

Abstract
Glioblastoma, one of the most fatal brain tumors, is associated with a dismal prognosis and an extremely short overall survival. We previously reported that the overexpressed transient receptor potential channel TRPM7 is an essential glioblastoma regulator. Accumulating evidence suggests that long noncoding RNAs (lncRNAs) play an important role in glioma’s initiation and progression. However, the function of lncRNA, HOX transcript antisense intergenic RNA (HOTAIR) mediated by TRPM7 in glioma remains unclear. In this study, HOTAIR expression was found to be positively regulated by TRPM7, significantly upregulated in glioma tissues, and is a poor prognosis factor for glioma patients. Moreover, reduced HOTAIR expression impeded the proliferation and invasion of glioma cells. Mechanistically, HOTAIR directly interacted with miR-301a-3p, and downregulation of miR-301a-3p efficiently reversed FOSL1 suppression induced by siRNA HOTAIR, which implied that HOTAIR positively regulated FOSL1 level through sponging miR-301a-3p and played an oncogenic role in glioma progression. In contrast to HOTAIR’s role, miR-301a-3p alone served as a tumor suppressor to decrease glioma cell viability and migration/invasion. In agreement with HOTAIR’s role, FOSL1 functioned as a tumorigenic gene in glioma pathogenesis, which was highly expressed in glioma tissues, and was shown to be an unfavorable prognostic factor for glioma patients. Mechanically, FOSL1 inhibition by siRNA FOSL1 efficiently rescued the oncogenic-like phenotypes caused by the miR-301a-3p inhibitor in glioma pathogenesis.
Keywords: TRPM7, HOTAIR, miR-301a-3p, FOSL1, Glioma
1. Introduction
Glioblastoma (GBM, World Health Organization grade IV glioma) remains highly lethal due to a lack of effective therapy and its inevitable recurrence [1,2]. Despite current aggressive first-line therapies such as maximal surgical resection, concurrent radiotherapy, adjuvant chemotherapy with temozolomide (TMZ), as well as immunotherapy, GBM recurrence is inevitable after extending survival to a median of only 14.6 months [1,3,4]. The poor prognosis and recurrence emphasize a necessity to understand the molecular characteristics of GBM and develop effective targeted molecular therapies.
The studies on protein-coding genes have identified commonly changed signaling pathways in GBM, including IDH, EGFR, TERT, ATRX loss, TP53 mutation, and CDKN2A/2B [5,6]. However, the targeted therapies for GBM focusing on the above pathways have not been effective, and results have been continuously disappointing. Recent studies showed that the human genome encompasses many long non-coding transcripts, long non-coding RNAs (lncRNAs), which may be more common than protein-coding genes [7]. These lncRNAs contain long non-encoding RNA transcripts >200 nucleotides in length, many of which show cell type-specific expression [8]. They are localized either in the nucleus or cytoplasm and play many different roles in the cancer cells, in part through chromatin remodeling, regulation of transcription/posttranscription, mRNA stability, mRNA translation, and post-translational modification [8,9]. In glioblastoma, non-coding RNAs, including lncRNAs, regulate numerous oncogenic phenotypes such as glioma cell proliferation, invasiveness, and glioblastoma stem cells (GSC)’ stemness [8,9].
