

Abstract
Two new fluorescence imaging probes have been synthesized by incorporating a versatile alkyne-substituted boron difluoride formazanate precursor with peptides through copper-catalyzed alkyne–azide cycloaddition. The formazanate dye was appended to a C-terminal amino acid of ghrelin for imaging the growth hormone secretagogue receptor (GHSR-1a). To demonstrate versatile bioconjugation chemistry, the formazanate dye was added to the N-terminus of bombesin for targeting the gastrin releasing peptide receptor (GRPR). These are the first examples of using this emerging class of dyes, boron difluoride formazanates, for the labelling of biomolecules.
Conjugation of a boron difluoride formazanate dye to receptor targeting peptides provides cancer imaging agents for fluorescence microscopy.
Introduction
Fluorescently labelled compounds have been developed and utilized in a variety of applications in biology and biotechnology. In particular, these types of compounds have garnered significant attention for use as molecular probes which find application in microscopy, cellular imaging, flow cytometry, and genotyping assays.1–5 To this end, several classes of naturally occurring dyes have been recognized and used as starting points for families of synthetic dyes, including porphyrins, coumarins, xanthenes, and cyanines.6–9 With respect to their use as molecular imaging agents, ideal dyes should exhibit high fluorescence quantum yields (ΦF), minimal photobleaching as well as tunable wavelengths of maximum absorbance (λabs) and emission (λem). In addition, they should be easily conjugated with targeting moieties to allow specificity with a biomolecular target of interest. Common targeting moieties for fluorescence imaging have been designed around peptides, antibodies, small molecules and nucleic acids.10
With increased advances in genome screening, new receptor targets are being found as promising markers for disease states. To go along with these receptor targets, novel molecular library screening strategies have accelerated the process of identification of ligands possessing high affinity for the protein targets. As a consequence, these advancements have led to a gap between the development of novel ligands found for receptor targets and the development of assays for quick and efficient screening for biomarker analysis. It is in this gap that development of both novel fluorophores and strategies for tagging ligands that bind specifically a receptor of interest, can assist in providing quick and efficient assays for biomarker evaluation.
Many of the targeting moieties used for designing imaging probes are based on small to medium size molecules, antibodies being the exception, which are typically large. A significant challenge encountered during the development of fluorescent imaging agents based on targeted small molecules is the disproportionately large size of the imaging tag compared to the targeting or receptor binding piece. Typically, fluorescent probes are designed with polycyclic aromatic rings and charged functional groups to push the absorbance and emission into the visible, far red and even infra-red wavelengths for imaging compatibility.11 These constraints inherently change the solubility, charge, size, and most importantly, can alter the binding and specificity of the targeting portion of the probe. The negative characteristics of typical fluorescent constructs have led to the research and development of small, organic, charge neutral dyes. In this regard, we have seen the increased development of dyes based on boron-containing scaffolds, in particular 4,4′-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) (Fig. 1).12,13 In general, BODIPY dyes have excellent photophysical characteristics including narrow absorption and emission bands, high molar extinction coefficients and good fluorescence quantum yields.14 However, given the synthetic challenges associated with BODIPY dyes and their conjugates, there is a need to explore alternative dye scaffolds for creating fluorescent reagents.
Fig. 1. General structure of a BODIPY dye and BF2 formazanate dye 1 used to produce dye-labelled peptide products via CuAAC chemistry.

A new class of fluorescent dyes that are promising for use in microscopy applications are the boron difluoride (BF2) formazanates (Fig. 1).15–17 Much like BODIPY dyes, BF2 formazanates are comprised of a “BF2” fragment bound by a chelating N-donor ligand, creating a stable six-membered heterocyclic ring. Despite their structural similarities, BF2 formazanates can be prepared from commercially available starting materials in two straightforward synthetic steps, often in higher yields than BODIPY derivatives. Furthermore, BF2 formazanate dyes exhibit tunable photophysical properties through structural variation,18,19 as well as large molar extinction coefficients and high ΦF's which are typically in the far-red or near-infrared region which is considered beneficial for many imaging applications.17 We recently described the synthesis of an asymmetric 3-cyano BF2 formazanate dye 1 which bears a terminal alkyne.20 We now describe the preparation of targeted fluorescent probes by using this alkyne functionalized dye, through copper-assisted alkyne–azide cycloaddition (CuAAC) chemistry.
