

Abstract
Traumatic spinal cord injury (SCI) above the major spinal sympathetic outflow (T6 level) disinhibits sympathetic neurons from supraspinal control, causing systems-wide “dysautonomia.” We recently showed that remarkable structural remodeling and plasticity occurs within spinal sympathetic circuitry, creating abnormal sympathetic reflexes that exacerbate dysautonomia over time. As an example, thoracic VGluT2+ spinal interneurons (SpINs) become structurally and functionally integrated with neurons that comprise the spinal–splenic sympathetic network and immunological dysfunction becomes progressively worse after SCI. To test whether the onset and progression of SCI-induced sympathetic plasticity is neuron activity dependent, we selectively inhibited (or excited) thoracic VGluT2+ interneurons using chemogenetics. New data show that silencing VGluT2+ interneurons in female and male mice with a T3 SCI, using hM4Di designer receptors exclusively activated by designer drugs (Gi DREADDs), blocks structural plasticity and the development of dysautonomia. Specifically, silencing VGluT2+ interneurons prevents the structural remodeling of spinal sympathetic networks that project to lymphoid and endocrine organs, reduces the frequency of spontaneous autonomic dysreflexia (AD), and reduces the severity of experimentally induced AD. Features of SCI-induced structural plasticity can be recapitulated in the intact spinal cord by activating excitatory hM3Dq-DREADDs in VGluT2+ interneurons. Collectively, these data implicate VGluT2+ excitatory SpINs in the onset and propagation of SCI-induced structural plasticity and dysautonomia, and reveal the potential for neuromodulation to block or reduce dysautonomia after severe high-level SCI.
SIGNIFICANCE STATEMENT In response to stress or dangerous stimuli, autonomic spinal neurons coordinate a “fight or flight” response marked by increased cardiac output and release of stress hormones. After a spinal cord injury (SCI), normally harmless stimuli like bladder filling can result in a “false” fight or flight response, causing pathological changes throughout the body. We show that progressive hypertension and immune suppression develop after SCI because thoracic excitatory VGluT2+ spinal interneurons (SpINs) provoke structural remodeling in autonomic networks within below-lesion spinal levels. These pathological changes can be prevented in SCI mice or phenocopied in uninjured mice using chemogenetics to selectively manipulate activity in VGluT2+ SpINs. Targeted neuromodulation of SpINs could prevent structural plasticity and subsequent autonomic dysfunction in people with SCI.
Introduction
The sympathetic and parasympathetic arms of the autonomic nervous system coordinate involuntary functions throughout the body. Whereas the parasympathetic nervous system is composed of both cranial (brainstem) and spinal (sacral) nerves, the projection neurons that comprise the sympathetic nervous system [i.e., the sympathetic preganglionic neurons (SPNs)], originate mostly in the thoracic spinal cord. SPNs project outside the spinal cord where they innervate postganglionic neurons or the adrenal gland (Ulrich-Lai and Herman, 2009).
Normally, reflex activation of SPNs by somatic and visceral stimuli is modulated by presympathetic supraspinal neurons with origins in the brain and brainstem and spinal cord interneurons (SpINs). However, after high-level spinal cord injury (SCI; i.e., at or above spinal level T6), most or all inhibitory supraspinal control over SPNs is lost (Weaver et al., 2006). As a result, in both SCI humans and animals, spinal sympathetic reflexes become hyperexcitable, ultimately causing pathological dysautonomia that manifests as an array of systemic comorbidities, including immune suppression (Guttmann and Whitteridge, 1947; Zhang et al., 2013; Brennan et al., 2021).
Immunological dysfunction develops after severe high-level SCI and worsens as a function of time post-injury because of the onset of aberrant intraspinal autonomic reflexes (Lucin et al., 2007, 2009; Zhang et al., 2013; Prüss et al., 2017; Mironets et al., 2020). Notably, we discovered that the onset of SCI-induced immune suppression could be blocked by silencing VGluT2+ SpINs located within the thoracic spinal cord (Ueno et al., 2016). However, it is unknown whether other pathological autonomic reflexes that develop after SCI also are controlled by these same thoracic VGluT2+ SpINs.
Autonomic dysreflexia (AD) is a condition of severe paroxysmal hypertension that is triggered when common somatic or visceral stimuli (e.g., bladder or bowel filling) activate spinal sympathetic networks that control peripheral vasomotor tone. Although the onset of AD occurs within days or weeks after SCI (Mayorov et al., 2001; Rabchevsky et al., 2012; Zhang et al., 2013; Brennan et al., 2021), the frequency of AD increases at later times post-injury. A delayed increase in AD frequency has been attributed to dynamic remodeling and sprouting of intraspinal circuitry, notably calcitonin gene-related peptide-positive (CGRP+) sensory afferents and multisegmental propriospinal neurons projecting from lumbar spinal cord (Krenz et al., 1999; Cameron et al., 2006; Hou et al., 2008, 2009; Brennan et al., 2021). Here, using chemogenetics and anatomic tracing techniques, we tested whether thoracic VGluT2+ SpINs, which regulate post-injury immune dysfunction, also regulate AD after SCI.
New data show that after SCI, thoracic VGluT2+ SpINs are key cellular determinants of newly formed multisegmental spinal sympathetic networks that amplify neurotransmission and cause maladaptive functional activity throughout the thoracic and lumbar spinal cord. Specifically, chemogenetic silencing of thoracic VGluT2+ SpINs prevents the temporal progression of AD after SCI, in part by inhibiting the remodeling of spinal autonomic circuitry. Also, silencing thoracic VGluT2+ SpINs blocks injury-induced synaptic and axonal plasticity and the maladaptive functional activity that emerges within neuronal networks distributed throughout the thoracic and lumbar spinal cord. Since the motor restorative effects of epidural or intraspinal stimulation can be linked to modulation of SpINs (Mushahwar et al., 2004; van den Brand et al., 2012; Formento et al., 2018; Skinnider et al., 2021), data in the present report indicate that similar interventional strategies may be used to target thoracic SpINs to prevent or overcome various SCI-induced comorbidities including immune suppression (Zhang et al., 2013; Ueno et al., 2016), primary hypercortisolism (Prüss et al., 2017), and AD.
Materials and Methods
Mice
Adult VGluT2-cre mice (n = 103) on a C57BL/6; FVB; 129S6 genetic background (Slc17a6tm2(cre)Lowl/J; stock no. 016963, The Jackson Laboratory) were purchased and bred for use in this study. Mice were weaned at postnatal day 14 (P14) for intraspinal injections, after which littermates of the same sex were housed together. Animals were housed in individually ventilated cages with HEPA (high efficiency particulate air)-filtered air on a 12 h light/dark cycle with ad libitum access to food and water. Mice were housed in groups of three to five except for telemetry studies, where individual housing was necessary. At the time of SCI or telemetry surgery where used, 50% of each group was male and 50% of each group was female.
For telemetry studies comparing the effects of inhibitory (Gi) DREADDs in VGluT2-cre mice (Figs. 2, 4), four mice from the saline group died before the end of the study, one as a result of complications with a telemetry probe, two related to complications with bladder management [23 and 25 d post-injury (dpi)], and one for unknown reasons (28 dpi). Two mice in the clozapine-N-oxide (CNO) group died; one because of complications with bladder expression (20 dpi) and one for unknown reasons (26 dpi). All procedures were performed in accordance with The Ohio State University Institutional Animal Care and Use Committee and the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines. Upon recovery from anesthetic, one mouse was found to have incomplete paralysis of the hindlimbs and was removed from the study.
Figure 2.

The frequency of spontaneous AD is reduced by silencing spinal glutamatergic interneurons. Radiotelemetry devices were implanted 3 d before mice received a complete T3 SCI. Heart rate and blood pressure were recorded from −3 to 30 d post-SCI. To silence Gi DREADD-transduced VGluT2 interneurons, mice were injected with CNO (1 mg/kg, i.p.) 2–3×/d starting at 14 dpi. Control mice were injected with saline. A, Time post-injury versus the number of daily AD events. Data pooled from two replicate experiments and analyzed using a mixed-effect model to compare slopes; n = 7–8 mice/group in each replicate study (total saline, n = 15; CNO, n = 16); **p < 0.01 comparing slopes; values are the mean ± SEM. B, The average number of daily spontaneous AD events grouped by week after T3 SCI. Mixed-effects model with Bonferroni post hoc test; n = 7–8 mice/group in each replicate study (total saline, n = 15; CNO, n = 16); ap < 0.01 comparing groups at matched time points; values are the mean ± SEM. C, The maximum number of daily AD events recorded between 14 and 30 dpi is reduced by silencing VGluT2+ interneurons with CNO. Mann–Whitney U test comparing groups; n = 15–16 mice/group; bp < 0.0001; values are the mean ± SEM.
Figure 4.

Silencing spinal interneurons blocks experimentally induced AD. Radiotelemetry was used to record blood pressure and heart rate before, during, and after CRD. A, B, Representative BP recordings from a Gi DREADD-injected SCI mouse during CRD experiments before (A) and after (B) CNO injection. C, D, CNO reduces CRD-induced hypertension in mice injected with Gi DREADDS (C), but CNO does not affect CRD-induced hypertension in mice without Gi DREADDs (D). The dashed line indicates a net zero change in BP. The BP change is the average of three consecutive CRD experiments in each mouse (see Materials and Methods). E, F, The average raw BP (E) and HR (F) values before and after CNO injection shows no effect of CNO on cardiovascular parameters in mice without Gi DREADDs. Paired Student's two-sided t tests comparing before versus after CNO: n = 15–16 mice/group (C); n = 5 mice/group (D–F). ap < 0.0001.
To ensure that individuals involved in data analysis remained blind to group designation, they had access only to the mouse ID and not the experimental condition. A key, specifying group designation was maintained by an individual not involved in data collection or analysis. Given limitations in the litter size of transgenic mice, the number of telemetry devices available, and the need to appropriately power these experiments, we were forced to prioritize and focus on the controls we found most likely to interfere with our outcome measures. Previous studies have shown that CNO has no effect on heart rate (HR) or blood pressure (BP) in mice without DREADDs (Agulhon et al., 2013). For this reason, a “T3 SCI + Gi DREADDs” control group is included in all figures, but T3 SCI + CNO is not.
Treatment groups
Before injecting adeno-associated virus (AAV)-Gi DREADDs, mice were randomized into groups, controlling for age, weight, and sex: Gi DREADDs or no DREADD controls. Within each group, mice were randomly assigned to SCI or sham surgery groups, then further subdivided into groups receiving CNO or saline injections. This generated four groups in total (T3 SCI + Gi DREADDs, T3 SCI + Gi DREADDs + CNO, sham + Gi DREADDs, sham + Gi DREADDs + CNO). Beginning at 14 dpi and always after bladder expression, mice were injected daily (intraperitoneally) with 100 μl of saline or CNO (1 mg/kg body weight/injection; catalog #C0832-5MG, Sigma-Aldrich). Since the biological effects of CNO on DREADD-expressing mice lasts for 6–10 h (Wess et al., 2013), our goal was to inject mice with CNO 3×/d, but this was not always possible. For one experimental run, CNO was injected 2×/d. For the second experimental run, for the 16 d spanning 15–30 dpi, mice were injected 3×/d for 9 d and then 2×/d for 7 d. Since there was no significant effect of CNO injection frequency on AD frequency (2.00 ± 0.235 events/d with 2 CNO injections vs 1.736 ± 0.206 events/d with three CNO injections, p = 0.399), data from both runs were combined as seen in Figure 2. To evaluate the role that increasing neuronal activity alone plays in driving intraspinal plasticity, we injected AAVs encoding excitatory DREADDs (Gq DREADDs) into uninjured mice. Before Gq DREADD injections, mice were randomized into two groups, controlling for age, weight, and sex: Gq DREADDs or no DREADD controls. All mice received sham surgery, then were randomly assigned to receive CNO or saline. This generated four groups in total (sham + Gq DREADDs, sham + Gq DREADDs + CNO, sham + CNO, sham). For these experiments, mice were injected (intraperitoneally) with 100 μl of saline or CNO 1×/d in the evening (5:00–6:00 P.M.), before the onset of the typical active period of the animals. This reduction in CNO injection frequency was necessary because the activation of intraspinal Gq DREADDs with a single CNO injection triggered pronounced hindlimb spasticity for ∼8 h. By reducing the frequency of CNO, we hoped to limit stress and sleep interruption in this cohort of mice.
Chemogenetic manipulation of intraspinal VGluT2+ interneurons
Six weeks before spinal surgery, AAV8-hSyn-DIO-hM4D(Gi)-mCherry (Gi DREADDs) or AAV8-hSyn-DIO-hM3D(Gq)-mCherry (Gq DREADDs) was injected on one side of the spinal cord at both the T4 and T7 spinal segment of VGluT2-Cre mice age P14. Injections were targeted to the intermediate gray matter of the thoracic spinal cord (1 μl/site/spinal segment, 0.25 mm lateral, 0.5 mm in depth; Ueno et al., 2016) and consistently labeled neurons throughout the T4–T9 thoracic spinal cord ipsilateral to the site of injection (Fig. 1). In an independent study, bilateral injections at T7 were given (Extended Data Fig. 3-1). Mice received a T3 SCI or sham surgery at P56, and then, starting at 14 dpi or sham surgery, CNO or saline was injected as described above. Independent pilot studies with unilateral T4 and bilateral T7 injections of AAV8-Gq DREADDs at T7 spinal level using the above coordinates and injection volumes indicate that this protocol transduces VGluT2+ neurons located predominantly in spinal laminae III–VIII and X, and in response to CNO injections, neurons in spinal laminae VII and X are preferentially activated. Moreover, 60–90% of activated neurons are also DREADD+ (Extended Data Fig. 3-1).
