

Abstract
A new series of 2-aminothiazole derivatives was designed and prepared as phosphodiesterase type 5 (PDE5) regulators and COX-1/COX-2 inhibitors. The screening of the synthesized compounds for PDE5 activity was carried out using sildenafil as a reference drug. Strikingly, compounds 23a and 23c were found to have a complete inhibitory effect on PDE5 (100%) at 10 μM without causing hypotension and the limited side effect of PDE5 inhibitors, suggest a distinctive therapeutic role of these derivatives in erectile dysfunction. On the other hand, compounds 5a, 17, 21 and 23b increased the PDE5 activity (PDE5 enhancers) at 10 μM. In addition, the study includes the screening of the COX-1/COX-2 inhibition induced by the synthesized compounds. All tested compounds have an inhibitory effect against COX-1 activity (IC50 = 1.00–6.34 μM range) and COX-2 activity (IC50 = 0.09–0.71 μM range). Moreover, a molecular docking study was implemented to reveal the binding interactions of potent compounds in the binding sites of PDE5 (PDB ID 2H42), COX-1 and COX-2 (PDB ID 3LN1) enzymes. For the interaction with the PDE5 enzyme, activator compounds had a strong binding mode (HB with Gln817:A) than inhibitory derivatives. Both types of compounds are considered as PDE5 regulators. This novel finding will encourage us to discover a new pharmacological application of small chemical entities as the PDE5 enhancer, or will lower side effects as PDE5 inhibitors. All active compounds adopted the Y-shape along the COX-2 active site.
A new series of 2-aminothiazole derivatives was designed and prepared as phosphodiesterase type 5 (PDE5) regulators and COX-1/COX-2 inhibitors.
1. Introduction
Phosphodiesterase 5 inhibitors and the COX activity have recently emerged and the phosphodiesterase type 5 inhibitors (PDE5-Is) have been reported as potential therapy for neuroinflammatory and neurodegenerative diseases.1 PDE5-Is produce an anti-inflammatory and neuroprotective action by enhancing the expression of nitric oxide synthases and in the accumulation of cGMP, in addition to activating protein kinase G (PKG), which play an important role in the development of several neurodiseases, such as Alzheimer's disease (AD), Parkinson's disease (PD), and multiple sclerosis (MS).2 Moreover, clinical experiments indicate that chronic inflammation is the main factor in benign prostatic hyperplasia (BPH) progression. The use of anti-inflammatory agents such as steroids and cyclooxygenase-2 (COX-2) inhibitors have been reported as treatment for prostate diseases.3 It has been reported that PDE5 inhibitors (PDE5-Is) relax the smooth muscle fibers of the bladder and prostate by NO/cGMPc signaling, and then reduce the hyperactivity of the autonomic nervous system.4 As a result, we can say that phosphodiesterase-5 inhibitors can be used to treat prostatic inflammation. The literature survey revealed that sildenafil (Fig. 1) (as a PDE5 inhibitor) has been shown to be beneficial in the early phases of inflammation and vascular remodeling in a pulmonary arterial hypertension (PAH). The administration of sildenafil significantly reduced the inflammation in the acute stage of the disease and prevented pulmonary arterial remodeling. These results revealed that sildenafil has a direct anti-inflammatory effect, in addition to its vasodilatation and anti-proliferative effects.5 The anti-inflammatory effect of PDE5 inhibitors was confirmed by another study in the rat model of colitis.6 On the other hand, PDE activators (biochemicals that activate PDE) have many applications in biochemical and physiological research. For example, calmodulin (a multifunctional intermediate calcium-binding messenger protein expressed in all eukaryotic cells) is an activator of NOS, PDE and CaMK.7 Another example of PDE activators is UCR1C, a novel activator of PDE4 long isoforms, where PDE4 activation by UCR1C attenuates cardiomyocyte hypertrophy.8 Regulatory molecules are chemical entities that either increase or inhibit enzyme activity. Compounds that enhance the activity of an enzyme are called activators, while those that decrease the activity of an enzyme are called inhibitors.
Fig. 1. Structures of PDE5 inhibitors and COX-2 inhibitory reference drugs.

Different modifications in sildenafil pharmacophoric features were performed in our previous studies for the design of new pyrazolo[3,4-d]pyrimidin-4-(5H)-one derivatives as inhibitors of phosphodiesterase enzyme type 5 (PDE5) as sildenafil analogues.9,10 Most of the explored tested compounds showed the same binding mechanism as vardenafil, and some of these compounds showed promising activity in comparison to sildenafil in the pharmacological studies (Fig. 1). The results indicated that compounds with a styryl moiety or those with a longer chain alkyl group are the most active derivatives.9,10
Based on these biological interests, in addition to our successful synthesis of fused moieties using 3-oxobutanamide derivatives as starting materials,11–14 it was of interest to prepare a series of 2-aminothiazole derivatives (2, 5a,b, 7, 11, 13, 16–19, 22, 23a–c) using 3-oxobutanamides as a precursor. We investigated their PDE5 regulatory activity, in addition to the COX-1 and COX-2 inhibitory activity (Fig. 2). Furthermore, a docking study was carried out for all synthesized compounds to predict their binding mode with the PDE5 compared to sildenafil and avanafil (Fig. 1) as standard PDE5 inhibitors. In the case of COX-1 and COX-2, the binding mode of the compounds was compared to celecoxib as the COX inhibitor (Fig. 1).
Fig. 2. Rational for the design of more potent PDE5 inhibitors and selective COX-2.

In order to find a new drug candidate with selective COX-2 inhibition and PDE5 inhibitors, a comparative shape alignment was carried out to synthesize a new lead candidate. Regarding PDE5 inhibition, the comparative shape alignment between sildenafil and avanafil with enamide 23c showed that the replacement of the methyl group with an aryl moiety is suggested. In the comparison of 23b with celecoxib (also illustrated), the replacement of methyl group with aryl moiety is suggested.
Finally, we hypothesize the synthesis of new derivatives of aminothiazole, especially with the general formula in Fig. 2.
2. Results and discussion
2.1. Chemistry
As we previously reported,11–14 3-oxobutanamide scan be utilized as a precursor in the synthesis of several heterocyclic substances. In the current work, we explored the reaction of 3-oxobutanamides 1 with some electrophilic and nucleophilic reagents. Thus, the treatment of 1 with sodium nitrite in acetic acid at 0 °C afforded the 2-hydroxyimino-3-oxo-N-thiazole-2-yl-butyramide 2.15 The structure of compound 2 was confirmed by spectroscopic data. Moreover, the reduction of compound 2 with zinc dust in acetic acid furnished the intermediate 2-amino-3-oxo-N-thiazole-2yl- butyramide 3 (not isolated), which converted directly to the pyrrole derivatives 5a,b on treatment with an equimolar amount of ethyl acetoacetate and acetyl acetone, respectively, through the intermediate 4.15 Compound 5 may proceed by condensation of the carbonyl group in active methylene to amino function in compound 3 to form the Schiff's base intermediate 4, which under intramolecular cyclization by loss of water, forms the final product 5a,b (Scheme 1).
Scheme 1.

