

Abstract
To evaluate a novel lamellar structural biomaterial as a potential biomaterial for guided bone regeneration, we describe the preparation of a collagen membrane with high mechanical strength and anti-enzyme degradation ability by using the multi-level structure of Ctenopharyngodon idella scales. The physical and chemical properties, in vitro degradation, biocompatibility, and in vivo osteogenic activity were preliminarily evaluated. In conclusion, it was shown that the multi-layered collagen structure material had sufficient mechanical properties, biocompatibility, and osteogenic ability. Meanwhile, it is also shown that there is a gap in current clinical needs, between the guided tissue regeneration membrane and the one being used. Therefore, this study provides useful insights into the efforts being made to design and adjust the microstructure to balance its mechanical properties, degradation rate, and osteogenic activity.
To evaluate a novel lamellar structural biomaterial for guided bone regeneration, we describe the preparation of a collagen membrane with high mechanical strength and anti-enzyme degradation ability using Ctenopharyngodon idella scales.
1. Introduction
When Nyman used a semipermeable membrane to treat the periodontium, he invented the guided tissue regeneration technology (GTR) in 1982, and then extended it to the treatment of bone defects.1 In 1993, Buser et al. proposed the concept of guided bone regeneration (GBR), which involves placing the membrane in the bone defect area, using the barrier function of the membrane to prevent non-osteoblasts from growing into the defect and forming a space under the membrane, and allowing the osteoblasts to migrate and grow preferentially, so as to achieve bone regeneration and repair.2 The regenerative effects of the GBR membrane make it useful in the treatment of bone defects, and oral craniomaxillofacial defect repair and implants are widely used. Therefore, GBR membranes have attracted a lot of research attention, both at home and abroad.3–5
The membrane material is the key factor in GBR technology's effectiveness. The membranes used for bone regeneration can be divided into absorbable and non-absorbable membranes. Polytetrafluoroethylene (PTFE) membranes have the ability to perfectly maintain the cavity shape and size, and its mechanical properties are stable. However, as a typical non-absorbable membrane, it needs to be removed by secondary surgery, and the risk of infection is very high, which limits its applications.6 The most common absorbable material is collagen, which can be modified through various collagen cross-linking processing techniques to change the resorption rate. Because of the presence of collagen in alveolar bone and periodontal ligament, using a collagen membrane might impart some additional advantages for GBR purposes by augmenting its native properties. Bio-Gide (BG; Geistlich, Wolhusen, Switzerland) is a typical membrane derived from pig collagen with a double-layer structure. Many studies have shown that it has a good ability to guide bone regeneration.7,8 However, BG has a major defect, that is, the degradation in vivo is too fast, and the mechanical properties of the wet state are poor, which cannot provide a good shielding and spatial structure in the bone regeneration cycle.9,10 To date, the mechanical properties of the degradable collagen membrane prepared by the reconstructed collagen materials seem to be unable to meet the requirements of soft tissue pressure during the healing process.11,12
Comparing the non-absorbable PTFE membrane and the absorbable collagen membrane, their advantages and disadvantages are very obvious. Although clinicians and patients prefer degradable membranes, the excellent wet mechanical properties of PTFE membranes cannot be achieved in clinical use. Therefore, it is key to improve the wet mechanical properties of the collagen membrane. Fish scales are mainly composed of calcium phosphorus compounds and collagen, which have attracted attention owing to their highly ordered multilayer structure.13,14 Generally, hydroxyapatite, collagen, and derivatives are extracted from fish scales. The collagen and derivatives prepared in this way destroy the original collagen hierarchical structure in fish scales. Even after cross-linking or modification, the mechanical strength and anti-degradation performance of the prepared guided tissue regeneration membranes are still poor.12,15 Wang et al.16 found in researching bionics that keeping the original multi-level structure of biological tissue can obtain better biomechanical properties than single-level structure materials. In our previous work, it was shown that a fish scale collagen matrix has a layered structure, special physical parameters, and biological characteristics, which can ensure sufficient time and space for bone regeneration.17
In this study, based on the fact that Ctenopharyngodon idella is one of the largest freshwater fishes in China and its scale utilisation rate is low, we described the preparation of a collagen membrane (FS) with high mechanical strength and antienzyme degradation ability by using the multi-level structure of fish scales. It is intended to be used as a guided tissue regeneration material, and its physical and chemical properties, in vitro degradation, biocompatibility, and in vivo osteogenic activity were preliminarily evaluated.
