

Abstract
Three new triazole meso-arylporphyrins (4a–c) were synthesized by the copper(i)-catalyzed azide alkyne cycloaddition (CuAAC) “click” reaction in high yield. The corresponding zinc(ii) coordination compounds (5a–c) have also been prepared. All 4a–c and 5a–c porphyrin species were fully characterized by elemental analysis, electrospray ionization and MALDI-TOF mass spectrometry, infrared spectroscopy, proton nuclear magnetic resonance, UV-visible, fluorescence and cyclic voltammetry. The zinc(ii) 5a–c complexes have been tested as detectors for Cl− and Br− anions. UV-visible titrations reveal that these host systems exhibit strong anion binding affinities. The efficiency of the adsorption of the malachite green dye (MG) dye on the 4a–c free base porphyrins and the corresponding zinc(ii) complexes 5a–c was investigated by a kinetic study using these synthetic porphyrin derivatives as adsorbents. The use of our triazole Zn(ii) complexes in the catalytic degradation of the MG dye is the first example where a metalloporphyrin is involved in the MG dye decolorization reaction. The degradation reactions were carried out using an ecological oxidant (H2O2), where the efficiency of the decolorization has been characterized by UV-visible spectroscopic analysis. Several factors affecting the degradation phenomenon have been studied. The energetic parameters concerning the degradation process have also been determined.
Three triazole porphyrins and there corresponding zinc complexes are described as well as their spectroscopic and electrochemical data. The chloride and bromide sensing efficiency and the degradation of the malachite green dye are also reported.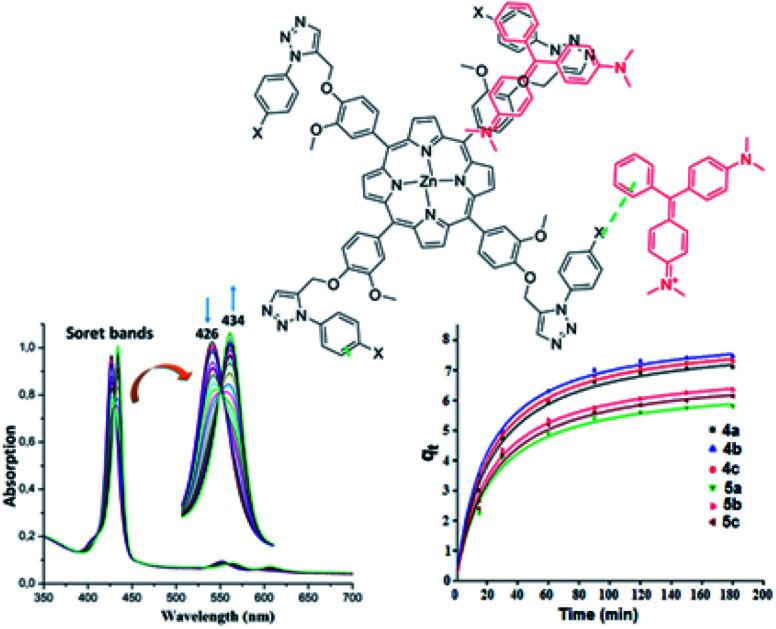
1. Introduction
In the fields of chemistry, biochemistry and material sciences, porphyrins and metalloporphyrins have been extensively studied due to their vast range of potential applications such as: photodynamic therapy, catalysis,1,2 as building blocks,3 in artificial photosynthetic systems4 and as sensors.5,6 The chemical modifications at the meso positions of porphyrins give rise to a large number of porphyrin derivatives with unique electronic and optical properties. Phosphines, pyridines, imidazoles and crown ethers, to name a few, have been used to functionalize meso-arylporphyrins.7–10 Since its introduction in 2001 by Sharpless et al.,11 the copper catalyzed azide–alkyne cycloaddition (CuAAC) known as “click chemistry” has been widely used to link moieties together in a very efficient and reliable way to prepare new functionalized meso-arylporphyrins. Beer et al.,12,13 reported the synthesis and the characterization of several functionalized meso-porphyrins such as the “porphyrin-cage” and the tetra-triazole-appended “picket-fence” zinc(ii) coordination compounds. These species were prepared to be tested as receptors for halide and other inorganic salts. Thus, the aim is to investigate the role of various substituents at the porphyrin meso-positions for receptor efficiency.
Interestingly, porphyrins and metalloporphyrins have been tested effectively in the degradation of chemical dyes. Indeed, these species constitute a large part of water pollutants due to the sewage discharge containing carcinogenic and chronically toxic organic dyes,14 which are extensively used in textile, printing and photographic industries. During the late decade, several free bases porphyrins have been used as catalysts in presence of TiO2, in the photocatalytic degradation of dyes, e.g. the meso-arylporphyrins substituted by phenyl carboxylic acid groups and di-tert-butyl-substituted phenyl rings have been used in the degradation of the methylene blue (MB) dye.15 Metal porphyrins such as the manganese(iii) metalloporphyrin based polymers have also been used in the methylene blue dye (MB) degradation in an aqueous solution of hydrogen peroxide.16 We also reported the use of two 4-cyanopyridine–cobaltous-porphyrin complexes type [CoII(Porph)(4-CNpy)] (Porph = meso-tetrakis(para-methoxyphenyl)porphyrinato and meso-tetra(para-chlorophenyl)porphyrin) in the same MB dye degradation in presence of H2O2.17 In 2018, we reported the catalytic degradation of the Calmagite dye using an aqueous solution of H2O2 with the (4,4′-bipyridine)[(tetrakis(ethyl-4(4-butyryl)oxyphenyl)porphyrinato)]zinc(ii) complex.18
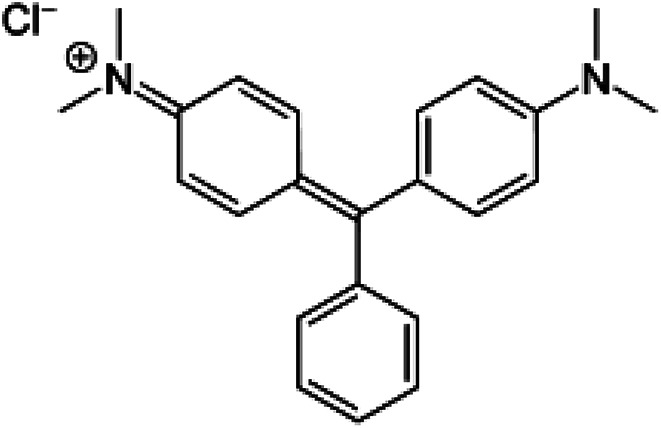
Among dyes, the malachite green (MG) dye, with the formula [C6H5C(C6H4N(CH3)2)2]Cl (Scheme 1) is widely used in the textile and paper industry. The [C6H5C(C6H4N(CH3)2)2]+ cation which gives the very intense green color to this dye exhibits a very strong absorption band at 621 nm.
Scheme 1. Structure of the malachite green (MG) dye.
During the past two decades, reports on the enduring carcinogenic and mutagenic characteristics of MG dye have increased.19 Since 2000, the MG dye has been listed as a priority chemical for carcinogenicity assessment by the U.S. Food and Drug Administration.20 However, due to its efficacy and low cost, it is still used in many countries. Therefore, many treatment technologies have been applied to decolorize MG from aqueous medium.21
Here, we describe the preparation of three new substituted meso-arylporphyrins using the “click chemistry” strategy and their zinc(ii) corresponding complexes namely: the meso-tetrakis(3-methoxy-4-((1-phenyl-1H-1,2,3-triazol-4-yl)methoxy)phenyl)porphyrin (4a) [H2(TAzP-HVP)], the meso-tetrakis(4-((1-(4-chlorophenyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-methoxyphenyl)porphyrin (4b) [H2(TAzP-ClVP)], and the meso-tetrakis(4-((1-(4-iodinephenyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-methoxyphenyl)porphyrin (4c) [H2(TAzP-IVP)] and the corresponding zinc(ii) metalloporphyrins: 5a–c with the formulas [Zn(TAzP-HVP)], [Zn(TAzP-ClVP)] and [Zn(TAzP-IVP)], respectively (Scheme 2).
Scheme 2. Structure of [Zn(TAzP-XVP)] complexes. 5a: X = H, 5b: X = Cl and 5c: X = I.