TRPM7, a member of the transient receptor potential channel TRP superfamily, is a cation channel permeable to divalent cations Mg2+, Ca2+, and Zn2+ fused to a C-terminal serine/threonine protein kinase domain [10–15]. TRPM7 is indispensable for cell growth, proliferation, apoptosis, senesce, migration, and immune cell function. Increased TRPM7 expression and/or activity has been correlated with malignant growth and cancer progression in various cancers [16–18]. Work that was done previously in our group demonstrated that altered TRPM7 expression and activity is not only required for glioma cell proliferation and migration/invasion [19,20] but also drives GSC plasticity through Notch and STAT3 activities [21,22]. In addition, previously, our in-depth data analysis from miRNA microarray data revealed that a panel of miRNAs is significantly changed in response to TRPM7 silencing [20]. Moreover, we reported that TRPM7 negatively regulates miR-28–5p to promote proliferation and invasion of glioma cells through upregulating target Rap1b gene [20]. As lncRNAs generally network with miRNAs via the “sponge effect,” by which lncRNAs may sequester miRNAs and compete with miRNA to consequently inhibit the miRNA’s inhibitory ability on target gene expression [23]; this prompted us to examine the possible altered lncRNA profiles caused by TRPM7 deletion. Our data analysis from lncRNA array data revealed a list of 10 downregulated and 7 upregulated lncRNAs whose transcripts are statistically significant changed by TRPM7 knockout in A172 glioma cells, while lncRNA HOX transcript antisense intergenic RNA (HOTAIR) was most positively affected by TRPM7 depletion. HOTAIR is a well-studied lncRNA that plays a pro-oncogenic role in several human cancers. In gliomas, HOTAIR mainly contributes to tumorigenesis by the inhibition of apoptosis [24], promotion of growth and invasion, and enhancement of angiogenesis [25]. It may also play a role in drug delivery in glioma by affecting the blood-tumor barrier (BTB) permeability [26]. The aim of the present study is to investigate the functional roles of HOTAIR, and the mechanisms by which HOTAIR networks with miRNA(s) to promote glioma tumorigenesis.
2. Materials and methods
2.1. Antibody and reagents
The following primary antibodies were used in this study. Anti-TRPM7 was purchased from Abcam (Cambridge, MA, cat. no. ab232455). A mouse monoclonal antibody that detects protein argonaute-2 (Ago2, cat. no. 04–642-I) and mouse anti-β-actin (cat. no. A3854) were purchased from Sigma-Aldrich (St. Louis, MO). Mouse monoclonal FOSL1 antibody was purchased from Santa Cruz Biotechnology (Dallas, TX, cat. no. sc-28,310). All secondary antibodies used for Western blot were purchased from Calbiochem (La Jolla, CA). TurboFectin was purchased from OriGene (Rockville, MD, cat. no. TF81001).
2.2. Plasmid, siRNA, and miRNAs
Control scrambled siRNA (cat. no. D-001810–01-05) and ON-TARGETplus SMARTpool siRNA targeting HOTAIR (cat.no. R-187951–00-0005) were purchased from Dharmacon (Lafayette, CO). Control scrambled siRNA and siRNA targeting FOSL1 (siRNA ID # s15585), hsa-miR-301a-3p mimic (Product ID: MC10978), hsa-miR-301a-3p inhibitor (Product ID: MH10978), and negative control (cat. no. 4464076) were purchased from Thermo Fisher Scientific (Waltham, MA). The scrambled siRNAs, with no homology to any known sequence, were used as controls. The TRPM7 KN2.0 human gene knockout via CRISPR was done using a non-homology mediated kit that was purchased from OriGene (Rockville, MD, cat. no. KN418043).
2.3. Cell culture
Human glioblastoma cell lines, A172 (RRID: CVCL_0131), U87MG (HTB-14), a glioblastoma of unknown origin (RRID: CVCL_0022), HTB-12, HTB-15, HTB-16, HTB-138, CRL-2611, M059K, and M059J were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). T98G and LN18 glioma cell lines were obtained from Dr. Erwin van Meir’s laboratory at the University of Alabama at Birmingham (UAB). All cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Life Technologies, Waltham, MA) plus 10% fetal bovine serum (FBS), 50 units/ml penicillin, and 50 μg/ml streptomycin at 37 °C. CRL-3417 was purchased from ATCC and cultured in neurocult basal medium with 20 ng/ml epidermal growth factor (EGF), 20 ng/ml fibroblast growth factor (β-FGF), and 2 μg/ml heparin sulfate. Patient derived xenoline (PDX)-tumor tissue cubes stored at liquid nitrogen were provided by Dr. Yancey G. Gillespie at UAB. PDX lines (PDX-L14 and PDX-L11) were generated by implanting PDX-tumor tissue cubes subcutaneously into the flanks of male or female 6–8 weeks old nude mice under anesthesia (ketamine/Xylazin 90/6 mg/kg BW). Briefly, cryopreserved tumor tissues were thawed at 37 °C and washed with phosphate-buffered saline (PBS) before subcutaneous implantation. To prepare a single-cell suspension of viable tumor cells, the xenograft tumor tissues were harvested and minced with scalpel blades followed by passes through cell strainers. The cells were then grown in DMEM/F-12 media plus 10% FBS, 50 units/ml penicillin, and 50 μg/ml streptomycin for future use. All experiments were performed with mycoplasma-free cells. PDX-L14 cells with proneural subtype have wild-type genes including EGFR, PTEN, CDKN2A, NF-κB, CDK4/MDM2, and amplified gene of CSNK2A, deleted TP53, and expression of CD133. PDX-L11 cells with classical subtype have wild-type genes including EGFR, PTEN, NF-κB, CDK4, and TP53, amplified MDM2, deletion of CDKN2A, gain of CSNK2A, and expression of CD133.
