

Abstract
10–23 DNAzyme has been extensively explored as a therapeutic and biotechnological tool, as well as in DNA computing. Faster cleavage or transformation is always needed. The present research displays a rational modification approach for a more efficient DNAzyme. In the catalytic core, amino, guanidinium and imidazolyl groups were introduced for its chemical activation through the adenine base. Among the six adenine residues, A9 is the unique residue that realizes all the positive effects; the 6-amino and 8-position of adenine and the 7-position of 8-aza-7-deaza-adenine could be used for the introduction of the functional groups. A12 is a new choice for catalytic improvement with an 8-substituent. Therefore, more active DNAzymes could be expected by this nucleobase-modified activation approach.
Chemical activation of 10–23 DNAzyme was realized at A9 modified with active functional groups amino, guanidinium, and imidazolyl groups.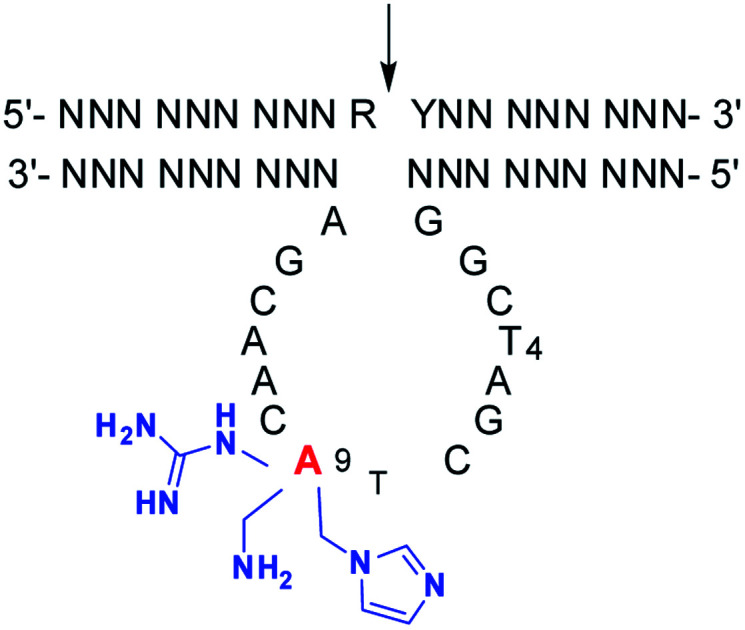
1. Introduction
Nucleic acids have been widely recognized as important modulating factors in biological systems. Both the natural (ribozymes, siRNA and non-coding RNAs)1–3 and synthetic functional nucleic acids (antisense, selected ribozymes, DNAzymes and aptamers)4–6 are being explored as therapeutics and biosensors, based on their interactions with specific target molecules. Optimization of the catalytic efficiency of ribozymes and DNAzymes, the silencing ability of siRNA and miRNA, and the binding affinity of aptamers are critically needed to meet the practical requirements. DNAzymes 10–23 and 8–17 are the most outstanding examples of DNA catalysts to date.7 Their great potential applications are being extensively explored, either as therapeutics to downregulate various RNAs (mRNA, miRNA),8,9 or as potential biosensor design platforms,10 by combining with PCR11 and nanoparticles12 as well as various photo-modulating and reporting groups13 for the analytes of interest, including RNA, proteins and metal ions.14 Therefore, many efforts have been devoted towards more efficient DNAzymes.15–18
The structural basis of these functional nucleic acids is essentially the same as that of protein–ligand interactions.19 Functional groups from nucleobases and metal ions have been demonstrated to be involved in the catalytic reactions of ribozymes20 and DNAzymes,21 and in binding interactions of aptamers,22 through their proton-transfer, hydrogen bonding, electrostatic and hydrophobic interactions. Learning from the extreme diversity of functions and structures of proteins, additional functional groups have been introduced for the expansion of the chemistry and functions of nucleic acids.23,24 Modified nucleoside triphosphates have been applied, as mimics of amino acids, in initial in vitro selection for new DNAzymes and ribozymes,25 towards simulating protein functions, although with limited success. For selected functional nucleic acids, post-selection chemical activation has been extensively practiced on DNAzymes and aptamers for better performance.7,26
Due to catalytic properties, small size and convenient synthesis of 10–23 DNAzyme, a faster cleavage or transformation has been pursued to meet its great potential applications. In order to find the structural basis for the functional improvement of 10–23 DNAzyme, attempts for the resolution of the molecular structure of 10–23 DNAzyme have been made with crystallization27 and theoretical simulation.28 It was suggested that three Mg2+ ions are responsible for the construction of the catalytic pocket as neutralizing species in the catalytic loop, and for the chemical reaction as general acid and base. Three stretches of nucleobases were supposed. The first six nucleotides are highly conserved, and the central C7–A12 is a backbone support for the active conformation, while the other conserved C13–A15 stretch locates near the cleavage site. Point mutations in the catalytic loop change the distance of three Mg2+ ions from the scissile phosphate at the target RNA, which is supposed to be the reason for the activity changes. Especially, point mutation at one nucleobase was demonstrated to induce local perturbations and a “domino” effect on the overall folding of the catalytic loop. However, little is known about the role and stacking mode of each nucleobase. Actually, each nucleobase has its own base-stacking interactions within the catalytic loop, as demonstrated by the effect of canonical residue replacement. Based on these calculations and experimental results, an effect of a random modification in the catalytic loop of 10–23 DNAzyme is not expected, without a rational structural basis.
The drastic negative effect from the deletion of functional groups (6-amino and 7-nitrogen atom) around adenine indicates that hydrogen-bonding interactions of the functional groups are extremely important in modulating the catalytically active conformation supported by the base-stacking and Mg2+-mediated electrostatic interactions. At the level of functional groups, all the adenine residues are also critically conserved.
The positive effect of the native functional groups around adenines might be strengthened by more active functional groups. Therefore, 2′-deoxyadenosine analogues with various functional groups were designed for adenine modifications. On the other hand, only six adenine residues were modified, and the interruption on the original base-stacking of adenines was thus minimized. As exemplified by compounds 1 and 2 with modifications on the 6-amino and 7-position of 8-aza-7-deaza-adenine,15,18 a positive effect was observed when they were located at A9. Here, further chemical activation of 10–23 DNAzyme was conducted, not only by the diversity of both functional groups and the linkages, but also with a new position, to explore the potential of A9 or other adenine residues as a positive modulating residue in the catalytic core. Amino, guanidinium, and imidazolyl groups, which are known to be active in protein enzymes, were introduced around adenine at its 6-amino or 8-position, as well as the 7-position of 8-aza-7-deaza-adenine with different linkages, respectively, as shown by compounds 3–9 (Scheme 1). Different from five other adenine residues, A9 was found to be a unique residue; its multiple-positioned functional groups were capable of improving the catalytic activity of 10–23 DNAzyme, and a guideline for more active DNAzyme was formed.
Scheme 1. 10–23 DNAzyme targeting VEGFR2 mRNA and the 2′-deoxyadenosine analogues for the modifications in the catalytic core. The bold letters in the substrate represents the RNA residues, and the arrow indicates the RNA phosphodiester linkage to be cleaved. 2′-Deoxyadenosine analogues 3–9 with multiple positioned functional groups to be used for the chemical modifications of 10–23 DNAzyme.