In the development of novel receptor-targeted imaging constructs it is imperative to choose a targeting moiety that is not only specific for the receptor of choice, but also allows for conjugation to the fluorescent probe through means that do not interfere with the receptor binding. One such approach is to begin with the naturally occurring peptide for a given receptor, followed by a series of structural activity relationship studies to increase stability, binding, specificity and determine an appropriate conjugation site that does not interfere with receptor binding.21 Of interest to the Luyt group are two such peptides, ghrelin and bombesin. In brief, ghrelin is a 28 amino acid peptide that has been shown to activate the growth hormone secretagogue receptor (GHSR-1a), a G protein-coupled receptor (GPCR). The GHSR-1a is of particular interest as it plays a significant role in release of growth hormone, regulation of appetite and food intake, modulation of gastrointestinal motility and secretion, managing cell proliferation and survival, regulation of pancreatic secretion and energy homeostasis.22–24 Analogues of ghrelin(1–8), with modifications of Dpr3(n-octanoyl) and Thr8, have provided small peptides with affinity significantly improved compared to the natural ghrelin(1–28).25 Another naturally occurring peptide, bombesin, is a 14 amino acid peptide that activates the gastrin releasing peptide receptor (GRPR), which has been shown to be highly expressed in a variety of cancers including prostate, breast, gastrointestinal and lung.26–29 To date a number of bombesin imaging constructs have been reported for the application in fluorescence imaging of GRPR using conjugated quantum dots or Alexa Fluor as fluorescent tags.30,31 Both GHSR and GRPR are of importance as they are overexpressed in various cancers and the development of targeted fluorescent entities will benefit rapid and efficient screening of cell receptor densities to distinguish between benign and malignant tumors. Furthermore, these two peptides require differing conjugation strategies when incorporating a dye, with ghrelin requiring conjugation in the C-terminal region and bombesin at the N-terminus.
Herein we report the synthesis and in vitro evaluation of two novel fluorescent probes utilizing a BF2 formazanate dye 1 coupled to peptides via CuAAC within the synthetic pathway. The two peptides selected have been previously reported to have good binding affinity for GHSR-1a or GRPR with modifications at their specific attachment sites (Fig. 2). In brief, peptide 2 is a previously reported ghrelin(1–8) analogue that was shown to have excellent affinity for GHSR-1a.25 A modified version of the ghrelin(1–8), compound 2, was designed with a lysine azide residue added at the C-terminus, to which the azide side chain could then be used for CuAAC. The GRPR peptide is based on a modified nine amino acid bombesin(6–14) (3) with the addition of an azide-PEG linker at the N-terminus (Fig. 2).32 The selection of these two peptides demonstrates the feasibility to conjugate dye 1 on either the C or N terminal regions of short peptides, which demonstrates the potential to synthesize any short peptide BF2 formazanate construct.
Fig. 2. Structure of the peptides [Dpr(octanoyl)3,Thr8]ghrelin(1–8) amide 2 and [DTyr6,βAla11,Phe13,Nle14]bombesin(6–14) amide 3.

Results and discussion
Peptides were synthesized using automated Fmoc solid-phase synthesis, purified by preparative high-performance liquid chromatography (HPLC), and characterized by liquid chromatography-mass spectrometry (LCMS) with electrospray ionization (ESI+) mass spectrometry (Scheme 1 and ESI†). For formazanate-labelled peptide 5 [Dpr(octanoyl)3,Thr8,Lys(triazole-formazanate)9]ghrelin(1–9), the BF2 formazanate was added directly to the peptide through a Lys(N3) added as the C-terminal amino acid (Scheme 1a). This conjugation was done in a yield of 7% after purification with a purity of 98%. In contrast, bombesin conjugates typically require a spacer at the N-terminus in order to position the imaging moiety away from the binding region of the peptide. Thus, a short poly(ethylene glycol) (PEG) chain was first added to the BF2 formazanate using CuAAC, prior to amide formation at the N-terminus of the peptide (Scheme 1b). The intermediate compound 6 was isolated in 35% yield and 99% purity after reverse-phase HPLC. The final conjugate 7 was also purified by HPLC (25% yield) and characterized by LCMS.
Scheme 1. Synthetic routes to BF2 formazanate–labelled peptides: (a) copper assisted “click” reaction of compound 4 with formazanate 1. (b) Synthesis of compound 6 through copper assisted “click” reaction of PEG azide with formazanate 1, followed by coupling with 3 to give compound 7.

The photophysical properties of compounds 1, 5 and 7 were evaluated and reported in Table 1. All compounds are strongly absorbing (ε = 31 900–34 500 L mol−1 cm−1) with absorption maxima between 546 and 560 nm. In addition, compounds 1, 5, and 7 are emissive with λem between 665–690 nm and ΦF ranging from 4 to 21%. The increase in ΦF upon peptide conjugation may be attributed to the large size of the peptide construct compared to the parent BF2 formazanate (1), which likely inhibits rotation of the functionalized N-aryl substituent and attenuates the associated non-radiative decay pathway. A notable property of BF2 formazanate dyes is that they exhibit large Stokes shifts (νST), superior to that of BODIPY and other commercial dyes.16 Upon conjugation, the νST increases from 119 nm (3278 cm−1) for 1, to 128 nm (3322 cm−1) for 5 and 135 nm (3525 cm−1) for 7.