Figure 1.

Intraspinal injections of Gi DREADDs transduce VGluT2+ spinal interneurons. A, Experimental timeline. Mice received intraspinal DREADD injections at T4 and T7 at P14. Six weeks later, mice were implanted with radiotelemetry probes, followed by T3 laminectomy (sham) or T3 SCI. Mice received 1–3×/d CNO or saline injections from day 14 to the end of the study. Mouse cohorts were injected with intrasplenic PRV at day 24, intra-adrenal PRV at day 28, or intraperitoneal Fluoro-Gold at day 28, then were perfused at day 28, 32, and 34, respectively (see figure legends for individual experiment details). B, Representative image of T4–T6 horizontal spinal cord section at the level of the central canal (IML level), showing VGluT2+ interneurons that have been induced by Gi DREADD (red profiles). Scale bar, 200 μm. C, D, High-magnification representative images of an injection site (C) and an injection border site (D), showing the density of DREADD-induced VGluT2+ (red) interneurons relative to total neurons (NeuN+ profiles). Scale bar, 100 μm. E, Quantification of DREADD-induced neurons as a percentage of total NeuN+ neurons in the intermediate gray matter of the thoracic spinal cord. Transduction efficiency decreases slightly away from the injection site. n = 6 mice/group; values are the mean ± SEM. F–H, Diagram showing IML neuron location and representative images of T7 spinal cord coronal section showing dorsal–ventral transduction of Gi DREADD. Transduced neurons are primarily in spinal laminae III–VIII and X. Scale bars: G, 200 μm; H, 40 μm.
SCI and postoperative care
SCI.
Mice were anesthetized by injecting a mixture of ketamine (80 mg/kg, i.p.) and xylazine (40 mg/kg, i.p.). During surgery, mouse body temperature was maintained at ∼37.5°C using a feedback-controlled heating pad (Harvard Apparatus). The third thoracic vertebra (T3) was identified based on anatomic landmarks (i.e., T2 spinous process and supraspinal vasculature), then a #4 Dumont forceps (with a tip of 0.4–0.2 mm; catalog #11241-30, Fine Science Tools) was inserted into the vertebral column on both sides of the spinal cord between T3 and T4 until the tips touched the ventral surface of the vertebral column, before laterally compressing the spinal cord for 3 s. This produced a severe crush injury but without breaking the dura (Faulkner et al., 2004; Brennan et al., 2021). The muscle was sutured with 5.0 polyglactin dissolvable sutures, and the skin was closed with wound clips. Sham-operated mice underwent anesthesia and surgical procedures exactly as above without a spinal crush injury. End point histology and visual confirmation of complete hindlimb paralysis at 1 dpi was used to confirm that each mouse received crush injuries of similar magnitude/duration. As noted above, on recovery from anesthetic, one mouse was found to have incomplete paralysis of the hindlimbs and was removed from the study.
Postoperative care.
Following surgery, animals were injected with saline (2 ml, s.c.) and antibiotics (1 mg/kg, s.c.; Gentocin) then placed into warmed cages (∼34°C). Bladders from SCI animals were manually voided twice daily for the duration of the experiments. Body weight and urinary pH were monitored weekly. To prevent dehydration, mice were supplemented with saline (1–2 ml, s.c., daily). Antibiotic coverage was achieved by administration of gentamicin (1 mg/kg, s.c., daily for 5 dpi). Mice showing alkaline urine were presumed to have mild urinary tract infections and were treated with antibiotics until the infection resolved (10 mg/kg, s.c.; Baytril).
Spontaneous AD
Surgical implantation of telemetry probes.
Telemetry transmitters were implanted into mice as described previously (Zhang et al., 2013). Briefly, 3 d before SCI, mice were anesthetized by injecting a mixture of ketamine (80 mg/kg) and xylazine (40 mg/kg) into the peritoneum. PA-C10 transmitters (Data Sciences International) were implanted via cannulation of the left common carotid artery, and the battery was positioned in a skin pocket under the left flank. The neck skin was sutured closed. Mice were supplemented with 1 ml of saline and allowed to recover from anesthesia in warmed cages (∼34°C) overnight.
Detection of AD events.
Dataquest data acquisition software (Data Sciences International) was used to continuously record BP and HR. BP and HR were recorded for 2 d before SCI (baseline) and from 5 to 30 dpi, with each data point representing the mean of one 5 s interval. MATLAB was used to create an algorithm to detect spontaneous AD events. The algorithm was based on the semiautomated method developed by Zhang et al. (2013) but incorporates validation steps to improve objectivity and efficiency. Each detected episode of AD was then manually verified by eye, excluding any false positives resulting from inconsistent or variable signal quality. The fully automated MATLAB AD detection algorithm is available on request. For telemetry experiments in this study, a positive AD event was registered if BP increased >20 mmHg above baseline, HR decreased >30 beats/min below baseline, and these events overlapped for >66% of a time period lasting at least 30 s. In vivo telemetry experiments to determine the effect of Gi DREADDs were repeated twice, each time with n = 7–8 mice/group. To eliminate any potential confound of systemic inflammation on spinal cord structure caused by a foreign object (i.e., telemetry probes), tissues from mice that were used for telemetry monitoring were not used for anatomic analyses.
Induced AD
To measure cardiovascular responses elicited by visceral stimulation, colorectal distention (CRD) was performed on the day of kill (28–34 dpi) in SCI mice implanted with telemetry probes and given saline or CNO (see Fig. 4). CRD was accomplished using a 4 French, 60 mm balloon-tipped catheter (Swan-Ganz monitoring catheter; model 116F4, Edwards Life Sciences). The catheter line was inserted into the rectum, and the balloon was positioned 1.5 cm from the anal opening. The catheter was secured to the tail with surgical tape. After securing the catheter, mice were left to acclimate for at least 20 min. After ensuring that the HR and BP traces were stable for 10 min, the balloon was inflated with 0.3 ml of air. Distention was maintained for 1 min before the balloon was deflated; the probe remained in the rectum. The procedure was repeated two more times, with a 10 min rest between stimulations. Prestimulation data were defined as the average HR and BP recorded in the 1 min immediately preceding stimulation. The poststimulation data were defined as the maximum HR and minimum BP recorded during and in the 3 min following CRD. The prestimulation and poststimulation values for each mouse were averaged over three trials (see Fig. 4).
Fluoro-Gold tracing
To retrogradely label all SPNs, mice were injected with 100 μl of 1% Fluoro-Gold (Fluorochrome) 7 d before perfusion (Schmued and Fallon, 1986). Successful labeling was confirmed by bilateral presence of bright, Fluoro-Gold+ neurons in the thoracic intermediolateral cell column (IML) and spinal lamina VII. Since systemic Fluoro-Gold has off-target effects (Mi et al., 2019), mice injected with Fluoro-Gold were not used for physiological analyses (i.e., AD readouts).
Pseudorabies virus tracing
Viral delivery.
Four days before perfusion (i.e., 24 or 28 dpi), the random number generator in Microsoft Excel was used to randomly choose a subset of mice from each group (n = 4–5/group) to receive pseudorabies virus (PRV) injections into the spleen or adrenal gland. These mice were anesthetized by isoflurane inhalation (2% isoflurane in oxygen during induction, 1.5% for maintenance). The left flank was shaved and sterilized, and a 1 cm diagonal incision was made into the skin and muscle overlying the spleen or the kidney. The spleen was carefully extracted from the peritoneal cavity and rested atop sterile gauze with forceps for injection. For adrenal injections, the suprarenal fat was held with forceps for stabilization and the adrenal gland was injected. The spleen or adrenal gland was injected with a GFP-expressing PRV (Bartha strain PRV152; 4.9 × 109 pfu/ml) using pulled glass pipettes (catalog #1B150F-4, World Precision Instruments) attached to the Nanoject system (catalog #PV830 Pneumatic PicoPump, World Precision Instruments) positioned under a dissecting microscope. A total of 3 μl of virus was delivered into the adrenal gland in one injection site, or 10 μl of virus was injected into 10 injection sites along the spleen (1 μl/site), as described previously (Cano et al., 2001; Ueno et al., 2016; Brennan et al., 2021). Any potential virus leaking from the injection sites was absorbed with a sterile cotton swab. The injected organs were then carefully repositioned back into the peritoneal cavity. The peritoneal cavity and skin were sutured closed. Mice were placed into warmed cages (34°C) and given 1 ml of saline and antibiotics (1 mg/kg, s.c., Gentocin) during anesthesia recovery.
Tissue processing.
At 28–34 dpi, mice were transcardially perfused as described below. Spinal cords were dissected and the T3 and T10 levels were marked in situ with a fine permanent marker. These landmarks were used to help define the other spinal levels during image acquisition. Samples were postfixed in 4% PFA overnight at 4°C, washed in 0.2 m phosphate buffer overnight, then cryopreserved for 48 h in 30% sucrose in 0.1 m PBS. Spinal levels C3–T3, T3–T10, and T10–L5 were embedded in Tissue-Tek optimal cutting temperature compound and stored at −80°C until sectioning. Horizontal floating sections (80 μm thick) were cut using a digital cryostat (model HM 505E, Microm). Floating slices were collected into a 24-well plate containing 0.1 m PBS. Samples were stored at 4°C in the dark until staining and imaging.
Imaging.
Horizontal sections at the depth of the central canal containing GFP+ neuronal cell bodies (i.e., the IML cell column) were transferred from a 96-well plate and mounted with VECTASHIELD antifade mounting medium (catalog #H-1000, Vector Laboratories) onto ColorFrost slides (catalog #9951L-006, Thermo Fisher Scientific). To visualize the spinal–splenic and spinal–adrenal networks in the spinal cord, 10-step Z-stacks of 8 μm increments spanning 80 μm were acquired using a confocal microscope (FV1000 Filter, Olympus) with 10× magnification. High-resolution images of the entire horizontal section were created using tiling and stitching. Maximum intensity projections (MIPs) of representative fluorescent images were imported into Adobe Photoshop, and a levels adjustment layer was applied to reveal the full tonal range of visible pixels in the RGB histogram.
Quantification of PRV+ neurons.
The thoracic gray matter was divided into the ipsilateral (left) and contralateral (right) sides relative to the location of the spleen or injected adrenal gland. Each side was then subdivided into the medial zone where the central autonomic nucleus is located (within ∼80 μm lateral to the central canal), the intermediate zone where the intercalated nucleus is located (between the medial and lateral zones), and the lateral zone where the IML column is located (within ∼80 μm medial to the gray/white matter interface; Watson et al., 2009). The number of GFP+ neuronal cell bodies in each region of interest (ROI) was counted with ImageJ. Briefly, Z-stacks were compressed to create one MIP. Positive staining in a region of interest was selected using a custom recipe in MIPAR, and then PRV+ neurons were counted in each region, with size limits of 10–400 μm (see Figs. 7, 8).
Figure 7.