2. Materials and methods
2.1. Preparation of fish scales (FS)
The preparation method was based on our previous research.17C. idella fish scales about 2.5 cm-diameter and 1000 g weight, were collected (Chengdu, Sichuan Province, China), and then washed at 15 °C in turn with distilled water, mixed solvents, and buffers to remove mucopolysaccharides, proteins, and fats. The scales were then immersed in 10% EDTA solution at 4 °C for decalcification for about 4 h, then cleaned with water and incubated in acetic acid solution at 4 °C for 1 h to obtain the basal plates from the scales. The basal plates were evenly placed on an etching disc with the osseous layer of the basal plates facing upwards, and the etching device was started. The etching solution was uniformly sprayed on the surface of the basal plates once every hour in the etching device, which was maintained at 4 °C. Only one side of the osseous layer was etched, and the inner layer was not etched in this experiment. The etched samples were rinsed with 75% ethanol and double-distilled water, and then stored in sterile phosphate-buffered saline (PBS) at 4 °C (Fig. 1).
Fig. 1. Schematic diagram of the process to prepare FS.

2.2. Characterisation of BG and FS
2.2.1. Morphological observation
The sectional morphologies of the BG and FS were analysed using scanning electron microscope (SEM, Hitachi, Japan). The resulting freeze-dried samples were cut into small pieces, and all detected surfaces were coated with gold before measurement.
2.2.2. Mechanical tests
The average thickness of the BG and FS was observed using a micrometer caliper (Mitutoyo, Japan). The tensile strength and elongation at break of the rectangular samples (2 cm × 2 cm) were determined using a universal testing machine (Shimadzu, Japan) at 25 °C under dry and wet conditions, respectively.
2.2.3. Swelling ratio
BG and FS (1 cm × 1 cm) were weighed and soaked in distilled water at room temperature for 3 h. Finally, we weighed and measured the wet scaffold according to the following equation: swelling ratio 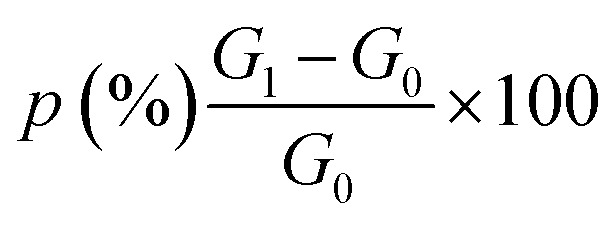 , where G1 represents the weight of the wet scaffold, and G0 represents the weight of the dry scaffold.
, where G1 represents the weight of the wet scaffold, and G0 represents the weight of the dry scaffold.
2.2.4. Water contents
The water contact angles of the BG and FS (1 cm × 1 cm) were measured with a flat surface. When the droplet was connected to the surface of the scaffold, its final surface shape was dependent upon the internal cohesion and adhesive force on the surface of the scaffold. Specifically, 20.0 μL of liquid was dropped onto the surface of the scaffold, photographed, and the contact angle was recorded using a drop analyser (Zhongchen, China).
2.2.5. Fourier transform infra-red spectroscopy
The BG and FS (1 cm × 1 cm) were analysed using an infra-red spectrometer under the following conditions: scan range of 450–4000 cm−1, sample scan time of 32 s, resolution of 4 cm−1, and indoor temperature of 25 °C.
2.2.6. Degradation rate in vitro
To evaluate the degradation rate of the BG and FS, each 1 cm × 1 cm rectangular membrane was determined as M0 after lyophilisation. Approximately 18 samples of each group, then the BG was immersed in 6 mL 0.4 mg mL−1 collagenase type I solution, and the FS was immersed in 6 mL 1.5 mg mL−1 collagenase type I solution, all incubated under 37 °C. Each timepoint (1, 2, 3, 6, 9, and 15 days), three samples from each group were determined after rinsing with distilled water and freeze-drying, separately, and weighed as M1. Residual weight 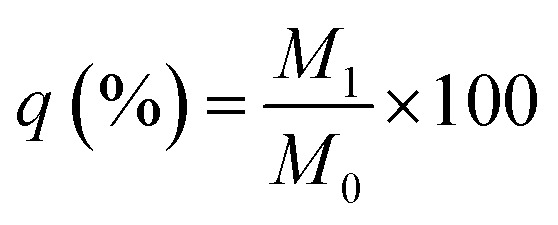 .