Photophysical data for BF2 formazanate 1 and dye-labelled peptides 5 and 7. All measurements were performed in DMSOa.
| Compound | λ abs (nm) | ε (M−1 cm−1) | λ em (nm) | ν ST (nm) | Φ F (%) |
|---|---|---|---|---|---|
| 1 | 546 | 34 500 | 665 | 119 | 4 |
| 5 | 560 | 32 200 | 688 | 128 | 21 |
| 7 | 555 | 31 900 | 690 | 135 | 12 |
In order to determine the binding affinity of probe 5 for GHSR-1a receptor and probe 7 for GRPR, radioligand competitive binding assays were completed. This was achieved using HEK293 cells transiently transfected with GHSR-1a for compound 5, and PC-3 cells with natural GRP receptor density for probe 7. A decrease in receptor affinity for both compounds 5 and 7 was observed. The affinity (IC50) for 5 was determined to be 89.0 nM compared to 3.3 nM for the parent peptide 2,25 while the affinity (IC50) for 7 was measured at 16.3 nM which was an increase from 0.7 nM for the parent peptide 3 (Table 2).32 These results indicate the formazanate dye has some influence on binding, however these affinity values are within a reasonable range to act as a fluorescent reporter during in vitro cell assays and microscopy studies.
IC50 (nM) for parent compounds 2 and 3 and dye-labelled peptides 5 and 7. Studies for compounds 2 and 5 were performed on HEK293 cells transfected with GHSR-1a whereas parental PC-3 cells were used for studies using compounds 3 and .7.
| Compound | IC50 (nM) |
|---|---|
| 2 | 3.3 |
| 3 | 0.7 |
| 5 | 89.0 |
| 7 | 16.3 |
The specific binding of 5 for GHSR-1a and analogue 7 for GRPR was demonstrated through confocal fluorescence microscopy (Fig. 3). For analyzing the ghrelin probe 5, OVCAR-8 cells stably transfected with GHSR-1a were incubated with 5 at 0.1 μM and confocal fluorescence micrographs were obtained after washing and fixing the cells. The images validated the cellular uptake of 5 as was evident from the red emission (Fig. 3a). The applicability of 5 for orthogonal imaging was demonstrated by co-staining the cells with nuclear stain DAPI (4′,6-diamidino-2-phenylindole) (in blue) exhibiting the ability to differentiate the cytoplasm and nuclei while being imaged simultaneously (Fig. 3b). To obtain a detailed view of cellular uptake, higher magnification images were also obtained (Fig. 3c and d). Parental OVCAR-8 cells were used as a negative control and displayed minimal uptake of 5, as parental OVCAR-8 cells have low expression of GHSR-1a (Fig. 3e and f). To further validate that our fluorescent peptide 5 was binding specifically to the GHSR-1a, a blocking study was done using a known small molecule antagonist,358 (Fig. 4), with GHSR-1a stably transfected OVCAR-8 cells (Fig. 3g and h).
Fig. 3. Confocal fluorescence micrographs obtained using the ghrelin–formazanate probe 5. Red images (from 5): excitation at 559 nm and emission collected between 598–698 nm. Blue images (DAPI): excitation at 405 nm and emission collected between 425–475 nm. (a) Imaging 5 with OVCAR-8 cells transfected with GHSR-1a (40× objective). (b) Imaging DAPI with OVCAR-8 cells transfected with GHSR-1a and overlay of image (a). (c) Imaging 5 with OVCAR-8 cells transfected with GHSR-1a using 60× objective. (d) Imaging DAPI with OVCAR-8 cells transfected with GHSR-1a and overlay of image (c), using 60× objective. (e) Imaging 5 with parental OVCAR-8 cells (40× objective). (f) Imaging DAPI with parental OVCAR-8 cells and overlay of image (e). (g) Imaging 5 OVCAR-8 cells transfected with GHSR-1a and blocked with antagonist 8 (40× objective). (h) Imaging DAPI with OVCAR-8 cells transfected with GHSR-1a and blocked with antagonist 8 and overlay of image (g). Scale bar 20 μm except (c) and (d) 10 μm.

Fig. 4. Small molecule antagonist 8 used in blocking studies of GHSR-1a.
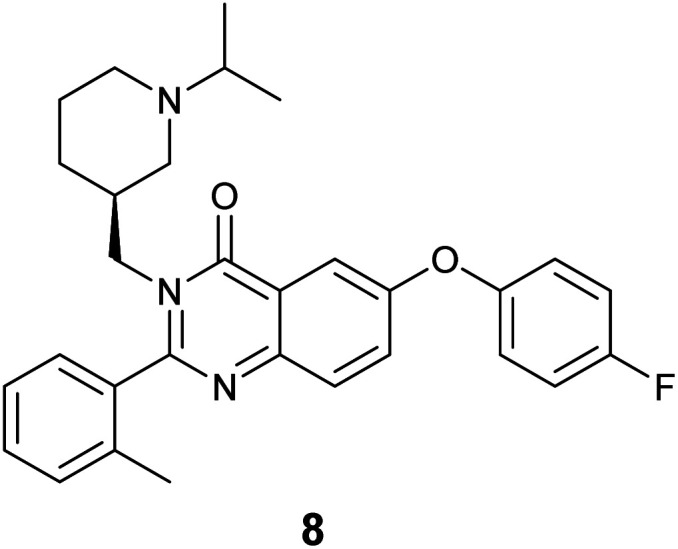
For analyzing analogue 7, PC-3 cells which have natural expression of GRPR, were used. The confocal fluorescence micrographs demonstrated that analogue 7 was internalized by these cells with uptake seen in the cytoplasm (Fig. 5a–d). Further, specific binding of analogue 7 to GRPR was depicted through blocking studies that were performed using 10-fold excess of the parent peptide 3 which was determined to have very high affinity for GRPR. Blocking studies demonstrated the displacement of 7 by 10-fold molar excess of 3 (Fig. 5e and f).