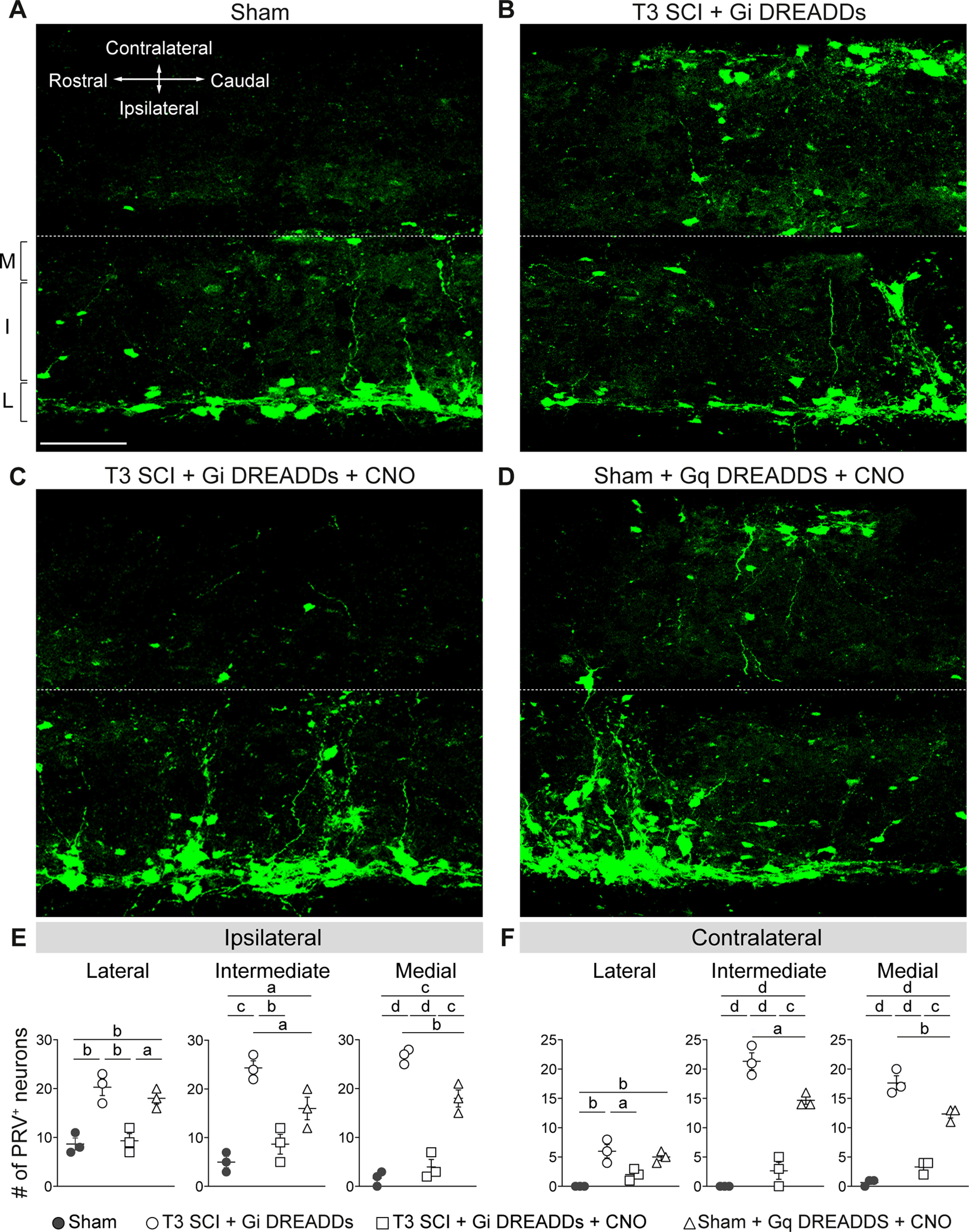
Silencing VGluT2 interneurons prevents expansion of the sympathetic spinal–splenic circuitry. At 24 dpi, PRV-GFP was injected into the spleen for retrograde labeling of the spinal–splenic sympathetic innervation. At 4 d postinjection (28 dpi), animals were perfused and spinal cords analyzed for PRV+ neurons. M, Medial; I, intermediate; L, lateral gray matter. A–D, Representative horizontal sections of the thoracic spinal cord (T7). Scale bar, 100 μm. E, F, Quantification of PRV+ neurons in the ipsilateral (E) and contralateral (F) lateral, intermediate, and medial zones of T7 gray matter. One-way ANOVA with Tukey's post hoc tests; n = 3 mice/group. ap < 0.05; bp < 0.01; cp < 0.001; dp < 0.0001. Values are the mean ± SEM.
Figure 8.

DREADD modulation of spinal interneurons alters the size of sympathetic adrenal-correlated circuitry. At 28 dpi, PRV-GFP was injected into the left adrenal gland for retrograde labeling of spinal–adrenal sympathetic innervation. At 4 d postinjection (32 dpi), animals were perfused and spinal cords analyzed for PRV+ neurons. M, Medial; I, intermediate; L, lateral gray matter. A–D, Representative horizontal sections of the thoracic spinal cord (T8–T9): images are representative of sham mice (A), SCI control mice with Gi DREADDs but without CNO (B), SCI mice given Gi DREADDs and CNO to silence VGluT2+ SpINs (C), or sham mice given Gq DREADDs and CNO to excite VGluT2+ SpINs (D). Scale bar, 100 μm. E, F, Quantification of PRV+ neurons in the ipsilateral (E) and contralateral (F) lateral, intermediate, and medial zones of T8–T9 gray matter. One-way ANOVA with Tukey's post hoc test; n = 3–4 mice/group. G, Representative horizontal sections of the lumbar spinal cord. Scale bar, 400 μm. H, Quantification of PRV+ neurons in lumbar gray matter. One-way ANOVA with Tukey's post hoc test, n = 3–4 mice/group. ap < 0.05; bp < 0.01; cp < 0.001; dp < 0.0001 compared with unlettered groups (E) or indicated groups (F). Values are the mean ± SEM.
Perfusion and tissue processing
At 28–34 dpi, mice were anesthetized with 1.5× the surgical dose of ketamine/xylazine (see above) and perfused transcardially with 0.1 m PBS, until blanching of the liver to a light brown color and the perfusate ran clear (∼60 s), followed by 4% PFA for 6 min. Spinal cords were dissected and postfixed in 4% PFA overnight at 4°C, then incubated in 0.2 m phosphate buffer overnight. Tissue for cryostat cutting was cryopreserved for 48 h in 30% sucrose in 0.1 m PBS, and then embedded in Tissue-Tek optimal cutting temperature compound and stored at −80°C until sectioning. Spinal cord sections were cut along the coronal axis in 1:10 series at 16 μm thickness, or along the longitudinal axis at 80 μm thickness, using a digital cryostat (model HM 505E, Microm). Sections were collected onto SuperFrost Plus slides and stored at −20°C until immunostaining.
Immunohistochemistry
For immunofluorescent staining, sections were dried for at least 30 min on a slide warmer, washed in 0.1 m PBS (3 × 4 min), then blocked for 1 h in 0.1 m PBS containing 1% bovine serum albumin and 0.1% Triton X-100 (BP+). Sections were incubated at 4°C overnight in primary antibody solutions made in BP+ (Table 1). After washing in 0.1 m PBS (3 × 4 min), slides were incubated in fluorophore-conjugated secondary antibodies (Table 1) with or without DRAQ5 nuclear dye (1:4000; catalog #ab108410, Abcam) for 1 h at room temperature. Slides underwent a final round of washing before being coverslipped with Immu-Mount media.
Table 1.
Antibodies used for immunochemistry
| Antigen | Host, dilution | RRID | Vendor, catalog # |
|---|---|---|---|
| Primary antibodies | |||
| GFP | Chicken, 1:500 | AB_10000240 | Aves Labs, GFP-1020 |
| CGRP | Rabbit, 1:500 | AB_259091 | Sigma-Aldrich, C8198 |
| FosB | Rabbit, 1:500 | AB_2721123 | Abcam, ab184938 |
| NeuN | Rabbit, 1:10,000 | AB_10711153 | Abcam, ab104225 |
| VGAT | Guinea pig, 1:500 | AB_887873 | Synaptic Systems, 131004 |
| VGluT2 | Guinea pig, 1:1000 | AB_2665454 | Millipore, AB2251-l |
| Secondary antibodies | |||
| Alexa Fluor 488-rabbit Ig | Goat, 1:500 | AB_143165 | Thermo Fisher Scientific, A-11008 |
| Alexa Fluor 546-rabbit Ig | Goat, 1:1000 | AB_143051 | Thermo Fisher Scientific, A-11035 |
| Alexa Fluor 488-guinea pig IgG | Goat, 1:500 | AB_2534117 | Thermo Fisher Scientific, A-11073 |
| Alexa Fluor 568-guinea pig IgG | Goat, 1:1000 | AB_141954 | Thermo Fisher Scientific, A-11075 |
| Alexa Fluor 488-chicken IgY | Goat, 1:500 | AB_142924 | Thermo Fisher Scientific, A-11039 |
NeuN+ and DREADD+ cell quantification
To quantify the induction efficiency of the DREADD virus, a randomly selected set of spinal cords from each group were sectioned along the longitudinal orientation and stained with NeuN to label all neurons. A region of interest was defined on the longitudinal section through the IML as any region with DREADD+/mCherry+ expression. This region corresponded to the gray matter on the ipsilateral side of the injection, spreading one full spinal segment both rostral and caudal to the injection site (e.g., the T4 injection site labels the ipsilateral gray matter from T4 to T6). For the quantification in Figure 1E, NeuN+ soma that were also DREADD+ were identified and quantified with a custom MIPAR recipe modified from “Fluorescence Co-localization” on the MIPAR website (https://www.mipar.us/recipe-store.html#!/Fluorescence-Co-localization/p/159344774/category=25315756).
FosB+ neuron analysis
FosB immunostaining is often used to define endogenous neuronal activity (Lyons and West, 2011). Therefore, to obtain a readout for neuronal activity, sections cut at spinal level T7 were immunostained for FosB, imaged, and quantified. The 16-step Z-stacks of 1 μm increments spanning the 16 μm were acquired using a confocal microscope (Filter FV1000, Olympus) with 20× magnification. Tiling and stitching was used to generate a high-resolution image covering the whole section. Z-stacks were compressed to create one MIP and converted to a binarized image (see Fig. 3). Using the Allen Mouse Spinal Cord Atlas, spinal laminae I–IV and V–X were outlined on each image using the selection tool in MIPAR. Positive staining in each region of interest was selected using the “basic threshold” tool. The image was then binarized, and the number of FosB+ neurons per region was quantified with size limit of 10–400 μm.
Figure 3.

Silencing VGluT2+ interneurons reduces neuron activity in the thoracic spinal cord. A–D, Representative binarized spinal cord cross sections (spinal level T7) immunostained for FosB, a marker of activated neurons. FosB+ cells were marked with circles of uniform size for clarity. Scale bar: A–D, 200 μm. E, F, Quantification of FosB+ cells in laminae I–IV (E) and laminae V–X (F). One-way ANOVA with Tukey's post hoc tests; n = 4–5 mice/group. ap < 0.05; bp < 0.01; cp < 0.001; values are the mean ± SEM. G, H, The resting blood pressure (G) and heart rate (H) of VGluT2-cre mice injected with Gi DREADDs before and after CNO injection. Paired Student's two-sided t tests, n = 16 mice/group. ap < 0.05; cp < 0.001 versus before CNO injection. Anatomical mapping of FosB+ and DREADD+ neurons is shown in Extended Data Figure 3-1.
VGluT2 and VGAT puncta analysis
For VGluT2 and vesicular GABA transporter (VGAT) puncta analysis, spinal cord sections from all groups between spinal levels T6 and T9 were immunostained for the presynaptic proteins VGluT2 and VGAT (see Fig. 5). Stained sections were imaged and analyzed using a modified protocol from Ippolito and Eroglu (2010). Briefly, 15-step Z-stacks of 0.33 μm increments spanning 5 μm were acquired on a confocal microscope (Filter FV1000, Olympus) with 40× magnification and 2× optical zoom (80× final magnification). Imaging parameters were consistent to minimize intensity variability. Z-stack steps were compressed serially in groups of three (i.e., steps 1–3, 4–6, 7–9, 11–12, 13–15) to create five MIPs. A MIPAR recipe was generated to identify puncta within the expected size range, and was positive for the fluorescent markers in the expected intensity range (Brennan et al., 2021). The MIPAR recipes were compared with manual counting to confirm accuracy and consistency, and the recipes are available on request.
Figure 5.

Silencing VGluT2+ interneurons restores the balance of excitatory–inhibitory input in the IML after SCI. Excitatory (VGluT2+) and inhibitory (VGAT+) presynaptic vesicles were labeled in the T6–T9 IML (34 d post-spinal surgery). A–H, Representative images showing VGluT2 (A–D) and VGAT (E–H) staining in each group. Scale bar: A–H, 20 μm. I–K, Quantification of the number of VGluT2+ (I) and VGAT+ (J) puncta, and the ratio of excitatory to inhibitory puncta (K) shows that SCI increases excitatory and decreases inhibitory puncta; this is returned to normal by silencing VGluT2 interneurons and can be recapitulated in sham mice by activating VGluT2 interneurons. I–K, One-way ANOVA with Tukey's post hoc tests; n = 3–4 mice/group. ap < 0.05. Values are the mean ± SEM.
CGRP+ axon analysis
To visualize nociceptive primary afferent fibers below the major sympathetic output, spinal level L4 was immunostained for CGRP and DRAQ5 nuclear dye. Ten-step Z-stacks of 1 μm increments spanning 16 μm were acquired using an confocal microscope (Filter FV1000, Olympus) with 20× magnification. Tiling with stitching was used to generate a high-resolution image covering the whole section. Imaging parameters were kept consistent to minimize intensity variability. Z-stacks were compressed to create one MIP and converted to a grayscale image (see Fig. 6A–H). Using the Allen Mouse Spinal Cord Atlas and the DRAQ5+ channel, spinal laminae I–IV and X were outlined on each image using the freehand selection tool in ImageJ. The image was converted to 8 bit format. The area of CGRP+ staining in each region of interest was selected using the threshold tool in ImageJ and expressed as positive number of pixels. The area of CGRP+ axons in laminae I–V was the average of the left and right dorsal horn (Fig. 6I). The line tool in ImageJ was used to trace the longest axon in the left and right dorsal horn, and the lengths were averaged between sides to obtain one data point per section (Fig. 6K).
Figure 6.

