

Abstract
In patients infected with the human immunodeficiency virus (HIV), the HIV-Tat protein may be continually produced despite adequate antiretroviral therapy. As the HIV-infected population is aging, it is becoming increasingly important to understand how HIV-Tat may interact with proteins such as amyloid β and Tau which accumulate in the aging brain and eventually result in Alzheimer’s disease. In this review, we examine the in vivo data from HIV-infected patients and animal models; and the in vitro experiments that show how protein complexes between HIV-Tat and amyloid β occur through novel protein-protein interactions and how HIV-Tat may influence the pathways for amyloid β production, degradation, phagocytosis and transport. HIV-Tat may also induce Tau phosphorylation through a cascade of cellular processes that lead to formation of neurofibrillary tangles, another hallmark of Alzheimer’s disease. We also identify gaps in knowledge and future directions for research. Available evidence suggests that HIV-Tat may accelerate Alzheimer’s like pathology in patients with HIV infection which cannot be impacted by current antiretroviral therapy.
Keywords: Alzheimer’s disease, AIDS, dementia, neurodegeneration, HIV Tat, protein misfolding, aggregation, brain
Introduction
Neurocognitive dysfunction is detected in almost thirty percent of human immunodeficiency virus (HIV)-infected patients, despite antiretroviral therapy (Heaton et al. 2011), with increased incidence in older people (Valcour et al. 2004; Becker et al. 2004; Heaton et al. 2011). While Alzheimer’s disease can occur in HIV-infected individuals (Hellmuth et al. 2018, Morgello et al. 2018), it is likely that HIV infection can modulate the key pathological features associated with Alzheimer’s disease. HIV-infected individuals have increased deposition of amyloid β (Aβ) plaques in the brain which are often perivascular and present both in plaques and inside neurons as opposed Alzheimer’s disease (AD) where the amyloid depositions are largely parenchymal and extracellular (Esiri et al. 1998, Green et al. 2005, Achim et al. 2009). Amyloid plaques and neurofibrillary tangles are hallmarks of Alzheimer’s disease (AD) and their role in disease pathogenesis remains an area of intense investigation. In the HIV-infected brain, the processes are even more complicated.
Many mechanisms have been proposed as contributing factors to HIV-associated neurocognitive disorders, including induction of oxidative stress in the central nervous system, chronic microglial-mediated neuroinflammation, Aβ deposition, hyperphosphorylation of Tau protein, and toxic effects of combination antiretroviral therapy (Ferrell and Giunta 2014). It has been shown that certain antiretroviral medications particularly reverse transcriptase inhibitors may have additive amyloidogenic effects in macrophages. However, these effects were seen at very high concentrations which cannot be achieved in the brains of HIV-infected individuals (Giunta et al. 2011).
Even when antiretroviral therapy successfully suppresses viral replication, HIV-trans-activator of transcription (Tat) protein can be produced from proviral DNA (Johnson et al. 2013) in HIV reservoirs in the brain. This is evidenced by detection of Tat in the cerebrospinal fluid of HIV-infected patients on antiretroviral therapy (Johnson et al. 2013). Tat is the first protein to be produced during viral replication and is released extracellularly in large amounts (Li et al. 2009). Therefore, Tat may be an important contributor to the AD amyloid production and neurotoxicity. This review focuses on the connections between Tat protein and the Aβ and Tau pathology of the AD in HIV-infected brain.
2. HIV-Tat protein – structure and properties
Tat is actively released from HIV-infected cells (infiltrating macrophages and glial cells (Nath and Steiner 2014, Mattson et al. 2005)) and is a key activator for HIV transcription (Bagashev et al. 2013). It is a protein of variable length that can be composed of 86 to 101/104 amino acids (aa) (Debaisieux et al. 2012, Li et al. 2009) It is encoded by 2 exons. First exon encodes the first 72 aa, which makes the most active part of the protein, and the second exon encodes the remaining aa; 73–101/104. This second part has large sequence heterogeneity amongst HIV clades and its complete biological function is not completely determined (Guo et al. 2003, Smith et al. 2003, Avraham et al. 2004). However the conserved Arg Gly Asp motif found in the region 73–101, the so called “cell adhesion motif” was found to bind to the integrin receptors of the cell membrane (Mattson et al. 2005). Tat has six functional regions, including a proline-rich region (aa 1–21), a cysteine rich region (aa 22–37), a basic region (aa 49–59), and a glutamine rich region (aa 60–72), all important for its functions (Li et al. 2009). Aa 1–48 make a minimal domain necessary for LTR activation, whereas the basic domain 49–72 confers TAR RNA binding and is important for nuclear localization and uptake of Tat by the cell (Jeang et al. 1999).
As determined by its electric charge and hydropathy, Tat qualifies as an unstructured protein (PONDR: Garner et al. 1999, Dunker et al. 2001, Peng et al. 2005), and has at least two predicted hot spots of aggregation (Aggrescan: Conchilo-Solet et al. 2007, de Groot et al. 2012). These predictions have been confirmed by several experimental observations which show the self-aggregation of the cationic fragment of Tat 47–57, involving a dimeric predominant step (Machi et al. 2017) and self-aggregation of Tat 1–72 (Hategan et al. 2017).
Nuclear magnetic resonance studies of Tat confirmed that Tat is an unstructured protein (Shojania et al. 2006, 2010). No stable conformation and fast dynamics are consistent with the ability of Tat to interact with a large number of molecules and support the concept of a natively unfolded protein (Shojania et al. 2006). A common mechanism of action for natively unfolded proteins involves folding upon interaction with a binding partner (Uverski 2002). The crystal structure of Tat complexed with the positive transcription elongation factor b (Tahirov et al. 2010) (Figure 1a) shows that, under milder crystallization conditions, the protein changes conformation dramatically and presents a well folded portion of 42 aa, held together by two Zn+2 ions and coordinated by most of the cysteine residues from the cysteine-rich region (Tahirov et al. 2010).
Figure 1. High resolution structures of Tat protein, Aβ 1–40 and 1–42 fibrils and Tau fibrils in cross-section.
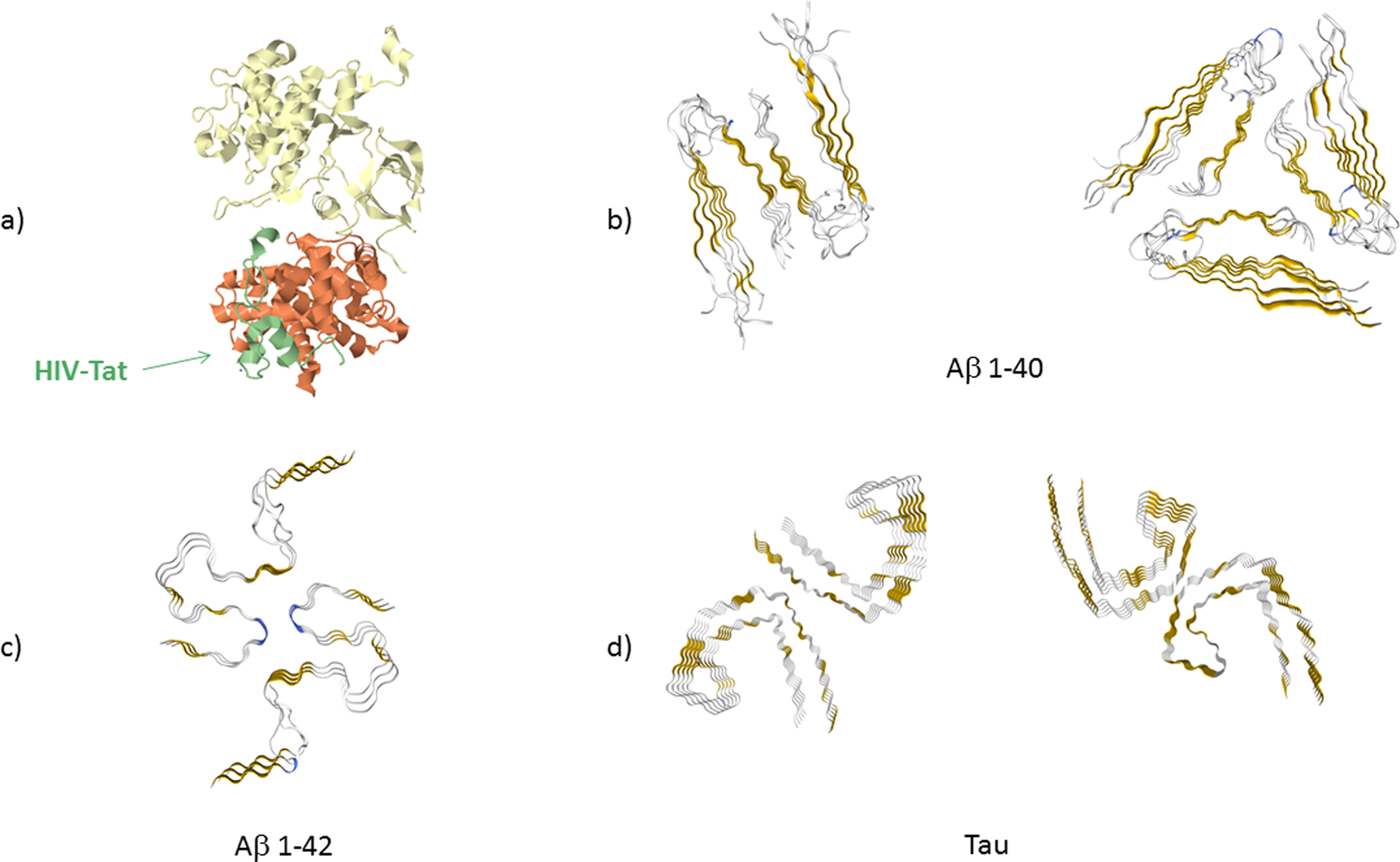
(a) Tat (colored in green) in bound state to the protein transcription elongation factor PTEFb (consisting of cell division protein kinase 9 (yellow) and cyclin T1 (orange)) (PDB: 3MI9) obtained by crystalography (Tahirov et al. 2010). Amino acids (aa) 5–50 of Tat are shown. The remaining structure of Tat could not be resolved. (b) Cross-section of Aβ 1–40 fibrils formed in agitated (PDB: 2LMO) and quiescent conditions (PDB: 2LMQ) obtained by nuclear magnetic resonance (Paravastu et al. 2008). Aa 9–40 are shown. (c) Cross-section of Aβ 1–42 fibrils as obtained by solid state, nuclear magnetic resonance, spectroscopy and electron microscopy (PDB: 2NAO) (Walti et al. 2016). Aa 1–42 are shown. (d) Cross-section of Tau protein fibrils for the straight fibrils (PDB: 5O3B) and helical fibrils (PDB: 5O3O), as obtained by cryo electron microscopy (Fitzpatrick et al. 2017). Aa 306–378 are shown (Fitzpatrick et al. 2017). In all cross-section figures of the fibrils, gold color indicates β−sheet structure and blue color indicates β-turn.