2. Experimental section
All commercially available reagents and solvents were used without further purification. TLC and silica gel (200–300 mesh) for flash column chromatography were purchased from Qingdao Chemicals Co. (Qingdao, China). NMR data were obtained on a JNM-ECA-400, with 1H NMR spectra recorded at 400 MHz, 13C NMR at 200 MHz, and 31P NMR at 160 MHz. All chemical shifts are reported in parts per million (ppm), and coupling constant (J) values in hertz (Hz), with DMSO-d6 or CDCl3 as the solvent. HRMS were measured on an Agilent TOF G6230A (Agilent Technologies, USA).
2.1. 8-(3-Trifluoroacetylaminopropylamino)-2′-deoxyadenosine (7a)
To the solution of 8-bromo-2′-deoxyadenosine (3.3 g, 10 mmol) in DMF (70 mL) was added a solution of 1,3-diaminopropane (7.4 g, 100 mmol) in isopropanol (50 mL). The reaction mixture was heated at 60 °C for 12 h. After cooling to r.t., triethylamine (2.0 g, 20 mmol) and ethyl trifluoroacetate (14 mL) were added to the reaction mixture, and it was stirred for 12 h. The product was purified by flash chromatography as a white solid (3.2 g, 76.2%). Rf (CH2Cl2 : CH3OH, 9 : 1) 0.4. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 1.82 (m, 2H, CH2CH2CH2), 1.99, 2.72 (2m, 2H, C2′-H), 3.26–3.45 (m, 4H, C5′-H, CH2CH2CH2), 3.66 (m, 2H, CH2CH2CH2), 3.91 (m, 1H, C4′-H), 4.42 (m, 1H, C3′-H), 5.33 (d, J = 3.6, 1H, C3′-OH), 5.82 (t, J = 4.9, 1H, C5′-OH), 6.35 (m, 1H, C1′-H), 6.52 (s, 2H, 6-NH2), 7.03 (t, J = 5.5, 1H, NH), 7.90 (s, 1H, C2-H), 9.46 (t, J = 5.3, 1H, NH). 13C NMR (200 MHz, DMSO-d6): δ (ppm) 29.9, 39.1, 39.5, 63.5, 73.3, 84.9, 89.3, 118.8, 150.4, 151.1, 152.8, 154.2. HRMS for C15H20F3N7O4 + H+, calcd: 420.1602; found: 420.1602 [M + H+].
2.2. 5′-O-(4,4′-Dimethoxytrityl)-8-(3-trifluoroacetylaminopropylamino)-2′-deoxyadenosine (7b)
Compound 7a (1.2 g, 2.8 mmol) was co-evaporated with dried pyridine three times, and the residue was dissolved in dried pyridine (2 mL). DMT-Cl (1.8 g, 5.6 mmol) was added in portions. After the reactant disappeared, the reaction was stopped by adding methanol (10 mL). The solution was concentrated for flash chromatography. The product was obtained as a white solid (0.7 g, 45%). Rf (CH2Cl2 : CH3OH, 9 : 1) 0.6. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 1.79 (m, 2H, CH2CH2CH2), 2.34 (m, 1H, C2′-H), 3.07–3.46 (m, 5H, C2′-H, CH2CH2CH2), 3.68–3.75 (m, 8H, C5′-H, 2OCH3), 3.91 (m, 1H, C4′-H), 4.42 (m, 1H, C3′-H), 5.90 (br, 1H, C3′-OH), 6.35 (m, 1H, C1′-H), 6.61 (s, 2H, 6-NH2), 6.93 (m, 4H, arom. H), 7.26–7.48 (m, 9H, arom. H), 7.90 (s, 1H, C2-H), 8.59 (m, 1H, NH), 9.48 (t, J = 5.3, 1H, NH). 13C NMR (200 MHz, DMSO-d6): δ (ppm) 29.3, 35.2, 37.1, 55.2, 55.9, 64.1, 64.6, 71.7, 83.4, 86.2, 87.9, 105.5, 113.9, 125.7, 127.5, 128.6, 130.5, 130.7, 136.5, 136.7, 145.9, 148.7, 153.2, 154.0, 155.0, 157.9, 158.8, 158.9. HRMS for C36H38F3N7O6 + H+, calcd: 722.2908; found: 722.2908 [M + H+].
2.3. 5′-O-(4,4′-Dimethoxytrityl)-N6-(di-n-butylaminomethylidenyl)-8-(3-trifluoroacetylaminopropylamino)-2′-deoxyadenosine (7c)
To the solution of 7b (0.5 g, 0.7 mmol) in methanol (3 mL) was added diacetyl N,N-di-n-butylformamide (0.84 mL, 0.7 mmol). After stirring for 4 h at r.t., the reaction mixture was concentrated for flash chromatography, and the product was obtained as a white solid (0.4 g, 66%). Rf (CH2Cl2 : CH3OH, 20 : 1) 0.6. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 0.91 (m, 6H, 2CH3), 1.32 (m, 4H, 2CH2), 1.56 (m, 4H, 2CH2), 1.83 (m, 2H, CH2), 2.13 (m, 1H, C2′-H), 1.79 (m, 2H, CH2CH2CH2), 2.34 (m, 1H, C2′-H), 3.18–3.58 (m, 11H, C5′-H, C2′-H, 2CH2, CH2CH2CH2), 3.71, 3.72 (2s, 6H, 2OCH3), 3.94 (m, 1H, C4′-H), 4.64 (m, 1H, C3′-H), 5.35 (d, J = 4.4, 1H, C3′-OH), 6.19 (t, J = 6.8, 1H, C1′-H), 6.76 (m, 4H, arom. H), 7.15–7.31 (m, 9H, arom. H), 7.90 (s, 1H, C2-H), 8.85 (m, 1H, NH), 9.48 (t, J = 5.7, 1H, NH). 13C NMR (200 MHz, DMSO-d6): δ (ppm) 14.5, 14.7, 20.1, 20.6, 29.2, 29.6, 31.5, 37.1, 38.0, 44.8, 51.4, 55.8, 55.9, 64.6, 71.7, 83.3, 86.1, 86.2, 113.9, 126.5, 127.4, 128.6, 130.4, 130.6, 136.5, 136.6, 245.9, 148.8, 153.1, 154.0, 158.2, 158.8, 158.9. HRMS for C45H55F3N8O6 + H+, calcd: 861.4269; found: 861.4269 [M + H+].