All these porphyrin derivatives have been characterized by UV-visible, fluorescence, IR and 1H NMR spectrometry, as well as by mass spectrometry and cyclic voltammetry. The second part of this paper concerns the study of the chloride and bromide ions receptor by the zinc(ii) chloro–triazole and iodine–triazole porphyrin complexes 5b–c. The other important goal of the present work is to study the efficiency of the free base porphyrins 4a–c and the corresponding zinc(ii) complexes 5a–c in the adsorption of the malachite green (MG) dye. The catalytic oxidative degradation efficiency of the MG dye using the three triazole meso-arylporphyrin zinc(ii) compounds 5a–c have also been described which is the first example of the use of a porphyrinic compound for the degradation of this dye.
2. Results and discussion
2.1. Synthesis
Scheme 3 depicts the overall synthetic route for the preparation of meso-tetrakis(3-methoxy-4-((1-phenyl-1H-1,2,3-triazol-4-yl)methoxy)phenyl)porphyrin (4a) [H2(TAzP-HVP)], the meso-tetrakis(4-((1-(4-chlorophenyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-methoxyphenyl)porphyrin (4b) [H2(TAzP-ClVP)] and the meso-tetrakis(4-((1-(4-iodinephenyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-methoxyphenyl)porphyrin (4c) [H2(TAzP-IVP)], as well as the corresponding zinc(ii) metalloporphyrins 5a–c with the formulas [Zn(TAzP-HVP)], [Zn(TAzP-ClVP)] and [Zn(TAzP-IVP)], respectively (see synthetic procedures in the ESI†). The first step is the preparation of the 3-methoxy-4-(prop-2-ynl-yloxy)benzaldehyde (2). This derivative was prepared by reacting the 3-hydroxy-3-methoxy-benzaldehyde (1) with K2CO3 and the propargyl bromide in acetone under reflux for 4 hours. The second step is the synthesis of the three substituted [1,2,3](triazol-4-ylmethoxy)-benzaldehyde: 3-methoxy-4-(3-phenyl-3H-[1,2,3]triazol-4-ylmethoxy)-benzaldehyde (3a), the 4-[3-(4-chloro-phenyl)-3H-[1,2,3]triazol-4-ylmethoxy]-3-methoxy-benzaldehyde (3b) and the (4-iodine-phenyl)-3H-[1,2,3]triazol-4-ylmethoxy]-3-methoxy-benzaldehyde (3c). These compounds were prepared using the so-called “click reaction” (CuAAC) by reacting the 3-methoxy-4-(prop-2-ynl-yloxy)benzaldehyde (2) with the 1-azidobenzene or the 1-azido-4-chlorobenzene or the 1-azido-4-iodinebenzene and the diisopropylethylamine (DIPEA) reagents in the presence of copper(i) (Cui) catalyst.22 The success of the click reaction is verified in IR by the disappearance of the two absorption bands of the azido group (νas(N3) ∼ 2130 and ∼2080 cm−1) and the alkyne group of compound 2 and the appearance of a new band at 1726 cm−1 assigned to ν(C N, N N) of the triazole group (Fig. SI-2†). The three triazole-meso-porphyrins 4a–c were prepared using a slightly modified Adler–Longo procedure.23 The metalation by the zinc(ii) of the three 4a–c porphyrins were made according to the literature method24 using the zinc(ii) acetate salt Zn(OAc)2·2H2O leading to the three zinc(ii) metalloporphyrins [Zn(TAzP-HVP)] (5a), [Zn(TAzP-ClVP)] (5b) and [Zn(TAzP-IVP)] (5c). The three free base porphyrins 4a–c and the corresponding zinc(ii) metalloporphyrins 5a–c were characterized by UV-visible, fluorescence, IR and 1H NMR spectroscopies as well as by mass spectrometry.
Scheme 3. Synthesis of aldehydes 2–3a–c, the free base porphyrins 4a–c and the zinc(ii) porphyrin complexes 5a–c.

2.2. Mass spectrometry
All synthetic compounds were characterized by ESI or MALDI-TOF mass spectrometry (see ESI†). The MALDI-TOF spectra of our three zinc(ii) metalloporphyrins [Zn(TAzP-HVP)], [Zn(TAzP-ClVP)] and [Zn(TAzP-IVP)] (5a–c), shown in Fig. SI-1,† were recorded in the tetrahydrofuran solvent using the trans-2-[3-(4-tert-butylphenyl)-2-propenylidene]malonitrile (DCTB) as matrix. For all three zinc(ii) porphyrin species, the [M]+ and [M + nH]n+ fragments were observed which is a clear indication of the stability of the 5a–c compounds in THF solutions.
2.3. IR and 1H NMR investigation
The IR spectra of the 4a–c free base porphyrins and the corresponding zinc(ii) metallated species 5a–c confirm the formation of the triazole meso-arylporphyrins as well as the corresponding zinc(ii) coordination compound. Indeed, the values of the ν(CH) stretching frequencies of the C__H bonds of the triazole and the aryl groups of H2(TAzP-HVT) (4a), H2(TAzP-ClVT) (4b) and H2(TAzP-IVT) (4c) are ∼3070 cm−1 and in the [2955–2854] cm−1 range, respectively (Fig. SI-3†). For the zinc(ii) corresponding complexes 5a–c (Fig. 1), the ν(CH) stretching frequencies values are practically the same as those of the free base porphyrins 4a–c. The presence of the triazole fragment is confirmed by a strong absorption band at ∼1725 cm−1 for both free base porphyrins 4a–c and the corresponding zinc(ii) complexes 5a–c. This band is attributed to ν(N N) and ν(C N) of the triazole group.25 The metallation of 4a–c porphyrins are confirmed by (i) the disappearance of the absorption band correspondent to the ν(N__H) stretching frequency of the free base porphyrins (∼3280 cm−1) and (ii) by the shift of the deformation frequency δ(CCH) from ∼965 cm−1 (4a–c) to ∼1000 cm−1 for the zinc(ii) complexes 5a–c (Fig. SI-3† and 1).
Fig. 1. IR spectra (solid state) of [Zn(TAzP-HVP)] (5a) (red color), [Zn(TAzP-ClVP)] (5b) (black color) and [Zn(TAzP-IVP)] (5c) (blue color).

The proton NMR spectra of the three zinc(ii) complexes 5a–c are shown in Fig. SI-4–6.† The β-pyrrolic protons of the three porphyrinates in complexes 5a–c resonate at about 9 ppm while the phenylic protons resonate between 7.19 and 7.71 ppm. For these three 5a–c species, the chemical shift values of the triazole protons are ∼8 ppm and those of the O–CH2-triazole groups are ∼5.4 ppm. The methoxy groups for 5a–c presents singlets at about 4.0 ppm.
2.4. Photophysical properties
The electronic absorption spectra of the free base porphyrins 4a–c and the corresponding zinc(ii) metallated porphyrin species (5a–c) are depicted in Fig. SI-7 and SI-8† while the UV-visible data are given in Table 1. The λmax values of the Soret band of the free base porphyrins 4a–c are ∼420 nm while those of the Q bands are ∼518, ∼550, ∼590 and ∼650 nm which are typical for meso-arylporphyrins. For the tetracoordinated zinc(ii) coordination compounds 5a–c type [Zn(Porph)] (Porph = TAzP-HVP or TAzP-ClVP or TAzP-IVP porphyrinates), the Soret and the Q bands of these metalloporphyrins are, as expected, slightly redshifted compared to the corresponding free base porphyrins.
UV-visible data of the three triazole meso-arylporphyrins (4a–c) and a selection of several meso-arylporphyrins and zinc(ii) tetracoordinated metalloporphyrins. The solvent used is the dichloromethane.
| Free base meso-arylporphyrins | |||||||
|---|---|---|---|---|---|---|---|
| Compound | λ max (nm) (ε × 10−3 L mmol−1 cm−1) | E gap-opt (eV) | Ref. | ||||
| Soret band | Q bands | ||||||
| H2(TPP)a | 416(419) | 513(20) | 550(20) | 590(6) | 646(6) | 1.89 | 26 |
| H2(TTP)b | 420 | 516 | 552 | 594 | 640 | 1.86 | 27 |
| H2(TPBP)c | 420(513) | 516(17) | 552(7) | 591(5) | 646(4) | 1.82 | 4 |
| H2(TEBOP)d | 422(295) | 517(9) | 554(8) | 593(5) | 651(7) | 1.85 | 18 |
| H2(TAzP-HVP) 4a | 420(565) | 517(42) | 554(19) | 593(15) | 650(13) | 1.88 | This work |
| H2(TAzP-ClVP) 4b | 422(551) | 518(35) | 555(26) | 594(15) | 651(17) | 1.87 | This work |
| H2(TAzP-IVP) 4c | 424(576) | 520(46) | 555(29) | 595(24) | 652(18) | 1.86 | This work |
TPP = meso-tetraphenylporphyrinato.
TTP = meso-tetratolylporphyrinato.
TPBP = meso-tetrakis-[4-(benzoyloxy)phenyl]porphyrinato.
TEBOP = meso-tetrakis(ethyl-4(4-butyryl)oxyphenyl)porphyrinato.
TMP = meso-tetrakis(2,4,6-trimethylphenyl)porphyrinato.
| Zinc(ii) meso-arylporphyrin complexes | |||||
|---|---|---|---|---|---|
| Compound | λ max (nm) (ε × 10−3 L mmol−1 cm−1) | E gap-opt (eV) | Ref. | ||
| Soret band | Q bands | ||||
| [Zn(TPP)]a | 421(524) | 550(21) | 591(25) | 1.91 | 28 |
| [Zn(TMP)]e | 420 | 550 | 586 | — | 29 |
| [Zn(TPBP)]c | 425(546) | 554(24) | 596(9) | 1.99 | 24 |
| [Zn(TEBOP)]d | 424(219) | 552(11) | 594(5) | 2.03 | 18 |
| [Zn(TAzP-HVP)] 5a | 424(530) | 551(26) | 592(10) | 2.04 | This work |
| [Zn(TAzP-ClVP)] 5b | 426(523) | 552(31) | 594(15) | 2.02 | This work |
| [Zn(TAzP-IVP)] 5c | 426(532) | 558(29) | 597(12) | 2.01 | This work |
Notably, the UV-visible data of our three free bases and the corresponding zinc metallated porphyrins are very close to those of the reported zinc(ii) related species (Table 1). The optical gap values (Eg-op) of our synthetic compounds 4a–c and 5a–c were determined using the Tauc's relation (eqn (1)):30,31
| (αhν)2 = A[hν − Eg-op] | 1 |
where A is a constant parameter depending on transition probability, hν is the incident photon energy and α is the optical absorption coefficient deduced from absorbance data. The intercepts between the solid lines and x-axis allow the determination of Eg-op values (Fig. SI-9†). The optical gap energy values of the free bases 4a–c are 1.88, 1.87 and 1.86 eV, respectively, while those of the corresponding zinc(ii) complexes 5a–c are 2.05, 2.02 and 2.01 eV, respectively. The fact that the Eg-op values of the metallated porphyrins are higher than those of the free bases is mainly due to the higher flexibility of the non metallated porphyrins which leads to the destabilization of the HOMO–LUMO orbital energies. Thus, the net result is the reduction of the gap energy values of these meso-porphyrins. However, the zinc metallation of these tetradentate ligands decreases the distortion of the porphyrin core. Therefore, the values of the optical energy of the metalloporphyrins are usually higher than those of the non metallated porphyrins.32 We notice that both 4a–c porphyrins and 5a–c Zn(ii) metalloporphyrins present Eg-op values very close to the related meso-arylporphyrins and zinc(ii) meso-arylporphyrins. Fig. 2 illustrates the fluorescence spectra of 4a–c and 5a–c, while the photoluminescence data of these porphyrin species are given in Table 2.
Fig. 2. The emission spectra of compounds 4a–c and 5a–c. The spectra were recorded in dichloromethane solvent with a concentration ∼10−6 M. The excitation wavelength value is 430 nm.