Similarly, when compound 1 is allowed to react with hydroxylamine hydrochloride in refluxing ethanol in the presence of sodium acetate, the pyrazole derivative 7 was obtained through the unisolated oxime 6. The synthetic potential of 1 toward a variety of nucleophilic regents was also investigated. Thus, the boiling of compound 1 in sodium ethoxide solution following the addition of an equimolar amount of ethyl chloroacetate did not yield the expected 9, and only ethyl 4-oxo-4-(thiazol-2-ylamino) butanoate 11 was obtained.16 Compound 11 is assumed to proceed by condensation of ethyl chloroacetate to the sodium salt of compound 1, forming the (not isolated) intermediate 10 by loss of a HCl molecule and subsequent loss of an acetaldehyde molecule to form 11. Compound 11 was confirmed based on its compatible spectroscopic data (IR, 1H NMR, 1H,1H COSY, 13C NMR, DEPT-135 of 13C NMR, NOE of 13C NMR). 1H NMR revealed a triplet signal at δ = 1.23 ppm, J = 7.2 Hz assigned to CH3, triplet signals at δ = 2.78 ppm, J = 6.4 Hz, δ = 2.87 ppm, J = 6.4 Hz assigned for 2CH2, quartet signal at δ = 4.13 ppm, J = 7.2 Hz assigned to CH2 ester, doublet signals at δ = 6.96 ppm, δ = 7.47 ppm assigned to 2 thiazole H-4, H-5, and a singlet signal at δ = 11.12 ppm for the NH group. In the 1H,1H COSY spectrum for compound 11, triplet protons for the methyl group at δ = 1.23 ppm showed J3 correlation with quartet methylene protons at δ = 4.13 ppm to indicate an ethyl moiety. At the same time, the methylene protons at δ = 2.78 ppm exhibited J3 coupling with methylene protons at δ = 2.87 ppm, In the 1H,1H COSY spectrum for compound 11, the methylene at δ = 4.3 ppm showed a correlation to the aromatic proton at δ = 7.47 ppm to indicate these two protons in the same direction. The 13C NMR of compound 11 showed a signal at δ = 14.17 ppm (q) for CH3, signals at δ = 28.84 ppm (t), δ = 30.74 ppm (t), δ = 60.95 ppm (t), for the 3CH2 carbons, signals at δ = 113.50 ppm (d), δ = 136.36 ppm (d), for the 2CH carbons, signals at δ = 159.95 ppm (s), δ = 169.64 ppm (s, C O), and δ = 172.39 ppm (s, C O). The (DEPT-135) of 13C NMR showed the disappearance of one quaternary carbon atom signal at δ = 159.95 ppm and the presence of the sp2 and sp3 carbons at δ = (+)14.11 ppm (q), δ = (−)28.77 ppm (t), δ = (−)30.66 ppm (t), δ = (−)60.91 ppm (t), δ = (+)113.45 ppm (d), and δ = (+)136.30 ppm (d). In the NOE (Nuclear Overhauser Effect) of 13C NMR, a quartet methyl carbon at δ = 12.17–16.04 ppm and a triplet methylene carbon at δ = 59.49–62.55 ppm were observed. At the same time, 2 methylene groups at δ = 27.49–31.95 ppm were overlapped together. In the aromatic region, a doublet carbon at δ = 112.39–114.84 ppm was overlapped with a doublet carbon in the range of δ = 135.63–137.47 ppm for the thiazole ring, and the quaternary carbon at δ = 169.57 ppm (s), and δ = 172.32 ppm (s) for the (2C O) group.
Similarly, the addition of an equimolar amount of ethyl chloroformate to the boiling solution of compound 1 in sodium ethoxide gave the unexpected thiazol-2-yl-carbamic acid ethyl ester 13 rather than the expected 3-oxo-2-(thiazole-2-yl-carbamoyl)-butyric acid ethyl ester 12. Compound 13 was established on the basis of the spectroscopic data. 1H NMR revealed a triplet signal at δ = 1.37 ppm, J = 6.8 Hz assigned to CH3, quartet signal at δ = 4.30 ppm, J = 6.8 Hz, doublet signal at δ = 6.91 ppm, δ = 7.39 ppm assigned to 2 thiazole-H, and a singlet signal at δ = 12.81 ppm assigned to the NH group. 1H,1H COSY showed a strong J3 coupling between the triplet methyl protons at δ = 1.36 ppm and quartet methylene protons at δ = 4.30 ppm to confirm the ethyl group. 13C NMR showed signals at δ = 14.47 ppm (q), δ = 62.13 ppm (t), δ = 112.33 ppm (d), δ = 136.71 ppm (d), δ = 154.02 ppm (s), and δ = 161.94 ppm (C O) group. (DEPT-135) of 13C NMR recorded the appearance of only the sp3 and sp2 carbons at δ = (+)14.46 ppm (q), δ = (−)62.13 ppm (t), δ = (+)112.31 ppm (d), and δ = (+)136.70 ppm (d). The NOE (Nuclear Overhauser Effect) of 13C NMR spectra showed two doublet aromatic methene carbons at δ = 111.31–113.18 ppm and δ = 135.74–137.60 ppm. The coupling constant difference indicates that they are geminal protons. At the same time, the triplet methylene carbon at δ = 60.66–63.61 ppm and quartet methyl carbon at δ = 12.57–16.37 ppm were observed and the quaternary carbon at δ = 154.02 ppm (s), and at δ = 162.05 (s) for the (C O) group. Structural elucidation of compound 13 came from its synthesis by another method, conducting the believes 2-aminothiazole and ethyl chloroformate in boiling solution of ethanolic sodium ethoxide (mp; mx.mp and TLC). We suggest that compound 13 proceeds by the addition of ethyl chloroformate to 8 (the unisolated sodium salt of compound 1). The structure was broken and freed from 2-amino-thiazole, which react directly with ethylchloroformate present in the reaction medium to give compound 13 (Scheme 2).
Scheme 2.

On the other hand, a reaction of compound 1 with α-chloroacetylchloride afforded the pyrrole derivative 16 through the intermediates 14 and 15. In the same manner, the reaction of compound 1 with acetyl chloride in benzene under refluxing gave the adduct 17. Compound 17 was confirmed by its reaction with nucleophilic reagents (such as hydrazine hydrate) and urea to obtain some azole and azine derivatives. So, the treatment of compound 17 with hydrazine hydrate in ethanol under reflux afforded the corresponding pyrazole derivative 18. In the same way, the pyrimidine derivative 19 was synthesized by reaction of compound 17 and urea in ethanol under reflux (Scheme 3).
Scheme 3.

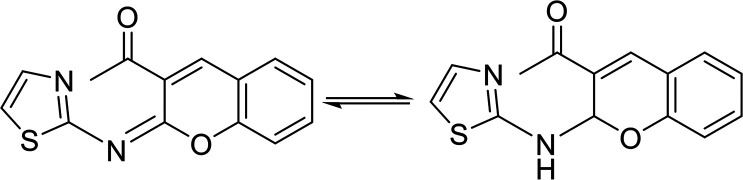
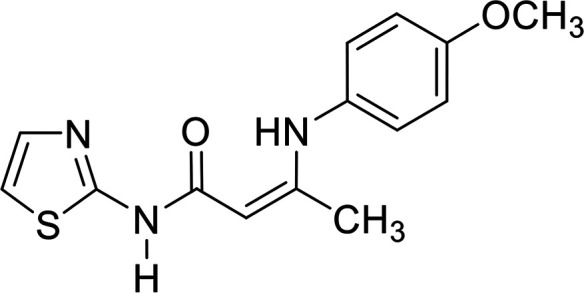
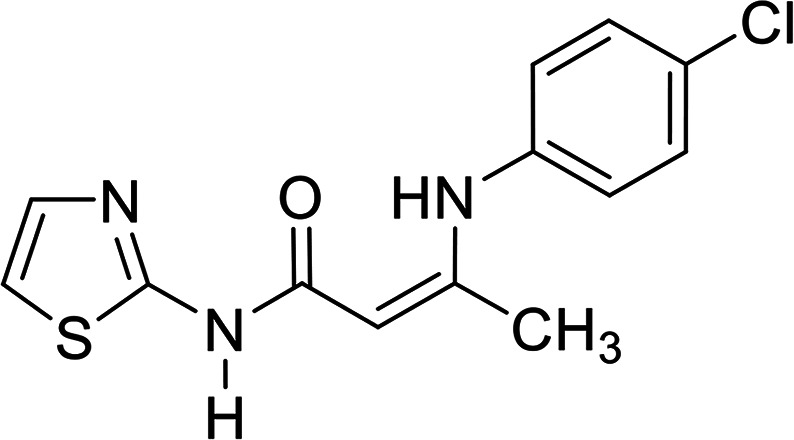
In contrast, the behavior of compound 1 with 2-hydroxy benzaldehyde in pyridine solution under reflux gave the benzopyrane derivative 21 through the intermediate 20. However, under the reaction conditions, the quinoline derivative 22 was obtained through a Dimroth rearrangement of 21. On the other hand, the reaction of compound 1 with aromatic amines was also investigated. So, it reacts with aromatic amines (like aniline, p-anisidine and p-chloroaniline) to give the adducts 23a–c with the loss of water. Structures 23a–c were confirmed on the basis of spectroscopic data. In the 1H,1H COSY spectrum, the protons A–B system showed a strong J3 coupling. At the same time, a doublet proton H4 at δ = 6.86 ppm showed a strong J3 correlation with a doublet proton at δ = 7.37 ppm for the thiazole ring (Scheme 4).
Scheme 4.