2.4. 5′-O-(4,4′-Dimethoxytrityl)-N6-(di-n-butylaminomethylidenyl)-8-(3-trifluoroacetylaminopropylamino)-2′-deoxyadenosine 3′-O-(2-cyanoethyl-N,N-diisopropylphosphoramidite) (7d)
Dried compound 7c (0.45 g, 0.54 mmol) was dissolved in CH2Cl2 containing diisopropylethylamine (1 mL), then 2-cyanoethyl N,N,N′,N′-tetraisopropylphosphoramidite (0.55 g, 1.1 mmol) and diisopropylammonium tetrazolide (0.17 g, 1.1 mmol) were added in sequence. The reaction mixture was diluted with CH2Cl2, washed with cold 5% aq. NaHCO3 and brine, and dried with anhydrous Na2SO4. The dried solution was concentrated for flash chromatography, and the product was obtained as a white foam (0.3 g, 52%). Rf (CH2Cl2 : CH3OH, 20 : 1) 0.8. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 0.82–1.58 (m, 26H, N(CH2CH2CH2CH3)2, N[CH(CH3)2]), 2.18–2.36 (m, 2H, C2′-H, CH2CH2CH2), 2.53–2.80 (m, 3H, C2′-H, OCH2CH2CN), 2.94–3.85 (m, 20H, CH2CH2CH2, C5′-H, OCH2CH2CN, N[CH(CH3)2], 2N(CH2CH2CH2CH3)2, 2OCH3), 4.04 (m, 1H, C4′-H), 4.79 (m, 1H, C3′-H), 5.74 (m, 1H, 6-NH), 6.51 (m, 1H, C1′-H), 6.78 (m, 4H, arom. H), 7.14–7.30 (m, 9H, arom. H), 8.26 (d, J = 1.7, C2-H), 8.64 (d, J = 2.2, CH), 9.15 (m, 1H, NH). 13C NMR (200 MHz, DMSO-d6): δ (ppm) 13.9, 14.1, 20.0, 20.2, 24.6, 24.9, 29.3, 29.6, 31.2, 36.1, 37.9, 38.8, 43.4, 43.5, 44.5, 51.5, 55.5, 62.0, 72.5, 72.7, 83.0, 84.7, 85.0, 87.0, 113.7, 117.5, 117.7, 123.8, 127.7, 128.2, 128.7, 128.8, 130.6, 130.7, 134.8, 143.7, 149.6, 152.2, 153.6, 155.4, 156.9, 159.1. 31P NMR (160 MHz, CDCl3): δ (ppm) 149.31, 149.81. HRMS for C54H72F3N10O7P + H+, calcd: 1061.5348; found: 1061.5347.
2.5. N 6-Benzoyl-5′-O-(4,4′-dimethoxytrityl)-8-[2-(imidazol-4-yl)ethylamino]-2′-deoxyadenosine (8b)
According to a published procedure,29 compound 8a (2.72 g, 3.7 mmol) was dissolved in anhydrous ethanol (30 mL), and histamine dihydrochloride (0.8 g, 4.4 mmol) and triethylamine (6 mL, 44.4 mmol) were added to the solution. The reaction mixture was stirred at 80 °C for 3 h, then it was concentrated for flash chromatography. The product was obtained as a white solid (2.05 g, 72.5%). Rf (CH2Cl2 : CH3OH, 20 : 1) 0.18. 1H NMR (400 MHz, DMSO-d6): δ 11.12 (br, 1H, 6-NH), 10.71 (br, 1H, 8-NH), 8.19 (s, 1H, C2-H), 8.02, 7.63–6.72 (m, 21H, arom. H, Im-H), 6.29 (t, J = 6.2 Hz, 1H, C1′-H), 5.42 (d, J = 4.0 Hz, 1H, C3′-OH), 4.61 (m, 1H, C3′-H), 4.01 (m, 1H, C4′-H), 3.72 (2s, 6H, OCH3), 3.58 (m, 2H, NCH2), 3.43–3.10 (m, C2′-H, C5′-H), 2.84 (m, 2H, CH2-Im), 2.18 (m, 1H, C2′-H). 13C NMR (200 MHz, DMSO-d6): δ 165.7, 158.5, 154.6, 154.1, 147.4, 145.5, 143.8, 136.2, 136.1, 135.1, 134.3, 132.5, 130.2, 128.9, 128.8, 128.2, 127.8, 127.1, 113.6, 85.9, 85.8, 71.3, 64.3, 55.5, 45.8, 42.9, 36.7. HRMS for C43H42N8O6 + H+, calcd: 767.3300; found: 767.3299.
2.6. N 6-Benzoyl-5′-O-(4,4′-dimethoxytrityl)-8-{2-[1-(tert-butoxycarbonyl)imidazol-4-yl]ethylamino}-2′-deoxyadenosine (8c)
To the solution of 8b (0.83 g, 1.1 mmol) in methanol (10 mL) was added di-tert-butyl dicarbonate (0.26 g, 1.2 mmol). The reaction mixture was stirred at r.t. and monitored by TLC. It was then concentrated for flash chromatography, and the product was obtained as a white solid (0.54 g, 58.3%). Rf (CH2Cl2 : CH3OH, 20 : 1) 0.32. 1H NMR (400 MHz, DMSO-d6): δ 10.69 (br, 1H, 8-NH), 8.19 (s, 1H, C2-H), 8.07, 8.02 (m, 3H, Im-H), 7.64–7.14, 6.80 (m, 18H, arom. H), 6.27 (t, J = 6.8 Hz, 1H, C1′-H), 5.40 (d, J = 4.2 Hz, 1H, C3′-OH), 4.61 (m, 1H, C3′-H), 3.99 (m, 1H, C4′-H), 3.72 (s, 6H, 2OCH3), 3.59 (m, 2H, NCH2), 3.42–3.12 (m, 3H, C5′-H, C2′-H), 2.83 (t, J = 7.2 Hz, 2H, CH2-Im), 2.18 (m, 1H, C2′-H), 1.52 (s, 9H, 3CH3). 13C NMR (200 MHz, DMSO-d6): δ 165.6, 158.5, 154.5, 154.1, 147.5, 147.2, 145.5, 143.9, 141.4, 137.2, 136.2, 136.1, 134.4, 132.5, 130.2, 128.9, 128.8, 128.2, 127.8, 127.1, 114.1, 113.6, 86.0, 85.8, 85.5, 71.3, 64.2, 55.5, 46.2, 42.3, 36.7, 27.9. HRMS for C48H50N8O8 + H+, calcd: 867.3824; found: 867.3821.