Fig. 5. Confocal fluorescence micrographs obtained using the bombesin–formazanate probe 7. Red images (from 7): excitation at 559 nm and emission collected between 605–705 nm. Blue images (DAPI): excitation at 405 nm and emission collected between 425–475 nm. (a) PC-3 cells stained with 7 (40× objective). (b) Overlay of PC-3 cells stained with DAPI (blue) and image (a). (c) PC-3 cells stained with 7 at 60× objective. (d) Overlay of PC-3 cells stained with DAPI (blue) and image (c) at 60× objective. (e) PC-3 cells blocked with 10-fold molar excess of 3 (40× objective). (f) Overlay of PC-3 cells stained with DAPI, blocked with 10-fold molar excess of 3 and image (e). Scale bar for (a), (b), (e) and (f) 30 μm, for (c) and (d) 10 μm.

Conclusions
The development of novel fluorescence imaging agents for analysis of receptor expression has led to target specific analysis of cells and tissues through fluorescence microscopy or flow cytometry methods. In an effort to assist in the development of fluorescent biomolecules, we demonstrated for the first time that BF2 formazanate dyes, with their ideal photophysical properties, can be conjugated to peptides through either their C or N terminal amino acid using copper-assisted alkyne–azide chemistry as an integral part of the synthetic pathway. Through the preparation of formazanate–peptide conjugates we developed imaging probes that are suitable for specific receptor reporting for GHSR-1a and GRPR. While the affinity for the receptor decreased for both fluorescent probes compared to their parent ligand, the conjugates maintained sub-micromolar affinities and were determined to be suitable for cellular imaging.
Experimental
Materials and methods
All common solvents were purchased from Fisher Scientific. All Fmoc-protected amino acids, coupling agents and resins were purchased from Chem Impex, Peptides International and Novabiochem and were used without further purification unless otherwise stated. All reagents were purchased from Sigma Aldrich. BF2 formazanate dye 1 was synthesized and fully characterized according to the previously reported procedure.20 CuAAC reactions were performed under a N2 atmosphere, using Schlenk techniques. [125I]-ghrelin was purchased from PerkinElmer. For analysis of samples, an analytical Agilent RP-C18 4.6 × 150 mm, 5 μm column was utilized. The flow rate was 1.5 mL min−1 over 25 min. For purification of samples, a reverse phase preparative HPLC column (Agilent RP-C18 19 × 150 mm, 5 μm) was employed. The flow rate in this case was 20 mL min−1 over 15 min. The gradient solvent system used were 0.1% trifluoroacetic acid (TFA) in CH3CN and 0.1% TFA in H2O. This system was provided with a Waters 600 controller, Waters Prep degasser, Waters Mass Lynx software (version 4.1). For the studies on UHPLC-MS, a Waters Inc. Acquity UHPLC H-Class system was used in combination with a Xevo QT mass spectrometer (ESI+, cone voltage 30 V). For analytical studies, a Waters Acquity UHPLC BEH C18 2.1 × 50 mm, 1.7 μm column was utilized. The gradient solvent system employed was 0.1% formic acid in CH3CN and 0.1% formic acid in H2O.
Cell imaging was carried out on an Olympus FluoView FV 1000 confocal microscope.
General synthesis of peptides
Peptides were prepared manually using solid-phase peptide synthesis chemistry. Peptides were synthesized on 0.1 mmol scale using Rink amide MBHA resin (0.52 mmol g−1). Fmoc deprotection was carried out using 2 mL of 20% piperidine/DMF for two cycles (10 min, 15 min). Activation of amino acids was carried out using 3 eq. of HCTU (O-(1H-6-chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate), and 6 eq. of N,N-diisopropylethylamine (DIPEA) in DMF (2 mL). The mixture was then added to the resin and vortexed for 60 min. These cycles were repeated until all N-terminal amino acids were coupled to the resin. The deprotection of the allyloxycarbonyl group of diaminopropionic acid was carried out under a N2 atmosphere. For this anhydrous CH2Cl2 (2 mL) was added to the resin, followed by 24 eq. of phenylsilane (PhSiH4) and finally 0.1 eq. of Pd(PPh3)4 and the solution was allowed to shake under N2 for 5 min. The peptide vessel was removed from inert conditions and allowed to shake for 30 min. Full deprotection and cleavage of the peptide was accomplished by adding 3 mL of a mixture comprised of 95% TFA, 2.5% triisopropylsilane (TIPS), and 2.5% H2O to the resin and shaking for 4 h. The cleaved peptide was precipitated using ice-cold tert-butyl methyl ether (TBME) and centrifuged at 3000 rpm for 15 min. The supernatant was removed and the peptide pellet was dissolved in 40% CH3CN in H2O, frozen at −78 °C and lyophilized to obtain a brown solid.