2. 2. Pharmacological evaluation
2. 2. 1. Phosphodiesterase 5 interaction and antihypertensive activity
The vasorelaxant effects of the tested compounds are presented in Table 1. The data showed that the MABP values of compounds 2, 5a, 21 and 23b are significantly higher than those of the control, while the values of compounds 5b, 23a and 23c are higher than the control values in a nonsignificant manner. In addition, the values of compounds 11 and 17 are significantly higher than the sildenafil values, but insignificantly lower than the control values. In contrast, the value of compound 7 is significantly lower than that of the control, and insignificantly higher than the sildenafil one. In addition, the results of the PDE5 regulatory activity of the tested compounds against sildenafil are summarized in Table 1, and graphically presented in Fig. 3. The compounds are categorized as three groups based on their action against PDE5: (a) activators, which are compound 21 (PDE5 4290% enhancement), compound 17 (PDE5 371% enhancement), and compound 5a (PDE5 222% enhancement); (b) weak regulators (activators), which are compounds 5b and 23b (PDE5 15% enhancement), and compound 11 (PDE5 3.5% enhancement); and finally (c) inhibitors, which are compounds 23a and 23b (PDE5 100% inhibition), compound 7 (PDE5 75% inhibition), and compound 2 (PDE5 53% inhibition).
Effect of tested compounds on PDE5 (10 μM) and MABP, and consensus scores against PDE5a,b,c.
| Comp. no. | Comp. structure | PDE5 activity (consensus scores) | MABP |
|---|---|---|---|
| 2 |

|
53↓ (122) | 115## |
| 5a |
|
222↑ (28) | 108## |
| 5b |
|
15↑ (49) | 98 |
| 7 |
|
75↓ (126) | 81# |
| 11 |

|
3.5↑ (100) | 85& |
| 17 |
|
371↑ (110) | 87& |
| 21 |
|
4290↑ | 118## |
| 23a |

|
100↓ (57) | 108 |
| 23b |
|
15↑ (50) | 110## |
| 23c |
|
100↓ (48) | 100 |
| Control (DMSO) | — | — | 93.10 ± 1.55 |
| Nitroprusside | — | 52.10 ± 1.11### | |
| Sildenafil | Fig. 1 | 100↓ | 72.50 ± 2.9## |
Data were expressed as means ± SE (n = 4).
Multiple comparisons were done using the one way ANOVA, followed by Tukey–Kramer as the post-ANOVA test.
#: Significantly different from the control group at P < 0.05. &: Significantly different from the sildenafil group at P < 0.05.
Fig. 3. Line chart representing the PDE5 regulatory activity (either increase or decrease in PDE5%) of the tested compounds.

The mean arterial blood pressure (MABP) results and that of PDE5 revealed that compounds 23a and 23c have a good chance to be used in erectile dysfunction without the development of the common side effect, hypotension, that is associated with the use of such drugs to manage erectile dysfunction. In contrast, compounds 5a, 7, and 21 illustrated the enhancement in PDE5 activity (Table 1). Other compounds, such as 5b, 11, 23b, showed very low activity (either in inhibition or enhancements). In a comparison study between compounds 5a and 5b or 23a and 23c to 23b, it is clear that any change in pharmacophoric feature in such types of compounds has a great effect on the PDE5 activity. Notably, compound 21 has a remarkable effect on PDE5 as a strong activator. This behavior could be attributed to the presence of a fused heterocyclic system (chromene ring) linked with the 2-aminothiazole moiety, and the capability to form an amine–imine tautomerism. This, of course, will increase the compound binding with the specific amino acid clefts inside the active site domains. We speculate that, according to the strength of the compound interaction with PDE5, a conformational change of the enzyme will occur. Therefore, the activity fluctuated from enhancement to inhibition.
2. 2. 2. In vitro anti-inflammatory activity
The prepared compounds were assayed against COX-1 and COX-2 using a colorimetric enzyme immunoassay (EIA) kit (Cayman Chemicals Inc., Ann Arbor, MI, USA) to screen the isozyme-specific inhibition. The potency of the testing compounds was estimated as the concentration causing 50% enzyme inhibition (IC50). The in vitro selective index (SI) COX-1/COX-2 isozyme inhibition studies determined the ability of all synthesized compounds in inhibiting ovine COX-1 and human recombinant COX-2.17
The data (Table 2) showed that the target compounds have higher inhibitory activities against both COX-1 isozyme (IC50 = 1.00–6.34 μM range) and COX-2 isozyme (IC50 = 0.09–0.71 μM range), with COX-2 selectivity indexes in the range of 3.03 to 16 in comparison with the COX-2 selective reference drug celecoxib (COX-1 IC50 = 7.21 μM, COX-2 IC50 = 0.83 μM and S.I. = 8.68). In addition, the COX-2 inhibitory activity of the tested compounds against celecoxib is graphically represented in Fig. 4.
In vitro COX-1 and COX-2 inhibitory activity of the target compounds and reference drugsa,b.
| Comp. no. | COX-1 IC50a (μM) | COX-2 IC50a (μM) | COX-2 S.I.b |
|---|---|---|---|
| 2 | 1.5 | 0.11 | 13.64 |
| 5a | 1.6 | 0.10 | 16 |
| 5b | 1.3 | 0.24 | 5.4 |
| 7 | 6.34 | 0.63 | 10.06 |
| 11 | 1.7 | 0.21 | 8.1 |
| 17 | 1.4 | 0.09 | 15.56 |
| 21 | 1.0 | 0.33 | 3.03 |
| 23a | 6.30 | 0.71 | 8.87 |
| 23b | 5.74 | 0.56 | 10.25 |
| 23c | 6.33 | 0.61 | 10.38 |
| Celecoxib | 7.21 | 0.83 | 8.68 |
The concentration of the test compound that produces 50% inhibition of COX-1, COX-2 enzyme activity. The result is a mean of two values obtained by the assay of enzyme kits.
The in vitro COX-2 selectivity index (COX-1/COX-2).
Fig. 4. Chart representing the COX-2 inhibitory activity of the tested compounds against celecoxib as a reference.

In the comparison between the compound activities on PDE5 and anti-inflammatory effect, all compounds have COX-2 selectivity whether they are an activator or inhibitor to PDE5. However, an urge need for further study in the future to describe the exact molecular mechanism that underlies the anti-inflammatory effect and selectivity index to the COX-2 isoenzyme.
2.3. Docking study
2. 3. 1. PDE5 interaction
All molecular docking studies were implemented using OpenEye scientific software academic license.18 All compounds were generated in the most stable conformer as performed by the Omega application. The study implemented different human phosphodiesterase-5 (PDE5) X-ray structures. Each was complexed with its co-crystallized ligand sildenafil, vardenafil or tadalafil (ID: 1TBF, 1XP0, 1XOZ, and 2H42), and were retrieved from the Protein Data Bank.19 However, the study focused on PDB ID 2H42.20
In order to validate our docking study, the docking protocol was continued with the redocking of the reported PDE5 inhibitor drugs sildenafil, vardenafil, tadalafil and avanafil with the catalytic domain receptor of PDE5 (PDB: ID: 2h42).20 All drugs showed a similar binding mode and pose, in particular, the co-crystallized ligand drug, sildenafil. Sildenafil forms two HBs with Gln: 817:A through its lactam functionality (Fig. 5A). This docking pattern is similar to the reported co-crystallized pose. Both enamides (23a and 23c), with 100% PDE5 inhibition, interact with the receptor with a similar pose. However, compound 23a forms a HB with the key amino acid Gln: 817:A through its carbonyl group (acceptor), as shown in Fig. 5B. Compound 23c forms a hydrophobic–hydrophobic interaction with the receptor. Both compounds adopted a complete overlay with the same orientation of the thiazole moiety. The sulfur atom was oriented towards the Gln: 817, as shown in Fig. 5C. This mode is different from that of the analogue 23b, which will be presented later. Both compounds 2 and 7 showed a low consensus score among the tested compounds (48, 51, respectively), and formed HBs and hydrophobic interactions, respectively. The shape alignment between 23c and sildenafil, as represented in Fig. 5D, or vardenafil emphasize the required modification of the synthesized compounds to get more potent derivatives at a lower dose.
Fig. 5. Visual representation by Vida. (A) Sildenafil in the catalytic domain of PDE5 (PDB code: 2H24); (B) 23a in the catalytic domain of PDE5 (PDB code: 2H24); (C) 23a, 23c overlay; (D) shape alignment of 23c with sildenafil.