2.7. N 6-Benzoyl-5′-O-(4,4′-dimethoxytrityl)-8-{2-[1-(tert-butoxycarbonyl)imidazole-4-yl]ethylamino}-2′-deoxyadenosine 3′-O-(2-cyanoethyl-N,N-diisopropylphosphoramidite) (8d)
With the same procedure for compound 7d, compound 8c (0.54 g, 0.71 mmol) was converted to compound 8d, with the 2-cyanoethyl N,N,N′,N′-tetraisopropylphosphoramidite (0.12 mL) in the presence of diisopropylammonium tetrazolide (0.18 g, 1.06 mmol). After stirring for 40 min at r.t., another portion of the phosphoramidite reagent (0.5 mL) was added. After flash chromatography, the product was obtained as a white foam (0.53 g, 80%). 1H NMR (400 MHz, CDCl3): δ 9.05 (br, 1H, 8-NH), 8.54 (s, 1H, C2-H), 8.07 (m, 2H, arom. H), 7.89 (m, 1H, Im-H), 7.58–7.48 (m, 3H, arom. H), 7.33 (m, 1H, H-Im), 7.28–7.18 (m, 9H, arom. H), 6.79 (m, 4H, arom. H), 6.24 (m, 1H, C1′-H), 4.82 (m, 1H, C3′-H), 4.28–4.09 (m, 3H, C4′-H, C5′-H), 3.30–3.90 (m, 13H, 2OCH3, OCH2CH2CN, C2′-H, 2NH, NCH2), 2.81–2.56 (m, 5H, CH2-Im, OCH2CH2CN, C2′-H), 1.60 (s, 9H, tBu), 1.16–1.01 (m, 12H, 4CH3). 13C NMR (200 MHz, CDCl3): δ 165.0, 158.7, 153.5, 153.4, 152.5, 148.9, 147.0, 144.4, 144.0, 140.9, 136.6, 135.4, 134.7, 132.2, 130.3, 130.2, 128.7, 128.4, 128.0, 127.2, 122.0, 117.6, 117.5, 117.0, 113.7, 113.2, 86.7, 85.5, 85.3, 85.2, 83.7, 83.6, 73.7, 73.1, 73.0, 62.8, 62.7, 58.6, 58.4, 58.2, 55.3, 46.2, 45.7, 45.4, 43.4, 42.5, 37.4, 28.0, 27.6, 24.7, 24.6, 23.1, 23.0, 22.3, 20.4, 20.2. 31P NMR (CDCl3): 149.68, 149.25. HRMS for C57H67N10O9P + H+, calcd: 1067.4903; found: 1067.4898.
2.8. 5′-O-(4,4′-Dimethoxytrityl)-7-(3-aminopropyl)-8-N-7-deaza-2′-deoxyadenosine (9b)
The solution of 9a (2.72 g, 3.85 mmol)25 in conc. aq. ammonia (15 mL) and aq. methylamine (15 mL, 40%) was sealed in a stainless steel bottle. It was heated at 55 °C for 1 h. The solution was cooled and concentrated to obtain a colourless solid 9b (2.06 g, 88%). It was used in the next step without further purification.
2.9. 5′-O-(4,4′-Dimethoxytrityl)-7-[3-(N,N′-di-tert-butoxycarbonylguanidinopropyl)]-8-N-7-deaza-2′-deoxyadenosine (9c)
To a solution of N,N′-di-Boc-N′′-triflylguanidine (0.28 g, 0.72 mmol) in CH2Cl2 (10 mL) containing triethylamine (0.1 mL) at 0 °C was added 9b (0.43 g, 0.71 mmol). The mixture was stirred at 0 °C for 1 h, followed by stirring at r.t. for 1 h. Then, it was diluted with CH2Cl2, washed with sat. aq. NaHCO3 and brine, and dried with anhydr. Na2SO4. The solution was concentrated for flash chromatography to obtain 9c as a white foam (0.45 g, 75.1%). Rf (CH2Cl2 : CH3OH, 9 : 1) 0.65. 1H NMR (400 MHz, DMSO-d6): δ 11.55 (s, 1H, NH-Boc), 8.21 (s, 1H, C2-H), 8.20 (m, 1H, NH), 7.29, 7.17–7.08, 6.74–6.64 (m, 13H, arom. H), 6.58 (m, 1H, C1′-H), 5.23 (d, J = 4.8 Hz, 1H, C3′-OH), 4.55 (m, 1H, C4′-H), 3.96 (m, 1H, C3′-H), 3.70, 3.69 (2s, 6H, 2OCH3), 3.40–2.80 (m, 5H, NCH2CH2CHNH, C2′-H), 2.74 (m, 1H, C2′-H), 2.28 (m, 1H, C2′-H), 1.58 (m, 2H, NCH2CH2CH2NH), 1.44, 1.29 (2s, 18H, 2C(CH3)3). 13C NMR (200 MHz, DMSO-d6): δ 163.8, 158.6, 158.4, 156.4, 155.8, 155.4, 152.6, 145.6, 145.4, 136.3, 136.0, 130.1, 128.1, 126.9, 113.4, 99.2, 86.0, 85.6, 83.7, 83.4, 78.7, 71.9, 65.7, 55.4, 46.2, 38.9, 28.5, 28.1, 27.9, 25.7, 12.2. HRMS for C45H56N8O9 + Na+, calcd: 875.4062; found: 875.4060.
2.10. 5′-O-(4,4′-Dimethoxytrityl)-N6-(dimethylaminomethylidenyl)-7-[3-(N,N′-di-tert-butoxycarbonylguanidinopropyl)]-8-N-7-deaza-2′-deoxyadenosine (9d)
Diacetyl N,N-dimethylformamide (1.75 mL, 1.47 mmol) was added to the solution of 9c (1.13 g, 1.3 mmol) in methanol (40 mL). After stirring at r.t. for 2 h, the solution was concentrated for flash chromatography. The product 9d was obtained as a white foam (1.06 g, 92.1%). Rf (CH2Cl2 : CH3OH, 20 : 1) 0.26. 1H NMR (400 MHz, DMSO-d6): δ 11.55 (s, 1H, NH-Boc), 8.98 (s, 1H, CH), 8.46 (s, 1H), 8.23 (t, J = 5.8 Hz, 1H), 7.29–7.08 (m, 9H, arom. H), 6.74–6.56 (m, 5H, arom. H, C1′-H), 5.24 (d, J = 5.2 Hz, 1H, C3′-OH), 4.57 (m, 1H C4′-H), 3.96 (dt, J = 7.0, 4.4 Hz, 1H, C3′-H), 3.69 (m, 6H, 2-OCH3), 3.34 (m, 2H, C5′-H), 3.10–2.75 (m, 8H, C5′-H, CH2CH2CH2N), 2.45–2.40 (m, 6H, 2CH3), 2.30 (m, 1H, C2′-H), 1.72 (m, 1H, C2′-H), 1.43–1.39 (m, 18H, 6CH3). 13C NMR (200 MHz, DMSO-d6): δ 207.1, 163.8, 162.8, 158.4, 158.3, 157.9, 156.0, 155.8, 152.6, 147.2, 145.7, 136.1, 130.2, 130.0, 128.1, 126.9, 113.4, 113.3, 106.5, 86.0, 85.6, 83.8, 83.5, 78.7, 71.9, 65.8, 55.4, 55.3, 46.2, 41.4, 38.9, 35.2, 31.2, 28.5, 28.2, 28.1, 26.2, 12.2. HRMS for C48H61N9O9 + Na+, calcd: 930.4484; found: 930.4493.
