

Abstract
Objectives
Adult hepatocytes are quiescent cells that can be induced to proliferate in response to a reduction in liver mass (liver regeneration) or by agents endowed with mitogenic potency (primary hyperplasia). The latter condition is characterized by a more rapid entry of hepatocytes into the cell cycle, but the mechanisms responsible for the accelerated entry into the S phase are unknown.
Materials and methods
Next generation sequencing and Illumina microarray were used to profile microRNA and mRNA expression in CD‐1 mice livers 1, 3 and 6 h after 2/3 partial hepatectomy (PH) or a single dose of TCPOBOP, a ligand of the constitutive androstane receptor (CAR). Ingenuity pathway and DAVID analyses were performed to identify deregulated pathways. MultiMiR analysis was used to construct microRNA‐mRNA networks.
Results
Following PH or TCPOBOP we identified 810 and 527 genes, and 102 and 10 miRNAs, respectively, differentially expressed. Only 20 genes and 8 microRNAs were shared by the two conditions. Many miRNAs targeting negative regulators of cell cycle were downregulated early after PH, concomitantly with increased expression of their target genes. On the contrary, negative regulators were not modified after TCPOBOP, but Ccnd1 targeting miRNAs, such as miR‐106b‐5p, were downregulated.
Conclusions
While miRNAs targeting negative regulators of the cell cycle are downregulated after PH, TCPOBOP caused downregulation of miRNAs targeting genes required for cell cycle entry. The enhanced Ccnd1 expression may explain the more rapid entry into the S phase of mouse hepatocytes following TCPOBOP.
Keywords: Cyclin D1, hepatomitogens, MiRNAs, partial hepatectomy, transcriptomics
A balance of pro‐ and anti‐proliferative signals is regulated by miRs in the priming phase of hepatocytes following pH‐induced liver regeneration, while miR deregulation leads only to pro‐proliferative signals in primary hyperplasia. This justifies the more rapid entry of hepatocytes into the cell cycle after TCPOBOP treatment.
1. INTRODUCTION
Adult liver is normally quiescent and shows a very low level of hepatocyte division. However, most hepatocytes rapidly proliferate in response to a reduction in liver mass caused by physical, chemical, nutritional, vascular or virus‐induced liver injury. 1 Although the mechanisms responsible for the exit from the quiescent state and the re‐entry into the cell cycle remain unknown, it has been proposed that the essential circuitry required for liver regeneration is encompassed by pathways activated by cytokines, growth factors and metabolic networks. 1 , 2 , 3
More recently, several studies investigated the role of microRNAs (miRs) in liver regeneration following 2/3 partial hepatectomy (PH). 4 , 5 , 6 Indeed, while miRs post‐transcriptionally regulate genes that orchestrate proliferation in development and cancer, their role in the proliferation of fully differentiated hepatocytes is still largely unknown. In this context, the finding that hepatocyte‐specific Dicer knockout transgenic mice developed normally, but exhibited enlarged livers compared to controls, strongly support the role for miRNAs in the control of hepatocyte proliferation. 7 This study, together with the discovery that the association of miRNAs with different polysome fractions was altered during liver regeneration, 8 raised the intriguing possibility that miRNAs might regulate the regeneration of this organ.
As shown by Shu et al., 9 upregulation of a cluster of miRNAs takes place between 0 and 4 h after PH, a time corresponding to the so‐called priming of hepatocytes, 10 , 11 characterized by refractoriness to DNA synthesis; conversely, downregulation of the vast majority of miRNAs associates with the transition G1‐S of the cell cycle and the recovery of liver mass. Accordingly, the expression of most cell cycle‐related genes is repressed for several hours after surgery. Similar findings were reported by Yin et al., who identified in rat liver transcription factors inhibiting the cell cycle, as early as 2 h after PH in rat liver. 12 After the priming phase, hepatocyte DNA synthesis peaks at 24 or 36 h in rats and mice, respectively. 11
Hepatocyte proliferation can be induced not only after cell death/loss but also following treatment with several xenobiotics or endogenous molecules (direct/primary mitogens), able to induce the entry of hepatocytes into the cell cycle in the absence of previous liver cell damage. 13 Among the broad spectrum of chemical mitogens, it is remarkable that many of them are ligands of nuclear receptors of the steroid/thyroid superfamily, including 1,4‐bis[2‐(3,5‐dichloropyridyloxy)]benzene TCPOBOP (abbreviated thereafter as TCP). Studies with knockout mice have shown that the initial signalling elicited by liver regeneration and direct mitogens is different. 1 , 2 , 3 , 14 , 15 Moreover, no change in the activation of transcriptional factors implicated in rat liver regeneration (such as, NF‐kB, AP‐1, STAT3) has been observed in nuclear receptor‐mediated hepatocyte proliferation. 13 In this context, it should be also mentioned that the entry of hepatocytes into the S phase of the cell cycle in mice is robustly anticipated in mitogen‐treated animals (18 h instead of the 30–36 h required after 2/3 PH, with mitotic figures being evident at 24 vs. 48 h). 16
Although existing studies analysed early responses of the mouse liver transcriptome at early times after treatment with TCP—an agonist of the constitutive androstane receptor (CAR)— 17 , 18 or soon after PH, 12 the involvement of miRNAs as critical regulators in the priming phase of hepatocytes in these two proliferative conditions, has not been studied so far.
In an attempt to investigate whether deregulation of miRNA expression could play an essential role in the priming phase of hepatocytes and whether differences might exist between the two proliferative conditions (compensatory regeneration and direct hyperplasia), we performed a transcriptomic and miRNomic analysis on the liver of mice sacrificed 1, 3 and 6 h after 2/3 PH (liver regeneration) or after a single dose of TCP (direct hyperplasia).
2. MATERIALS AND METHODS
2.1. Animals and treatments
Guidelines for the Care and Use of Laboratory Animals were followed during the investigation. All animal procedures were approved by the Ethical Commission of the University of Cagliari and the Italian Ministry of Health. Three‐month‐old CD‐1 female mice (30 g) were fed a laboratory chow diet provided by Ditta Mucedola (Settimo Milanese, Italy) with free access to food and water. All experiments were performed in a temperature‐controlled room with alternating 12‐h dark‐light cycles. TCP (Sigma‐Aldrich), was dissolved in dimethyl sulphoxide/corn oil. A single dose of 3 mg/kg body weight was administered by gavage. PH was performed by removal of 70% of the liver mass as originally described by Higgins and Anderson. 19 In the first set of experiments, mice were sacrificed 24, 36 and 48 h after PH or TCP. Bromodeoxyuridine (BrdU) (100 mg/kg) was injected intraperitoneally 2 h before sacrifice. To investigate the role of miRs in the priming of hepatocytes, mice were sacrificed 1, 3 and 6 h after each treatment. Three mice were used per group at each time point. Liver segments were fixed in formalin for histology or snap‐frozen in liquid nitrogen and kept at –80°C until use.
