

Abstract
Copy number variants (CNVs) have provided a reliable entry point to identify the structural correlates of atypical cognitive development. Hemizygous deletion of human chromosome 22q11.2 is associated with impaired cognitive function; however, the mechanisms by which the CNVs contribute to cognitive deficits via diverse structural alterations in the brain remain unclear. This study aimed to determine the cellular basis of the link between alterations in brain structure and cognitive functions in mice with a heterozygous deletion of Tbx1, one of the 22q11.2-encoded genes. Ex vivo whole-brain diffusion-tensor imaging (DTI)–magnetic resonance imaging (MRI) in Tbx1 heterozygous mice indicated that the fimbria was the only region with significant myelin alteration. Electron microscopic and histological analyses showed that Tbx1 heterozygous mice exhibited an apparent absence of large myelinated axons and thicker myelin in medium axons in the fimbria, resulting in an overall decrease in myelin. The fimbria of Tbx1 heterozygous mice showed reduced mRNA levels of Ng2, a gene required to produce oligodendrocyte precursor cells. Moreover, postnatal progenitor cells derived from the subventricular zone, a source of oligodendrocytes in the fimbria, produced fewer oligodendrocytes in vitro. Behavioral analyses of these mice showed selectively slower acquisition of spatial memory and cognitive flexibility with no effects on their accuracy or sensory or motor capacities. Our findings provide a genetic and cellular basis for the compromised cognitive speed in patients with 22q11.2 hemizygous deletion.
Subject terms: Autism spectrum disorders, Neuroscience
Introduction
Although copy number variants (CNVs) are rare and occur in <1% of patients with any psychiatric sample population, they are robustly and consistently associated with psychiatric disorders [1, 2]. Moreover, CNVs affect specific cognitive functions independent of clinically defined mental illness [3, 4] and cognitive impairments are more severe among CNV carriers with psychiatric diagnosis [5]. The currently available pharmaceutical medications do not significantly improve cognitive deficits associated with many mental disorders due to our lack of understanding of the mechanisms underlying these deficits.
Despite their robust association with cognitive impairments and psychiatric disorders, CNVs pose a challenge when attempting to understand the composition of contributory genes, as accurate identification of CNV-encoded genes contributing to human phenotypes remains difficult. Recent large-scale genome-wide exome screening studies have identified ultra rare protein-truncating variants of genes encoded in several large-sized CNVs linked with autism-spectrum disorder (ASD) [6] and schizophrenia [7]. However, failure to detect similar variants of other CNV-encoded genes may be attributable to their rarity, as larger sample sizes identify more gene variants than smaller-scale analyses [6]. Variants in promoters and enhancers may contribute to phenotypes [8]. Moreover, variants of CNV-encoded single genes may simply not exist, and single-gene hemizygosity or duplication, as part of a CNV, may play the role of a driver gene. Thus, there is a need to utilize complementary approaches to identify driver genes encoded by large CNVs.
There are more human and mouse studies of human chromosome 22q11.2 deletion than other CNVs, given that it was found to be associated with mental illness much earlier than other CNVs [9]. Hemizygous deletion of 22q11.2 is robustly associated with diverse neurodevelopmental disorders, including ASD, attention-deficit/hyperactivity disorder, schizophrenia, and intellectual disability (ID) [10]. Moreover, individuals with 22q11.2 hemizygosity exhibit deterioration in specific cognitive functions, including the accuracy and speed of memory acquisition, executive functions, and social cognition [11–13]. Cognitive impairment may be an integral component of late-onset neurodevelopmental disorders, as it precedes and predicts the onset of schizophrenia among 22q11.2 hemizygosity carriers [14, 15].
Recent large-scale imaging studies have demonstrated altered white matter integrity in the brains of 22q11.2 hemizygous deletion carriers [16–18]. However, since many regions show altered white matter integrity and this CNV contains a minimum of 30 protein-coding genes, the exact causative associations among encoded genes, structural alterations, and atypical cognitive development remain unclear.
Rare loss-of-function variants (e.g., frameshift deletion) of TBX1, a gene encoded by a 22q11.2 CNV, have been associated with ASD, ID, and seizures [19–22]. However, these TBX1 variant carriers also exhibit single-nucleotide variants (SNVs) in other genes [21], and only a few cases/families with those variants have been identified. A recent large-scale exome sequencing identified an ultra rare protein-truncating variant of TBX1 among individuals with schizophrenia [7], but its statistical significance is unclear due to power limitation. The causative structural substrates in the brain mediating the impacts of deficiency of this 22q11.2-encoded gene on cognitive impairment remain unknown.
Mouse studies have provided a complementary means to address limitations of these human studies by systematically examining the roles of small chromosomal segments and individual genes in behaviors against a homogeneous genetic background [10, 22–32]. These studies have demonstrated that some, but not all, 22q11.2-encoded single genes contribute to select behavioral targets [10, 28, 31]. Our results have revealed that Tbx1 heterozygosity impairs social interaction and communication [26, 29, 30, 32]. No DTI–MRI analysis of mouse models of 22q11.2 hemizygosity or Tbx1 heterozygosity has been reported. The present study aimed to determine the structural and cellular basis underlying the effects of Tbx1 heterozygosity on specific cognitive functions in a congenic mouse model.
Methods and materials
Animal handling and use followed the protocols that were approved by the Animal Care and Use Committees of Albert Einstein College of Medicine and the University of Texas Health Science Center at San Antonio, in accordance with NIH guidelines.
All experimental details are provided in Supplementary Information (SI). Briefly, congenic Tbx1 heterozygous mice and their wild-type littermates were used. We characterized the myelin in the brains, using ex vivo DTI–MRI, Black Gold II staining, and electron microscopy (EM) analyses. Genes involved in oligodendrocytes and their precursor cells were evaluated by qRT-PCR. The production capacity of oligodendrocyte precursor cells was evaluated in vitro using precursor cells derived from the subventricular zone of postnatal (P21) mice. The accuracy and speed of spatial memory and of cognitive flexibility were evaluated in the Morris water maze and attentional set shifting, respectively. Moreover, nonspecific deficits of visual and olfactory senses and general motivation to approach an object were examined using olfactory responses to nonsocial and social cues. The minimal sample size was determined by power analyses based on our previous study [26].
We compared group means using analysis of variance (ANOVA), followed by Newman–Keuls post hoc tests. Two-sided t-tests were used when there were only two groups. A probability of ≤0.05 was considered significant. When either homogeneity of variance or normality was violated, data were analyzed by a generalized linear mixed model; for comparisons of a pair of data, nonparametric tests were used. When multiple tests were applied to a dataset, the significance level was adjusted using Benjamini–Hochberg’s correction.
Results
There are alterations in white matter microstructures in many brain regions of 22q11.2 hemizygosity carriers [16–18]. However, little is known regarding the exact nature of altered white matter microstructures and driver genes that affect both the white matter and cognitive functions in humans and mice.
Analysis of white matter structures
Tbx1 deficiency decreases fractional anisotropy (FA) signals in the fimbria
Tbx1 +/− mice and their +/+ littermates underwent ex vivo DTI–MRI. We analyzed 19 brain regions (Fig. S1), as defined by the standard regional classification of the mouse brain [33]. The FA value is the most histologically validated DTI–MRI metric [34]. Since FA signals of <0.3 are not reliably correlated with the degree of myelination [35] and cannot be accurately aligned across individual animals [36], we selected regions with FA values ≥0.3. The corpus callosum, anterior commissure, internal capsule, and fimbria met this criterion (Fig. S2a). The fimbria was the only region exhibiting a significant change: FA values were lower in +/− mice than in +/+ mice (Fig. 1). Consistently, the fimbria exhibited the largest effect size for genotype-dependent differences in FA values (Fig. S2b). There were no significant differences in axial diffusivity (AD) (Fig. S3a, b), radial diffusivity (RD) (Fig. S4a, b), or mean diffusivity (MD) (Fig. S5a, b) values between +/+ and +/− mice.