Emission parameter values of several meso-arylporphyrins and a selection of zinc(ii) meso-metalloporphyrins.
| Compound | λ max (nm) | Φ f a | τ f b (ns) | Ref. | |
|---|---|---|---|---|---|
| Q(0, 0) band | Q(0, 1) band | ||||
| Free base meso-arylporphyrins | |||||
| H2(TAzP-HVP) 4a | 655 | 719 | 0.091 | 8.82 | This work |
| H2(TAzP-ClVP) 4b | 651 | 715 | 0.076 | 7.18 | This work |
| H2(TAzP-IVP) 4c | 650 | 714 | 0.071 | 7.12 | This work |
| H2(TPP)c | 656 | 717 | 0.09 | — | 33 |
| H2(TPP)c | 653 | 712 | 0.12 | 9.6 | 34 |
| H2(TMPP)d | 656 | 719 | 0.082 | 7.16 | 35 |
| H2(TClPP)e | 651 | 714 | 0.089 | 7.42 | 26 |
| Tetracoordinated zinc( ii ) meso-metalloporhyrins | |||||
| [Zn(TAzP-HVP)] 5a | 601 | 649 | 0.051 | 1.96 | This work |
| [Zn(TAzP-ClVP)] 5b | 600 | 648 | 0.035 | 1.78 | This work |
| [Zn(TAzP-IVP)] 5c | 599 | 647 | 0.032 | 1.51 | This work |
| [Zn(TPP)]c | 597 | 647 | 0.033 | 1.90 | 35 |
| [Zn(TTP)]f | 600 | 648 | 0.030 | 1.60 | 36 |
Φ f = fluorescence quantum yield.
τ f = fluorescent lifetime.
H2(TPP) = meso-tetraphenylporphyrin.
H2(TMPP) = meso-tetra(para-methoxy)phenylporphyrin.
H2(TClPP) = meso-tetra(para-chlorophenyl)porphyrin.
TTP = meso-tetratolylporphyrinato.
Importantly, it should be noticed that porphyrin derivatives exhibit interesting photophysical properties especially due to the significant aromaticity of the porphyrin macrocycle. Indeed, these species present one very weak emission transition S2 → S0 of the Soret band between the second excited singlet state S2 and the ground state S0. The second emission transition of porphyrin and metalloporphyrins, which is much stronger than the first transition, is the S1 → S0 of the Q bands between the first excited singlet state S1 to the ground state S0. Therefore, only the following two S1 → S0 transitions are observed for the free base porphyrins and metalloporphyrin complexes: S1[Q(0, 0)] → S0 and S1[Q(0, 1)] → S0. For the three free base porphyrin species 4a–c, the emission spectra show two bands: the weak band at λmax ∼ 715 nm is assigned as Q(0, 1), and the stronger band at λmax ∼ 650 nm is assigned as Q(0,0) (Fig. 2). The values of each λmax of our three derivatives are very close to those of the reported meso-arylporphyrins (Table 2). The hypochromic shifts of the Q(0, 0) and the Q(0, 1) of the zinc(ii) metalloporphyrins 5a–c compared to those of the corresponding free bases 4a–c are important for consideration. The λmax values of the Q(0, 0) band are ∼600 nm, while the λmax values of the Q(0, 1) bands are ∼650 nm. These shifts are mainly due to the metallation of the porphyrins.37 The Stocks shifts values of the free bases 4a–c and the 5a–c zinc(ii) metalloporphyrins are small, indicating a fluorescence emission with no significant conformational change between the fundamental and excited states.
The fluorescence properties were studied by both steady-state and time-resolved fluorescence techniques. As indicated in Table 2, the fluorescence quantum yield (Φf) value of the H2TAzP-HVP (4a) is slightly higher than those of the chloro–triazole and iodine–triazole free base derivatives H2TAzP-ClVP (4b) and H2TAzP-IVP (4c) with Φf values of 9.1%, 7.6% and 7.1%, respectively, which could be attributed to the higher molar mass of the iodine (in 4c) and chlorine (in 4b) atoms compared to that of the hydrogen atom (in 4a). Notably, the Φf value of the 4a porphyrin is comparable with that of the meso-tetraphenylporphyrin H2TPP (Φf ∼ 10) (Table 2). As expected the Φf values of our three zinc(ii) porphyrins 5a–c which are 5.1%, 3.5 and 3.1%, respectively, are quite smaller than those of the related free base porphyrins 4a–c. We also notice that the iodine–triazole and chloro–triazole zinc(ii) derivatives exhibit slightly higher fluorescence quantum yields than that of the H-triazole porphyrin species. The lifetime of singlet excited state (τf) was measured by the single photon counting technique, and the fluorescence decays were fitted to single exponentials. The representative fluorescence decays (τf) of these four derivatives are similar (see Fig. SI-10†). Our [Zn(Porph)] complexes (5a–c) display, as expected, τf values between 7.2 and 8.82 ns which are much higher than the corresponding free bases (4a–c) with values in the range 1.51 and 1.96 ns.
2.5. Cyclic voltammetry
Cyclic voltammograms (CV) of our porphyrin derivatives 4a–c and 5a–c were recorded at room temperature in a mixture of dichloromethane and acetonitrile (4/1) under an argon atmosphere with tetrabutylammonium hexafluorophosphate (TBAPF6) as the supporting electrolyte (0.1 M). The CV of the free base porphyrins 4a–c are illustrated in Fig. SI-11,† while those of the corresponding zinc(ii) complexes 5a–c are depicted in Fig. 3. The electrochemical data of our synthetic porphyrin species and several related compounds are given in Table 3. The free bases 4a–c exhibit two reversible one-electron reduction waves and two reversible one-electron oxidation waves, which correspond to the reduction and the oxidation of the porphyrin ring. The half potential values (E1/2) of the three triazole meso-arylporphyrins 4a–c are much smaller than those of the unsubstituted meso-tetraphenylporphyrin (H2TPP) (Table 3). Neya et al.,38 reported that the second reduction of the porphyrin ring in metalloporphyrins, with non-redox-active cationic metal centers such as Zn(ii) and Mg(ii), is usually not observed in the electrochemical window of the solvent which is the case for our zinc(ii) derivatives 5a–c. It was also reported that the zinc metallation of the meso-arylporphyrins leads to the shifts of the characteristic reduction and oxidation potentials to more negative values.29,39 In our case, only the E1/2 values of the first reduction waves for [Zn(TazP-HVP)], [Zn(TazP-ClVP)] and [Zn(TazP-IVP)] (5a–c) are shifted to more negative values (Table 3).
Fig. 3. Cyclic voltammograms of complexes 5a–c. The solvent is a mixture of dichloromethane and acetonitrile (4/1) and the concentration is ca. 10−3 M in 0.1 M TBAPF6, 100 mV s−1, vitreous carbon working electrode (∅ = 2 mm).