The novelty in this work arose from some compounds with an enhancement effect on PDE5. The docking protocol was applied to understand this action and subsequently, the development of new drugs as PDE enhancers, which will open the gate for new pharmacological applications.
Concerning other compounds with an enhancement activity on PDE5, these compounds showed strong HBs formation with the same catalytic site of PDE5. Compound 21 has the highest enhancement value for PDE5 activity with a consensus score of 23. It forms a strong HB with Gln: 81A, as shown in Fig. 6A. In addition, it has high similarity with compound 5a in its binding mode and pose, as shown in Fig. 6B. For the small molecule, compound 17, it forms two strong HBs with Gln: 817A. The enamide 23b has a hydrophobic–hydrophobic interaction with the receptor in which the nitrogen atom of the thiazole moiety is oriented toward Gln: 817, as shown in Fig. 6C. This behavior differs from the other enamide 23a. Both compounds 5a and 21 occupied the receptor domain with high similarity in mode and pose, as shown in Fig. 6D.
Fig. 6. Visual representation as visualized by Vida. (A) Compound 21 in the catalytic domain of PDE5 (PDB code: 2H24); (B) compound 5a in the catalytic domain of PDE5 (PDB code: 2H24); (C) compound 23b with a hydrophobic–hydrophobic interaction without the formation of a HB; (D) overlay between compounds 5a and 21.

2. 3. 2. COX-2 inhibition
The docking started point was determined by examining the interaction of the standard drug celecoxib with the COX-2 enzyme. Celecoxib was found to interact with the receptor of PDB ID: 3LN1,21 taking its known pose with the Y shape (pyrazole core with vicinal diaryl pharmacophores), as shown in Fig. 7A, with a best consensus score of 0.00.
Fig. 7. (A) Celecoxib with the COX-2 active site (PD: ID 3IN1); (B) enamide 23c; (C) 23a and 23b overlay; (D) Y shape of enamide (blue) inside the receptor overlay with celecoxib (pink colour).