2.2. RNA and miR isolation
Total RNA was isolated with the RNeasy Plus Mini isolation kit (Qiagen) from 3 livers of untreated and treated mice, subjected to PH or TCP treatment. RNA was quantified by Nanodrop spectrophotometer (Thermo Scientific) and its integrity was evaluated by Agilent Bioanalyzer 2100. Only RNA samples with a RIN (RNA Integrity Number) ≥7 were included in the study.
2.3. Deep sequencing
For miR sequencing experiments, indexed libraries were prepared using 100 ng of total RNA as starting material, with a TruSeq Stranded Total RNA Sample Prep Kit and QIAseq miRNA Library Kit (Illumina Inc.), respectively. Libraries were sequenced (single‐end, 75 cycles) at a concentration of 8 pM/lane on the HiSeq 3000 platform (Illumina Inc.). Raw miRNA reads were preprocessed using FASTQC (Andrews, S. (2010) for quality control. Further, reads with Unique Molecular Identifiers (UMI) and low‐quality base calls were trimmed off using UMI‐tools 20 and Trim Galore, 21 respectively. Processed reads were mapped to the reference mouse genome build (mm10) downloaded from UCSC (https://genome.ucsc.edu/index.html) using Bowtie. 22 The R/Bioconductor package “DESeq2” was used to identify differentially expressed genes and miRs. Data were filtered according to read count value (threshold ≥6 reads). Only miRs having an adjusted P‐value of ≤0.05) and fold change value of 1.3 were considered for further analysis.
2.4. Microarray
For time‐course expression profiling, total liver RNA was extracted and purified from the liver of three animals before (t=0) or 1, 3 or 6 h after treatment. For the gene expression profile, RNA was amplified (TotalPrep RNA Amplification Kit; Illumina Inc., San Diego, CA), labelled and hybridized on Illumina microarray Mouse WG‐6 v2.0 Gene Expression BeadChip (Illumina Inc., San Diego, CA, USA), including 45,281 specific oligonucleotide probes.
The intensity files were loaded into the Illumina BeadStudio software version 3.0.19.0 (Illumina Inc.) and BRB Array Tools version 4.6.0 for quality control and gene expression analysis. First, the quantile normalization algorithm was applied to the data set. Only genes whose expression differed by at least 1.5‐fold from the median in at least 20% of the arrays and characterized by the 50th percentile of intensities >300 were retained. The false discovery rate–adjusted P‐values were calculated using the Benjamini‐Hochberg procedure. To identify the differentially expressed genes, F‐test and Multivariate Permutation Test were applied. Further, the genes were filtered based on their fold change values (±1.5).
2.5. miR Target gene Prediction
The R package multiMiR 23 was used to predict and validate miR‐mRNA target interactions. List of DE genes and miRs passing the cut‐off value was used as input. Among the databases in the multiMiR package, the validated databases (miRecords, miRTarBase and TarBase) and top 10% results of the predicted database (DIANA‐microT ElMMo, Microcosm, miRanda, miRDB, Pictar, PITA, TargetScan) were used for analysis.
2.6. qRT‐PCR analysis
The same cDNA used for gene sequencing was used also for qRT‐PCR analysis. Total RNA was retro‐transcribed using the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Gene expression analysis of Ccnd1, Cdkn1a, Cyp2b10, Socs3, Gadd45a and Gadd45b was performed using specific TaqMan probes (Thermo Fisher Scientific, 4369016). Each sample was run in triplicate and all measurements were normalized to β‐actin. Relative mRNA expression analysis for each gene was calculated by using the 2−ΔΔCt method.
Analysis of miRNA expression. cDNA was synthesized using the TaqMan® MicroRNA ReverseTranscription Kit (4366596). qRT‐PCR amplification was performed with the reverse transcription product, TaqMan® 2X Universal PCR Master Mix, No AmpErase ®UNG (4324018) and miR specific primers. The endogenous control sno202 was used to normalize miRNA expression levels.
2.7. Histology and Immunohistochemistry
Liver sections were fixed in 10% of buffered formalin and processed for immunohistochemistry (IHC). For BrdU detection, paraffin‐embedded 4 µm sections were deparaffinized, treated with HCl 2N for 1 h and then with 0.1% trypsin at 37°C. Sections were sequentially incubated with goat serum (Abcam), mouse monoclonal anti‐BrdU antibody (Becton Dickinson) and with Dako EnVision+® System Labelled Polymer‐HRP anti‐mouse (Dako). Peroxidase binding sites were detected by Vector NovaRED Peroxidase (HRP) Substrate Kit (Vector Laboratories). Harris haematoxylin solution (Sigma‐Aldrich) was used to counterstain liver sections. Labelling index (L.I.) was expressed as the number of BrdU‐positive nuclei/field (at x40 magnification). Ten to 50 fields per liver were scored. A segment of the duodenum, an organ with a high rate of cell proliferation, was used as a positive control for BrdU incorporation.
2.8. Statistical analysis
All data were expressed as the mean ± SD. Differences between groups were compared using one‐way analysis of variance ANOVA with the use of GraphPad Software Inc., San Diego, CA. A value of p < 0.05 was considered as a significant difference between groups.
2.9. Cell culture and in vitro experiments
HepG2 (ATCC, Manassas, VA, USA) and Mahlavu (kindly provided by Dr. N. Atabey) human liver cancer cells were cultured in DMEM complete medium with 10% foetal bovine serum (Lonza, Basel, Switzerland) in a 5% CO2 atmosphere. 50×103 cells were transfected using Lipofectamine2000 (Thermo Fisher, Waltham, MA, USA) in a 6 wells plate. Transfection reagents plus miRNA/negative control (hsa‐miR‐106b‐5p #MC10067, Negative Control #4464058) at final concentration of 20nM were used following standard protocols. Seventy‐two hours after transfection, total RNA was extracted with Maxwell® RSC miRNA Tissue Kit (Promega, Madison, WI, USA) according to manufacturer protocol. Total RNA was retro‐transcribed starting from 0.25μg RNA/sample using the High Capacity Kit (Thermo Fisher). Gene expression analysis was performed using the specific TaqMan probes (Thermo Fisher) hCCND1 (hs00765553_m1), and hACTIN (hs99999903_m1) as endogenous control. MiRNA expression was evaluated using the specific Taqman miRNA assay kits (Thermo): hsa‐miR‐106b‐5p #000442 and RNU48 #01006 (as endogenous control). PCR runs were performed with ABI Prism 7900HT (Applied Biosystems).