Fig. 1. Box-and-whisker plots of fractional anisotropy (FA) values of the four regions with FA >0.3.
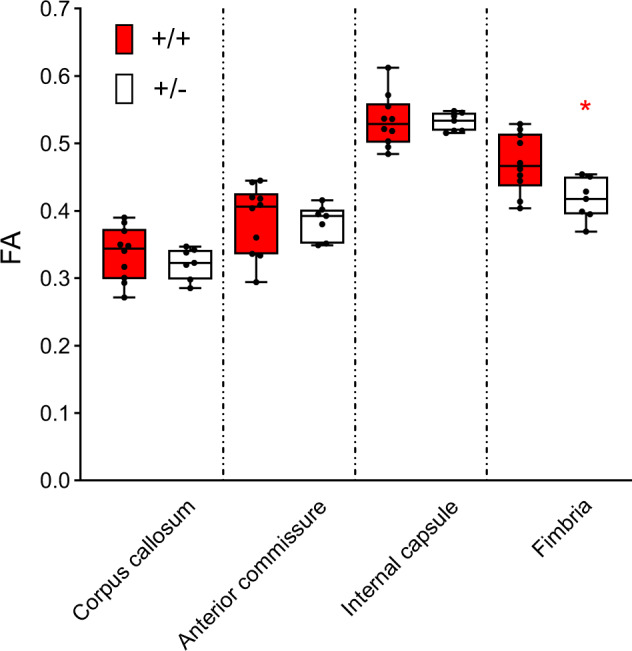
Analysis using a generalized linear mixed model revealed a region-dependent differential effect of genotype on FA values (Genotype × Region, F(3,45) = 7.337, P < 0.001). Mann–Whitney U-tests revealed a significant between-genotype difference in the fimbria only (*, U = 11, p = 0.0185). +/+, n = 10; +/−, n = 7.
Tbx1 deficiency reduces myelination in the fimbria
DTI–MRI analysis of the mouse brain has limited spatial resolution, as well as technical and interpretative limitations [34, 37]. Our resolution (150 μm isotropic voxel) may not have been sufficient for detecting subtle alterations. The structural classifications of the mouse brain by Ma et al. [33] include the fimbria, fornix, stria terminalis, and hippocampal commissure in the “fimbria”. To circumvent these limitations and histologically validate the DTI–MRI findings, we used the non-hydroscopic gold-phosphate complex Black-Gold II [38]. This method provides more consistent staining than hydroscopic gold chloride staining and higher contrast and resolution than lipid-soluble dyes (e.g., Luxol Fast Blue). Black-Gold II also directly stains myelin, unlike markers of myelin components (e.g., myelin basic protein [MBP]), which may not perfectly correlate with the degree of myelination. We examined regions with the largest and second-largest effect sizes among FA values >0.3 (see Fig. S2b): the fimbria and corpus callosum (Fig. S6). The intensity of gold staining was lower in the anterior fimbria of +/− mice than in that of +/+ mice (Fig. S7a). There was no statistically detectable between-genotype difference in the posterior fimbria or anterior/posterior isthmus of the corpus callosum (Fig. S7b,c,d).
Tbx1 deficiency reduces large myelinated axons in the fimbria
We used electron microscopy (EM) to characterize the myelination of axons in the fimbria and corpus callosum at the ultrastructural level. Myelination appeared thicker and thinner in the fimbria and corpus callosum, respectively, of +/− mice than in that of +/+ mice (Fig. 2a, b). We compared g-ratios (i.e., the ratio of axon diameter to the axon + myelin diameter) to quantitatively evaluate relative myelin thickness (Fig. 2c, d). The g-ratios of +/+ mice plateaued slightly above 0.8, which is an expected value for the optimal efficiency of axon myelination in the central nervous system (CNS) [39]. The g-ratios of +/− mice were smaller (i.e., relatively thicker myelin sheath) between 700-nm and 1600-nm-diameter axons in the fimbria (Fig. 2c; Fig. S8a). The overall g-ratios did not differ in the corpus callosum between +/+ and +/− mice (Fig. 2d), but +/− mice had relatively thinner myelin sheath between 1000-nm and 1300-nm-diameter axons in this region (Fig. S8b). Volume is a limiting factor in the CNS: The myelination efficiency steeply decreases when myelin thickness deviates from the optimal g-ratios (~0.8) [39], regardless of whether it is hyper- or hypomyelination. Therefore, this gene deficiency results in a functionally suboptimal population of axons in the fimbria.
Fig. 2. EM analysis of myelinated axons.

EM images of myelin in the fimbria (a) and corpus callosum (b) are shown. We analyzed 300 and 200 axons in the fimbria of both hemispheres in three +/+ and two +/− mice, respectively. We analyzed 260 and 200 axons in the corpus callosum of both hemispheres in three +/+ and two +/− mice, respectively. Ten images were obtained from the fimbria or corpus callosum of each mouse, except for one +/+ mouse that had six available images of the corpus callosum. Ten randomly chosen myelinated axons were analyzed from each image. Scale bar = 800 nm. G ratios increased as a logarithmic function of axon diameter in the fimbria (c, +/+, R = 0.845, p < 0.001; +/−, R = 0.686, p < 0.001) and corpus callosum (d, +/+, R = 0.804, p < 0.001; +/−, R = 0.749, p < 0.001). The degree of these increases differed in the fimbria (Fisher’s z = 4.3319, p < 0.0001), but not in the corpus callosum (Fisher’s z = 1.4695, p = 0.1417), between +/− and +/+ mice (see Fig. S8 for detailed analyses).
A further in-depth analysis revealed that there were proportionally fewer myelinated axons >1600 nm in diameter in the fimbria of +/− mice than of +/+ mice (Fig. S9a; Table S1) and that there were no detectable myelinated axons >1700 nm in diameter in the fimbria of +/− mice (Fig. S9a; Table S1). In the corpus callosum, there were no statistically significant alterations in relative proportion of axons in the corpus callosum of +/− mice (Fig. S9b; Table S1).
Tbx1 heterozygosity impacts a molecule critical for early oligodendrogenesis
Oligodendrocytes and their precursor cells are present locally in the fimbria and, to a lesser extent, in the corpus callosum. The molecular steps through which Tbx1 impacts oligodendrogenesis and myelination remain unknown. Given that Tbx1 mRNA is reduced in the fimbria and corpus callosum of Tbx1 +/− mice compared with +/+ mice (Fig. 3), we examined the impact of a gene-dose reduction of Tbx1 mRNA on genes functionally critical for each step of oligodendrogenesis and myelination in 2- to 3-month-old Tbx1 +/− and +/+ littermates, using qRT-PCR. The myelinating process—and expression of genes involved in each step of oligodendrocytes—of the fimbria starts in the second neonatal week and continues its peak levels from postnatal day 24 to 37 in rodents [40, 41].
Fig. 3. Effect of Tbx1 heterozygosity on myelin-related genes.

