R. Engel L. Fleischer S. Malki M. Gal I. Shapira A. Shacham-Diamand Y. Dvir T. Nat. Mater. 2016;15:679–685. doi: 10.1038/nmat4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu J. Zhao X. Liang Y. P. Zhang T. L. Ma P. X. Guo B. L. Biomaterials. 2018;183:185–199. doi: 10.1016/j.biomaterials.2018.08.044. [DOI] [PubMed] [Google Scholar]
- Zhou F. F. Hong Y. Zhang X. Z. Yang L. Li J. Jiang D. M. Bunpetch V. Hu Y. J. Ouyang H. W. Zhang S. F. Applied Materials Today. 2018;13:32–44. doi: 10.1016/j.apmt.2018.08.005. [DOI] [Google Scholar]
- Liang S. Zhang Y. Y. Wang H. B. Xu Z. Y. Chen J. R. Bao R. Tan B. Y. Cui Y. L. Fan G. W. Wang W. X. Wang W. Liu W. G. Adv. Mater. 2018;30:1704235. doi: 10.1002/adma.201704235. [DOI] [PubMed] [Google Scholar]
- Annabi N. Zhang Y. N. Assmann A. Sani E. S. Cheng G. Lassaletta A. D. Vegh A. Dehghani B. Ruiz-Esparza G. U. Wang X. C. Gangadharan S. Weiss A. S. Khademhosseini A. Sci. Transl. Med. 2017;9:eaai7466. doi: 10.1126/scitranslmed.aai7466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakalopoulos K. A. Wu Z. Q. Kroese L. Kleinrensink G. J. Jeekel J. Vendamme R. Dodou D. Lange J. F. Ann. Surg. 2015;261:323–331. doi: 10.1097/SLA.0000000000000599. [DOI] [PubMed] [Google Scholar]
- Hong Y. Zhou F. F. Hua Y. J. Zhang X. Z. Ni C. Y. Pan D. H. Zhang Y. Q. Jiang D. M. Yang L. Lin Q. N. Zou Y. W. Yu D. S. Arnot D. E. Zou X. H. Zhu L. Y. Zhang S. F. Ouyang H. W. Nat. Commun. 2019;10:2060. doi: 10.1038/s41467-019-10004-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett D. G. Bushnell G. G. Messersmith P. B. Adv. Healthcare Mater. 2013;2:745–755. doi: 10.1002/adhm.201200316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L. Yan L. W. Wang K. F. Fang L. M. Zhang H. P. Tang Y. H. Ding Y. H. Weng L. T. Xu J. L. Weng J. Liu Y. J. Ren F. Z. Lu X. NPG Asia Mater. 2017;9:e372. doi: 10.1038/am.2017.33. [DOI] [Google Scholar]
- Kamata H. Li X. Chung U. I. Sakai T. Adv. Healthcare Mater. 2015;4:2360–2374. doi: 10.1002/adhm.201500076. [DOI] [PubMed] [Google Scholar]
- Freedman B. R. Mooney D. J. Adv. Mater. 2019;31:1806695. doi: 10.1002/adma.201806695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buwalda S. J. Boere K. W. M. Dijkstra P. J. Feijen J. Vermonden T. Hennink W. E. J. Controlled Release. 2014;190:254–273. doi: 10.1016/j.jconrel.2014.03.052. [DOI] [PubMed] [Google Scholar]
- Zhu J. M. Marchant R. E. Expert Rev. Med. Devices. 2011;8:607–626. doi: 10.1586/erd.11.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding B. B. Gao H. C. Song J. H. Li Y. Y. Zhang L. N. Cao X. D. Xu M. Cai J. ACS Appl. Mater. Interfaces. 2016;8:19739–19746. doi: 10.1021/acsami.6b05302. [DOI] [PubMed] [Google Scholar]
- Lee K. Y. Mooney D. J. Prog. Polym. Sci. 2012;37:106–126. doi: 10.1016/j.progpolymsci.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I. L. Mauck R. L. Burdick J. A. Biomaterials. 2011;32:8771–8782. doi: 10.1016/j.biomaterials.2011.08.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. Yang Y. H. Yao J. R. Shao Z. Z. Chen X. ACS Sustainable Chem. Eng. 2014;2:1318–1324. doi: 10.1021/sc500154t. [DOI] [Google Scholar]
- Cheng N. C. Lin W. J. Ling T. Y. Young T. H. Acta Biomater. 2017;51:258–267. doi: 10.1016/j.actbio.2017.01.060. [DOI] [PubMed] [Google Scholar]
- Bao Z. T. Xian C. H. Yuan Q. J. Liu G. T. Wu J. Adv. Healthcare Mater. 2019;8:1900670. doi: 10.1002/adhm.201900670. [DOI] [PubMed] [Google Scholar]
- Dimatteo R. Darling N. J. Segura T. Adv. Drug Delivery Rev. 2018;127:167–184. doi: 10.1016/j.addr.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyles D. A. Castro L. D. Silva J. O. C. Ribeiro-Costa R. M. Eur. Polym. J. 2017;88:373–392. doi: 10.1016/j.eurpolymj.2017.01.027. [DOI] [Google Scholar]