3. RESULTS
3.1. Hepatocyte proliferation following PH or TCP treatment.
According to previous reports, 16 the measurement of labelling index of hepatocytes from mice subjected to TCP and PH showed that while a high number of hepatocytes was in an active S phase as early as 24 h after TCP treatment, almost no BrdU‐positive cells could be observed at the same time point after PH (Figure 1A, B). However, the peak of DNA synthesis was observed in both groups 36 h after treatment (Figure 1A, B) with a trend towards a return to quiescence at 48 h. Since these data suggest that different molecular events are responsible for the accelerated entry of hepatocytes into S phase observed after TCP treatment, we investigated the possible involvement of miRs in the priming phase of liver cells. To this aim, we analysed the expression profiles of mouse hepatic mRNA and miR at 1, 3 and 6 h after PH or TCP. Identification of miR and mRNA expression abundance was evaluated by NGS (miR) and Illumina microarray (mRNA) in the same samples.
FIGURE 1.
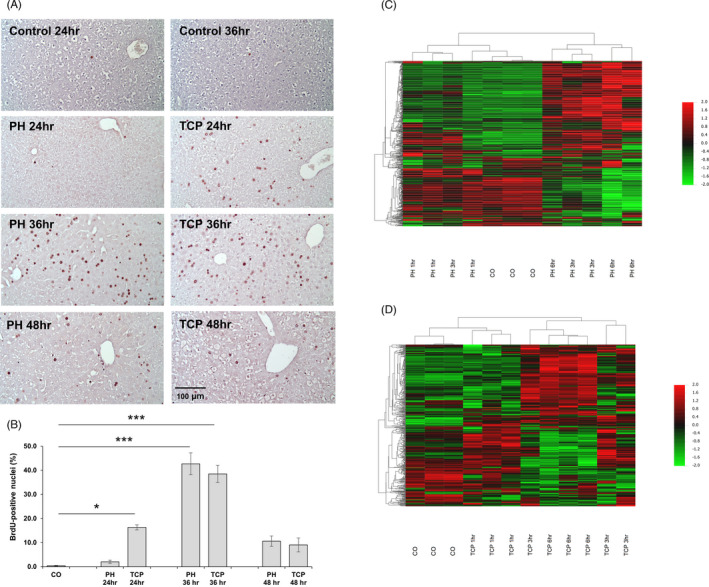
Hepatocyte proliferation and global gene expression profile following PH or TCP treatment. (A) Microphotographs illustrating the presence of BrdU‐positive hepatocyte nuclei (magnification ×20). CD‐1 female mice were subjected to 70% PH or TCP (3 mg/kg body weight) treatment and sacrificed 24, 36 or 48 h thereafter. All animals received BrdU (100 mg/kg) 2 h before the sacrifice; (B) Labelling Index (LI). LI was expressed as number of BrdU‐positive hepatocyte nuclei/100 nuclei. *p < 0.05; ***p < 0.001; (C) Hierarchical clustering of genes at the baseline (CO), 1, 3 and 6 h post‐PH. The red and green colours indicate upregulation and downregulation, respectively. Each row represents the expression of a gene and each column a sample. (D) Heatmap of differentially expressed genes at the baseline (CO), 1, 3 and 6 h post‐TCP. The red and green colours indicate upregulation and downregulation, respectively
3.2. Global gene expression profiles in regenerating livers after PH and TCP.
Global transcriptome changes at 1, 3 and 6 h revealed a total of 810 and 527 genes differentially expressed (DE) in the PH and TCP groups, respectively (Table S1 and S2). Hierarchical clustering analysis of the PH differentially expressed genes stratified them into two major clusters: (1) control liver and PH 1 h and (2) PH 3 and 6 h (Figure 1C). Similarly, it also stratified differentially expressed genes after TCP into two major clusters: (1) control liver and TCP 1 h and (2) TCP 3 and 6 h (Figure 1D).
As shown in Figure 2A TCP induced deregulation of a lower number of genes (Figure 2A). The highest number of genes was found to be deregulated at 6 h after PH (395 genes), while a much lower number of genes was deregulated after TCP treatment—148 and 225 genes—at 3 and 6‐h, respectively. Ninety‐six genes resulted commonly altered at all the time points after PH, while 48 were commonly altered at all the time points upon TCP (Figure 2B). Our analysis also showed that 20 genes were commonly deregulated upon both treatments (11 were upregulated, 8 downregulated and 1 exhibited an inconsistent pattern of expression; Figure S1).
FIGURE 2.
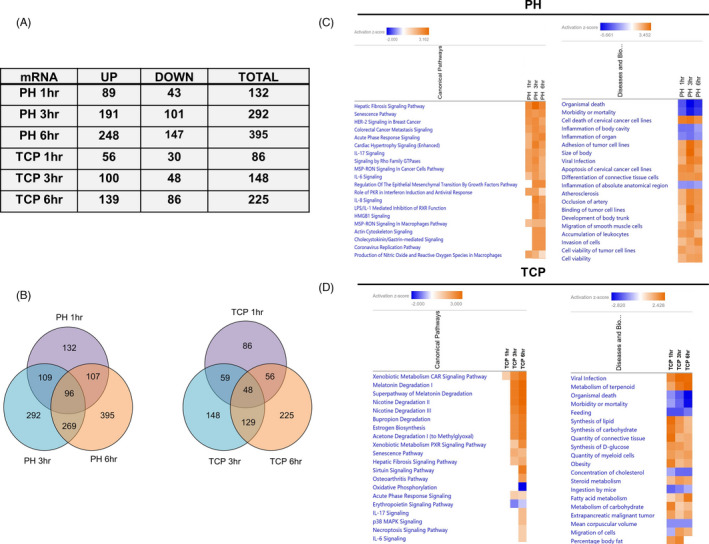
Ingenuity Pathway Analysis of deregulated genes after PH or TCP treatment. (A) Table showing the number of genes up‐ or downregulated at 1, 3 and 6 h post‐PH and TCP treatment; (B) Venn diagrams showing the number of deregulated and overlapping genes at 1, 3 and 6 h post‐PH or TCP treatment; (C) Heatmap for top 20 canonical pathways and diseases and biological function at 1, 3 and 6 h post‐PH. Colour is determined by Z‐score; the Z‐score >2 and <−2 is considered significant. Blue colour indicates suppressed disease /biological function or canonical pathways; orange indicates activated disease/biological function or canonical pathways; (D) Heatmap for top 20 canonical pathways and diseases and biological functions at 1, 3 and 6 h post‐TCP. Colour is determined by Z‐score; the Z‐score >2 and <−2 are considered significant. Blue colour indicates suppressed disease /biological function or canonical pathways; orange indicates activated disease/biological function or canonical pathways
Ingenuity Pathway Analysis (IPA) of DE genes 1, 3 and 6 h after PH versus control liver revealed their involvement in pathways related to Hepatic Fibrosis Signaling Pathway and Senescence Pathway (Figure 2C). The functional investigation also underlined common modifications between 1, 3 and 6 h post‐PH (Figure 2C), but none of them was directly related to cell cycle/cell proliferation.
