

Abstract
It is well-known that Ag–Au bimetallic nanoplates have attracted significant research interest due to their unique plasmonic properties and surface-enhanced Raman scattering (SERS). In recent years, there have been many studies on the fabrication of bimetallic nanostructures. However, controlling the shape, size, and structure of bimetallic nanostructures still has many challenges. In this work, we present the results of the synthesis of silver nanoplates (Ag NPls), and Ag–Au bimetallic core/shell and alloy nanostructures, using seed-mediated growth under green LED excitation and a gold salt (HAuCl4) as a precursor of gold. The results show that the optical properties and crystal structure strongly depend on the amount of added gold salt. Interestingly, when the amount of gold(x) in the sample was less than 0.6 μmol (x < 0.6 μmol), the structural nature of Ag–Au was core/shell, in contrast x > 0.6 μmol gave the alloy structure. The morphology of the obtained nanostructures was investigated using the field emission scanning electron microscopy (FESEM) technique. The UV–Vis extinction spectra of Ag–Au nanostructures showed localized surface plasmon resonance (LSPR) bands in the spectral range of 402–627 nm which changed from two peaks to one peak as the amount of gold increased. Ag–Au core/shell and alloy nanostructures were utilized as surface enhanced Raman scattering (SERS) substrates to detect methylene blue (MB) (10−7 M concentration). Our experimental observations indicated that the highest enhancement factor (EF) of about 1.2 × 107 was obtained with Ag–Au alloy. Our detailed investigations revealed that the Ag–Au alloy exhibited significant EF compared to pure metal Ag and Ag–Au core/shell nanostructures. Moreover, the analysis of the data revealed a linear dependence between the logarithm of concentration (log C) and the logarithm of SERS signal intensity (log I) in the range of 10−7–10−4 M with a correlation coefficient (R2) of 0.994. This research helps us understand better the SERS mechanism and the application of Raman spectroscopy on a bimetallic surface.
It is well-known that Ag–Au bimetallic nanoplates have attracted significant research interest due to their unique plasmonic properties and surface-enhanced Raman scattering (SERS).
1. Introduction
Recently, silver and gold nanoparticles have attracted considerable attention due to their interesting properties, such as high catalytic activities,1 and unique plasmonic,2,3 photothermal,4 and electronic properties.5,6 Moreover, noble metal nanoparticles have shown antimicrobial and antibacterial activities,7 and essential to applications such as surface-enhanced Raman scattering (SERS), bioimaging, and optical sensing properties.8,9 The magnitude of the SERS signal can be significantly enhanced in the case of the scatterer being placed in very close proximity to the surface. Ag nanoparticles can produce intense electromagnetic field enhancements due to localized surface plasmon resonance (LSPR) properties.10 García de Abajo and many other researchers have demonstrated that the electronic mechanism (EM) enhancement of Ag is approximately three orders of magnitude stronger than that of Au.11 However, compared to gold, the individual demerit of silver is less chemically stable in ambient condition so it often suffers oxidation effects toward many chemical reactions,12 this demerit often limits its use in practical applications.13 The individual demerits of silver and gold can be circumvented through the hybridization process, and substantial research efforts have been directed toward the rational design of two-component bimetallic Ag–Au materials in a core–shell fashion.14 Ag–Au nanostructures have applications in solar cell, microelectronics and sensor fabrication.15 The hybridized nanomaterials exhibited the chemically stable and thriving optically responsive properties such as the position and bandwidth of SPR peaks, which primarily depends upon the individual proportion of the metals in the composite.16 The surface plasmon absorption tunability in the absorption bands of individual Ag and Au nanoparticles results from the hybridization of silver and gold17 and the optical field enhancements mediated by surface plasmon are exploited in many applications.18 Shahjamali et al. succeeded in fabricating bimetallic Ag–Au by creating a gold layer coating silver to protect inner silver atoms from being oxidized, this material showed the improvement in the Raman signal.19 The gold atoms created a few nanometer thin layer at sharp, rough sites through the epitaxial growth.20,21 To evaluate the chemical stability of the samples, several authors have used H2O2 to test this oxidation through UV-Vis measurements.22,23 In general, to create the core/shell structure, silver (Ag) is used as a seed template and gold (Au3+) ions are reduced to Au0 on the surface of silver seeds using reducing agents such as ascorbic acid,24 hydroxylamine…23 When Au was deposited onto Ag template surface, Au favorably deposited on the edges of Ag template because of the surface energy of the edges is higher than that of the facets.21 Besides, many other studies have focused on finding a bimetallic Ag–Au alloy structure.25 The gold atoms replaced the silver atoms with galvanic replacement reaction between the Ag seeds and Au precursors leading to the formation of hollow nanostructures.25–28 The earlier reports revealed that the Ag–Au alloy was fabricated based on post irradiation of colloidal mixtures by laser pulses and that alloying process occurred through core–shell intermediates.15,29,30 However, the reasonable explanations for the different Ag–Au structures have not been well deciphered yet, and they are also the motivation of our work. In the present study, Ag–Au bimetallic core/shell and alloy nanostructures have been fabricated using seed-mediated growth under green LED excitation instead of laser pulses. The prepared Ag–Au nanostructures were characterized using field emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM), high-resolution transmission electron microscopy (HRTEM) techniques, ultraviolet-visible (UV-Vis) absorption spectrum, X-ray diffraction analysis and energy X-ray diffraction spectroscopy (EDS). Specificantly, both the core/shell and alloy nanostructure of Ag–Au were utilized as surface enhanced Raman scattering (SERS) targets for trace level detection of methylene blue (MB) molecules. The Raman signal was substantially enhanced for the Ag–Au alloy compared to Ag–Au core/shell.
SERS has become a highly sensitive powerful analytical tool to detect trace concentration of molecules based on electromagnetic mechanism (EM) and chemical mechanism (CM).31,32 For the Ag–Au SERS substrate, the EM mechanism plays an essential role from the contribution of the surface of a metallic substrate due to the localized surface plasmon resonance (LSPR) and the production of many “hot spots” at the sharpened edges and nanogaps between Ag and Au.33,34 The sensitivity of the SERS signal can be improved by coupling the LSPR with the molecular vibrations.35,36 The EM enhancement can be realized by the surface plasmon excitation of noble metal particles by visible radiation. Moreover, the performances of the SERS substrate depend on various factors, including enhancement factor, physical or chemical state, and the stability of the surface as well. Some of the major factors that influence Raman scattering enhancement degree are the surface charge on a substrate, orientation of chemical structure of analyte molecules, and excitation laser wavelength, which might produce the resonant Raman signals.37 On the other hand, thanking to anisotropic crystalline nanostructures, Ag–Au gives more advantages in SERS applications.38 In this case, the enhancement of the electromagnetic field (EM) dominates the chemical mechanism due to the surface plasmon resonance (SPR). As a result, they create many “hot spots”. So at their sharp corners, the strong electromagnetic field enhancement can drastically increase the Raman scattering cross-sections of molecules for SERS signal. Furthermore, a recent article indicated that the CM enhancement could exceed a factor of 100 when gold coating on silver nanoparticles surface thanks to charge transfer between a Au surface and adsorbed molecules, thus leads to increase EF in total.39
In terms of SERS application, bimetallic structures have been used to detect many dyes, such as crystal violet,24,40 rhodamine 6G (R6G),41 methylene blue (MB)42,43 or secondary explosive molecules such as 1,1-diamino-2,2-dinitroethene (FOX-7) and 1-nitro pyrazole (1NPZ).16 In particular, for the MB detection, Manish Kumar Singh et al. used bimetallic Au–Cu to detect MB with the detecting limitation concentraion of 10−6 M.43 William et al. fabricated bimetallic Ag–Au in potato-like shape, and it showed a high sensitive single-molecule detection by the SERS spectra.44 As witnessed by the vibrant research, it is apparent that the closer to the exciting laser wavelength SPR of the Ag–Au nanopaticles is, the higher the enhancement factor will be.45,46 In another respect, Oscar Olea-Mejía et al. showed that Ag–Au alloy nanoparticles, which were fabricated by laser ablation in liquid, achieved the detecting limitation concentraion of 10−7 M for MB sensing application.47
Although many works have studied on bimetallic Ag–Au and its applications for SERS, investigation of anisotropic Ag–Au core/shell, and alloy structures for MB detecting application seems to be paid little attention. For this reason, here we report the synthesis and investigation of the effect of gold precursor on the formation of bimetallic Ag–Au anisotropic nanostructures as well as the ability to detect MB molecules by SERS technique. Additionally, the sensitivity and the limitation of detection of SERS are also evaluated. Our results show that with the low content of Au, the surface energy of the edge crystal surface (100) of Ag NPls is higher than that of surface (111); thus Au atoms preferably coat on the (111) surfaces through a galvanic reaction-free deposition to form the core/shell structure. In contrast, when the content of Au increases, AuCl4− ions concentration increases, Ag ions are replaced by Au atoms with galvanic mechanism. Thereby the anisotropic Ag–Au alloy structure is formed. They are the main reasons for the difference in the formation of either core/shell or alloy structure. The quality of SERS substrates based on pure silver nanoplates and bimetallic Ag–Au nanostructures in probing MB molecules were compared. The result revealed important information in controlling the shape and structure of the bimetallic Ag–Au substrate for application as chemical sensor in analysis and detection of trace concentration of MB.
2. Experiment
2.1. Chemicals and materials
Silver nitrate (AgNO3, 99.98%), chloroauric acid (HAuCl4·4H2O, 99.9%) were used as precursors of Ag and Au, respectively. Trisodium citrate dihydrate (TSC, Na3C6H5O7·2H2O, 99%), sodium borohydride (NaBH4, 99%), l-ascorbic acid (l-AA, C6H8O6, 99.96%) were used as reducing agents. Polyvinyl pyrrolidone (PVP, molar weight 40 000 g mol−1) was used as the stabilizer. Sodium hydroxide (NaOH, 99%), nitric acid (HNO3) were used to control the pH value of the reactant solutions and methylene blue (MB, 95.6%) was used as dye in the SERS study. All of these chemicals were purchased from Merck Co. (Germany) and used as received from the supplier. Milli-Q water was used as solvent for solution preparation and glassware rinsing.
