

Abstract
The G-protein coupled receptors (GPCRs) superfamily comprise similar proteins arranged into families or classes thus making it one of the largest in the mammalian genome. GPCRs take part in many vital physiological functions making them targets for numerous novel drugs. GPCRs share some distinctive features, such as the seven transmembrane domains, they also differ in the number of conserved residues in their transmembrane domain. Here we provide an introductory and accessible review detailing the computational advances in GPCR pharmacology and drug discovery. An overview is provided on family A-C GPCRs; their structural differences, GPCR signalling, allosteric binding and cooperativity. The dielectric constant (relative permittivity) of proteins is also discussed in the context of site-specific environmental effects.
The G-protein coupled receptors (GPCRs) superfamily comprise similar proteins arranged into families or classes thus making it one of the largest in the mammalian genome.
Background
The G-protein coupled receptor (GPCR) superfamily consists of structurally similar proteins arranged into families (classes), and is one of the most abundant protein classes in the mammalian genome.1–5 GPCRs undertake a plethora of essential physiological functions and are targets for numerous novel drugs.4,5 Their ligands are structurally heterogenous, including natural odorants, nucleotides, amines, peptides, proteins, and lipids.4 The conserved structure of GPCRs consists of seven TMD of approximately 25–35 successive amino acid residues that express moderately high levels of hydrophobicity4 and are characterised by α-helices which span the plasma membrane.4 The primary function of GPCRs is the transduction of extracellular stimuli into intracellular signals.2 Currently, approximately thirty to forty percent of marketed pharmaceuticals target GPCRs.1,6–10 Hence, there is enormous potential for the development of new drugs targeting these receptors.3 Examples of drugs targeting GPCRs include histamine receptor blockers, opioid agonists, β-blockers and angiotensin receptor blockers.5 Computational biology methods are currently being employed to understand GPCRs as such drug targets.6,11,12 Breakthroughs in GPCR crystallography has facilitated novel discovery through virtual screening as well as better off-target rationalisation.6 Recently, the Tikhonova group developed a computational protocol which combines concepts from statistical mechanics and cheminformatics to explore the flexibility of the bioamine receptors as well as to identify the geometrical and physicochemical properties which characterise the conformational space of the bioamine family.13 Multiple-microsecond timescale molecular dynamics (MD) simulations have been used in capturing the process of several drugs binding to β1- and β2-adrenergic receptors.14 Molecular docking is one of the most commonly used methods in GPCR structure-based drug design (SBDD).14 Esguerra et al. developed GPCR-ModSim, a web-based portal designed specifically for the homology modelling and MD simulation of GPCRs.15
It was historically assumed that GPCRs exist in two conformations: active and inactive.16–18 The long-established extended ternary-complex model of GPCR-driven signalling was based on this concept.16,19,20 This model suggested that the active GPCR conformation opted for by G-protein-coupled receptor kinases (GRKs), arrestins and G proteins is uniform.16 Nevertheless, biophysical investigations with a refined fluorescent-labelled β2-adrenergic receptor (β2AR) demonstrated that a receptor can exist in numerous conformations and that the conformational equilibrium is influenced both by the bound ligand and the proximity to the related G protein.16
The human genome alone contains approximately 800 GPCRs making it the largest family of membrane proteins.5,21 GPCRs have been classified based on structural and physiological features.4 Some systems of classification have grouped these based on location of the ligand binding pocket, while some have utilised both the structural and physiological properties.4,22 The A–F classification system was the first system of classification to be introduced.23 This was first introduced in 1994 as A–F, and O for the (now obsolete) GCRDb database by Kolakowski.23 The defunct GCRDb system was further developed, leading to the GPCRDB24,25 database by Horn et al. with the rhodopsin family (class A) being the largest and consisting of four main groups: α, β, γ, and δ, and 13 sub-branches.4,23,24 All GPCRs comprise of seven TMD helices (Fig. 1), alongside an eight helix and a palmitoylated cysteine at the C terminal tail.26
Fig. 1. A schematic representation of a GPCR showing the transmembrane domains, N-terminus, C-terminus, the intracellular and extracellular loops (generated using GPCRDB Tools, https://gpcrdb.org/).27.

The diversity of GPCRs has resulted in a perceived difficulty in developing a comprehensive classification system.5 The A–F system orders the GPCRs into six classifications on the basis of their sequence homology and functional similarity, namely: family A (rhodopsin-like receptors), family B (secretin receptor family), family C (metabotropic glutamate receptors), family D (parasitic mating pheromone receptors), family E (cyclic AMP receptors) and family F (frizzled and smoothened receptors).5 Based on phylogenetic studies, human GPCRs have been classified under a system called “GRAFS”, this system comprises of five main families namely; glutamate (G), rhodopsin (R), adhesion (A), frizzled/taste2 (F), and secretin (S).4,21,26 The major difference between the two systems concerns the additional division of family B into the adhesion and secretin families within GRAFS.26 This division was based on early findings describing a distinctive evolutionary history between both families.26
Family A (rhodopsin-like receptors)
The rhodopsin receptor family (RRF) is the largest of the GPCR families, comprising of approximately 680 members, and accounts for 80% of receptors in humans.4,28 The RRF is classified into four groups (α, β, γ, δ) and 13 main subdivisions,4,29 and it has numerous characteristics which indicate a common ancestry.4,29 These characteristics include the DRY motif situated at the border between TM3 and intracellular loop (IL) 2 and NSxxNPxxY motif in TM7 (Fig. 2).4,29 The N-terminal region of the family A GPCR receptors are situated extracellularly,29,30 while the C-terminal is located within the cytoplasm (Fig. 3).29,30 The ligand binding site is located within the extracellular region of the TMD bundle.29
Fig. 2. Schematic diagram showing the structure of family A GPCRs generated using ClustalW.31 Reprinted with permission from Springer Nature: Springer Nature, Nature Reviews Drug Discovery, Structural diversity of G protein-coupled receptors and significance for drug discovery, M. C. Lagerström and H. B. Schiöth, Copyright (2008). The upper section of shows the differences in the secondary structure of the N termini of the family A receptors.31 The scissor image indicates the cleavage site of the protease activated receptors whilst in the lower part of the image, the schematic TMD regions show the consensus of an alignment generated using ClustalW 1.82.31 In addition, the area circled in red describes the elliptical orientation.31 Residues conserved in all eight sequences are displayed as circles in which conserved aromatic residues are shown in purple, polar in orange, aliphatic residues are shown in beige, positively charged in red and negatively charged in blue.31.