IPA of the genes differentially expressed at 1, 3 and 6 h after TCP vs. control livers displayed completely different pathways and included Xenobiotic Metabolism CAR, Superpathway of Melatonin Degradation as well as Nicotine Degradation (Figure 2D). Functional investigation mainly underlined metabolic pathway modifications such as Synthesis of lipid, Feeding and Synthesis of Carbohydrate (Figure 2D).
We also performed DAVID functional analysis using Gene Ontology annotation. As shown in Figure 3A, among the genes upregulated after PH, many were related to apoptosis, cell cycle regulation and cell cycle arrest. Interestingly, genes classified as negative regulators of cell proliferation (Sox9, Cdkn1a, Jun, Trp53inp1, Agt, Gja1, Tgif1) or of ERK1/ERK2 cascade (Ats3, Dusp1, Dusp6, Timp3, Ptpn1) were upregulated following surgery (Figure S2). On the other hand, genes related to cellular metabolism were among the most deregulated after TCP (Figure 3B). Interestingly, only genes positively related to the cell cycle (Gadd45b, Sgk1, and Ccnd1) were observed following xenobiotic treatment (Figure S2). No evidence of increased expression of negative regulators of cell proliferation, cell cycle arrest or of ERK1/ERK2 cascade was observed after TCP (Figure 3B).
FIGURE 3.
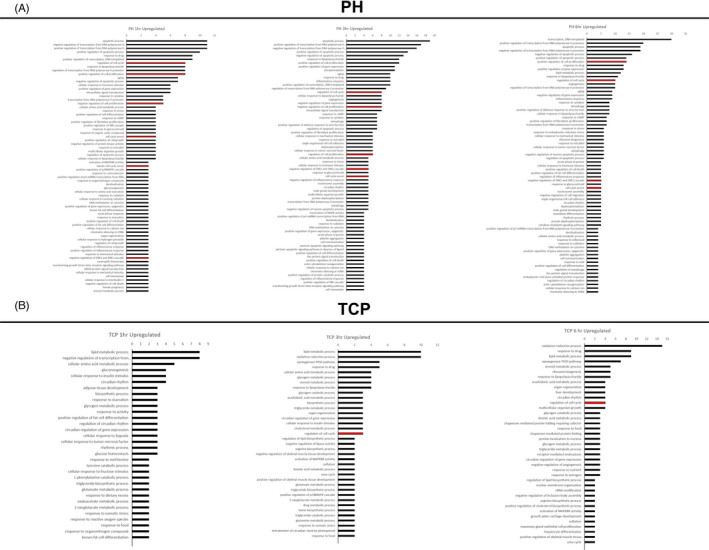
DAVID functional analysis using Gene Ontology annotation. (A) Gene Ontology enrichment analysis of biological processes for upregulated genes at 1, 3 and 6 h post‐PH; (B) Gene Ontology enrichment analysis of biological processes for upregulated genes at 1, 3 and 6 h post‐TCP treatment. Red colour indicates pathways positively/negatively related to regulation of cell proliferation
The most downregulated pathways in both experimental conditions are listed in Figure S3. While most of them involved metabolic pathways, none was directly related to cell cycle/cell proliferation.
3.3. Transcription Factors‐Dependent Pathways
Next, we analysed transcription factor (TF)‐dependent pathways differentially activated in the livers of PH and TCP mice. By examining the top 20 TFs in each group, we found striking differences between the two proliferative stimuli. Indeed, while RB1 was the most significantly downregulated TF in the PH livers at all the analysed time points it was not listed among the first 20 TFs after TCP (Figure 4A, B). In addition, while STAT3 was among the most significantly upregulated TF after PH (Figure 4A), it was profoundly downregulated 1 h after TCP treatment (Figure 4B). Furthermore, C/EBPβ, a TF that initiates a cascade of gene expression responsible for proliferation, 24 was strongly upregulated by TCP at all the examined time points, whereas it was not modified within the first 6 h after surgery. As shown in Figure 4C, several CEBPβ‐controlled genes progressively increased in the liver of mice treated with TCP 1, 3 and 6 h after treatment.
FIGURE 4.
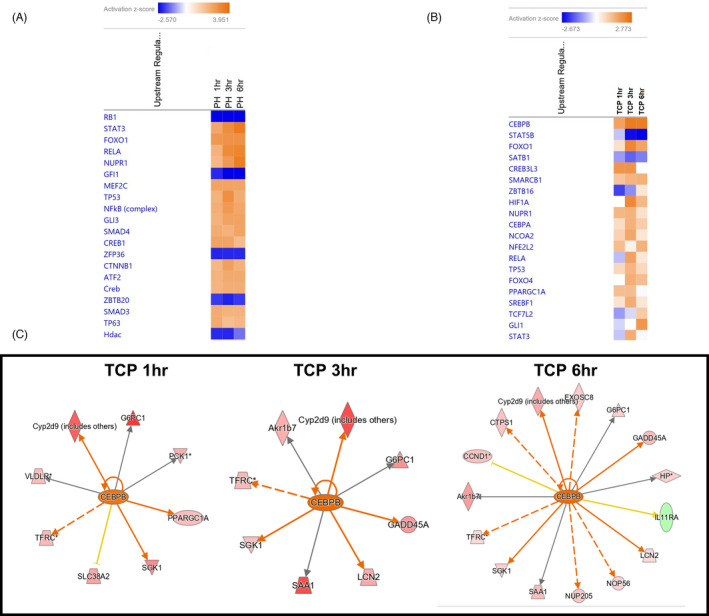
Transcription factor analysis following PH or TCP treatment. (A, B) Heatmaps for top 20 upstream regulators at 1, 3 and 6 h post‐PH (A) or TCP (B). Colour is determined by Z‐score; the Z‐score >2 and <−2 is considered significant. Blue colour indicates suppressed disease/biological function or canonical pathways; orange indicates activated disease/biological function or canonical pathways; (C) Gene interaction networks regulated by the transcription factor CEBPβ in mice 1, 3 and 6 h after treatment with TCP
Five TFs (STAT3, RELA, NUPR1, FOXO1, TP53) were deregulated in both PH and TCP‐treated mice (compare Figure 4A, B). STAT3 and RELA were strongly upregulated following PH at all the analysed time points, while they were profoundly downregulated 1 h following TCP treatment. These results confirm previous observations showing that no change in the activation of transcription factors implicated in liver regeneration such as, NF‐kB, AP‐1 and STAT3 has been observed in nuclear receptor‐mediated hepatocyte proliferation. 13 As to FOXO1—strongly upregulated after PH and unchanged or only slightly upregulated post‐TCP—it is interesting to note that forced expression or conditional activation of FOXO factors led to reduced Cyclin D1 expression. 25 , 26
3.4. miR expression in PH or TCP‐treated mice
To investigate miRs differentially expressed in the liver following the two proliferative stimuli, we applied time‐course analysis using the R/Bioconductor package “DESeq2”. Hierarchical clustering analysis in the PH group stratified the three time points into 2 major clusters: 1) Controls (CO), PH 1 and 3 h and 2) PH 6 h. Hierarchical clustering analysis on TCP samples did not display a clear separation of the different time points (Figure 5A, B).