Tat can enter most cells (Frankel et al. 1988). In the brain, it is taken up by astrocytes and neurons (Ma and Nath 1997). Several membrane proteins interact with Tat, such as the low-density lipoprotein receptor-related protein (LRP), postsynaptic density protein 95 (PSD95), n-methyl-D-aspartic acid (NMDA), chemokine receptor CXCR4, and importantly, heparan sulfate proteoglycans (Li et al. 2009) which are present in large amounts on the external side of the cell membranes. Some of these structures facilitate its internalization (Liu et al. 2000). Tat also directly penetrates lipid membranes, without the help of proteins, by inducing pore formation (Zeitler et al. 2015. Brooks et al. 2005). The Tat protein transduction domain was shown to penetrate lipid membranes by inducing a Saddle-Splay curvature of the membrane that leads to pore formation (Mishra et al. 2008). Also, the uptake of Tat 52–62 peptide proceeds via two pathways which differ from macropinocytosis (Ben-Dov and Korenstein 2015).
We may think of Tat simply as an opportunistic peptide, adhering strongly to the cell surface on the basis of its charge to any negative entity, such as lipids or proteins, and then being internalized through natural cell membrane recycling of microdomains, presumably captured by any type of endocytic vesicle (Brooks et al. 2005). This is due to a constant cell plasma membrane turnover that leads to all cell’s surface to be internalized nonspecifically in less than an hour, notwithstanding the faster receptor mediated or stimulated routes of uptake (Brooks et al. 2005).
Tat also perturbs glial and monocyte/macrophage functions, promoting the release of neurotoxic agents including matrix metalloproteinases (MMPs) (Mattson et al. 2005). The exact role of these agents in Tat-mediated neurotoxicity remains to be determined. MCP-1/CCL2 and MMPs released by Tat may also increase the permeability of the blood-brain barrier and promote the transmigration of monocytes across the BBB (Matson et al. 2005, Huang et al. 2014).
Most of the Tat that enters the cell ends up finally in the nucleus (Liu et al. 2000). In vivo, it was shown that Tat alters gene expression in the mouse brain, following intranasal exposure (Pulliam et al. 2007). Five genes of interest in the Tat-treated mice were significantly elevated: Toll-like receptor 9, Fas, two cysteine-rich cytokines Ccl2 and Ccl17and sestrin. These genes are involved in innate immunity, inflammatory and apoptotic reactions (Pulliam et al. 2007). Also a gene that accounts for Tau protein was reported to be affected (Kadri et al. 2015).
Nanomolar concentrations of Tat1-72, Tat1-86, and Tat1-101 have similar potency in terms of their neurotoxic effects (Aksenov et al. 2009, Nath et al. 1996). Tat levels in sera of HIV-1 infected patients have been reported in the nanomolar range, but these levels are very likely underestimated considering how reactive Tat is to proteins and cells (Westendorp et al. 1995).
The brain regions that are particularly susceptible to the toxic effects of Tat include the striatum, hippocampal dentate gyrus and the CA3 region of the hippocampus (Everall et al. 1999, Maragos et al. 2003, Hayman et al. 1993).
3. Aβ peptides – structure and properties
Generally, the term amyloid refers to abnormal fibrillar, extracellular proteinaceous deposits formed in organs and tissues, that are insoluble and are structurally dominated by β-sheet structure (Rambaran and Serpell 2008). Even though formed from different proteins or peptides, they all share a β-sheet structure as the backbone, in which hydrogen bonding occurs along the length of the fiber and the β strands run perpendicular to the fiber axis (Serpell 2000). More than 20 proteins, most from plasma, have been identified to form amyloids (Rambaran and Serpell 2008). The amyloid formation is likely to be a general behavior for disordered proteins and peptides (Kayed et al. 2003, Nguyen and Hall 2004). It is likely that the common β-sheet conformation of the backbone is the one that gives the fibrillar, proteolytic resistant and insoluble characteristics to all forms of amyloids ((Rambaran and Serpell 2008). The β-sheet conformation is tightly linked to fibrilogenessis (Lansbury 1999, Chiti et al. 1999). Aβ β-sheet content is linked to insolubility (Halverston et al. 1990) and related to neurotoxicity (Fraser et al. 1992).
While some studies have previously shown that neurons are a major source of Aβ in the brain (Zhao et al. 1996, Rossner et al. 2005), recent research shows also that blood derived Aβ peptide may contribute to Alzheimer disease pathophysiology (Bu et al. 2018). The amyloid precursor protein (APP) is a transmembrane protein with a large extracellular domain. Its 695 aa isoform is expressed predominantly in the central nervous system (Bayer et al. 1999). The physiological function of APP is not completely known and remains one of the vexing issues in the field (O’Brien and Wong 2011). However, it has a role in the neuroprotection against excitotoxic injuries (Masliah et al. 1997). APP is produced in large quantities in neurons and is rapidly metabolized (Lee et al. 2008). After sorting in the endoplasmic reticulum and Golgi bodies, APP reaches the axon, and is transported by fast axonal transport to synaptic terminals (Koo et al. 1990). Once on the cell surface, APP can be proteolyzed directly by α-secretase and then γ-secretase, which is a process that does not generate Aβ. The other possibility is that APP is reinternalized in clathrin-coated pits into another endosomal compartment containing the proteases BACE1(β secretase) and γ-secretase. BACE1 initiates the Aβ generation (Li et al. 2015) and the processes here result in Aβ production, which is then released into the extracellular space following vesicle recycling or degradation in lysosomes. Although APP must pass through the cell surface as part of its processing, this step is likely very fast, since little APP is on the surface at any point in time (O’Brien and Wong 2011). Why some surface APP is internalized into endosomes and some proteolyzed directly by α -secretase is unclear, although segregation of APP and BACE1 into lipid rafts may be a crucial element (Ehehalt et al. 2003). The standard model suggests that little Aβ is generated outside of endosomal pathways, from which the Aβ is externalized from the cell (O’Brien and Wong 2011).
Aβ 1–40 is found in the amyloid plaques in the brain, and it is the most abundantly secreted amyloid peptide from the cells (Sisodia et al. 1990). The structure of Aβ fibrils has been extensively studied (Ball et al. 2014) and their molecular structure, determined by solution NMR, electron microscopy or atomic force microscopy (Petkova et al. 2005, Kodali et al. 2010, Moores et al. 2011).The structure is largely dependent on the polymerization conditions, and there are significant differences between fibrils formed in quiescent or agitated conditions (Petkova et al 2005, Paravastu et al 2008) (Figure 1b). Likely the quiescent conditions better simulate the in vivo conditions (Lu et al. 2013).
Aβ 1–42 is the major component of amyloid plaques (Walti et al. 2016). This may be due to its larger reactivity, increased stability once it aggregates or decreased clearance from the brain. Amyloid fibrils were extensively studied, and the atomic resolution structure has been resolved by NMR, spectroscopy and electron microscopy (Walti et al. 2016, Colvin et al. 2016) (Figure 1c). The transition of amyloid oligomers to fibrils has also been studied (Ahmed et al. 2010). Aβ 1–42 oligomers are believed to be the principal neurotoxic species. Aβ 1–42 oligomers can decrease synapse number, inhibit long-term potentiation and enhance long-term synaptic depression in rodent hippocampus, and injecting them into healthy rats impairs memory. Experiments in mice suggest that oxidative damage, inflammation and inhibition of neurogenesis are all a downstream consequence of Aβ oligomer formation and aggregation (Parthsarathy et al. 2013, Selkoe and Hardy 2016).
4. Tau protein – structure and properties
Tau is the major microtubule associated protein in mature neurons. An established function is the interaction with tubulin and promotion of its assembly into microtubules and stabilization of the microtubule network. The microtubule assembly promoting activity of Tau, a phosphoprotein, is regulated by its degree of phosphorylation. Hyperphosphorylation of Tau depresses its biological activity. In AD brain, Tau is ~three to four-fold more hyperphosphorylated than in the normal adult brain and this hyperphosphorylated state induces self-assembly of Tau into tangles containing paired helical filaments (PHF) mixed with straight filaments (SF) (Iqbal et al. 2010), in this way losing its ability to sequester normal microtubule associated proteins. Some of the Tau in AD brain is truncated which also promotes its self-assembly (Iqbal et al. 2010.) Fitzpatrick et al. (2017) resolved the structure of Tau fibrils by cryo electron microscopy (Figure 1d). These fibers, like all other amyloids (Rambaran and Serpell 2008), present a β-sheet backbone of the fiber, which is surrounded by the rest of the unstructured polypeptide chains, that constitute a fuzzy coat for the fibers (Fitzpatrick et al. 2017).