- Drury J. L. Mooney D. J. Biomaterials. 2003;24:4337–4351. doi: 10.1016/S0142-9612(03)00340-5. [DOI] [PubMed] [Google Scholar]
- Braun M. Menges M. Opoku F. Smith A. M. J. Exp. Biol. 2013;216:1475–1483. doi: 10.1242/jeb.077149. [DOI] [PubMed] [Google Scholar]
- Sun J. Y. Zhao X. H. Illeperuma W. R. K. Chaudhuri O. Oh K. H. Mooney D. J. Vlassak J. J. Suo Z. G. Nature. 2012;489:133–136. doi: 10.1038/nature11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal D. Matsuhiro B. Rossi M. Caruso F. Carbohydr. Res. 2008;343:308–316. doi: 10.1016/j.carres.2007.10.016. [DOI] [PubMed] [Google Scholar]
- Biswal D. R. Singh R. P. Carbohydr. Polym. 2004;57:379–387. doi: 10.1016/j.carbpol.2004.04.020. [DOI] [Google Scholar]
- Kim I. Choi J. S. Lee S. Byeon H. J. Lee E. S. Shin B. S. Choi H. G. Lee K. C. Youn Y. S. J. Controlled Release. 2015;214:30–39. doi: 10.1016/j.jconrel.2015.07.012. [DOI] [PubMed] [Google Scholar]
- Bu Y. Zhang L. Liu J. Zhang L. Li T. Shen H. Wang X. Yang F. Tang P. Wu D. ACS Appl. Mater. Interfaces. 2016;8:12674–12683. doi: 10.1021/acsami.6b03235. [DOI] [PubMed] [Google Scholar]
- Lien S. M. Ko L. Y. Huang T. J. Acta Biomater. 2009;5:670–679. doi: 10.1016/j.actbio.2008.09.020. [DOI] [PubMed] [Google Scholar]
- Annabi N. Nichol J. W. Zhong X. Ji C. D. Koshy S. Khademhosseini A. Dehghani F. Tissue Eng., Part B. 2010;16:371–383. doi: 10.1089/ten.teb.2009.0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies K. L. Jones L. Optom. Vis. Sci. 2010;87:387–399. doi: 10.1097/OPX.0b013e3181da863e. [DOI] [PubMed] [Google Scholar]
- Jeon I. Cui J. X. Illeperuma W. R. K. Aizenberg J. Vlassak J. J. Adv. Mater. 2016;28:4678–4683. doi: 10.1002/adma.201600480. [DOI] [PubMed] [Google Scholar]
- Wang P. Deng G. H. Zhou L. Y. Li Z. Y. Chen Y. M. ACS Macro Lett. 2017;6:881–886. doi: 10.1021/acsmacrolett.7b00519. [DOI] [Google Scholar]
- Yu Y. Yuk H. Parada G. A. Wu Y. Liu X. Y. Nabzdyk C. S. Youcef-Toumi K. Zang J. F. Zhao X. H. Adv. Mater. 2019;31:1807101. doi: 10.1002/adma.201807101. [DOI] [PubMed] [Google Scholar]
- Ghobril C. Grinstaff M. W. Chem. Soc. Rev. 2015;44:1820–1835. doi: 10.1039/C4CS00332B. [DOI] [PubMed] [Google Scholar]
- Gao L. Zhou Y. L. Peng J. L. Xu C. Xu Q. Xing M. Chang J. NPG Asia Mater. 2019;11:66. doi: 10.1038/s41427-019-0168-0. [DOI] [Google Scholar]
- Sun X. M. Lang Q. Zhang H. B. Cheng L. Y. Zhang Y. Pan G. Q. Zhao X. Yang H. L. Zhang Y. G. Santos H. A. Cui W. G. Adv. Funct. Mater. 2017;27:1604617. doi: 10.1002/adfm.201604617. [DOI] [Google Scholar]
- Li S. Q. Dong S. J. Xu W. G. Tu S. C. Yan L. S. Zhao C. W. Ding J. X. Chen X. S. Adv. Sci. 2018;5:1700527. doi: 10.1002/advs.201700527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X. Li P. Guo B. L. Ma P. X. Acta Biomater. 2015;26:236–248. doi: 10.1016/j.actbio.2015.08.006. [DOI] [PubMed] [Google Scholar]
- Qin C. Q. Xiao Q. Li H. R. Fang M. Liu Y. Chen X. Y. Li Q. Int. J. Biol. Macromol. 2004;34:121–126. doi: 10.1016/j.ijbiomac.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Li J. Tang X. X. Zhu P. Zhao Y. H. Ling J. Yang Y. M. J. Biomater. Tissue Eng. 2018;8:1725–1734. doi: 10.1166/jbt.2018.1936. [DOI] [Google Scholar]
- Drury J. L. Mooney D. J. Biomaterials. 2003;24:4337–4351. doi: 10.1016/S0142-9612(03)00340-5. [DOI] [PubMed] [Google Scholar]
- Zhang Y. N. Avery R. K. Vallmajo-Martin Q. Assmann A. Vegh A. Memic A. Olsen B. D. Annabi N. Khademhosseini A. Adv. Funct. Mater. 2015;25:4814–4826. doi: 10.1002/adfm.201501489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno H. Mori T. Fujinaga T. Adv. Drug Delivery Rev. 2001;52:105–115. doi: 10.1016/S0169-409X(01)00189-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.