Relative mRNA expression levels (mean ± standard error of the mean [SEM]) for Tbx1, Ng2, Pdgfr2, myelin basic protein (MBP), and myelin oligodendrocyte glycoprotein (MOG) in the fimbria (a) and corpus callosum (b) of Tbx1+/+ (n = 5) and +/− (n = 4) mice are shown. Tbx1 mRNA levels were lower in the fimbria (a, t(7) = 4.081, p = 0.0047, ***) and corpus callosum (b, t(7) = 5.221, p = 0.0012, **) of +/− mice than in those of +/+ mice. In the fimbria, levels of Ng2 (a, t(7) = 3.394, p = 0.0115, *) were lower in +/− mice than in +/+ mice. These significant differences survived Benjamini–Hochberg’s correction at the false-discovery rate (FDR) of 5%. There were no other significant differences in the fimbria or corpus callosum (a,b, p > 0.05).
Ng2 (Cspg4) and Pdgfr2, markers of oligodendrocyte precursor cells, are functionally required for the production of oligodendrocyte precursor cells [42, 43]. We found that mRNA levels of Ng2, but not of Pdgrf2, were selectively lower in the fimbria of +/− mice than in that of +/+ mice (Fig. 3a). There was no detectable difference in Ng2 or Pdgfr2 mRNA levels in the corpus callosum between +/+ and +/− mice (Fig. 3b). MBP is essential for the maintenance of myelin and is involved in the adhesion and compaction of the cytosolic membrane leaflets that form the structural basis of multilayered myelin [44, 45]. Myelin oligodendrocyte glycoprotein (MOG) is a marker of mature oligodendrocytes and myelin, although it is not functionally critical for myelin formation or maintenance [46]. No differences in MBP or MOG levels were observed in the fimbria or corpus callosum between +/+ and +/− mice (Fig. 3ab).
Tbx1 heterozygosity reduces oligodendrocyte generation
Another source of oligodendrocytes in the fimbria is the population of adult neural progenitor cells in the subventricular zone [47–50], which is distinct from those generating neurons [51]. Given the enrichment of Tbx1 protein in the adult subventricular zone (SVZ) [26], we aimed to determine whether Tbx1 heterozygosity affects the cell-autonomous capacity of this oligodendrocyte population. Progenitor cells were taken from the lateral ventricular wall, including the subventricular zone, of 3-week-old Tbx1 +/− and +/+ littermates and cultured and differentiated into oligodendrocytes in vitro. Progenitor cells derived from the subventricular zone of Tbx1 +/− mice produced fewer O4-positive immature and mature oligodendrocytes than those derived from +/+ mice (Fig. 4ab). This in vitro assay demonstrated that Tbx1 heterozygosity reduced oligodendrocyte production from progenitor cells of the SVZ in a cell-autonomous manner.
Fig. 4. Effect of Tbx1 heterozygosity on oligodendrocytes.

Representative images (a) and box-and-whisker plots (b) of O4-positive (red) oligodendrocytes among all DAPI-positive (blue) cells in culture are shown. Since the assumption of normality was not met (Shapiro–Wilk tests: +/+, W(35) = 0.888, p = 0.002; +/−, W(19) = 0.898, p = 0.045), we applied a generalized linear mixed model of log-transformed data. Progenitor cells derived from the lateral ventricular walls of P21 Tbx1 +/− mice produced consistently fewer O4-positive oligodendrocytes than those of +/+ mice across the cultures (Genotype, F(1,11.451) = 12.841. p = 0.004, *** Image field, F(3, 33.978) = 0.609, p = 0.614; Genotype x Image field, F(3,33.978) = 0.134, p = 0.939). Scale bar = 200 μm. +/+, n = 9; +/−, n = 5.
Although decreased FA values and reduced net myelin signals are suggestive of less myelin, axonal degeneration, reduced axonal density, or changes in axonal organization [52], our EM analyses complemented these assessments of the net signal intensities by demonstrating a loss of large myelinated axons in the fimbria. Our qRT-PCR and in vitro analyses further indicated that Tbx1 heterozygosity reduced levels of the molecule needed for the generation of local oligodendrocyte precursor cells and the production capacity of oligodendrocytes in the lateral ventricular wall.
Analysis of cognitive functions
Individuals with 22q11.2 hemizygous deletions exhibit lower scores on measures of attention, executive function, processing speed, visual memory, visuospatial skills, and social cognition [11, 13]. However, the link between structural alterations caused by single 22q11.2 genes and changes in cognitive function remains unknown. In addition, although human studies have reported an association between loss-of-function TBX1 variants and neurodevelopmental disorders [7, 19–22], their effects on cognitive function remain uncharacterized. Since we observed that Tbx1 heterozygosity leads to myelin alterations in the fimbria, we examined its effects on cognitive capacities known to rely on the fimbria.
Tbx1 heterozygosity slows the acquisition of spatial reference memory
The spatial reference memory version of the Morris water maze requires an intact fimbria, whereas the visual cued version depends on the dorsal striatum in rodents [53, 54]. Humans with 22q11.2 hemizygosity exhibit impaired spatial processing and memory [11, 55–57]. While our congenic Tbx1 heterozygous mice are normal in motor capacities [26], the effect of Tbx1 deficiency on spatial memory has not been examined in mice.
Tbx1 +/− mice exhibited delayed spatial memory acquisition in the Morris water maze (Fig. 5a–c). In contrast, there was no between-genotype difference in the probe test (Fig. 5d) or during visual cue memory acquisition (Fig. 5e). These data indicate that Tbx1 heterozygosity impairs the acquisition speed of fimbria-dependent spatial reference memory, but not its retention or recall, or fimbria-independent visual cued memory.
Fig. 5. Performance in the Morris water maze test.

a Experimental design. b The mean (± standard error of the mean [SEM]) escape latency in seconds (s) to the platform during acquisition is plotted against days. Compared with +/+ mice, +/− mice exhibited delayed acquisition (Genotype, F(1,26) = 4.643, p = 0.041, ∗[; Day, F(4,104) = 55.490, p < 0.001; Genotype × Day, F(4,104) = 2.329, p = 0.061). The overall genotype effect was primarily due to robust differences on Day 2 (*, p < 0.05) and Day 3 (**, p < 0.01), as determined by Newman–Keuls post hoc tests. +/+, n = 14; +/–, n = 14. c Representative swim paths of a +/+ mouse and +/− mouse on the third training day. The target quadrant included the hidden platform. d The mean (± SEM) time spent during recall probe tests before training on Day 4 (left) and Day 8 (right). Regardless of the quadrant, there were between-genotype differences on Day 4 (Genotype, F(1,26) = 5.597, p = 0.026; Quadrant, F(3,78) = 14.259, p < 0.001; Genotype × Quadrant, F(3,78) = 0.295, p = 0.829) and Day 8 (Genotype, F(1,26) = 10.207, p = 0.004; Quadrant, F(3,78) = 24.031, p < 0.001; Genotype × Quadrant, F(3,78) = 0.562, p = 0.642). The significant main effects of genotype on both days primarily resulted from the generally lower amounts of time spent in three out of the four quadrants in +/− mice (Day 4, Quadrants 1, 3, and 4; Day 8, Quadrants 2, 3, and 4). e The mean (± SEM) escape latency in the visible cue task. A separate set of mice underwent examination using this version of the Morris water maze. +/+ and +/− mice equally acquired this task (Genotype, F(1,17) = 1.861, p = 0.190; Day, F(2,34) = 52.313, p < 0.001; Genotype × Day, F(2,34) = 1.229, p = 0.305). +/+, n = 8; +/−, n = 11.