All examined compounds interacted with the COX-2 enzyme and docked inside the receptor. Some of these compounds exhibited binding poses that were similar to that of celecoxib. Enamide 23c has the highest consensus score value (12) with the formation of the hydrophobic–hydrophobic interaction, as shown in Fig. 7B. Enamides 23a and 23b with consensus scores of 19 and 14, respectively, adopted binding poses with the formation of HB with Tyr 341:A, as shown in Fig. 7C. From a medicinal chemistry perspective, the enamides adopted the Y shape inside the receptor, as illustrated in Fig. 7D. Enamide 23b and celecoxib interacted through HBs with the amino acids cleft of Tyr 341:A, Leu 338:A, and Hist 75:A.
3. Conclusion
New aminothiazole derivatives having different functionalities were synthesized and their activity with PDE5 and COX-1/COX-2 was examined. The synthesized compounds adopted two different patterns against PDE5. Compounds 23a and 23b exhibited 100% inhibition of PDE5 at 10 μM without a notable decrease in MABP. This kind of compound has the benefit over current drugs and PDE5 inhibitors, which have a hypotensive effect as a drawback. In contrast, compounds 5a, 21, 17 and 23b increased the PDE5 activity (PDE5 enhancers) at 10 μM. Both types of compounds are considered PDE5 regulators. To the best of our knowledge, this is the first report of small molecules as PDE5 enhancers with the hope of finding a pharmacological application. All compounds showed anti-inflammatory activity based on the COX-1/COX-2 Ic50% values.
All tested compounds have an inhibitory effect against the COX-1 activity (IC50 = 1.00–6.34 μM range) and COX-2 activity (IC50 = 0.09–0.71 μM range). The selectivity indexes of these compounds to the COX-2 isoenzyme fall in the range of 3.03 to 16 in comparison with celecoxib, a selective COX-2 inhibitor (COX-1 IC50 = 7.21 μM, COX-2 IC50 = 0.83 μM and S.I. = 8.68).
We envisioned that there is a relationship between the strength of the compound interaction and their effect on the enzyme from inhibition, ranging from nothing to activation.
4. Experimental
4.1. Chemistry
Melting points were uncorrected and determined on a Stuart melting point apparatus (Stuart Scientific, Redhill, UK). Elemental analyses of C, H and N were performed on a Perkin-Elmer 2400 analyzer (Perkin-Elmer, Norwalk, CT, USA). The IR spectra (KBr) were measured on a Shimadzu IR 435 spectrophotometer and the values are represented in cm−1. 1H NMR (400 MHz) and 13C NMR (75 MHz) spectra were carried out using a Varian Gemini 300-BB spectrophotometer (Bruker, Munich, Germany). Splitting patterns were designated as follows: s: singlet; d: doublet; t: triplet; m: multiplet, and chemical shift values were recorded in ppm on a scale. Mass spectra were run on a Shimadzu Qp-2010 plus. The progress of the reactions was monitored by TLC using TLC sheets pre-coated with UV florescent silica gel (Merck 60 F 254), and was visualized using a UV lamp. The chemicals used were supplied from Acros (New Jersey, USA).
4.1.1. Preparation of 2-(hydroxyimino)-3-oxo-N-(thiazol-2-yl)butanamide (2)
A mixture of 3-oxobutanamides 1 (0.03 mol) in 100 ml of glacial acetic acid was cooled in an efficient freezing mixture to 5 °C, and a cold solution of (0.0147 mole) sodium nitrite in 5 ml water was added dropwise with vigorous stirring at such a rate that the temperature remained between 5–7 °C. With efficient cooling, about one-half hour was required to add the nitrite. The mixture was stirred for one-half hour longer, and then allowed to stand for 4 h. During this time, it warmed up to room temperature. Finally, the solid product formed was collected by filtration and recrystallized from aqua's ethanol to give 2.
Brown crystals; yield, 65%; mp 182 °C; 1H NMR (DMSO-d6): δ2.40 (s, 3H, CH3), 7.28 (d, 1H, CH-thiazole, J = 0.8 Hz), 7.50 (d, 1H, CH-thiazole, J = 1.6 Hz), 12.54 (s, 1H, NH), 13.06 (s, 1H, N–OH); 13C NMR (DMSO-d6): δ 25.28, 114.17, 137.85, 152.16, 156.77, 160.18, 194.35; Dept 135 NMR: δ 25.22, 114.10, 137.85; MS (relative intensity) m/z: 214.11 (M+, 7.8%); anal. calcd for C7H7N3O3S (213.21): C, 39.43; H, 3.31; N, 19.71; S, 15.04%, found: C, 39.41; H, 3.33; N, 19.72; S, 15.05%.
4.1.2. General procedure for the synthesis of compounds 5a,b
To a solution of 2 (0.01 mol) in 50 ml of glacial acetic acid, portions of (0.5 gram atoms) of zinc dust were added quickly. It was stirred until the liquid boiled, and then frequently enough to keep it boiling. 0.01 mol of ethyl acetoacetate and acetyl acetone were added. After the addition was completed, the mixture was heated by a burner and refluxed for one hour. While still hot, the contents of the flask were decanted from the remaining zinc into a beaker containing 500 ml of water, which was vigorously stirred. The zinc residue was washed with two 50 ml portions of hot glacial acetic acid, which were also decanted into the water. After standing overnight, the crude product was filtered by suction, washed on the filter with two 500 ml portions of water, and dried in air to a constant weight to give 5a,b.
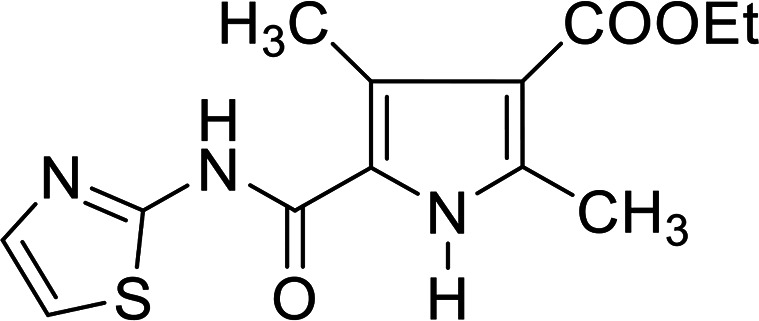
4.1.3. Ethyl 2,4-dimethyl-5-(thiazol-2-ylcarbamoyl)-1H-pyrrole-3-carboxylate 5a
Dark red crystals from aqua's ethanol; yield 45%; mp 147 °C; IR (KBr) ν cm−1 = 3450 (2NH), 3081 (CH arom.), 2921 (CH aliph.), 1657 (C O); 1H NMR (CDCl3-d1): δ 1.33 (t, 3H, CH3, J = 7.2 Hz), 2.58 (s, 3H, CH3), 2.69 (s, 3H, CH3), 4.24 (q, 2H, CH2, J = 7.2 Hz), 7.07 (d, 1H, CH-thiazole, J = 4.4 Hz), 7.51 (d, 1H, CH-thiazole, J = 4.4 Hz), 11.12 (s, 2H, NH); 13C NMR (CDCl3-d1): δ 12.17, 14.36, 14.75, 59.77, 113.17, 114.81, 117.12, 125.34, 137.30, 142.41, 156.39, 162.86, 164.83; Dept 135 NMR δ 12.17, 14.36, 14.75, 59.81, 113.15, 125.34; anal. calcd for C13H15N3O3S (293.34): C, 53.23; H, 5.15; N, 14.32; S, 10.93%, found: C, 53.25; H, 5.12; N, 14.30; S, 10.91%.
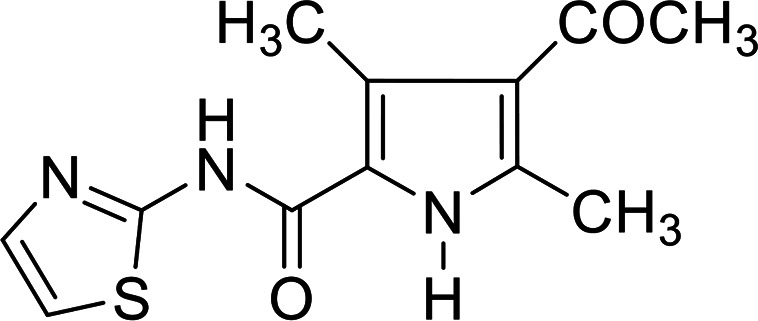
4.1.4. 4-Acetyl-3,5-dimethyl-N-(thiazol-2-yl)-1H-pyrrole-2-carboxamide 5b
Brown crystals from aqua's ethanol; yield 56%; mp 220 °C; IR (KBr) ν cm−1 = 3404 (NH), 3086 (CH arom.), 2918 (CH aliph.); 1687 (C O); 1H NMR (DMSO-d6): δ 2.39 (s, 3H, CH3), 2.51 (s, 3H, CH3), 2.57 (s, 3H, CH3), 7.22 (d, 1H, CH-thiazole, J = 3.6 Hz), 7.51 (d, 1H, CH-thiazole, J = 3.6 Hz), 11.79 (s, 2H, 2NH); 13C NMR (DMSO-d6): δ 12.43, 14.79, 31.21, 113.30, 119.01, 122.68, 129.03, 137.14, 137.88, 158.56, 194.40; MS (relative intensity) m/z: 265.9 (M2+, 93.3%); anal. calcd for C12H13N3O2S (263.32): C, 54.74; H, 4.98; N, 15.96; S, 12.18%, found: C, 54.76; H, 4.95; N, 15.94; S, 12.16%.
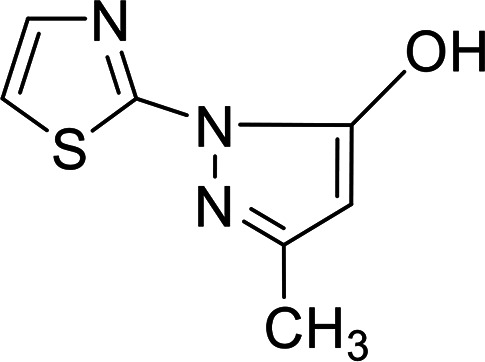
4.1.5. Preparation of 3-methyl-1-(thiazol-2-yl)-1H-pyrazol-5-ol 7