Electrochemical dataa for the three free base triazole meso-arylporphyrins 4a–c and the corresponding zinc(ii) complexes 5a–c and a selection of several related porphyrins species.
| Compound | Oxidations | Reductions | Ref. | ||
|---|---|---|---|---|---|
| First oxid. | Second oxid. | First red | Second red. | ||
| (O1, R1) | (O2, R2) | (R3, O3) | (R4, O4) | ||
| E 1/2 b | E 1/2 | E 1/2 | E 1/2 | ||
| H2(TAzP-HVP) 4a | 0.86 | 1.09 | −0.95 | −1.36 | This work |
| H2(TAzP-ClVP) 4b | 0.85 | 1.08 | −0.76 | −1.40 | This work |
| H2(TAzP-IVP) 4c | 0.78 | 1.13 | −0.82 | −1.37 | This work |
| H2(TPP)c | 1.02 | 1.26 | −1.20 | −1.55 | 40 |
| H2(TBPP)d | 0.95 | 1.36 | −1.12 | −1.53 | 24 |
| [Zn(TAzP-HVP)] 5a | 0.72 | 1.08 | −1.36 | — | This work |
| [Zn(TAzP-ClVP)] 5b | 0.71 | 1.09 | −1.37 | — | This work |
| [Zn(TAzP-IVP)] 5c | 0.72 | 1.05 | −1.37 | — | This work |
| [Zn(TPP)]c | 0.89 | 1.18 | −1.26 | −1.70 | 41 |
| [Zn(TBPP)]d | 0.81 | 1.11 | −1.48 | −1.76 | 24 |
| [Zn(DTPP)]e | 0.72 | 1.15 | −1.42 | — | 40 |
The potentials are reported versus SCE.
E 1/2 = half wave potential.
TPP = meso-tetraphenylporphyrinato.
H2TBPP = meso-{tetrakis-[4-(benzoyloxy)phenyl]porphyrin.
DTPP = 5,15-p-ditolyl-10-phenylporphyrinato.
In short, the CV investigation on our synthetic 4a–c and 5a–c porphyrin species show that the nature of the substituted groups on the phenyls of the meso-arylporphyrins do not have a very significant effect on the electrochemical properties of this type of species.
Ghosh et al.,42 estimated the electrochemical gap energy (Eg-el) as the difference between the potential of the first oxidation potential and the first reduction potential of the porphyrin. The estimated energy values of the HOMO and LUMO orbitals as well as the value of the Eg-el can be calculated using the following equations (eqn (2)–(4)):43
| EHOMO = −(Vonset-ox − Vref + 4.8) eV | 2 |
| ELUMO = −(Vonset-red − Vref + 4.8) eV | 3 |
| Eg-el = (ELUMO − EHOMO) eV | 4 |
The EHOMO and the ELUMO are also known as the ionization potential (IP) and the electron affinity (EA), respectively. The Vonset-ox and the Vonset-red are the oxidation onset and the reduction onset, respectively and the Vref is the reference half-wave potential. The values of EHOMO,ELUMO and Eg-el were calculated using equations eqn (2)–(4) and are reported in Table 4 along with those of several related compounds. The energy level of the HOMO and LUMO orbitals as well as the Eg-el of 4a–c and 5a–c are illustrated in Fig. SI-12.† From Table 4 we can deduce: (i) except for the case of the H2(TPBP) porphyrin, the electrochemical gap energy values of the zinc metalloporphyrins are higher than those of the free base porphyrins; (ii) as expected, the values of the Eg-el of 4a–c and 5a–c are higher than those of the Eg-op (optical gap energy) of the same species, which is also the case for all known meso-arylporphyrins and the corresponding [Zn(Porph)] complexes24 and (iii) the iodine–triazole fee base porphyrin 4c exhibits the smallest Eg-el gap value (1.77 eV) compared to those of 4a–b (1.95 and 1.87 eV, respectively) while the three zinc(ii) metallated porphyrins 5a–c presents very close values of Eg-el ∼ 2.14 eV.
Values of the HOMO, LUMO orbitales energies and the electrochemical gap energy for a selection of porphyrins species.
| Compound | E HOMO (eV) | E LUMO (eV) | E g-el (eV) | Ref. |
|---|---|---|---|---|
| Free base meso-arylporphyrins | ||||
| H2(TPP)a | −5.50 | −3.49 | 2.01 | 44 |
| H2(TPBP)b | −5.87 | −3.73 | 2.14 | 24 |
| H2(TAzP-HVP) 4a | −5.40 | −3.45 | 1.95 | This work |
| H2(TAzP-ClVP) 4b | −5.53 | −3.66 | 1.87 | This work |
| H2(TAzP-IVP) 4c | −5.32 | −3.56 | 1.77 | This work |
| Zinc( ii ) meso-arylporphyrin tetracoordinated complexes | ||||
| [Zn(TPP)]a | −5.58 | −3.46 | 2.12 | 45 |
| [Zn(TPBP)]b | −5.64 | −3.27 | 2.37 | 24 |
| [Zn(TAzP-HVP)] 5a | −5.24 | −3.09 | 2.15 | This work |
| [Zn(TAzP-ClVP)] 5b | −5.23 | −3.09 | 2.14 | This work |
| [Zn(TAzP-IVP)] 5c | −5.24 | −3.11 | 2.13 | This work |
TPP = meso-tetraphenylporphyrinato.
TBPP = meso-tetrakis-[4-(benzoyloxy)phenyl]porphyrinato.
2.6. Anion binding studies
The zinc(ii) complexes 5b–c with the chloro–triazole and the iodine–triazole meso-porphyrinates were tested as Cl− and Br− anions detector by UV-visible titration. The two halide ions titrations, carried out in dichloromethane solvent, revealed significant perturbation of the Soret and the Q bands of the zinc(ii) 5b–c receptors as a function of the concentration of the tert-butyl ammonium chloride and bromide salts (TBACl and TBABr). In the literature, several UV-visible titrations involving the tetracoordinated zinc(ii) meso-porphyrins type [Zn(Porph)] (Porph = meso-arylporphyrin) and neutral N-donor ligands are reported.18 These investigations indicate the presence of a redshift of the Soret and the Q bands, upon addition of the axial ligand, of about 10 nm and 5 nm for the two bands, respectively, and the formation of a 1 : 1 coordination complex type [Zn(Porph)(L)] (L = N-neutral axial ligand).
The UV-visible titration spectra of [Zn(TAzP-ClVP)] (5b) (concentration about 10−6 M) with Cl−, in the Soret region, are shown in Fig. 4 and 5. The gradual addition of chloride to 5b leads to a bathochromic shift of the Soret band from 426 nm to 436 nm (Δλmax = 10 nm) with one distinct isosbestic point at 431 nm. In the Q region, the titration of the same complex 5b with Cl− exhibits also a redshift of the Q(0, 0) and Q(0, 1) bands. The titration of the same complex 5b with the bromide anion, using the same solvent and concentration as the Cl− titration, gives a comparable result with a redshift of the Soret and the Q bands (Fig. 4). The isosbestic points are 431 nm for the Soret region and 561 and 594 nm for the Q bands region. These values are practically the same as those of the Cl− titration.
Fig. 4. Changes in the absorption spectra in the Soret band region of complex 5b (∼10−6 M) recorded in dichloromethane, upon addition of Cl− (0–500 equiv.). The inset shows enlarged view.

Fig. 5. Changes in the absorption spectra in the Soret band region of complex 5b (∼10−6 M) recorded in dichloromethane, upon addition of Br− (0–600 equiv.). The inset shows enlarged view.

For the zinc iodine–triazole complex [Zn(TAzP-IVP)] (5c), the titrations with both the Cl− and Br− halide ions are very similar to each other and to those of the zinc chloro–triazole complex 5b (Fig. 6 and 7). As mentioned above, the presence of isosbestic points is an indication of formation of a 1 : 1 receptor/anion adduct, i.e., leading to a pentacoordinate complex type [Zn(Porph)(L)]. The redshift of the absorption bands of metalloporphyrins is due to the narrowing of the HOMO–LUMO and the energy gap is explained by the deformation of the porphyrin core.46 This bathochromic effect is also related to the electron-withdrawing groups at the meso and β-pyrrolic positions of the porphyrin macrocycle and to the nature of the metal ion.47,48
Fig. 6. Changes in the absorption spectra in the Soret band region of complex 5c (∼10−6 M) recorded in dichloromethane, upon addition of Cl− (0–30 equiv.). The inset shows enlarged view.

Fig. 7. Changes in the absorption spectra in the Soret band region of complex 5c (∼10−6 M) recorded in dichloromethane, upon addition of Br− (0–30 equiv.). The inset shows enlarged view.