Silencing VGluT2+ interneurons prevents SCI-induced sprouting of CGRP+ lumbar primary afferents. A–H, Representative grayscale images of CGRP immunostaining in the lumbar dorsal horn (A–D) and lamina X (E–H). Arrows in F and H point to CGRP+ fibers sprouting into deep laminae. Scale bars: (in A) A–D, 200 μm; (in E) E–H, 400 μm. I–K, Quantification of CGRP+ areas in laminae I–V (I), lamina X (J), and the longest axon (K) in the lumbar spinal cord show that SCI increases CGRP+ area and longest length. This phenotype is reversed by silencing VGluT2+ interneurons. I–K, One-way ANOVA with Tukey's post hoc tests; n = 4–5 mice/group. ap < 0.05; bp < 0.01. Values are the mean ± SEM.
Statistics
A mixed-effect model was conducted in SAS 9.4 (SAS Institute), and other data were analyzed in GraphPad Prism version 9.1.1 (GraphPad Software). Heart rate and blood pressure data from the two radiotelemetry experiments testing the effect of inhibitory DREADDs on spontaneous AD were pooled and analyzed using a mixed-effect model, incorporating repeated measures for each mouse, followed by slope comparison. We included “group” as a fixed factor, “mouse” as a random factor, and “experiment” as an adjusting factor in this model (Fig. 2A). Two-way ANOVA with repeated measures was used to compare daily AD events per week post-SCI (Fig. 2B). A Mann–Whitney U test was used to compare the maximum number of AD events between 14 and 30 dpi (Fig. 2C). One-way ANOVA with Tukey's post hoc test was used to compare data from four independent groups (Fig. 3E,F; see also Figs. 5–8). Paired t tests were used to compare BP and HR before and after CNO administration (Figs. 3G,H, 4). Two-way ANOVA with Tukey's post hoc test was used to compare two anatomic locations (i.e., IML and lamina II) from three independent groups (see Fig. 9D,E). Repeated-measures one-way ANOVA with Tukey's post hoc test was used to compare AD events before and after CNO washout (see Fig. 9F,G). Statistical significance was determined at p < 0.05. Group sizes were determined a priori to produce a power of 0.8 to reduce type II errors.
Figure 9.
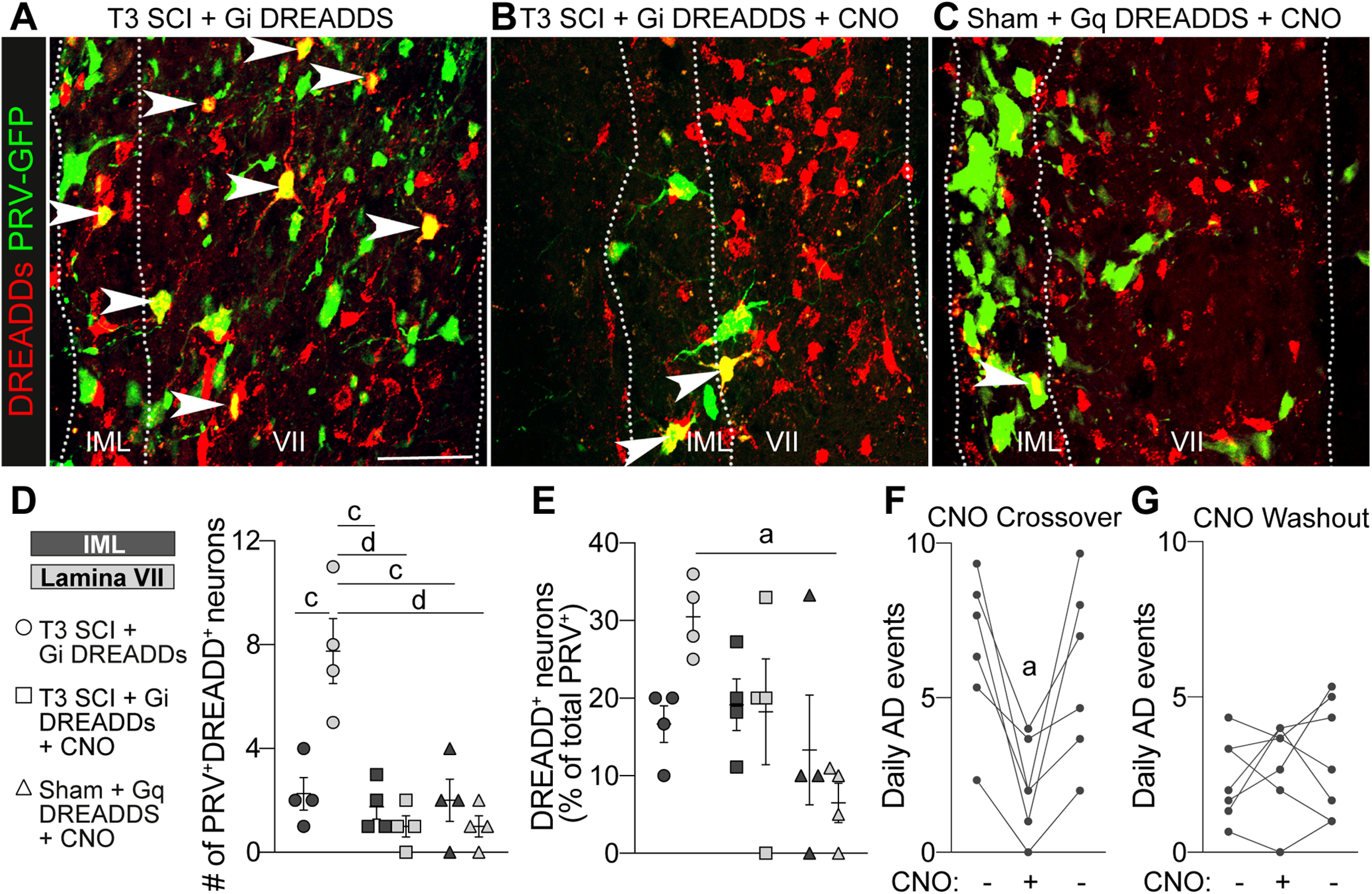
Sympathetic networks expand through VGluT2+ interneurons after SCI. A–C, Representative images of DREADD+PRV+ neurons in the T7–T8 spinal cord. Three microliters of PRV-GFP was injected into the left adrenal gland. Double-positive neurons are marked by white arrowheads. Scale bar, 50 μm. D, Quantification of the number of double-positive neurons in the IML (dark gray data symbols) and lamina VII (light gray data symbols). E, Quantification of the double-positive neurons expressed as a percentage of total PRV+ neurons. D, E, Two-way ANOVA comparing groups and gray matter regions with Tukey's post hoc test; n = 4 mice/group. ap < 0.05; cp < 0.001; dp < 0.0001. Values are the mean ± SEM. F, G, CNO crossover and washout experiments in VGluT2-Cre mice injected with Gi DREADDS. The first and third data points in each graph are the average of 3 consecutive days during chronic SCI (see Materials and Methods), while the middle data point represented 1 d. Administering CNO to crossover mice reduced AD (F), but the acute withdrawal of CNO did not affect AD in mice previously given CNO (G). F, G, One-way repeated-measures ANOVA with Tukey's post hoc test; n = 5–6 mice/group. ap < 0.05 versus before CNO; each data point represents the mean of one animal.
Results
Chemogenetic silencing of thoracic spinal VGluT2 interneurons prevents autonomic dysreflexia after SCI
Previously, we discovered that intentionally triggering spinal sympathetic reflexes simultaneously elicits AD and immune suppression in SCI mice and humans (Zhang et al., 2013). In SCI mice, it is possible to abolish immune suppressive reflexes by silencing VGluT2+ thoracic (T4–T8) SpINs using chemogenetics (Ueno et al., 2016). Here, to determine whether the aberrant cardiovascular reflexes that cause AD also involve the activation of thoracic SpINs, in vivo telemetry was used to continuously monitor HR and BP in T3 SCI mice, with or without chemogenetic silencing of thoracic VGluT2+ SpINs beginning at 14 dpi (Fig. 1A).
To transduce VGluT2+ SpINs throughout the thoracic spinal cord, unilateral intraspinal injections of AAV8-hSyn-DIO-hM4D(Gi)-mCherry (Gi DREADDs) were made into the T4 and T7 spinal segments of VGluT2-Cre mice at P14 (Fig. 1A; 1 μl/site/spinal segment, 0.25 mm lateral, 0.5 mm in depth; also see Materials and Methods). Immunohistochemical analyses indicate that 20–35% of all NeuN+ neurons ipsilateral to the AAV injection, mostly in the dorsal and intermediate gray matter (spinal laminae III–VIII and X) throughout the T4–T9 spinal cord, were transduced (Fig. 1B–H). To silence VGluT2+ SpINs transduced with Gi DREADDs, SCI mice were injected with CNO 2–3×/d (intraperitoneally; see Materials and Methods), starting at 14 dpi (i.e., when the frequency of AD normally begins to increase). Spontaneous daily episodes of AD (sADs) were quantified in awake and freely moving animals at baseline (−3 to −1 dpi) then daily from 3 to 30 dpi. Consistent with published data (Mayorov et al., 2001; Rabchevsky et al., 2012; Zhang et al., 2013; Brennan et al., 2021), sADs develop in T3 SCI mice within the first week post-injury, then increases in frequency throughout the evaluation period (Fig. 2A,B). Chemogenetic silencing of thoracic VGluT2+ SpINs, beginning at 14 dpi, blocks the delayed increase in sAD frequency (overall test, **p = 0.0054, saline vs CNO; mixed-effects model; Fig. 2A). Indeed, the number of daily AD events was not different between groups at 1 or 2 weeks post-injury but was significantly reduced after injecting CNO (average number of daily AD events in CNO vs saline group: 2.350 ± 0.4137 vs 6.238 ± 0.9792 during week 3 post-injury, p = 0.0068; and 2.305 ± 0.3756 vs 8.019 ± 1.2689 during week 4 post-injury, p = 0.002; Fig. 2B). This blockade of delayed sADs resulted in a significant reduction in the maximum number of dysreflexic events between 14 and 30 dpi (CNO vs saline, p < 0.0001; Fig. 2C).
We hypothesized that the reduction in sAD frequency in SCI mice injected with CNO was caused by silencing thoracic VGluT2+ SpIN networks that control splanchnic vascular tone. If true, then neuronal activity should be reduced in spinal gray matter regions containing sympathetic neurons, and the resting systemic BP should decrease in SCI animals after injecting CNO to silence SpINs (SCI + Gi DREADDs + CNO group). To test this hypothesis, spinal cord cross-sections were immunostained for FosB, a marker of neuronal activity (Lyons and West, 2011). FosB+ neurons were counted throughout laminae I–IV and V–X (Niraula et al., 2018; Brennan et al., 2021). SCI did not affect baseline neuronal activity in laminae I–IV (Fig. 3A,B, quantified in E; p = 0.8196); but, within laminae V–X, where sympathetic preganglionic neurons and most sympathetic interneurons reside, neuronal activity consistently increased after SCI (Fig. 3A,B, quantified in F; sham vs T3 SCI + Gi DREADDs without CNO, p = 0.0065). When thoracic VGluT2+ SpINs were silenced (i.e., after CNO injection), the number of active neurons was significantly reduced compared with SCI controls (Fig. 3C,F; T3 SCI + Gi DREADDs without CNO vs T3 SCI + Gi DREADDs with CNO, p = 0.0002).
Injecting CNO in sham-injured mice activates Gq DREADD transduced VGluT2+ thoracic spinal cord neurons in intermediate gray matter. A, B, Representative images of Gq DREADD (red) and FosB (green) labeling in spinal cord (T7) from mice that received bilateral intraspinal injections of AAV8-Gq DREADDs then were injected systemically with saline (A) or CNO (B). Inset squares (yellow) for each image are magnified on the right. C–E, Lamina-specific quantification of DREADD+ cells (C), FosB+ cells (D), and DREADD+FosB+ double-positive cells (E). F, Quantification of the percentage of FosB+ cells that are also DREADD+. C–F, Two-way ANOVA with Bonferroni post hoc tests; n = 3 mice/group. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Data are related to Figure 3. Download Figure 3-1, TIF file (29.7MB, tif) .
We also sought to characterize the phenotype, location, and function of VGluT2 neuronal populations that were targeted by DREADDs. However, technical and logistical barriers precluded further anatomic studies with the original tissue samples used in Figure 3. Instead, we analyzed tissues from a separate cohort of mice that were prepared as part of an independent research project. In those mice, bilateral injections of AAV8-Gq DREADDs were made at the T7 spinal level only, although the coordinates and injection volumes were identical to those used in Figure 3. These data illustrate that targeting intraspinal injections in the intermediate thoracic gray matter (0.25 mm lateral from the spinal midline and 0.5 mm deep) transduced VGluT2+ neurons located mostly in spinal laminae III–VIII and X. Also, CNO injections in these mice preferentially activated VGluT2+ neurons in spinal laminae VII and X, and 60–90% of activated (FosB+) VGluT2+ neurons were DREADD+ (Extended Data Fig. 3-1).
Silencing VGluT2+ SpINs consistently reduced resting blood pressure (Fig. 3G; after vs before CNO, p = 0.0008) with a corresponding drop in resting HR (Fig. 3H; after vs before CNO, p = 0.028). Importantly, we were able to phenocopy the neuronal activation that occurs within laminae V–X after SCI in sham mice by activating VGluT2 SpINs using Gq DREADDS (Fig. 3D, quantified in E, F; sham vs sham + Gq DREADDS + CNO, p = 0.0092). Together, the data in Figures 2 and 3 show that after SCI, thoracic VGluT2+ SpINs control activity within the spinal sympathetic neuronal networks that regulate vasomotor tone and the subsequent development of maladaptive cardiovascular reflexes.