We used the TRPM7 KN2.0 human gene knockout kit via CRISPR, a non-homology mediated kit from OriGene to knockout all the splicing variants of the TRPM7 gene according to the manufacturer’s instruction. Briefly, A172 cells were seeded at 3 × 105 in a 6-well plate to obtain 50–70% confluence, and 1 μg of TRPM7 gRNA and 1 μg of the Donor DNA were transfected into A172 cells using TurboFectin. 48 h post-transfection, the cells were split 1:10 and grown for an additional 3 days followed by continuous split for 4 times. Then, the cells at passage 5 (P5) were grown directly in the growth medium containing 1 μg/ml of puromycin. Two single-cell clones (A172 KO1 and A172 KO2) were generated after puromycin selection.
2.4. Transfection of siRNA and miRNA
When glioma cells grew to reach about 50–75% confluency, the appropriate amount of specific siRNAs or miRNAs, along with corresponding controls with a final concentration of 50 nM, were transfected using Lipofectamine RNAiMAX reagent in serum-free OptiMEM-1 medium (Invitrogen, Carlsbad, CA) according to manufacturer’s instruction. 48 or 72 h post-transfection, target knockdowns or increases were assessed by quantitative real-time RT-PCR (qRT-PCR) analysis, stem-loop pulsed RT-PCR, or Western blot, accordingly. All studies were done in triplicates.
2.5. Proliferation and cell viability assay
All glioma cells were seeded at 2.5 × 104 cells in 100 μl of medium per well into 96-well plates and transfected with specific siRNA, miRNA, or controls accordingly using Lipofectamine RNAiMAX reagents for the indicated times. 10 μl of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagent (Sigma-Aldrich, the ratio of MTT reagent to medium is 1:10) was added into each well and incubated in the dark at 37 °C for 2 to 4 h. Absorbance at 570 nm was measured using 690 nm as the reference using the CytoFluorTM 2300 plate reader.
2.6. Clonogenic assay
After transfection, cells (1000/well) were seeded into 6-well plates and cultured for an additional two weeks. Cells were fixed with 4% paraformaldehyde and stained with 0.5% crystal violet. Colonies of more than 50 cells were counted to determine survival and the number of colonies obtained from three replicates was averaged for each treatment.
2.7. Cell migration and invasion assay
The migration and invasion potential were assessed as we previously described [20,22]. Briefly, cell culture chambers with 8 μm pore size polycarbonate membrane filters (Corning, USA) were used for cell invasion assays with the filters pre-coated with Matrigel (50 μl, 1.25 mg/ml). Each of the glioma cell lines/PDXs line was transfected with or without siRNA or miRNA for 48 h, harvested, and seeded with 1% FBS medium in upper chambers that were soaked in bottom chambers filled with 500 μl whole medium (DMEM and 10% FBS). After another 24 h of incubation at 37 °C, Matrigel and cells on the upper surface of the filter were wiped off thoroughly with Q-tips. Cells attached on the lower surface of the membrane filters were fixed with 4% paraformaldehyde/PBS for 10 min and stained with 0.5% crystal violet/methanol for 10 min. The cells were then counted under light microscopy with 10× magnification in 3–4 random fields. Cell numbers under different treatments were normalized to appropriate controls. Assays were done in triplicate samples and performed in three independent experiments.