A mixture of 1 (0.01 mol) and hydroxyl amine hydrochloride (0.01 mol) was refluxed in ethanol containing fused sodium acetate for 8 h, and then 30 ml of glacial acetic acid was stirred and portions of (0.5 gram atoms) of zinc dust were added. After the addition was completed, the mixture was heated by a burner and refluxed for one hour. While still hot, the contents of the flask were decanted from the remaining zinc into a beaker containing 500 ml water, which was vigorously stirred. The zinc residue was washed with two 50 ml portions of hot glacial acetic acid, which were also decanted into the water. After standing overnight, the crude product was filtered by suction, washed on the filter with two 500 ml portions of water, and dried in air to a constant weight, and then left to cool. The solid product formed was filtered off and recrystallized from aqua's ethanol to give 7.
Dark red crystals; yield: 48%, mp 290 °C; IR (KBr) ν cm−1 = 3426 (OH), 3027 (CH arom.), 2949 (CH aliph.); 1H NMR (DMSO-d6): δ 2.15 (s, 3H, CH3), 5.65 (s, 1H, CH pyrazole), 7.05 (d, 1H, CH-thiazole, J = 1.6 Hz), 7.38 (d, 1H, CH-thiazole, J = 1.6 Hz), 11.77 (br. S, 1H, OH); anal. calcd for C7H7N3OS (181.21): C, 46.40; H, 3.89; N, 23.19; S, 17.69%, found: C, 46.41; H, 3.88; N, 23.20; S, 17.66%.
4.1.6. Preparation of ethyl 4-oxo-4-(thiazol-2-ylamino)butanoate 11
To a solution of compound 1 (0.01 mol) in 30 ml of absolute ethanol containing 0.01 mol of clean sodium metal, 0.01 mol of ethyl chloroacetate was added slowly. The reaction mixture was refluxed for 6 h and left to cool, then poured onto ice/water containing a few drops of HCl. The formed solid product was collected by filtration and recrystallized from aqua's ethanol to give 11.
White crystals; yield: 56%, mp = 154 °C; IR (KBr) ν cm−1 = 3435 (NH), 3082 (CH arom.), 2922 (CH aliph.), 1726, 1688 (2C O); 1H NMR (CDCl3-d1): δ 1.23 (t, 3H, CH3, J = 7.2 Hz), 2.78 (t, 2H, CH2, J = 6.4 Hz), 2.87 (t, 2H, CH2, J = 6.4 Hz), 4.13 (q, 2H, CH2, J = 7.2 Hz), 6.96 (d, 1H, CH-thiazole, J = 3.2 Hz), 7.47 (d, 1H, CH-thiazole, J = 4 Hz), 11.12 (s, 1H, NH); 13C NMR (CDCl3-d1): δ 14.17, 28.84, 30.74, 60.95, 113.50, 136.36, 159.95, 169.64, 172.39; Dept 135 NMR δ 14.11, 28.77, 30.66, 60.91, 113.45, 136.30; NOE NMR δ 12.17-16.04 (q, CH3), 27.49–31.95 (m, 2CH2), 59.49 (t, CH2), 112.39 (d, CH), 135.63 (d, CH); 159.7 8 (s), 169.57 (s), 172.32 (s); anal. calcd for C9H12N2O3S (228.27): C, 47.35; H, 5.30; N, 12.27; S, 14.05%, found: C, 47.33; H, 5.31; N, 12.29; S, 14.04%.
4.1.7. Preparation of ethyl thiazol-2-ylcarbamate 13
A mixture of 3-oxobutanamide 1 (0.01 mol), ethyl chloroformate (0.01 mol) in sodium ethoxide (20 ml) was refluxed for 12 h, and then left to cool. It was poured onto ice/water, which was vigorously stirred. The solid product formed was collected by filtration and recrystallized from aqua's ethanol to give 13.
Brown crystals; yield; 58%, mp 142 °C; IR (KBr) ν cm−1 = 3423 (NH), 3080 (CH arom.), 2977 (CH aliph.), 1722, 1644 (2C O); 1H NMR (CDCl3-d1): δ 1.37 (t, 3H, CH3, J = 6.8 Hz), 4.30 (q, 2H, CH2, J = 6.8 Hz), 6.91 (d, 1H, CH-thiazole, J = 3.6 Hz), 7.39 (d, 1H, CH-thiazole, J = 4 Hz), 12.81 (s, 1H, NH); 13C NMR (CDCl3-d1): δ 14.47, 62.13, 112.33, 136.71, 154.02, 161.94; Dept 135 NMR δ 14.46 (q), 62.13, 112.31, 136.70; NOE NMR δ 12.57 (q, CH3); 60.66 (t, CH2); 111.31 (d, CH); 135.74 (d, CH); 154.02 (s); 162.05 (s); anal. calcd for C6H8N2O2S (172.2): C, 41.85; H, 4.68; N, 16.27; S, 18.62%, found: C, 41.88; H, 4.67; N, 16.29; S, 18.60%.
4.1.8. Preparation of 1-(2,4-dihydroxy-1-(thiazol-2-yl)-1H-pyrrol-3-yl)ethanone 16
A mixture of 1 (0.01 mol), 2-chloroacetyl chloride (0.01 mol) in ethanol (30 ml) was treated with a few drops of piperidine and refluxed for 6 h, and then left to cool. The solid product formed was collected by filtration and recrystallized from ethanol to give 16.
Brown crystals; yield: 48%, mp 270 °C; 1H NMR (DMSO-d6): δ 2.41 (s, 3H, CH3), 5.001 (s, 2H, 2OH), 6.08 (s, 1H, CH-pyrrole), 7.30 (d, 1H, CH-thiazole, J = 4.8 Hz), 7.76 (d, 1H, CH-thiazole, J = 5.2 Hz); 13C NMR (DMSO-d6): δ 18.18, 109.66, 109.86, 123.37, 146.57, 153.17, 165.13, 167.27, 196.28 (s, C O); Dept 135 NMR δ 18.04, 109.55, 109.73, 123.24; anal. calcd for C9H8N2O3S (224.24): C, 48.21; H, 3.60; N, 12.49; S, 14.30%, found: C, 48.22; H, 3.63; N, 12.50; S, 14.31%.
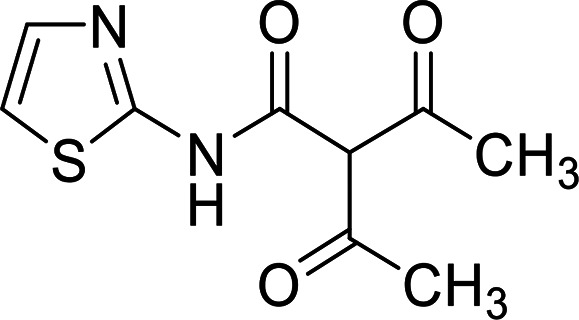
4.1.9. Preparation of 2-acetyl-3-oxo-N-(thiazol-2-yl)butanamide 17
A mixture of (0.01 mol) of magnesium turnings, (0.01 mol) of 3-oxobutanamide 1, 20 ml of benzene (dried over sodium), and (0.015 mol) of acetyl chloride was heated under reflux for 12 h in a 100 ml round-bottomed flask. This flask was provided with a condenser closed by a calcium chloride tube and supported in an oil bath (85–90 °C). The reaction mixture was cooled in an ice bath, and the liquid portion was decanted into a separating funnel. The residue left in the flask after decantation was washed twice with 10 ml portions of ether, and the ethereal solution was poured over ice. The ether–water mixture was then added to the benzene solution in the separating funnel, and the mixture was shaken thoroughly. The aqueous layer was drawn off and discarded. The benzene–ether solution was washed once with 10 ml of 5% sodium bicarbonate solution, and once with 50 ml of water. Finally, it was dried over calcium chloride. The ether and benzene were removed by distillation from a water bath. The solid product formed was recrystallized from ethanol to give 17.
Brown crystals; yield: 46%, mp 187 °C; 1H NMR (DMSO-d6): δ 2.31 (s, 3H, CH3), 2.59 (s, 3H, CH3), 6.43 (s, 1H, CH), 7.29 (d, 1H, CH-thiazole, J = 3.2 Hz), 7.52 (d, 1H, CH-thiazole, J = 3.6 Hz), 13.20 (s, 1H, NH); 13C NMR (DMSO-d6): δ 19.01, 20.09, 113.76, 114.18, 137.54, 161.56, 166.37, 171.82, 177.62; Dept 135 NMR δ 18.99, 20.07, 113.75, 114.16, 137.54; NOE NMR: δ 18.15-20.75, 112.92, 113.19, 136.65, 161.60, 166.41, 171.75, 177.62; anal. calcd for C9H10N2O3S (226.25): C, 47.78; H, 4.45; N, 12.38; S, 14.17%, found: C, 47.76; H, 4.47; N, 12.39; S, 14.15%.
4.1.10. Preparation of 3,5-dimethyl-N-(thiazol-2-yl)-1H-pyrazole-4-carboxamide 18
A mixture of 17 (0.01 mol) and an excess of hydrazine hydrate was refluxed for 6 h, and then left to cool. The solid product formed was collected by filtration and recrystallized from ethanol to give 18.
Brown crystals; yield: 57%, mp 210 °C; 1H NMR (DMSO-d6): δ 2.31 (s, 3H, CH3), 2.59 (s, 3H, CH3), 7.29 (d, 1H, CH-thiazole, J = 3.2 Hz), 7.52 (d, 1H, CH-thiazole, J = 3.6 Hz), 9.69 (s, 1H, NH), 12.67 (s, 1H, NH); 13C NMR (DMSO-d6): δ 19.01, 20.09, 114.18, 137.52, 157.14, 161.56, 166.07, 171.82, 177.62; Dept 135 NMR δ 18.99, 20.07, 114.16, 137.59; MS (relative intensity) m/z: 223.23 (M1+, 2.8%); anal. calcd for C9H10N4OS (222.27): C, 48.63; H, 4.53; N, 25.21; S, 14.43%, found: C, 48.61; H, 4.50; N, 25.24; S, 14.46%.