In order to compare the Cl− and Br− detecting properties of our zinc complexes 5a–b with those of the unsubstituted zinc(ii)-meso-tetraphenylporphyrin ([Zn(TPP)]) we also performed a UV-visible titration in the Soret band region of this complex with Cl− and Br− halide ions. The spectra concerning these titrations are shown in Fig. SI-13.† The association constants values of the 1 : 1 complexes 5a–b : X (X = Cl− and Br−) as well as those of the [Zn(TPP)X] compounds are reported in Table SI-1.† To calculate these associate constants Kas (and log Kas) we use the so-called “strong interactions” method49 (see the ESI† for details).
A close inspection of Table SI-1† indicates that the iodine–triazole zinc derivative 5c presents a better binding affinity than the chloro–triazole zinc metalloporphyrin 5b for both Cl− and Br− anions. This could be explained by the higher donor effect of the chlorine in the para position of the TAzP-ClVP porphyrinate than that of the iodine atom in the TAzP-IVP moiety leading to a higher electron density on the zinc(ii) center metal for 5b. Therefore, the bonding affinity of zinc(ii) in 5b for the two halides is smaller than that of the iodine–triazole zinc TAzP-IVP receptor. In contrast, the value of the log Kas for the 5b receptor for Cl− is higher than that for Br− which are 4.0245 and 3.5512, respectively. This is not the case for the 5c receptor for which log Kas value, in the case of the Br− anion, is higher than that for the Cl− anion; i.e. log Kas is 4.6179 and 5.0566, respectively.
Notably, our two receptors present much better affinity for Cl− and Br− than the zinc(ii) unsubstituted meso-tetraphenylporphyrin complex ([Zn(TPP)]) used as reference (Table SI-1†).
Beer et al.,12 reported the use of a zinc(ii) cage porphyrin complex [Zn(PC)] (PC = meso-tetrayltetrakis(carbonyloxymethanediyl-1H-1,2,3-triazole-4,1-diylmethanediyl)tetraphenyl)porphyrin) as receptor of several anions (such as F−, Cl−, Br−, I− and SO42−). This later complex presents a high binding affinity for Cl− (log Kas = 4.0860), but a very small association constant Kas value (<50) for Br−. This can be explained by the small size of the porphyrin cage which is not accommodated to host the highly diffuse bromine ion. It is interesting to note (i) that our zinc(ii) chlorine porphyrin derivative (5b) has a similar bonding affinity for Cl− ion as the [Zn(PC)] related receptor with very close log Kas values (4.0224 and 4.0860, respectively) and (ii) our iodine–triazole zinc derivative (5c) exhibits much better bonding affinity for Cl− anion than the zinc-porphyrin cage species ([Zn(PC)] with log Kas value of 4.6179 for our 5c complex compared to log Kas value of 4.0860 [Zn(PC)].
2.7. Adsorption
The adsorption process is generally due to several physicochemical forces that occur at the solid–liquid interface such as van der Waals forces,50,51 hydrogen bonds52 and hydrophobic interactions.53,54 To determine the efficiency of adsorption of the MG dye on the 4a–c free base porphyrins and the corresponding zinc(ii) complexes 5a–c, we carried out a kinetic study using our synthetic porphyrin derivatives as adsorbents. The adsorption spectrum of MG dye as a function of time in presence of 4a–c and 5a–c species are shown in Fig. SI-14.† The adsorption efficiency (R%) of the MG dye on 4a–c and 5a–c porphyrin compounds are depicted in Fig. 8. The R% values for the 4a–c free base porphyrins are 35.5, 37.25 and 36.5%, while those for the 5a–c complexes are 29, 31.75 and 30.75%, respectively. The fact that the adsorption efficiency of the free base porphyrins are higher than those of the corresponding [Zn(Porph)] complexes 5a–c could be related to the pyrrole hydrogen atoms in 4a–c leading to hydrogen bond type interactions with the nitrogens of the MG dye molecules. Notably, the chloro–triazole and iodine–triazole 4b–c free base meso-arylporphyrin derivatives present better adsorption than that of the H-triazole TAzP-HVP species (4a). This trend is also present in the corresponding zinc(ii) porphyrin complexes (5a–c). This might result from the fact that the chlorine and iodine atoms in the case of the 4b–c and 5b–c species are involved in a n–π type interactions with the MG molecules, which is not possible in the case of the H-triazole 4a and the chloro–triazole 5a porphyrin species (Fig. SI-15†).55 It should also be noticed that all 4a–c and 5a–c porphyrin compounds present π–π type interactions with the MG dye molecules (Fig. SI-16†). Furthermore, the nitrogen atom of the –(CH3)2N– group of the malachite green molecule is most likely coordinated to the center Zn2+ ion as in the case of N-donor ligands (L) which react with the tetra-coordinated [Zn(Porph)] species leading to penta-coordinated complexes type [Zn(Porph)(L)].
Fig. 8. Adsorption efficiency curves of malachite green dye (C0 = 20 mg L−1) as a function of time of the porphyrin compounds 4a–c and 5a–c (m = 5 mg, pH = 8).

2.8. Adsorption kinetic
Adsorption kinetic determines the time required to reach equilibrium between the solute and the adsorbent and it also gives an idea of the adsorption mechanism and the mode of transfer between the liquid and solid phases. Several kinetic models have been developed to describe the adsorption kinetics and to specify the nature of the interactions at the solid–liquid interface. In this work, four kinetic models have been selected to study the kinetic behavior of the MG dye at the surface of our porphyrinic compounds, namely: the pseudo first order kinetic model, the pseudo second order kinetic model, the intraparticle diffusion model and the Elovich model.56 The adsorption capacity curves as function of time for our six porphyrinic compounds (4a–c and 5a–c) are given in Fig. 9.
Fig. 9. Adsorption capacity of the MG dye (C0 = 20 mg L−1) as a function of time for the 4a–c and 5a–c porphyrin species (m = 5 mg, pH = 8). Dots represents the experimental data and the curves where plotted using the pseudo second-order kinetic model.

Fig. SI-17† illustrates the experimental data of the adsorption of MG dye on the 4a–c and 5a–c compounds. The data were fitted using the four kinetic models and the results are given in Table SI-2.† The values of the amount of MG adsorption calculated from the pseudo second order model are the closest to those determined experimentally for all 4a–c and 5a–c porphyrin species, which indicates the adequacy of the use of this model to describe the adsorption phenomenon. The correlation coefficient (R2) values of the pseudo second order model are close to 0.99. In the case of the pseudo first order model, the R2 parameters are between 0.86 and 0.90 for all compounds. For the intraparticle diffusion model, the correlation coefficient does not exceed 0.91 and in the case of Elovich model, the R2 values are between 0.93 and 0.97. These data indicate that the pseudo second order kinetic model is the most appropriate for describing the adsorption of MG dye by our porphyrin derivatives.
The second order model equation used is as follows (eqn (5)):
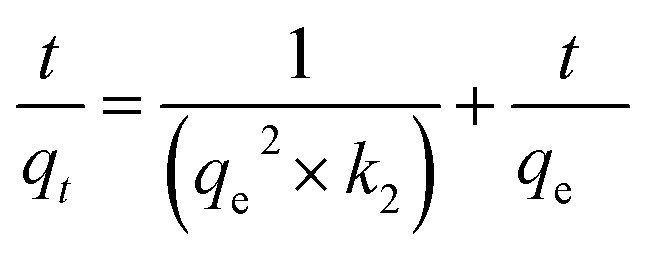 |
5 |
where, qt and qe represent the amounts of the adsorbent adsorbed at time t and equilibrium, respectively, while k2 is the rate constant for the pseudo second order model (g mg−1 min−1). The values of k2 and qcal were calculated for each of the six porphyrinic compounds from the slope and the intersection of the corresponding plot, respectively.
2.9. Degradation of MG dye
2.9.1. Effect of the initial MG concentration
In order to obtain the optimal condition of the initial MG dye concentration, only the 5a derivative was tested. The degradation procedure of the MG dye consists of reacting the dye with H-triazole meso-arylporphyrin zinc(ii) (5a) (m = 5 mg) in the presence of an aqueous solution of H2O2 (6 mg L−1) at pH = 8. Fig. 10 shows the evolution of the color removal against initial MG dye concentration which shows that the degradation kinetics of MG is slow when the initial dye concentration is increased. This indicates that the increase of the initial concentration of the dye leads to an increase in the number of malachite green molecules, while the number of hydroxyl radicals remains constant. As a consequence, there is a decrease in the kinetics of the degradation reaction as well as the discoloration efficiency. These trends are consistent with those reported in the literature.57
Fig. 10. Evolution of the color removal against initial dye concentration. The hydrogen peroxide concentration, Co is 6 mg L−1, 5a catalyst weight m = 5 mg, pH = 8 and the MG mass, mo is 5 mg.