2.2. Synthesis of colloidal silver nanoparticle (AgNPs) seeds
In a typical synthesis, colloidal spherical silver nanoparticles (AgNPs) seeds were synthesized by the reduction process of AgNO3 in the presence of NaBH4 as a reducing agent. The synthesis of colloidal spherical AgNPs seeds involved a simple aqueous phase mixing of AgNO3 to TSC, PVP, and NaBH4 solutions.48 Briefly, 1.5 ml of silver nitrate (AgNO3) solution (0.01 M), 0.225 ml of trisodium citrate (TSC) solution (0.3 M) and 0.3 ml of polyvinyl pyrrolidone (PVP) solution (0.005 M) were mixed, and the total volume of the reaction solution was fixed at 120 ml by adding deionized (DI) water. The solution mixture was magnetically stirred vigorously at room temperature for 15 min and 0.5 ml of sodium borohydrate (NaBH4) solution (0.05 M) was dropwise injected into the reaction mixture to initiate the reduction. Finally, 2 μl of NaOH (0.3 M) was added to the solution to adjust pH above 7. A change in color of the reaction mixture from colorless to yellow was observed within 2 min of reaction. Then, after the stirring process was stopped, the generated solution was maintained for 2 h at room temperature in dark before employing for further experimentation without any additional modifications.
2.3. Synthesis of Ag nanoplate seeds
The preparing the colloidal suspension of Ag spheres with diameter smaller than 10 nm was followed by conversion of Ag seeds to the triangular silver nanoplates with visible light in the photoinduced synthesis. In a typical experiment, colloidal silver seed solutions were irradiated with a narrow band light source using the green LED (light emitting diode) with a light output ∼0.5 mW cm−2 and centre wavelength 520 nm. Eight obtained colloidal spherical AgNPs solutions (each 20 ml) were irradiated with the green LED for different periods of time from 25 min to 95 min named as Table 1 below.
Symbols of AgNPs solutions irradiatated with the green LED for different times.
| Symbol of the obtained silver nanoplates | The period of time of green LED irradiation |
|---|---|
| Ag NPl1 | 25 min |
| Ag NPl2 | 35 min |
| Ag NPl3 | 45 min |
| Ag NPl4 | 55 min |
| Ag NPl5 | 65 min |
| Ag NPl6 | 75 min |
| Ag NPl7 | 85 min |
| Ag NPl8 | 95 min |
The setup for the photochemical reactions is similar to our previous study and other authors.49 The convertion the spherical nanoparticles into nanoplates with various sizes was carried out at room temperature. Monitoring the ultraviolet-visible (UV-Vis) optical absorption spectrum during the entire photochemical synthesis process (Fig. 1) showed that a sequential change in color of the solution was observed in the following manner, from bright yellow to moss green, blue, and finally dark blue when the irradiating time increased, suggesting the formation of Ag nanoplates.
Fig. 1. (a) UV-Vis absorbance spectra of silver nanoplates as a function of exposure time to green LED. (b) TEM image of AgNPs seeds and the inset is the size distribution, SEM images of silver product after exposing to green LED for 25 min (Ag NPl1) (c), 35 min (Ag NPl2) (d), 45 min (Ag NPl3) (e), 55 min (Ag NPl4) (f), 65 min (Ag NPl5) (g), 75 min (Ag NPl6) (h) and 95 min (Ag NPl8) (i). Photographs of dispersions of silver nanospheres (yellow) (inset in (b)), the color of the nanoplate solutions turned from yellow to moss green, blue, and dark blue when the irradiating time increased (insets in (c–i)).

2.4. Preparation of bimetallic Ag–Au nanostructure
We chose a sample of Ag NPl4 to make a nano template for further experiments. The Ag–Au nanostructure was prepared by the seed-mediated growth process at room temperature using Ag NPls as the template, PVP as a capping agent, and l-AA as a reducing agent.24 In a typical experiment, 12 solutions (50 ml) of Ag NPl4 were prepared in 12 separate tubes. The deposition of Au shells onto the triangular Ag nanoplates seeds was initiated by injection of aqueous HAuCl4 at a relatively slow rate and vigorously stirring on a magnetic stirrer for one hour. 12 different amounts of HAuCl4 precursor of 0.1, 0.2, 0.4, 0.6, 0.8, 1, 1.2, 1.6, 2, 2.4, 2.8, and 3.4 μmol was chosen corresponding to 12 solutions of Ag NPl4 above. These samples were denoted as Table 2 below. The Au atoms were deposited onto the surface of Ag NPls by the extremely slowly reducing of l-AA in separate tubes on a mechanical syringe pump. The concentration of l-AA is optimized (10 mM) to just enough amount of HAuCl4. For three initial samples (Ag–Au1, Ag–Au2, Ag–Au3), the solutions were added 150 μl of aqueous NaOH solution (0.3 M) under magnetic stirring at room temperature to adjust the pH value to 10.2.
Symbols of bimetallic Ag–Au using different amounts of HAuCl4 precursor.
| Symbol of bimetallic Ag–Au nanoplates | HAuCl4 amount added to 50 ml Ag NPl4 solution |
|---|---|
| Ag–Au1 | 0.1 μmol |
| Ag–Au2 | 0.2 μmol |
| Ag–Au3 | 0.4 μmol |
| Ag–Au4 | 0.6 μmol |
| Ag–Au5 | 0.8 μmol |
| Ag–Au6 | 1 μmol |
| Ag–Au7 | 1.2 μmol |
| Ag–Au8 | 1.6 μmol |
| Ag–Au9 | 2 μmol |
| Ag–Au10 | 2.4 μmol |
| Ag–Au11 | 2.8 μmol |
| Ag–Au12 | 3.4 μmol |
2.5. Materials characterization
Ultraviolet-visible (UV-Vis) absorbance spectra were recorded using a Jasco V-770 UV-Vis spectrophotometer in the range of 250–1000 nm. The particle size distribution of the individual well-dispersed AgNPs seeds in the medium was evaluated by particle size analyzer (PSA, Delta Nano C, Beckman) using the dynamic light scattering (DLS) technique. The light is scattered on particles due to Brownian motion and is collected in different angles which are inversely related to the particle size. The particle size distribution is resulted from the dynamic scattering intensity of red laser having wavelength 750 nm.50 The XRD spectra were collected using an X-ray diffractometer (Bruker D8 Advance, Germany) operated at 30 kV with Cu-Kα radiation (wavelength of λ = 0.154 056 nm) with parallel-beam geometry between 30 to 80° range. The morphology of the as-prepared nanostructures was analyzed in a JEOL JEM-1010 transmission electron microscope (TEM) operating at 80 kV and in a Hitachi S4800 scanning electron microcope (SEM) operating at 10 kV. The TEM images were obtained by drop casting the nanostructure dispersions on carbon coated Cu-grids and drying at room temperature. SEM technique can image and directly measure size of nanoparticles and infer shape information. SEM uses a high energy electron beam scanning over the sample surface and looks the back scattering electrons. The electrons interact with the atoms and produce signals containing information about the surface topography. The elemental composition in the nanostructures was examined through energy dispersive X-ray spectroscopy (EDS) mapping in a Hitachi SU 8020 at an accelerating voltage of 200 kV. EDS line-scan profiles were taken by using a probe with diameter of ca. 0.5 nm and the acquisition time of 5 s for each spectrum. For the SERS measurements, MB dye with a concentration in the range of 10−7 ÷ 10−4 M was used as molecular Raman probe. Raman spectra were collected on a Raman spectrometer (Raman Horiba XploRa plus Raman microprobe, France). The SERS spectra were collected using a laser beam with excitation wavelength of 532 nm, 4 μm in diameter, laser power of 3.2 mW and a signal acquisition time of 8 s. In a typical procedure, we deposited 50 μl suspension containing Ag–Au colloid (0.5 mg ml−1) onto a 1 cm2 glass substrate which has been cleaned several times with ethanol and acetone. After that, we dropped 2 μl of an aqueous MB solution with the desired concentration over these metallic films and dried at room temperature.51 We took five scans of laser exposure for each measurement then averaged them.
3. Results and discussion
3.1. Preparation of silver nanoparticle (AgNPs) seeds
Fig. 1a shows the UV-Vis spectrum of the AgNPs (black line) with only one specific plasmon resonant (PR) band at around 400 nm and the full width at half maximum of 65 nm which indicates the well stabilized particles without precipitation or aggregation. This characteristic band agrees with Mie's theory for spherical particles.52–54 The TEM analysis in Fig. 1b showed that most of the particles were spherical or nearly spherical with the average size of 8 nm. The size distribution histogram is the inset in Fig. 1b together with the representative TEM image. Silver ions (Ag+) were reduced in aqueous solution by NaBH4 in an ice bath to form colloidal silver seeds. The chemical reaction between silver nitrate and sodium borohydide could be expressed as follows:
| 2AgNO3 + 2NaBH4 → 2Ag + H2↑ + 2BH3 + 2NaNO3 | 1 |
Ag+ ions were reduced by NaBH4 to the zerovalent silver atoms and oxidized NaBH4 to hydrogen and borohydride (BH3) as by-products. The silver atoms aggregated to form the nanoscale silver particles. This process is attributable to the Ostwald ripening phenomenon, where small particles tend to attach to large particles. The formation of AgNPs was observed by the change in the color of the solution from colorless to yellow during the reaction (photograph in Fig. 1b). The addition of alkaline solution such as NaOH increased the pH of the solution (i.e., the increase in the alkalinity of the solution) and formed a precipitate at the bottom of the reacting solution, leading to large nanoparticles. This may be caused by diffusion of hydroxyl (OH−) ions which attracted the collision of Ag atoms. The formed AgNPs was a spherical shape and well dispersive under assistance of the capping agent (PVP molecules). PVP molecules play an important role in stabilizing the reaction process and in the formation of colloidal silver, where PVP formed a self-assembly monolayer on the silver seeds, protected the surface from dissolution, and avoided shape transformation.55 The PVP structure has a polyvinyl skeleton with polar groups containing nitrogen and oxygen atoms. The PVP molecules bond to the surface of the AgNPs through the nitrogen atom in their molecules. Therefore, PVP can be firmly adsorbed on the surface of the silver atoms which hinders the diffusion of silver and reduces the growth of silver grain, and leads to the production of small silver seeds.56 Thanks to the physical barrier provided by PVP, the size of the silver particles decreased. This physical barrier helps to prevent particle agglomeration and increases the solubility of the nanosystem.57 PVP molecules bind to the nanoparticles surface via covalent bonds or chemical interactions and separate the silver seeds from each other, which lowers the formation of large aggregates.