5. Tat - Aβ interaction
Tat can affect Aβ genesis and deposition through several mechanisms. It has been suggested that differences between HIV and AD in the patterns of Aβ and Tau biomarkers from cerebrospinal fluid of patients indicate that HIV-associated neurocognitive disorders and AD may not share some of the same mechanisms of neuronal injury (de Almeida et al. 2018). One needs to consider the complexity of the HIV-infected brain environment. Tat derived from HIV clade B was shown to have synergistic effect in the presence of the HIV envelope protein, gp120by inducing neuronal cell death at subtoxic concentrations of both proteins (Nath et al. 2000). Importantly, Tat and gp120 promote the secretion of Aβ 1–42 in primary rat fetal hippocampal cell cultures (Aksenov et al. 2010) and the Tat from HIV clade B specifically induces the release of Aβ 1–42 and the accumulation of cell bound Aβ aggregates (Aksenov et al. 2010).
To better understand the impact of Tat protein on Aβ pathology, one has to consider the locations at which these interactions may occur. We present a step by step approach to discuss how these interactions occur including interactions with the cellular structures where these interactions take place.
5.1. In the extracellular space.
Once released in the extracellular space, Tat can directly interact with the Aβ molecules and deposits present there. We found that the Tat-Aβ complex occurs in vitro and in vivo and is more neurotoxic than Aβ alone (Hategan et al. 2017). In the presence of Tat, Aβ fibrils bind to each other, forming double twisted fibrils. At higher concentrations they form populations of thick unstructured filaments and aggregates. Tat binds to the exterior surfaces of the Aβ fibrils and increases β-sheet formation and lateral aggregation of the fibrils resulting in thick multifibrillar structures. These fibers with increased rigidity and mechanical resistance, which, coupled with stronger adhesion induced by the presence of Tat in the fibrils may account for increased damage, potentially through pore formation in membranes (Hategan et al. 2017). Direct interaction between Aβ and Tat can occur with Tat molecule or with Tat aggregates (Figure 2a,b). Tat has propensity for self-aggregation (Hategan et al. 2017, Zeitler et al. 2015, Li et al. 2009) however, this does not interfere with its capacity to bind Aβ, even in an aggregated state.
Figure 2. Tat - Aβ direct interaction.
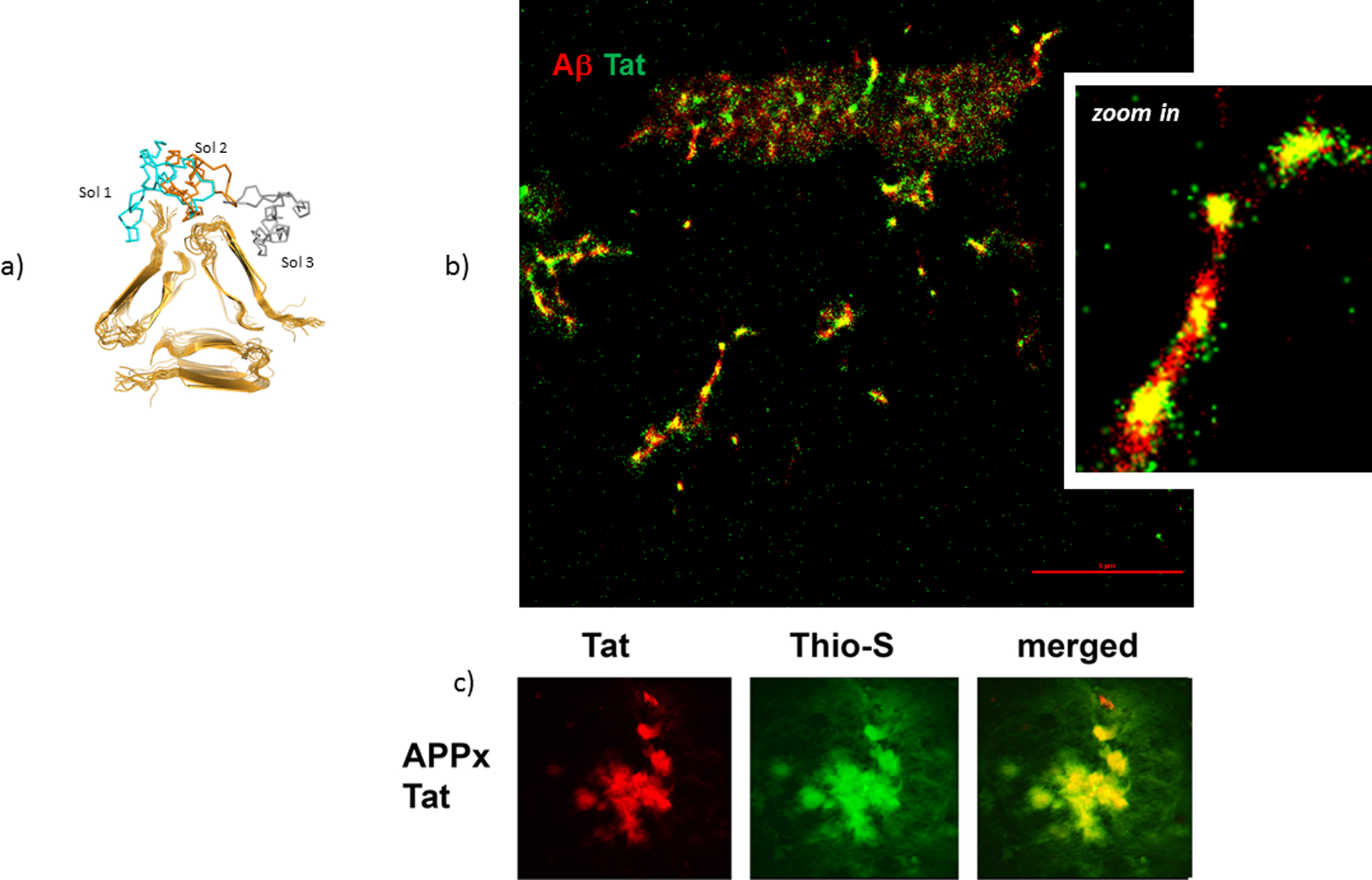
(a) The binding site of Tat to the Aβ 1–40 fibril involves an interaction between a negatively charged region from the terminal regions and the hairpin turn of the Aβ molecule on one side, the positively charged residues of Tat mainly from the basic region on the other side (Hategan et al. 2017). Shown in blue, orange and grey are the first 3 positions of Tat binding to Aβ. (b) STORM super resolution image of large Tat aggregates bound to Aβ 1–40 structures, showing that Tat aggregates bind to Aβ in vitro. (c) In double transgenic APP/Tat mice brains, colocalization of Aβ plaques with Tat deposits is seen. The diameter of Aβ plaques is about 20 μm in the confocal images (900x magnification). Aβ plaques were identified with Thioflavin S and Tat by using a fluorescent Tat antibody.
When the Tat transgenic mice were cross-bred with the PSAPP mouse model of AD, these mice showed significantly more Aβ deposition, neurodegeneration, neuronal apoptotic signaling and phosphorylated Tau when compared to PSAPP mice. This shows that Tat contributes to AD-like pathology (Giunta et al. 2009). In another study, where we cross-bred the Tat transgenic mice (Fields et al. 2015) with the mThy1-APP line 41 transgenic mice (Rockenstein et al. 2001), we show that Aβ deposition colocalizes with Tat protein in the mice brains (Figure 2c). This shows that Tat directly interacts/contributes in vivo with the Aβ deposits.
Both Tat (Rahimian et al. 2016) and Aβ (Pulliam et al. 2019) were found to be carried by exosomes, however, it remains to be determined if Tat and Aβ can be found in the same exosomes. Exosomes containing Nef have been isolated from plasma of individuals with HIV-associated dementia and shown to induce Ab 1–42 secretion in SY5Y neural cells (Khan et al. 2016). Similar studies with exosomes containing Tat have yet to be performed.
5.2. At the cellular level.
Once secreted by the HIV-infected cells, Tat can reach adjacent cells. It can bind to their cellular membranes, via a multitude of structures. Some of the transmembrane proteins from the plasma membrane have large extracellular domains that can easily interact with Tat. Some of these interactions affect Aβ production and processing. In cell-based studies, Tat has been shown to increase the extracellular levels of Aβ (Aksenov et al. 2010, Giunta et al. 2008, Rempel and Pulliam, 2005).
5.2.1. At the plasma membrane.
Interaction with APP.
Tat can interact directly with APP and stimulate Aβ production intracellularly (Kim et al. 2013). It interacts with APP both in vitro and in vivo and increases the level of Aβ 1–42 by recruiting APP into lipid rafts. Tat colocalizes with APP in the cytosol in cells that express high levels of Tat. In the presence of Tat, APP gets redistributed into lipid rafts, a site of increased β- and γ-secretase activity. In vitro, Tat enhanced the cleavage of APP by β-secretase, resulting in 5.5-fold higher levels of Aβ 1–42. Stereotaxic injection of a lentiviral Tat expression construct into the hippocampus of APP/presenilin-1 (PS1) transgenic mice resulted in increased production and processing of Aβ with increased levels of Aβ 1–42, and an increase in the number and size of Aβ plaques (Kim et a. 2013).
Interaction with neprilysin.
Neprilysin is a transmembrane zinc metalloprotease which consists of three domains: a short, intracellular 27 aa domain, a short 22 aa in-membrane domain and a large 699 aa extracellular domain (Moss et al. 2018). The extracellular domain contains a large central cavity which presents a conserved zinc binding motif (Moss et al. 2018). Neprilysin can degrade several biologically active peptides, including insulin, enkephalin, substance P, endothelin-1, neurotensin and Aβ (Moss et al. 2018). But importantly, neprilysin is the dominant Aβ–degrading enzyme in the brain (Iwata et al. 2001). Neprilysin cleaves both Aβ 1–40 and Aβ 1–42 peptides (Shirotani et al. 2001). In an in vitro assay, Tat inhibited neprilysin by 80% and Tat added directly to brain cultures showed an increase in soluble Aβ (Rempel and Pulliam 2005). The cysteine rich domain of Tat was essential for neprilysin inhibition (Rempel and Pulliam 2005, Pulliam 2009). Neprilysin can cleave Tat into small peptides, initiating a positive feedback mechanism for inhibition of neprilysin. (Daily et al. 2006). The mechanism of neprilysin inhibition involves: chelation of zinc at the active site of neprilysin, binding of Tat to the active site of neprilysin with the cysteine residues of Tat forming a tight complex with the zinc or by formation of a covalent dimer by reaction of the Tat cysteines with the cysteine of neprilysin (Nath and Hersh 2005). This would convert the homodimeric neprilysin into either a neprilysin-Tat structure or produce monomeric neprilysin via a neprilysin-Tat intermediate (Nath and Hersh 2005).