2.9.2. Effect of the initial H2O2 concentration
The 5a triazole zinc(ii) derivative was also used to obtain the optimal condition for the initial hydrogen peroxide concentration. Thus, the effect of the initial hydrogen peroxide concentration Co on the malachite green dye degradation was studied using an initial MG concentration of 20 mg L−1 at room temperature using 5a compound as a catalyst. The Co initial H2O2 aqueous concentrations used were 0, 3, 6 and 9 mg L−1. The increase in H2O2 concentration from 0 to 6 mg L−1 leads to an increase in the degradation efficiency (Fig. 11). However, for a Co concentration of 9 mg L−1, the discoloration efficiency of the MG decreases. This phenomenon could be explained by the fact that at high concentration, hydrogen peroxide is a powerful OH˙ scavenger58 (Equations (eqn (6)–(8)).
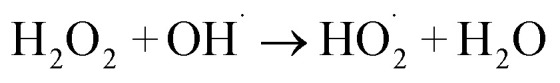 |
6 |
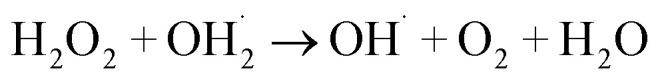 |
7 |
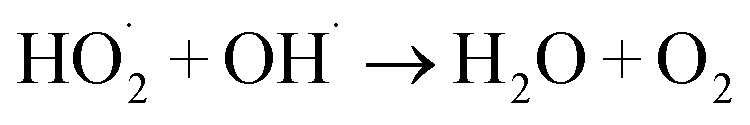 |
8 |
Fig. 11. Change in the degradation efficiency of the MG dye on varying initial H2O2 concentration using 5a triazole zinc(ii) derivative as a catalyst.

2.9.3. Effects of pH
To investigate the effects of pH on the degradation of the MG dye by complexes 5a–c, aqueous solutions with pH from 4 to 8 were adjusted by 0.1 M HCl and 0.1 NaOH. The influence of pH on the dye oxidation was studied using five solutions with pH values determined initially (4, 6, 8, 10 and 12) and without any modifications or control of the pH during the process. The results obtained for dye removal as a function of the initial pH of the solution at various reaction times are presented in Fig. SI-18.† The maximum conversion was achieved after 3 h from the start of the reaction for a pH of 8.
2.9.4. Degradation with optimal conditions
The degradation study of the MG dye was carried out using an aqueous H2O2 solution under optimal conditions, found for the 5a derivative, and applied to the two other species 5b–c (Fig. SI-19b and c†). Fig. SI-19a† represents the absorption curve of the MG dye in presence of H2O2 without catalyst (blank experiment) versus time where it can be seen that the absorption remains practically the same for a period of 180 min. By using the same mass of the three catalysts 5a–c, the obtained degradation yields are 54.0, 52.1 and 48.9% for 5a, 5b and 5c, respectively (Fig. SI-20a†). The degradation reactions were achieved after 180 min. Fig. SI-20b† shows the degradation removal efficiency for complexes 5a–c using the same number of moles (3.35 × 10−6 mol.) for the three species. The obtained degradation yields are 54.0, 53.88 and 53.94% for 5a, 5b and 5c, respectively. These results show that using the same number of moles for our three 5a–c catalysts, the degradation efficiency is virtually the same (∼54%). This confirms the fact that the central metal is the main site of degradation of the dye58 and that the nature of the substituent X (X = H, Cl and I) on the para-position of the terminal phenyl of the three triazole meso-aryporphyrins 4a–c, practically has no effect on this degradation.
2.9.5. Effect of temperature
For the zinc(ii) porphyrin derivative 5a, the effect of temperature on the degradation of MG dye has been investigated. The temperatures used were 25, 35 and 45 °C (Fig. 12). As expected, the degradation efficiency of MG dye increased from 54.0% to 64.2% as a consequence of increasing the temperature from 25 to 45 °C. This is due to the fact that higher temperatures increase the reaction rate between hydrogen peroxide and the zinc metal center. This in turn increases the rate of generation of the OH˙ radical oxidizing species.59
Fig. 12. Change of the degradation efficiency of the MG solution as function of time using 5a used as catalyst at 298, 308 and 318 K.