3.2. Preparation of Ag NPls
The formation mechanism of Ag NPls was well-known in literatures.58,59 In this work, we fabricated the Ag NPls by using plasmon-mediated growth method under green LED exposure.48 The nucleation process of silver seeds was a prior trend at the inceptive chemical reaction stage. These tiny seeds served as nuclei core for the following growth, they were gradually consumed as the reaction proceed.60 After the solution of AgNPs seeds was exposed to LED radiation, the accumulation of tiny silver seeds occured to form larger nanoplates via the Ostwald ripening mechanism,61 where small particles joined together to form various morphologies depending on the duration of LED radiation. While the role of PVP was to generate well-dispersed and small Ag seeds, citrate ions promote the morphological transition from spherical to triangular particles, where the citrate ions preferentially interact with the {111} facets of silver nanocrystals and inhibit the growth of the {111} plane, leading to the formation of platelike morphology.10 Silver ions were reduced on silver seeds by a mild reducing agent (ascorbic acid) and the result was the formation of truncated triangular nanoplates. The UV-Vis spectra determined the structure of the AgNPs based on their plasmon oscillations of free surface electrons.62,63 The spectra of AgNPs and Ag NPls in Fig. 1a exhibit peaks at 400 nm and 610 nm, each peak represents the size and shape of silver nanostructures in surface plasmon resonance (SPR) bands.64 The bandwidth increases as the particle size increases and the position of the plasmon band as a function of the irradiation time. The intensity of the 400 nm peak decreases as the green LED irradiating time increases, which is accompanied by a shift in the position of the peak toward higher wavelength values, indicating an increase in the size of the silver particles because of aggregation. The decrease in the intensity of the 400 nm peak in the aggregation process showed that the number of silver seeds decreased, namely, original silver seeds were consumed. During 25 minutes under LED illumination, the absorption band at 400 nm of the seed solutions decreased in intensity along with the simultaneous increase of the absorption band at 603 nm, indicating the growth of silver triangular plates agglomerated from silver seeds induced by LED irradiation.65 The absorption intensity of this secondary peak increases gradually when prolonging the exposure time to 95 minutes. This increase is due to the increase of the particle size. In addition, the light shift of absorption spectrum maximum can be neglected. The interaction of photons with the sharp corners of Ag NPls is preferred due to their higher energy. As a result, corrosion at these sharp vertices results in the creation of some truncated triangular silver plates and hexagons, as well as some circular silver disks (SEM images in Fig. 1(g–i)). Especially after a green LED illumination time of 95 minutes, the specific absorption peak (400 nm) decreases minimum. That is the clear evidence of the incomplete agglomeration of the nanoparticles. Of seed solutions exposed to green LED irradiation for long period of time over 35 minutes, a sharp peak appears about 339 nm. In contrast, almost no peak at 339 nm can be observed in the Ag NPl1 and Ag NPl2 spectra (corresponding to 25 minutes and 35 minutes) and AgNPs seeds due to the small size of these particles. These results indicate that the key products from Ag NPl3 to Ag NPl8 synthesized for 45 min to 95 min of green LED illumination were silver nanoplates. This is confirmed in SEM images in Fig. 1(e–i).
Detailed SEM analysis allowed us to measure the thickness and demonstrated the flat nature of the Ag nanoplates. Fig. 1c displays the SEM image of the Ag NPls after exposure to a green LED for 25 min (Ag NPl1) with the morphology of regular triangular nanoplates (edge average length of about 40 nm and thickness of 10 nm, aspect ratio AR = 4). When the irradiation time was increased to 35 min, the product was the mixture of triangular and truncated triangular nanoplates, where the triangular shape was the predominant product (Ag NPl2, Fig. 1d). If the irradiation time was further extended to 45 min, the main product was the silver truncated triangular nanoplates with round vertices and a small population of trapezoid nanoplates (Ag NPl3, Fig. 1e). Increasing the irradiation time to 55 min, 65 min, 75 min and 95 min, the silver truncated triangular nanoplates were gradually transformed into a hexagonal shape and finally into silver nanodisks with round edges (Ag NPl4, Ag NPl5, Ag NPl6, Ag NPl7, Ag NPl8, Fig. 1f–i). Photographs were also collected for the samples of Ag NPls and the insets to Fig. 1(c–f) with the green LED irradiating time increasing from 25 min to 95 min. The Ag particles are a yellow color and the Ag nanoplates are a green color. The corners of hexagons became rounder than they were at the former stages. The major products were hexagons and by-products were truncated triangles and nanodisks with an average diameter of 50 nm. The lateral dimension of Ag nanoplates is notably larger than their thickness, so the degree of anisotropy of silver nanoplates is high.
3.3. Preparation of Ag–Au core/shell and alloy
3.3.1. Morphology and UV-Vis absorption spectrum
Fig. 2 shows a series of TEM images of the synthesized Ag and Ag–Au NPs. Fig. 2a shows the SEM image for Ag NPl4 which serves as the cores for further coating with Au using HAuCl4 as the gold precursor in the presence of l-AA (l-ascorbic acid). The mophologies contained the triangular, hexagonal and disklike Ag–Au core/shell nanoplates. The TEM images in Fig. 2(e–h) reveal a triangular Ag core/Au shell morphology, in which the triangular Ag seed template shape is preserved, Ag elements in the core and Au elements are located around the Ag nanoplates template. Fig. 2(b and c) indicates the lighter triangular or hexagonal center and darker outside layer, the diameter of the core ∼50 nm and the thickness of the shell ∼9 nm. However, Fig. 2(f, g and h) displays no darker outside layer and light center at all due to the formation of an incomplete Au shell on the Ag particle surface or the formation of Ag–Au alloy nanostructures. The deposition of Au on the Ag nanoplates depends on the added amount of HAuCl4, where Au3+ ions are reduced by L-AA to form Au atoms and deposite at the edges of the triangular Ag nanoplates. The coordination number is lowest at the tips of the Ag nanoplates at which the reduced reaction of Au3+ occurs. However, Au atoms preferentially nucleate and grow on the edges of the Ag nanoplates which are the high-energy {110} facets, therefore the triangular shape was maintained. The deposited Au atoms form uniform Ag–Au core/shell nanoplates with uniform Au–shell thickness around Ag-core.13 The Au shell thickness increases along with the amount of HAuCl4 gold precursor used in the synthesis. Because of the close lattice match of the two metals (Ag and Au), an alloy structure is easy to be formed at the interface of the Ag and Au metals, so the absolute shell thickness is difficult to determine and the relative shell thickness is a function of amount of HAuCl4 used. These samples were all stable for several months of storage at ambient conditions with no precipitation of color change of the solution. The Au acts as a barrier to the oxidation of the Ag core, so the Au coating is expected as the protection increasing the stability of Ag NPs.66
Fig. 2. (a) SEM image of Ag NPl4 used as template seeds. The typical TEM images of (b) Ag–Au1 (x = 0.1 μmol), (c) Ag–Au2 (x = 0.2 μmol), (d) Ag–Au3 (x = 0.4 μmol), (e) Ag–Au4 (x = 0.6 μmol), (f) Ag–Au6 (x = 1.0 μmol), (g) Ag–Au9 (x = 2.0 μmol) and (h) Ag–Au12 (x = 3.4 μmol). Insets in each image are photographs of the corresponding bimetallic Ag–Au core/shell or alloy solutions.

The reduction power of the reducing agents such as ascorbic acid can be boosted by increasing the pH of the solution. We added 150 μl NaOH (0.3 M) to adjust the pH value of 3 samples (AgAu1, AgAu2, AgAu3) to 10.2. Au3+ ions were reduced faster, the growth sites of Au at the edges of Ag nanoplates were formed more, thus allowing more uniform deposition of Au (Fig. 2b–d). The surfactant PVP preferentially binds with the reduced Au atoms to achieve homogenous core/shell nanostructures while preserving the original triangular Ag seed nanoplate shape.67,68
The UV-Vis spectroscopy was used to study the optical properties of the as-prepared core–shell nanoplates. Fig. 3a shows the UV-Vis spectra collected for each sample of Ag and Ag–Au NPs, and strong absorption band is observed around 600 nm. The UV-Vis spectrum is for the as-synthesized Ag NPl4 sample with surface plasmon resonance (SPR) peak maximum of 402 nm and the second peak of 627 nm. Spectrum for AgAu1 with HAuCl4 amount of 0.1 μmol also has two main bands, the 410 nm peak intensity has decreased as a result of the suppression of the SPR band by the coating with Au. Increasing the HAuCl4 amount, the primary band at 410 nm has shifted to higher wavelength (433 nm) than that of AgNPs and the intensity of this band is drastically decreased from that of the earlier sample. Spectrum for AgAu4 shows that the original 400 nm peak is now completely obscured, and only long wavelength band at ∼600 nm remained. The 600 nm peak shifted to higher wavelength as the Au amount increases (Fig. 3b) due to the large variation in the collective oscillations of the surface free electrons after Au-shell formation.69 These spectra present the effect of the HAuCl4 amount on the plasmonic resonance peak of Ag–Au samples. The peak position and intensity of the SPR depend on the shape, size and composition of the sample,70 which is dependent on the amount of Au added in the coating procedure. Thus, the shifting of the Ag–Au LSPR may be interpreted in terms of the electron cloud oscillation of Au atoms. Spectra for AgAu9–AgAu12 alloy samples show the characteristics different from the other spectra, illustrating the unique optical properties of the Ag–Au NPs. The alloy nanoparticle samples show only one peak, where the Ag–Au core/shell samples show two distinct peaks, which agrees with the previous report.17,71,72 A silver core is coated completely by a thin gold shell at the surface with a uniform thickness and intimate contact between the Ag and Au. The peak maxima for the alloy case are lower than that of pure Ag sample and lie in between that for the monometallic NPs, which has been observed previously.72 The first peak ranging between ∼400–500 nm is attributed to the Ag core component of the structure and the second peak around 600 nm is attributed to the gold shell component. The structure and optical property gradually change as more Au is added in the coating experiment. The relative reduction potential of Au is higher than that of Ag, so Au ions are reduced to metallic state and Ag metal is oxidized which often causes the formation of alloyed Ag–Au NPs and some Ag is inevitably etched away.
Fig. 3. (a) UV-Vis spectra of Ag NPl4 and Ag–Au samples with different amounts of HAuCl4. (b) The dependence of maximum wavelength of LSPRs (blue line) and maximum intensity (red line) of dipolar LSPR mode as a function of concentration of HAuCl4.

The alloy structure of Ag–Au will be further analyzed in structural transition (powder X-ray diffraction – XRD) and through high-resolution TEM (HRTEM) images. Remarkably, the maximum LSPR is redshifted as the amount of Au gradually increases (Fig. 2b). The position of the plasmon band redshifted during Au deposition can be explained by an increase in the aspect ratio of the plate structure. The redshift of the plasmonic peak suggests that the reaction is dominated by deposition, which is completely suitable with previous works in which the plasmonic peak shifts to the blue since the plates made were thinner during galvanic replacement. This redshift is evident that there is a large contribution of gold in the nanoparticle materials.