.
2.3. Animal experiments
The study protocol was approved by the Ethics Review Committee of the West China School of Stomatology, Sichuan University (WCHSIRB-D-2017-217). The study group consisted of 36 3 month-old Sprague-Dawley (SD) male rats, each weighing an average of 300 g. Animals were fed standard pellets and water.
2.3.1. Subcutaneous implantation and surgical procedures
According to the protocols of Deliormanli et al.,12 six male SD rats were used for the subcutaneous implantation procedures. After the surgical area was aseptically scraped, 10% chloral hydrate (3 mL kg−1) was intraperitoneally injected. We had made six subcutaneous incisions through the cutaneous tissue using a surgical scissor per rat, with each incision spacing 20 mm, then FS and BG were implanted into the dorsal flank of the same rat, respectively, and the remaining two incisions were the same as those of the blank group. Finally, the skin was sutured for primary closure. They were administered 10% chloral hydrate anaesthesia (3 mL kg−1) at the first week after surgery, for the following tests.
2.3.2. Cranial bone defect implantation and surgical procedures
30 SD rats were randomly divided into the blank, BG, and FS groups, with 10 rats in each group. Group 1 (n = 10): the blank group, no biomaterial was applied; group 2 (n = 10): the BG group, BG were applied; group 3 (n = 10): the FS group, FS was applied at the cranial defect, respectively.
After the surgical area was scraped aseptically, 10% chloral hydrate (3 mL kg−1) was intraperitoneally injected, and a V-shaped incision was created on the rat cranial bone with a blade. After the cranial bone surface was exposed, a 5 mm-diameter surgical trephine was used to form two bilateral symmetric full-thickness defects that reached the dura mater surface. The BG and FS (5 mm × 5 mm) were then laid above the cranial bone defect. Finally, the skin was sutured for primary closure. Five rats in each group were administered 10% chloral hydrate anaesthesia (3 mL kg−1) at the 4th and 8th week after surgery, separately, for the following tests.18
2.3.3. Micro CT analysis
The rat cranial bone was cautiously removed, cleaned, fixed in 4% polyoxymethylene for 24 h, and finally stored in 75% alcohol at 4 °C. After the samples were fixed in the scan tube, they were scanned and reconstructed using MicroCT50 (SCANCO Medical AG, Switzerland). Simultaneously, bone volume/total volume (BV/TV), trabecular number (Tb.N), and trabecular space (Tb.Sp) were also measured.
2.3.4. Histological staining and analysis
The cranial bone samples were decalcified in 10% ethylene diamine tetraacetic acid (EDTA) solution for 2 months, dehydrated in absolute alcohol, cleared with xylene, and finally embedded in paraffin. 5 mm-thick slices of each bone sample were stained with the Haematoxylin & Eosin (H&E) and toluidine blue in accordance with protocols.
2.4. Statistical analysis
SPSS 20.0 and Origin 8.5 software were used for data analysis. Three parallel experiments were set up in each experiment, and then the experimental data were analysed by variance (p < 0.05) and represented as X ± S.
3. Results
3.1. Morphological observation
FS was transparent and tough, with uniform thickness and smooth surface (Fig. 2A). The longitudinal section of the FS had a multi-layered structure, with a parallel arrangement of fibres (Fig. 2B). The fibres were arranged in parallel (Fig. 2C), while the other part of the FS treated with pepsin was denser (Fig. 2D). BG presented as pale white, translucent, as rectangular patches with a smooth surface (Fig. 2E). The arrangement of fibres in the longitudinal section of BG was regular but scattered in the gap (Fig. 2F). In the cross section, the spongy layer displayed a dynamic distribution of fibres (Fig. 2G). The compact layer consisted of adjacent fibres, imparting a smooth and uniform surface with small collagenous spaces (Fig. 2H).
Fig. 2. SEM morphological observations. FS: (A) general view; (B) profile view; (C) positive view; (D) negative view. BG: (E) general view; (F) profile view; (G) positive view; (H) negative view; (B–D and F–H). Magnification: 2000×.