4.1.11. Preparation of 4,6-dimethyl-2-oxo-N-(thiazol-2-yl)-1,2-dihydropyrimid-ine-5-carboxamide 19
A mixture of 17 (0.01 mol) and urea (0.01 mol) in ethanol (30 ml) was refluxed for 6 h, and then left to cool. The solid product formed was collected by filtration and recrystallized from ethanol to give 19.
Brown crystals; yield: 53%, mp 235 °C; 1H NMR (DMSO-d6) (δ ppm); δ 2.07 (s, 3H, CH3), 2.36 (s, 3H, CH3), 7.28 (d, 1H, CH-thiazole, J = 0.8 Hz), 7.50 (d, 1H, CH-thiazole, J = 1.6 Hz), 8.76 (s, 1H, NH), 12.81 (s, 1H, NH); 13C NMR (DMSO-d6): δ 17.07, 20.07, 114.25, 137.53, 157.35, 161.52, 166.39, 171.96 (s), 177.48, 196.29; anal. calcd for C10H10N4O2S (250.28): C, 47.99; H, 4.03; N, 22.39; S, 12.81%, found: C, 47.96; H, 4.07; N, 22.40; S, 12.78%.
4.1.12. Preparation of 3-acetyl-1-(thiazol-2-yl)quinolin-2(1H)-one 22
A mixture of 1 (0.01 mol) and 2-hydroxy benzaldehyde (0.01 mol) was refluxed for 5 h in pyridine. The solid product formed was collected by filtration and recrystallized from benzene to give 22.
Yellow crystals; yield: 45%, mp 110 °C; 1H NMR (CDCl3-d1): δ 2.70 (s, 3H, CH3), 7.30 (d, 1H, CH-thiazole), 7.33 (d, 1H, CH-thiazole), 7.35 (d, 1H, Ar–H), 7.61–7.62 (m, 1H, Ar-H), 7.63–7.65 (m, 1H, Ar-H), 7.65 (s, 1H, Ar-H), 8.49 (s, 1H, CH-quinoline); 13C NMR (CDCl3-d1): δ 30.74, 114.24, 117.26, 124.83, 129.80, 131.17, 138.57, 139.90, 142.34, 143.97, 156.05, 161.36, 177.05, 198.71; anal. calcd for C14H10N2O2S (270.31): C, 62.21; H, 3.73; N, 10.36; S, 11.86%, found: C, 62.24; H, 3.76; N, 10.32; S, 11.83%.
4.1.13. General procedure for the synthesis of compounds 23a–c
In a round-bottomed flask attached to a Dean and Stark constant water separator, which was connected to a reflux condenser, was placed a mixture of 3-oxobutanamide 1 (0.05 mol), 0.05 mol of aromatic amines, 100 ml of benzene, and 1 ml of glacial acetic acid. The flask was heated in an oil bath at about 125 °C, and the water which distilled out of the mixture with the refluxing benzene was removed at intervals. Refluxing was continued until no more water separated, and then for an additional 30 minutes. The benzene was then distilled under reduced pressure. The solid product formed was filtered off and recrystallized from aqua's ethanol to give 23a–c.
4.1.14. Preparation of (Z)-3-(phenylamino)-N-(thiazol-2-yl)but-2-enamide 23a
Yellow crystals; yield 74%; mp 220 °C; IR cm−1ν = 3192, 3136 (2NH), 3008 (CH arom.), 2914 (CH aliph.), 1644 (C O); 1H NMR (DMSO-d6) (δ ppm); δ 2.03 (s, 3H, CH3), 5.05 (s, 1H, Ali CH), 7.11 (d, 1H, CH-thiazole, J = 4 Hz), 7.17–7.14 (m, 5H, Ar-H), 7.45 (d, 1H, CH-thiazole, J = 3.6 Hz), 11.03 (s, 1H, NH), 11.65 (s, 1H, NH); 13C NMR (DMSO-d6) δ 20.20, 87.88, 112.41, 123.60, 124.58, 129.21, 137.50, 138.93, 157.94, 158.55, 166.74; Dept 135 NMR δ 20.15, 87.83, 112.38, 123.55, 124.55, 129.16, 137.47; MS (relative intensity) m/z: 261.46 (M2+, 3.6%); anal. calcd for C13H13N3OS (259.33): C, 60.21; H, 5.05; N, 16.20; S, 12.36%, found: C, 60.22; H, 5.07; N, 16.19; S, 12.35%.
4.1.15. Preparation of (Z)-3-((4-methoxyphenyl)amino)-N-(thiazol-2-yl)but-2-en-amide 23b
Green crystals; yield 60%; mp = 235 °C; IR (KBr) cm−1ν = 3208, 3124 (2NH), 3011 (CH arom.), 2955 (CH aliph.), 1652 (C O); 1H NMR (DMSO-d6) (δ ppm): δ 1.90 (s, 3H, CH3), 3.73 (s, 3H, OCH3), 4.93 (s, 1H, Ali CH), 6.89 (d, 2H, Ar–H, J = 8.8 Hz), 7.04 (d, 1H, CH-thiazole, J = 3.6 Hz), 7.11 (d, 2H, Ar–H, J = 8.8 Hz), 7.38 (d, 1H, CH thiazole, J = 3.6 Hz), 10.38 (s, 1H, NH), 11.51 (s, 1H, NH); 13C NMR (DMSO-d6): δ 19.95, 55.26, 86.51, 112.26, 114.25, 126.10, 131.63, 137.45, 156.84, 158.91, 166.77; Dept 135 NMR δ 19.90, 55.21, 86.46, 112.21, 114.25, 126.05, 137.40; anal. calcd for C14H15N3O2S (289.35): C, 58.11; H, 5.53; N, 14.52; S, 11.08%, found: C, 58.13; H, 5.55; N, 14.50; S, 11.09%.
4.1.16. Preparation of (Z)-3-((4-chlorophenyl)amino)-N-(thiazol-2-yl)but-2-enamide 23c
Yellow crystals; yield 68%; mp = 190 °C; IR (KBr) ν cm−1 = 3242, 3208 (2N–H), 3056 (CH arom.), 2923 (CH aliph.), 1649 (C O); 1H NMR (CDCl3-d1) (δ ppm): δ 1.97 (s, 3H, CH3), 4.88 (s, 1H, Ali CH), 6.86 (d, 1H, thiazole, J = 2.8 Hz), 6.99 (d, 2H, Ar-H, J = 8 Hz), 7.24 (d, 2H, Ar-H, J = 8 Hz), 7.37 (d, 1H, thiazole, J = 2.8 Hz), 10.86 (s, 1H, NH); 11.96 (s, 1H, NH); 13C NMR (CDCl3-d1) (δ ppm): δ 20.45, 87.83, 112.34, 125.90, 129.24, 131.04, 133.73, 137.34, 159.78, 161.03, 166.36; Dept 135 NMR δ C 20.45, 87.81, 112.34, 126.00, 129.23, 133.62; NOE NMR δ 19.81–22.37 (q), 86.97–88.58 (d), 111.34–113.36 (d), 159.86–161.05 (d), 125.19–137.24 (m, s) 166.31 (s, C O); anal. calcd for C13H12ClN3OS (293.77): C, 53.15; H, 4.12; N, 14.30; Cl, 12.07; S, 10.91%, found: C, 53.11; H, 4.12; N, 14.31; Cl, 12.09; S, 10.90%.
4.2. Pharmacological assessment
4.2.1. Phosphodiesterase 5 enzyme activity assay
The standard enzymatic reaction mixture (total volume of 200 μl) contained 100 ml M Tris–HCl buffer (pH 8.3), 10 mM MgCl2, 10 mM KCl at 37 °C. Alfa casein (2 mg) was used as a carrier for protein precipitation when the protein concentration was low. The PDE A1 enzyme sample had a final protein concentration of 0.5 mg ml−1.22 A concentration of 10 μM of the agents under study (sildenafil analogues) was prepared in dimethyl sulfoxide (DMSO) and pre-incubated in the enzymatic mixture for 5 min at room temperature. The reaction was initiated by the addition of the substrate cGMP (5 μM) at 35 °C for 30 min. The reaction was finished by transferring the reaction mixture tubes in a boiling water bath for 3 min. The sample was then centrifuged and filtered through a nylon-66 filter, 0.2 mm (Rainin Corporation). The clear filtrate obtained may be used directly for HPLC assay or stored at −20 °C. A blank with protein, denaturated by boiling water bath for 3 min, with and without substrate was performed. Both incubation time and enzyme concentration were adjusted so that no more than 25% of the substrate was hydrolyzed under the assay conditions. The chromatographic system was an Agilent Technologies 1200 Series, G1315D DAD. The column used was a Zorbax Eclipse. A rapid resolution 4.69 × 150 mm 3.5 μM particle size. The mobile phase employed for the separation (isocratic elution) consisted of 200 mM ammonium acetate at pH 6.0 with 2% acetonitrile (v/v). The flow rate was 1.5 cm3 min−1 and the detector DAD was at 254 nm. The injection volume was 30 mm3. Peak identities were confirmed by co-elution with standards, and all assays were done in duplicate.
4.2.2. Arterial blood pressure measurement
4.2.2.1. Experimental animals