Fig. 3. Illustration showing the modification of rhodopsin and its orientation in membranes.30 Reprinted with permission from Annual Reviews: Annual Reviews, Annual review of biochemistry, G protein–coupled receptor rhodopsin, K. Palczewski, Copyright (2006). (a) Two-dimensional illustration of rhodopsin. The polypeptide of rhodopsin is seen to cross the membrane seven times with C-I, C-II, C-III comparable to the cytoplasmic loops and E-I, E-II, E-III to the extracellular loops. The yellow cylinders represent the transmembrane region (b) depicts the location of the chromophore and the charges on the extracellular and cytoplasmic surface of rhodopsin. Red and blue colours represent negative and positive charged residues respectively, while the location of the chromophore is revealed by deleting fragments of the transmembrane helices.30.

According to Palczewski, the arrangement of the seven TMD helices which vary in length from 20 to 30 residues is responsible for the overall elliptic, cylindrical shape of rhodopsin (Fig. 3).30 The family A GPCRs vary greatly when their ligand preference and primary structure are considered.31 However, there is homogeneity in the N-termini of family A GPCRs, but heterogeneity within the TMD regions.31 However, some of the family A GPCRs share specific sequence motifs within the TMD region.31
Palczewski reported the dimensions of rhodopsin as an ellipsoid of approximately 35 × 48 × 75 Å, with the long axis perpendicular to the membrane in the standard view.30 The surface area of the section protruding from the membrane is approximately 1200 Å2, with cytoplasmic projection being larger in surface area and volume than the extracellular surface (Fig. 2b).30 The TMD helices of rhodopsin are irregularly shaped due to the conformational changes associated with the Gly–Pro residues; they also incline at several angles in correspondence to the anticipated membrane surface.32 Teller et al. reported that helix 1 tilted from the membrane plane at 25° and contains a 12° kink within it as a result of Pro53 residues being present.32 Helix 2 kinked at an angle of 30° around Gly89 and Gly90 and the most significant bend being at Helix 6 at angle of 36° due to the presence of Pro267.32
Family B (secretin receptor family)
The family B GPCRs form a small group, and with an extracellular hormone-binding site, they bind to large peptides.31 The family name “secretin” derives from the secretin receptor, which was the first to be cloned in this family.3 In 1975, Sasaki et al.33 solved the first X-ray crystal structure of glucagon, a family B GPCR.34 The family corresponds to group B of the A–F system of classification,3 and comprises 15 members including: vasoactive intestinal peptide receptors (vIPR1, vIPR2), glucagon-like peptide receptors (GLP1R, GLP2R), adenylate cyclase activating polypeptide receptor (PAC1/ADCYAP1R1), growth-hormone-releasing hormone receptor (GHRHR), calcitonin and calcitonin-like receptors (CALCR, CALCRL), gastric inhibitory polypeptide receptor (GIPR), secretin receptor (SCTR), corticotropin-releasing hormone receptors (CRHR1, CRHR2), glucagon receptor (GCGR), and parathyroid hormone receptors (PTHR1, PTHR2).3,31 These 15 receptors share between 21 and 67% sequence identity, and a large portion of the dissimilarity is identified in the N-terminal sequence.31,35 These receptors contain conserved cysteine residues in the first and second extracellular loops of the TMD regions (Fig. 3).31 However, the majority of the receptors within this family contain conserved cysteine residues that make up a cluster of cysteine bridges in the N-terminus31 The binding profile of the secretin receptors is outlined by three binding domains comprising of the proximal region and the juxtamembrane region of the N-terminus, as well as the extracellular loops, together with TM6 (Fig. 4).31 The ligand is thought to activate the receptor by spanning the N-terminal and the TMD extracellular loops, this way mediating the active conformation of the receptor, which increases the probability of activation of the signalling units.36
Fig. 4. Schematic diagram showing the structure of family B GPCRs generated using ClustalW.31 Reprinted with permission from Springer Nature: Springer Nature, Nature Reviews Drug Discovery, Structural diversity of G protein-coupled receptors and significance for drug discovery, M. C. Lagerström and H. B. Schiöth, Copyright (2008). The residues conserved in all 15 sequences are displayed as circles, the conserved polar residues are shown in orange, the aromatic residues in purple, the aliphatic residues in beige, the positively and negatively charged residues are shown in red and blue respectively.31 The uppercase letters show the completely conversed positions, the lowercase letters show the well-conserved positions (>50%) while the letter “x” show the variable positions. The conserved sequence motifs which are found in the TMD of the family B GPCRs are surrounded by red boxes.31 The conserved cysteine residues are depicted as yellow circles, the cysteine bridges between EL1 and EL2 are shown as two straight lines while the N-terminal cysteine bridges are drawn as lines.31.