3.2. Physicochemical and mechanical tests
As shown in Table 1, BG was 444 ± 2.65 μm, while FS was 344 ± 5.74 μm. Furthermore, FS displayed a higher average tensile strength (16.5 ± 0.43 MPa) compared with that of BG (8.5 ± 1.52 MPa), and slightly lower elongation at break (15.66 ± 2.34%) compared with that of BG (28.25 ± 3.50%) under dry condition; similar results could also be obtained under wet condition. Usually, the decline of tensile strength in wet conditions stems from the absorption of water. The water contact angles of BG and FS were 38.0 ± 4.04° and 60.5 ± 5.62°, respectively, and the water absorption rates were 232.9 ± 15.94% and 143.3 ± 17.34%, respectively (p < 0.05) (Fig. 3).
The physicochemical properties of BG and FS.
| BG | FS | |
|---|---|---|
| Thickness (μm) | 444 ± 2.65 | 344 ± 5.74 |
| Tensile strength (MPa) dry condition | 8.5 ± 1.52 | 16.5 ± 0.43 |
| Elongation at break (%) dry condition | 28.25 ± 3.50 | 15.66 ± 2.34 |
| Tensile strength (MPa) wet condition | 6.3 ± 0.92 | 15.0 ± 0.32 |
| Elongation at break (%) wet condition | 38.37 ± 2.00 | 20.97 ± 3.20 |
| Contact angle (°) | 38.0 ± 4.04 | 60.5 ± 5.62 |
| Water absorption (%) | 232.9 ± 15.94 | 143.3 ± 17.34 |
Fig. 3. (A) Tensile strength testing; (B) water contents.

As shown in Fig. 4A, the spectra of FS were similar to that of BG. They both had five distinctive absorption bands of type I collagen: amide A, amide B, amide I, amide II, and amide III. The absorption peaks of the amide A band appeared at wavelengths 3308.72 cm−1 and 3322.26 cm−1, separately. The amide B band peaks appeared at wavelengths 2953.59 cm−1 and 2956.40 cm−1, separately, which were related to the stretching vibration of CH2. The C O stretching vibration of the polypeptide backbone leads to the amide I band with a distinctive absorption peak within the range of 1600–1700 cm−1, while the amide II band was produced by the CN stretching vibration and out-of-phase NH in-plane bending vibration within the range of 1500–1600 cm−1. The amide III band, related to the triple helices of collagen, was a combined absorption peak produced by in-phase NH bending vibration and the CN stretching vibration, appearing within the range of 1200–1360 cm−1.
Fig. 4. (A) FTIR; (B) degradation rate in vitro.

The degradation rate showed that FS degraded more slowly at each timepoint compared with that of BG. Specifically, the residual weight of FS was 61.61% on the first day, and it degraded completely on the 15th day at a concentration of 1.5 mg mL−1 collagenase, while the residual weight of BG was 62.35% on the first day, and it degraded completely on the 6th day at a concentration of 0.4 mg mL−1 collagenase (Fig. 4B).
3.3. Host immune response in vivo
As shown in Fig. 5, one week after the operation, the rats were in good condition, and no abnormal phenomena, such as swelling, mass, infection, or material excretion were found in the incision. H&E and Masson staining results showed that both BG and FS maintained relatively intact morphology, the fibrous capsule was thin, and a small number of neutrophils were scattered around, whereas no statistical differences were observed in all groups.
Fig. 5. H&E and Masson staining of the rat's subcutaneous tissue. Magnification: left 20×; right: 200×. ★ fibrous capsule; ※ implanted biomaterials.

3.4. Examination of repaired bone defects
3.4.1. Radiographic analysis
In accordance with the three-dimensional reconstruction results of micro-CT (Fig. 6(A–F) – RRB), the FS and BG groups, compared with the blank group, displayed more new bone formation along the cranial bone defects at the 4th week. Over time, the newly formed bone completely filled in the bone defects in the BG group, which was higher than that in the FS group at the 8th week. Quantitative analysis further indicated that the values of BV/TV and Tb.N in the BG group were higher than those in the FS group at the 4th week, while the difference was not statistically significant (p > 0.05). At the 8th week, there were statistically significant differences (p < 0.05) (Fig. 6(G and H) – RRB). Moreover, a higher value of Tb.Sp (Fig. 6(I) – RRB) was displayed in the blank group compared with those of the BG and FS groups at the 4th week (p < 0.05), there was a difference in Tb.Sp between any two groups at the 8th week (p < 0.05).