2.8. qRT-PCR
Total RNA isolation, cDNA synthesis, and PCR amplification were performed as we previously described [20]. Total RNA was isolated from cells using a miRNeasy Kit (Qiagen, Valencia, CA) and quantified using the Nanodrop N-1000 by Agilent Biosystems (Santa Clara, CA). Purified total RNA (0.75 μg) was reverse transcribed using the iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Inc., Hercules, CA) according to manufacturer’s protocol. Reverse transcription was performed by using random hexamers at 25 °C for 5 min, 42 °C for 30 min, and 85 °C for 5 min. After diluting ten times, the cDNA was then amplified using iQ SYBR Green Supermix (Bio-Rad Laboratories, Inc.) according to manufacturer’s protocol under the following conditions: activation of the Taq DNA polymerase at 95 °C for 3 min, 40 cycles at 95 °C for 10 s (denaturation), and 61 °C for 45 s (combined annealing and extension). The quantitative gene analysis utilized the CFX Connect Real-Time PCR Detection System. Each condition was conducted in biological triplicates, and each biological replicate was amplified in technical triplicates. Relative expression for each gene was evaluated using the 2−ΔΔCt Livak method, and GAPDH was used as the reference gene (20).
2.9. Stem-loop pulsed RT-PCR
The miRNA detection was performed using stem-loop pulsed RT-PCR with some modifications as described before [27]. The RT primer for miR-301a-3p reverse transcription, forward and reverse primers for RT product amplification were designed based on miR-301a-3p’s sequence: 5’-CAGUGCAAUAGUAUUGUCAAAGC-3′ (http://www.mirbase.org/). For each reaction, the “no RNA” master mix comprised of 10 mM dNTP, 5 μM RT primer (see Table 1), and appropriate water, was heated at 65 °C for 5 min and incubated on ice for 2 min. Then, the “no RNA” master mix was combined with the RT master mix containing first-strand buffer, 0.1 M DTT, 4 units RNaseOUT, and 50 units of Super-Script III-RT. Then, the pulsed RT was performed under the following conditions: load thermal cycler and incubate for 30 min at 16 °C, pulsed RT of 60 cycles at 30 °C for 30 s, 42 °C for 30 s, and 50 °C for 1 s and incubate at 85 °C for 5 min to inactivate the reverse transcriptase. Finally, the RT product was amplified using iQ SYBR Green Supermix (Bio-Rad) as described above, and U6 was used as the reference gene.
Table 1.
List of primers used in the study.
| Primer set | Forward 5′−3′ | Reverse 5′−3′ |
|---|---|---|
| TRPM7 | CTTTGACCAAGAGGGAATGTG | GACCAAGCGACCACAAAAAC |
| HOTAIR | GAGTCTGATGTTTACAAGACC | CAACGAGCTTATAAGGAAGG |
| FOSL1 | CTCCAGGGGTACGTCGAAG | TCAGTTCCTTCCTCCGGTTC |
| GAPDH | GAAGGTGAAGGTCGGAGTC | GAAGATGGTGATGGGATTTC |
| U6 | CAAGGATGACACGCAAATTC | AAAAATGGAACGCTTCACGA |
| miR-301a-3p RT primer | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACGCTTTG | |
| miR-301a-3p | GTATACCAGTGCAATAGTATT | GTGCAGGGTCCGAGGT |
| hFOSL1 3’UTR | ATCGACGCGTACCCTCCTCGCTTTGTGAG | ATCGAGATCTGTGGGGCTGGTGAGTTAGTG |
| hFOSL1 3’UTR mutant | CACAATAACCACAAAATCAGAGACAAAAT | CTAGAGAGGCCAGCTCAAGAGAAACAGTGGG |
2.10. Western blotting
Cells were lysed with lysis buffer of M-PER™ Mammalian Protein Extraction Reagent (Thermo Fisher Scientific cat. no. 78501) supplemented with Halt™ Protease and Phosphatase Inhibitor Single-Use Cocktail (100×) (Thermo Fisher Scientific cat. no. 78442). SDS/PAGE separated samples, and separated proteins were transferred to nitro-cellulose membranes and identified by immunoblotting. Primary antibodies were obtained from commercial sources and were diluted to the ratio of 1:500 or 1:1000 according to manufacturer’s instruction. Blots were developed with Supersignal Pico or Femto substrate (Pierce). Densitometric analysis of the bands was performed with the ImageQuant program (Bio-Rad).