Synthesis of 5, [Dpr(octanoyl)3,Thr8,Lys(triazole-formazanate)9]ghrelin(1–9)
Pentamethyldiethylenetriamine (PMDETA) (4.5 μL, 0.02 mmol) was dissolved in anhydrous THF (2.5 mL). In a separate flask compound 4 (125 mg, 0.10 mmol) was dissolved in anhydrous THF (2.5 mL). The solutions were degassed via three freeze–pump–thaw cycles. Dry CuI (4 mg, 0.02 mmol) was then added to the PMDETA solution and stirred at room-temperature for 20 min. Compound 1 (37 mg, 0.10 mmol) and the freshly prepared CuI–PMDETA solution were then added to the THF solution of compound 4 and the reaction mixture was heated at 40 °C for 4 h. THF was removed using a rotary evaporator and the crude compound was then dissolved in CH3CN (0.1% TFA) and H2O (0.1% TFA) and purified by preparative HPLC-MS. The fractions were combined and frozen at −78 °C and lyophilized to yield 11.7 mg (7.2%, 0.0076 mmol), purity 98%. HRMS (ESI+): m/z calculated for C66H93BF2N19O14, [M + H]+ = 1424.7132; found [M + H]+ = 1424.7237.
Synthesis of 6, 32-(4-(4-(6-cyano-3,3-difluoro-4-(4-methoxyphenyl)-3,4-dihydro-1,2λ4,4,5,3λ4-tetrazaborinin-2-yl)phenyl)-1H-1,2,3-triazol-1-yl)-5-oxo-3,9,12,15,18,21,24,27,30-nonaoxa-6-azadotriacontanoic acid
PMDETA (4.5 μL, 0.02 mmol) was dissolved in anhydrous THF (2.5 mL). In a separate flask compound (PEG)7N3–OH (95 mg, 0.17 mmol) was dissolved in 2.5 mL THF and both solutions were degassed via three freeze–pump–thaw cycles. Anhydrous CuI (6.5 mg, 0.034 mmol) was then added to the PMDETA solution and stirred at room-temperature for 20 min. Compound 1 (60 mg, 0.17 mmol) and CuI–PMDETA solution were then added to the THF solution of (PEG)7N3–OH and reaction mixture was heated at 40 °C for 4 h. THF was removed using a rotary evaporator and the crude compound was then dissolved in CH3CN (0.1% TFA) and H2O (0.1% TFA) and purified by preparative HPLC-MS. The fractions were combined and frozen at −78 °C and lyophilized to yield 54 mg (35%, 0.041 mmol), purity 99%. HRMS (ESI+): m/z calculated for C66H93BF2N19NaO14, [M + Na]+ = 928.3800; found [M + Na]+ = 928.3817.
Synthesis of 7, Formazanate-PEG7-[DTyr6,βAla11,Phe13,Nle14]bombesin(6–14) amide
Compound 6 (12 mg, 0.013 mmol) was dissolved in dry DMF (0.5 mL). Compound 3 (18 mg, 0.013 mmol), 1-hydroxybenzotriazole hydrate (HOBt·H2O) (0.35 mg, 0.0026 mmol) and DIPEA (5 μL, 0.029 mmol) were added and the reaction mixture was cooled to 0 °C in ice bath. After 15 min N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl) (3 mg, 0.0026 mmol) was added and the reaction mixture was left on an ice bath for another 15 min. The reaction was then removed and stirred at r.t. for 16 h. The reaction mixture was diluted with CH2Cl2 (20 mL) and washed with deionised H2O (2 × 5 mL). The organic layer was isolated and dried over Na2SO4 and the solvent was removed on a rotary evaporator. The crude compound was then dissolved in CH3CN (0.1% TFA) and H2O (0.1% TFA) and purified by preparative HPLC-MS. The fractions were combined and frozen at −78 °C and lyophilized to yield 7 mg (25%, 0.0031 mmol), purity 98%. HRMS (ESI+): m/z calculated for C96H129BF2N23O22, [M + 2H]2+ = 1003.4950; found [M + 2H]2+ = 1003.5022.