2.11. 5′-O-(4,4′-Dimethoxytrityl)-N6-(dimethylaminomethylidenyl)-7-[3-(N,N′-di-tert-butoxycarbonylguanidinopropyl)]-8-N-7-deaza-2′-deoxyadenosine (9e)
Compound 9d (0.53 g, 0.58 mmol) was dissolved in CH2Cl2 (10 mL), followed by the addition of 2-cyanoethyl N,N,N′,N′-tetraisopropylphosphoramidite (0.4 mL) and diisopropylammonium tetrazolide (0.18 g, 1.06 mmol). The reaction mixture was stirred at r.t. for 0.5 h, then it was washed with cold 5% NaHCO3 and brine, then dried with anhydr. Na2SO4. The solution was concentrated for flash chromatography to obtain the product 9e as a white foam (0.55 g, 82.5%). Rf (CH2Cl2 : CH3OH, 20 : 1) 0.67. 1H NMR (400 MHz, CDCl3): δ 11.51 (s, 1H, NH-Boc), 8.84 (s, 1H, C8-H), 8.49 (s, 1H, C2-H), 8.27 (m, 1H, NH), 7.29–7.08 (m, 12H, arom. H, CH), 6.74–6.56 (m, 5H, arom. H, C1′-H), 4.85 (m, 1H, C4′-H), 4.25 (m, 1H, C3′-H), 3.92–3.40 (m, 12H, 2OCH3, C5′-H, OCH2CH2CN, CH2CH2CH2NH), 3.30–2.94 (m, 10H, OCH2CH2CN, CH2CH2CH2N, 2CH3), 2.62 (m, 1H, C2′-H), 2.46 (m, 1H, C2′-H), 1.92 (m, 2H, CH2CH2CH2N), 1.50, 1.47 (18H, 2s, tBu), 1.18 (m, 14H, 2CH(CH3)2). 13C NMR (200 MHz, CDCl3): δ 163.8, 162.8, 158.3, 157.0, 156.2, 155.7, 153.4, 147.3, 145.0, 136.2, 130.2, 128.3, 127.7, 126.6, 117.6, 113.0, 107.2, 86.0, 85.4, 85.2, 84.4, 83.1, 79.3, 64.6, 64.4, 58.7, 58.5, 55.2, 43.3, 41.3, 41.0, 35.3, 28.4, 28.1, 26.6, 24.8, 24.7, 24.6, 24.5, 20.5, 20.5, 20.3, 20.2. 31P NMR (160 MHz, CDCl3): δ (ppm) 149.30, 149.15. HRMS for C57H78N11O10P + Na+, calcd: 1130.5563; found: 1130.5556.
2.12. Oligonucleotides
Phosphoramidites of canonical and modified nucleosides were used for DNA synthesis on a 392 DNA/RNA synthesizer (Biosystem, USA). The coupling time for the modified residues was extended to 3 min. The sequences were cleaved from CPG and deprotected with conc. aq. ammonia by incubation in a sealed tube at 55 °C for 16 h. The solution was concentrated for denaturing PAGE (20%, 8 M urea). The product was extracted from the gel band with 0.5 M NH4OAc, and further desalted with Sep-Pak column. The oligonucleotides were lyophilized and stored at −30 °C. Characterization was performed with ESI-MS on a HTCS Oligo LC/MS system (Thermo Finnigan, USA). The sequence was 5′-d(tgc tct cca GGC TAG CTA CAA CGA cct gca cct)-3′ (lowercase letters for the two recognition arms and capital letters for the catalytic core). The modified DNAzyme (DZ) was named with the modified position and the residue (Table S1†).
2.13. Cleavage kinetics under single-turnover conditions
Cleavage reaction of DNAzymes was designed against the DNA–RNA–DNA substrate 3′-d(aac gag agg)-r(ua)-d(gga cgt gga)-5′ (purchased from Takara, Dalian, China), with the two RNA residues as the cleavage site of DNAzyme. The substrate was 32P-labeled as the reporting method of the reaction. Briefly, the substrate was incubated with γ-32P-ATP and T4 polymerase in the buffer at 37 °C for one hour. On a Sep-Pak column, the labelled DNAzyme was extracted from the reaction mixture and washed with sterilized and deionized water, then it was eluted from the column with methanol/water (70/30, v/v) and lyophilized. All the radio-labelled DNAzymes were quantitated and stored at −30 °C.
The DNAzyme–substrate (100 : 1) was mixed in a buffer solution (50 mM Tris–HCl, pH 7.5). The reaction was initiated with the addition of Mg2+ (4 mM, in 50 mM Tris–HCl, pH 7.5). The reaction was monitored by pipetting samples from the reaction mixture at certain time intervals. All the samples from one reaction were analysed with denaturing PAGE (20%, 8 M urea). The substrate and cleaved product were imaged on a PhosphoImager, and cleavage percentage was quantitated. The observed rate constants were calculated with the single-exponential decay function: P(t) = P∞ × exp(−kobst), where P(t) is the percentage of product at time t, and P∞ is the limiting extent of the product, with background cleavage extracted.
3. Results and discussion
3.1. Chemistry
Phosphoramidites of compounds 3–6 were prepared according to the literature.30 8-Substituents of 2′-deoxyadenosine were realized by replacement of the 8-Br atom of 2′-deoxyadenosine with amines (Scheme 2). In the case of nucleoside analogue 7, the end amino group was protected with a trifluoroacetyl group, and the 6-amino group was later protected with a di-n-butylformamidine group.31 For the introduction of 8-histaminyl group in compound 8, the synthesis was based on the literature,29 and Boc was used for the protection of the imidazolyl group. The synthesis of the phosphoramidite of compound 9 started from compound 9a17 (Scheme 2); its 7-amino group was deprotected and transferred to a Boc-protected guanidinyl group with N,N′-di-Boc-N′′-triflylguanidine as a guanidinylation reagent.32,33 Phosphitylation at 3′-OH was performed under standard conditions. All these phosphoramidites were applied for DNA solid-phase synthesis by standard protocol, except the coupling time was extended to 3 min. All the modified DNAzymes were characterized from their ESI-MS spectra (Table S1 and Fig. S2†).
Scheme 2. 8-Substituted 2′-deoxyadenosine and phosphoramidites. Conditions: (i) 1,3-diaminopropane in isopropanol, 60 °C; ethyl trifluoroacetate, triethylamine, at r.t.; (ii) DMTCl, in pyridine, at r.t.; (iii) (nBu)2NCH(OMe2)2, in MeOH, 2 h; (iv) (NCCH2CH2O)[(iPr)2N]2P, (iPr)2EtN tetrazolium, in CH2Cl2, at r.t.; (v) histamine, in ethanol, 80 °C; (vi) (Boc)2O, in MeOH, at r.t.; (vii) aq. ammonia/methylamine (1 : 1); (viii) N,N′-di-Boc-N′′-triflylguanidine, Et3N, CH2Cl2, 0 °C to r.t.; (ix) Me2NCH(OMe2)2, in MeOH, 2 h.

3.2. Effects of substituents at the 6-amino group of adenine
It has been previously reported that deletion of the 6-amino group of A9 led to a 17-fold decrease in the kobs value, and this amino group could be modified for a positive effect, such as DNAzyme DZ-A9-1 with residue 1 at A9.18 This observation means that the 6-amino group of A9 is a conservative functional group, but its interactions in the catalytic core can be modulated for a greater contribution using additional functional groups. First, the extra amino group of residue 1 is linked by a longer linkage in residue 3, and a slightly more positive effect was obtained at A9 of DNAzyme DZ-A9-3 (Table 1). When two amino groups were added by residue 4 for stronger interactions, a four-fold increase in the positive effect of DNAzyme DZ-A9-4 was obtained. Secondly, new functional groups were introduced with the same propyl linkage; guanidinium (residue 5) in DZ-A9-5 and imidazolyl (residue 6) in DZ-A9-6 were more active in inducing a favourable effect on the catalytic reaction; a four-fold increase of kobs was observed with DZ-A9-6. It seems that stronger interactions from the 6-amino group of A9 led to a more positive effect on the DNAzyme.