In addition to the presence of an extracellular N-terminal domain (ECD) of 120–160 residues, three intracellular (IL) and extracellular (EL) loops interconnect seven TMD (TM1-TM7) of 310–420 residues that are structurally similar and are thus members of the family B GPCR.37,38 According to Parthier et al. hormonal recognition in family B GPCRs is believed to follow the ‘two-domain’ binding mode, the N- and C-terminal regions of the peptides interact with the J- and N-domains of the receptors respectively, i.e. the C terminus of the peptide initiates a peptide recognition with the ECD, thus allowing the peptide N terminus to bind the TMD ligand-binding pocket activating the receptor and prompting a downstream signalling cascade.34,38–40 The presence of a conserved ECD structure and the ‘two-domain’ binding mode across the family B GPCRs suggest a similar receptor activation across the GPCR family.38
The secretin receptors have immense potential in drug discovery due to their importance in fundamental homeostatic functions.31,38 To date, three of these hormones (glucagon, parathyroid hormone and calcitonin) are used clinically for the treatment of hypoglycaemia, osteoporosis and hypercalcaemia individually.31 GLP1-R and GLP2-R are particularly relevant targets, as a result of their part in appetite control and the treatment of type 2 diabetes.31
Family C (metabotropic glutamate receptors)
The family C GPCRs comprise of the two γ-aminobutyric acidB receptors (GABAB receptors), odorant receptors in fish, eight metabotropic glutamate receptors (mGlu receptors or GRM), pheromone receptors, Ca2+-sensing receptors (CaS receptors or CASR), sweet and umami taste receptors (TAS1R1-3), GPCR Class C Group 6 Member A (GPRC6A) and seven orphan receptors.3,4,31,41 The taste receptors in this GPCR family are targeted by the taste additives used in the food industry.41 The CaS, mGlu and GABAB receptors belong to a novel category of drug targets that are essential for considering conditions which affect the central nervous system and calcium homeostasis.42 Currently, family C GPCRs are targeted by two therapeutic drugs in the market. One is Cinacalcet,41–45 the first GPCR allosteric modulator to be marketed, which targets the CaS receptor. The other is Baclofen (now sold under the brand names Lioresal, Liofen, Gablofen, etc.), which is a GABAB agonist used in the treatment of muscle spasms.41,42,44–46
The family C GPCRs differ from others by possessing a large extracellular domain, distal to the TMD receptors, and containing the orthosteric sites; they also form constitutive dimers with unique activation systems in comparison with other GPCR families.41 Similarly, to their related families, family C GPCRs exhibit a typical motif of seven TMD helices however differ structurally from other GPCR families in their possession of an unusually large extracellular domain, an intracellular carboxyl-terminal (C-terminal) domain and a heptahelical TMD (Fig. 5A).41 The family C GPCRs are structurally distinct from other GPCR families as a result of their extracellular domain including a cysteine rich domain (CRD, with the exception of GABAB receptor) and Venus flytrap module (VFT).31,41 The TM domain of family C GPCRs contain only the allosteric binding sites differing from other families with their TM domains conserved while the orthosteric sites are situated in the VFT module.41,44 Domains present in the family C GPCRs provide numerous ligand sites of action, bar the intracellular C-terminal domain; this is highly variable and plays an essential role in signalling protein coupling and scaffolding.41 The family C GPCRs are unique due to their compulsory dimerization, either as heterodimers (GABAB receptor and TIRs) or homodimers (mGlu and CaS receptors) (Fig. 5B).41,47,48
Fig. 5. Graphical illustration of family C GPCR structure.41 Reprinted with permission from Springer Nature: Springer Nature, Acta Pharmacologica Sinica, Structure and ligand recognition of class C GPCRs, L. Chun, W.-h. Zhang and J.-f. Liu (2012). (A) Represents the structural organisation of family C GPCRs. Family C GPCRs have a peculiar structure which comprises of VFT with two lobes separated by an orthosteric binding pocket, a CRD and a TMD except for GABAB receptor. (B) Graphical illustration of two members family C GPCRs; GABAB receptor (heterodimer) and mGlu receptor (homodimer). There is a direct link between VFT and TMD in the GABAB receptors and the two subunits, GABAB1 and GABAB2 make an obligatory heterodimer while the VFT connects to TMD using CRD in the mGlu receptors. The mGlu receptors form homodimers which can potentially offer two other orthosteric binding pocket per dimer.41.

Structural differences
GPCRs share a common structural characteristic, the TMD region, with its intracellular C-terminus and extracellular N-terminus, which exhibits the greatest homology.21,28,49,50 The intracellular loops which span TM5 and 6, the amino terminus and the carboxyl terminus are among the most irregular structures in GPCRs with a substantial variation observed in the amino terminus (N-terminus).21,51 The sequence is relatively short for peptide and monoamine receptors comprising of about 10–50 amino acids,21,51 and larger for glutamate family receptors and glycoprotein hormone receptors (350–600 amino acids).21,51 The largest amino terminal domains were observed in the adhesion family receptors.21,51
Bortolato et al. compared crystal structures of family B and family A GPCRs using receptors in the various classes (glucagon receptors, corticotropin-releasing factor receptor 1 (CRF1) and dopamine D3 receptor).52 The comparison of the CRF1 and glucagon receptor crystal structure to dopamine D3 receptor, a family A GPCR, showed that their cytoplasmic regions superimposed well.52 However, the TM6 regions of both glucagon receptors and CRF1 extend outwardly while the cytoplasmic moieties are situated in proximity to the TM3 regions in sites similar to the dopamine, as well as other class A receptors.52 The family B GPCRs lack the direct connectivity between TM3 and TM6 which is regarded as the classical ‘ionic lock’, playing an important role in family A GPCR activation.52,53 The family C GPCRs structurally differ from family A and B due to their remarkably large extracellular domain which comprises of a cysteine-rich domain and VFT; an intracellular carboxyl-terminal (C-terminal) domain. The TMD regions in family A and B GPCRs are conserved however family C GPCRs have the allosteric binding site within the TMD region (Fig. 4).41Table 1 shows some of the characteristics of the GPCR families discussed in this review.
Table showing some characteristics of family A–C GPCRsa.
| Feature | Family A | Family B | Family C | Reference |
|---|---|---|---|---|
| Transmembrane domains | All families possess seven transmembrane domains | 31, 54 and 55 | ||
| Orthosteric binding site | TM region | Extracellular loops, extracellular N-terminus, TM6 | Extracellular N-terminus (VFTM, SUSHI) | 12, 41, 55 and 56 |
| Number of approved and marketed drugs | 33 | 16 | 22 | 57 |
| Motifs | All GPCRs share the D/E-R-Y/W motifs | 30, 49 and 54 | ||
| Number of conserved residues in TMD regions | 25 | 33 | 94 | 55 |
| Type of ligand | Small molecules, proteins, peptides | Proteins, peptides | Small molecules, cations, amino acids | 31 |
| Suitable as drug targets? | Yes, except the sensory receptors | Yes | Yes, except the sensory receptors | 31 |
TM: transmembrane, GPCR(s): G-protein coupled receptor(s), VFTM: venus fly trap module, SUSHI: short consensus repeats.