Tbx1 heterozygosity slows the acquisition of discrimination and cognitive flexibility
Individuals with 22q11.2 hemizygous deletion also exhibit impairments in executive functions [11]. Congenic mouse models of 22q11.2 hemizygosity require an increased number of trials to reach the criteria for simple discrimination and reversal learning [58] or extradimensional shifting (EDS) [59]. However, the individual 22q11.2 genes contributing to impairments in executive functions remain unclear. In humans, prefrontal cortical lesions increase the number of trials required to reach the criterion of attentional set shifting; on the other hand, lesions of the hippocampus and its connections affect the latency for completing each trial [60]. In rodents, orbitofrontal cortical lesions increase the number of trials required to achieve reversal of the intradimensional set (IDS-IV rev) [61], but the rodent brain regions critical for the speed to complete each trial of attentional set shifting are not clear.
Tbx1 +/− mice lacked detectable white matter alterations in the basal forebrain or cortex (see Fig. S2–S5) but exhibited altered myelination in the fimbria. Thus, we reasoned that Tbx1 +/− mice may exhibit altered latency in completing attentional set shifting but may be unaffected in terms of the aspect of attentional set-shifting task requiring the prefrontal cortex (number of trials needed to reach a criterion).
There was no between-genotype difference in the number of trials required to complete each phase of attentional set shifting (Fig. 6a). In contrast, +/− mice were slower in completing each trial of attentional set shifting, most significantly in simple discrimination (SD) and IDS-IV rev (Fig. 6b).
Fig. 6. Attentional set shifting.
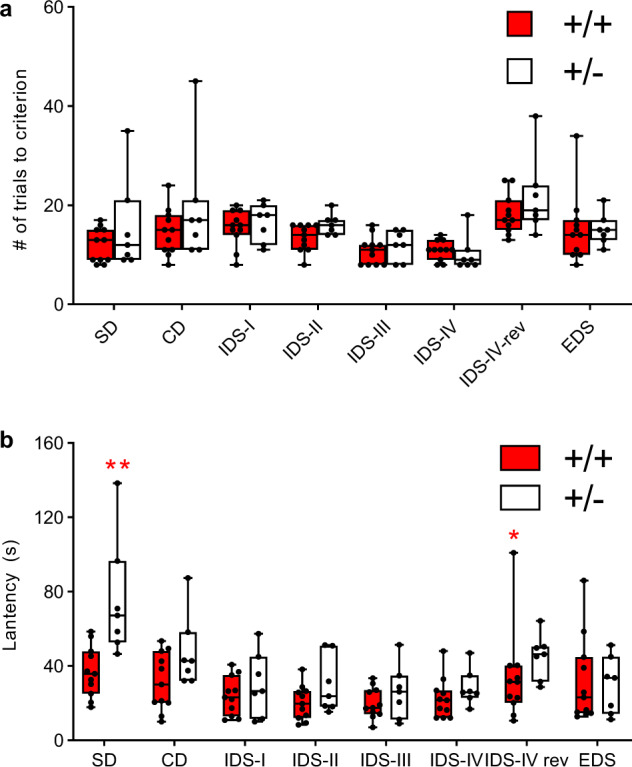
Box-and-whisker plots of (a) the number of trials required to reach the criterion (i.e., eight consecutive correct choices) and (b) latency to complete each trial during the first five correct choices. Since the normality assumption was violated (a, p = 0.002, at IDS-IV of +/−; b, p = 0.001, at IDS-IV rev of +/+), we analyzed both sets of data using a generalized linear mixed model. There was no between-genotype difference in the number of trials taken to reach the criterion (Genotype, F(1,16) = 1.965, p = 0.180; Genotype × Phase, F(7,112) = 0.824, p = 0.569). SD simple discrimination, CD compound discrimination, IDS intradimensional shift, rev reversal, EDS extradimensional shift. +/− mice were consistently slow in completing this task in a phase-dependent manner (Genotype, F(1,16) = 10.010, p = 0.006; Genotype × Phase, F(7, 112) = 2.566, p = 0.017). Mann–Whitney nonparametric post hoc comparisons revealed a significant between-genotype difference in latency to completing the two phases of SD (**, p < 0.01) and IDS-IV rev (*, p < 0.05). +/+ = 11, +/− = 7.
Tbx1 heterozygosity has no detectable effects on olfactory responses
Consistent with the lack of detectable alterations in the white matter integrity of the neocortex, amygdala, and olfactory bulb (Fig. S2–S5), there was no between-genotype difference in responses or habituation to nonsocial and social olfactory cues (Fig. S10). This observation suggests that Tbx1 heterozygosity does not exert nonspecific effects on visual or olfactory perception or on the general motivation to approach an object or odorants.
In sum, our behavioral analysis identified a highly demarcated deficit in the acquisition speed of fimbria-dependent cognitive tasks in Tbx1 +/− mice.
Discussion
The cellular, structural, or cognitive consequences of loss-of-function TBX1 variants in humans remain unclear. Our parallel analysis of structural and behavioral measures in a congenic mouse model of Tbx1 heterozygosity indicated that Tbx1 deficiency caused highly demarcated changes in the structural features of the brain, including reduced production of oligodendrocytes, suboptimal composition of myelination in the fimbria and corpus callosum, and loss of large myelinated axons in the fimbria. These structural alterations impacted fimbria-dependent cognitive functions: Tbx1 heterozygous mice exhibited increased latency to acquire spatial memory and simple discrimination and reversal of intradimensional shift. These effects of Tbx1 heterozygosity on structures and functions are likely due, at least in part, to its enriched expression in adult progenitor/stem cells in the mouse brain [26]. Our single-gene analysis provides a valid first step for deconstruction and reconstruction of the mechanistic composition of CNV-encoded genes in terms of their association with specific behavioral and structural dimensions. As individuals with 22q11.2 hemizygosity exhibit impairments in cognitive speed [11, 13, 62] as well as altered white matter integrity in the hippocampal-projection fibers [16–18, 63], our analyses of a single 22q11.2-encoded gene offer insight into the genetic and cellular substrates of these structural and behavioral alterations in carriers of 22q11.2 hemizygosity. There are inherent limitations in assessing the cognitive dimensions in mouse models due to species-specific differences between mice and humans. The ultimate validation of our mouse data would be achieved by testing therapeutic interventions based on our hypothetical mechanisms underlying defective cognitive speed in humans.
The absence of large myelinated axons in the fimbria of +/− mice may be attributable to a reduced number of oligodendrocytes. Our in vivo data indicated that Tbx1 heterozygosity impacts Ng2, a molecule required for the production of oligodendrocyte precursor cells, in the fimbria. Our in vitro analysis further revealed that fewer oligodendrocytes are produced from postnatal progenitor cells derived from +/− mice. There are several possible reasons for the selective loss of large myelinated axons in the fimbria. Given their higher need for metabolic support from myelin and oligodendrocytes [64–66], large axons may degenerate. Alternatively, but not mutually exclusive, Tbx1 heterozygosity may lead to selective inactivation of large-diameter axons and consequently reduced myelination of those axons, as oligodendrocytes tend to myelinate electrically active axons [67]. In either case, the remaining oligodendrocytes may have instead myelinated medium axons in the fimbria, which would explain the hypermyelination of medium axons observed in the present study. While little is known about the functional contribution of large myelinated axons in the fimbria to cognitive functions, our data provide a potential cellular basis for cognitive speed for further investigation.