Fig. 6. Three-dimensional reconstruction of micro-CT at the (A–C) 4th week and (D–F) 8th week. (G) BV/TV, (H) Tb.N, and (I) Tb.Sp values in the blank group, BG group, and FS group at the 4th week and the 8th week after surgery. *p < 0.05.

3.4.2. Histological analysis
H&E staining was the most used histological method. Masson staining was used to observe new collagen fibres, and toluidine blue staining was used to observe osteocytes in new bone tissue. By the 4th week, the results indicated that a small number of new strip-shaped bone formations (marked for ‘▲’) related to the old bone (marked for ‘★’) and implanted scaffolds (marked for ‘*’) displayed at the edge of the bone defects in the BG and FS group, and inflammatory cell infiltration and fibrous connective tissue were observed in the blank group, but without new bone tissue formed. At the 8th week, inflammatory cell infiltration in the blank group was reduced, while there was a small amount of newly formed bone along the border of the cranial defects. In the FS group, the bone defects were almost filled with new strip-shaped bone formations at the edge of the bone defects, and residual FS was observed. Meanwhile, new massive and mature-like bone tissue could be observed in the BG group, and the BG degraded completely (Fig. 7A–C).
Fig. 7. (A) H&E staining of the cranial bone. Magnification: left 20×; right: 200×. ▲ new bone; ★ old bone; ※ implanted biomaterials. (B) Masson staining of the cranial bone. Magnification: left 20×; right: 200×. ▲ new bone; ★ old bone; ※ implanted biomaterials. (C) Toluidine blue staining of the cranial bone. Magnification: left 20×; right: 200×. ▲ new bone; ★ old bone; ※ implanted biomaterials.

4. Discussion
Biomimetic materials are becoming a promising alternative to better mimic the hierarchical structure of natural bone, as the absence of regular structures often leads to poorly functioning tissues.19 Traditional fish-scaled collagen extraction methods, such as acid and enzyme processing, are usually complicated, time-consuming, expensive, and sometimes harmful to the environment.20 The unmodified fish-scaled collagen shows remarkable biological capabilities, but the lack of control of biodegradability and poor mechanical properties have always been a challenge when using it directly as a scaffolding material. In this study, we developed a new strategy to remove mucopolysaccharides, proteins, and fats from fish scales, which was different from the established method for a novel collagen biomaterial preparation.21
Mechanical loading plays a critical role in bone remodelling and development due to the promotion of osteogenic major genes and mineral deposition.22 The FS was 0.34 ± 0.05 mm in thickness, without curvature. The results showed that the tensile strength of the FS was significantly higher than that of the BG, while the elongation of the FS was lower than that of the BG, closer to the natural bone.23 The reason may be related to the fact that the lamellar structure of the FS could withstand greater stress tension and was less prone to plastic deformation and stress fatigue than the BG.24 The mechanical properties of collagen biomaterials should match those of natural bone to play a supporting role in the initial stage of bone regeneration. Otherwise, the mismatch of the mechanical properties between biomaterials and natural bone can cause stress shielding, leading to local bone resorption and low bone regeneration, which affects the growth of bone tissue.22,25
According to our previous research, FS is composed of multilayered collagen fibers.17 The FTIR spectra recorded for FS showed typical bands for type I collagen: amide A, amide B, amide I, amide II, and amide III. We observed that the band amide A has been moved to lower frequencies due to the loss of binding water in collagen and the conformational changes of collagen molecules. And the amide II bands appeared at wavelengths of 1552.21 cm−1 and 1552.03 cm−1, respectively, which are consistent with the FTIR absorbance bands for collagen in the amide II wavelength range. These are also higher than those of gelatin in the amide II wavelength range. Therefore, this new method did not destroy the triple helical structure of the collagen molecules. In general, the scaffold degradation rate should match the bone regeneration rate. A very fast degradation rate may result in an inflammatory response due to the accumulation of amino saccharides produced from the degradation.26 The collagenase solutions increased the degradation rate by digesting the fibrous structure of the FS and BG, but the introduction of methyl groups may have caused some spatial hindrance to the interactions between the collagenase and collagen molecules.27 The FS was found to degrade more slowly than the BG because the multi-layered structure of the FS increased connections between collagen molecules, which in turn led to a reduction in hydrolysis reaction and collagenase digestion. However, the degradation of biological scaffolds resembles a double-edged sword: on the one hand, biological scaffolds needed to support bone defect area for a certain time and provide attachment points for osteoblasts to adhere, proliferate, differentiate, or secrete extracellular matrix; on the other hand, biological scaffolds need to be degraded to vacate enough space for the growth of new bone tissue.28,29