The neprilysin dysfunction induced by Tat was observed in neurons, (Rempel and Pulliam 2005), in cerebral microvascular endothelial cells (Jiang et al. 2017), and in astrocytes (Martinez-Bonet et al. 2018). Astrocytes are the most numerous cells in the brain, therefore a small change in astrocytic Aβ metabolism could make a significant contribution to brain pathology. HIV infected astrocytes showed more extracellular Aβ compared to controls, whereas the intracellular Aβ deposits were unchanged, processes accompanied by reduced expression of neprilysin and significant reduction of neprilysin activity (Martinez-Bonet et al. 2018).
Interaction with the low-density lipoprotein receptor-related protein (LRP).
LRP is a large endocytic and signaling receptor that is widely expressed in tissues, including neuronal cells. It is synthesized as a 600 kDa protein and is cleaved by furin into a light chain that consists of an 85 kDa subunit containing the transmembrane and intracellular domain and a non-covalently bound 515 kDa amino-terminal fragment. The extracellular domain contains four clusters to which ligands bind. Tat binds to LRP specifically at domains III and IV and to lesser extent to domain II, by its core domain (aa 37 to 48) (Liu et al. 2000). This binding promotes efficient uptake of Tat into neurons (Liu et al. 2000). Tat initially binds heparan sulfate proteoglycans, which are widely expressed on the cell surface (Tyagi et al. 2001) which is followed by LRP-mediated endocytosis (Liu et al. 2000). Tat binding to LRP results in significant inhibition of binding, uptake and degradation of its physiological ligands: alpha2-macroglobulin, apolipoprotein E4, and importantly APP and Aβ (Liu et al. 2000).
5.2.2. Inside cells.
Processes at the endolysosome.
The endocytic delivery of macromolecules from the cell surface for degradation by lysosomal acid hydrolases requires their traffic through early endosomes to late endosomes followed by transient or complete fusions between late endosomes and lysosomes. The fusion of these vesicles results in the formation of endolysosomes, from which eventually lysosomes are reformed (Bright et al. 2016). It is unclear which is the predominant process by which Tat is internalized into neurons. Once Tat reaches the cytosol, it can react with a multitude of structures. Most of the Tat (90%) will be finally found in the nucleus (Liu et al. 2000). From the membrane, Tat may directly reach the endolysosome through an endocytic pathway. Tat also gets internalized in T cells: it is found in early endosomes after 3 hours and in late endosomes after 6 hours and can result in T cell activation (Vendeville et al. 2004, Johnson et al. 2013). Tat enlarges endolysosomes, elevates endolysosome pH, and disturbs endolysosome function in neurons (Hui, et al. 2012). This may alter Aβ metabolism, since endolysosome dysfunction is one of the earliest pathological features of AD which precedes detectable extracellular deposition of Aβ in brain (Cataldo et al. 2000). This is also consistent with the observation that there is increased Aβ intracellularly in neurons of patients with HIV encephalitis particularly in autophagosomes suggesting that the clearance of Aβ is impaired (Achim et al. 2009). Tat-induced changes in endolysosome function in neurons preceed Tat-induced increases in Aβ extracellular levels (Chen et al. 2013). Tat also increases endolysosome accumulation of APP and Aβ, as well as of Aβ converting enzyme (BACE-1) and enhances BACE-1 activity. Together, these findings suggest that Tat increases neuronal Aβ extracellular levels and thereby contributes to the development of AD-like pathology in HIV-infected individuals by disturbing endolysosome structure and function. Further, due to the Tat-induced disfunction of endolysosomes, Tat may be released from endolysosomes into the cytoplasm (Chen et al. 2013). The mechanism by which Tat escapes endolysosomes is not entirely clear. Membrane integrity of endolysosomes can be disrupted by Tat (Hui et al. 2012). Endolysosomes have low pH. At this pH, Tat inserts itself into lipid membranes by a Trp 11 dependent mechanism (Yezid et al. 2009). Tat also directly penetrates lipid membranes by inducing pore formation (Zeitler et al. 2015, Brooks et al. 2005). Tat peptide 47–57 was shown to escape the endolysosome into the cytosol by using the high transmembrane proton gradient (Potocky et al. 2003). Interestingly, caffeine blocks Tat-induced endolysosome dysfunction and also neuronal Aβ production (Soliman et al. 2017).
5.2.3. Tat inhibits microglial phagocytosis of Aβ.
Microglia, which are resident brain macrophages, have a critical role in Aβ plaque clearance (Rogers and Lue, 2001, Rogers et al. 2002). Tat inhibits the uptake of Aβ by microglial cells, suggesting that Tat also regulates the extracellular levels of Aβ by inhibiting microglial phagocytosis (Giunta et al. 2008). Additionally, Tat disrupts apolipoprotein-3 promoted microglial Aβ uptake, which suggests that, similarly to neurons, microglial LRP may be a site of Tat-binding (Giunta et al. 2008).
5.2.4. Tat interacts with cerebral endothelial cells and affects Aβ clearance from the brain.
The blood brain barrier (BBB) prevents the unregulated exchange of substances between brain and blood. A major component of BBB are the brain microvascular endothelial cells joined by tight junctions (Andras and Toborek 2013). A balance between the lipoprotein receptor-related protein (LRP1), which transports Aβ from the brain into the blood (Jaeger et al. 2009) and the receptor for advanced glycation end products (RAGE), which transports Aβ into the brain (Andras et al. 2010) has been proposed to regulate Aβ levels (Deane et al. 2004). In cerebral endothelial cells, Tat causes a decrease in expression of tight junction proteins zonula occludens 1 (ZO-1) (Pu et al. 2005, Chen et al. 2016, Jiang et al. 2017), claudin-1 (Andras et al. 2003) claudin-5 (Andras et al. 2005, Andras et al. 2011), and ZO-2 expression (Andras et al. 2003), leading to increased permeability of the BBB. Conversely Tat also induces p-glycoprotein expression in brain microvascular endothelial cells (Hayashi et al 2005), and upregulates expression of multidrug resistance protein 1 (Hayashi et al. 2006). Of these, the last two proteins are known to be involved in Aβ translocation across the BBB (Andras and Toborek 2013). Tat also decreases LRP expression while increasing the expression of RAGE in endothelial cells (Chen et al. 2016). This is consistent with observations that transendothelial transfer of Aβ and intracellular reactive oxygen species were also increased by Tat (Jiang et al. 2017). Aβ and Tat can synergistically potentiate the expression of inflammatory genes in human brain microvascular endothelial cells (Andras et al. 2008). When Tat was injected into cerebral vasculature of mice with amyloid deposits, Tat induced enhanced cerebrovascular toxicity, as evidenced by permeability across cerebral capillaries, enhanced disruption of ZO-1 protein, and elevated levels of matrix metalloproteinase 9 (Chen et al. 2012). These studies show that Tat has the capacity to dysregulate the expression of various proteins with important functions in the BBB, thus affecting the clearance of Aβ from the brain.
6. Tau phosphorylation induced by Tat
Tau, a microtubule associated protein, is involved in stabilization of the neuronal cytoskeleton and ensures vesicular and protein transport (Kadri et al. 2015). Tau has differential ability to polymerize and stabilize microtubules, which depends on the availability of different Tau isoforms, produced as a result of alternative splicing. The ratio of Tau with four binding domains (generated by exon 10) versus Tau with three binding domains (generated by exons 2 and 3) is relevant to pathology (Kadri et al. 2015). Tat induces Tau protein phosphorylation in Tat transgenic mice (Giunta et al. 2009). Multiple mechanisms have been proposed by which Tat causes Tau phosphorylation. In neuronal cell cultures and in Tat transgenic mice Tat causes phosphorylation and therefore alters the structure of SC35 protein nuclear speckle domains. This affects the Tau 10 exon leading to altered ratios of Tau protein with three to four binding domains (Kadri et al. 2015). Tat can also bind directly to Tau RNA (Kadri et al. 2015). Tat nuclear interactions lead to gene alteration and Tau protein is one of the affected products.
Tat, via calcium dysregulation promotes calpain-1 cleavage of p35 to p25, which in turn hyperactivates cyclin dependent kinase CDK5 resulting in abnormal phosphorylation of Tau and other downstream targets (Fields et al. 2015). Additionally, Tat interferes with the trafficking of CDK5 between the nucleus and the cytoplasm, leading to its prolonged presence in the cytoplasm which leads to accumulation of aberrantly phosphorylated cytoplasmic targets including Tau protein (Fields et al. 2015). It is unknown if Tat interacts directly with Tau.
In a human neuroblastoma cell line overexpressing wild-type APP, Tat significantly increased secreted and intracellular levels of Aβ as well as cellular protein levels of phosphorylated Tau (Soliman et al. 2017). Caffeine significantly decreased not only Aβ production but also Tau phosphorylation (Soliman et al. 2017).
7. Model of interaction of Tat with Aβ and Tau
Multiple interactions of Tat with neurons, astrocytes, microglia and brain endothelial cells lead the growth of Aβ deposits and increased neurofibrillary tangles which both ultimately lead to neurodegeneration.