3.3.2. Structure analysis of Ag–Au
Powder X-ray diffraction (XRD) has been proved to be an advantageous method to study the structural evolution. The bimetallic phase of the samples was confirmed by XRD peaks. Fig. 4 depicts the XRD patterns of Ag NPl4 and Ag–Au (x = 0.1, 0.2, 0.4, 1.0 and 3.4 μmol). Four peaks in each spectrum are assigned to the diffraction of {111}, {200}, {220} and {311} planes of fcc silver corresponding to 2θ angles 37.9°, 44.07°, 63.9° and 76.7°, respectively. In addition, X-ray diffraction (XRD) result verified the face-centered cubic (fcc) structure of obtained Ag NPl4 (ICDD no. 01-071-3752)73 as well as Ag–Au core/shell or alloyed nanoplates. The planes observed in the bimetallic phase of Ag–Au are similar to that of individual Ag, since their lattice constants are similar [Au (0.408 nm) and Ag (0.409 nm)] [109]. This similarity of Ag and Au lattice constants enable their easier amalgamation, so Ag–Au system can be easily prepared due to their miscibility. With the main peak at 37.9° of all samples, the ratio between the intensities of (111) and (200) diffraction peaks is higher than the literature value (2.8 versus 2.5).74 This indicates that the silver nanocrystals are {111} oriented, thus diffraction intensity of the {111} plane is enhanced compared with that of other planes. The nanoplates are single crystals with {111} planes as the basal planes and the top and bottom surfaces of the nanoplates are bounded by {111} atomic planes.73 This means that this plane may possess the lowest surface tension, and the atom density of (100) crystal surface in the fcc lattice structure was 91.04%,74,75 so the silver and gold atoms favor to locate along the [111]direction. Besides, as can be seen in Fig. 4a (from Ag–Au1 to Ag–Au12), the relatively higher peak formed at 37.9°, the preferential alignment of the (111) orientation was generated in Ag–Au plate.12 The average crystalline size is calculated using Debye–Scherrer formula,76
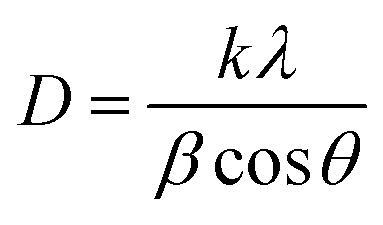 |
2 |
where k = 0.893 is the geometric factor or Scherrer constant, λ = 0.154 056 nm is the wavelength of X-ray radiation, β is the angular full-width at half maximum (FWHM) of the XRD peak (rad), θ is the diffraction angle of the corresponding peak. Fig. 5 describes the crystal size evaluation of Ag–Au samples (x = 0–3.4 μmol HAuCl4) and FWHM as a function of the HAuCl4 amount in all crystal facets (111), (200), (220) and (311). As a result, when a small amount of HAuCl4 is added to the Ag NPl4 solution in the first stage, the crystal size decreases rapidly for all crystal faces from 14.2 nm down 6.8 nm for the face (111), 24.1 nm down 7.02 nm for (220) plane, 19.08 nm down 7.02 nm for (311) plane, except for the case of the (200) crystal facet whose crystal size has increased from 8.2 nm (sample Ag NPl4, x = 0) to 9.9 nm (x = 0.1 μmol) and then decreases down 2.8 nm (x = 0.4 μmol).
Fig. 4. (a) XRD analysis of the silver nanoplate (Ag NPl4) and bimetallic Ag–Au nanoplates (Ag–Au1, Ag–Au2, Ag–Au3, Ag–Au6, and Ag–Au12) (b-d) the diffraction peaks at around 37.9° (111), 44.07° (200), 63.9° (220), and 76.7° (311).

Fig. 5. (a) Crystalline size, and (b) FWHM as a function of HAuCl4 amount at all crystal facets (111), (200), (220) and (311).

The core/shell structure was more closely examined with a transmission electron microscopy (TEM) analysis, the Au shell seems slightly darker than the Ag core region (Fig. 6a) because the Au atom is heavier than Ag atom. Fig. 6b shows the high-resolution transmission electron microscopy (HRTEM) image of the Ag–Au1 (x = 0.1 μmol) core/shell nanostructures. The measured lattice distance to [111] direction of single crystalline Au is d111(Au) = 0.232 nm, verifying the presence of a Au outer shell. The Au atoms covered the surface of AgNPs to form a thin Au outer shell with the deposition thickness of about 9 nm. The lattice distance of d111(Ag) = 0.234 nm corresponding to [111] direction of single crystalline Ag. The calculated lattice constants according to the spacing distance dhkl of the {111} planes as the following equation:
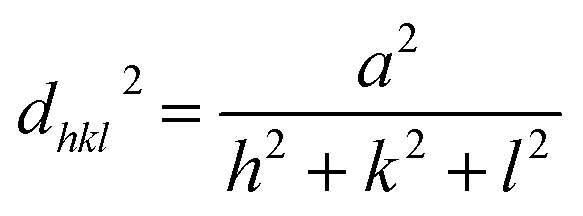 |
3 |
Fig. 6. (a) TEM image of a typical Ag–Au core/shell nanoplate. (b) HRTEM image of Ag–Au core/shell nanoplate, (c) A typical TEM image of an individual Ag–Au core/shell (d and e) EDS elemental maps. (f) EDS spectrum of a typical Ag–Au core/shell nanoplate, (g and h) line profiles of the Ag–Au core/shell nanoplate.

Thus, the lattice constants of Au and Ag crystallines are equal to 0.405 and 0.401, respectively, which approach the previous report.77
In terms of addressing the core/shell structure of the Ag–Au nanoplates, the EDX mapping technique was employed. The compositional analysis by energy-dispersive X-ray spectroscopy (EDX)-elemental mapping result for a single Ag–Au nanoplate (Ag–Au4) shown in Fig. 6(d and e) indicates the presence of Ag elements in the green color and Au elements in the red color. Fig. 6c shows the line scan profile of a typical TEM image of an individual Ag–Au core/shell. The formation of core/shell morphology is evident, a majority of the Ag is located at the center of the particle while the periphery of the nanoplate has a heightened amount of Au. The uniform deposition may be the consequence of higher lattice match between Ag and Au elements. The EDX mapping data illustrates the core/shell nature of the nanoplates, while the compositional data shows that the various samples all contain silver and gold. Fig. 6f indicates that there are 21.93 at% Ag and 78.07 at% Au in a single Ag–Au4 nanoplate. The bimetallic alloy nanoplates are formed when Ag+ and AuCl4− ions are simultaneously reduced.78 According to TEM image of Ag–Au12 (x = 3.4 μmol) sample (Fig. 7a), the average size of Ag–Au nanoplates of alloy type was of 50 nm. The HRTEM image in Fig. 7b shows that the finally formed Ag–Au nanoplates have high crystalinity. The motion of Ag atoms facilitates the diffusion of Au atoms into the Ag seeds in the Au deposition process which leads to the growth of alloy Ag–Au nanoplates. Interestingly, as the concentration of coated gold increases (x = 0.6–3.4 μmol), the HRTEM image shows the overlap of 2 elements Ag and Au (Fig. 7c). As well known, the atomic radius, the lattice constant of Ag and Au crystals are quite similar, so that they easily substitute each other. We used EDS elemental analysis to check the content of metals in obtained nanoplates. For the sample Ag–Au12, the Ag : Au ratio shown in atomic% ratio was 15.82 at% Ag and 84.18 at% Au (Fig. 7d). Thus, the initial pure Ag NPls core must have been partly replaced by Au atoms to form the Ag–Au alloy. The HRTEM image in Fig. 7c gives the information of the measured lattice plane separation of the Miller plane {111} of 0.234 nm for Ag and 0.232 nm for Au corresponding to crystal sizes of 0.405 and 0.401 nm, respectively.77 The HRTEM image clearly indicates the miscible nature of Ag and Au metals at all proportions. Therefore, we can estimate the lattice constant of Ag–Au alloys following Vegard's law:79,80
| aAu1−nAgn(alloy) = (1 − n)aAu + naAg | 4 |
where n is determined from the EDS spectrum in Fig. 7d, it was found to be 0.158 in our case. This value is quite reasonable when the atomic ratio of Au/Ag is approximately 5.3 times in a single Ag–Au12 alloy nanoplate. The calculated result is aalloy = 0.2323 nm, which demonstrated that the change in the unit cell size of Ag–Au alloy is typically less than 1% for all fractions of Ag and Au with respect to pure Ag and Au.
Fig. 7. (a) Low-resolution TEM image of Ag–Au alloy nanoplate. (b) HRTEM of a typical synthesized single Ag–Au alloy and (c) HRTEM image of a part of this alloy, which indicated the lattice plane constants of Ag (0.234 nm) and Au (0.232 nm). (d) EDS spectrum of (b) which shows the contribution of Ag and Au atoms in the Ag–Au alloy (x = 3.4 μmol).

3.3.3. Growth mechanisms
Recently, a variety of attempts have been made to try to understand clearly the synthesis mechanism of Ag–Au core/shell and alloy nanocrystal.13,23 We hypothesize that the mechanism of Au deposition on the Ag nanoplates after adding HAuCl4 as follows. The redox reaction between Ag atoms and Au3+ ions occurs at the surface atoms with the lowest metal coordination number such as the vertexes of the AgNPs. The reduced Au atoms are initially deposited at the edges of the triangular AgNPs due to the higher surface energy of these facets. The inside Ag atoms gain the chemical stability provided by the deposited Au atoms, so the oxidation dissolution is resisted and therefore the triangular shape would be maintained.19 The relatively higher reduction potential of AgCl4−/Au (0.99 V vs. SHE, standard hydrogen electrode) compared to that of Ag+/Ag (0.8 V vs. SHE)23 leads to the oxidation of Ag by Au3+. l-ascorbic acid (l-AA) reducing agent controlled the galvanic exchange between Ag and Au3+ and deposition of Au onto the Ag template.
Fig. 8 shows the schematic illustration of the formation mechanism of Ag–Au core/shell nanoplates. In the step 1, l-AA was injected into the Ag NPls colloidal solution. In the step 2, Au precursor solution (HAuCl4) was slowly added to the seed solution containing l-AA. The reduction power of the l-AA was raised significantly, Au3+ ions were reduced and formed the growth sites at the edges of Ag nanoplates, allowing uniform deposition of Au around Ag NPls. The chemical reactions involved in the gold coating process occur as follows (eqn (5)–(9)):
| HAuCl4(aq) → H(aq)+ + AuCl4(aq)− | 5 |
| 3x|C6H8O6(aq) − 2e → C6H6O6(aq) + 2H(aq)+, | 6 |
| 2x|AuCl4(aq)− + 3e → Au(s) + 4Cl(aq)− | 7 |
| 2AuCl4(aq)− + 3C6H8O6(aq) → 2Au(s) + 3C6H6O6(aq) + 6H(aq)+ + 8Cl(aq)− | 8 |
| (Agn)(s) + 2pHAuCl4(aq) + 3pC6H8O6(aq) → (AgnAu2p)(s) + 3pC6H6O6(aq) + 8pH(aq)+ + 8pCl(aq)− | 9 |
Fig. 8. Schematic illustration of the formation mechanism Ag–Au nanoplate core/shell and alloy.