The biocompatibility of collagen biomaterials is closely related to their collagen sources, structures, fabrication methods, and physicochemical properties.30–32 The amino acid composition of the fish-scaled protein is similar to that of the porcine protein. To date, there is no strong evidence that fish collagen was associated with any mutagenicity and carcinogenicity in humans. Essen et al.21 demonstrated that a fish scale-derived artificial cornea displayed no cytotoxicity, is not immunogenic, and is well-tolerated in the cornea. Immunogenicity is a critical factor for the biological safety of type I collagen, implantation of the FS subcutaneously in rats allowed us to compare the immune response to the FS with that to the BG that has been safely used in humans as well as with sham surgery. We demonstrated that the FS was biocompatible and had a low immunogenicity, as there was no specific immune response and no signs of sensitisation upon reintroducing the FS subcutaneously in the same animal, accounting from their components, which were type I collagen, regardless of their difference between microstructure, mechanical strength, and contact angle. However, its biocompatibility in vivo requires further observation.
The calvarial bone originates from intramembranous ossification, and it includes two layers of cortical bone on the surface and cancellous bone in the middle of the two layers of cortical bone, similar to the mandibular bone. Moreover, there is a lack of blood supply and self-regeneration. Therefore, the calvarial bone defect is more suitable for bone regeneration testing.33–35 When the FS was implanted in rat critical-sized calvarial defects, there was no obvious inflammation or suppuration in the operation area from the beginning to the end, confirming that the FS had good biocompatibility. Furthermore, the FS showed good osteogenic activity in the early stage of osteogenesis, but the osteogenic activity still needed to be further improved in the late stage. Fish-scaled collagen has good biocompatibility and low immunogenicity, so it could be used as a substitute for mammalian collagen. Tang36 also proved that fish-scaled collagen had good osteogenic activity, similar to that of mammalian collagen. Moreover, the FS had a multi-layered structure, which could better simulate the arrangement of collagen fibres of natural bone, with good mechanical properties, beneficial to maintain osteogenic space, and promote osteoblast differentiation.37 It has been proven that the BG degradation was obviously faster than that of the FS in vivo, so the BG had been completely degraded in the late osteogenic stage, leaving enough osteogenic space for new bone to grow. Meanwhile, the FS still remained in the late stage, the undegraded scaffolds that remain in bone defects may delay the bone regeneration process. Owing to its dense structure, it could quench cells but might block nutrient transportation, and its osteogenic activity still needs to be improved further in the late stage of osteogenesis. Meanwhile, its dense structure may have considerable advantages in the retention of bacteria in comparison to porous membranes.38
Therefore, an ideal membrane does not exist to date; the FS has low porosity, and the attachment of bacteria and cells to the surface of the membrane is hindered, allowing the exposition to the oral environment. In addition, the FS demonstrated good mechanical properties, biocompatibility, osteogenic ability, easy handling, and low cost.
Some limitations existed in our study. First, the degradation rate of the FS was not quantitatively determined in vivo. Another limitation is that the FS needs to be modified to improve its osteogenic activity. Therefore, we are working to adjust the FS using new technologies, such as electrospinning and 3D biological printing, achieving a balance between its ultra-structure, degradation rate, mechanical properties, and osteogenic activity. In the future, the modified FS can be used in alveolar ridge preservation, repair of bone wall defects after jaw cysts, and so on.
5. Conclusions
In this study, through the characterisation of a multi-layered collagen material from fish scales, it was proven that the multi-layered collagen structure material has sufficient mechanical properties, biocompatibility, and osteogenic ability. At the same time, it also shows that there is a gap in clinical needs between the guided tissue regeneration membrane and the one being used. This study provides useful insights into the efforts being made to design and adjust the microstructure to balance its mechanical properties, degradation rate, and osteogenic activity.
Conflicts of interest
The authors declare no conflicts of interest.