Chemogenetic silencing of thoracic spinal VGluT2+ interneurons blocks autonomic dysreflexia triggered by activation of pelvic visceral afferents
After high-level SCI, episodes of AD are triggered when innocuous or nociceptive stimuli originating below the level of injury reflexively activate sympathetic vasomotor neurons leading to vasoconstriction and a rise in blood pressure. As an example, CRD activates CGRP+ nociceptive afferents in the lumbar spinal cord, which in turn activate at-level dorsal gray commissure (DGC) propriospinal axons that project rostrally to innervate thoracic sympathetic preganglionic neurons (Hou et al., 2008). To test whether thoracic VGluT2+ SpINs are involved in the intersegmental relay of visceral afferent volley between the lumbar spinal cord and sympathetic preganglionic neurons in the thoracic spinal cord, CRD was used to trigger AD in SCI mice in which thoracic VGluT2+ SpINs were transduced with Gi DREADDs (Figs. 1–3). Cardiovascular responses were measured in the same mice before and after CNO injection. Consistent with published data (Cameron et al., 2006; Inskip et al., 2009; Zhang et al., 2013; Mironets et al., 2018), CRD triggered exaggerated and prolonged hypertensive episodes in SCI mice (Fig. 4A, representative BP trace, C, change in BP from baseline); however, the ability of CRD to elicit AD was abolished when thoracic VGluT2+ SpINs were silenced (Fig. 4B, representative BP recording, quantified in C; after vs before CNO, ap < 0.0001). Importantly, CRD-induced hypertension was not affected when CNO was injected into VGluT2-cre SCI mice without intraspinal Gi DREADDs (Fig. 4D), nor was their resting BP or HR altered by CNO (Fig. 4E,F), confirming that CNO does not exert off-target effects on the heart, vasculature, or cardiovascular neural networks.
VGluT2+ thoracic spinal interneurons control the synaptic balance within spinal autonomic networks after SCI
After high-level SCI, structural remodeling of sensory and propriospinal circuitry (Cameron et al., 2006; Zinck et al., 2007; Hou et al., 2009; Ueno et al., 2016; Mironets et al., 2018; Brennan et al., 2021) and changes in the ratio of excitatory and inhibitory synapses (Llewellyn-Smith and Weaver, 2001, Llewellyn-Smith et al., 2006; Brennan et al., 2021) occur throughout the spinal cord below the level of injury. This structural plasticity correlates with an increase in the frequency and magnitude of activation of reflexes that control cardiovascular and immunologic functions (Cameron et al., 2006; Hou et al., 2008; Brennan et al., 2021). Since blockade of excitatory neurotransmission in the brain can prevent the formation of new dendritic spines and axon sprouting (Engert and Bonhoeffer, 1999; Toni et al., 1999; Wibrand et al., 2006), we tested whether chemogenetic silencing of VGluT2+ thoracic SpINs would block the post-injury induction of axonal and synaptic plasticity within spinal autonomic networks.
First, the number of excitatory (VGluT2) and inhibitory (VGAT) presynaptic puncta were quantified in the IML of sham control and SCI mice. Recent data show a progressive increase in excitatory synaptogenesis in the IML beginning 2 weeks after a severe high-level SCI (Brennan et al., 2021). Consistent with those data, we observed that at 34 dpi, significantly more VGluT2+ puncta exist throughout the thoracic IML in SCI mice compared with sham-operated control mice (Fig. 5A,B, quantified in I; p = 0.034), while the number of VGAT+ puncta are reduced in the same regions of IML (Fig. 5E,F, quantified in J; p = 0.0337). Collectively, this caused the ratio of excitatory to inhibitory puncta to increase more than twofold (Fig. 5K; p = 0.0152).
Simultaneously, a group of SCI mice that had been transduced with Gi DREADDs received CNO injections beginning at 14 dpi. Chemogenetic silencing of thoracic VGluT2+ SpINs in these mice blocked the post-injury increase in the number of excitatory puncta (Fig. 5A,C, quantified in I; sham vs T3 SCI + Gi DREADDS, p = 0.0442; sham vs T3 SCI + Gi DREADDS + CNO, p = 0.9982) and partially reversed the SCI-dependent decrease in VGAT+ inhibitory puncta (Fig. 5E,G, quantified in J; sham vs T3 SCI + Gi DREADDs, p = 0.0337; sham vs T3 SCI + Gi DREADDS + CNO, p = 0.2793). Overall, chemogenetic silencing of VGluT2+ thoracic SpINs in the injured spinal cord preserved normal ratios of excitatory to inhibitory puncta in the IML (Fig. 5K; T3 SCI + Gi DREADDs vs T3 SCI + Gi DREADDs + CNO, p = 0.0432; sham vs T3 SCI + Gi DREADDs + CNO, p = 0.0905).
After SCI, diverse visceral and somatic stimuli cause daily episodic activation of intersegmental thoracic neuronal networks. In the absence of any tonic descending modulation, this recurrent activation becomes exaggerated and the increased neuronal activity may be sufficient to induce structural plasticity within spinal autonomic networks. To determine whether enhanced plasticity within these networks is the result of enhanced neurotransmission through thoracic VGluT2+ SpINs, a separate cohort of uninjured (i.e., sham) VGluT2-Cre mice received intraspinal injections of excitatory AAV8-hSyn-DIO-hM3D(Gq)-mCherry DREADDs. To phenocopy the effects of exaggerated reflex activation after SCI, recurrent episodic activation of intersegmental thoracic SpINs was achieved by 1×/d injection of CNO for 2 weeks. As described above, more activated neurons were observed in laminae V–X of sham + Gq DREADDs + CNO mice compared with sham controls (Fig. 3D, FosB+ cells, quantified in F). This increase in neuronal activation was associated with a significant increase in the number of VGluT2+ puncta in the IML (Fig. 5A,D, quantified in I; sham vs T3 SCI + Gi DREADDs, p = 0.0385); however, VGAT puncta were unchanged (Fig. 5E,H, quantified in J; sham vs T3 SCI + Gq DREADDs + CNO, p = 0.5011). Accordingly, the ratio of excitatory to inhibitory puncta was unchanged relative to sham controls (Fig. 5K).
Post-injury sprouting of lumbar CGRP+ sensory fibers is inhibited after chemogenetic silencing of thoracic VGluT2+ spinal interneurons
After severe high-level SCI, structural plasticity also occurs in lumbar spinal circuitry. Notably, CGRP+ fibers sprout in dorsal and intermediate gray matter of the lumbar spinal cord (Ondarza et al., 2003; Zinck et al., 2007; Mironets et al., 2018; Brennan et al., 2021), and the magnitude of this sprouting correlates with the severity of an AD event elicited by triggering visceral–sympathetic reflexes (Weaver et al., 2001; Cameron et al., 2006; Brennan et al., 2021). Since chemogenetic silencing of thoracic VGluT2+ SpINs reduced AD (Figs. 2, 4), we tested whether this approach would also reduce lumbar CGRP+ primary afferent sprouting after SCI.
Consistent with published data, SCI significantly increased the sprouting of CGRP+ fibers into superficial and deep laminae of the lumbar dorsal horn (Fig. 6A,B, quantified in I, K; T3 SCI + Gi DREADDs vs sham, p = 0.0489 and 0.0145, respectively) and also into the DGC in lamina X of the L4 lumbar spinal cord (Fig. 6E,F, quantified in J; T3 SCI + Gi DREADDs vs sham, p = 0.0069). In contrast, after silencing thoracic VGluT2+ SpINs in the T4–T8 spinal cord, lumbar CGRP+ fibers were primarily restricted to laminae I–II (Fig. 6C,G, quantified in I,J,K; T3 SCI + Gi DREADDs + CNO vs T3 SCI + Gi DREADDs, p = 0.0212, 0.0476, and p = 0.0247 for laminae I–V, lamina X, and the longest axon, respectively).
Although selective activation of Gq DREADD-transduced thoracic VGluT2+ SpINs in uninjured spinal cord increased neuronal activation and structural remodeling in autonomic networks in thoracic spinal cord (Figs. 3F, 5I), this technique did not cause lumbar CGRP+ fibers to sprout in either laminae I–V (sham = 4931 ± 1045.78 pixel units vs sham + Gq DREADDs + CNO = 6892.25 ± 1390.26 pixel units; p = 0.6810; Fig. 6D,I) or lamina X (sham = 74 ± 21.82 pixel units vs sham + Gq DREADDs + CNO = 182.75 ± 20.20 pixel units; p = 0.0667; Fig. 6H,J). Collectively, data in Figures 4–6 indicate that after SCI, the increase in structural and synaptic plasticity that occurs throughout spinal autonomic networks is modulated in part via “gating” of excitatory neurotransmission between lumbar and thoracic spinal cord segments by thoracic VGluT2+ SpINs. However, boosting neuronal activity selectively in these neurons in an uninjured spinal cord is insufficient to promote lasting neuroplasticity of lumbar primary afferent nerve fibers, indicating that additional mechanisms contribute to lasting plasticity after SCI.
Chemogenetic silencing of thoracic VGluT2+ spinal interneurons limits the post-injury remodeling of spinal autonomic networks
An increase in the number of excitatory synapses within autonomic regions of thoracic spinal cord gray matter implies expanded connectivity within these regions. Using PRV, a retrograde neurotropic transsynaptic viral tracer, we previously showed that the receptive field and connectivity of organ-specific spinal autonomic networks increases after SCI (Ueno et al., 2016; Noble et al., 2018; Brennan et al., 2021). Here, we evaluated the role played by thoracic VGluT2+ SpINs in regulating structural remodeling of spinal autonomic networks. To do so, PRV was used to trace spinal–splenic neuronal connectivity in sham-injured and SCI mice, with and without post-injury silencing of thoracic VGluT2+ SpINs. At 24 dpi, PRV-GFP was injected into the spleen for retrograde labeling of the spinal–splenic sympathetic innervation. At 4 d postinjection (28 dpi), animals were perfused and spinal cords were analyzed for PRV+ neurons.
Consistent with previous data from our laboratory (Ueno et al., 2016; Brennan et al., 2021), in the intact spinal cord, PRV labeling was mostly found in SPNs located in the lateral horn of the thoracic gray matter (Fig. 7A). However, after SCI, the number and distribution of PRV+ neurons increased; PRV+ labeling extended into neurons located in intermediate and medial gray matter throughout the mid-thoracic spinal cord, both ipsilateral and contralateral to the spleen (left side of the mouse; Fig. 7B, quantified in E,F). After SCI, the number of PRV+ neurons in the ipsilateral lateral, intermediate, and medial zones, increased 236%, 486%, and 1606%, respectively. Notably, chemogenetic silencing of thoracic VGluT2+ interneurons reduced the number of PRV+ neurons (Fig. 7C, quantified in E,F) by 54%, 64%, and 85% in the ipsilateral lateral, intermediate, and medial zones, respectively, compared with injured levels without DREADD silencing. In summary, DREADD inhibition in the injured spinal cord blocks structural remodeling of spinal–splenic autonomic networks.
To determine whether the activation of excitatory thoracic VGluT2+ SpINs is sufficient to induce structural plasticity within spinal–splenic neuronal networks, a group of uninjured (i.e., sham) VGluT2-Cre mice received intraspinal injections of excitatory Gq DREADDs (as in Figs. 4, 5). Again, selective chemogenetic activation of thoracic VGluT2+ SpINs in uninjured spinal cord partially recapitulated the effects of SCI; PRV labeling increased 207%, 320%, and 1084% in the ipsilateral lateral, intermediate, and medial zones, respectively, relative to sham (Fig. 7D, quantified in E,F).
To determine whether thoracic VGluT2+ interneurons influence structural remodeling in spinal sympathetic networks that control other organs, PRV was used to label spinal–adrenal circuitry. Consistent with previous reports in uninjured rats (Strack et al., 1988), retrograde PRV labeling from the adrenal gland was concentrated in SPNs and interneurons in the lateral thoracic gray matter and intermediate gray matter at the T8 spinal segment (Fig. 8A, quantified in E,F), although PRV+ neurons were detected as far rostral as T4 and as far caudal as L3. As in the spinal–splenic circuit, 236%, 384%, and 644% more neurons were labeled with PRV after SCI, in the ipsilateral lateral, intermediate, and medial gray matter, respectively (Fig. 8B, quantified in E,F). More PRV+ neurons also were found rostral and caudal to T8, including in the lumbar spinal cord, a region where few adrenal-related PRV+ neurons exist in sham mice (Fig. 8G, quantified in H).