Tat protein is expressed in HIV-infected macrophages/microglia (Nath and Steiner 2014, Mattson et al. 2005) and released into the extracellular space, where it can interact directly with Aβ. The resultant Aβ-Tat complex is more neurotoxic than Aβ alone (Hategan et al. 2017). Tat molecules that do not bind to extracellular structures, reach the cell membrane of nearby cells, where they interact with a multitude of structures (Li et al. 2009). First might be the heparan sulfate proteoglycans that are abundant on the cell surface (Tyagi et al. 2001). From this point further Tat can interact with the membrane lipids and penetrate the bilayer (Zeitler et al. 2015, Brooks et al. 2005) or bind to a multitude of proteins on the cell surface (Li et al 2009). Interaction with LRP transports Tat inside the cell and the mechanism of uptake involves LRP interaction with the heparan sulfate proteoglycans (Liu et al. 2000, Tyagi et al. 2001). The transmembrane uptake of Tat seems to be mediated through calveolar endocytosis (Fittipaldi et al. 2003, Ferrari et al. 2003) and clathrin-dependent endocytosis (Venderville et al. 2004). Tat membrane penetration through lipids was observed at 1–10 μM Tat concentrations (Zeitler et al. 2015) and takes minutes (Zeitler et al. 2015, Brooks et al. 2005). Tat can reach the endolysosome (Chen at al. 2013), from where it may further reach the cytosol and finally accumulate into the nucleus (Liu et al. 2000). Tat also modifies LRP interaction with its ligands, including APP (Liu et al. 2000). Tat interaction with the neprilysin’s large extracellular domain inhibits neprilysin’s capacity to cleave Aβ (Rempel and Puliam 2005, Daily et al. 2006, Pulliam 2009), leading to accumulation of extracellular Aβ deposits. Tat can interact directly with APP on the cell membrane and recruit it in lipid rafts (Kim et al. 2013). APP is present for a short time on the surface of the membrane, likely that is why only a small amount of Aβ is produced there (O’Brien and Wong 2011). Once recruited in lipid rafts, APP is transported to the endolysosome, where the majority of the Aβ is produced (O’Brien and Wong 2011). Tat can increase Aβ production at the endolysosome level (Kim et al. 2013, Chen et al. 2013). Tat also inhibits microglial phagocytosis of Aβ (Giunta et al. 2008). Lastly, by interaction with the endothelial cells within the BBB, Tat affects the barrier properties and the clearance of Aβ deposits in the brain is impeded (Chen et al. 2016, Jiang et al. 2017). Tat induces also Tau phosphorylation in neurons (Giunta et al. 2009) through a cascade of cellular processes involving CDK5 (Fields et al. 2015) and the endolysosome (Soliman et al. 2017). Tau phosphorylation leads to formation of neurofibrillary tangles (Alonso et al 2001).
In conclusion, Tat, a small, unstructured, positively charged, opportunistic protein has multiple interaction partners. To date it is unknown which of these interactions are predominant at the cellular level, however the nucleus is its main final target. Tat and its aggregates influence the production of Aβ, inhibit its degradation and directly interact with it. Tat also influences the phosphorylation of Tau, and thus contributes to pathological hallmarks in the HIV-infected brain. These Tat interactions lead to increased neurotoxicity.
Future studies should determine the relevance of Tat-Aβ in vivo by detection and quantification of these complexes in brain and CSF of HIV-infected patients and determine the degree to which these may corelate with markers of neuronal injury or inflammation. Development of therapeutic approaches to block Tat-Aβ interaction, or Tat activity by itself needs to be considered.
Figure 3. Model ofinteraction of Tat with Aβ and Tau.
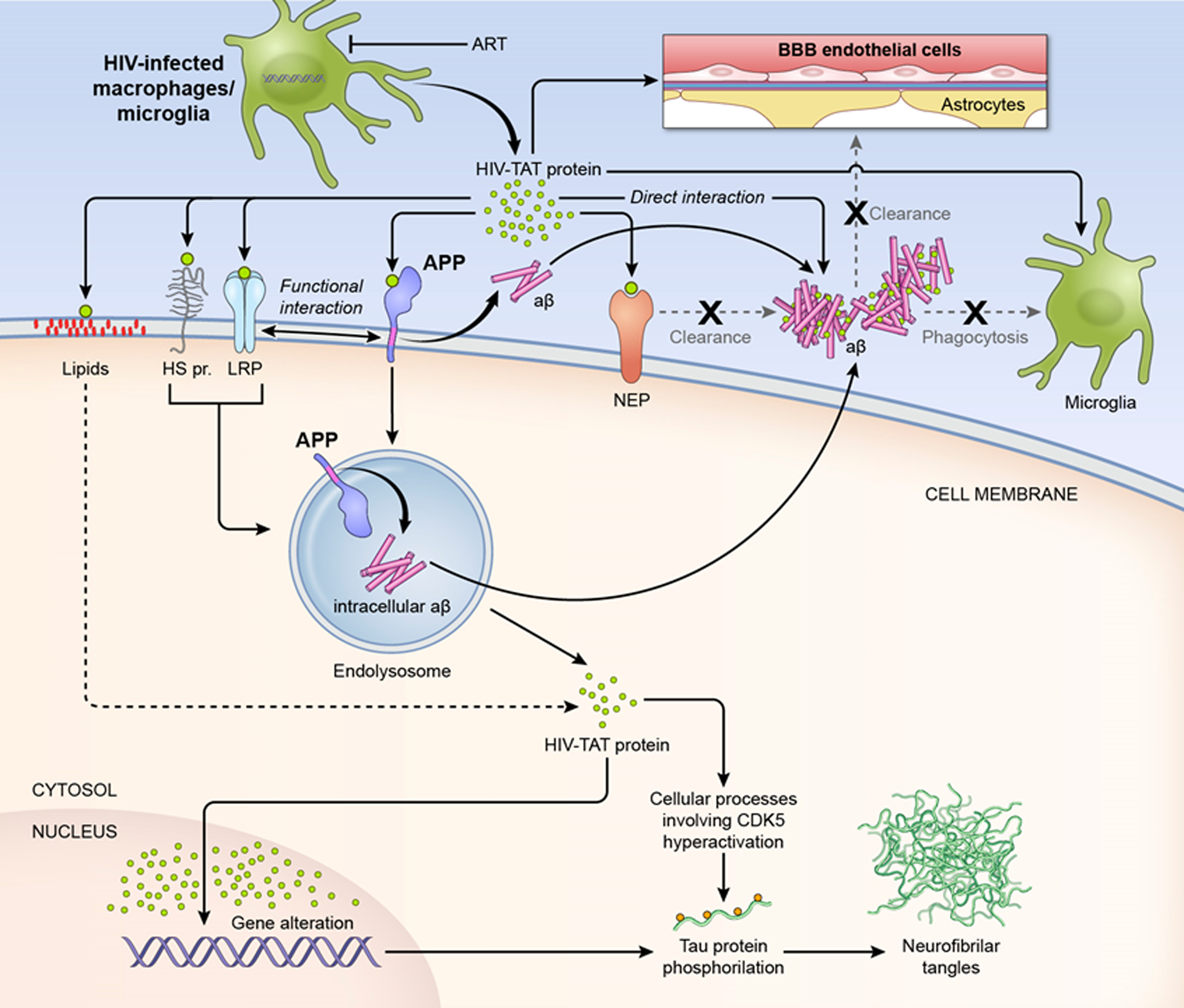
Tat protein is expressed in HIV-infected macrophages/microglia (Nath 2014, Mattson et al. 2005) and released into the extracellular space, even under antiretroviral therapy (AT). Here it can interact directly with Aβ. The resultant Aβ - Tat complex is more neurotoxic then Aβ alone (Hategan et al. 2017). Tat that does not bind to extracellular structures, can reach the cell membranes of nearby cells, where it can interact with a multitude of structures. It may interact with the heparan sulfate proteoglycans (HS pr) that are abundant on the cell surface (Tyagi et al. 2001). Interaction with LRP transports Tat inside the cell and the mechanism of uptake involves LRP complexing with HS pr (Liu et al. 2000). The transmembrane uptake of Tat is mediated through calveolar endocytosis (Fittipaldi et al. 2003) and clathrin-dependent endocytosis (Venderville et al. 2004), depending on the cell type. Tat can reach the endolysosome (Chen at al. 2013), from which it may be released into the cytosol and finally accumulate in the nucleus (Liu 2000). Tat also modifies LRP interaction with its ligands, including APP (Liu et al. 2000). Tat can interact directly with the lipids and translocate across the bilayer through pore formation (Zeitler et al. 2015) on a time scale of minutes (Zeitler et al. 2015, Brooks et al. 2005). Tat’s interaction with the large extracellular domain of neprilysin (NEP) inhibits NEP’s capacity to cleave Aβ (Rempel and Puliam 2005), therefore the extracellular Aβ deposits will grow. Tat can interact directly with amyloid precursor protein (APP) on the cell membrane and recruit it to the lipid rafts (Kim et al. 2013). APP is present for a short time on the surface of the membrane, likely that is why only a small amount of Aβ is produced there (O’Brien and Wong 2011). Once recruited in lipid rafts, APP is transported to the endolysosome, where the majority of Aβ is produced (O’Brien and Wong 2011). Tat can increase Aβ production at the endolysosome level (Kim et al. 2013, Chen et al. 2013). Tat also inhibits microglial phagocytosis of Aβ (Giunta et al. 2008). Lastly, by interaction with the endothelial cells within the blood brain barrier, Tat impedes the clearance of Aβ deposits from the brain (Chen et al. 2016, Jiang et al. 2017). Tat also induces Tau phosphorylation in neurons (Giunta et al. 2009) through a cascade of cellular processes involving CDK5 (Fields et al. 2015) and the endolysosome (Soliman et al. 2017). Tau phosphorylation leads to neurofibril tangles formation (Alonso et al 2001), another hallmark of AD.