2.9.6. Comparison of our procedure with other reported methods
Many investigations concerning the MG degradation have previously been reported in the literature.19 The most important of which are: the chemical methods, the photocatalysis methods and the biodegradation methods (Table 5).
Selection of several methods used for MG degradation with the optimal reaction conditions and yields.
| Method of degradation | Degradation system and optimal reacting conditions | Degradation yield, time reaction | Ref. |
|---|---|---|---|
| Chemical method: Fenton's reagent | Fe(ii)/aqueous H2O2, pH = 3.4, [H2O2]o = 0.50 mM, [Fe2+] = 0.10 mM, [MG]0 = 20 mg L−1, 30 °C | 99.25% (60 min) | 60 |
| Chemical method: mesoporous carbon adsorbent | Mesoporous silica (MCM-48), pH = 8.5, [silica] = 5.10−3 g/25 mL, [MG]0 = 50 mg L−1, T = 25 °C | 98.2% (30 min) | 61 |
| Photocatalysis method | Using naked niobium oxide (Nb2O5) as a photocatalyst. weight of catalyst = 0.1 g, [MG]o = 5 ppm; T = 25 °C, 3 mV cm−2 light intensity, pH = 6 | 53.25% (60 min) | 62 |
| Photocatalysis method | Ag doped TiO2, pH = 9, [MG]0 = 5 × 10−6 M, [catalyst] = 0.12 g, 60 mW cm−2 light intensity | 99.5% | 63 |
| Biodegradation method | System used: bacterium Enterobacter asburiae strain XJUHX-4TM, semi-synthetic medium having the following composition was used: MG: 0.100–1.00 g L−1, KH2PO4: 1 g L−1, KCl: 0.5 g L−1, NaNO3: 2 g L−1, MgSO4·7 H2O: 0.5 g L−1, FeSO4·7H2O: 0.01 g L−1, sucrose: 15 g L−1, and beef extract: 3 g L−1 | Maximum decolorization (>98%) in semi-synthetic media | 64 |
| Biodegradation method | System used: Pseudomonas sp. strain DY1, [MGo] = 100–1000 mg L−1, pH = 6.6, T = 28–30 °C | Effective decolorization: 78.9–84.3% | 65 |
| Use of our three zinc(II) complexes 5a–c | |||
|---|---|---|---|
| Complex | System used | Yield | |
| [Zn(TAzP-HVP)] 5a | [H2O2] = Co = 6 mg L−1, [MG] = 20 mg L−1, weight of catalyst (5a) = mo = 5 mg, pH = 8, T = 25 °C | 54.0% (180 min) | |
| [Zn(TAzP-ClVP)] 5b | [H2O2] = Co = 6 mg L−1, [MG] = 20 mg L−1, weight of catalyst (5b) = mo = 5 mg, pH = 8, T = 25 °C | 52.0% (180 min) | |
| [Zn(TAzP-IVP)] 5c | [H2O2] = Co = 6 mg L−1, [MG] = 20 mg L−1, weight of catalyst (5c) = mo = 5 mg, pH = 8, T = 25 °C | 48.9% (180 min) | |
As depicted in Table 5, the degradation yields of the MG dye obtained using our zinc(ii) 5a–c (∼50%) porphyrin zinc(ii) complexes are lower than those obtained using the other methods shown in this table. It is expected that the use of other metal ions instead of Zn(ii), such as Cu(ii) or Fe(ii)60 with our three triazole porphyrins in the presence of H2O2 will lead to an increase in the degradation efficiency of the dye. This is due to the fact that ferrous and cuprous ions are more effective than Zn2+ ion in giving 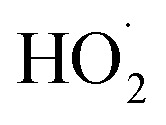 and OH˙ radicals.
and OH˙ radicals.
Nevertheless, the obtained yields are quite acceptable for a first reported investigation on the MG degradation using a porphyrin derivative. Thus, these encouraging results suggest that our zinc(ii) 5a–c can be further improved and can be concomitantly used with other degradation techniques such as the photodegradation where the expected yields are higher than those reported here.
2.9.7. Activation energy and thermodynamic variables
To better understand the degradation phenomenon, the pseudo first order model was used to calculate the kinetic parameters. Arrhenius law (eqn (9)) was used to determine the activation energy (Ea). The thermodynamic activation parameters of enthalpy (ΔH*), entropy (ΔS*) and Gibbs free energy (ΔG*) were calculated using Eyring's equation (eqn (10) and (11)).66
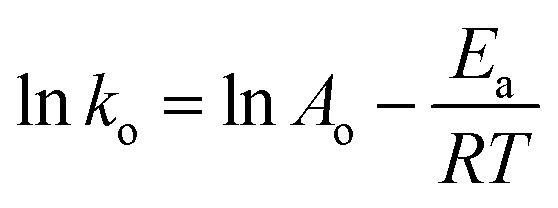 |
9 |
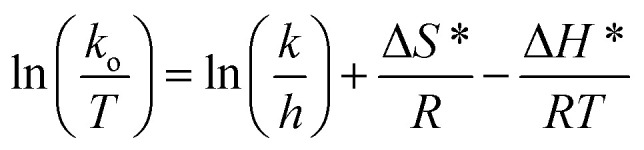 |
10 |
| ΔG* = ΔH* − T ΔS* | 11 |
where ko is the kinetic rate, h and k are the Plank's and Boltzmann's constants, respectively. Ao is the Arrhenius constant and R is the universal gas constant (8.314 J mol−1 K−1). The obtained thermodynamic parameters and activation energy values are summarized in Table 6. From these results, it can be seen that the free energy is positive in all the cases. This indicates that the degradation of MG dye using H2O2 is a non-spontaneous reaction regardless of temperature. The free enthalpy is positive, meaning the degradation process is endothermic and a higher temperature facilitates degradation. The negative value of the entropy suggests the diminution of the disorder during the degradation process. The activation energy value is 9.867 kJ mol−1. Since the thermodynamic parameters found for both 5b–c complexes are very close to those of complex 5a, only the results founds for the latter zinc(ii) derivative are reported.
Thermodynamic parameters (ΔS*, ΔH*, ΔG*) and activation energy (Ea) for complex 5a.
| T (°C) | 25 | 35 | 45 |
| k o (min−1) | 0.0042 | 0.0046 | 0.0054 |
| E a (kJ mol−1) | 9.867 | 9.867 | 9.867 |
| ΔS* (J mol−1 K−1) | −265.983 | −265.983 | −265.983 |
| ΔH* (kJ mol−1) | 7.308 | 7.308 | 7.308 |
| ΔG* (kJ mol−1) | 86.570 | 89.230 | 91.890 |
3. Conclusion
We have successfully synthesized three novel triazole meso-arylporphyrins, the: H-triazole, chloro–triazole and iodine–triazole meso-arylporphyrins with the respective formulas H2(TAzP-HVP) (4a), H2(TAzP-ClVP) (4b) and H2(TAzP-IVP) (4c) as well as their corresponding zinc(ii) porphyrins 5a–c. The chloride and bromide binding properties of the chloro–triazole and the iodine–triazole zinc(ii) triazole meso-arylporphyrins derivatives 5b–c have been monitored by UV-visible technique. Both 5b–c receptors exhibit strong Cl− and Br− anion binding affinities, forming 1 : 1 stoichiometric complexes. The adsorption efficiency of the malachite green dye (MG) on all three base porphyrins and their corresponding zinc(ii) coordination compounds revealed that: (i) the adsorption efficiency of the free base porphyrins 4a–c are higher than those of the corresponding zinc(ii) porphyrin complexes 5a–c, (ii) the free base chloro–triazole and iodine–triazole 4b–c porphyrin derivatives show better adsorption than that of the H-triazole TAzP-HVP species (4a), which is also the case for the corresponding zinc(ii) porphyrin complexes (5a–c) and (iii), the pseudo second order kinetic model is the most appropriate to describe the adsorption of MG dye by our free porphyrin compounds. The degradation investigation of the MG dye, using complexes 5a–c as catalysts, carried out using an aqueous H2O2 solution under optimal conditions, shows that the degradation yield using the H-triazole meso-arylporphyrin zinc(ii) derivative 5a is higher than those of the chloro–triazole and the iodine–triazole meso-arylporphyrin zinc(ii) complexes 5b–c. The degradation yields using our 5a–c species are ∼54% which are much smaller than several reported MG dye degradations using other methods. Nevertheless, this is an encouraging result as this is the first time a porphyrin derivative has been used for the degradation of this dye and studies are underway to test our zinc(ii) derivatives in photochemical degradations of the malachite green and other dyes. The thermodynamic parameters were determined and show that the free energy is positive in all the cases indicating that the degradation of MG dye using H2O2 is a non-spontaneous, temperature-dependent reaction. Furthermore, the enthalpy is positive which implies that the degradation process is endothermic, while the negative value of the entropy suggests the diminution of disorder during the degradation process.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The authors extend their appreciation to the deanship of Scientific Research at Majmaah University, Saudi Arabia.
Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03070h
References
- Pineiro M. Carvalho A. L. Pereira M. M. Gonsalves A. M. d'A. R. Arnaut L. G. Formosinho S. J. Chem.–Eur. J. 1998:2299–2307. doi: 10.1002/(SICI)1521-3765(19981102)4:11<2299::AID-CHEM2299>3.0.CO;2-H. [DOI] [Google Scholar]
- Liang X. I. Li X. Yue X. Angew. Chem., Int. Ed. 2011;50:11622–11627. doi: 10.1002/anie.201103557. [DOI] [PubMed] [Google Scholar]
- Lin S. Diercks C. S. Zhang Y.-B. Kornienko N. Nichols E. M. Zhao Y. Paris A. R. Kim D. Yang P. Yaghi O. M. Chang C. J. Science. 2015;349:1208–1213. doi: 10.1126/science.aac8343. [DOI] [PubMed] [Google Scholar]
- Imahori H. Hayashi S. Hayashi H. Oguro A. Eu S. Umeyama T. Matano Y. J. Phys. Chem. C. 2009;113:18406–18413. doi: 10.1021/jp907288h. [DOI] [Google Scholar]
- Florescu M. David M. Sensors. 2017;17:1–16. doi: 10.3390/s17061314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombelli A. Manera M. G. Borovkovb V. Giancanec G. Valli L. Rell R. Sens. Actuators, B. 2017;246:1039–1048. doi: 10.1016/j.snb.2017.01.192. [DOI] [Google Scholar]