Regardless of sex factor, White New Zealand rabbits weighing 2–2.5 kg were obtained from the animal house of the Faculty of Medicine, Assiut University. The animals were housed for one week to adapt to the environmental conditions. Rabbits were fed a standard diet and allowed free access to water. “All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Faculty of Pharmacy, Minia University and approved by the Animal Ethics Committee” of the Faculty of Pharmacy, Minia University.
4.2.2.2. Experimental design
Fifty-two of normotensive white New Zealand rabbits (2–2.5 kg) were selected randomly, and used in this study (four rabbits for each group) to investigate the possible vasorelaxant effect of the selected compounds. Using pentobarbital sodium (30 mg kg−1, i.v.), normotensive rabbits were anesthetized. Then, they were held on their backs with legs fixed and heads pinned down, and the trachea was exposed to cut before proper cannulation was done.
4.2.2.3. Data recoding
After separation, the two muscle bundles of the sternomastoid and the sternothyroid, the common carotid artery lying in the neck between the lateral bundles of muscle (longus capitis), and the trachea was exposed and carefully freed from the nerves, veins and connective tissue. The heparinized cannula was carefully inserted and firmly tied via fine thread. Then, the cannula was connected to the pressure transducer using a universal oscillograph (Harvard apparatus, ser. no. K10542) for blood pressure recording. The external jugular vein was also tightly cannulated for administration of tested compounds (dissolved in DMSO), followed by heparinized saline (0.90% w/v NaCl).
The mean arterial blood pressure was calculated using the formula:MABP = DBP + 1/3(SBP − DBP).where MDP = mean arterial blood pressure, DBP = diastolic blood pressure, SBP = systolic blood pressure.
4.2.2.4. Statistical analysis and data interpretation
Statistical analysis was done using GraphPad Prism 5.0 software (GraphPad, San Diego, CA, USA). Data were represented as the mean ± SE. Multiple comparisons were carried out using the one-way ANOVA with the Tukey–Kramer test as a multiple comparison post-ANOVA test. P < 0.05 was selected to refer to statistical significance.
4.2.3. In vitro anti-inflammatory activity screening
The in vitro COX-1/COX-2 inhibition was detected using an enzyme immunoassay (EIA) kit (Cayman Chemical, Ann Arbor, MI) as the reported procedure for screening the isozyme-specific inhibition. The potency of the tested compounds was determined as the concentration causing 50% inhibition of enzyme activity (IC50). Also, the COX-2 selectivity indexes (S.I. values) were calculated as IC50 (COX-1)/IC50 (COX-2), and compared with that of the standard drug, celecoxib, which is a selective COX-2 inhibitor.17
4.3. Docking study
The study was performed according to our previous method using OpenEye software.23
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05561a
References
- Zhang L. Zhang Z. Zhang R. L. Cui Y. LaPointe M. C. Silver B. Chopp M. Brain Res. 2006;1118(1):192–198. doi: 10.1016/j.brainres.2006.08.028. [DOI] [PubMed] [Google Scholar]
- Zhao S. Zhang L. Lian G. Wang X. Zhang H. Yao X. Yang J. Wu C. Int. Immunopharmacol. 2011;11(4):468–474. doi: 10.1016/j.intimp.2010.12.017. [DOI] [PubMed] [Google Scholar]
- Morelli A. Filippi S. Comeglio P. Sarchielli E. Chavalmane A. K. Vignozzi L. Fibbi B. Silvestrini E. Sandner P. Gacci M. Carini M. Vannelli G. B. Maggi M. J. Sex. Med. 2010;7:107–120. doi: 10.1111/j.1743-6109.2009.01558.x. [DOI] [PubMed] [Google Scholar]
- Morelli A. Sarchielli E. Comeglio P. Filippi S. Mancina R. Gacci M. Vignozzi L. Carini M. Vannelli G. B. Maggi M. J. Sex. Med. 2011;8(27):46–60. doi: 10.1111/j.1743-6109.2011.02416.x. [DOI] [PubMed] [Google Scholar]
- Bogdan S. Seferian A. Totoescu A. Dumitrache-Rujinski S. Ceausu M. Coman C. Ardelean C. M. Dorobantu M. Bogdan M. Maedica. 2012;7:109–116. [PMC free article] [PubMed] [Google Scholar]
- Margonis G. A. Christoloukas N. Antoniou E. Arkadopoulos N. Theodoropoulos G. Agrogiannis G. Pikoulis E. Patsouris E. S. Zografos G. C. Papalois A. E. J. Surg. Res. 2015;193:667–674. doi: 10.1016/j.jss.2014.08.064. [DOI] [PubMed] [Google Scholar]
- Stevens F. C. Can. J. Biochem. Cell Biol. 1983;61(8):906–910. doi: 10.1139/o83-115. [DOI] [PubMed] [Google Scholar]
- Wang L. Burmeister B. T. Johnsona K. R. Baillie G. S. Karginov A. V. Skidgel R. A. O'Bryan J. P. Carnegie G. K. Cell. Signalling. 2015;27:908–922. doi: 10.1016/j.cellsig.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaaban M. A. Radwan A. A. Elshaier Y. A. M. M. Abdelwahab B. A. Saudi Pharm. J. 2009;17:109–129. [Google Scholar]
- Elshaier Y. A. M. M., ACS Midwest Regional, St. Joseph, MO, October 2015; 21–24, Missouri, USA, ABSTRACT ID: 2361553, oral presentation, 10 [Google Scholar]
- Hussein A. M. Othman M. M. Gad-Elkareem M. A. M. El-Adasy M. Khames A. A. Elixir Org. Chem. 2017;106:46634–46643. [Google Scholar]
- Hussein A. M. El-Adasy M. Khames A. A. Atalla A. A. Abdel-Rady M. ChemistrySelect. 2017;2:1625–1629. doi: 10.1002/slct.201601424. [DOI] [Google Scholar]
- Hussein A. M. Khames A. A. Abu-Bakr A. A. El-Adasy M. Gad-Elkareem M. A. M. Othman M. M. Int. J. Pharm. Sci. 2015;5(1):864–874. [Google Scholar]
- Hussein A. M. Gad-ElkareemA M. A. M. Abu-Bakr A. El-Adasy M. Khames A. A. Othman M. M. Int. J. Org. Chem. 2012;2:341–351. doi: 10.4236/ijoc.2012.24047. [DOI] [Google Scholar]
- Fischer H. Org. Synth. 1941;21:67. doi: 10.15227/orgsyn.021.0067. [DOI] [Google Scholar]
- Adkins H. Isbell N. Wojcik B. Org. Synth. 1934;14:38. doi: 10.15227/orgsyn.014.0038. [DOI] [Google Scholar]
- Roschek Jr B. Fink R. C. Li D. McMichael M. Tower C. M. Smith R. D. Alberte R. S. J. Med. Food. 2009;12:615–623. doi: 10.1089/jmf.2008.0133. [DOI] [PubMed] [Google Scholar]
- https://www.eyesopen.com/, 15-6-2020
- https://www.rcsb.org/search, 15-6-2020
- Wang H. Liu Q. Huai Q. Cai J. Zoraghi R. Francis Sh H. Corbin J. D. Robinson H. Xin Z. Lin G. Hengming Ke. J. Biol. Chem. 2006;281(30):21469–21479. doi: 10.1074/jbc.M512527200. [DOI] [PubMed] [Google Scholar]
- Wang J. L. Limburg D. Graneto M. J. Springer J. Hamper J. R. B. Liao S. Pawlitz J. L. Kurumbail R. G. Maziasz T. Talley J. J. Bioorg. Med. Chem. Lett. 2010;20(23):7159–7163. doi: 10.1016/j.bmcl.2010.07.054. [DOI] [PubMed] [Google Scholar]
- Spoto G., Berardi S. and AjerbaGand De Laurentiis V., A reverse-phase HPLC method for cyclic nucleotide phosphodiesterases activity and classification, in Purine and pyrimidine metabolism in man, Springer, US, 1995, vol. 8, pp. 815–820 [DOI] [PubMed] [Google Scholar]
- Elshaier Y. A. M. M. Shaaban M. A. AbdEl-Hamid M. K. Abdelrahman M. H. Abou-Salim M. A. Elgazwi S. M. Halaweish F. Bioorg. Med. Chem. 2017;25(12):2956–2970. doi: 10.1016/j.bmc.2017.03.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.