Allosteric binding and cooperativity
Allostery is a widespread biological process, which is defined as the ability of interactions occurring at a particular site on a molecule to modulate actions on a different binding site on the same molecule.58,59 For example, the binding of an allosteric modulator on a molecule allosterically changes the conformation of its binding pocket as shown in Fig. 6. Currently, there are two types of marketed pharmaceuticals: allosteric modulators, which bind at the allosteric binding site on the receptor and allosterically change the structural conformation of the receptor binding site, and orthosteric modulators, which bind at the active site of the receptor.60 Orthosterically-binding drugs must overcome a major challenge in mediating the potential side effects arising from binding to homologous proteins sharing similar binding sites.60 Hence an orthosterically-binding drug must have a very high affinity for its target, in order for a small dose to selectively achieve the goal of target-only binding.60 The binding of transcription factors (TFs) to DNA regulatory elements (REs) provides a good example illustrating the specificity in orthosteric drugs.60
Fig. 6. Mechanism of action of allosteric modulators.63 Reprinted with permission from Springer Nature: Springer Nature, Nature Reviews Drug Discovery, Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders, P. J. Conn, A. Christopoulos and C. W. Lindsley, Copyright (2009). (a) Allosteric ligands bind to an alternative binding site on a receptor to modulate the activities of an orthosteric ligand efficacy (blue) and/or affinity (red). A number of allosteric ligands can also directly disrupt signalling in their own right (green). (b) Results from simulation show the effects on the function (right) or binding (left) of an orthosteric agonist mediated by three allosteric potentiators depicted in red, blue and green; red enhanced orthosteric agonist affinity only, blue enhanced only the efficacy, green was observed to modestly enhance both efficacy and affinity, as well as showing allosteric agonism.63.

The process of GPCR signaling initiates when an endogenous extracellular signal interacts with the orthosteric binding site of a GPCR, resulting in a conformational change which passes on the signal through the plasma membrane traversing the TMD region, and eventually activating intracellular signaling cascades through heterotrimeric G proteins and other adjunct proteins.58,61,62 A different approach, demonstrated for ligand-gated particle channels, is the advancement of allosteric modulators of the receptor subtypes, these small molecules do not bind to the traditional orthosteric binding site, instead interacting with the allosteric binding site to either enhance or inhibit receptor activation.63
Allosteric GPCR modulators show at least one of the outlined pharmacological properties (Fig. 6). Agonism/reverse agonism: the allosteric modulator disrupts receptor signaling in either a positive (agonism) or negative (antagonism) manner, notwithstanding the presence or absence of an orthosteric ligand.63 Efficacy modulation: the effect of allosterism causes changes in intracellular responses, leading to alterations in the inherent efficacy of an orthosteric ligand.63 Affinity modulation: conformational change influences the orthosteric binding pocket, resulting in dissociation or association rate (or sometimes both) of the ligand being modified (Fig. 6).63 Some known allosteric modulators of family B GPCRs include NovoNordisk compounds 1–6:63 T-0632, which blocks the GLP-1 induced cAMP production63,64 (GLP 1 receptor); DMP696, which blocks the CRF-stimulated adenylyl cyclase activity in cell line expressing CRF1 receptor;63,65 NBI 27914, which blocks the CRF1 receptor;63,66 NBI 35965;63 antarlamin63 (CRF 1 receptor).63
Cooperativity is a thermodynamic term which has varying meanings in different biochemical contexts.67,68 It is used to explain the complex interactions of identical ligands with a receptor at multiple binding sites.67 Cooperativity also describes the thermodynamics of macromolecular conformational transitions, which include nucleic acid helix–coil transitions and protein folding.67 Positive cooperativity is defined as the increase of binding affinity at one site of a receptor when a ligand is bound elsewhere.69 A classic example of positive cooperativity is the binding of oxygen to haemoglobin; the binding of one oxygen molecule to the ferrous iron of the heme molecule increases the affinity of deoxyhaemoglobin for oxygen.69 Negative cooperativity is observed when 2,3-bisphosphoglycerate binds to an allosteric binding site of haemoglobin and the affinity for oxygen is reduced.67,69
GPCR signalling via G-proteins
G-proteins consist of several families of varied cellular proteins which perform several cellular functions, such as contractility and angiogenesis, learning and memory.70,71 These proteins bind to the guanine nucleotides (guanine diphosphate (GDP) and guanine triphosphate (GTP)) and also have inherent GTPase activity.71 They play a principal role in a many cellular processes, including protein synthesis and cell development, vesicular transport, and cytoskeleton assembly, in addition to signal transduction.71 G-proteins are trimers comprising of two functional components: a beta-gamma dimer (35 and 8 kDa) which closely relates with the alpha subunit upon binding with GDP, and an alpha subunit (39–52 kDa) which is a catalyst for GTPase activity.72 Human G proteins are classified into two classes, namely small (monomeric), and heterotrimeric G proteins.71,72
GPCRs are the largest superfamily of cell-surface receptors involved in TMD signalling, usually transmitting signals into cells via their response to a range of extracellular stimuli, such as glycoproteins, polypeptides and ions, and hence regulating a wide variety of physiological and developmental functions.73 The intracellular-signalling cascades activated by GPCRs have been proven to be remarkably complex.73,74 The binding of a ligand to the GPCR binding site leads to a conformational change in the receptor, in turn promoting the binding of the heterotrimeric G proteins, consisting of Gα-GDP and Gβγ-subunits, within the intracellular moiety of the receptor.74 The exchange of GTP for GDP on the Gα-subunit results in the reversible dissociation of the G protein subunits, initiating a downstream signalling via Gα-GTP and Gβγ.73,74
Dielectric constant
The most effective way of correlating the structure and function of macromolecules is through the examination of its electrostatic energies.75 The intermolecular interactions present are affected by the effective dielectric constant (relative permittivity, εr),76 which differs according to the size and composition of the protein.77 The accuracy of the method of determination is important in understanding various biochemical interactions such as protein–ligand and protein–protein interactions, charge separation, ion channel selectivity and electron and proton transfer signal transduction and macromolecular assembly;77,78 these interactions are influenced by the electrostatic potential of the protein surface.77–79 The dielectric constant of dry proteins ranges from 2.5 to 3.5 obtained from direct measurement.78 The theoretical calculation of local dielectric constant of lone proteins based on their amino acid composition yielded an average of 2.7.80 The polarity of the residues which make up the structural motifs within a protein have been shown to affect its dielectric constant values, these findings were based on computational studies based on continuum electrostatics and molecular dynamics simulations.77,78
According to Warshel and Åqvist, the value of the dielectric constant of proteins is dependent on the property used to define it. They highlighted several possible ways of defining the dielectric constant in proteins, as outlined in Table 2,81 where Q1 and Q2 are charges on ionisable groups separated by distance r, μ is a group dipole moment (in units of electron Ångström), ΔG is the electrostatic Gibbs free energy, ā is the effective radius of charge, and εB is the effective dielectric constant associated with a given interaction.