Supplementary Material
Acknowledgments
This study was supported by the Science Foundation of Sichuan Provincial Department of Science and Technology, China (2019YFG0260 and 2020YJ0242).
References
- Nyman S. Lindhe J. Karring T. Rylander H. J. Clin. Periodontol. 1982;9:290–296. doi: 10.1111/j.1600-051X.1982.tb02095.x. [DOI] [PubMed] [Google Scholar]
- Buser D. Dula K. Belser U. Hirt H. P. Berthold H. International Journal of Periodontics and Restorative Dentistry. 1993;13:29–45. [PubMed] [Google Scholar]
- Tatakis D. N. Promsudthi A. Wikesjö U. M. Periodontology. 1999;19:59–73. doi: 10.1111/j.1600-0757.1999.tb00147.x. [DOI] [PubMed] [Google Scholar]
- Bottino M. C. Thomas V. Schmidt G. Vohra Y. K. Chu T. M. Kowolik M. J. Janowski G. M. Dent. Mater. 2012;28:703–721. doi: 10.1016/j.dental.2012.04.022. [DOI] [PubMed] [Google Scholar]
- Omar O. Elgali I. Dahlin C. Thomsen P. J. Clin. Periodontol. 2019;46:103–123. doi: 10.1111/jcpe.13068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florjanski W. Orzeszek S. Olchowy A. Grychowska N. Wieckiewicz W. Malysa A. Smardz J. Polymers. 2019;11:782. doi: 10.3390/polym11050782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Maawi S. Herrera-Vizcaíno C. Orlowska A. Willershausen I. Sader R. Miron R. J. Choukroun J. Materials. 2019;12:3993. doi: 10.3390/ma12233993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunyaratavej P. Wang H. L. J. Periodontol. 2001;72:215–229. doi: 10.1902/jop.2001.72.2.215. [DOI] [PubMed] [Google Scholar]
- Ferreira A. M. Gentile P. Chiono V. Ciardelli G. Acta Biomater. 2012;8:3191–3200. doi: 10.1016/j.actbio.2012.06.014. [DOI] [PubMed] [Google Scholar]
- Kozlovsky A. Aboodi G. Moses O. Tal H. Artzi Z. Weinreb M. Nemcovsky C. E. Clin. Oral Implants Res. 2009;20:1116–1123. doi: 10.1111/j.1600-0501.2009.01740.x. [DOI] [PubMed] [Google Scholar]
- Pati F. Datta P. Adhikari B. Dhara S. Ghosh K. Das Mohapatra P. K. J. Biomed. Mater. Res., Part A. 2012;100:1068–1079. doi: 10.1002/jbm.a.33280. [DOI] [PubMed] [Google Scholar]
- Deliormanli A. M. Liu X. Rahaman M. N. Biomater. Appl. 2014;28:643–653. doi: 10.1177/0885328212470013. [DOI] [PubMed] [Google Scholar]
- Arola D. Murcia S. Stossel M. Pahuja R. Linley T. Devaraj A. Ramulu M. Ossa E. A. Wang J. Acta Biomater. 2018;67:319–330. doi: 10.1016/j.actbio.2017.12.011. [DOI] [PubMed] [Google Scholar]
- Qiu Z. Y. Cui Y. Tao C. S. Zhang Z. Q. Tang P. F. Mao K. Y. Wang X. M. Cui F. Z. Materials. 2015;8:4733–4750. doi: 10.3390/ma8084733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kara A. Tamburaci S. Tihminlioglu F. Havitcioglu H. Int. J. Biol. Macromol. 2019;130:266–279. doi: 10.1016/j.ijbiomac.2019.02.067. [DOI] [PubMed] [Google Scholar]
- Wang J. Cheng Q. Tang Z. Chem. Soc. Rev. 2012;41:1111–1129. doi: 10.1039/C1CS15106A. [DOI] [PubMed] [Google Scholar]
- Feng H. H. Li X. Deng X. M. Li X. L. Guo J. T. Ma K. Jiang B. RSC Adv. 2020;10:875. doi: 10.1039/C9RA08189E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spicer P. P. Kretlow J. D. Young S. Jansen J. A. Kasper F. K. Mikos A. G. Nat. Protoc. 2012;7:1918–1929. doi: 10.1038/nprot.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao W. He J. Han Q. Sang F. Wang Q. Chen L. Cui S. Ding B. Mater. Sci. Eng., C. 2016;67:599–610. doi: 10.1016/j.msec.2016.05.081. [DOI] [PubMed] [Google Scholar]