Optical analysis
Studies were carried out in DMSO. UV absorption data were acquired using a Varian Carry 300 Bio UV-Vis spectrophotometer. Molar extinction coefficients were determined from the slope of a plot of absorbance against concentration using seven different solutions of known concentrations. Excitation and emission spectra were acquired using a Photon Technology International QM-4 SE spectrometer. Fluorescence quantum yields were determined using the comparative method described by Fery-Forgues and coworkers33 using the standard [Ru(bpy)3][PF6]2.34
Cell imaging studies
For 5: OVCAR-8 cells stably transfected with GHSR-1a (as previously described36) were released from the tissue culture flask by trypsin free dissociation buffer (Fischer Scientific) and seeded onto coverslips in a 12-well tissue culture plate at a cell density of 50 000 cells per well. The cells were incubated overnight in Roswell Park Memorial Institute medium (RPMI) containing 10% fetal bovine serum (FBS) at 37 °C with 5% CO2. The serum containing RPMI in each well was removed and replaced with serum free RPMI containing a concentration of 0.1 μM of compound 5 and incubated at 37 °C for 1 h. Cells were then washed three times with phosphate buffered saline (PBS), fixed with 4% paraformaldehyde in PBS and mounted onto slides containing Pro-Long Gold Antifade mounting medium with DAPI. For the control experiments, parental OVCAR-8 cells without GHSR-1a were incubated with 5.
Blocking studies with 8: OVCAR-8 cells with GHSR-1a were released from the tissue culture flask by trypsin free dissociation buffer and seeded onto coverslips in a 12-well tissue culture plate at a cell density of 50 000 cells per well. The cells were incubated overnight in RPMI containing 10% FBS at 37 °C with 5% CO2. The serum containing RPMI in each well was removed and replaced with serum free RPMI. Cells were then incubated with 0.1 μM of compound 5 at 37 °C for 1 h. 10-fold molar excess of compound 7 was used as blocking compound for these studies. Cells were then washed three times with PBS, fixed with 4% paraformaldehyde in PBS and mounted onto slides containing Pro-Long Gold Antifade mounting medium with DAPI.
For 7: PC-3 cells were released from the tissue culture flask by trypsin free dissociation buffer and seeded onto coverslips in a 12-well tissue culture plate at a cell density of 50 000 cells per well. The cells were incubated overnight in F-12K medium containing 10% fetal bovine serum (FBS) at 37 °C with 5% CO2. The serum containing F-12K in each well was removed and replaced with serum free F-12K containing a concentration of 0.5 μM of compound 7 respectively and incubated at 37 °C for 1 h. Cells were then washed three times with PBS, fixed with 4% paraformaldehyde in PBS and mounted onto slides containing Pro-Long Gold Antifade mounting medium with DAPI.
Blocking Studies with 3: PC-3 cells were released from the tissue culture flask by trypsin free dissociation buffer and seeded onto coverslips in a 12-well tissue culture plate at a cell density of 50 000 cells per well. The cells were incubated overnight in F-12K containing 10% FBS at 37 °C with 5% CO2. The serum containing F-12K in each well was removed and replaced with serum free F-12-K. Cells were then incubated with 0.5 μM of compound 7 respectively at 37 °C for 1 h. 10-fold molar excess of 3 was used as blocking compound for these studies. Cells were then washed three times with PBS, fixed with 4% paraformaldehyde in PBS and mounted onto slides containing Pro-Long Gold Antifade mounting medium with DAPI.
Competitive binding assay IC50
For 5: The affinity for GHSR-1a was determined using a radioligand binding assay. Assays were performed using HEK293 cells transiently transfected with GHSR-1a as receptor source and human [125I]-ghrelin(1–28) (PerkinElmer Inc.) as radioligand. Human ghrelin(1–28) was used as reference to ensure the validity of the results. Compound 5 (at concentrations of 10−5 M, 10−6 M, 10−7 M, 10−8 M, 10−9 M, 10−10 M, 10−11 M) and [125I]-ghrelin (15 000 cpm per assay tube) were mixed in binding buffer (25 mM HEPES, 5 mM MgCl2, 1 mM CaCl2, 2.5 mM EDTA, and 0.4% BSA, pH 7.4). A suspension of membranes from GHSR-1a transfected HEK293S cells (50 000 cells per assay tube) was added to the assay tube containing test peptides and [125I]-ghrelin(1–28). The resulting suspension was incubated for 20 min under shaking (550 rpm). Unbound [125I]-ghrelin was removed and the amount of [125I]-ghrelin bound to the membranes was measured on a gamma counter. The IC50 was determined by nonlinear regression analysis. All binding assays were performed in triplicate.
For 7: The affinity for GRPR was determined using a radioligand binding assay. Assays were performed using PC-3 cells and [125I]Tyr4-bombesin (PerkinElmer Inc.) as GRPR specific radioligand. PC-3 cells were grown in Ham's F-12K medium supplemented with 10% fetal bovine serum. Compound 7 (30 μL, at concentrations ranging from 10−12 to 10−6 M) and 20,0000 cpm of [125I]Tyr4-bombesin were mixed with the binding buffer (25 mM HEPES, 0.4% BSA, 5 mM MgCl2, 1 mM CaCl2, 2.5 mM EDTA in H2O, pH 7.4) in 1.5 mL Eppendorf vials. A suspension of 500 000 PC-3 cells in 50 μL binding buffer was added to each vial to give a final volume of 300 μL. The vials were shaken at 550 rpm for 1 h at 37 °C. Immediately after the incubation, the vials were centrifuged at 13 000 rpm, and the supernatant removed. The cell pellet was washed with 500 μL of 50 mM Tris buffer (pH 7.4), centrifuged again, and the supernatant was removed. The amount of [125I]Tyr4-bombesin bound to the cells was measured using a gamma counter (PerkinElmer). The IC50 was determined by nonlinear regression analysis. All binding assays were performed in triplicate.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
Funding gratefully received from the Natural Science and Engineering Research Council of Canada (NSERC).