- Aguilar-Ortíz E. Lévaray N. Vonlanthen M. Morales-Espinoza E. G. Rojas-Aguirre Y. Zhu X. X. Rivera E. Dyes Pigm. 2016;132:110–120. doi: 10.1016/j.dyepig.2016.04.047. [DOI] [Google Scholar]
- Suijkerbuijk B. M. J. M. Gebbink R. J. M. Angew. Chem., Int. Ed. Engl. 2008;47:7396–7421. doi: 10.1002/anie.200703362. [DOI] [PubMed] [Google Scholar]
- Auwärter W. Écija D. Klappenberger F. Barth J. V. Nat. Chem. 2015;7:105–120. doi: 10.1038/nchem.2159. [DOI] [PubMed] [Google Scholar]
- Gottfried J. M. Surf. Sci. Rep. 2015;70:259–279. doi: 10.1016/j.surfrep.2015.04.001. [DOI] [Google Scholar]
- Kolb H. C. Finn M. G. Sharpless K. B. Angew. Chem., Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Gilday L. C. White N. G. Beer P. D. Dalton Trans. 2012;41:7092–7097. doi: 10.1039/C2DT30124E. [DOI] [PubMed] [Google Scholar]
- Gilday L. C. White N. G. Beer P. D. Dalton Trans. 2013;42:15766–15773. doi: 10.1039/C3DT52093E. [DOI] [PubMed] [Google Scholar]
- Ismail M. Akhtar K. Khan M. I. Kamal T. Khan M. A. Asiri A. M. Seo J. Khan S. B. Curr. Pharm. Des. 2019;25:3653–3671. doi: 10.2174/1381612825666191021142026. [DOI] [PubMed] [Google Scholar]
- Su Min K. Satish Kumar R. Lee J.-H. Kim K. S. Lee S. G. Son Y.-A. Dyes Pigm. 2019;160:37–47. doi: 10.1016/j.dyepig.2018.07.045. [DOI] [Google Scholar]
- Li Y. Wang L. Gao Y. Yang W. Lia Y. Guo C. RSC Adv. 2018;8:7330–7339. doi: 10.1039/C7RA12701D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guergueb M. Nasri S. Brahmi J. Loiseau F. Molton F. Roisnel T. Guerineau V. Turowska-Tyrk I. Aouadi K. Nasri H. RSC Adv. 2020;10:6900–6918. doi: 10.1039/C9RA08504A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soury R. Jabli M. Saleh T. A. Abdul-Hassan W. S. Saint-Aman E. Loiseau F. Philouze C. Nasri H. RSC Adv. 2018;8:20143–20156. doi: 10.1039/C8RA01134F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raval P. N. P. Shah P. U. Shah N. K. Appl. Water Sci. 2017;7:3407–3445. doi: 10.1007/s13201-016-0512-2. [DOI] [Google Scholar]
- Srivastava S. Sinha R. Roy D. Aquat. Toxicol. 2004;66:319–329. doi: 10.1016/j.aquatox.2003.09.008. [DOI] [PubMed] [Google Scholar]
- An S.-Y. Min S.-K. Cha I.-H. Choi Y.-L. Cho Y.-S. Kim C.-H. Lee Y.-C. Biotechnol. Lett. 2002;24:1037–1040. doi: 10.1023/A:1015610018103. [DOI] [Google Scholar]
- Brahmi J. Aouadi K. Msaddek M. Praly J.-P. Vidal S. C. R. Chim. 2016;19:933–935. doi: 10.1016/j.crci.2016.03.003. [DOI] [Google Scholar]
- Adler A. D. Longo F. R. Finarelli J. D. Goldmacher J. Assour j. Korsakoff L. J. Org. Chem. 1967;32:476. doi: 10.1021/jo01288a053. [DOI] [Google Scholar]
- Nasri S. Zahou I. Turowska-Tyrk I. Roisnel T. Loiseau F. Saint-Amant E. Nasri H. Eur. J. Inorg. Chem. 2016:5004–5019. doi: 10.1002/ejic.201600575. [DOI] [Google Scholar]
- Görlitzer K. Huth S. Jones P. G. Pharmazie. 2005;60:269–272. [PubMed] [Google Scholar]
- Ezzayani K. Denden Z. Najmudin S. Bonifácio C. Saint-Aman E. Loiseau F. Nasri H. Eur. J. Inorg. Chem. 2014:5348–5361. doi: 10.1002/ejic.201402546. [DOI] [Google Scholar]
- Mak C. C. Bampos N. Sanders J. K. M. Angew Chem. Int. Ed. 1998;37:3020–3023. doi: 10.1002/(SICI)1521-3773(19981116)37:21<3020::AID-ANIE3020>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Zeyada H. M. Makhlouf M. M. Ali M. A. Jpn. J. Appl. Phys., Part 1. 2016;55:022601. doi: 10.7567/JJAP.55.022601. [DOI] [Google Scholar]
- Lin C.-I. Fang M.-Y. Cheng S.-H. J. Electroanal. Chem. 2002;531:155–162. doi: 10.1016/S0022-0728(02)01056-2. [DOI] [Google Scholar]
- Tao P. Viswanath A. Schadler L. S. Benicewicz B. C. Siegel R. W. ACS Appl. Mater. Interfaces. 2011;3:3638–3645. doi: 10.1021/am200841n. [DOI] [PubMed] [Google Scholar]
- Singh P. Kaushal A. Kaur D. J. Alloys Compd. 2009;471:11–15. doi: 10.1016/j.jallcom.2008.03.123. [DOI] [Google Scholar]
- Barkigia K. M. Berber M. D. Fajer J. Medforth C. J. Renner M. W. Smith K. M. J. Am. Chem. Soc. 1990;112:8851–8854. doi: 10.1021/ja00180a029. [DOI] [Google Scholar]
- Zhang J. Zhang P. Zhang Z. Wei X. J. J. Phys. Chem. A. 2009;113:5367–5374. doi: 10.1021/jp811209k. [DOI] [PubMed] [Google Scholar]
- Gouterman M. J. Mol. Spectrosc. 1961;6:138–163. doi: 10.1016/0022-2852(61)90236-3. [DOI] [Google Scholar]
- Mak C. C. Bampos N. Sanders J. K. M. Angew. Chem., Int. Ed. 1998;37:3020–3022. doi: 10.1002/(SICI)1521-3773(19981116)37:21<3020::AID-ANIE3020>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Denden Z. Ezzayani K. Saint-Aman E. Loiseau F. Najm S. Bonifácio Daran J.-C. Nasri H. Eur. J. Inorg. Chem. 2015:2596–2610. doi: 10.1002/ejic.201403214. [DOI] [Google Scholar]
- Kim D. Shin E. J. Bull. Korean Chem. Soc. 2003;24:1490–1494. doi: 10.5012/bkcs.2003.24.10.1490. [DOI] [Google Scholar]
- Neya S. Funasaki N. Tetrahedron Lett. 2002;43:1057–1058. doi: 10.1016/S0040-4039(01)02324-3. [DOI] [Google Scholar]
- Quimby D. J. Longo F. R. J. Am. Chem. Soc. 1975;97:5111–5117. doi: 10.1021/ja00851a015. [DOI] [Google Scholar]
- Kadish K. M. Morrison M. M. J. Am. Chem. Soc. 1976;98:3326–3328. doi: 10.1021/ja00427a046. [DOI] [PubMed] [Google Scholar]
- Lin C.-I. Fang M.-Y. Cheng S.-H. J. Electroanal. Chem. 2002;531:155–162. doi: 10.1016/S0022-0728(02)01056-2. [DOI] [Google Scholar]
- Ghosh A. Mobin S. M. Fröhlich R. Butcher R. J. Maity D. K. Ravikanth M. Inorg. Chem. 2010;49:8287–8297. doi: 10.1021/ic1008522. [DOI] [PubMed] [Google Scholar]
- Bredas J. L. Silbey R. Bordeaux D. S. Chance R. R. J. Am. Chem. Soc. 1983;105:6555–6559. doi: 10.1021/ja00360a004. [DOI] [Google Scholar]
- Lyons D. M. Kesters J. Maes W. Christopher W. Bielawski J. Sessler L. Synth. Met. 2013;178:56–61. doi: 10.1016/j.synthmet.2013.06.013. [DOI] [Google Scholar]
- Kadish K. M. Chen P. Enakieva Y. Yu. Nefedov S. E. Gorbunova Y. G. Tsivadze A. Y. Bessmertnykh-Lemeune A. Stern C. Guilard R. J. Electroanal. Chem. 2011;656:61–71. doi: 10.1016/j.jelechem.2011.01.011. [DOI] [Google Scholar]
- Barkigia K. M. Chantranupong L. Smith K. M. Fajer J. J. Am. Chem. Soc. 1988;110:7566–7567. doi: 10.1021/ja00230a063. [DOI] [Google Scholar]
- Shelnutt J. A., in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2000, vol. 7, pp. 167–223 [Google Scholar]
- Ozarowski A. Lee H. M. Balch A. L. J. Am. Chem. Soc. 2003;125:12606–12614. doi: 10.1021/ja030221f. [DOI] [PubMed] [Google Scholar]
- Polster J. and Lachmann H.Spectrometric Titrations, Verlag Chemie, Weinheim, 1989, p. 292 [Google Scholar]
- Khan S. U., Fundamental aspects of pollution control and environmental Science, Elsevier, New York, 1980 [Google Scholar]
- Senesi N. and Chen Y., Ecological Studies, ed. Z. Gerstl, Y. Chen, U. Mingelgril and B. Yaron, Sringer verglag, Berlin, 1989, pp. 37–90 [Google Scholar]
- Chowdhury S. Das P. Sep. Sci. Technol. 2011;46:1966–1976. doi: 10.1080/01496395.2011.584930. [DOI] [Google Scholar]
- Dzombak D. A. Luthy R. G. Soil Sci. 1984;137:292–308. doi: 10.1097/00010694-198405000-00002. [DOI] [Google Scholar]
- Chiou C. T. Peters L. J. Freed V. H. Science. 1979;206:831–832. doi: 10.1126/science.206.4420.831. [DOI] [PubMed] [Google Scholar]
- Swan N. B. Zaini M. A. A. Ecol. Chem. Eng. S. 2019;26:119–132. [Google Scholar]
- Qui H. Lv L. Pan B.-C. Zhang Q.-J. Zhang W.-M. Zhang Q.-X. J. Zhejiang Univ., Sci., A. 2009;10:716–724. doi: 10.1631/jzus.A0820524. [DOI] [Google Scholar]
- Hameed B. H. Lee T. W. J. Hazard. Mater. 2009;164:468–472. doi: 10.1016/j.jhazmat.2008.08.018. [DOI] [PubMed] [Google Scholar]
- Aravindhan R. Fathima N. N. Rao J. R. Nair B. U. J. Hazard. Mater. 2006;B138:152–159. doi: 10.1016/j.jhazmat.2006.05.052. [DOI] [PubMed] [Google Scholar]
- Sun J. H. Sun S. P. Wang G. L. Qiao L. P. Dyes Pigm. 2007;74:647–652. doi: 10.1016/j.dyepig.2006.04.006. [DOI] [Google Scholar]
- Hameed B. H. Lee T. W. J. Hazard. Mater. 2009;164:468–472. doi: 10.1016/j.jhazmat.2008.08.018. [DOI] [PubMed] [Google Scholar]
- Anbia M. Ghaffari A. J. Iran. Chem. Soc. 2012;8:67–76. doi: 10.1007/BF03254283. [DOI] [Google Scholar]
- Hussein A. S. Fairooz N. Y. Journal of University of Babylon for Pure and Applied Sciences. 2016;9:2510–2518. [Google Scholar]
- Khan H. R. Choudhary M. A. Mirza M. A. Ahmed Z. Khan Y. Asian J. Chem. 2017;29:257–260. doi: 10.14233/ajchem.2017.20092. [DOI] [Google Scholar]
- Mukherjee T. Das M. Clean: Soil, Air, Water. 2014;42:849–856. [Google Scholar]
- Du L.-N. Wang S. Li G. Wang B. Jia X.-M. Zhao Y.-H. Chen Y.-L. Ecotoxicology. 2011;20:438–446. doi: 10.1007/s10646-011-0595-3. [DOI] [PubMed] [Google Scholar]
- Fil B. A. Yilmaz M. T. Bayar S. Elkoca M. T. Braz. J. Chem. Eng. 2014;31:171–182. doi: 10.1590/S0104-66322014000100016. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.