FIGURE 5.
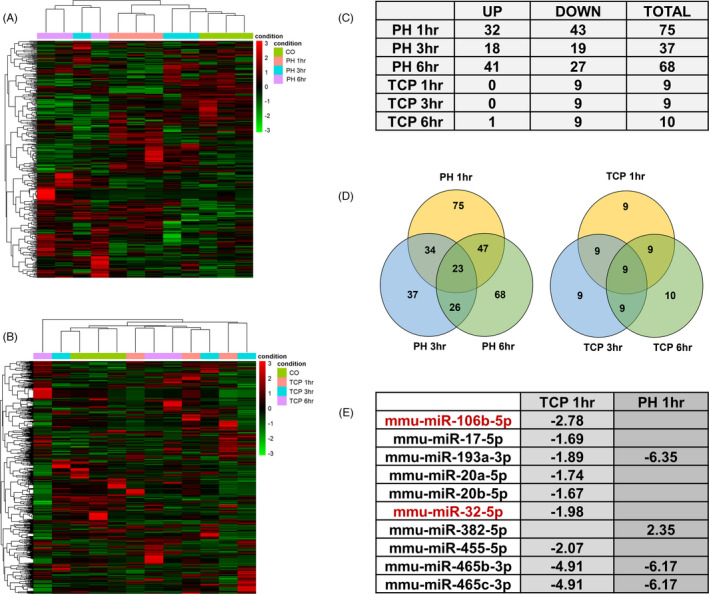
TCP and PH modify the global miR expression profile. (A) Heatmap of differentially expressed miRs at 1, 3 and 6‐h post‐PH. Red and green colours indicate miR upregulation and downregulation, respectively; (B) Heatmap of differentially expressed miRNAs at 1, 3 and 6 h post‐TCP. Red and green colours indicate upregulation and downregulation, respectively; (C) Table showing the number of miRs up or downregulated at 1, 3 and 6 h post‐PH or TCP treatment; (D) Venn diagrams showing the number of deregulated and overlapping miRs at 1, 3 and 6 h post‐PH or TCP treatment; (E) Table indicating miRs deregulated by TCP at 1 h, in common with PH. In red are indicated the miR deregulated exclusively in the TCP‐treated mice
A striking difference in the number of differentially expressed miRs was found between the two proliferative conditions (Figure S4 and S5). Indeed, while the expression of 102 miRs was significantly modified after PH, only 10 miRs were found differentially expressed in the TCP group. As to PH, the Venn diagram showed that the highest number of miRs was deregulated at 1 h (75 miRs) and 6 h (68 miRNAs) post‐surgery, whereas 23 miRs resulted commonly altered at all the time points (Figure 5C, D). Notably, the three most upregulated miRs 1 h after PH, miR‐124‐3p, miR‐9‐3p and miR‐9‐5p (Figure S4) act as inhibitors of proliferation in several cell types. 27 , 28
Similar to what was observed with mRNA expression, a much lower number of deregulated miRs was found after TCP treatment at all the analysed time points, with only 9 miRs being commonly altered at all the time points. Differently from PH, TCP caused downregulation of all the miRs at each time point, with the only exception of miR‐382‐5p that was upregulated 6 h after administration of the drug (Figure 5E, Figure S5). Interestingly, out of the 9 miRs deregulated 1 h after TCP, only 3 were altered in PH livers (Figure 5E). Notably, while most miRs were altered in both experimental groups (Figure S4 and S5), only 2 miRs—miR‐106b‐5p and miR‐32‐5p—were exclusively deregulated in TCP‐treated mice (Figure 5E).
3.5. miR‐mRNA interactions
To identify a possible link between differentially expressed miRs and genes, among the identified dysregulated genes, we selected multiMiR validated/predicted miR targets. In both experimental groups, several genes associated to the cell cycle and differentially expressed in both experimental groups were indeed targets of deregulated miRs. Among the most downregulated miRs post‐PH, we found 4 miRs (miR‐106a‐5p, miR 340‐5p, miR‐19b‐3p and miRNA‐455‐5p), targeting Socs3, a known tumour suppressor and an inhibitor of cell cycle. Indeed, Socs3 expression was already significantly upregulated 1 h after PH and remained elevated until 6 h post‐surgery (Table S3).
After PH, we also found upregulated both genes considered to be positively correlated to induction of proliferation (ie c‐fos and Ccnl1), as well as negative regulators of cell cycle (Gadd45a and Cdkn1a). In particular, Gadd45a was upregulated at all the time points and its upregulation was paralleled by downregulation of miR‐301b‐3p, miR‐484 and miR‐19b‐3p (validated) and miR‐130a‐3p, miR‐130b‐3p and miR‐301‐3p (predicted) to target it, as early as 1‐h post‐surgery. Moreover, downregulation of miR‐301 was associated with the upregulation of its target gene Cdkn1a (Figure 6A and Table S3).
FIGURE 6.
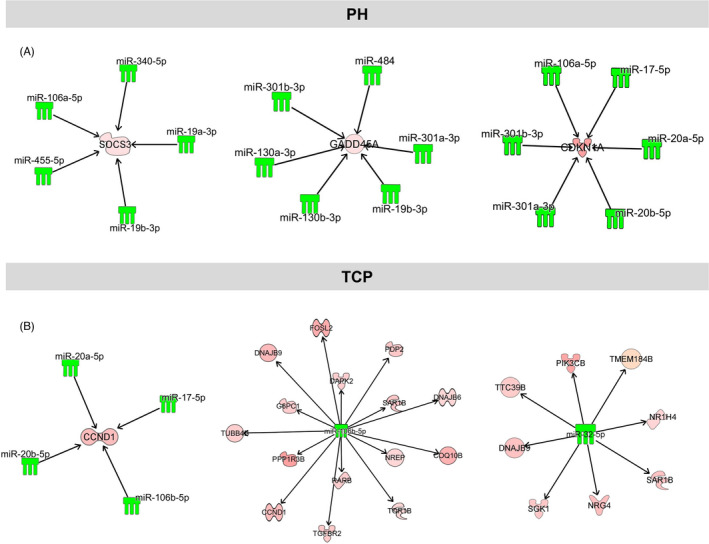
mRNA‐miR interaction networks of cell cycle‐related genes in mice subjected to PH or treated with TCP. (A) mRNA‐miR networks of Socs3, Gadd45a and Cdkn1a in mice subjected to PH showing that downregulation of several miRNAs is associated with the upregulation of their target genes; (B) mRNA‐miR networks of Ccnd1‐miR‐106b‐5p and Sgk1‐Pik3cb‐miR‐32‐5p in TCP‐treated mice. The panel shows miRs predicted/validated to target Ccnd1 and downregulated after TCP (left side), and all the predicted target genes of miR‐106b‐5p and miR‐32‐5p –the only two miRs differentially expressed only in TCP mouse liver (middle and right side)
Nfkbiz—another negative regulator of the cell cycle 12 , 29 was upregulated 3 and 6 h after PH. Interestingly, in rat liver Nfkbiz is a target of miR‐376b. 30 In our study, however, the role of miR‐376b is unclear as it was upregulated at 1 and 3 h after PH and downregulated at 6 h (Table S3). Whether mouse Nfkbiz is a target of miRs other than miR‐306b will require further studies.