- Huang C. Y. Kuo J. M. Wu S. J. Tsai H. T. Food Chem. 2016;190:997–1006. doi: 10.1016/j.foodchem.2015.06.066. [DOI] [PubMed] [Google Scholar]
- van Essen T. H. van Zijl L. Possemiers T. Mulder A. A. Zwart S. J. Chou C. H. Lin C. C. Lai H. J. Luyten G. P. M. Tassignon M. J. Zakaria N. El-Ghalbzouri A. Jager M. J. Biomaterials. 2016;81:36–45. doi: 10.1016/j.biomaterials.2015.11.015. [DOI] [PubMed] [Google Scholar]
- Hung B. P. Hutton D. L. Grayson W. L. Stem Cell Res. Ther. 2013;4:10. doi: 10.1186/scrt158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou F. Li R. Jiang J. Mo X. Gu G. Guo Z. Chen Z. J. Biomater. Sci., Polym. Ed. 2017;28:2255–2270. doi: 10.1080/09205063.2017.1392672. [DOI] [PubMed] [Google Scholar]
- Zadpoor A. A. J. Mech. Behav. Biomed. Mater. 2017;70:1–6. doi: 10.1016/j.jmbbm.2017.03.018. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee P. Kundu B. Naskar D. Kim H. W. Maiti T. K. Bhattacharya D. Kundu S. C. Acta Biomater. 2017;63:1–17. doi: 10.1016/j.actbio.2017.09.027. [DOI] [PubMed] [Google Scholar]
- Hu X. Park S. H. Gil E. S. Xia X. X. Weiss A. S. Kaplan D. L. Biomaterials. 2011;32:8979–8989. doi: 10.1016/j.biomaterials.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet I. D. Shreiber D. I. Biointerphases. 2012;7:25. doi: 10.1007/s13758-012-0025-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseti L. Parisi V. Petretta M. Cavallo C. Desando G. Bartolotti I. Grigolo B. Mater. Sci. Eng., C. 2017;78:1246–1262. doi: 10.1016/j.msec.2017.05.017. [DOI] [PubMed] [Google Scholar]
- Shi R. Huang Y. Ma C. Wu C. Tian W. Front. Med. 2019;13:160–188. doi: 10.1007/s11684-018-0629-9. [DOI] [PubMed] [Google Scholar]
- Meyer M. Biomed. Eng. Online. 2019;18:24. doi: 10.1186/s12938-019-0647-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozdzen L. C. Vucetic A. Harley B. A. C. J. Mech. Behav. Biomed. Mater. 2017;66:28–36. doi: 10.1016/j.jmbbm.2016.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters B. D. Stegemann J. P. Acta Biomater. 2014;10:1488–1501. doi: 10.1016/j.actbio.2013.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel K. A. Lang F. J. Donath K. Kulow J. T. Wiltfang J. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontology. 2006;102:7–13. doi: 10.1016/j.tripleo.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Su Y. Su Q. Liu W. Lim M. Venugopal J. R. Mo X. Ramakrishna S. Al-Deyab S. S. El-Newehy M. Acta Biomater. 2012;8:763–771. doi: 10.1016/j.actbio.2011.11.002. [DOI] [PubMed] [Google Scholar]
- Zeng J. H. Qiu P. Xiong L. Liu S. W. Ding L. H. Xiong S. L. Li J. T. Xiao Z. B. Zhang T. Int. J. Artif. Organs. 2019;42:325–337. doi: 10.1177/0391398819834944. [DOI] [PubMed] [Google Scholar]
- Tang J. Saito T. BioMed Res. Int. 2015;2015:139476. doi: 10.1155/2015/139476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuiyan D. B. Middleton J. C. Tannenbaum R. Wick T. M. J. Biomater. Sci., Polym. Ed. 2016;27:1139–1154. doi: 10.1080/09205063.2016.1184121. [DOI] [PubMed] [Google Scholar]
- Monteiro A. S. Macedo L. G. Macedo N. L. Balducci I. Med. Oral Patol. Oral Cir. Bucal. 2010;15:e401–e406. doi: 10.4317/medoral.15.e401. [DOI] [PubMed] [Google Scholar]