2.11. RT2 lncRNA PCR arrays
Total RNA, from A172 or A172 KO glioma cells, were extracted and subjected to Human Cancer Pathway Finder, RT2 LncRNA PCR Array (Qiagen, cat. no. LAHS-002ZD). The difference at mRNA transcript levels between A172 cells and A172 KO was analyzed using an online software https:/dataanalysis2.qiagen.com/lncRNA. Fold changes and p values were calculated using Student’s t-test. A p-value <0.05 with a fold change greater than 2 was considered to be a significant dysregulation.
2.12. Reporter constructs and luciferase reporter assays
DNA was extracted from HTB-138 cells using the Blood & Cell Culture DNA Mini Kit (Qiagen) according to manufacturer’s instruction. Full-length 3’UTR of FOSL1 gene was amplified from HTB-138 DNA and cloned into pGL3-Promoter vector (Promega, cat. no. E1761) between MluI [New England Biolabs Inc. (NEB), Ipswitch, MA, NEB, cat. no. R0198L] and BglII (NEB, cat. no. R0144L) sites. Primers for amplification are listed in Table 1. For mutated construct, mutant FOSL1 or mut-FOSL1, miR-301a-3p binding sites was mutated using Q5 site-directed mutagenesis kit (NEB, cat. no. E0554S). For the reporter assays, 5 × 104 cells were seeded in a 24-well plate and co-transfected with wild type (Wt-FOSL1) or mut-FOSL1 into U87MG and PDX-L14 cells together with miR-301a-3p or control. Firefly and Renilla luciferase activities were measured 24 h post-transfection using a Dual-Luciferase Reporter Assay system (Promega, cat. no. E1910). The firefly luminescence was normalized to Renilla luminescence as an internal control for transfection efficiency. Experiments were performed three times.
2.13. RNA immunoprecipitation (RIP) assay
The Magna RNA-Binding Protein Immunoprecipitation Kit (Sigma Aldrich Inc., cat. No 17–700) was used for RIP experiments. The RIP assay was used to explore the binding activity between endogenous HOTAIR and miR-301a-3p in glioma cells according to manufacturer’s instructions. Briefly, U87MG and PDX-L14 cells transfected with miR-301a-3p, and control were collected and lysed in RIP lysis buffer, 100 μl of cell lysate was incubated with a human anti-Ago2 antibody or negative control. The samples were incubated with proteinase K with shaking to digest protein and then the precipitation of RNA was obtained. Purified RNA was analyzed to qRT-PCR for HOTAIR expression.
2.14. Bioinformatics analysis
Kaplan-Meier analysis of overall survival (OS) rate according to the HOTAIR was obtained from The Cancer Genome Atlas (TCGA) survival data to the lncRNA expression levels in OncoLnc (http://www.oncolnc.org). The dataset contained information on 152 glioblastoma patients, classified based on WHO classification as GBM (WHO grade IV glioma). The potential miR-301a-3p binding sites in HOTAIR and FOSL1 were predicted through the bioinformatics database (LncBase Predicted v.2) and public prediction algorithms (miRbase.org and Starbase v2.0), respectively. The TCGA database, which contained information on 454 glioblastoma patients, classified based on WHO classification as GBM was used to analyze for FOSL1 mRNA expression and its co-expression with the lncRNA HOTAIR (http://www.betastasis.com/glioma/tcga_gbm/). Kaplan-Meier analysis of OS rate for GBM patients according to the FOSL1 was determined by the Human Protein Atlas (https://www.proteinatlas.org).
2.15. Statistical analysis
The results obtained in this work were expressed as mean ± S.D. of at least 3 independent experiments done in triplicate. Paired Student t-test or one-way ANOVA tests were performed for data analysis, and a significant difference was defined as p < 0.05.