Observed rate constants of modified DNAzymes under single-turnover conditions in the presence of 2 mM Mg2+a.
| DNAzyme | k obs (min−1) | DNAzyme | k obs (min−1) | DNAzyme | k obs (min−1) |
|---|---|---|---|---|---|
| DZ01 | 0.0055 ± 0.0001 | ||||
| DZ-A9-1 | 0.0118 ± 0.0007 | ||||
| DZ-A9-3 | 0.0124 ± 0.0008 | DZ-A5-3 | —b | DZ-A11-3 | 0.0034 ± 0.0001 |
| DZ-A9-4 | 0.0234 ± 0.00005 | DZ-A5-4 | —b | DZ-A11-4 | 0.0029 ± 0.00005 |
| DZ-A9-5 | 0.0174 ± 0.0008 | DZ-A5-5 | —b | DZ-A11-5 | 0.0045 ± 0.00017 |
| DZ-A9-6 | 0.0220 ± 0.0015 | DZ-A5-6 | —b | DZ-A11-6 | 0.0039 ± 0.0001 |
| DZ-A12-3 | —b | DZ-A15-3 | 0.0008 ± 0.0001 | DZ-A0-3 | 0.0028 ± 0.0001 |
| DZ-A12-4 | —b | DZ-A15-4 | —b | DZ-A0-4 | —b |
| DZ-A12-5 | —b | DZ-A15-5 | 0.0057 ± 0.0009 | DZ-A0-5 | 0.0014 ± 0.00005 |
| DZ-A12-6 | 0.0006 ± 0.00001 | DZ-A15-6 | 0.0017 ± 0.0001 | DZ-A0-6 | 0.0017 ± 0.0001 |
Kinetic measurement under single-turnover conditions. kobs is the averaged results of two to four experiments.
Values of kobs < 0.0001 min−1.
As shown in Fig. 1, for the other five adenine residues (A0, A5, A11, A12, and A15), all the modified 2′-deoxyadenosine analogues at these positions led to a partial or complete loss of activity, compared to DZ01. These results demonstrated that A9 is a unique residue; its 6-amino could be used as starting point for promoting the catalytic reaction. Further enhancement of its positive effect is expected with the optimization of both the functional group and linkage.
Fig. 1. A comparison of the position-specific effect of modified residues 3–9. Each modified residue was replaced in the six residues A5, A9, A11, A12, A15 and A0, respectively, and the effect was evaluated under single-turnover conditions.

3.3. Effects of 8-substituents of adenine
The 8-position of purine nucleoside triphosphates is often applied for the introduction of functional groups in in vitro selection of functional nucleic acids.25 Here, amino and imidazolyl groups were introduced at the 8-position of 2′-deoxyadenosine with residues 7 and 8, respectively. At A9 and A12, DZ-A9-7 and DZ-A12-7 showed the same two-fold kobs increase (Table 2). The imidazolyl group of residue 8 induced negative effects at all six positions, but it was noticeable that DZ-A9-8 and DZ-A12-8 were more active than the other four DNAzymes with residue 8 at A5, A11, A15 and A0, and DZ-A12-8 was even slightly more active than DZ-A9-8. The similar position-dependent effect of residues 7 and 8 is clearly shown in Fig. 1. Based on the positive effect of residue 7, the 8-substituent of A9 and A12 could be used to search for more effective DNAzymes.
Observed rate constants of DNAzymes under single-turnover conditions in the presence of 2 mM Mg2+.
| DNAzyme | k obs (min−1) | DNAzyme | k obs (min−1) |
|---|---|---|---|
| DZ-A9-7 | 0.0110 ± 0.0006 | DZ-A5-8 | —a |
| DZ-A9-8 | 0.0035 ± 0.0001 | DZ-A11-8 | — |
| DZ-A9-2 | 0.0450 ± 0.004 | DZ-A12-8 | 0.0045 ± 0.0001 |
| DZ-A9-9 | 0.0351 ± 0.0015 | DZ-A15-8 | 0.0008 ± 0.00001 |
| DZ-A9-10 | 0.0089 ± 0.0005 | DZ-A0-8 | 0.0012 ± 0.0001 |
| DZ-A9-11 | 0.0128 ± 0.0018 | ||
| DZ-A9-12 | 0.0079 ± 0.0006 | ||
| DZ-A5-7 | — | DZ-A5-9 | — |
| DZ-A11-7 | — | DZ-A11-9 | 0.0035 ± 0.0002 |
| DZ-A12-7 | 0.0111 ± 0.0003 | DZ-A12-9 | 0.0023 ± 0.0001 |
| DZ-A15-7 | 0.0009 ± 0.0001 | DZ-A15-9 | 0.0010 ± 0.0001 |
| DZ-A0-7 | 0.0010 ± 0.0001 | DZ-A0-9 | 0.0015 ± 0.0005 |
k obs < 0.0001.
3.4. Effect of 7-substituents of 8-aza-7-deaza-adenine
For the five-membered ring moiety of adenine, 7-substituted 8-aza-7-deaza-adenine was demonstrated to be an effective modification approach when located at A9.17 Based on the performance of residue 2, its extra amino group was transferred to a guanidinium group in residue 9 for stronger interactions. As shown in Table 2, the stronger interaction of the 7-substituent was not always favourable for the catalytic activity of DZ-A9-9, but a similar position-dependent effect of residue 9 was still kept (Fig. 1).
3.5. Divalent metal ion dependence of DNAzymes
The catalytic reaction of 10–23 DNAzyme is divalent metal ion dependent. Under the present single-turnover conditions, four other divalent metal ions, Mn2+, Pb2+, Ca2+, and Zn2+, were evaluated for their promotion effect on the modified DNAzymes. A similar influence of these metal ions on DZ01 and all the modified DNAzymes was observed, following the order of Mn2+ ≈ Pb2+ ≫ Mg2+ ≈ Ca2+ ≫ Zn2+. The Mn2+ (2 mM) and Pb2+ (2 mM)-mediated reactions run very fast under the same conditions. Fig. 2 shows the Ca2+ and Zn2+-induced catalytic reactions of several modified DNAzymes and DZ01. These results demonstrate that the modified A9 might have little direct influence on the coordination environment of the catalytic metal ions, but the residues in the active site might be driven to form a more favourable catalytic conformation for the trans-esterification reaction.
Fig. 2. The time course of catalytic reactions of modified DNAzymes in the presence of 2 mM Ca2+ (top) and 2 mM Zn2+ (bottom).