References
- Achim CL, Adame A, Dumaop W, Everall IP, Masliah E; Neurobehavioral Research Center (2009). Increased accumulation of intraneuronal amyloid beta in HIV-infected patients. J Neuroimmune Pharmacol 4(2):190–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, Elliott JI, Van Nostrand WE, Smith SO (2010). Structural conversion of neurotoxic amyloid-beta (1–42) oligomers to fibrils. Nat Struct Mol Biol 17(5):561–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K (2001). Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A 98(12):6923–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida SM, Ribeiro CE, Rotta I, Piovesan M, Tang B, Vaida F, Raboni SM, Letendre S, Potter M, Batistela Fernandes MS, Ellis RJ; HIV Neurobehavioral Research Center (HNRC) Group (2018). Biomarkers of neuronal injury and amyloid metabolism in the cerebrospinal fluid of patients infected with HIV-1 subtypes B and C. J Neurovirol 24(1):28–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksenov MY, Aksenova MV, Mactutus CF, Booze RM (2009). Attenuated neurotoxicity of the transactivation-defective HIV-1 Tat protein in hippocampal cell cultures. Exp Neurol 219(2): 586–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksenov MY, Aksenova MV, Mactutus CF, Booze RM (2010). HIV-1 protein-mediated amyloidogenesis in rat hippocampal cell cultures. Neurosci Lett 475(3):174–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- András IE, Pu H, Deli MA, Nath A, Hennig B, Toborek M (2003). HIV-1 Tat protein alters tight junction protein expression and distribution in cultured brain endothelial cells. J Neurosci Res 74(2):255–65. [DOI] [PubMed] [Google Scholar]
- András IE, Pu H, Tian J, Deli MA András IE, Pu H, Tian J, Deli MA, Nath A, Hennig B, Toborek M (2005). Signaling mechanisms of HIV-1 Tat-induced alterations of claudin-5 expression in brain endothelial cells. J Cereb Blood Flow Metab 25(9):1159–70. [DOI] [PubMed] [Google Scholar]
- András IE, Rha G, Huang W, Eum S, Couraud PO, Romero IA, Hennig B, Toborek M (2008). Simvastatin protects against amyloid beta and HIV-1 Tat-induced promoter activities of inflammatory genes in brain endothelial cells. Mol Pharmacol 73(5):1424–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- András IE, Eum SY, Huang W, Zhong Y, Hennig B, Toborek M (2010). HIV-1-induced amyloid beta accumulation in brain endothelial cells is attenuated by simvastatin. Mol Cell Neurosci 43(2):232–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- András IE, Toborek Michal (2011). HIV-1-Induced Alterations of Claudin-5 Expression at the Blood–Brain Barrier Level. Methods Mol Biol 762: 355–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- András IE, Toborek M (2013). Amyloid beta accumulation in HIV-1-infected brain: The role of the blood brain barrier. IUBMB Life 65(1):43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avraham HK, Jiang S, Lee TH, Prakash O, Avraham S (2004). HIV-1 Tat-mediated effects on focal adhesion assembly and permeability in brain microvascular endothelial cells. J Immunol 173:6228–6233. [DOI] [PubMed] [Google Scholar]
- Bagashev A, Sawaya BE (2013). Roles and functions of HIV-1 Tat protein in the CNS: an overview. Virol. J 10:358–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball KA, Wemmer DE, Head-Gordon T (2014). Comparison of structure determination methods for intrinsically disordered amyloid-β peptides. J. Phys. Chem. B 118:6405–6416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer TA, Cappai R, Masters CL, Beyreuther K, Multhaup G (1999). It all sticks together--the APP-related family of proteins and Alzheimer’s disease. Mol Psychiatry 4(6):524–8. [DOI] [PubMed] [Google Scholar]
- Becker JT, Lopez OL, Dew MA, Aizenstein HJ (2004). Prevalence of cognitive disorders differs as a function of age in HIV virus infection. AIDS 18:S11–S18. [PubMed] [Google Scholar]
- Ben-Dov N, Korenstein R (2015). The uptake of HIV Tat peptide proceeds via two pathways which differ from macropinocytosis. Biochim Biophys Acta 1848(3):869–77. [DOI] [PubMed] [Google Scholar]
- Bright NA, Davis LJ, Luzio JP (2016). Endolysosomes Are the Principal Intracellular Sites of Acid Hydrolase Activity. Curr Biol 26(17):2233–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks H, Lebleu B, Vivès E (2005). Tat peptide-mediated cellular delivery: back to basics. Adv Drug Deliv Rev 57(4):559–77. [DOI] [PubMed] [Google Scholar]
- Bu XL, Xiang Y, Jin WS, Wang J, Shen LL, Huang ZL, Zhang K, Liu YH, Zeng F, Liu JH, Sun HL, Zhuang ZQ, Chen SH, Yao XQ, Giunta B, Shan YC, Tan J, Chen XW, Dong ZF, Zhou HD, Zhou XF, Song W, Wang YJ (2018). Blood-derived amyloid-β protein induces Alzheimer’s disease pathologies. Mol Psychiatry 9:1–9. [Google Scholar]
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA (2000). Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’ s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol 157(1):277–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Choi JJ, Choi YJ, Hennig B, Toborek M (2012). HIV-1 Tat-induced cerebrovascular toxicity is enhanced in mice with amyloid deposits. Neurobiol Aging 33(8):1579–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Hui L, Geiger NH, Haughey NJ, Geiger JD (2013). Endolysosome involvement in HIV-1 transactivator protein-induced neuronal amyloid beta production. Neurobiol Aging 34(10):2370–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Huang W, Jiang W, Wu X, Ye B, Zhou X (2016). HIV-1 Tat regulates occludin and Aβ transfer receptor expression in brain endothelial cells via Rho/ROCK signaling pathway. Oxid. Med. Cell. Longev 2016:4196572. [Google Scholar]
- Chiti F, Webster P, Taddei N, Clark A, Stefani M, Ramponi G, Dobson CM (1999). Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc Natl Acad Sci USA 96(7):3590–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin MT, Silvers R, Ni QZ, Can TV, Sergeyev I, Rosay M, Donovan KJ, Michael B, Wall J, Linse S, Griffin RG (2016). Atomic Resolution Structure of Monomorphic Aβ42 Amyloid Fibrils. J Am Chem Soc 138(30):9663–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conchillo-Solé O, de Groot NS, Avilés FX, Vendrell J, Daura X, Ventura S (2007). AGGRESCAN: a server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinformatics 2007, 8:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daily A, Nath A, Hersh L (2006). Tat peptides inhibit neprilysin. J. Neurovirol 12:153 – 160. [DOI] [PubMed] [Google Scholar]
- Deane R, Wu Z, Zlokovic BV (2004). RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke 35(11 Suppl 1):2628–31. [DOI] [PubMed] [Google Scholar]
- Debaisieux S, Rayne F, Yezid H, Beaumelle B (2012). The ins and outs of HIV-1 Tat. Traffic 13:355–363. [DOI] [PubMed] [Google Scholar]
- Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Aussio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, and Obradovic Z (2001). Intrinsically disordered protein. J. Mol. Graphics and Modeling 19:26–59. [Google Scholar]
- Ehehalt R, Keller P, Haass C, Thiele C, Simons K (2003). Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol 160:113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esiri MM, Biddolph SC, Morris CS (1998). Prevalence of Alzheimer plaques in AIDS. J. Neurol. Neurosurg. Psychiatry 65:29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everall IP, Heaton RK, Marcotte TD, Ellis RJ, McCutchan JA, Atkinson JH, Grant I, Mallory M and Masliah E (1999). Cortical synaptic density is reduced in mild to moderate human immunodeficiency virus neurocognitive disorder. HNRC Group. HIV Neurobehavioral Research Center. Brain Pathol 9: 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari A, Pellegrini V, Arcangeli C, Fittipaldi A, Giacca M, Beltram F (2003). Caveolae-mediated internalization of extracellular HIV-1 tat fusion proteins visualized in real time. Mol Ther 8(2):284–94. [DOI] [PubMed] [Google Scholar]
- Ferrell D, Giunta B (2014). The impact of HIV-1 on neurogenesis: implications for HAND. Cell Mol Life Sci 71(22):4387–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields J, Dumaop W, Eleuteri S, Campos S, Serger E, Trejo M, Kosberg K, Adame A, Spencer B, Rockenstein E, He JJ, Masliah E (2015). HIV-1 Tat alters neuronal autophagy by modulating autophagosome fusion to the lysosome: implications for HIV-associated neurocognitive disorders. J Neurosci 35(5):1921–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields JA, Dumaop W, Crews L, Adame A, Spencer B, Metcalf J, He J, Rockenstein E, Masliah E (2015). Mechanisms of HIV-1 Tat neurotoxicity via CDK5 translocation and hyper-activation: role in HIV-associated neurocognitive disorders. Curr HIV Res 13(1):43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fittipaldi A, Ferrari A, Zoppé M, Arcangeli C, Pellegrini V, Beltram F, Giacca M. (2003). Cell membrane lipid rafts mediate caveolar endocytosis of HIV-1 Tat fusion proteins. J Biol Chem 278(36):34141–9. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, Scheres SHW (2017). Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547(7662):185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel AD, Pabo CO (1988). Cellular uptake of the tat protein from human immunodeficiency virus. Cell 55(6):1189–93. [DOI] [PubMed] [Google Scholar]