The observations that Ng2 mRNA was reduced in the fimbria (see Fig. 3a), but markers of mature oligodendrocytes were not (see Fig. 3a), are seemingly difficult to reconcile. Given that the fimbria of +/- mice contained hypermyelinated medium axons but was devoid of large myelinated axons, it is possible that the effects of these positive and negative alterations on the net amount of MBP and MOG mRNA cancel out in the fimbria. Additionally, as myelin was selectively reduced in the anterior fimbria only (see Fig. S7ab), such a regionally limited effect may be difficult to detect in the whole fimbria tissue used for qRT-PCR.
It is striking that, among all the brain regions scanned, the fimbria was the only region that showed a statistically significant reduction in FA values in Tbx1 +/− mice. The reason for this selectivity remains unclear, but may be due to the postnatal onset of myelination in the rodent brain [40] and highly limited expression of Tbx1 protein in the subventricular zone and granule cell layer of the hippocampus in the postnatal mouse brain [26]. Oligodendrocytes in the subventricular zone postnatally migrate to the fimbria and corpus callosum [50]. Reduced myelination of medium-diameter axons in the corpus callosum—as well as a lack of large myelinated axons in the fimbria—may have also occurred due to reduced postnatal migration of oligodendrocytes from the subventricular zone of Tbx1 +/− mice. More work is needed to determine how local oligodendrocyte precursor cells in the fimbria and oligodendrocytes postnatally provided from the subventricular zone contribute to the myelination of axons of different sizes.
The fornix is an extension of the fimbria and shows one of the largest reductions in FA values among all the brain regions in carriers of 22q11.2 hemizygous deletion [17, 18]. The reduction in FA is more severe in patients with psychosis than without psychosis [17]. FA reductions are larger in idiopathic cases of schizophrenia than in bipolar disorder, major depression, PTSD, or OCD [16–18]. The fimbria carries efferent fibers, via the fornix, to the anterior thalamic nuclei, mammillary body, nucleus accumbens, lateral septum, and afferents from the medial septum in rodents [68]. Our work offers a basis for future studies to delineate the precise axons, targets, and neuronal types underlying slow cognitive speed.
A previous study reported that acquisition speed, retention, and recall of spatial reference memory in the Morris water maze were normal in a mouse model of 22q11.2 hemizygosity [69]. It seems inconsistent that the deletion of Tbx1, one of the 22q11.2 genes, slowed the acquisition speed of spatial memory in the Morris water maze, but a mouse model of 22q11.2 deletion, including Tbx1, did not. There are several potential reasons for this. First, many mouse models of CNVs have failed to recapitulate behavioral deficits, while models of single CNV genes exhibit robust phenotypes [31, 70, 71]. Why mouse models of the entire 22q11.2 hemizygous deletion are normal in many cognitive tasks remains unclear. Second, unlike the 22q11.2-deletion mouse model used by Drew et al., our Tbx1 +/− mice were fully congenic with homogeneous genetic backgrounds in +/+ and +/−. Substantial allelic differences are expected between the control and mutant non-congenic mice near the deletion region in noncongenic mice, which could potentially contribute to the apparent absence of phenotypic differences [70]. Third, in the study of Drew et al., the same mice were first trained and tested in the visual cue version and subsequently in the spatial memory version without any interval. The prior training in the visual task might have ameliorated the potential deficit in subsequent testing in the hidden-platform version [72] In contrast, we used different sets of mice for the visual and spatial memory versions of the Morris water maze to avoid a carryover effect of the prior training on subsequent performance.
The structural alterations and cognitive deficits observed in the present study are not unique to Tbx1 heterozygosity or 22q11.2 CNVs. Lower FA values have been reported in the fimbria/fornix of individuals with idiopathic ASD [73] and schizophrenia [18, 74]. Slow processing speed in individuals with idiopathic ASD is correlated with low FA values, but not with MD, RD, or AD values, in the whole brain [75]. Individuals with idiopathic ASD also exhibit impairments in difficult cognitive tasks [76]. A selective loss of extralarge myelinated axons has been observed in the brains of humans with ASD [77]. Moreover, patients with idiopathic schizophrenia exhibit impaired processing speed across numerous cognitive dimensions, including attention, memory, spatial processing, emotional identification, and sensorimotor capacity [78, 79]. Previous studies have also reported that other oligodendrocyte-related genes are dysregulated in brain samples from individuals with ASD and genetic mouse models for ASD [80–84]. Taken together, our findings open a new window for investigating the potential substrates of altered cognitive speed in carriers of TBX1 SNVs, 22q11.2, and other CNVs, and in idiopathic cases of ASD and schizophrenia.
Supplementary information
Acknowledgements
General: We thank Dr. Bernice Morrow for providing the original breeders of Tbx1+/- mice and Dr. E. M. Powell for providing her protocol of attentional set shifting.
Author contributions
TH and NH designed the study and analyzed all the data. AS, RR, HN, and RK designed and performed the DTI–MRI study and analyzed the data. TY performed gold staining immunohistochemistry and qRT-PCR and analyzed the data. GK maintained and genotyped the mouse colony of Tbx1+/− mice and performed behavioral studies, except for the Morris water maze and attentional set-shifting tasks. TH performed the Morris water-maze test and analyzed the data. TH, SE, and TI performed attentional set shifting and analyzed the data. KT performed in vitro cell cultures of oligodendrocytes and analyzed the data. TH, AS, RK, TY, GK, KT, and NH wrote the paper.
Funding
This study was partly supported by the National Institutes of Health (R01MH099660, R01DC015776, and R21HD105287). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Data availability
All data are available upon request. Mice are available through the Material Transfer Agreement.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Takeshi Hiramoto, Akira Sumiyoshi, Takahira Yamauchi.
Supplementary information
The online version contains supplementary material available at 10.1038/s41380-021-01318-4.
References
- 1.Malhotra D, Sebat J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell. 2012;148:1223–41. doi: 10.1016/j.cell.2012.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kirov G, Rees E, Walters JT, Escott-Price V, Georgieva L, Richards AL, et al. The penetrance of copy number variations for schizophrenia and developmental delay. Biol Psychiatry. 2013;75:378–85. doi: 10.1016/j.biopsych.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stefansson H, Meyer-Lindenberg A, Steinberg S, Magnusdottir B, Morgen K, Arnarsdottir S, et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature. 2014;505:361–6. doi: 10.1038/nature12818. [DOI] [PubMed] [Google Scholar]
- 4.Kendall KM, Rees E, Escott-Price V, Einon M, Thomas R, Hewitt J, et al. Cognitive performance among carriers of pathogenic copy number variants: analysis of 152,000 UK Biobank subjects. Biol Psychiatry. 2017;82:103–10. doi: 10.1016/j.biopsych.2016.08.014. [DOI] [PubMed] [Google Scholar]
- 5.Hubbard L, Rees E, Morris DW, Lynham AJ, Richards AL, Pardinas AF, et al. Rare Copy Number Variants Are Associated With Poorer Cognition in Schizophrenia. Biol Psychiatry. 2021;90:28–34. doi: 10.1016/j.biopsych.2020.11.025. [DOI] [PubMed] [Google Scholar]
- 6.Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. 2020;180:568–584 e523. doi: 10.1016/j.cell.2019.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh T, Neale BM, Daly MJ, SCHEMA. Exome sequencing identifies rare coding variants in 10 genes which confer substantial risk for schizophrenia. MedRxiv 2020.