Some rules for the definition of dielectric constants in proteins.
| Definition | Value | Comments |
|---|---|---|
| Polar = ε large | ε = large | Protein sites are always polar near small radii ions. |
| Nonpolar = ε small | ||
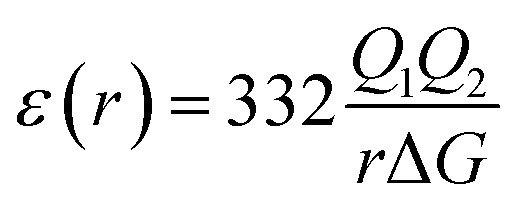
|
ε(r) > 10 often ε(r) ≥ 40 | The value of ε is large for charge–charge interactions. |
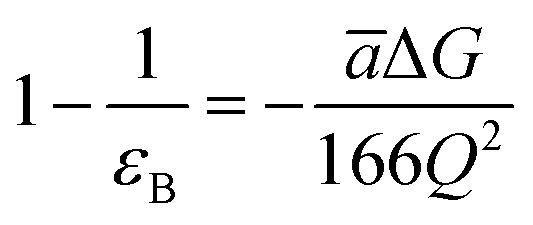
|
ε B > 10 | Proteins can provide as much solvation as water for ionised groups with small radii. |
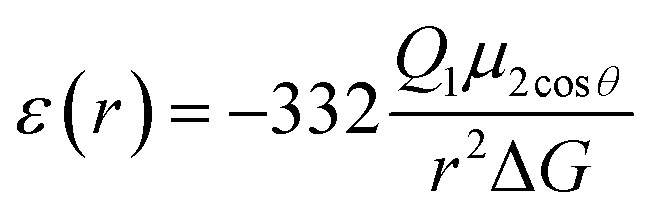
|
ε ≥ 4 | For functionally important charge–dipole interactions, the value of ε could be as small as 4. Such a low value, however, requires relatively fixed dipoles with little energy for reorganisation. |
Li et al. reported that the average dielectric constant inside a protein is relatively low, about 6–7, but this figure reaches about 20–30 on the surface of the protein.82 The high average local dielectric constant values are often linked to the charged residues while the low values are assigned automatically to the regions comprised of mostly hydrophobic residues.82
According to Wilson et al. solvent effects on mechanisms of reactions have been established, but its effect on kinetic isotope effects (KIEs) are rather well less comprehended.83 A change in solvent can alter the KIE indirectly by changing the transition-state (TS) structure. It can also affect KIE by affecting isotopically sensitive vibrational frequencies directly, notwithstanding the TS structure or identity of the rate-determining step.83 Wilson et al. investigated the medium effects on KIE for SN2 methyl transfer using UFF or UAO cavity method within the polarized continuum model (PCM) and a hybrid quantum mechanical/molecular mechanical (QM/MM) method.83 Their findings showed that the majority of variation in the equilibrium isotope effects (EIE) occur within the same range of dielectric constants (1 ≤ ε ≤ 10) as is considered to occur with enzyme active sites and proteins.83 There is a possibility that any reaction which involves separation, neutralisation or charge distribution within an enzyme active site could indicate variations in KIEs, between a wildtype and mutant form of an enzyme, which originates as a result of changes in the local dielectric response within the diverse protein environment.83 The use of UFF or UAO cavity method within the polarized continuum model (PCM) and a hybrid QM/MM method to characterise ligand binding in GPCRs would further assist in understanding the interactions which occur in the both the active and inactive states of GPCRs, as well the changes which occur during the transition from inactive state to active state upon ligand activation.