In contrast, the formation mechanism of Ag–Au alloy nanoplates obeys different 2 steps as illustrated in Fig. 8. In the step 1, the HAuCl4 solution was added to the seed solution. In the step 2, the reducing agent (l-AA) was injected to above mixture. A galvanic replacement reaction can be described as an oxidation-reduction (redox) process, where electrons are transferred from silver atoms to gold ions in solution.81 The driving force of this process is the difference in the reduction potentials of Ag and Au, in which the reduction potential of the Au precursor is higher than that of Ag template. The galvanic process contains the two half equations as shown in (10) and (11) below.
| 3x|Ag(s) − 1e → Ag(aq)+ | 10 |
| 1x|AuCl4(aq)−+ 3e → Au(s) + 4Cl(aq)− | 11 |
This replacement reaction could be described according to the eqn (10) and (11).82
| (8), (9) => 3Ag(s) + AuCl4(aq)− → 3Ag(aq)+ + Au(s) + 4Cl(aq)− | 12 |
In general, we can write the equation as follows:
| (Agn)(s) + mHAuCl4(aq) → (Ag(n−3m)Aum)(s) + 3mAg(aq)+ + 4mCl(aq)− + mH(aq)+ | 13 |
The composition and structure of Ag–Au alloy nanoplates obtained by the galvanic replacement reaction can be controlled by adjusting the amount of HAuCl4 precursor added into the reaction mixture. When Ag atoms contact with an aqueous HAuCl4 solution, they are oxidized and dissolved by AuCl4− ions, producing Ag+ ions into the solution. At the same time, Au atoms are deposited at the surface of Ag nanoplates due to the reduction of the AuCl4− ions precursor.83 Au and Ag have the similar face-centered cubic (fcc) crystal structure and lattice constants, resulting in a great miscibility of these two metals. As more Ag is replaced by Au as increasing the amount of Au precursor, alloying process leads to the formation of a homogenous distribution of Ag and Au in the final product.
3.4. SERS application for MB detection
Fig. 9a presents the Raman spectrum of MB solid only on the bare glass substrate. Several prominent peaks at 445, 501, 772, 1037, 1070, 1154, 1302, 1395, 1625 cm−1 correspond to the characteristic peak of MB molecules. The assignments of primary characteristic peaks were provided in ref. 84. The characteristic peaks at 445 cm−1 and 501 cm−1 are attributed to the C–N–C skeletal deformation mode. The peaks at 772 cm−1, 1037 cm−1, 1070 cm−1, 1154 cm−1 are assigned to the in-plane bending mode of C–H. The peak at 1302 cm−1 is responsible for the in-plane ring deformation mode of C–H. Two prominent peaks at around 1395 cm−1 and 1625 cm−1 are assigned to C–N symmetrical stretching and C–C ring stretching, respectively.85Fig. 9b shows the typical absorption spectra of pure MB (10−4 M) and of MB adsorbed onto Ag–Au alloy nanoplates. The main absorption bands of MB adsorbed on the surface of the Ag–Au nanoplates at 663 nm, and a shoulder at 610 nm are characteristic of MB solid. Interestingly, the change in FWHM of the absorption spectrum of MB on the Ag–Au nanoplate proves that there is an interaction between MB and the electrons of the nanoplate surfaces.86 MB anions adsorb onto Ag–Au alloy nanoplates with Coulomb attraction and are neutralized by positively charged surface, leading to the decrease of surface electron density and the half-peak width of the plasmon adsorption is broadened.
Fig. 9. (a) Raman spectra of only MB (10−4 M) and chemical structure (inset). (b) Absorption spectra of pure MB and the mixture of MB and Ag–Au alloy nanoplates.

The performance of fabricated Ag–Au bimetallic core/shell or alloy nanoplates was evaluated by recording the Raman spectra of methylene blue (MB) molecules (the chemical formula C16H18ClN3S, molar mass of 319.851 g mol−1). MB was dissolved in pure water to achieve the desired concentration and was adsorbed on Ag, Ag–Au NPls drop casted on a glass slide. The SERS measurement principle is illustrated schematically in Fig. 10. The glass slide was placed underneath an objective lens (100×) of a micro-Raman spectrometer utilizing a continuous wave laser at 532 nm. The laser beam was focused onto the analyte molecules through the objective lens and the Raman signals were collected in back scattering geometry. The acquisition time used for recording spectra was 0.5 s for each sample. The Raman spectra of MB were recorded at 10−4 M concentration from Ag–Au4 core/shell (Fig. 9a). 2 μl of MB solution was adsorbed on the portion of the target which was exposed to the laser. The analyte solution was allowed to dry and later, the Raman spectra were recorded. The efficacy of SERS activity was enhanced by the chemical stability provided the combination of Ag–Au metals. SERS enhancement depends on the Ag : Au ratio and the chemical nature of the analyte molecules.87 The charge transfer between Au and Ag atoms in alloy NPs provoke positively charged regions in Ag atoms and negatively charged regions in Au atoms which influence the attachment of analyte molecules on the nanomaterials. In addition, sharp tips of anisotropic bimetallic nanostructures can serve as active “hot spots” for enhancing the SERS signals.88 Strong SERS signals will be obtained only if dye molecules adsorb onto metal surfaces effectively.37 The enhancement of the Raman signals near metal surfaces when molecules adsorb onto metal surfaces is based on the surface selection rules of SERS as follows. Perpendicular vibrational modes of molecules will be enhanced greatly, parallel vibrational modes cannot get large enhancement, short-range vibrational modes can be enhanced much more than long-range vibrational modes.89 Metal particles not only can capture free electrons on the surfaces, but may also produce counterelectrons inside the metal. Metal surface acts as active adsorption locus and energy transfer channel.90,91
Fig. 10. Schematic illustration of SERS measurement principle.

We now focus on the SERS measurement of the MB on the Ag–Au nanoplates. It is interesting to compare the SERS signal of Ag–Au core/shell and Ag–Au alloy nanoplate structures with Ag NPls seeds. The 10−4 M MB aqueous solution was prepared and various concentrations of MB solution such as 5 × 10−5 M, 10−5 M, 5 × 10−6 M, 10−6 M, 5 × 10−7 M, 10−7 M can be obtained by diluting it with deionized water. Fig. 11a shows the SERS spectra of MB (10−4 M) dispersed onto the glass substrate and of MB (10−5 M) adsorbed on the different Ag–Au bimetallic nanoplates. Characteristic spectral Raman bands of MB were observed at 445, 501, 772, 1037, 1070, 1154, 1302, 1395, 1437 and 1624 cm−1, where the strongest peak was 1624 cm−1. Meanwhile, many earlier works have only found some significant Raman peaks at 445, 501, 772, 1154, 1302, 1395, and 1624 cm−1 while using other metal substrates.42,92 In this study, we found two more bands at 1037 and 1070 cm−1 appearing in almost substrates. These results are in good agreement with the previous works, and the standard literatures.84,85 Among twelve Ag–Au nanoplates samples, Ag–Au12 alloy nanoplates led to the strongest enhancement effect and was selected for further study. Fig. 11b shows the intensity of Raman peak at 1624 cm−1 corresponding to C–C ring stretching with the variant of Au; it is obvious that the SERS signal of MB molecules is proportional with HAuCl4 concentration. The intensity of SERS signal increases with increasing Au content in the Ag–Au SERS substrate. This increase might be due to several factors, such as composition, distribution of sprouts, and the possibility of a molecule locating at a “hot spot”. There were no red- or blue-shift in the peak position, but there were changes in the relative intensities for different Ag–Au samples. The change in relative intensity of characteristic spectral features is due to interaction of analyte molecules with substrate surface. This interaction is affected by the adsorption sites, the adsorption orientation and the vibrational modes of the adsorbed molecule on the active metal surface.37,84 The SERS signal intensity was also affected by the surface morphology of substrate, location and direction of the targeted molecules adsorbed onto the surface of the substrate. The highest SERS intensity was found for the case of the Ag–Au12 substrate in comparison with the other products. This observation demonstrated that SERS enhancement was obvious when the analyte molecules adsorbed on the Ag–Au system of appropriate proportions. It indicates an excellent enhancement capability for SERS application. This enhanced Raman signal could come from the roughness or defect of the SERS substrate surface.93 The SERS intensities gradually decline along with the decrease of MB concentration absorbed to substrates from 10−4 M to 10−7 M (Fig. 11c). The characteristic peak of MB at 1624 cm−1 could be identified at concentration as low as 10−7 M with the use of Ag–Au12 alloy nanoplates substrate. No peak could be detected for MB solution with concentration below 10−7 M. The Ag–Au12 substrate can detect MB at several orders of magnitude lower concentration than the silver nanoplates.
Fig. 11. (a) The SERS spectra of MB (10−4 M) on glass substrate and MB (10−5 M) on diverse Ag–Au nanomaterials-based substrates, (b) the variation of SERS (1624 cm−1) intensity originated from different Ag–Au nanomaterials, (c) SERS spectra of Ag–Au12 sample for different MB concentrations (from 10−4 to 10−7 M), (d) SERS spectra of MB at the lowest concentration of 10−7 M adsorbed on Ag–Au12 alloy nanoplates substrate, (e) the linear relationship between log I of the band peaking at 1624 cm−1 and log C, (f) comparation SERS intensity between as-prepared substrate and substrate tested after 8 months.

As can be seen from this figure, almost major Raman peaks are possible observable for Ag–Au12 alloy nanoplates. The intensity of the SERS signal increases as the concentration of MB solution increases. An average of 3 spectra acquired at different positions of a substrate was used to achieve reproducible results. The average intensity of the major peak at 1624 cm−1 from SERS spectra for each MB concentration was used to determine if there was linear relationship between the MB concentration and the intensity of this characteristic peak. Part (e) in Fig. 11 gives the linear relationship between the logarithmic intensity (log I) of the peak centered at 1624 cm−1 and the logarithmic concentration (log C) within the concentration range from 10−7 M to 10−4 M. This linear relationship on a log–log plot allows calibration of our substrate and determination of unknown concentrations of MB solutions using the equation:
| log(I) = 0.51 log(C) + 6.2 | 14 |
The result indicates that this bimetallic nanoplates can be used as chemical sensor to quantify MB in the environment. In addition, Fig. 11d reveals that the SERS detection level of MB for the Ag–Au12 alloy nanoplates substrate was almost 10−7 M, that is the detection limit or the limit of detection (LOD) for Ag–Au12 alloy substrate. Obviously, the performance of Ag–Au12 substrate is better than other substrates (Ag NPls, Ag–Au core/shell and other Ag–Au alloys). The quality of SERS substrate is evaluated based on the important parameters such as the enhancement, sensitivity, stability and reproducibility of SERS substrate. Fig. 11f shows the stability evaluation of Ag–Au12 alloy substrate. The Raman spectra of 10−7 M MB adsorbed on the Ag–Au12 alloy substrate corresponding as-obtained substrate and substrate stored for 8 months. As we can see, the SERS performance deteriorates slightly owing to the oxidation of silver, characteristic peaks of the MB molecules can be easily observed and the enhancement effect of Ag–Au alloy substrate is considerable. The intensity of Raman signal at 1624 cm−1 decreases of about 11.1% after storing time of 8 months. The stability of Ag–Au structure can be improved by limiting its surface oxidation. The SERS peak intensity and position do not substantially change over the 8 months, indicating that it has quite good time-stability.