Chemogenetic manipulation of thoracic VGluT2+ SpINs produced effects similar to those described above for the spinal–splenic circuit. Specifically, silencing VGluT2+ thoracic SpINs after SCI limited the PRV labeling from the adrenal gland to SPNs and interneurons in the lateral and intermediate thoracic gray matter, much like in sham mice (Fig. 8C,E). Overall, silencing VGluT2+ SpINs reduced PRV+ labeling in the ipsilateral lateral, intermediate, and medial zones 56%, 62%, and 69%, respectively, compared with SCI control mice (T3 SCI +Gi DREADDs). Notably, PRV labeling at remote spinal levels (Fig. 8G, quantified in H), far removed from the location of Gi DREADD-transduced thoracic VGluT2+ interneurons, was reduced by silencing thoracic VGluT2+ SpINs; 63% fewer PRV+ neurons were found in lumbar spinal cord after silencing VGluT2+ thoracic SpINs. Conversely, selective activation of these same VGluT2+ SpINs in uninjured mice (using Gq DREADDs) caused both the number and relative distribution of PRV+ neurons to increase in the thoracic and lumbar spinal cord (Fig. 8D,G, quantified in G,H). Thus, repeat activation of thoracic VGluT2+ interneurons partially recapitulated the effects of SCI, indicating that the enhanced structural plasticity within autonomic networks after SCI is dependent, in part, on enhanced neurotransmission by excitatory SpINs.
VGluT2+ thoracic spinal interneurons become structurally and functionally integrated with remodeled spinal autonomic circuitry after SCI
When considered together, data in this manuscript indicate that after SCI, thoracic VGluT2+ SpINs regulate the development of structural plasticity and the exaggerated intraspinal sympathetic reflexes that cause systemic pathologies including AD and immune suppression. Indeed, silencing VGluT2+ SpINs inhibited SCI-induced structural and synaptic remodeling (Figs. 5–8) and blocked the progressive worsening of AD (Figs. 2, 4). Also, repeat chemogenetic activation of thoracic VGluT2+ SpINs in uninjured spinal cord was sufficient to provoke structural remodeling of spinal autonomic circuitry, even though this form of activity-dependent plasticity was consistently less and more variable than that observed after SCI. This was particularly obvious with respect to neuroplastic changes induced in the uninjured lumbar spinal cord (i.e., CGRP sprouting and PRV labeling of spinal–adrenal networks; Figs. 6, 8). Could the more robust effects of thoracic VGluT2+ SpINs observed in the injured spinal cord (relative to intact/sham spinal cord) indicate that these spinal neurons become structurally and functionally integrated with other spinal neurons that undergo structural remodeling and form new connections after SCI? To answer this question, PRV+VGluT2+ double-positive neurons were counted within the intermediate gray matter at the T7–T8 spinal level in injured spinal cord, with or without chemogenetic silencing. If after SCI, thoracic VGluT2+ SpINs become integrated into remodeling spinal circuitry in an activity-dependent manner, then silencing these interneurons after SCI should block a post-injury increase in the number of PRV+VGluT2+ double-positive neurons found throughout the intermediate gray matter (relative to sham).
Data in Figure 9A–C show intraspinal labeling of green PRV+ neurons retrogradely labeled from the adrenal gland, and red, thoracic VGluT2+ SpINs that were transduced with DREADDs at P14 but do not become integrated into the remodeling PRV+ spinal–adrenal circuitry. PRV+DREADD+ double-positive neurons are thoracic VGluT2+ SpINs that have become integrated with and synaptically connected to neurons within this remodeling spinal autonomic network. Consistent with our published data (Brennan et al., 2021; Fig. 7), after SCI, PRV labeling extends beyond the boundaries of the IML; many PRV+ neurons are found in both the lateral and the intermediate gray matter (approximately lamina VII). Many of these neurons are PRV+VGluT2+ double positive [Fig. 9A, quantified in D (2.25 ± 0.6292 neurons and 7.75 ± 1.25 neurons/ROI in the IML and lamina VII, respectively) and in E (16.67 ± 2.357% and 30.5 ± 2.466% of neurons/ROI in the IML and lamina VII, respectively)]. However, when VGluT2+ interneurons are silenced beginning at 14 dpi, PRV labeling is limited to lateral gray matter, and the number of PRV+DREADD+ double-positive neurons is significantly reduced compared with identical regions of the injured spinal cord without chemogenetic silencing (Fig. 9B, quantified in D; number of double-positive neurons in lamina VII compared with SCI without silencing was 1 ± 0.41 vs 7.75 ± 1.25, p < 0.0001). Chemogenetic activation of Gq DREADDs failed to recapitulate the SCI-induced increase in the number of PRV+VGluT2+ double-positive neurons within the T7–T8 lamina VII of the uninjured spinal cord [Fig. 9C, quantified in D (number of double positive neurons in lamina VII of Gq DREADD group vs SCI group, 1 ± 0.41 vs 7.75 ± 1.25, p < 0.0001) and E (percentage of PRV+ neurons that are also DREADD+ in lamina VII vs SCI, 6.5 ± 2.53% vs 30.5 ± 2.47%, p = 0.0161)]. Considered together, these data show that thoracic VGluT2+ SpINs become integrated with remodeling intraspinal circuitry in an activity-dependent manner. However, enhancing neuronal activity alone is insufficient to cause the formation of synaptic contacts between DREADD+VGluT2+ SpINs and neurons within the spinal–adrenal sympathetic networks.
If the enhanced integration of thoracic VGluT2+ SpINs into spinal autonomic circuitry is indeed critical for regulating the pathologic consequences of exaggerated spinal sympathetic reflexes, then silencing these same thoracic SpINs at later post-injury periods, after structural plasticity and AD worsening have developed, should have little or no lasting effect on AD. To test this hypothesis, we performed a CNO crossover/washout experiment. Two cohorts of mice were generated using a design identical to that shown in Figure 2. In cohort 1 (CNO Crossover), SCI mice received CNO injections beginning at 25 dpi (i.e., when frequent daily episodes of AD and structural plasticity are already firmly established). In cohort 2 (CNO Washout), SCI mice that had been receiving daily CNO injections from 14 to 24 dpi, stopped receiving CNO injections for 24 h at 25 dpi. A 3 d average of daily AD events, both before and after CNO crossover or washout, was tallied for each group. In the CNO Crossover group, where mice were experiencing frequent bouts of daily AD, silencing VGluT2+ interneurons for a single day reduced AD in every mouse (n = 6 mice; Fig. 9F; before CNO, 6.56 ± 1.02 events/d; vs with CNO, 2.11 ± 0.62 events/d; p = 0.0110). Daily episodes of AD returned to the preceding 3 d average after removing CNO (before CNO, 6.56 ± 1.02 events/d; vs after CNO, 5.83 ± 1.18 events/d; p = 0.348). Conversely, in the CNO Washout cohort, where chemogenetic silencing of thoracic VGluT2+ SpINs limits structural plasticity and reduces daily episodes of AD, stopping CNO injections for 1 d had no effect on daily episodic AD, either during the period of washout or after restoration of CNO (Fig. 9G; with CNO, 2.38 ± 0.50 events/d; vs without CNO, 2.86 ± 0.55 events/d; p = 0.8385). Thus, we can conclude that, within the first 4 weeks after SCI, thoracic VGluT2+ SpINs become functionally integrated into spinal autonomic circuitry, and that this integration is, in part, activity dependent.
Discussion
After SCI, the loss of supraspinal control over spinal sympathetic reflexes allows normally innocuous visceral or somatic stimuli to elicit exaggerated sympathetic reflexes. We and others have shown that these exaggerated reflexes become progressively worse over time and occur alongside structural remodeling of spinal autonomic circuitry (Krassioukov and Weaver, 1995; Weaver et al., 1997; Krenz and Weaver, 1998a,b; Llewellyn-Smith and Weaver, 2001; Weaver et al., 2001; Cameron et al., 2006; Llewellyn-Smith et al., 2006; Ackery et al., 2007; Hou et al., 2009; Ueno et al., 2016; Brennan et al., 2021). The mechanisms underlying this structural remodeling are not well defined.
Most preclinical data implicate lesion-derived cytokines and growth factors as the primary stimuli that promote maladaptive autonomic plasticity after SCI (Krenz et al., 1999; Cameron et al., 2006; Mironets et al., 2018, 2020). Recent data indicate that glial-derived matricellular proteins and their receptors may also promote maladaptive plasticity and pathologic dysautonomia after severe high-level SCI. Indeed, treating SCI mice with the Food and Drug Administration-approved drug gabapentin (GBP), which binds neuronal α2δ−1 calcium channel subunits (Eroglu et al., 2009), prevents multisegmental excitatory synaptogenesis and sprouting of the autonomic and sensory neurons that cause cardiovascular and immune dysfunction (Brennan et al., 2021). These benefits of GBP are likely explained by the ability of the drug to block astrocyte-secreted thrombospondin binding to neuronal α2δ−1 subunits (Christopherson et al., 2005; Eroglu et al., 2009). However, by binding voltage-gated neuronal α2δ−1 subunits, GBP also inhibits excitatory neurotransmission. Thus, GBP may also prevent structural remodeling of autonomic circuitry by blocking injury-induced activity-dependent plasticity.
Data in the current report provide new evidence that activity-dependent plasticity contributes to post-injury remodeling of autonomic circuitry, and that thoracic VGluT2+ SpINs are key cellular determinants that coordinate this process. Specifically, using chemogenetics, we show that silencing VGluT2+ SpINs prevents aberrant cardiovascular reflexes and the delayed onset of widespread SCI-induced structural plasticity. Also, simply activating VGluT2+ SpINs in uninjured mice, once daily for 3 weeks, phenocopies the anatomic and molecular changes that occur in spinal autonomic networks after SCI. However, these effects were not consistent or as robust as those caused by SCI, suggesting that additional mechanisms, including activated glia, likely participate in promoting autonomic plasticity after SCI.
Although chemogenetic silencing of VGluT2+ SpINs consistently reduced structural and functional plasticity within autonomic circuitry, these effects might be enhanced using newer viral vectors or controlled electrical stimulation. As our original intent was to preferentially target the sympathetic networks that we and others have predicted would comodulate inflammatory, neuroendocrine, and cardiovascular reflexes (Zhang et al., 2013; Ueno et al., 2016; Prüss et al., 2017), we injected DREADDs at only two spinal segments (T4 and T7) and only on one side of the spinal cord. Using this approach, we transduced ∼25–30% of ipsilateral glutamatergic SpINs between the T4 and T8 spinal levels. Perhaps more efficient transduction of both ipsilateral and contralateral SpINs over a greater rostrocaudal extent of the spinal cord would produce even greater or longer-lasting structural and functional changes in the spinal autonomic network.
Attempts to phenocopy injury-induced and activity-dependent plasticity by activating Gq DREADDs in uninjured mice produced modest effects, but technical limitations constrained the robustness of this approach. In uninjured mice, activating intraspinal Gq DREADDs caused significant hindlimb spasticity, often lasting up to 8 h after a single injection of CNO. Because we were concerned that repeat injections of CNO would stress mice and interrupt their sleep, mice were only injected with CNO 1×/d beginning at the onset of their “awake” phase. Although this single-injection paradigm still triggered activity in SpINs (and connected neurons) along with evidence of structural plasticity, the results likely underestimate the frequency and magnitude of recurrent spinal autonomic reflexes that are triggered each day by naturally occurring visceral and somatic stimuli. Since completing these studies, newer viral vector technology was developed that can more effectively transduce SpINs (Brommer et al., 2021). Future studies should incorporate these new vectors to see whether improving DREADD transduction efficiency can yield more robust effects than those described herein, especially with respect to activation-induced plasticity in uninjured spinal cord.
We must also consider a potential effect of age on our results. In this study, mice were injected with DREADDs and received spinal cord injuries between the ages of P56 and P70, corresponding with the earliest stages of sexual maturity and a period when some indices of spinal cord development may still be incomplete (Taft et al., 2006; Fu et al., 2013; Dutta and Sengupta, 2016). The corresponding age range in humans would be ∼15–27 years old (Dutta and Sengupta, 2016). Thus, data in the current report were generated on the background of a young and presumably resilient nervous system. Since outcomes from SCI and the efficacy of rehabilitation therapy decline with age (Scivoletto et al., 2003), it would be of interest in future studies to understand whether activity and neural plasticity in the autonomic nervous system can be modulated as effectively at more advanced ages.
It may be possible to limit neurotransmission more effectively within SpINs and subsequently their ability to promote circuit remodeling using intraspinal or epidural stimulation. When combined with physical rehabilitation, epidural stimulation promotes significant spinal reorganization with restoration of voluntary locomotion (van den Brand et al., 2012; Asboth et al., 2018). It is likely that sympathetic preganglionic neurons and their surrounding interneurons are similarly susceptible to epidural stimulation and that modulating their activity in real time could influence autonomic plasticity. In fact, a recent report showed that after severe high-level SCI in rats, excitatory spinal interneuronal relays from the lumbar spinal cord gate exaggerated hemodynamic reflexes and these reflexes can be controlled using epidural electrical stimulation (EES; Squair et al., 2021). This may explain why, in clinical studies, the reversal of abnormal vasopressor responses in SCI patients have been reported during EES of rostral lumbar spinal segments (Harkema et al., 2018). Thus, EES may be able to restore more than motor function. If epidural electrodes were implanted or transcutaneous electrodes were applied in/at the level of the thoracic spinal cord early after SCI in patients with high-level severe SCI, EES might effectively prevent structural remodeling in autonomic circuitry and the subsequent development of pathologic dysautonomia. Preserving normal autonomic function after SCI by selectively targeting the mechanisms underlying injury-induced plasticity in excitatory SpINs, could mitigate or prevent various post-SCI complications linked to dysautonomia including autonomic dysreflexia, immune suppression, gut dysbiosis, and metabolic disease (Zhang et al., 2013; Kigerl et al., 2016, 2020; Wallace et al., 2019; Carpenter et al., 2020; Brennan et al., 2021).