In the TCP experimental group, no cell cycle negative regulator—other than Gadd45a—was significantly modified compared to control liver. Remarkably, Ccnd1, the gene encoding for cyclin D1, responsible for the G0‐G1 transition, was upregulated 6 h after TCP treatment. Such upregulation was paralleled by downregulation of cyclin D1 targeting miRs (miR‐20a‐5p, miR‐20b‐5p and miR‐17‐5p) as early as 1 h after TCP (Figure 6B). Notably, after PH, downregulation of the same miRs was observed only at 6 h (Figure S4). Only 2 miRs resulted exclusively deregulated in the liver of TCP‐treated mice: miR‐106b‐5p, predicted to target Ccnd1, which was downregulated at all the time points, and miR‐32‐5p targeting other positive regulators of the cell cycle, namely Sgk1 and Pik3cb 31 , 32 (Figure 6B; Table S3). To validate the NGS results, we performed qRT‐PCR analysis on a selected set of genes and miRNAs. As shown in Figure S6A, the expression of all the investigated genes (Ccnd1, Cdkn1a, Cype2b10, Socs3, Gadd45b and Gadd45a) was deregulated similar to what observed by NGS.
QRT‐PCR analysis was also performed to validate the changes in the levels of miR‐106b‐5p predicted to target Ccnd1 and whose expression was downregulated after TCP (Figure 5E) and of miR‐301b and miR‐455—predicted to target Cdkn1a and Socs3, respectively and found upregulated after PH (Figure S4). As shown in Figure S6B, the expression of these miRs was deregulated in accord to NGS results.
Next, to further validate the effect of miR‐106b‐5p on Ccnd1 we transduced two human liver carcinoma cell lines (Mahlavu and HepG2) with this miRNA and measured the mRNA levels of its target gene. As shown in Figure S7, expression of miR‐106b‐5p led to a significant downregulation of Ccnd1.
4. DISCUSSION
In the present study, we unveiled the miR‐mRNA networks involved in the priming phase of hepatocyte proliferation elicited by stimuli of different nature: PH, which stimulates liver regeneration, and TCP, which induces direct hyperplasia. To this aim, we evaluated the expression profiles of mRNAs and miRs at 1, 3 and 6 h after surgery or treatment with TCP.
Several studies reported early response of the mouse liver transcriptome following treatment with TCP 17 , 18 or after PH, 12 but none of the researches conducted so far correlated gene expression changes with miR profile in the same samples in both the experimental conditions. Indeed, previous studies in the literature reported deregulation of some miRs in the priming, proliferative and termination phases upon PH, but the results were often contradictory due to differences in strain, age and timing of observation. 4 , 5 , 6 , 33 , 34 , 35 , 36 As to TCP, few studies have investigated the role of miRs in hepatocyte proliferation, but none of them analysed early time points post‐treatment. 37 , 38 Thus, the key strength of our study is that these two experimental protocols (liver regeneration post‐PH and direct hyperplasia following TCP treatment) were performed in parallel on mice of the same strain, gender and age and at the same time. Moreover, we analysed both miRs and mRNAs in the same samples to generate a network of interactions, possibly explaining the transcriptomic modifications driving the two different proliferative modalities.
A much higher number of genes and miRNAs was found deregulated after PH when compared to TCP. The difference was particularly striking for miRNAs (102 post‐PH vs. 10 after TCP). In this context, it is important to stress that liver regeneration after 2/3 PH is part of a complex interplay of distinct sets of rapidly evolving changes, such as those caused by the metabolic and circulatory perturbations consequent to the removal of 2/3 of the liver and mitogenic changes. This makes difficult to discriminate genes and miRNAs directly related to the entry into the cell cycle from those responsible for the adjustments of essential hepatic functions. Such a massive metabolic rearrangement is clearly not required by the liver following a single treatment with TCP, as the liver does not have to compensate for a reduced size. It is possible that while the vast majority of miRNAs play a relevant role in the metabolic rearrangement of the remnant liver post‐PH and in the quite stressful condition associated to the surgical procedure, this is not the case after TCPOBOP, where only few miRNAs—some of which targeting cell cycle genes—are downregulated.
The most important findings of the present work are: 1) many genes functioning as negative regulators of the cell cycle were upregulated after PH, but not after TCP; 2) miRs negatively controlling cell cycle genes were downregulated only after surgery; 3) miRs predicted to target Ccnd1, such as miR‐106b‐5p, were significantly downregulated only in TCP‐treated mice.
Previous studies demonstrated that, in mice, the entry of hepatocytes into the S phase of the cell cycle is robustly anticipated in TCP‐treated animals compared to PH. 16 Indeed, an active DNA synthesis takes place 24 h after TCP, a time when virtually no dividing hepatocytes can be seen in livers undergone PH. In this context, it is interesting to note that Cyclin D1 induction occurs early after treatment with TCP, but not after PH. 16 For this reason, the identification of Ccnd1 targeting miRs already downregulated 2 h after TCP may explain the increase of cyclin D1 and the consequent faster entry into the cell cycle. Importantly, miR‐106b‐5p, predicted to target Ccnd1, was indeed exclusively downregulated after TCP treatment, at all the time points analysed. A second miR, miR‐32‐5p, was similarly downregulated only after TCP; this miR targets other positive regulators of the cell cycle, such as Sgk1 and Pik3cb, whose expression was increased in the same samples following TCP treatment. In agreement with Yin et al. 12 following surgery we observed downregulation of miRs targeting Socs3, a known tumour suppressor and a cell cycle inhibitor. In addition, we also found increased levels of the cell cycle inhibitor Cdkn1a, preceded by downregulation of miR‐301, targeting Cdkn1a. Furthermore, we observed a strong upregulation of the oncosuppressor miR‐34 family. Of note, the increased expression of miR‐34 observed during the termination phase of liver regeneration has been reported as a potential ‘stop’ signal. 39 This finding together with the report of Sun 40 showing that miR‐34a targets the 3’ untranslated mRNA region of Ccnd1, supports the concept that an active control on mitogenic signals operates after PH, thus leading to a delay in the entry into the cell cycle. Conversely, no evidence of miR‐34 deregulation was observed at any time point in TCP‐treated livers (Figure S4).