Computational biology techniques in GPCR research
The first major breakthrough in human GPCR structural biology took place in 2007 as the solving of the β2-adrenergic receptor (β2AR with a diffusible ligand) using a modified lipidic cubic phase (LCP) produce to produce β2AR-TCL crystals which diffracted to a resolution of 2.2 Å, the structure was further refined at a 2.4 Å resolution.12 Presently 64 structures of unique GPCRs with varying resolutions have been solved using spectroscopic methods such as fluorescence, electron paramagnetic resonance (EPR) and nuclear magnetic resonance (NMR) spectroscopy and structural techniques such as cryogenic electron microscopy (cryo-EM), this provides opportunities in employing computational biology techniques such as molecular modelling, and molecular docking in drug discovery research.84,85 The milestones achieved in GPCR structural studies have provided insights on the arrangements of the transmembrane domains,1–5,11,12 the location of the orthosteric,12,31,41 allosteric,12,31,41 bitopic,12 as well as biased ligand binding sites,12 the homo- or hetero-oligomerization of receptors12 and the structural rearrangements associated with conformational changes upon GPCR activation and inactivation.12 This base of structural information on GPCRs is vital for SBDD,12,86 ligand-based drug design (LBDD),12 and integrated models which complement drug discovery efforts.12
In 2012, Sosei Heptares published a detailed account on the use of A2AR structure in identifying series of agents as potential antagonists, this became the first published GPCR SBDD discovery.87 In a research carried out by de Graaf et al. using structure based virtual screening (SBVS), they identified allosteric modulators of two family B receptors namely; glucagon receptor and glucagon-like peptide receptor.88 SBDD approaches has also lead to the development of new agonists of the A3 adenosine receptor (A3AR).89
Conclusion and future prospects
GPCRs are multifaceted proteins which exist in varying conformations, and that the conformational equilibrium of these group of receptors is influenced both by the bound ligand and the proximity to the related G protein. Their structure is highly conserved comprising of seven TMD. These receptors possess different binding domains, namely; allosteric and orthosteric binding domains. The progress in GPCR structural biology has substantially accelerated our understanding of GPCRs as potential drug targets using SBDD and LBDD approaches. Further computational studies assessing nuclear quantum effects on ligand receptor binding, as well as hybrid QM/MM and empirical valence bond theory in the mechanistic studies of GPCRs would allow for further insight into the interactions which occur in both the active and inactive states of GPCRs, as well the changes which occur during the transition from these states upon ligand activation. This review has aimed to provide an accessible and introductory perspective on advances in GPCR-based drug discovery approaches; many reviews on the topic highlighted herein are indeed highly detailed and authoritative but may not provide as accessible an account for a less specialised or more general audience in the chemical sciences.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
References
- Dijkman P. M. Castell O. K. Goddard A. D. Munoz-Garcia J. C. de Graaf C. Wallace M. I. Watts A. Nat. Commun. 2018;9:1710. doi: 10.1038/s41467-018-03727-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeze W. K. Sheffler D. J. Roth B. L. J. Cell Sci. 2003;116:4867. doi: 10.1242/jcs.00902. [DOI] [PubMed] [Google Scholar]
- Fredriksson R. Lagerström M. C. Lundin L.-G. Schiöth H. B. Mol. Pharmacol. 2003;63:1256. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Schiöth H. B. Fredriksson R. Gen. Comp. Endocrinol. 2005;142:94–101. doi: 10.1016/j.ygcen.2004.12.018. [DOI] [PubMed] [Google Scholar]
- Ghosh E. Kumari P. Jaiman D. Shukla A. K. Nat. Rev. Mol. Cell Biol. 2015;16:69–81. doi: 10.1038/nrm3933. [DOI] [PubMed] [Google Scholar]
- Hauser A. S. Attwood M. M. Rask-Andersen M. Schiöth H. B. Gloriam D. E. Nat. Rev. Drug Discovery. 2017;16:829–842. doi: 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X.-l. Wang Y. Li D.-l. Luo J. Liu M.-y. Acta Pharmacol. Sin. 2012;33:363–371. doi: 10.1038/aps.2011.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. R. Duc N. M. Chung K. Y. Biomol. Ther. 2018;26:101–108. doi: 10.4062/biomolther.2017.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram K. Insel P. A. Mol. Pharmacol. 2018;93:251–258. doi: 10.1124/mol.117.111062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel P. A. Sriram K. Gorr M. W. Wiley S. Z. Michkov A. Salmerón C. Chinn A. M. Trends Pharmacol. Sci. 2019;40:378–387. doi: 10.1016/j.tips.2019.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K. A. Costanzi S. Paoletta S. Trends Pharmacol. Sci. 2014;35:658–663. doi: 10.1016/j.tips.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basith S. Cui M. Macalino S. J. Y. Park J. Clavio N. A. B. Kang S. Choi S. Front. Pharmacol. 2018;9:128. doi: 10.3389/fphar.2018.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heifetz A. Schertler G. F. X. Seifert R. Tate C. G. Sexton P. M. Gurevich V. V. Fourmy D. Cherezov V. Marshall F. H. Storer R. I. Moraes I. Tikhonova I. G. Tautermann C. S. Hunt P. Ceska T. Hodgson S. Bodkin M. J. Singh S. Law R. J. Biggin P. C. Naunyn-Schmiedeberg's Arch. Pharmacol. 2015;388:883–903. doi: 10.1007/s00210-015-1111-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X. Xu Y. Int. J. Mol. Sci. 2018;19:2105. doi: 10.3390/ijms19072105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esguerra M. Siretskiy A. Bello X. Sallander J. Gutiérrez-de-Terán H. Nucleic Acids Res. 2016;44:W455–W462. doi: 10.1093/nar/gkw403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich V. V. Gurevich E. V. Int. J. Mol. Sci. 2017;18:2519. doi: 10.3390/ijms18122519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park P. S. H. Curr. Med. Chem. 2012;19:1146–1154. doi: 10.2174/092986712799320619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provasi D. Artacho M. C. Negri A. Mobarec J. C. Filizola M. PLoS Comput. Biol. 2011;7:e1002193. doi: 10.1371/journal.pcbi.1002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samama P. Cotecchia S. Costa T. Lefkowitz R. J. J. Biol. Chem. 1993;268:4625–4636. [PubMed] [Google Scholar]