3.6. Unique positive effect of the active functional groups at A9
All six adenine residues (A0, A5, A9, A11, A12, and A15) are conserved for the catalytic activity of 10–23 DNAzyme when evaluated at the level of functional group. Further activation with extra protein-like functional groups was only realized at A9. These dramatically different behaviours reflected that each adenine has its own specific interactions in the catalytic core, although modified DNAzymes and DZ01 had similar conformation when forming a complex with the substrate, as shown by the very similar thermal stability (Table S2†) and CD spectra (Fig. S1†). In the case of amino group, it was most effective when located at the 7-position of 8-aza-7-deaza-adenine by propyl linkage at A9 (residue 2). This position was also appropriate for the guanidinium group, as revealed when the positive effects of residues 5 and 9 were compared. For the imidazolyl group, both the position and linkage were compared with residues 6 and 8, as well as 10,1611 and 1234 (Scheme 3 and Table 2). It was found that residue 6 at A9 was the most effective introduction approach, with a 4-fold increase of the observed rate constant.
Scheme 3. 2′-Deoxyadenosine analogues with imidazolyl group.

The consistent position-dependent effects of the extra substituents at all adenine residues demonstrate that the catalytic core forms a relatively static conformation, in which the extra functional groups from each adenine residue could have its own location and interactions, resulting in a stable position-specific effect.
A9 is supposed to be located in the central stretch, offering a supportive backbone to the conserved residues, and it is far from the catalytic Mg2+ and the active site. Around A9, the new substituents of the modified residues 1 and 3–9 induced a favourable change in the active site. The modified DNAzymes had very similar divalent metal ion-dependence as the original DNAzyme. This demonstrates that the additional functional groups might be little related to the coordination of the catalytic metal ion, and an indirect influence on the conformation of the active site was thus suggested. For the substituents at the 6-amino group, it seems that the stronger interactions induced more positive effects. The single amino group is less active than the groups with multiple heads for hydrogen-bonding. In the catalytic conformation of 10–23 DNAzyme, at the site defined by these extra functional groups from A9, further improvement could be expected by new linkage and functional groups. Therefore, a rational modification was achieved with A9 and 2′-deoxyadenosine analogues.
This kind of modification based on nucleobases could be the guidance for further improvement of 10–23 DNAzyme. Three other canonical nucleobases could be modified similarly.15 When 2′-deoxyguanosine analogues were introduced at guanine residues in the catalytic loop of 10–23 DNAzyme, the extreme conservation of G2 and G14 was broken, and the added functional groups had a positive effect.35
2′-Deoxyadenosine analogues have also been applied for adenine modification of 8–17 DNAzymes. When a specific adenine was modified, a positive effect was also obtained,30 and even the metal ion dependence of DNAzyme was changed, depending on the position of the modified residue in the catalytic core.
4. Conclusions
Successful activation of 10–23 DNAzyme was achieved at A9 through 2′-deoxyadenosine analogues, with amino, guanidinium and imidazolyl groups at the 6-amino of adenine, or the 7-position of 8-aza-7-deaza-adenine. A9 is the unique position to realize the positive effect. It is interesting that the 8-substituent of adenine in residue 7 is also favourable for A12. It offers another choice, a combination of A12 and its 8-substituent, for the improvement of 10–23 DNAzyme. Considering the negative effect of 8-imidazolyl group in residue 8 at A12, the linkage and functional group need to be further optimized.
These modified 2′-deoxyadenosine analogues are being applied for modifications of adenine residues in other DNAzymes and aptamers in our research group.
As little is known about the role of each nucleobase in the catalytic loop of 10–23 DNAzyme, it is a challenge to start chemical evolution on the highly conserved residue or the less conserved residue. In the present study, hydrogen bonding interactions of nucleobases were emphasized and studied, along with active functional groups.
From the consistent effects of all the extra functional groups at all the adenine residues, we learned that the active site is constructed not only by base stacking and Mg2+, but also by numerous hydrogen bonds. More importantly, from this study, the positive effect of extra functional groups at A9 is predictable, by the 6-substituent of adenine (compounds 1, 3–6) or the 7-substituent of 8-aza-7-deaza-adenine (compounds 2 and 9).
This study confirmed that functional group modification on a nucleobase is a feasible approach for the activation of functional nucleic acids. Compared to this kind of nucleobase modification, when cross-nucleobase modifications were used, it was more difficult to find a favourable modification, as much more interactions related to base stacking and hydrogen bonding are involved, like a “domino” effect.
More efficient DNAzymes are always needed for practical applications. As a potential therapeutic agent, more efficient cleavage of DNAzyme could be less dependent on divalent metal ion concentration in vivo, as there is not a sufficient divalent metal ion for the fast cleavage of target RNA. In addition, the modification sites around A9 could be explored as a photo or chemical switch in the design of biosensors without any loss of activity.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We give thanks for the financial support from the National Natural Science Foundation of China (No. 21572268) and the National Science and Technology Major Project of China (No. 2018ZX09711003-003-001).
Electronic supplementary information (ESI) available: Tm and CD spectra, MS characterization results and MS spectra. See DOI: 10.1039/d0ra02226h
Notes and references
- Jimenez R. M. Polanco J. A. Lupták A. Trends Biochem. Sci. 2015;40:648. doi: 10.1016/j.tibs.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelini F. Jalihal A. P. Francia S. Meers C. Neeb Z. T. Rossiello F. Gioia U. Aguado J. Jones-Weinert C. Luke B. Biamonti G. Nowacki M. Storici F. Carninci P. Walter N. G. d'Adda di Fagagna F. Chem. Rev. 2018;118:4365. doi: 10.1021/acs.chemrev.7b00487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theil E. C. Westhof E. Acc. Chem. Res. 2011;44:1255. doi: 10.1021/ar200261e. [DOI] [PubMed] [Google Scholar]