Electronic supplementary information (ESI) available: UHPLC chromatograms, mass spectra, absorbance and emission spectra, IC50 binding curves. See DOI: 10.1039/d0ra02104k
Notes and references
- Ioffe V. M. Gorbenko G. P. Kinnunen P. K. Tatarets A. L. Kolosova O. S. Patsenker L. D. Terpetschnig E. A. J. Fluoresc. 2007;17:65–72. doi: 10.1007/s10895-006-0142-4. [DOI] [PubMed] [Google Scholar]
- Sednev M. V. Belov V. N. Hell S. W. Methods Appl. Fluoresc. 2015;3:042004. doi: 10.1088/2050-6120/3/4/042004. [DOI] [PubMed] [Google Scholar]
- Li Q. Liu W. Wu J. Zhou B. Niu G. Zhang H. Ge J. Wang P. Spectrochim. Acta, Part A. 2016;164:8–14. doi: 10.1016/j.saa.2016.03.042. [DOI] [PubMed] [Google Scholar]
- Hyka P. Züllig T. Ruth C. Looser V. Meier C. Klein J. Melzoch K. Meyer H. P. Glieder A. Kovar K. Appl. Environ. Microbiol. 2010;76:4486–4496. doi: 10.1128/AEM.02475-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatsu K. Yokomaku T. Kurata S. Kanagawa T. Nucleic Acids Res. 2004;32:e60. doi: 10.1093/nar/gnh054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N. Zhang B. Yan J. Xue X. Peng X. Li Y. Yang Y. Ju C. Fan C. Feng Y. Renew. Energy. 2015;77:579–585. doi: 10.1016/j.renene.2014.12.066. [DOI] [Google Scholar]
- Li H. Cai L. Li J. Hu Y. Zhou P. Zhang J. Dyes Pigm. 2011;91:309–316. doi: 10.1016/j.dyepig.2011.05.011. [DOI] [Google Scholar]
- Shin D. G. Kim M. Y. Yi W. Han J. W. Bull. Korean Chem. Soc. 2015;36:1037–1039. [Google Scholar]
- Pisoni D. S. Todeschini L. Borges A. C. A. Petzhold C. L. Rodembusch F. S. Campo L. F. J. Org. Chem. 2014;79:5511–5520. doi: 10.1021/jo500657s. [DOI] [PubMed] [Google Scholar]
- Gao M. Yu F. Lv C. Choo J. Chen L. Chem. Soc. Rev. 2017;46:2237–2271. doi: 10.1039/C6CS00908E. [DOI] [PubMed] [Google Scholar]
- Yuan L. Lin W. Zheng K. Hea L. Huang W. Chem. Soc. Rev. 2013;42:622–661. doi: 10.1039/C2CS35313J. [DOI] [PubMed] [Google Scholar]
- Mendive-Tapia L. Subiros-Funosas R. Zhao C. Albericio F. Read N. D. Lavilla R. Vendrell M. Nat. Commun. 2016;7:10940. doi: 10.1038/ncomms10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur P. Singh K. J. Mater. Chem. C. 2019;7:11361. doi: 10.1039/C9TC03719E. [DOI] [Google Scholar]
- Frath D. Massue J. Ulrich G. Ziessel R. Angew. Chem., Int. Ed. 2014;53:2290–2310. doi: 10.1002/anie.201305554. [DOI] [PubMed] [Google Scholar]
- Barbon S. M. Reinkeluers P. A. Price J. T. Staroverov V. N. Gilroy J. B. Chem.–Eur. J. 2014;20:11340–11344. doi: 10.1002/chem.201404297. [DOI] [PubMed] [Google Scholar]
- Barbon S. M. Staroverov V. N. Gilroy J. B. J. Org. Chem. 2015;80:5226–5235. doi: 10.1021/acs.joc.5b00620. [DOI] [PubMed] [Google Scholar]
- Maar R. R. Zhang R. Stephens D. G. Ding Z. F. Gilroy J. B. Angew. Chem., Int. Ed. 2019;58:1052–1056. doi: 10.1002/anie.201811144. [DOI] [PubMed] [Google Scholar]
- Maar R. R. Barbon S. M. Sharma N. Groom H. Luyt L. G. Gilroy J. B. Chem.–Eur. J. 2015;21:15589–15599. doi: 10.1002/chem.201502821. [DOI] [PubMed] [Google Scholar]
- Melenbacher A. Dhindsa J. S. Gilroy J. B. Stillman M. J. Angew. Chem., Int. Ed. 2019;58:15339–15343. doi: 10.1002/anie.201908999. [DOI] [PubMed] [Google Scholar]
- Barbon S. M. Novoa S. Bender D. Groom H. Luyt L. G. Gilroy J. B. Org. Chem. Front. 2017;4:178–190. doi: 10.1039/C6QO00640J. [DOI] [Google Scholar]