- 8.Mitra I, Huang B, Mousavi N, Ma N, Lamkin M, Yanicky R, et al. Patterns of de novo tandem repeat mutations and their role in autism. Nature. 2021;589:246–50. doi: 10.1038/s41586-020-03078-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shprintzen RJ, Goldberg R, Golding-Kushner KJ, Marion RW. Late-onset psychosis in the velo-cardio-facial syndrome. Am J Med Genet. 1992;42:141–2. doi: 10.1002/ajmg.1320420131. [DOI] [PubMed] [Google Scholar]
- 10.Zinkstok J, Boot E, Bassett AS, Hiroi N, Butcher NJ, Vingerhoets C, et al. The 22q11.2 deletion syndrome from a neurobiological perspective. Lancet Psychiatry. 2019;6:951–60. doi: 10.1016/S2215-0366(19)30076-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gur RE, Yi JJ, Donald-McGinn DM, Tang SX, Calkins ME, Whinna D, et al. Neurocognitive development in 22q11.2 deletion syndrome: comparison with youth having developmental delay and medical comorbidities. Mol Psychiatry. 2014;19:1205–11. doi: 10.1038/mp.2013.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kendall KM, Bracher-Smith M, Fitzpatrick H, Lynham A, Rees E, Escott-Price V, et al. Cognitive performance and functional outcomes of carriers of pathogenic copy number variants: analysis of the UK Biobank. Br J Psychiatry. 2019;214:297–304. doi: 10.1192/bjp.2018.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chawner S, Owen MJ, Holmans P, Raymond FL, Skuse D, Hall J, et al. Genotype-phenotype associations in children with copy number variants associated with high neuropsychiatric risk in the UK (IMAGINE-ID): a case-control cohort study. Lancet Psychiatry. 2019;6:493–505. doi: 10.1016/S2215-0366(19)30123-3. [DOI] [PubMed] [Google Scholar]
- 14.Gur RC, Calkins ME, Satterthwaite TD, Ruparel K, Bilker WB, Moore TM, et al. Neurocognitive growth charting in psychosis spectrum youths. JAMA Psychiatry. 2014;71:366–74. doi: 10.1001/jamapsychiatry.2013.4190. [DOI] [PubMed] [Google Scholar]
- 15.Vorstman JA, Breetvelt EJ, Duijff SN, Eliez S, Schneider M, Jalbrzikowski M, et al. Cognitive decline preceding the onset of psychosis in patients with 22q11.2 deletion syndrome. JAMA Psychiatry. 2015;72:377–85. doi: 10.1001/jamapsychiatry.2014.2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thompson PM, Jahanshad N, Ching CRK, Salminen LE, Thomopoulos SI, Bright J, et al. ENIGMA and global neuroscience: A decade of large-scale studies of the brain in health and disease across more than 40 countries. Transl Psychiatry. 2020;10:100. doi: 10.1038/s41398-020-0705-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Villalon-Reina JE, Martinez K, Qu X, Ching CRK, Nir TM, Kothapalli D, et al. Altered white matter microstructure in 22q11.2 deletion syndrome: a multisite diffusion tensor imaging study. Mol Psychiatry. 2020;25:2818–31. doi: 10.1038/s41380-019-0450-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kochunov P, Hong LE, Dennis EL, Morey RA, Tate DF, Wilde EA et al. ENIGMA-DTI: Translating reproducible white matter deficits into personalized vulnerability metrics in cross-diagnostic psychiatric research. Hum Brain Mapp 2020. 10.1002/hbm.24998. Online ahead of print. [DOI] [PMC free article] [PubMed]
- 19.Gong W, Gottlieb S, Collins J, Blescia A, Dietz H, Goldmuntz E, et al. Mutation analysis of TBX1 in non-deleted patients with features of DGS/VCFS or isolated cardiovascular defects. J Med Genet. 2001;38:E45. doi: 10.1136/jmg.38.12.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hasegawa K, Tanaka H, Higuchi Y, Hayashi Y, Kobayashi K, Tsukahara H. Novel heterozygous mutation in TBX1 in an infant with hypocalcemic seizures. Clin Pediatr Endocrinol. 2018;27:159–64. doi: 10.1297/cpe.27.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ogata T, Niihori T, Tanaka N, Kawai M, Nagashima T, Funayama R, et al. TBX1 mutation identified by exome sequencing in a Japanese family with 22q11.2 deletion syndrome-like craniofacial features and hypocalcemia. PLoS ONE. 2014;9:e91598. doi: 10.1371/journal.pone.0091598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paylor R, Glaser B, Mupo A, Ataliotis P, Spencer C, Sobotka A, et al. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome. Proc Natl Acad Sci USA. 2006;103:7729–34. doi: 10.1073/pnas.0600206103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hiroi N, Zhu H, Lee M, Funke B, Arai M, Itokawa M, et al. A 200-kb region of human chromosome 22q11.2 confers antipsychotic-responsive behavioral abnormalities in mice. Proc Natl Acad Sci USA. 2005;102:19132–7. doi: 10.1073/pnas.0509635102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki G, Harper KM, Hiramoto T, Funke B, Lee M, Kang G, et al. Over-expression of a human chromosome 22q11.2 segment including TXNRD2, COMT and ARVCF developmentally affects incentive learning and working memory in mice. Hum Mol Genet. 2009;18:3914–25. doi: 10.1093/hmg/ddp334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki G, Harper KM, Hiramoto T, Sawamura T, Lee M, Kang G, et al. Sept5 deficiency exerts pleiotropic influence on affective behaviors and cognitive functions in mice. Hum Mol Genet. 2009;18:1652–60. doi: 10.1093/hmg/ddp086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hiramoto T, Kang G, Suzuki G, Satoh Y, Kucherlapati R, Watanabe Y, et al. Tbx1: identification of a 22q11.2 gene as a risk factor for autism spectrum disorder in a mouse model. Hum Mol Genet. 2011;20:4775–85. doi: 10.1093/hmg/ddr404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harper KM, Hiramoto T, Tanigaki K, Kang G, Suzuki G, Trimble W, et al. Alterations of social interaction through genetic and environmental manipulation of the 22q11.2 gene Sept5 in the mouse brain. Hum Mol Genet. 2012;21:3489–99. doi: 10.1093/hmg/dds180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hiroi N, Takahashi T, Hishimoto A, Izumi T, Boku S, Hiramoto T. Copy number variation at 22q11.2: from rare variants to common mechanisms of developmental neuropsychiatric disorders. Mol Psychiatry. 2013;18:1153–65. doi: 10.1038/mp.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takahashi T, Okabe S, Broin PO, Nishi A, Ye K, Beckert MV, et al. Structure and function of neonatal social communication in a genetic mouse model of autism. Mol Psychiatry. 2016;21:1208–14. doi: 10.1038/mp.2015.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boku S, Izumi T, Abe S, Takahashi T, Nishi A, Nomaru H, et al. Copy number elevation of 22q11.2 genes arrests the developmental maturation of working memory capacity and adult neurogenesis. Mol Psychiatry. 2018;23:985–92. doi: 10.1038/mp.2017.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hiroi N, Yamauchi T. Modeling and predicting developmental trajectories of neuropsychiatric dimensions associated with copy number variations. Int J Neuropsychopharmacol. 2019;22:488–500. doi: 10.1093/ijnp/pyz026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kato R, Machida A, Nomoto K, Kang G, Hiramoto T, Tanigaki K, et al. Maternal approach behaviors toward neonatal calls are impaired by mother’s experiences of raising pups with a risk gene variant for autism. Dev Psychobiol. 2021;63:108–13. doi: 10.1002/dev.22006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma Y, Hof PR, Grant SC, Blackband SJ, Bennett R, Slatest L, et al. A three-dimensional digital atlas database of the adult C57BL/6J mouse brain by magnetic resonance microscopy. Neuroscience. 2005;135:1203–15. doi: 10.1016/j.neuroscience.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 34.Lazari A, Lipp I. Can MRI measure myelin? Systematic review, qualitative assessment, and meta-analysis of studies validating microstructural imaging with myelin histology. Neuroimage 2021; 230: 117744. [DOI] [PMC free article] [PubMed]