3. Results
3.1. TRPM7 functions as a positive regulator of lncRNA HOTAIR
We recently observed that TRPM7 upregulation positively correlates with glioma tumorigenesis, doing so by promoting tumor cell proliferation and invasion via regulation of small non-coding RNAs (sncRNAs) [20]. The contribution of TRPM7-mediated sncRNA to glioma prompted us to determine the possible role of lncRNAs mediated by TRPM7 in glioma cell proliferation and invasion. Since emerging studies have shown that lncRNAs function to regulate cellular proliferation and differentiation, as well as tumor development [8], therefore, we investigated whether TRPM7 regulates lncRNAs and cross-talks with sncRNAs to lead to glioma progression. To this end, we used TRPM7 KN2.0 human gene knockout via CRISPR, a non-homology mediated technology from OriGene to knockout all the splicing variants of the TRPM7 gene. Two single-cell clones (A172 KO1 and A172 KO2) were generated after puromycin selection and further developed into two stable cell lines. Fig. 1A confirmed the knockout effect by GFP positive cells (Fig. 1A, upper panel), reduced TRPM7 mRNA expression (Fig. 1A middle panel), and reduced TRPM7 protein expression (Fig. 1A lower panel). A172 KO1 was the more efficient one of the two and was selected for further experiments and named A172 KO thereafter. Next, total RNA from A172 KO and parental A172 glioma cells were extracted and then subjected to Human Cancer Pathway Finder, RT2 lncRNA PCR Array. Data analysis from RT2 data resulted in a list of 10 downregulated and 7 upregulated lncRNAs whose transcripts are statistically significant with fold changes greater than 2.0 by TRPM7 knockout (Fig. 1, B–D). The IncRNAs with a fold change greater than 2.0 are listed in Fig. 1B, while the scatter plot is shown in Fig. 1C and heat map in Fig. 1D.
Fig. 1.
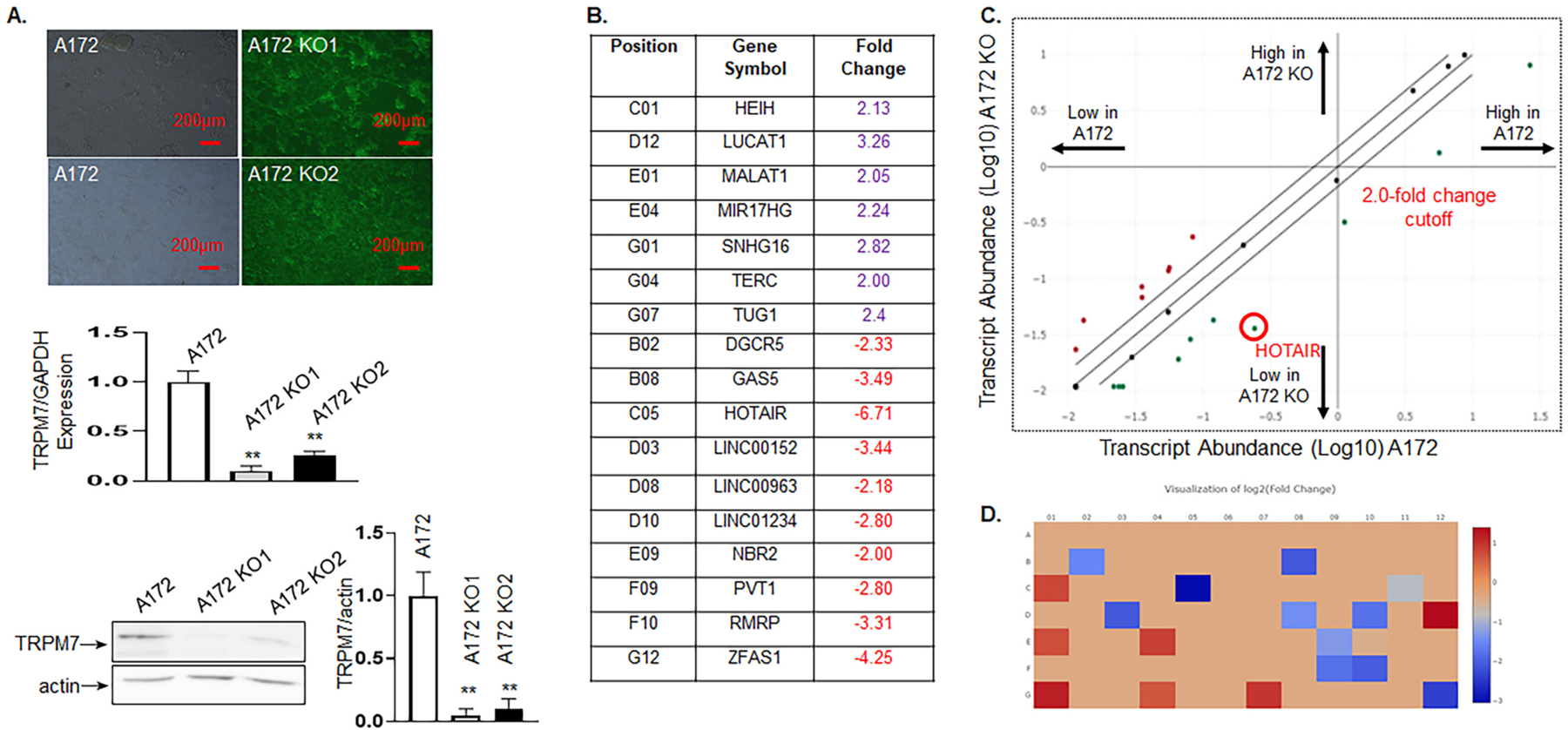
TRPM7 functions as a positive regulator of lncRNA HOTAIR. We used the TRPM7 KN2.0 human gene knockout via CRISPR, a non-homology mediated kit from OriGene to knockout all the splicing variants of the TRPM7 gene in A172 glioma cells. A, Two of A172 stable cell lines with TRPM7 deletion (A172 KO1 and A172 KO2) were generated after puromycin selection. TRPM7 knockout was confirmed by GFP positive cells (upper panel), reduced expression of TRPM7 mRNA by qPCR (middle panel), and reduced