- Fraser PE, Nguyen JT, Inouye H, Surewicz WK, Selkoe DJ, Podlisny MB, Kirschner DA (1992). Fibril formation by primate, rodent, and Dutch-hemorrhagic analogues of Alzheimer amyloid beta-protein. Biochemistry 31(44):10716–23. [DOI] [PubMed] [Google Scholar]
- Garner E, Romero P, Dunker AK, Brown C, and Obradovic Z (1999). Predicting binding regions within disordered proteins. Genome Informatics 10:41–50. [PubMed] [Google Scholar]
- Green DA, Masliah E, Vinters HV, Beizai P, Moore DJ, Achim CL (2005). Brain deposition of β-amyloid is a common pathologic feature in HIV positive patients. AIDS 19:407–411. [DOI] [PubMed] [Google Scholar]
- Giunta B, Zhou Y, Hou H, Rrapo E, Fernandez F, et al. (2008) HIV-1 Tat Inhibits Microglial Phagocytosis of Abeta Peptide. Int J Clin Exp Pathol 1: 260–275. [PMC free article] [PubMed] [Google Scholar]
- Giunta B, Hou H, Zhu Y, Rrapo E, Tian J, et al. (2009) HIV-1 Tat Contributes to Alzheimer’s Disease-like Pathology in PSAPP Mice. Int J Clin Exp Pathol 2:433–443. [PMC free article] [PubMed] [Google Scholar]
- Giunta B, Ehrhart J, Obregon DF, Lam L, Le L, Jin J, Fernandez F, Tan J, Shytle RD (2011). Antiretroviral medications disrupt microglial phagocytosis of β-amyloid and increase its production by neurons: implications for HIV-associated neurocognitive disorders. Mol Brain 4(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot NS, Castillo V, Graña-Montes R, Ventura S (2012) AGGRESCAN: Method, Application, and Perspectives for Drug Design. In: Baron R (eds) Computational Drug Discovery and Design. Methods in Molecular Biology (Methods and Protocols), vol 819. Springer, New York, NY. [Google Scholar]
- Guo X, Kameoka M, Wei X, Roques B, Gotte M, Liang C, Wainberg MA (2003). Suppression of an intrinsic strand transfer activity of HIV-1 Tat protein by its second-exon sequences. Virology 307:154–163. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Pu H, Tian J, Andras IE, Lee YW, Hennig B, Toborek M (2005). HIV-Tat protein induces P-glycoprotein expression in brain microvascular endothelial cells. J Neurochem 93(5):1231–41. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Pu H, Andras IE, Eum SY, Yamauchi A, Hennig B, Toborek M (2006). HIV-TAT protein upregulates expression of multidrug resistance protein 1 in the blood-brain barrier. J Cereb Blood Flow Metab 26(8):1052–65. [DOI] [PubMed] [Google Scholar]
- Hayman M, Arbuthnott G, Harkiss G, Brace H, Filippi P, Philippon V, Thomson D, Vigne R and Wright A (1993). Neurotoxicity of peptide analogues of the transactivating protein tat from Maedi-Visna virus and human immunodeficiency virus. Neuroscience 53: 1–6. [DOI] [PubMed] [Google Scholar]
- Halverson K, Fraser PE, Kirschner DA, Lansbury PT Jr (1990). Molecular determinants of amyloid deposition in Alzheimer’s disease: conformational studies of synthetic beta-protein fragments. Biochemistry 29(11):2639–44. [DOI] [PubMed] [Google Scholar]
- Hategan A, Bianchet MA, Steiner J, Karnaukhova E, Masliah E, Fields A, Lee MH, Dickens AM, Haughey N, Dimitriadis EK, Nath A (2017). HIV Tat protein and amyloid-β peptide form multifibrillar structures that cause neurotoxicity. Nat Struct Mol Biol 24(4):379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, Collier AC, Marra CM, Morgello S, Mindt MR, Taylor MJ, Marcotte TD, Atkinson JH, Wolfson T, Gelman BB, McArthur JC, Simpson DM, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I; CHARTER Group; HNRC Group (2011). HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol 17(1):3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmuth J, Milanini B, Masliah E, Tartaglia MC, Dunlop MB, Moore DJ, Javandel S, DeVaughn S, Valcour V (2018). A neuropathologic diagnosis of Alzheimer’s disease in an older adult with HIV-associated neurocognitive disorder. Neurocase 4:213–219. [Google Scholar]
- Huang W, Chen L, Zhang B, Park M, Toborek M (2014). PPAR agonist-mediated protection against HIV Tat-induced cerebrovascular toxicity is enhanced in MMP-9-deficient mice. J Cereb Blood Flow Metab 34(4):646–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui L, Chen X, Haughey NJ, Geiger JD (2012). Role of endolysosomes in HIV-1 Tat-induced neurotoxicity. ASN Neuro 4(4):243–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Liu F, Gong CX, Grundke-Iqbal I (2010). Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res 7(8):656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, Gerard C, Hama E, Lee HJ, Saido TC (2001). Metabolic regulation of brain Abeta by neprilysin. Science 292(5521):1550–2. [DOI] [PubMed] [Google Scholar]
- Jaeger LB, Dohgu S, Hwang MC, Farr SA, Murphy MP, Fleegal-DeMotta MA, Lynch JL, Robinson SM, Niehoff ML, Johnson SN, Kumar VB, Banks WA (2009). Testing the neurovascular hypothesis of Alzheimer’s disease: LRP-1 antisense reduces blood-brain barrier clearance, increases brain levels of amyloid-beta protein, and impairs cognition. J Alzheimers Dis 17(3):553–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TP, Patel K, Johnson KR, Maric D, Calabresi PA, Hasbun R, Nath A (2013). Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proc Natl Acad Sci U S A 10(33):13588–93. [Google Scholar]
- Jeang KT, Xiao H, Rich EA (1999). Multifaceted activities of the HIV-1 transactivator of transcription, Tat. J. Biol. Chem 274:28837–28840. [DOI] [PubMed] [Google Scholar]
- Jiang W, Huang W, Chen Y, Zou M, Peng D, Chen D (2017). HIV-1 Transactivator Protein Induces ZO-1 and Neprilysin Dysfunction in Brain Endothelial Cells via the Ras Signaling Pathway. Oxid Med Cell Longev 2017:3160360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadri F, Pacifici M, Wilk A, Parker-Struckhoff A, Del Valle L, Hauser KF, Knapp PE, Parsons C, Jeansonne D, Lassak A, Peruzzi F (2015). HIV-1-Tat Protein Inhibits SC35-mediated Tau Exon 10 Inclusion through Up-regulation of DYRK1A Kinase. J Biol Chem 290(52):30931–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG (2003). Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science 300:486–489. [DOI] [PubMed] [Google Scholar]
- Khan MB, Lang MJ, Huang MB, Raymond A, Bond VC, Shiramizu B, Powell MD (2016). Nef exosomes isolated from the plasma of individuals with HIV-associated dementia (HAD) can induce Aβ (1–42) secretion in SH-SY5Y neural cells. J Neurovirol 2:179–90. [Google Scholar]
- Kim J, Yoon JH, Kim YS (2013). HIV-1 Tat interacts with and regulates the localization and processing of amyloid precursor protein. PLoS One 8(11): e77972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodali R, Williams AD, Chemuru S, Wetzel R (2010). Abeta(1–40) forms five distinct amyloid structures whose β-sheet contents and fibril stabilities are correlated. J. Mol. Biol 401:503–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K, Fischer P, Masters CL, Price DL (1990). Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci USA 87:1561–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansbury PT Jr (1999). Evolution of amyloid: what normal protein folding may tell us about fibrillogenesis and disease. Proc Natl Acad Sci U S A 96(7):3342–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Retamal C, Cuitino L, Caruano-Yzermans A, Shin JE, van Kerkhof P, Marzolo MP, Bu G (2008). Adaptor protein sorting nexin 17 regulates amyloid precursor protein trafficking and processing in the early endosomes. J Biol Chem 283:11501–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Hou H, Mori T, Sawmiller D, Smith A, Tian J, Wang Y, Giunta B, Sanberg PR, Zhang S, Tan J (2015). Swedish mutant APP-based BACE1 binding site peptide reduces APP β-cleavage and cerebral Aβ levels in Alzheimer’s mice. Sci Rep 5:11322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Li G, Steiner J, Nath A (2009). Role of Tat protein in HIV neuropathogenesis. Neurotox Res 16(3):205–20. [DOI] [PubMed] [Google Scholar]
- Liu Y, Jones M, Hingtgen CM, Bu G, Laribee N, Tanzi RE, Moir RD, Nath A, He JJ (2000). Uptake of HIV-1 tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat Med 6(12):1380–7. [DOI] [PubMed] [Google Scholar]
- Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, Tycko R (2013). Molecular structure of β-amyloid fibrils in Alzheimer’s disease brain tissue. Cell 2013 154(6):1257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M, Nath A (1997). Molecular determinants for cellular uptake of Tat protein of human immunodeficiency virus type 1 in brain cells. J Virol 71(3):2495–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Westland CE, Rockenstein EM, Abraham CR, Mallory M, Veinberg I, Sheldon E, Mucke L (1997). Amyloid precursor proteins protect neurons of transgenic mice against acute and chronic excitotoxic injuries in vivo. Neuroscience 78(1):135–46. [DOI] [PubMed] [Google Scholar]
- Maragos WF, Tillman P, Jones M, Bruce-Keller AJ, Roth S, Bell JE and Nath A (2003). Neuronal injury in hippocampus with human immunodeficiency virus transactivating protein, Tat. Neuroscience 117: 43–53. [DOI] [PubMed] [Google Scholar]
- Macchi S, Nifosì R, Signore G, Di Pietro S, Boccardi C, D’Autilia F, Beltram F, Cardarelli F (2017). Self-aggregation propensity of the Tat peptide revealed by UV-Vis, NMR and MD analyses. Phys Chem Chem Phys 19(35):23910–23914. [DOI] [PubMed] [Google Scholar]
- Martínez-Bonet M, Muñoz-Fernández MÁ, Álvarez S (2018). HIV-1 increases extracellular amyloid-beta levels through neprilysin regulation in primary cultures of human astrocytes. J Cell Physiol doi: 10.1002/jcp.26462. [DOI] [Google Scholar]