Another interesting observation that could justify the different kinetics of S phase entry of the two proliferative stimuli is the finding that while after PH pathways related to inflammatory response were among the most deregulated, pathways involving upregulation of metabolic changes, especially lipid metabolism—required for sustaining cell proliferation—were the most modified after TCP.
In conclusion, the analysis of the microRNAs‐mRNA networks performed in the present study unveils on the one hand that different miRs are implicated in the early phase of hepatocyte proliferation induced by mitogenic stimuli of different nature, and on the other hand that miRs, such as miR‐106b‐5p are critical in regulating the levels of the main cyclin implicated in the G1‐S transition of the cell cycle.
A limitation of this study relies on the impossibility to functionally validate some of the present findings since primary hepatocytes in vitro do not express CAR and do not proliferate after TCP. 41 Nevertheless, the present work, performed in a strictly controlled experimental condition, contains a number of novel findings that can be helpful for a better comprehension of the molecular mechanisms responsible for deciphering proliferative signals in totally diverse conditions, such as those where hepatocyte replication is needed to replace cell loss (ie PH or chemically induced necrosis) or those where proliferation occurs in the intact liver (TCP as well as other hepatomitogens, such as T3 and PPAR ligands).
CONFLICT OF INTEREST
All authors declare that they have no conflict of interest.
AUTHOR CONTRIBUTION
Rajesh Pal and Marta Anna Kowalik conducted research and drafted the manuscript. Marina Serra provided assistance in the process of revised drafting manuscript and figure and tables construction. Cristina Migliore performed in vitro experiments. Amedeo Columbano, Andrea Perra and Silvia Giordano contributed to conceptual framework, supervised the study and revised the manuscript.
Supporting information
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7
Table S1
Table S2
Table S3
Supporting Information
ACKNOWLEDGEMENT
This work was supported by Associazione Italiana Ricerca sul Cancro (AIRC, Grants IG‐20176 to AC), Regione Autonoma Sardegna (RAS to AC), Associazione Italiana Ricerca sul Cancro (AIRC, Grant IG‐20210 to SG).
Pal R, Kowalik MA, Serra M, et al. Diverse MicroRNAs‐mRNA networks regulate the priming phase of mouse liver regeneration and of direct hyperplasia. Cell Prolif. 2022;55:e13199. doi: 10.1111/cpr.13199
Rajesh Pal and Marta Anna Kowalik equally contributed to the work.
Contributor Information
Amedeo Columbano, Email: columbano@unica.it, Email: andrea.perra@unica.it.
Andrea Perra, Email: andrea.perra@unica.it.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request. Raw data can be accessed in following IDs: GEO accession No (mRNA): GSE185316. SRA ID (miRNA): PRJNA769011.
REFERENCES
- 1. Michalopoulos GK, DeFrances MC. Liver regeneration. Science. 1997;276(5309):60‐66. doi: 10.1126/science.276.5309.60 [DOI] [PubMed] [Google Scholar]
- 2. Cressman DE, Greenbaum LE, DeAngelis RA, et al. Liver failure and defective hepatocyte regeneration in interleukin‐6‐deficient mice. Science. 1996;274(5291):1379‐1383. doi: 10.1126/science.274.5291.1379 [DOI] [PubMed] [Google Scholar]
- 3. Yamada Y, Kirillova I, Peschon JJ, Fausto N. Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc Natl Acad Sci U S A. 1997;94(4):1441‐1446. doi: 10.1073/pnas.94.4.1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Castro RE, Ferreira DMS, Zhang X, et al. Identification of microRNAs during rat liver regeneration after partial hepatectomy and modulation by ursodeoxycholic acid. Am J Physiol Gastrointest Liver Physiol. 2010;299(4):G887‐G897. doi: 10.1152/ajpgi.00216.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Song G, Sharma AD, Roll GR, et al. MicroRNAs control hepatocyte proliferation during liver regeneration. Hepatology. 2010;51(5):1735‐1743. doi: 10.1002/hep.23547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen X, Zhao Y, Wang F, Bei Y, Xiao J, Yang C. MicroRNAs in Liver Regeneration. Cell Physiol Biochem. 2015;37(2):615‐628. doi: 10.1159/000430381 [DOI] [PubMed] [Google Scholar]
- 7. Hand NJ, Master ZR, Le Lay J, Friedman JR. Hepatic function is preserved in the absence of mature microRNAs. Hepatology. 2009;49(2):618‐626. doi: 10.1002/hep.22656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kren BT, Wong PY‐P, Shiota A, Zhang X, Zeng Y, Steer CJ. Polysome trafficking of transcripts and microRNAs in regenerating liver after partial hepatectomy. Am J Physiol Gastrointest Liver Physiol. 2009;297(6):G1181‐G1192. doi: 10.1152/ajpgi.90636.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shu J, Kren BT, Xia Z, et al. Genomewide microRNA down‐regulation as a negative feedback mechanism in the early phases of liver regeneration. Hepatology. 2011;54(2):609‐619. doi: 10.1002/hep.24421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fausto N. Liver regeneration. J Hepatol. 2000;32(1 Suppl):19‐31. doi: 10.1016/s0168-8278(00)80412-2 [DOI] [PubMed] [Google Scholar]
- 11. Michalopoulos GK. Liver regeneration. J Cell Physiol. 2007;213(2):286‐300. doi: 10.1002/jcp.21172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yin L, Guo X, Zhang C, Cai Z, Xu C. In silico analysis of expression data during the early priming stage of liver regeneration after partial hepatectomy in rat. Oncotarget. 2018;9(14):11794‐11804. doi: 10.18632/oncotarget.24370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Columbano A, Shinozuka H. Liver regeneration versus direct hyperplasia. FASEB J off Publ Fed Am Soc Exp Biol. 1996;10(10):1118‐1128. doi: 10.1096/fasebj.10.10.8751714 [DOI] [PubMed] [Google Scholar]
- 14. Behrens A, Sibilia M, David J‐P, et al. Impaired postnatal hepatocyte proliferation and liver regeneration in mice lacking c‐jun in the liver. EMBO J. 2002;21(7):1782‐1790. doi: 10.1093/emboj/21.7.1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ledda‐Columbano GM, Curto M, Piga R, et al. In vivo hepatocyte proliferation is inducible through a TNF and IL‐6‐independent pathway. Oncogene. 1998;17(8):1039‐1044. doi: 10.1038/sj.onc.1202018 [DOI] [PubMed] [Google Scholar]