A better understanding of the molecular phenotype and function of autonomic-related SpINs may lead to the development of novel strategies to modulate pathologic autonomic output after SCI. Unfortunately, apart from their excitatory phenotype, we know little about the subtypes of SpINs that we silenced or the neurons that they form synapses with after SCI. SpINs comprise a vast range of transcriptionally defined neuronal types with unique properties and connectivity (Sathyamurthy et al., 2018). These include long and short propriospinal neurons, with ascending and descending projections, and local SpINs with ipsilateral or commissural projections (Gebber and McCall, 1976; Shik and Orlovsky, 1976; Zholudeva et al., 2021). To unequivocally identify the phenotype of thoracic VGluT2+ SpINs that we silenced, and their associated neuronal networks, would require extensive anatomic tracing/morphologic characterization together with electrophysiological and gene expression analyses. However, based on where we injected AAV (dorsal and dorsal–intermediate spinal cord gray matter) and the fact that the anatomic and functional effects of silencing VGluT2+ SPNs were robust and consistent despite only transducing neurons across two spinal segments (T4 and T7) on one side of the spinal cord, we predict that we preferentially targeted SpINs that originate from ventrally (V) derived progenitors, specifically a class of long multisegmental V2a glutamatergic interneurons (Laliberte et al., 2019).
V2a interneurons (Chx10+) are rhythmically active neurons classically known for propagating locomotor commands to commissural interneurons involved with left–right coordination (Laliberte et al., 2019). Two classes of V2a interneurons have been defined in the cervical and lumbar spinal cord (Hayashi et al., 2018). Type I (Nfib+) V2a neurons, enriched in the medial gray matter, consist of three genetically distinct subsets and form recurrent networks with neighboring spinal neurons in the medial gray matter. Conversely, type II (Zfhx3+) V2a neurons are enriched in the lateral gray matter and consist of eight genetically distinct subsets that downregulate Chx10 and project to supraspinal structures (Hayashi et al., 2018). Since lateral fibers project along the entire length of the spinal cord, whereas medial fibers have shorter projections (Petko and Antal, 2000), it is conceivable that by silencing long-range type II V2a SpINs we blocked lumbar sensory afferent signaling to thoracic SPNs and blocked local (intrasegmental) circuit integration by silencing short type I V2a SpINs. Since we silenced neurons on only one side of the spinal cord, it is possible that we also silenced excitatory commissural SpINs that project bilaterally within and between spinal segments (Maxwell and Soteropoulos, 2020). These could include subclasses of long and short V0 (DBX1+EXV1+; Pierani et al., 2001; Lanuza et al., 2004; Talpalar et al., 2013) and short V3 (SIM1+) SpINs (Lanuza et al., 2004; Laliberte et al., 2019). Given that most published data characterize the phenotype and function of cervical and lumbar SpINs, primarily in the context of locomotor or respiratory function (Zholudeva et al., 2017; Jensen et al., 2019), future studies will need to determine whether these same neuronal classifications apply to autonomic related interneurons.
In conclusion, we show that the activity of VGluT2+ SpINs orchestrates structural and functional plasticity over long distances in the injured spinal cord, and that silencing these neurons suppresses maladaptive plasticity and blocks autonomic dysfunction after SCI. Ongoing work to identify the unique subclasses of SpINs driving post-SCI plasticity will likely reveal novel ways to modulate spinal circuitry and stabilize autonomic output after SCI.
Footnotes
This work was supported by Department of Defense Grant W81XWH-13-1-0358 (to P.G.P.); Craig H. Neilsen Foundation Grant 457267 (to F.H.B.); the Wings for Life Foundation (to F.H.B., P.G.P.); National Institutes of Health (NIH) Grants R01-NS-099532, R01-NS-083942, and R35-NS-111582 (to P.G.P.), and NIH Core Facility Grant P30-NS-104177; and the Ray W. Poppleton Endowment (to P.G.P.). J.M.S. is supported by the National Institutes of Neurological Disorders of NIH Grant R01-NS-118200-01; the National Institute of Disability, Independent Living and Rehabilitation Research Grant 90SI5020; the European Union (EU Era Net – Neuron Program) SILENCE Grant 01EW170A; Craig H. Neilsen Foundation Grant 596764; the Wings for Life Foundation; and the Hunt and Curtis endowment (to J.M.S.). J.M.S. is a Discovery Theme Initiative Scholar (Chronic Brain Injury) of The Ohio State University. The pseudorabies virus used was supplied by the NIH Center for Neuroanatomy with Neurotropic Viruses, which is supported by NIH Virus Center Grant P40-OD-010996. We thank Lori Hudson for assistance with animal husbandry.
The authors declare no competing financial interests.
References
- Ackery AD, Norenberg MD, Krassioukov A (2007) Calcitonin gene-related peptide immunoreactivity in chronic human spinal cord injury. Spinal Cord 45:678–686. 10.1038/sj.sc.3102020 [DOI] [PubMed] [Google Scholar]
- Agulhon C, Boyt KM, Xie AX, Friocourt F, Roth BL, McCarthy KD (2013) Modulation of the autonomic nervous system and behaviour by acute glial cell Gq protein-coupled receptor activation in vivo. J Physiol 591:5599–5609. 10.1113/jphysiol.2013.261289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asboth L, Friedli L, Beauparlant J, Martinez-Gonzalez C, Anil S, Rey E, Baud L, Pidpruzhnykova G, Anderson MA, Shkorbatova P, Batti L, Pagès S, Kreider J, Schneider BL, Barraud Q, Courtine GC (2018) Cortico–reticulo–spinal circuit reorganization enables functional recovery after severe spinal cord contusion. Nat Neurosci 21:576–588. 10.1038/s41593-018-0093-5 [DOI] [PubMed] [Google Scholar]
- Brennan FH, Noble BT, Wang Y, Guan Z, Davis H, Mo X, Harris C, Eroglu C, Ferguson AR, Popovich PG (2021) Acute post-injury blockade of α2δ-1 calcium channel subunits prevents pathological autonomic plasticity after spinal cord injury. Cell Rep 34:108667. 10.1016/j.celrep.2020.108667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brommer B, He M, Zhang Z, Yang Z, Page JC, Su J, Zhang Y, Zhu J, Gouy E, Tang J, Williams P, Dai W, Wang Q, Solinsky R, Chen B, He Z (2021) Improving hindlimb locomotor function by Non-invasive AAV-mediated manipulations of propriospinal neurons in mice with complete spinal cord injury. Nat Commun 12:781. 10.1038/s41467-021-20980-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron AA, Smith GM, Randall DC, Brown DR, Rabchevsky AG (2006) Genetic manipulation of intraspinal plasticity after spinal cord injury alters the severity of autonomic dysreflexia. J Neurosci 26:2923–2932. 10.1523/JNEUROSCI.4390-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano G, Sved AF, Rinaman L, Rabin BS, Card JP (2001) Characterization of the central nervous system innervation of the rat spleen using viral transneuronal tracing. J Comp Neurol 439:1–18. 10.1002/cne.1331 [DOI] [PubMed] [Google Scholar]
- Carpenter RS, Marbourg JM, Brennan FH, Mifflin KA, Hall JCE, Jiang RR, Mo XM, Karunasiri M, Burke MH, Dorrance AM, Popovich PG (2020) Spinal cord injury causes chronic bone marrow failure. Nat Commun 11:3702. 10.1038/s41467-020-17564-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA (2005) Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120:421–433. 10.1016/j.cell.2004.12.020 [DOI] [PubMed] [Google Scholar]
- Dutta S, Sengupta P (2016) Men and mice: relating their ages. Life Sci 152:244–248. 10.1016/j.lfs.2015.10.025 [DOI] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T (1999) Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature 399:66–70. 10.1038/19978 [DOI] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA (2009) Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139:380–392. 10.1016/j.cell.2009.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV (2004) Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci 24:2143–2155. 10.1523/JNEUROSCI.3547-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formento E, Minassian K, Wagner F, Mignardot JB, Le Goff-Mignardot CG, Rowald A, Bloch J, Micera S, Capogrosso M, Courtine G (2018) Electrical spinal cord stimulation must preserve proprioception to enable locomotion in humans with spinal cord injury. Nat Neurosci 21:1728–1741. 10.1038/s41593-018-0262-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Rusznák Z, Herculano-Houzel S, Watson C, Paxinos G (2013) Cellular composition characterizing postnatal development and maturation of the mouse brain and spinal cord. Brain Struct Funct 218:1337–1354. 10.1007/s00429-012-0462-x [DOI] [PubMed] [Google Scholar]
- Gebber GL, McCall RB (1976) Identification and discharge patterns of spinal sympathetic interneurons. Am J Physiol 231:722–733. 10.1152/ajplegacy.1976.231.3.722 [DOI] [PubMed] [Google Scholar]
- Guttmann L, Whitteridge D (1947) Effects of bladder distension on autonomic mechanisms after spinal cord injuries. Brain 70:361–404. 10.1093/brain/70.4.361 [DOI] [PubMed] [Google Scholar]
- Harkema SJ, Legg Ditterline BL, Wang S, Aslan S, Angeli CA, Ovechkin A, Hirsch GA (2018) Epidural spinal cord stimulation training and sustained recovery of cardiovascular function in individuals with chronic cervical spinal cord injury. JAMA Neurol 75:1569–1571. 10.1001/jamaneurol.2018.2617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, Hinckley CA, Driscoll SP, Moore NJ, Levine AJ, Hilde KL, Sharma K, Pfaff SL (2018) Graded arrays of spinal and supraspinal V2a interneuron subtypes underlie forelimb and hindlimb motor control. Neuron 97:869–884.e5. 10.1016/j.neuron.2018.01.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou S, Duale H, Cameron AA, Abshire SM, Lyttle TS, Rabchevsky AG (2008) Plasticity of lumbosacral propriospinal neurons is associated with the development of autonomic dysreflexia after thoracic spinal cord transection. J Comp Neurol 509:382–399. 10.1002/cne.21771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou S, Duale H, Rabchevsky AG (2009) Intraspinal sprouting of unmyelinated pelvic afferents after complete spinal cord injury is correlated with autonomic dysreflexia induced by visceral pain. Neuroscience 159:369–379. 10.1016/j.neuroscience.2008.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inskip JA, Ramer LM, Ramer MS, Krassioukov AV (2009) Autonomic assessment of animals with spinal cord injury: tools, techniques and translation. Spinal Cord 47:2–35. 10.1038/sc.2008.61 [DOI] [PubMed] [Google Scholar]
- Ippolito DM, Eroglu C (2010) Quantifying synapses: an immunocytochemistry-based assay to quantify synapse number. J Vis Exp (45):2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen VN, Seedle K, Turner SM, Lorenz JN, Crone SA (2019) V2a neurons constrain extradiaphragmatic respiratory muscle activity at rest. eNeuro 6:ENEURO.0492-18.2019. 10.1523/ENEURO.0492-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, Hall JC, Wang L, Mo X, Yu Z, Popovich PG (2016) Gut dysbiosis impairs recovery after spinal cord injury. J Exp Med 213:2603–2620. 10.1084/jem.20151345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, Zane K, Adams K, Sullivan MB, Popovich PG (2020) The spinal cord-gut-immune axis as a master regulator of health and neurological function after spinal cord injury. Exp Neurol 323:113085. 10.1016/j.expneurol.2019.113085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krassioukov AV, Weaver LC (1995) Reflex and morphological changes in spinal preganglionic neurons after cord injury in rats. Clin Exp Hypertens 17:361–373. 10.3109/10641969509087077 [DOI] [PubMed] [Google Scholar]
- Krenz NR, Weaver LC (1998a) Changes in the morphology of sympathetic preganglionic neurons parallel the development of autonomic dysreflexia after spinal cord injury in rats. Neurosci Lett 243:61–64. 10.1016/S0304-3940(98)00101-3 [DOI] [PubMed] [Google Scholar]
- Krenz NR, Weaver LC (1998b) Sprouting of primary afferent fibers after spinal cord transection in the rat. Neuroscience 85:443–458. 10.1016/S0306-4522(97)00622-2 [DOI] [PubMed] [Google Scholar]