Now we focus on estimation the enhancement factor of SERS signal for some typical substrates. From the Raman spectra of MB in Fig. 11a, we calculated the SERS enhancement factor (EF) values for ten peaks are located at 445 cm−1, 501 cm−1, 772 cm−1, 1037 cm−1, 1070 cm−1, 1154 cm−1, 1302 cm−1, 1395 cm−1, 1437 cm−1 and 1624 cm−1. The SERS enhancement factors (EF) were calculated based upon the following equation:92,94
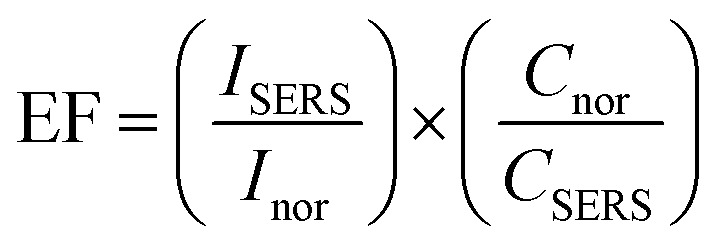 |
15 |
where ISERS is the SERS intensity of MB adsorbed on an Ag–Au nanostructure substrate, Inor represents the normal Raman intensity (non-SERS) of MB on the glass substrate without Ag–Au nanostructures collected under the same test condition. CSERS is the concentration of MB (10−5 M) in SERS spectrum, Cnor is the concentration of MB (10−4 M) in normal Raman spectrum (background line in Fig. 11a). Based on the strongest characteristic peak at 1624 cm−1, the EF for Ag–Au12 alloy nanoplates was estimated to be 1.5 × 105.
We can see that as the concentration of HAuCl4 increased, the EF coefficient increased correspondingly in all bands, and the detail results are described in Table 3. Fig. 11a and b show that SERS activity of Ag–Au samples is weaker for the core/shell structure than for alloy structure. Two SERS enhancement mechanisms are the electromagnetic enhancement and the chemical enhancement. Chemical enhancement involves charge transfer between the SERS substrate and the detected molecules. The high performance SERS signal intensity of Ag–Au alloy nanostructures could be assigned to the electromagnetic enhancement effect which is generated from the light induced localized surface plasmon resonance (LSPR) in the vicinity of the surface of noble metals.95,96 Many “hot-spots” are formed at the gaps, crevices, sharp vertices of the nanostructures, where the optical field intensity is much higher than that of other sites, and a number of highly active hot-spots may provide giant electromagnetic enhancement thus obtaining a higher enhancement factor value.94
EF values calculated at ten different peaks of MB (10−5 M) taking Ag–Au for SERS studies.
| Substrate used | Enhancement factor (EF) values at a peak position for MB (10−5 M) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1624 cm−1 | 1437 cm−1 | 1395 cm−1 | 1302 cm−1 | 1154 cm−1 | 1070 cm−1 | 1037 cm−1 | 772 cm−1 | 501 cm−1 | 445 cm−1 | |
| Ag NPls | 5.8 × 104 | 1.0 × 104 | 1.0 × 104 | 2.9 × 103 | 4.9 × 103 | 3.7 × 103 | 3.9 × 103 | 3.9 × 103 | 3.7 × 103 | 5.3 × 103 |
| Ag–Au1 | 6.4 × 104 | 1.1 × 104 | 1.0 × 104 | 3.2 × 103 | 5 × 103 | 4.2 × 103 | 4.3 × 103 | 4 × 103 | 3.7 × 103 | 5.7 × 103 |
| Ag–Au2 | 7.6 × 104 | 1.2 × 104 | 1.3 × 104 | 4.3 × 103 | 5.7 × 103 | 5.3 × 103 | 5.3 × 103 | 5.2 × 103 | 4.1 × 103 | 6.5 × 103 |
| Ag–Au5 | 7.7 × 104 | 2 × 104 | 1.3 × 104 | 3.7 × 103 | 7.0 × 103 | 6.1 × 103 | 7.2 × 103 | 5.3 × 103 | 4.5 × 103 | 6.6 × 103 |
| Ag–Au6 | 8.7 × 104 | 1.5 × 104 | 1.6 × 104 | 4.6 × 103 | 7.5 × 103 | 6.3 × 103 | 6.2 × 103 | 6.1 × 103 | 6 × 103 | 7.2 × 103 |
| Ag–Au7 | 1.3 × 105 | 3.3 × 104 | 2.6 × 104 | 7.0 × 103 | 1.0 × 104 | 1.1 × 104 | 1.2 × 104 | 1.0 × 104 | 8.5 × 103 | 1.3 × 104 |
| Ag–Au9 | 1.4 × 105 | 3.8 × 104 | 3.0 × 104 | 7.6 × 103 | 1.2 × 104 | 1.1 × 104 | 1.3 × 104 | 1.1 × 104 | 1.0 × 104 | 1.5 × 104 |
| Ag–Au12 | 1.5 × 105 | 3.9 × 104 | 3.2 × 104 | 7.3 × 103 | 1.3 × 104 | 1.2 × 104 | 1.3 × 104 | 1.2 × 104 | 1.0 × 104 | 1.6 × 104 |
To investigate the role of Au–Ag ratio on SERS signal, 10 μl of MB (the Raman probe molecules) solution of 10−5 M was drop casted on the SERS substrates (Ag NPls, Ag–Au1 to Ag–Au12). Fig. 11a shows the SERS spectra of MB molecules obtained with the bimetallic nanoplates of different Ag–Au ratios. It is apparent that the frequencies of all Raman peaks were same for all samples. The observed Raman peaks of MB molecules agree well with the standard literature and are described in detail the assignment of modes in.44 The Raman peak at 1624 cm−1 is relatively dominant in all ratios. Fig. 11b shows the Raman peak at 1624 cm−1 alone which indicates the variation in the intensity of Raman peak with different Au–Ag ratios. From Table 3, the maximum EF value of Raman peak at 1624 cm−1 of 1.5 × 105 for Ag–Au12 alloy nanoplates, 5–10 times stronger than that of the other Ag–Au nanoplates, and approximately 2.6 times greater than that of Ag NPls. The added Au atoms replacing Ag atoms form a lot of roughness and defect. Morever, the EF values at 1624 cm−1 peak quickly increase after adding Au atoms. Therefore, this band is used as fingerprint in the MB concentration detection limit in environment.8,31,84,97 Interestingly, almost EF values at all bands increase as amount of HAuCl4 increase (Fig. 12a).
Fig. 12. The variation of the EF values at different Raman peaks as a function of HAuCl4 amount with MB concentration of 10−5 M (a) and 10−7 M (b).

Remarkably, the Ag–Au2 sample (x = 0.2 μmol) has a core/shell structure, while Ag–Au12 (x = 3.4 μmol) has alloy structure nature. Thereby, SERS activity in the core/shell structure might be restricted by the outer Au shell layer (∼9 nm in thickness) which limits the characteristics of the Ag SPR sensor. Numerous previous studies have showed that the thinner the gold layer coated over silver, the more sensitive the plasmonic Ag is.98 In contrast, in the case of Ag–Au12 nanoplate alloy, several roughness and defects may appear in the exchange process Ag by Au atoms leading to the formation of numerous nanogaps, so SERS activity is enhanced (Table 4).
EF values calculated at ten different peaks of MB (10−7 M) taking Ag NPl4, Ag–Au2, Ag–Au5, and Ag–Au12 for SERS studies.
| Substrate used | Enhancement factor (EF) values at a peak position for MB (10−7 M) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1624 cm−1 | 1437 cm−1 | 1395 cm−1 | 1302 cm−1 | 1154 cm−1 | 1070 cm−1 | 1037 cm−1 | 772 cm−1 | 501 cm−1 | 445 cm−1 | |
| Ag NPl4 | 1.3 × 106 | 4.8 × 104 | 2.2 × 104 | No detect | No detect | No detect | 3.2 × 10 | No detect | No detect | No detect |
| Ag–Au2 | 8.3 × 106 | 2.5 × 105 | 2 × 104 | 6.8 × 104 | 8 × 104 | 6.4 × 104 | 1 × 105 | 6 × 104 | 5.4 × 104 | 9.2 × 104 |
| Ag–Au5 | 1.0 × 107 | 2.2 × 105 | 2.7 × 104 | 8.2 × 104 | 9.2 × 104 | 7.8 × 105 | 6.8 × 104 | 7.4 × 104 | 6 × 104 | 8.6 × 104 |
| Ag–Au12 | 1.2 × 107 | 2.2 × 105 | 2.4 × 105 | 7.2 × 104 | 7.8 × 104 | 9.2 × 104 | 6.4 × 104 | 8.2 × 104 | 7.8 × 104 | 1.1 × 105 |
5. Conclusions
In conclusion, with a simple approach of using seed-mediated growth we have successful fabricated and investigated the structural and optical properties of bimetallic Ag–Au and applied their SERS activities to detect MB. Changing the order of addition reducing agents and amount of gold precursor can create Ag–Au core/shell or alloy structure. Specifically, when a very small amount of chloroauric acid (HAuCl4) is injected very slowly into the seed solution, the received product was Ag–Au core/shell nanostructure, whereas the alloy structure is received if using the large amount of HAuCl4. XRD and HRTEM analysis confirms that both Ag and Ag–Au nanoplates (core/shell and alloy) have face-center-cubic structure. The obtained substrates exhibit homogeneity and high sensitivity to methylene blue (MB). Using MB as probe molecule, the SERS results imply that the Ag–Au alloy nanoplates substrate is superior to the chemical sensor. The estimated EF values for MB (10−7 M) were 1.2 × 105, 8.3 × 105 and 1.2 × 107 for Ag NPls, Ag–Au core/shell nanoplates and Ag–Au alloy nanoplates substrates, respectively. The lowest concentration of MB can be detected as 10−7 M. This promises for using the bimetallic Ag–Au substrates in biosensor, plasmonic and sensing applications.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This research was supported by Project of the TNU-University of Science in Vietnam under grant number CS2020-TN06-14.
Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04132g
References
- Jiang Z. J. Liu C. Y. Sun L. W. J. Phys. Chem. B. 2005;109:1730–1735. doi: 10.1021/jp046032g. [DOI] [PubMed] [Google Scholar]
- Cobley C. M. Skrabalak S. E. Plasmonics. 2009;4:171–179. doi: 10.1007/s11468-009-9088-0. [DOI] [Google Scholar]
- Lien N. T. H. Hoa V. X. Duong V. T. T. Van Tinh N. Nhung T. H. Commun. Phys. 2011;21:63. doi: 10.15625/0868-3166/21/1/96. [DOI] [Google Scholar]
- Vu X. H. Levy M. Barroca T. Tran H. N. Fort E. Nanotechnology. 2013;24:325501. doi: 10.1088/0957-4484/24/32/325501. [DOI] [PubMed] [Google Scholar]
- Diantoro M., Suprayogi T., Diantoro M., Suprayogi T., Sa’adah U., Mufti N., Fuad A., Sa’adah U., Mufti N., Fuad A., Hidayat A. and Nur H., Modification of Electrical Properties of Silver Nanoparticle, 2012, vol. 13, pp. 233–248, 10.5772/intechopen.75682 [DOI] [Google Scholar]
- Teranishi T. Compt. Rendus Chem. 2003;6:979–987. doi: 10.1016/j.crci.2003.08.012. [DOI] [Google Scholar]
- Vu X. H. Thanh T. Duong T. Thu T. Pham H. Trinh D. K. Adv. Nat. Sci. Nanosci. Nanotechnol. 2018;9:025019. doi: 10.1088/2043-6254/aac58f. [DOI] [Google Scholar]
- Abalde-Cela S. Aldeanueva-Potel P. Mateo-Mateo C. Rodríguez-Lorenzo L. Alvarez-Puebla R. A. Liz-Marzán L. M. J. R. Soc. Interface. 2010;7:S435–S450. doi: 10.1098/rsif.2010.0125.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W. Qiu P. Mao C. J. Mater. Chem. 2011;21:5190–5202. doi: 10.1039/C0JM03301D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastoriza-Santos I. Liz-Marzán L. M. J. Mater. Chem. 2008;18:1724–1737. doi: 10.1039/B716538B. [DOI] [Google Scholar]
- De Abajo F. J. G. Rev. Mod. Phys. 2007;79:1267–1290. doi: 10.1103/RevModPhys.79.1267. [DOI] [Google Scholar]
- Wang B. Liu M. Wang Y. Chen X. J. Phys. Chem. C. 2011;115:11374–11381. doi: 10.1021/jp201023x. [DOI] [Google Scholar]
- Kumar-Krishnan S. Estevez-González M. Pérez R. Esparza R. Meyyappan M. RSC Adv. 2017;7:27170–27176. doi: 10.1039/C7RA04301E. [DOI] [PubMed] [Google Scholar]
- Bu Y. Lee S. W. Int. J. Nanomedicine. 2015;10:47–54. doi: 10.2147/IJN.S88308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah A. Latif-Ur-Rahman Qureshi R. Zia-Ur-Rehman Rev. Adv. Mater. Sci. 2012;30:133–149. [Google Scholar]
- Podagatlapalli G. K. Hamad S. Rao S. V. J. Phys. Chem. C. 2015;119:16972–16983. doi: 10.1021/acs.jpcc.5b03958. [DOI] [Google Scholar]
- Kuladeep R. Jyothi L. Alee K. S. Deepak K. L. N. Rao D. N. Opt. Mater. Express. 2012;2:161. doi: 10.1364/OME.2.000161. [DOI] [Google Scholar]
- Intartaglia R. Das G. Bagga K. Gopalakrishnan A. Genovese A. Povia M. Di Fabrizio E. Cingolani R. Diaspro A. Brandi F. Phys. Chem. Chem. Phys. 2013;15:3075–3082. doi: 10.1039/C2CP42656K. [DOI] [PubMed] [Google Scholar]
- Shahjamali M. M. Salvador M. Bosman M. Ginger D. S. Xue C. J. Phys. Chem. C. 2014;118:12459–12468. doi: 10.1021/jp501884s. [DOI] [Google Scholar]
- Liu H. Liu T. Zhang L. Han L. Gao C. Yin Y. Adv. Funct. Mater. 2015;25:5435–5443. doi: 10.1002/adfm.201502366. [DOI] [Google Scholar]
- Aherne D. Charles D. E. Brennan-Fournet M. E. Kelly J. M. Gun'ko Y. K. Langmuir. 2009;25:10165–10173. doi: 10.1021/la9009493. [DOI] [PubMed] [Google Scholar]
- Zhang H. Xu L. Tian Y. Chen M. Liu X. Chen F. Opt. Express. 2017;25:29389. doi: 10.1364/OE.25.029389. [DOI] [Google Scholar]
- Shahjamali M. M. Bosman M. Cao S. Huang X. Saadat S. Martinsson E. Aili D. Tay Y. Y. Liedberg B. Loo S. C. J. Zhang H. Boey F. Xue C. Adv. Funct. Mater. 2012;22:849–854. doi: 10.1002/adfm.201102028. [DOI] [Google Scholar]
- Sanchez I. C. Krishnan S. K. Flores-Ruiz F. J. Luna-Bárcenas G. Esparza R. Pal U. Padilla-Ortega E. ACS Omega. 2018;3:12600–12608. doi: 10.1021/acsomega.8b02333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal U. Sánchez-Ramírez J. F. Nolasco-Hernández L. Mendoza-Álvarez J. Pescador-Rojas J. A. J. Nanomater. 2008:1–9. [Google Scholar]
- Tsai C. H. Chen S. Y. Song J. M. Haruta M. Kurata H. Nanoscale Res. Lett. 2015;10:1–9. doi: 10.1186/1556-276X-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K. Bai Y. Zhang L. Yang Z. Fan Q. Zheng H. Yin Y. Gao C. Nano Lett. 2016;16:3675–3681. doi: 10.1021/acs.nanolett.6b00868. [DOI] [PubMed] [Google Scholar]
- Moreau L. M. Schurman C. A. Kewalramani S. Shahjamali M. M. Mirkin C. A. Bedzyk M. J. J. Am. Chem. Soc. 2017;139:12291–12298. doi: 10.1021/jacs.7b06724. [DOI] [PubMed] [Google Scholar]
- Hajiesmaeilbaigi F. Motamedi A. Laser Phys. Lett. 2007;4:133–137. doi: 10.1002/lapl.200610083. [DOI] [Google Scholar]
- Compagnini G. Messina E. Puglisi O. Cataliotti R. S. Nicolosi V. Chem. Phys. Lett. 2008;457:386–390. doi: 10.1016/j.cplett.2008.04.051. [DOI] [Google Scholar]
- Zhang C. Jiang S. Z. Yang C. Li C. H. Huo Y. Y. Liu X. Y. Liu A. H. Wei Q. Gao S. S. Gao X. G. Man B. Y. Sci. Rep. 2016;6:4–11. doi: 10.1038/s41598-016-0011-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H. Cong S. Zheng Z. Wang Z. Chen Z. Zhao Z. J. Am. Chem. Soc. 2019;141:870–878. doi: 10.1021/jacs.8b09414. [DOI] [PubMed] [Google Scholar]
- Xuan T. Gao Y. Cai Y. Guo X. Wen Y. Yang H. Sens. Actuators, B. 2019;293:289–295. doi: 10.1016/j.snb.2019.05.017. [DOI] [Google Scholar]
- Sun M. Qian H. Liu J. Li Y. Pang S. Xu M. Zhang J. RSC Adv. 2017;7:7073–7078. doi: 10.1039/C6RA25956A. [DOI] [Google Scholar]
- Hou H. Wang P. Zhang J. Li C. Jin Y. ACS Appl. Mater. Interfaces. 2015;7:18038–18045. doi: 10.1021/acsami.5b04946. [DOI] [PubMed] [Google Scholar]
- Loganathan B. Chandraboss V. L. Senthilvelan S. Karthikeyan B. Phys. Chem. Chem. Phys. 2015;17:21268–21277. doi: 10.1039/C4CP05170J. [DOI] [PubMed] [Google Scholar]
- Zhang A. Fang Y. J. Colloid Interface Sci. 2007;305:270–274. doi: 10.1016/j.jcis.2006.09.068. [DOI] [PubMed] [Google Scholar]
- Yin H. J. Chen Z. Y. Zhao Y. M. Lv M. Y. Shi C. A. Wu Z. L. Zhang X. Liu L. Wang M. L. Xu H. J. Sci. Rep. 2015;5:1–9. doi: 10.1038/srep14502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton S. M. Jensen L. J. Am. Chem. Soc. 2009;131:4090–4098. doi: 10.1021/ja809143c. [DOI] [PubMed] [Google Scholar]
- Mandal M. Jana N. R. Kundu S. Ghosh S. K. Panigrahi M. Pal T. J. Nanoparticle Res. 2004;6:53–61. doi: 10.1023/B:NANO.0000023227.17871.0f. [DOI] [Google Scholar]
- Yang M. Zhang L. Chen B. Wang Z. Chen C. Zeng H. Nanotechnology. 2017;28:055301. doi: 10.1088/1361-6528/28/5/055301. [DOI] [PubMed] [Google Scholar]
- Bharati M. S. S. Byram C. Soma V. R. Front. Phys. 2018;6:1–13. doi: 10.3389/fphy.2018.00001. [DOI] [Google Scholar]
- Singh M. K. Chettri P. Basu J. Tripathi A. Mukherjee B. Tiwari A. Mandal R. K. Mater. Lett. 2019;249:33–36. doi: 10.1016/j.matlet.2019.04.065. [DOI] [Google Scholar]
- William R. V. Das G. M. Dantham V. R. Laha R. Sci. Rep. 2019;9:1–12. doi: 10.1038/s41598-018-37186-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilayurganapathy S., PhD thesis, Western Michigan University, 2013 [Google Scholar]
- Fleger Y. Rosenbluh M. Res. Lett. Opt. 2009;2009:1–5. doi: 10.1155/2009/475941. [DOI] [Google Scholar]
- Olea-Mejía O. Fernández-Mondragón M. Rodríguez-De La Concha G. Camacho-López M. Appl. Surf. Sci. 2015;348:66–70. doi: 10.1016/j.apsusc.2015.01.075. [DOI] [Google Scholar]