- de Munnik S. M. Smit M. J. Leurs R. Vischer H. F. Front. Pharmacol. 2015;6:40. doi: 10.3389/fphar.2015.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka B. K. Biochim. Biophys. Acta. 2007;1768:794–807. doi: 10.1016/j.bbamem.2006.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockaert J. Philippe Pin J. EMBO J. 1999;18:1723–1729. doi: 10.1093/emboj/18.7.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies M. N. Secker A. Freitas A. A. Mendao M. Timmis J. Flower D. R. Bioinformatics. 2007;23:3113–3118. doi: 10.1093/bioinformatics/btm506. [DOI] [PubMed] [Google Scholar]
- Horn F. Bettler E. Oliveira L. Campagne F. Cohen F. E. Vriend G. Nucleic Acids Res. 2003;31:294–297. doi: 10.1093/nar/gkg103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn F. Weare J. Beukers M. W. Hörsch S. Bairoch A. Chen W. Edvardsen Ø. Campagne F. Vriend G. Nucleic Acids Res. 1998;26:275–279. doi: 10.1093/nar/26.1.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G.-M. Mai T.-L. Chen C.-M. Sci. Rep. 2017;7:15495. doi: 10.1038/s41598-017-15707-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pándy-Szekeres G. Munk C. Tsonkov T. M. Mordalski S. Harpsøe K. Hauser A. S. Bojarski A. J. Gloriam D. E. Nucleic Acids Res. 2018;46:D440–D446. doi: 10.1093/nar/gkx1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum D. M. Rasmussen S. G. F. Kobilka B. K. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gacasan S. B. Baker D. L. Parrill A. L. AIMS Biophys. 2017;4:491–527. doi: 10.3934/biophy.2017.3.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K. Annu. Rev. Biochem. 2006;75:743–767. doi: 10.1146/annurev.biochem.75.103004.142743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerström M. C. Schiöth H. B. Nat. Rev. Drug Discovery. 2008;7:339–357. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- Teller D. C. Okada T. Behnke C. A. Palczewski K. Stenkamp R. E. Biochemistry. 2001;40:7761–7772. doi: 10.1021/bi0155091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K. Dockerill S. Adamiak D. A. Tickle I. J. Blundell T. Nature. 1975;257:751–757. doi: 10.1038/257751a0. [DOI] [PubMed] [Google Scholar]
- Parthier C. Reedtz-Runge S. Rudolph R. Stubbs M. T. Trends Biochem. Sci. 2009;34:303–310. doi: 10.1016/j.tibs.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Wheatley M. Wootten D. Conner M. T. Simms J. Kendrick R. Logan R. T. Poyner D. R. Barwell J. Br. J. Pharmacol. 2012;165:1688–1703. doi: 10.1111/j.1476-5381.2011.01629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace C. R. R. Perrin M. H. DiGruccio M. R. Miller C. L. Rivier J. E. Vale W. W. Riek R. Proc. Natl. Acad. Sci. U. S. A. 2004;101:12836–12841. doi: 10.1073/pnas.0404702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karageorgos V. Venihaki M. Sakellaris S. Pardalos M. Kontakis G. Matsoukas M.-T. Gravanis A. Margioris A. Liapakis G. Hormones. 2018;17:45–59. doi: 10.1007/s42000-018-0009-5. [DOI] [PubMed] [Google Scholar]
- de Graaf C. Song G. Cao C. Zhao Q. Wang M.-W. Wu B. Stevens R. C. Trends Biochem. Sci. 2017;42:946–960. doi: 10.1016/j.tibs.2017.10.003. [DOI] [PubMed] [Google Scholar]
- Wu F. Yang L. Hang K. Laursen M. Wu L. Han G. W. Ren Q. Roed N. K. Lin G. Hanson M. A. Jiang H. Wang M.-W. Reedtz-Runge S. Song G. Stevens R. C. Nat. Commun. 2020;11:1272. doi: 10.1038/s41467-020-14934-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenstein K. de Graaf C. Bortolato A. Wang M.-W. Marshall F. H. Stevens R. C. Trends Pharmacol. Sci. 2014;35:12–22. doi: 10.1016/j.tips.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun L. Zhang W.-h. Liu J.-f. Acta Pharmacol. Sin. 2012;33:312–323. doi: 10.1038/aps.2011.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondard P. Goudet C. Kniazeff J. Pin J.-P. Prézeau L. Neuropharmacology. 2011;60:82–92. doi: 10.1016/j.neuropharm.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Hellyer S. D. Albold S. Wang T. Chen A. N. Y. May L. T. Leach K. Gregory K. J. Mol. Pharmacol. 2018;93:504. doi: 10.1124/mol.117.111518. [DOI] [PubMed] [Google Scholar]
- Hans B.-O. Wellendorph P. Anders J. Curr. Drug Targets. 2007;8:169–184. doi: 10.2174/138945007779315614. [DOI] [PubMed] [Google Scholar]
- Tautermann C. S. Bioorg. Med. Chem. Lett. 2014;24:4073–4079. doi: 10.1016/j.bmcl.2014.07.009. [DOI] [PubMed] [Google Scholar]
- Roth B. L. Nat. Struct. Mol. Biol. 2019;26:535–544. doi: 10.1038/s41594-019-0252-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. C. Liu J. Jiang D. Protein Cell. 2014;5:492–495. doi: 10.1007/s13238-014-0062-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller T. C. Moreno-Delgado D. Pin J.-P. Kniazeff J. Biophys. Rep. 2017;3:57–63. doi: 10.1007/s41048-017-0036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D. Zhao Q. Wu B. Mol. Cells. 2015;38:836–842. doi: 10.14348/molcells.2015.0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong M. Koole C. Wootten D. Sexton P. M. Miller L. J. Br. J. Pharmacol. 2014;171:1085–1101. doi: 10.1111/bph.12293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orel M. Padrós E. Manyosa J. FEBS Journal. 2012;279:2357–2367. doi: 10.1111/j.1742-4658.2012.08614.x. [DOI] [PubMed] [Google Scholar]