- Krenz NR, Meakin SO, Krassioukov AV, Weaver LC (1999) Neutralizing intraspinal nerve growth factor blocks autonomic dysreflexia caused by spinal cord injury. J Neurosci 19:7405–7414. 10.1523/JNEUROSCI.19-17-07405.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laliberte AM, Goltash S, Lalonde NR, Bui TV (2019) Propriospinal neurons: essential elements of locomotor control in the intact and possibly the injured spinal cord. Front Cell Neurosci 13:512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanuza GM, Gosgnach S, Pierani A, Jessell TM, Goulding M (2004) Genetic identification of spinal interneurons that coordinate left-right locomotor activity necessary for walking movements. Neuron 42:375–386. 10.1016/S0896-6273(04)00249-1 [DOI] [PubMed] [Google Scholar]
- Llewellyn-Smith IJ, Weaver LC (2001) Changes in synaptic inputs to sympathetic preganglionic neurons after spinal cord injury. J Comp Neurol 435:226–240. 10.1002/cne.1204 [DOI] [PubMed] [Google Scholar]
- Llewellyn-Smith IJ, Weaver LC, Keast JR (2006) Effects of spinal cord injury on synaptic inputs to sympathetic preganglionic neurons. Prog Brain Res 152:11–26. [DOI] [PubMed] [Google Scholar]
- Lucin KM, Sanders VM, Jones TB, Malarkey WB, Popovich PG (2007) Impaired antibody synthesis after spinal cord injury is level dependent and is due to sympathetic nervous system dysregulation. Exp Neurol 207:75–84. 10.1016/j.expneurol.2007.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucin KM, Sanders VM, Popovich PG (2009) Stress hormones collaborate to induce lymphocyte apoptosis after high level spinal cord injury. J Neurochem 110:1409–1421. 10.1111/j.1471-4159.2009.06232.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons MR, West AE (2011) Mechanisms of specificity in neuronal activity-regulated gene transcription. Prog Neurobiol 94:259–295. 10.1016/j.pneurobio.2011.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell DJ, Soteropoulos DS (2020) The mammalian spinal commissural system: properties and functions. J Neurophysiol 123:4–21. 10.1152/jn.00347.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayorov DN, Adams MA, Krassioukov AV (2001) Telemetric blood pressure monitoring in conscious rats before and after compression injury of spinal cord. J Neurotrauma 18:727–736. 10.1089/089771501750357663 [DOI] [PubMed] [Google Scholar]
- Mi D, Yuan Y, Zhang Y, Niu J, Wang Y, Yan J, Yang Y, Hu W (2019) Injection of Fluoro-Gold into the tibial nerve leads to prolonged but reversible functional deficits in rats. Sci Rep 9:9906. 10.1038/s41598-019-46285-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironets E, Osei-Owusu P, Bracchi-Ricard V, Fischer R, Owens EA, Ricard J, Wu D, Saltos T, Collyer E, Hou S, Bethea JR, Tom VJ (2018) Soluble TNFα signaling within the spinal cord contributes to the development of autonomic dysreflexia and ensuing vascular and immune dysfunction after spinal cord injury. J Neurosci 38:4146–4162. 10.1523/JNEUROSCI.2376-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironets E, Fischer R, Bracchi-Ricard V, Saltos TM, Truglio TS, O'Reilly ML, Swanson KA, Bethea JR, Tom VJ (2020) Attenuating neurogenic sympathetic hyperreflexia robustly improves antibacterial immunity after chronic spinal cord injury. J Neurosci 40:478–492. 10.1523/JNEUROSCI.2417-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mushahwar VK, Aoyagi Y, Stein RB, Prochazka A (2004) Movements generated by intraspinal microstimulation in the intermediate gray matter of the anesthetized, decerebrate, and spinal cat. Can J Physiol Pharmacol 82:702–714. 10.1139/y04-079 [DOI] [PubMed] [Google Scholar]
- Niraula A, Wang Y, Godbout JP, Sheridan JF (2018) Corticosterone production during repeated social defeat causes monocyte mobilization from the bone marrow, glucocorticoid resistance, and neurovascular adhesion molecule expression. J Neurosci 38:2328–2340. 10.1523/JNEUROSCI.2568-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble BT, Brennan FH, Popovich PG (2018) The spleen as a neuroimmune interface after spinal cord injury. J Neuroimmunol 321:1–11. 10.1016/j.jneuroim.2018.05.007 [DOI] [PubMed] [Google Scholar]
- Ondarza AB, Ye Z, Hulsebosch CE (2003) Direct evidence of primary afferent sprouting in distant segments following spinal cord injury in the rat: colocalization of GAP-43 and CGRP. Exp Neurol 184:373–380. 10.1016/j.expneurol.2003.07.002 [DOI] [PubMed] [Google Scholar]
- Petko M, Antal M (2000) Propriospinal afferent and efferent connections of the lateral and medial areas of the dorsal horn (laminae I-IV) in the rat lumbar spinal cord. J Comp Neurol 422:312–325. [DOI] [PubMed] [Google Scholar]
- Pierani A, Moran-Rivard L, Sunshine MJ, Littman DR, Goulding M, Jessell TM (2001) Control of interneuron fate in the developing spinal cord by the progenitor homeodomain protein Dbx1. Neuron 29:367–384. 10.1016/S0896-6273(01)00212-4 [DOI] [PubMed] [Google Scholar]
- Prüss H, Tedeschi A, Thiriot A, Lynch L, Loughhead SM, Stutte S, Mazo IB, Kopp MA, Brommer B, Blex C, Geurtz LC, Liebscher T, Niedeggen A, Dirnagl U, Bradke F, Volz MS, DeVivo MJ, Chen Y, von Andrian UH, Schwab JM (2017) Spinal cord injury-induced immunodeficiency is mediated by a sympathetic-neuroendocrine adrenal reflex. Nat Neurosci 20:1549–1559. 10.1038/nn.4643 [DOI] [PubMed] [Google Scholar]
- Rabchevsky AG, Patel SP, Lyttle TS, Eldahan KC, O'Dell CR, Zhang Y, Popovich PG, Kitzman PH, Donohue KD (2012) Effects of gabapentin on muscle spasticity and both induced as well as spontaneous autonomic dysreflexia after complete spinal cord injury. Front Physiol 3:329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathyamurthy A, Johnson KR, Matson KJE, Dobrott CI, Li L, Ryba AR, Bergman TB, Kelly MC, Kelley MW, Levine AJ (2018) Massively parallel single nucleus transcriptional profiling defines spinal cord neurons and their activity during behavior. Cell Rep 22:2216–2225. 10.1016/j.celrep.2018.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Fallon JH (1986) Fluoro-Gold: a new fluorescent retrograde axonal tracer with numerous unique properties. Brain Res 377:147–154. 10.1016/0006-8993(86)91199-6 [DOI] [PubMed] [Google Scholar]
- Scivoletto G, Morganti B, Ditunno P, Ditunno JF, Molinari M (2003) Effects on age on spinal cord lesion patients' rehabilitation. Spinal Cord 41:457–464. 10.1038/sj.sc.3101489 [DOI] [PubMed] [Google Scholar]
- Shik ML, Orlovsky GN (1976) Neurophysiology of locomotor automatism. Physiol Rev 56:465–501. 10.1152/physrev.1976.56.3.465 [DOI] [PubMed] [Google Scholar]
- Skinnider MA, Squair JW, Kathe C, Anderson AJ, Gautier M, Matson KJE, Milano M, Hutson TH, Barraud Q, Phillips AA, Foster LJ, La Manno G, Levine AJ, Courtine G (2021) Cell type prioritization in single-cell data. Nat Biotechnol 39:30–34. 10.1038/s41587-020-0605-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squair JW, et al. (2021) Neuroprosthetic baroreflex controls haemodynamics after spinal cord injury. Nature 590:308–314. 10.1038/s41586-020-03180-w [DOI] [PubMed] [Google Scholar]
- Strack AM, Sawyer WB, Marubio LM, Loewy AD (1988) Spinal origin of sympathetic preganglionic neurons in the rat. Brain Res 455:187–191. 10.1016/0006-8993(88)90132-1 [DOI] [PubMed] [Google Scholar]
- Taft RA, Davisson M, Wiles MV (2006) Know thy mouse. Trends Genet 22:649–653. 10.1016/j.tig.2006.09.010 [DOI] [PubMed] [Google Scholar]
- Talpalar AE, Bouvier J, Borgius L, Fortin G, Pierani A, Kiehn O (2013) Dual-mode operation of neuronal networks involved in left-right alternation. Nature 500:85–88. 10.1038/nature12286 [DOI] [PubMed] [Google Scholar]
- Toni N, Buchs P-A, Nikonenko I, Bron CR, Muller D (1999) LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature 402:421–425. 10.1038/46574 [DOI] [PubMed] [Google Scholar]
- Ueno M, Ueno-Nakamura Y, Niehaus J, Popovich PG, Yoshida Y (2016) Silencing spinal interneurons inhibits immune suppressive autonomic reflexes caused by spinal cord injury. Nat Neurosci 19:784–787. 10.1038/nn.4289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich-Lai YM, Herman JP (2009) Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci 10:397–409. 10.1038/nrn2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Brand R, Heutschi J, Barraud Q, DiGiovanna J, Bartholdi K, Huerlimann M, Friedli L, Vollenweider I, Moraud EM, Duis S, Dominici N, Micera S, Musienko P, Courtine GC (2012) Restoring voluntary control of locomotion after paralyzing spinal cord injury. Science 336:1182–1185. 10.1126/science.1217416 [DOI] [PubMed] [Google Scholar]
- Wallace DJ, Sayre NL, Patterson TT, Nicholson SE, Hilton D, Grandhi R (2019) Spinal cord injury and the human microbiome: beyond the brain-gut axis. Neurosurg Focus 46:E11. 10.3171/2018.12.FOCUS18206 [DOI] [PubMed] [Google Scholar]
- Watson C, Paxinos G, Kayalioglu G, Heise C (2009) Atlas of the mouse spinal cord. In: The spinal cord (Watson C, Paxinos G, Kayalioglu G, eds), pp 308–379. New York: Academic. [Google Scholar]
- Weaver LC, Cassam AK, Krassioukov AV, Llewellyn-Smith IJ (1997) Changes in immunoreactivity for growth associated protein-43 suggest reorganization of synapses on spinal sympathetic neurons after cord transection. Neuroscience 81:535–551. 10.1016/S0306-4522(97)00151-6 [DOI] [PubMed] [Google Scholar]
- Weaver LC, Verghese P, Bruce JC, Fehlings MG, Krenz NR, Marsh DR (2001) Autonomic dysreflexia and primary afferent sprouting after clip-compression injury of the rat spinal cord. J Neurotrauma 18:1107–1119. 10.1089/08977150152693782 [DOI] [PubMed] [Google Scholar]
- Weaver LC, Marsh DR, Gris D, Brown A, Dekaban GA (2006) Autonomic dysreflexia after spinal cord injury: central mechanisms and strategies for prevention. Prog Brain Res 152:245–263. [DOI] [PubMed] [Google Scholar]
- Wess J, Nakajima K, Jain S (2013) Novel designer receptors to probe GPCR signaling and physiology. Trends Pharmacol Sci 34:385–392. 10.1016/j.tips.2013.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wibrand K, Messaoudi E, Håvik B, Steenslid V, Løvlie R, Steen VM, Bramham C (2006) Identification of genes co-upregulate with arc during BDNF-induced long-term potentiation in adult rat dentate gyrus in vivo. Eur J Neurosci 23:1501–1511. 10.1111/j.1460-9568.2006.04687.x [DOI] [PubMed] [Google Scholar]
- Zhang Y, Guan Z, Reader B, Shawler T, Mandrekar-Colucci S, Huang K, Weil Z, Bratasz A, Wells J, Powell ND, Sheridan JF, Whitacre CC, Rabchevsky AG, Nash MS, Popovich PG (2013) Autonomic dysreflexia causes chronic immune suppression after spinal cord injury. J Neurosci 33:12970–12981. 10.1523/JNEUROSCI.1974-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zholudeva LV, Abraira VE, Satkunendrarajah K, McDevitt TC, Goulding MD, Magnuson DSK, Lane MA (2021) Spinal interneurons as gatekeepers to neuroplasticity after injury or disease. J Neurosci 41:845–854. 10.1523/JNEUROSCI.1654-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zholudeva LV, Karliner JS, Dougherty KJ, Lane MA (2017) Anatomical recruitment of spinal V2a interneurons into phrenic motor circuitry after high cervical spinal cord injury. J Neurotrauma 34:3058–3065. 10.1089/neu.2017.5045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinck ND, Rafuse VF, Downie JW (2007) Sprouting of CGRP primary afferents in lumbosacral spinal cord precedes emergence of bladder activity after spinal injury. Exp Neurol 204:777–790. 10.1016/j.expneurol.2007.01.011 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Injecting CNO in sham-injured mice activates Gq DREADD transduced VGluT2+ thoracic spinal cord neurons in intermediate gray matter. A, B, Representative images of Gq DREADD (red) and FosB (green) labeling in spinal cord (T7) from mice that received bilateral intraspinal injections of AAV8-Gq DREADDs then were injected systemically with saline (A) or CNO (B). Inset squares (yellow) for each image are magnified on the right. C–E, Lamina-specific quantification of DREADD+ cells (C), FosB+ cells (D), and DREADD+FosB+ double-positive cells (E). F, Quantification of the percentage of FosB+ cells that are also DREADD+. C–F, Two-way ANOVA with Bonferroni post hoc tests; n = 3 mice/group. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Data are related to Figure 3. Download Figure 3-1, TIF file (29.7MB, tif) .