- 35.Chang EH, Argyelan M, Aggarwal M, Chandon TS, Karlsgodt KH, Mori S, et al. The role of myelination in measures of white matter integrity: Combination of diffusion tensor imaging and two-photon microscopy of CLARITY intact brains. Neuroimage. 2017;147:253–61. doi: 10.1016/j.neuroimage.2016.11.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sampaio-Baptista C, Khrapitchev AA, Foxley S, Schlagheck T, Scholz J, Jbabdi S, et al. Motor skill learning induces changes in white matter microstructure and myelination. J Neurosci. 2013;33:19499–503. doi: 10.1523/JNEUROSCI.3048-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soares JM, Marques P, Alves V, Sousa N. A hitchhiker’s guide to diffusion tensor imaging. Front Neurosci. 2013;7:31. doi: 10.3389/fnins.2013.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmued L, Bowyer J, Cozart M, Heard D, Binienda Z, Paule M. Introducing Black-Gold II, a highly soluble gold phosphate complex with several unique advantages for the histochemical localization of myelin. Brain Res. 2008;1229:210–7. doi: 10.1016/j.brainres.2008.06.129. [DOI] [PubMed] [Google Scholar]
- 39.Chomiak T, Hu B. What is the optimal value of the g-ratio for myelinated fibers in the rat CNS? A theoretical approach. PLoS ONE. 2009;4:e7754. doi: 10.1371/journal.pone.0007754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Downes N, Mullins P. The development of myelin in the brain of the juvenile rat. Toxicol Pathol. 2014;42:913–22. doi: 10.1177/0192623313503518. [DOI] [PubMed] [Google Scholar]
- 41.Thompson CL, Ng L, Menon V, Martinez S, Lee CK, Glattfelder K et al. A high-resolution spatiotemporal atlas of gene expression of the developing mouse brain. Neuron 2014;83:309–23. [DOI] [PMC free article] [PubMed]
- 42.de Vrij FM, Bouwkamp CG, Gunhanlar N, Shpak G, Lendemeijer B, Baghdadi M et al. Candidate CSPG4 mutations and induced pluripotent stem cell modeling implicate oligodendrocyte progenitor cell dysfunction in familial schizophrenia. Mol Psychiatry. 2019; 24: 757–71. [DOI] [PMC free article] [PubMed]
- 43.Fruttiger M, Karlsson L, Hall AC, Abramsson A, Calver AR, Bostrom H, et al. Defective oligodendrocyte development and severe hypomyelination in PDGF-A knockout mice. Development. 1999;126:457–67. doi: 10.1242/dev.126.3.457. [DOI] [PubMed] [Google Scholar]
- 44.Meschkat M, Steyer AM, Weil M-T, Kusch K, Jahn O, Piepkorn L, et al. White matter integrity requires continuous myelin synthesis at the inner tongue bioRxiv 2020; 10.1101/2020.09.02.279612. [DOI] [PMC free article] [PubMed]
- 45.Readhead C, Popko B, Takahashi N, Shine HD, Saavedra RA, Sidman RL, et al. Expression of a myelin basic protein gene in transgenic shiverer mice: correction of the dysmyelinating phenotype. Cell. 1987;48:703–12. doi: 10.1016/0092-8674(87)90248-0. [DOI] [PubMed] [Google Scholar]
- 46.Delarasse C, Daubas P, Mars LT, Vizler C, Litzenburger T, Iglesias A, et al. Myelin/oligodendrocyte glycoprotein-deficient (MOG-deficient) mice reveal lack of immune tolerance to MOG in wild-type mice. J Clin Invest. 2003;112:544–53. doi: 10.1172/JCI15861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aguirre A, Gallo V. Postnatal neurogenesis and gliogenesis in the olfactory bulb from NG2-expressing progenitors of the subventricular zone. J Neurosci. 2004;24:10530–41. doi: 10.1523/JNEUROSCI.3572-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jackson EL, Garcia-Verdugo JM, Gil-Perotin S, Roy M, Quinones-Hinojosa A, VandenBerg S, et al. PDGFR alpha-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron. 2006;51:187–99. doi: 10.1016/j.neuron.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 49.Belachew S, Chittajallu R, Aguirre AA, Yuan X, Kirby M, Anderson S, et al. Postnatal NG2 proteoglycan-expressing progenitor cells are intrinsically multipotent and generate functional neurons. J Cell Biol. 2003;161:169–86. doi: 10.1083/jcb.200210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Menn B, Garcia-Verdugo JM, Yaschine C, Gonzalez-Perez O, Rowitch D, Alvarez-Buylla A. Origin of oligodendrocytes in the subventricular zone of the adult brain. J Neurosci. 2006;26:7907–18. doi: 10.1523/JNEUROSCI.1299-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ortega F, Gascon S, Masserdotti G, Deshpande A, Simon C, Fischer J, et al. Oligodendrogliogenic and neurogenic adult subependymal zone neural stem cells constitute distinct lineages and exhibit differential responsiveness to Wnt signalling. Nat Cell Biol. 2013;15:602–13. doi: 10.1038/ncb2736. [DOI] [PubMed] [Google Scholar]
- 52.Jones DK, Cercignani M. Twenty-five pitfalls in the analysis of diffusion MRI data. NMR Biomed. 2010;23:803–20. doi: 10.1002/nbm.1543. [DOI] [PubMed] [Google Scholar]
- 53.Packard MG, McGaugh JL. Double dissociation of fornix and caudate nucleus lesions on acquisition of two water maze tasks: further evidence for multiple memory systems. Behav Neurosci. 1992;106:439–46. doi: 10.1037/0735-7044.106.3.439. [DOI] [PubMed] [Google Scholar]
- 54.Hannesson DK, Skelton RW. Recovery of spatial performance in the Morris water maze following bilateral transection of the fimbria/fornix in rats. Behav Brain Res. 1998;90:35–56. doi: 10.1016/S0166-4328(97)00081-8. [DOI] [PubMed] [Google Scholar]
- 55.Yi JJ, Weinberger R, Moore TM, Calkins ME, Guri Y, McDonald-McGinn DM, et al. Performance on a computerized neurocognitive battery in 22q11.2 deletion syndrome: A comparison between US and Israeli cohorts. Brain Cogn. 2016;106:33–41. doi: 10.1016/j.bandc.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goldenberg PC, Calkins ME, Richard J, McDonald-McGinn D, Zackai E, Mitra N, et al. Computerized neurocognitive profile in young people with 22q11.2 deletion syndrome compared to youths with schizophrenia and at-risk for psychosis. Am J Med Genet B Neuropsychiatr Genet. 2012;159B:87–93. doi: 10.1002/ajmg.b.32005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bearden CE, Woodin MF, Wang PP, Moss E, Donald-McGinn D, Zackai E, et al. The neurocognitive phenotype of the 22q11.2 deletion syndrome: selective deficit in visual-spatial memory. J Clin Exp Neuropsychol. 2001;23:447–64. doi: 10.1076/jcen.23.4.447.1228. [DOI] [PubMed] [Google Scholar]