- 16. Ledda‐Columbano GM, Pibiri M, Loi R, Perra A, Shinozuka H, Columbano A. Early increase in cyclin‐D1 expression and accelerated entry of mouse hepatocytes into S phase after administration of the mitogen 1, 4‐Bis[2‐(3,5‐Dichloropyridyloxy)] benzene. Am J Pathol. 2000;156(1):91‐97. doi: 10.1016/S0002-9440(10)64709-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Locker J, Tian J, Carver R, et al. A common set of immediate‐early response genes in liver regeneration and hyperplasia. Hepatology. 2003;38(2):314‐325. doi: 10.1053/jhep.2003.50299 [DOI] [PubMed] [Google Scholar]
- 18. Lodato NJ, Melia T, Rampersaud A, Waxman DJ. Sex‐differential responses of tumor promotion‐associated genes and dysregulation of novel long noncoding RNAs in constitutive androstane receptor‐activated mouse liver. Toxicol Sci. 2017;159(1):25‐41. doi: 10.1093/toxsci/kfx114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Higgins G, Anderson RE, Higgins G, Anderson R. Experimental pathology of liver: restoration of liver in white rat following partial surgical removal. Arch Pathol. 1931;12:186‐202. [Google Scholar]
- 20. Smith T, Heger A, Sudbery I. UMI‐tools: modeling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Res. 2017;27(3):491‐499. doi: 10.1101/gr.209601.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krueger FTG [Internet]. 2015. Available from: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
- 22. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ru Y, Kechris KJ, Tabakoff B, et al. The multiMiR R package and database: integration of microRNA‐target interactions along with their disease and drug associations. Nucleic Acids Res. 2014;42(17):e133. doi: 10.1093/nar/gku631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Taub R, Greenbaum LE, Peng Y. Transcriptional regulatory signals define cytokine‐dependent and ‐independent pathways in liver regeneration. Semin Liver Dis. 1999;19(2):117‐127. doi: 10.1055/s-2007-1007104 [DOI] [PubMed] [Google Scholar]
- 25. Schmidt M, Fernandez de Mattos S, van der Horst A, et al. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol Cell Biol. 2002;22(22):7842‐7852. doi: 10.1128/MCB.22.22.7842-7852.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pauta M, Rotllan N, Fernández‐Hernando A, et al. Akt‐mediated foxo1 inhibition is required for liver regeneration. Hepatology. 2016;63(5):1660‐1674. doi: 10.1002/hep.28286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gao M, Cui Z, Zhao D, Zhang S, Cai Q. MicroRNA‐9 inhibits proliferation and progression in retinoblastoma cells by targeting PTEN. Genes & Genomics. 2021;43(9):1023‐1033. doi: 10.1007/s13258-021-01043-w [DOI] [PubMed] [Google Scholar]
- 28. Ying M, Feng H, Zhang X, Liu R, Ning H. MiR‐9‐5p Inhibits the Proliferation, Migration and Invasion of Choroidal Melanoma by Targeting BRAF. Technol Cancer Res Treat. 2020;19:1533033820956987. doi: 10.1177/1533033820956987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Alexander E, Hildebrand DG, Kriebs A, et al. IκBζ is a regulator of the senescence‐associated secretory phenotype in DNA damage‐ and oncogene‐induced senescence. J Cell Sci. 2013;126(Pt 16):3738‐3745. doi: 10.1242/jcs.128835 [DOI] [PubMed] [Google Scholar]
- 30. Liu Z, Tang C, He L, et al. The negative feedback loop of NF‐κB/miR‐376b/NFKBIZ in septic acute kidney injury. JCI Insight. 2020;5(24): doi: 10.1172/jci.insight.142272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dituri F, Mazzocca A, Lupo L, et al. PI3K class IB controls the cell cycle checkpoint promoting cell proliferation in hepatocellular carcinoma. Int J Cancer. 2012;130(11):2505‐2513. doi: 10.1002/ijc.26319 [DOI] [PubMed] [Google Scholar]
- 32. Jia S, Liu Z, Zhang S, et al. Essential roles of PI(3)K‐p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454(7205):776‐779. doi: 10.1038/nature07091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Finch ML, Marquardt JU, Yeoh GC, Callus BA. Regulation of microRNAs and their role in liver development, regeneration and disease. Int J Biochem Cell Biol. 2014;54:288‐303. doi: 10.1016/j.biocel.2014.04.002 [DOI] [PubMed] [Google Scholar]
- 34. Yi P‐S, Zhang M, Xu M‐Q. Role of microRNA in liver regeneration. Hepatobiliary Pancreat Dis Int. 2016;15(2):141‐146. doi: 10.1016/S1499-3872(15)60036-4 [DOI] [PubMed] [Google Scholar]
- 35. Raschzok N, Sallmon H, Dame C, Sauer IM. Liver regeneration after partial hepatectomy: inconsistent results of expression screenings for human, mouse, and rat microRNAs. Am J Physiol Gastrointest Liver Physiol. 2012;302(4):G470‐G471. doi: 10.1152/ajpgi.00494.2011 [DOI] [PubMed] [Google Scholar]
- 36. Raschzok N, Werner W, Sallmon H, et al. Temporal expression profiles indicate a primary function for microRNA during the peak of DNA replication after rat partial hepatectomy. Am J Physiol Regul Integr Comp Physiol. 2011;300(6):R1363‐R1372. doi: 10.1152/ajpregu.00632.2010 [DOI] [PubMed] [Google Scholar]
- 37. Hao R, Su S, Wan Y, et al. Bioinformatic analysis of microRNA networks following the activation of the constitutive androstane receptor (CAR) in mouse liver. Biochim Biophys Acta. 2016;1859(9):1228‐1237. doi: 10.1016/j.bbagrm.2016.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Moriya N, Kataoka H, Nishikawa J, Kugawa F. Identification of candidate target Cyp genes for microRNAs whose expression is altered by PCN and TCPOBOP, representative ligands of PXR and CAR. Biol Pharm Bull. 2016;39(8):1381‐1386. doi: 10.1248/bpb.b16-00279 [DOI] [PubMed] [Google Scholar]
- 39. Chen H, Sun Y, Dong R, et al. Mir‐34a is upregulated during liver regeneration in rats and is associated with the suppression of hepatocyte proliferation. PLoS One. 2011;6(5):e20238. doi: 10.1371/journal.pone.0020238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sun F, Fu H, Liu Q, et al. Downregulation of CCND1 and CDK6 by miR‐34a induces cell cycle arrest. FEBS Lett. 2008;582(10):1564‐1568. doi: 10.1016/j.febslet.2008.03.057 [DOI] [PubMed] [Google Scholar]
- 41. Abe T, Amaike Y, Shizu R, et al. Role of YAP activation in nuclear receptor CAR‐mediated proliferation of mouse hepatocytes. Toxicol Sci. 2018;165(2):408‐419. doi: 10.1093/toxsci/kfy149 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7
Table S1
Table S2
Table S3
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. Raw data can be accessed in following IDs: GEO accession No (mRNA): GSE185316. SRA ID (miRNA): PRJNA769011.