- Lee S.-W. Chang S.-H. Lai Y.-S. Lin C.-C. Tsai C.-M. Lee Y.-C. Chen J.-C. Huang C.-L. Materials. 2014;7:7781–7798. doi: 10.3390/ma7127781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. W. Chang S. H. Lai Y. S. Lin C. C. Tsai C. M. Lee Y. C. Chen J. C. Huang C. L. Materials. 2014;7:7781–7798. doi: 10.3390/ma7127781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodha K. H. Jadav J. K. Gajera H. P. Rathod K. J. Int. J. Pharma Bio Sci. 2015;6:199–208. [Google Scholar]
- Araújo A. Caro C. Mendes M. J. Nunes D. Fortunato E. Franco R. Águas H. Martins R. Nanotechnology. 2014;25:415202. doi: 10.1088/0957-4484/25/41/415202. [DOI] [PubMed] [Google Scholar]
- Vincenzo A. Roberto P. Marco F. Onofrio M. M. Maria Antonia I. J. Phys. Condens. Matter. 2017;29:203002. doi: 10.1088/1361-648X/aa60f3. [DOI] [PubMed] [Google Scholar]
- Nehl C. L. Grady N. K. Goodrich G. P. Tam F. Halas N. J. Hafner J. H. Nano Lett. 2004;4:2355–2359. doi: 10.1021/nl048610a. [DOI] [Google Scholar]
- Jain P. K. Lee K. S. El-sayed I. H. El-sayed M. A. Nat. Rev. Mater. 2006;110:7238–7248. doi: 10.1021/jp057170o. [DOI] [PubMed] [Google Scholar]
- Koczkur K. M. Mourdikoudis S. Polavarapu L. Skrabalak S. E. Dalt. Trans. 2015;44:17883–17905. doi: 10.1039/C5DT02964C. [DOI] [PubMed] [Google Scholar]
- Zhang Z. Zhao B. Hu L. J. Solid State Chem. 1996;121:105–110. doi: 10.1006/jssc.1996.0015. [DOI] [Google Scholar]
- Wiley B. Sun Y. Mayers B. Xia Y. Chem.–Eur. J. 2005;11:454–463. doi: 10.1002/chem.200400927. [DOI] [PubMed] [Google Scholar]
- Cardoso-Avila P. E. Pichardo-Molina J. L. Murali Krishna C. Castro-Beltran R. J. Nanopart. Res. 2015;17:160. doi: 10.1007/s11051-015-2920-x. [DOI] [Google Scholar]
- Stamplecoskie K. G. Scaiano J. C. J. Am. Chem. Soc. 2010;132:1825–1827. doi: 10.1021/ja910010b. [DOI] [PubMed] [Google Scholar]
- Thanh N. T. K. Maclean N. Mahiddine S. Chem. Rev. 2014;114:7610–7630. doi: 10.1021/cr400544s. [DOI] [PubMed] [Google Scholar]
- Gentry S. T. Kendra S. F. Bezpalko M. W. J. Phys. Chem. C. 2011;115:12736–12741. doi: 10.1021/jp2009786. [DOI] [Google Scholar]
- Abou El-Nour K. M. M. Eftaiha A. Al-Warthan A. Ammar R. A. A. Arab. J. Chem. 2010;3:135–140. doi: 10.1016/j.arabjc.2010.04.008. [DOI] [Google Scholar]
- Pham T. T. H. Dien N. D. Vu X. H. Tran T. T. Ca N. X. Truong N. V. Tan P. M. Van H. T. Van Do P. J. Electron. Mater. 2020 doi: 10.1007/s11664-020-08240-5. [DOI] [Google Scholar]
- Martínez-Castañón G. A. Niño-Martínez N. Martínez-Gutierrez F. Martínez-Mendoza J. R. Ruiz F. J. Nanoparticle Res. 2008;10:1343–1348. doi: 10.1007/s11051-008-9428-6. [DOI] [Google Scholar]
- Wang H. Zheng X. Chen J. Wang D. Wang Q. Xue T. Liu C. Jin Z. Cui X. Zheng W. J. Phys. Chem. C. 2012;116:24268–24273. doi: 10.1021/jp304941b. [DOI] [Google Scholar]
- Mott D. Lee J. D. Thuy N. T. B. Aoki Y. Singh P. Maenosono S. Jpn. J. Appl. Phys. 2011;50:065004. doi: 10.1143/JJAP.50.065004. [DOI] [Google Scholar]
- Al-Saidi W. A. Feng H. Fichthorn K. A. Nano Lett. 2012;12:997–1001. doi: 10.1021/nl2041113. [DOI] [PubMed] [Google Scholar]
- Murshid N. Kitaev V. Chem. Commun. 2014;50:1247–1249. doi: 10.1039/C3CC48003H. [DOI] [PubMed] [Google Scholar]
- Wang X. Zhang Z. Hartland G. V. J. Phys. Chem. B. 2005;109:20324–20330. doi: 10.1021/jp054233u. [DOI] [PubMed] [Google Scholar]
- Haiss W. Thanh N. T. K. Aveyard J. Fernig D. G. Anal. Chem. 2007;79:4215–4221. doi: 10.1021/ac0702084. [DOI] [PubMed] [Google Scholar]
- Zhang C. Chen B. Q. Li Z. Y. Xia Y. Chen Y. G. J. Phys. Chem. C. 2015;119:16836–16845. doi: 10.1021/acs.jpcc.5b04232. [DOI] [Google Scholar]
- Ferrando R. Jellinek J. Johnston R. L. Chem. Rev. 2008;108:845–910. doi: 10.1021/cr040090g. [DOI] [PubMed] [Google Scholar]
- Yi Z. Zhang J. B. He H. Bin Xu X. Luo B. C. Li X. B. Li K. Niu G. Tan X. L. Luo J. S. Tang Y. J. Wu W. D. Yi Y. G. Trans. Nonferrous Metals Soc. China. 2012;22:865–872. doi: 10.1016/S1003-6326(11)61258-2. [DOI] [Google Scholar]
- Lin J. Y. Hsueh Y. L. Huang J. J. J. Solid State Chem. 2014;214:2–6. doi: 10.1016/j.jssc.2013.12.017. [DOI] [Google Scholar]
- Chen S. Carroll D. L. Nano Lett. 2002;2:1003–1007. doi: 10.1021/nl025674h. [DOI] [Google Scholar]
- Jyoti K. Baunthiyal M. Singh A. J. Radiat. Res. Appl. Sci. 2016;9:217–227. doi: 10.1016/j.jrras.2015.10.002. [DOI] [Google Scholar]
- Mao H. Feng J. Ma X. Wu C. Zhao X. J. Nanoparticle Res. 2012;14:887. doi: 10.1007/s11051-012-0887-4. [DOI] [Google Scholar]
- Shmarakov I. Mukha I. Vityuk N. Borschovetska V. Zhyshchynska N. Grodzyuk G. Eremenko A. Nanoscale Res. Lett. 2017;12:333. doi: 10.1186/s11671-017-2112-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempter C. P. Phys. Status Solidi. 1966;18:K117–K118. doi: 10.1002/pssb.19660180251. [DOI] [Google Scholar]
- King H. W. Lo Vegard Y. J. Mater. Sci. 1921;1:79–90. doi: 10.1007/BF00549722. [DOI] [Google Scholar]
- Geonmonond R. S. Da Silva A. G. M. Camargo P. H. C. An. Acad. Bras. Cienc. 2018;90:719–744. doi: 10.1590/0001-3765201820170561. [DOI] [PubMed] [Google Scholar]
- Zou X. Ying E. Dong S. J. Colloid Interface Sci. 2007;306:307–315. doi: 10.1016/j.jcis.2006.10.084. [DOI] [PubMed] [Google Scholar]
- Da Silva A. G. M. Rodrigues T. S. Haigh S. J. Camargo P. H. C. Chem. Commun. 2017;53:7135–7148. doi: 10.1039/C7CC02352A. [DOI] [PubMed] [Google Scholar]
- Li C. Huang Y. Lai K. Rasco B. A. Fan Y. Food Contr. 2016;65:99–105. doi: 10.1016/j.foodcont.2016.01.017. [DOI] [Google Scholar]
- Naujok R. R. Duevel R. V. Corn R. M. Langmuir. 1993;9:1771–1774. doi: 10.1021/la00031a026. [DOI] [Google Scholar]
- Uppal M. A. Ewing M. B. Parkin I. P. Eur. J. Inorg. Chem. 2011;29:4534–4544. doi: 10.1002/ejic.201100536. [DOI] [Google Scholar]
- Fan M. Lai F. J. Chou H. L. Lu W. T. Hwang B. J. Brolo A. G. Chem. Sci. 2013;4:509–515. doi: 10.1039/C2SC21191B. [DOI] [Google Scholar]
- Kelly K. L. Coronado E. Zhao L. L. Schatz G. C. J. Phys. Chem. B. 2003;107:668–677. doi: 10.1021/jp026731y. [DOI] [Google Scholar]
- Change S. G. J. Phys. Chem. 1988;2:6327–6329. [Google Scholar]
- Treguer M. Rocco F. Lelong G. Le Nestour A. Cardinal T. Maali A. Lounis B. Solid State Sci. 2005;7:812–818. doi: 10.1016/j.solidstatesciences.2005.01.017. [DOI] [Google Scholar]
- Mooradian A. Phys. Rev. Lett. 1969;22:185–187. doi: 10.1103/PhysRevLett.22.185. [DOI] [Google Scholar]
- Kundu S. Dai W. Chen Y. Ma L. Yue Y. Sinyukov A. M. Liang H. J. Colloid Interface Sci. 2017;498:248–262. doi: 10.1016/j.jcis.2017.03.058. [DOI] [PubMed] [Google Scholar]
- Lee H. K. Lee Y. H. Koh C. S. L. Phan-Quang G. C. Han X. Lay C. L. Sim H. Y. F. Kao Y. C. An Q. Ling X. Y. Chem. Soc. Rev. 2019;48:731–756. doi: 10.1039/C7CS00786H. [DOI] [PubMed] [Google Scholar]
- Pham T. B. Hoang T. H. C. Pham V. H. Nguyen V. C. Van Nguyen T. Vu D. C. Pham V. H. Bui H. Sci. Rep. 2019;9:1–10. doi: 10.1038/s41598-018-37186-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K. Clime L. Cui B. Veres T. Nanotechnology. 2008;19:145305. doi: 10.1088/0957-4484/19/14/145305. [DOI] [PubMed] [Google Scholar]
- Wang K. Li S. Petersen M. Wang S. Lu X. Nanomaterials. 2018;8:762. doi: 10.3390/nano8100762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng L. Zhou J. Liang Z. Zhang Y. Petti L. Jiang T. Gu C. Yang D. Mormile P. Anal. Methods. 2019;11:2960–2968. doi: 10.1039/C9AY00573K. [DOI] [Google Scholar]
- Gao C. Lu Z. Liu Y. Zhang Q. Chi M. Cheng Q. Yin Y. Angew. Chem., Int. Ed. 2012;51:5629–5633. doi: 10.1002/anie.201108971. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.