- Bortolato A. Doré A. S. Hollenstein K. Tehan B. G. Mason J. S. Marshall F. H. Br. J. Pharmacol. 2014;171:3132–3145. doi: 10.1111/bph.12689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culhane K. J. Liu Y. Cai Y. Yan E. C. Y. Front. Pharmacol. 2015;6:264. doi: 10.3389/fphar.2015.00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewatia P. Agrawal N. Gaur M. Sahi S. Biochimie. 2014;101:168–182. doi: 10.1016/j.biochi.2014.01.016. [DOI] [PubMed] [Google Scholar]
- Trzaskowski B. Latek D. Yuan S. Ghoshdastider U. Debinski A. Filipek S. Curr. Med. Chem. 2012;19:1090–1109. doi: 10.2174/092986712799320556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis W. I. Kobilka B. K. Annu. Rev. Biochem. 2018;87:897–919. doi: 10.1146/annurev-biochem-060614-033910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D. Stevens R. C. Roth B. L. Cell. 2017;170:414–427. doi: 10.1016/j.cell.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry P. R. Sexton P. M. Christopoulos A. J. Biol. Chem. 2015;290:19478–19488. doi: 10.1074/jbc.R115.662759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motlagh H. N. Wrabl J. O. Li J. Hilser V. J. Nature. 2014;508:331–339. doi: 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R. Chung-Jung T. Curr. Pharm. Des. 2012;18:1311–1316. doi: 10.2174/138161212799436377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuteja N. Plant Signaling Behav. 2009;4:942–947. doi: 10.4161/psb.4.10.9530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanlon C. D. Andrew D. J. J. Cell Sci. 2015;128:3533–3542. doi: 10.1242/jcs.175158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn P. J. Christopoulos A. Lindsley C. W. Nat. Rev. Drug Discovery. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibaduiza E. C. Chen C. Beinborn M. J. Biol. Chem. 2001;276:37787–37793. doi: 10.1074/jbc.M106692200. [DOI] [PubMed] [Google Scholar]
- Li Y.-W. Fitzgerald L. Wong H. Lelas S. Zhang G. Lindner M. D. Wallace T. McElroy J. Lodge N. J. Gilligan P. Zaczek R. CNS Drug Rev. 2005;11:21–52. doi: 10.1111/j.1527-3458.2005.tb00034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baram T. Z. Chalmers D. T. Chen C. Koutsoukos Y. De Souza E. B. Brain Res. 1997;770:89–95. doi: 10.1016/S0006-8993(97)00759-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson J. R. Nat. Chem. Biol. 2008;4:458–465. doi: 10.1038/nchembio.102. [DOI] [PubMed] [Google Scholar]
- Lenaerts T. Ferkinghoff-Borg J. Schymkowitz J. Rousseau F. BMC Syst. Biol. 2009;3:9. doi: 10.1186/1752-0509-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisov I. G. Sligar S. G. Arch. Biochem. Biophys. 2012;519:91–102. doi: 10.1016/j.abb.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jong Y.-J. I. Harmon S. K. O'Malley K. L. Br. J. Pharmacol. 2018;175:4026–4035. doi: 10.1111/bph.14023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariou V., Duman R. S. and Nestler E. J., in Basic Neurochemistry, ed. S. T. Brady, G. J. Siegel, R. W. Albers and D. L. Price, Academic Press, New York,8th edn, 2012, pp. 411–422 [Google Scholar]
- Schulman H., in From Molecules to Networks, ed. J. H. Byrne, R. Heidelberger and M. N. Waxham, Academic Press, Boston, 3rd edn, 2014, pp. 119–148 [Google Scholar]
- Doijen J. Van Loy T. Landuyt B. Luyten W. Schols D. Schoofs L. Biosens. Bioelectron. 2019;137:33–44. doi: 10.1016/j.bios.2019.04.041. [DOI] [PubMed] [Google Scholar]
- Augustine G. J., Neuroscience, ed. D. Purves, G. Augustine, D. Fitzpatrick, L. Katz, A.-S. LaMantia, J. McNamara and M. Williams, Sinauer Associates, Sunderland MA, 3rd edn, 2004 [Google Scholar]
- Warshel A. Papazyan A. Curr. Opin. Struct. Biol. 1998;8:211–217. doi: 10.1016/S0959-440X(98)80041-9. [DOI] [PubMed] [Google Scholar]
- Braslavsky S. E. Pure Appl. Chem. 2007;79:293–465. [Google Scholar]
- Amin M. Küpper J. ChemistryOpen. 2020;9:691–694. doi: 10.1002/open.202000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin M. and Küpper J., 2020, arXiv e-prints, arXiv:2001.07053
- Schutz C. N. Warshel A. Proteins: Struct., Funct., Bioinf. 2001;44:400–417. doi: 10.1002/prot.1106. [DOI] [PubMed] [Google Scholar]
- Alshami A. S. Tang J. Rasco B. Food Bioprocess Technol. 2017;10:1548–1561. doi: 10.1007/s11947-017-1920-5. [DOI] [Google Scholar]
- Warshel A. Aqvist J. Annu. Rev. Biophys. Biophys. Chem. 1991;20:267–298. doi: 10.1146/annurev.bb.20.060191.001411. [DOI] [PubMed] [Google Scholar]
- Li L. Li C. Zhang Z. Alexov E. J. Chem. Theory Comput. 2013;9:2126–2136. doi: 10.1021/ct400065j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson P. B. Weaver P. J. Greig I. R. Williams I. H. J. Phys. Chem. B. 2015;119:802–809. doi: 10.1021/jp505344a. [DOI] [PubMed] [Google Scholar]
- Jaiteh M. Rodríguez-Espigares I. Selent J. Carlsson J. PLoS Comput. Biol. 2020;16:e1007680. doi: 10.1371/journal.pcbi.1007680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilger D. Masureel M. Kobilka B. K. Nat. Struct. Mol. Biol. 2018;25:4–12. doi: 10.1038/s41594-017-0011-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakliang P. Lazim R. Chang H. Choi S. Biomolecules. 2020;10:631. doi: 10.3390/biom10040631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congreve M. de Graaf C. Swain N. A. Tate C. G. Cell. 2020;181:81–91. doi: 10.1016/j.cell.2020.03.003. [DOI] [PubMed] [Google Scholar]
- de Graaf C. Rein C. Piwnica D. Giordanetto F. Rognan D. ChemMedChem. 2011;6:2159–2169. doi: 10.1002/cmdc.201100317. [DOI] [PubMed] [Google Scholar]
- Ciancetta A. and Jacobson K. A., in Computational Methods for GPCR Drug Discovery, ed. A. Heifetz, Springer New York, New York, NY, 2018, pp. 45–72 [Google Scholar]