- Silverman S. K. Trends Biochem. Sci. 2016;41:595. doi: 10.1016/j.tibs.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L. Xing H. Zhang J. Lu Y. Acc. Chem. Res. 2019;52:2415. doi: 10.1021/acs.accounts.9b00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell E. M. Cozna I. Morrison D. Li Y. Anal. Chem. 2020;92:327. doi: 10.1021/acs.analchem.9b04868. [DOI] [PubMed] [Google Scholar]
- Santoro S. W. Joyce G. F. Proc. Natl. Acad. Sci. U. S. A. 1997;94:4262. doi: 10.1073/pnas.94.9.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greulich T. Hohlfeld J. M. Neuser P. Lueer K. Klemmer A. Schade-Brittinger C. Harnish S. Garn H. Renz H. Homburg U. Renz J. Kirsten A. Pedersen F. Müller M. Vogelmeier C. F. Watz H. Respir. Res. 2018;19:55. doi: 10.1186/s12931-018-0751-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy M. Aung-Htut M. T. Le B. T. Veedu R. N. Sci. Rep. 2017:1613. doi: 10.1038/s41598-017-01559-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H. Newbigging A. M. Wang Z. Tao J. Deng W. Le X. C. Zhang H. Anal. Chem. 2018;90:190. doi: 10.1021/acs.analchem.7b04926. [DOI] [PubMed] [Google Scholar]
- Zhao Y. Zhou L. Tang Z. Nat. Commun. 2013;4:1493. doi: 10.1038/ncomms2492. [DOI] [PubMed] [Google Scholar]
- Peng H. Li X.-F. Zhang H. Le X. C. Nat. Commun. 2017;8:14378. doi: 10.1038/ncomms14378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. Feng M. Xiao L. Tong A. Xiang Y. ACS Chem. Biol. 2016;11:444. doi: 10.1021/acschembio.5b00867. [DOI] [PubMed] [Google Scholar]
- Lin Y. Yang Z. Lake R. J. Zheng C. Lu Y. Angew. Chem., Int. Ed. 2019;58:17061. doi: 10.1002/anie.201910343. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wu Y. Huang J. Yang X. Yang Y. Quan K. Xie N. Li J. Ma C. Wang K. Anal. Chem. 2017;89:8377. doi: 10.1002/anie.201910343. [DOI] [PubMed] [Google Scholar]
- Smuga D. Majchrzak K. Sochacka E. Nawrot B. New J. Chem. 2010;34:934. doi: 10.1039/B9NJ00705A. [DOI] [Google Scholar]
- Nawrot B. Widera K. Wojcik M. Rebowska B. Nowak G. Stec W. J. FEBS J. 2007;274:1062. doi: 10.1111/j.1742-4658.2007.05655.x. [DOI] [PubMed] [Google Scholar]; Zaborowska Ż. Fürste J. P. Erdmann V. A. Kurreck J. J. Biol. Chem. 2002;277:40617. doi: 10.1111/j.1742-4658.2007.05655.x. [DOI] [PubMed] [Google Scholar]; Zaborowska Ż. Schubert S. Kurreck J. Erdmann V. A. FEBS Lett. 2005;579:554. doi: 10.1111/j.1742-4658.2007.05655.x. [DOI] [PubMed] [Google Scholar]; Peracchi A. Bonaccio M. Clerici M. J. Mol. Biol. 2005;352:783. doi: 10.1111/j.1742-4658.2007.05655.x. [DOI] [PubMed] [Google Scholar]
- He J. Zhang D. Wang Q. Wei X. Cheng M. Liu K. Org. Biomol. Chem. 2011;9:5728. doi: 10.1039/C1OB05065F. [DOI] [PubMed] [Google Scholar]
- Zhu J. Li Z. Wang Q. Liu Y. He J. Bioorg. Med. Chem. Lett. 2016;26:4462. doi: 10.1016/j.bmcl.2016.07.076. [DOI] [PubMed] [Google Scholar]
- Gelinas A. D. Davies D. R. Janjic N. Curr. Opin. Struct. Biol. 2016;36:122. doi: 10.1016/j.sbi.2016.01.009. [DOI] [PubMed] [Google Scholar]
- Grasby J. A. Mersmann K. Singh M. Gait M. J. Biochemistry. 1995;34:4068. doi: 10.1021/bi00012a025. [DOI] [PubMed] [Google Scholar]
- Silverman S. K. Acc. Chem. Res. 2015;48:1369. doi: 10.1021/acs.accounts.5b00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelinas A. D. Davies D. R. Janjic N. Curr. Opin. Struct. Biol. 2016;36:122. doi: 10.1016/j.sbi.2016.01.009. [DOI] [PubMed] [Google Scholar]
- Diafa S. Hollenstein M. Molecules. 2015;20:16643. doi: 10.3390/molecules200916643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro S. W. Joyce G. F. Sakthivel K. Gramatikova S. Barbas III C. F. J. Am. Chem. Soc. 2000;122:2433. doi: 10.1021/ja993688s. [DOI] [PubMed] [Google Scholar]
- Perrin D. M. Garestier T. Hélène C. J. Am. Chem. Soc. 2001;123:1556. doi: 10.1039/C7SC04491G. [DOI] [PubMed] [Google Scholar]; Sidorov A. V. Grasby J. A. Williams D. M. Nucleic Acids Res. 2004;32:1591. doi: 10.1039/C7SC04491G. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wang Y. Liu E. Lam C. H. Perrin D. M. Chem. Sci. 2018;9:1813. doi: 10.1039/C7SC04491G. [DOI] [PMC free article] [PubMed] [Google Scholar]; Takezawa Y. Nakama T. Shionoya M. J. Am. Chem. Soc. 2019;141:19342. doi: 10.1039/C7SC04491G. [DOI] [PubMed] [Google Scholar]; Hollenstein M. Hipolito C. J. Lam C. H. Perrin D. M. Nucleic Acids Res. 2009;37:1638. doi: 10.1039/C7SC04491G. [DOI] [PMC free article] [PubMed] [Google Scholar]; Rohloff J. C. D Gelinas A. Jarvis T. C. Ochsner U. A. Schneider D. J. Gold L. Janjic N. Mol. Ther.--Nucleic Acids. 2014;3:e201. doi: 10.1039/C7SC04491G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviñó A. Fàbrega C. Tintoré M. Eritja R. Curr. Pharm. Des. 2012;18:2036. doi: 10.2174/138161212799958387. [DOI] [PubMed] [Google Scholar]
- Nowakowski J. Shim P. J. Prasad G. S. Stout C. D. Joyce G. F. Nat. Biotechnol. 1999;6:151. doi: 10.1080/15257770.2016.1276291. [DOI] [PubMed] [Google Scholar]; Dolot R. Sobczak M. Mikołajczyk B. Nawrot B. Nucleosides, Nucleotides Nucleic Acids. 2017;36:292. doi: 10.1080/15257770.2016.1276291. [DOI] [PubMed] [Google Scholar]
- Plashkevych O. and Chattopadhyaya J., in Medicinal Chemistry of Nucleic Acids, ed. L. H. Zhang, Z. Xi and J. Chattopadhyaya, John Wiley & Sons. Inc., 1st edn, 2011, ch. 7, p. 272 [Google Scholar]
- Lermer L. Hobbs J. Perrin D. M. Nucleosides, Nucleotides Nucleic Acids. 2002;21:651. doi: 10.1081/NCN-120015723. [DOI] [PubMed] [Google Scholar]
- Du S. Li Y. Chai Z. Shi W. He J. Bioorg. Chem. 2020:103401. doi: 10.1016/j.bioorg.2019.103401. [DOI] [PubMed] [Google Scholar]
- Froehler B. C. Matteucci M. D. Nucleic Acids Res. 1983;11:8031. doi: 10.1093/nar/11.22.8031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feichtinger K. Zapf C. Sings H. L. Goodman M. J. Org. Chem. 1998;63:3804. doi: 10.1021/jo980425s. [DOI] [Google Scholar]
- Brzezinska J. D'Onofrio J. Buff M. C. R. Hean J. Ely A. Marimani M. Arbuthnot P. Engels J. W. Bioorg. Med. Chem. 2012;20:1594. doi: 10.1016/j.bmc.2011.12.024. [DOI] [PubMed] [Google Scholar]
- Li Z. Liu Y. Liu G. Zhu J. Zheng Z. Zhou Y. He J. Bioorg. Med. Chem. 2014;22:4010. doi: 10.1016/j.bmc.2014.05.070. [DOI] [PubMed] [Google Scholar]
- Liu Y. Li Z. Liu G. Wang Q. Chen W. Zhang D. Cheng M. Zheng Z. Liu K. He J. Chem. Commun. 2013;49:5037. doi: 10.1039/C3CC42067A. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.