- Charron C. L. Hickey J. L. Nsiama T. K. Cruickshank D. R. Turnbull W. L. Luyt L. G. Nat. Prod. Rep. 2016;33:761–800. doi: 10.1039/C5NP00083A. [DOI] [PubMed] [Google Scholar]
- Müller T. D. Nogueiras R. Andermann M. L. Andrews Z. B. Anker S. D. Argente J. Batterham R. L. Benoit S. C. Bowers C. Y. Broglio F. Casanueva F. F. D'Alessio D. Depoortere I. Geliebter A. Ghigo E. Cole P. A. Cowley M. Cummings D. E. Dagher A. Diano S. Dickson S. L. Diéguez C. Granata R. Grill H. J. Grove K. Habegger K. M. Heppner K. Heiman M. L. Holsen L. Holst B. Inui A. Jansson J. O. Kirchner H. Korbonits M. Laferrère B. LeRoux C. W. Lopez M. Morin S. Nakazato M. Nass R. Perez-Tilve D. Pfluger P. T. Schwartz T. W. Seeley R. J. Sleeman M. Sun Y. Sussel L. Tong J. Thorner M. O. van der Lely A. J. van der Ploeg L. H. T. Zigman J. M. Kojima M. Kangawa K. Smith R. G. Horvath T. Tschöp M. H. Mol. Metab. 2015;4:437–460. doi: 10.1016/j.molmet.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima M. Kangawa K. Physiol. Rev. 2005;85:495–522. doi: 10.1152/physrev.00012.2004. [DOI] [PubMed] [Google Scholar]
- Kojima M. Hosoda H. Matsuo H. Kangawa K. Trends Endocrinol. Metab. 2001;12:118–122. doi: 10.1016/S1043-2760(00)00362-3. [DOI] [PubMed] [Google Scholar]
- Charron C. L. Hou J. McFarland M. S. Dhanvantari S. Kovacs M. S. Luyt L. G. J. Med. Chem. 2017;60:7256–7266. doi: 10.1021/acs.jmedchem.7b00164. [DOI] [PubMed] [Google Scholar]
- Markwalder R. Reubi J. C. Cancer Res. 1999;59:1152–1159. [PubMed] [Google Scholar]
- Chao C. Ives K. Hellmich H. L. Townsend C. M. Hellmich M. R. J. Surg. Res. 2009;156:26–31. doi: 10.1016/j.jss.2009.03.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott N. Millward E. Cartwright E. J. Preston S. R. Coletta P. L. J. Clin. Pathol. 2004;57:189–192. doi: 10.1136/jcp.2003.10660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S. M. Grandis J. R. Wentzel A. L. Gooding W. E. Lui V. W. Y. Siegfried J. F. Neoplasia. 2005;7:426–431. doi: 10.1593/neo.04454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young S. H. Rozengurt E. Am. J. Physiol. Cell Physiol. 2006;290:C728–C732. doi: 10.1152/ajpcell.00310.2005. [DOI] [PubMed] [Google Scholar]
- Cai Q. Y. Yu P. Besch-Williford C. Smith C. J. Sieckman G. L. Hoffman T. J. Ma L. Prostate. 2013;73:842–854. doi: 10.1002/pros.22630. [DOI] [PubMed] [Google Scholar]
- Murrell E. Kovacs M. S. Luyt L. G. ChemMedChem. 2018;13:1625–1628. doi: 10.1002/cmdc.201800334. [DOI] [PubMed] [Google Scholar]
- Fery-Forgues S. Lavabre D. J. Chem. Educ. 1999;76:1260–1264. doi: 10.1021/ed076p1260. [DOI] [Google Scholar]
- Suzuki K. Kobayashi A. Kaneko S. Takehira K. Yoshihara T. Ishida H. Shiina Y. Oishic S. Tobita S. Phys. Chem. Chem. Phys. 2009;11:9850–9860. doi: 10.1039/B912178A. [DOI] [PubMed] [Google Scholar]
- Rudolph J. Esler W. P. O'Connor S. Coish P. D. G. Wickens P. L. Brands M. Bierer D. E. Bloomquist B. T. Bondar G. Chen L. Chuang C. Y. Claus T. H. Fathi Z. Fu W. Khire U. R. Kristie J. A. Liu X. G. Lowe D. B. McClure A. C. Michels M. Ortiz A. A. Ramsden P. D. Schoenleber R. W. Shelekhin T. E. Vakalopoulos A. Tang W. Wang L. Yi L. Gardell S. J. Livingston J. N. Sweet L. J. Bullock W. H. J. Med. Chem. 2007;50:5202–5216. doi: 10.1021/jm070071+. [DOI] [PubMed] [Google Scholar]
- Lalonde T. Shepherd T. G. Dhanvantari S. Luyt L. G. Pept. Sci. 2018:e24055. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.