- Mattson MP, Haughey NJ, Nath A (2005). Cell death in HIV dementia. Cell Death Differ Suppl 1:893–904. [Google Scholar]
- Mishra A, Gordon VD, Yang L, Coridan R, Wong GC (2008). HIV TAT forms pores in membranes by inducing saddle-splay curvature: potential role of bidentate hydrogen bonding. Angew Chem Int Ed Engl 47(16):2986–9. [DOI] [PubMed] [Google Scholar]
- Moores B, Drolle E, Attwood SJ, Simons J, Leonenko Z (2011). Effect of surfaces on amyloid fibril formation. PLoS One 6: e25954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgello S, Jacobs M, Murray J, Byrd D, Neibart E, Mintz L, Meloni G, Chon C, Crary J (2018). Alzheimer’s disease neuropathology may not predict functional impairment in HIV: a report of two individuals. J Neurovirol 5:629–637. [Google Scholar]
- Moss S, Subramanian V, Acharya KR (2018). High resolution crystal structure of substrate-free human neprilysin. J Struct Biol 204(1):19–25. [DOI] [PubMed] [Google Scholar]
- Nath A, Psooy K, Martin C, Knudsen B, Magnuson DS, Haughey N, Geiger JD (1996). Identification of a human immunodeficiency virus type 1 Tat epitope that is neuroexcitatory and neurotoxic. J Virol 70(3):1475–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A, Haughey NJ, Jones M, Anderson C, Bell JE, Geiger JD (2000). Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: protection by memantine. Ann Neurol 47(2):186–94. [PubMed] [Google Scholar]
- Nath A and Hersh LB (2005). Tat and amyloid: multiple interactions. AIDS 19(2):203–4. [DOI] [PubMed] [Google Scholar]
- Nath A, Steiner J (2014). Synaptodendritic injury with HIV-Tat protein: What is the therapeutic target? Exp Neurol 251:112–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HD and Hall CK (2004). Molecular dynamics simulations of spontaneous fibril formation by random-coil peptides. Proc Natl Acad Sci USA 101(46):16180–16185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien RJ, Wong PC (2011). Amyloid precursor protein processing and Alzheimer’ s disease. Annu Rev Neurosci 34:185–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paravastu AK, Leapman RD, Yau WM, Tycko R (2008). Molecular structural basis for polymorphism in Alzheimer’s β-amyloid fibrils. Proc. Natl. Acad. Sci. USA 105:18349–18354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthsarathy V, McClean PL, Hölscher C, Taylor M, Tinker C, Jones G, Kolosov O, Salvati E, Gregori M, Masserini M, Allsop D (2013). A novel retro-inverso peptide inhibitor reduces amyloid deposition, oxidation and inflammation and stimulates neurogenesis in the APPswe/PS1ΔE9 mouse model of Alzheimer’s disease. PLoS One 8(1): e54769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK, Obradovic Z (2005). Optimizing long intrinsic disorder predictors with protein evolutionary information. J Bioinform Comput Biol 1:35–60. [Google Scholar]
- Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R (2005). Self-propagating, molecular-level polymorphism in Alzheimer’s β-amyloid fibrils. Science 307:262–265. [DOI] [PubMed] [Google Scholar]
- Potocky TB, Menon AK, Gellman SH (2003). Cytoplasmic and nuclear delivery of a TAT-derived peptide and a beta-peptide after endocytic uptake into HeLa cells. J Biol Chem 278(50):50188–94. [DOI] [PubMed] [Google Scholar]
- Pu H, Tian J, Andras IE, Hayashi K, Flora G, Hennig B, Toborek M (2005). HIV-1 Tat protein-induced alterations of ZO-1 expression are mediated by redox-regulated ERK 1/2 activation. J Cereb Blood Flow Metab 25(10):1325–35. [DOI] [PubMed] [Google Scholar]
- Pulliam L, Sun B, Rempel H, Martinez PM, Hoekman JD, Rao RJ, Frey WH 2nd, Hanson LR (2007). Intranasal tat alters gene expression in the mouse brain. J Neuroimmune Pharmacol 2(1):87–92. [DOI] [PubMed] [Google Scholar]
- Pulliam LJ (2009). HIV regulation of amyloid β production. J Neuroimmune Pharmacol 4:213–127. [DOI] [PubMed] [Google Scholar]
- Pulliam L, Sun B, Mustapic M, Chawla S, Kapogiannis D (2019). Plasma neuronal exosomes serve as biomarkers of cognitive impairment in HIV infection and Alzheimer’ s disease. J. Neurovirol doi: 10.1007/s13365-018-0695-4. [DOI] [Google Scholar]
- Rahimian P, He JJ. Exosome-associated release, uptake, and neurotoxicity of HIV-1 Tat protein (2016). J Neurovirol 6:774–788. [Google Scholar]
- Rambaran RN, Serpell LC (2008). Amyloid fibrils: abnormal protein assembly. Prion 2(3):112–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempel HC, Pulliam L (2005). HIV-1Tat inhibits neprilysin and elevates amyloid beta. AIDS 19(2):127–35. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Hansen LA, Mallory M, Trojanowski JQ, Galasko D, Masliah E (2001). Altered expression of the synuclein family mRNA in Lewy body and Alzheimer’s disease. Brain Res 914(1–2):48–56 [DOI] [PubMed] [Google Scholar]
- Rogers J, Lue LF (2001). Microglial chemotaxis, activation, and phagocytosis of amyloid beta-peptide as linked phenomena in Alzheimer’s disease. Neurochem Int 39(5–6):333–40. [DOI] [PubMed] [Google Scholar]
- Rogers J, Strohmeyer R, Kovelowski CJ, Li R (2002). Microglia and inflammatory mechanisms in the clearance of amyloid beta peptide. Glia 2002 Nov;40(2):260–9. [DOI] [PubMed] [Google Scholar]
- Rossner S, Lange-Dohna C, Zeitschel U, Perez-Polo JR (2005). Alzheimer’s disease beta-secretase BACE1 is not a neuron-specific enzyme. J Neurochem 92(2):226–34. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Hardy J (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8(6):595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpell LC (2000). Alzheimer’s amyloid fibrils: structure and assembly. Biochim Biophys Acta 1502(1):16–30. [DOI] [PubMed] [Google Scholar]
- Shirotani K, Tsubuki S, Iwata N, Takaki Y, Harigaya W, Maruyama K, Kiryu-Seo S, Kiyama H, Iwata H, Tomita T, Iwatsubo T, Saido TC (2001). Neprilysin degrades both amyloid beta peptides 1–40 and 1–42 most rapidly and efficiently among thiorphan- and phosphoramidon-sensitive endopeptidases. J Biol Chem 276(24):21895–901. [DOI] [PubMed] [Google Scholar]
- Shojania S, O’Neil JD (2006). HIV-1 Tat is a natively unfolded protein: the solution conformation and dynamics of reduced HIV-1 Tat-(1–72) by NMR spectroscopy. J. Biol Chem 281:8347–8356. [DOI] [PubMed] [Google Scholar]
- Shojania S, O’Neil JD (2010). Intrinsic disorder and function of the HIV-1 Tat protein. Protein Pept. Lett 17:999–1011. [DOI] [PubMed] [Google Scholar]
- Sisodia SS, Koo EH, Beyreuther K, Unterbeck A, Price DL (1990). Evidence that beta-amyloid protein in Alzheimer’s disease is not derived by normal processing. Science 248:492–495. [DOI] [PubMed] [Google Scholar]
- Soliman ML, Geiger JD, Chen X (2017). Caffeine Blocks HIV-1 Tat-Induced Amyloid Beta Production and Tau Phosphorylation. J Neuroimmune Pharmacol 12(1):163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Pentlicky S, Klase Z, Singh M, Neuveut C, Lu CY, Reitz MS Jr, Yarchoan R, Marx PA, Jeang KT (2003). An in vivo replication-important function in the second coding exon of Tat is constrained against mutation despite cytotoxic T lymphocyte selection. J. Biol. Chem 278:44816–44825. [DOI] [PubMed] [Google Scholar]
- Tahirov TH, Babayeva ND, Varzavand K, Cooper JJ, Sedore SC, Price DH (2010). Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature 465(7299):747–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi M, Rusnati M, Presta M, Giacca M (2001). Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J Biol Chem 276(5):3254–61. [DOI] [PubMed] [Google Scholar]
- Uversky VN (2002). Natively unfolded proteins: a point where biology waits for physics. Protein Sci 11:739–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valcour VG, Shikuma CM, Watters MR, Sacktor NC (2004). Cognitive impairment in older HIV-1-seropositive individuals: prevalence and potential mechanisms. AIDS 18: S79–S86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendeville A, Rayne F, Bonhoure A, Bettache N, Montcourrier P, Beaumelle B (2004). HIV-1 Tat enters T cells using coated pits before translocating from acidified endosomes and eliciting biological responses. Mol Biol Cell 15(5):2347–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wälti MA, Ravotti F, Arai H, Glabe CG, Wall JS, Böckmann A, Güntert P, Meier BH, Riek R (2016). Atomic-resolution structure of a disease-relevant Aβ (1–42) amyloid fibril. Proc Natl Acad Sci USA 113(34): E4976–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westendorp MO, Frank R, Ochsenbauer C, Stricker K, Dhein J, Walczak H, Debatin KM, Krammer PH (1995). Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature 375(6531):497–500. [DOI] [PubMed] [Google Scholar]
- Yezid H, Konate K, Debaisieux S, Bonhoure A, Beaumelle B (2009). Mechanism for HIV-1 Tat insertion into the endosome membrane. J Biol Chem 284(34):22736–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitler M, Steringer JP, Müller HM, Mayer MP, Nickel W (2015). HIV-Tat Protein Forms Phosphoinositide-dependent Membrane Pores Implicated in Unconventional Protein Secretion. J Biol Chem 290(36):21976–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Paganini L, Mucke L, Gordon M, Refolo L, Carman M, Sinha S, Oltersdorf T, Lieberburg I, McConlogue L (1996). Beta-secretase processing of the beta-amyloid precursor protein in transgenic mice is efficient in neurons but inefficient in astrocytes. J Biol Chem 271(49):31407–11. [DOI] [PubMed] [Google Scholar]