- 58.Meechan DW, Rutz HL, Fralish MS, Maynard TM, Rothblat LA, LaMantia AS. Cognitive ability is associated with altered medial frontal cortical circuits in the LgDel mouse model of 22q11.2DS. Cereb Cortex 2015; [Epub ahead of print]: 1143–51. [DOI] [PMC free article] [PubMed]
- 59.Tripathi A, Spedding M, Schenker E, Didriksen M, Cressant A, Jay TM. Cognition- and circuit-based dysfunction in a mouse model of 22q11.2 microdeletion syndrome: effects of stress. Transl Psychiatry. 2020;10:41. doi: 10.1038/s41398-020-0687-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Owen AM, Roberts AC, Polkey CE, Sahakian BJ, Robbins TW. Extra-dimensional versus intra-dimensional set shifting performance following frontal lobe excisions, temporal lobe excisions or amygdalo-hippocampectomy in man. Neuropsychologia. 1991;29:993–1006. doi: 10.1016/0028-3932(91)90063-E. [DOI] [PubMed] [Google Scholar]
- 61.Hamilton DA, Brigman JL. Behavioral flexibility in rats and mice: contributions of distinct frontocortical regions. Genes Brain Behav. 2015;14:4–21. doi: 10.1111/gbb.12191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jalbrzikowski M, Carter C, Senturk D, Chow C, Hopkins JM, Green MF, et al. Social cognition in 22q11.2 microdeletion syndrome: relevance to psychosis? Schizophr Res. 2012;142:99–107. doi: 10.1016/j.schres.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deng Y, Goodrich-Hunsaker NJ, Cabaral M, Amaral DG, Buonocore MH, Harvey D, et al. Disrupted fornix integrity in children with chromosome 22q11.2 deletion syndrome. Psychiatry Res. 2015;232:106–14. doi: 10.1016/j.pscychresns.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Franklin RJ, ffrench-Constant C, Edgar JM, Smith KJ. Neuroprotection and repair in multiple sclerosis. Nat Rev Neurol. 2012;8:624–34. doi: 10.1038/nrneurol.2012.200. [DOI] [PubMed] [Google Scholar]
- 65.Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–8. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saab AS, Tzvetanova ID, Nave KA. The role of myelin and oligodendrocytes in axonal energy metabolism. Curr Opin Neurobiol. 2013;23:1065–72. doi: 10.1016/j.conb.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 67.Salzer JL, Zalc B. Myelination. Curr Biol. 2016;26:R971–5. doi: 10.1016/j.cub.2016.07.074. [DOI] [PubMed] [Google Scholar]
- 68.Senova S, Fomenko A, Gondard E, Lozano AM. Anatomy and function of the fornix in the context of its potential as a therapeutic target. J Neurol Neurosurg Psychiatry. 2020;91:547–59. doi: 10.1136/jnnp-2019-322375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Drew LJ, Stark KL, Fenelon K, Karayiorgou M, Macdermott AB, Gogos JA. Evidence for altered hippocampal function in a mouse model of the human 22q11.2 microdeletion. Mol Cell Neurosci. 2011;47:293–305. doi: 10.1016/j.mcn.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hiroi N. Critical reappraisal of mechanistic links of copy number variants to dimensional constructs of neuropsychiatric disorders in mouse models. Psychiatry Clin Neurosci. 2018;72:301–21. doi: 10.1111/pcn.12641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nilsson SR, Fejgin K, Gastambide F, Vogt MA, Kent BA, Nielsen V, et al. Assessing the cognitive translational potential of a mouse model of the 22q11.2 microdeletion syndrome. Cereb Cortex. 2016;26:3991–4003. doi: 10.1093/cercor/bhw229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Paylor R, Spencer CM, Yuva-Paylor LA, Pieke-Dahl S. The use of behavioral test batteries, II: effect of test interval. Physiol Behav. 2006;87:95–102. doi: 10.1016/j.physbeh.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 73.Koshiyama D, Fukunaga M, Okada N, Morita K, Nemoto K, Usui K, et al. White matter microstructural alterations across four major psychiatric disorders: mega-analysis study in 2937 individuals. Mol Psychiatry. 2020;25:883–95. doi: 10.1038/s41380-019-0553-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kelly S, Jahanshad N, Zalesky A, Kochunov P, Agartz I, Alloza C et al. Widespread white matter microstructural differences in schizophrenia across 4322 individuals: results from the ENIGMA Schizophrenia DTI Working Group. Mol Psychiatry 2018; 23: 1261–9. [DOI] [PMC free article] [PubMed]
- 75.Travers BG, Bigler ED, Tromp do PM, Adluru N, Froehlich AL, Ennis C, et al. Longitudinal processing speed impairments in males with autism and the effects of white matter microstructure. Neuropsychologia. 2014;53:137–45. doi: 10.1016/j.neuropsychologia.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Desaunay P, Briant AR, Bowler DM, Ring M, Gerardin P, Baleyte JM, et al. Memory in autism spectrum disorder: A meta-analysis of experimental studies. Psychol Bull. 2020;146:377–410. doi: 10.1037/bul0000225. [DOI] [PubMed] [Google Scholar]
- 77.Zikopoulos B, Barbas H. Changes in prefrontal axons may disrupt the network in autism. J Neurosci. 2010;30:14595–609. doi: 10.1523/JNEUROSCI.2257-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gur RC, Braff DL, Calkins ME, Dobie DJ, Freedman R, Green MF, et al. Neurocognitive performance in family-based and case-control studies of schizophrenia. Schizophr Res. 2015;163:17–23. doi: 10.1016/j.schres.2014.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Keefe RS. The longitudinal course of cognitive impairment in schizophrenia: an examination of data from premorbid through posttreatment phases of illness. J Clin Psychiatry. 2014;75:8–13. doi: 10.4088/JCP.13065su1.02. [DOI] [PubMed] [Google Scholar]
- 80.Phan BN, Bohlen JF, Davis BA, Ye Z, Chen HY, Mayfield B, et al. A myelin-related transcriptomic profile is shared by Pitt-Hopkins syndrome models and human autism spectrum disorder. Nat Neurosci. 2020;23:375–85. doi: 10.1038/s41593-019-0578-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Silva AI, Haddon JE, Ahmed Syed Y, Trent S, Lin TE, Patel Y, et al. Cyfip1 haploinsufficient rats show white matter changes, myelin thinning, abnormal oligodendrocytes and behavioural inflexibility. Nat Commun. 2019;10:3455. doi: 10.1038/s41467-019-11119-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marie C, Clavairoly A, Frah M, Hmidan H, Yan J, Zhao C, et al. Oligodendrocyte precursor survival and differentiation requires chromatin remodeling by Chd7 and Chd8. Proc Natl Acad Sci USA. 2018;115:E8246–55. doi: 10.1073/pnas.1802620115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kawamura A, Katayama Y, Nishiyama M, Shoji H, Tokuoka K, Ueta Y, et al. Oligodendrocyte dysfunction due to Chd8 mutation gives rise to behavioral deficits in mice. Hum Mol Genet. 2020;29:1274–91. doi: 10.1093/hmg/ddaa036. [DOI] [PubMed] [Google Scholar]
- 84.Berret E, Barron T, Xu J, Debner E, Kim EJ, Kim JH. Oligodendroglial excitability mediated by glutamatergic inputs and Nav1.2 activation. Nat Commun. 2017;8:557. doi: 10.1038/s41467-017-00688-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available upon request. Mice are available through the Material Transfer Agreement.