TRPM7 protein expression on Western blot (lower panel). B-D, A172 KO1 was the most efficient to be selected for further experiments and is labeled as A172 KO. Total RNA from glioma cells either with A172 or A172 KO cells were extracted and then subjected to Human Cancer Pathway Finder, RT2 lncRNA PCR Array. Data analysis from RT2 data resulted in a list of 10 downregulated and 7 upregulated lncRNAs whose transcripts are statistically significant with fold changes greater than 2.0 by TRPM7 knockout. B, A list of the IncRNAs with a fold change greater than 2.0. C, the scatter plot, and D, heat map.
3.2. Knockdown of HOTAIR inhibits glioma cell proliferation and migration/invasion
Among the lncRNAs regulated by TRPM7, we found that HOTAIR was most strongly affected in response to TRPM7 knockout, which was decreased 6.71 times compared to control (Fig. 1B). The finding that TRPM7 regulates HOTAIR was also supported by two previous works that TRPM7 modulates transcription factor NF-κB activation in macrophage [28] and that NF-κB directly transcriptionally regulates HOTAOR in ovarian cancer [29] (see Fig. 6). The TCGA database, which contained information on 454 glioblastoma patients, was first analyzed for the clinical significance of HOTAIR expression. HOTAIR expression levels in GBM patients were significantly higher than those in normal individuals as detected by Affymetrix HT HG U133A (Fig. 2A left panel). Furthermore, HOTAIR expression levels in mesenchymal and neural molecular subtypes in glioma tissue were higher than those in the normal brain (p < 0.05; Fig. 2A, right panel). Although a similar tendency was displayed in the classical and proneural subtype, they did not reach a significant difference (Fig. 2A, right panel). To evaluate the prognostic value of HOTAIR in GBM patients, we linked TCGA survival data to the lncRNA expression levels in OncoLnc (http://www.oncolnc.org). The dataset contained information of 152 glioblastoma patients, classified based on WHO classification as GBM. When all GBM patients were analyzed in a pooled setting, and the median value was selected as the cut-off point, the 10-year OS rate, as revealed by the Kaplan-Meier survival curve (Fig. 2B), was significantly higher in those with low HOTAIR levels (blue curve) compared to those with high expression (red curve) (p = 0.0401, log-rank test).
Fig. 6.
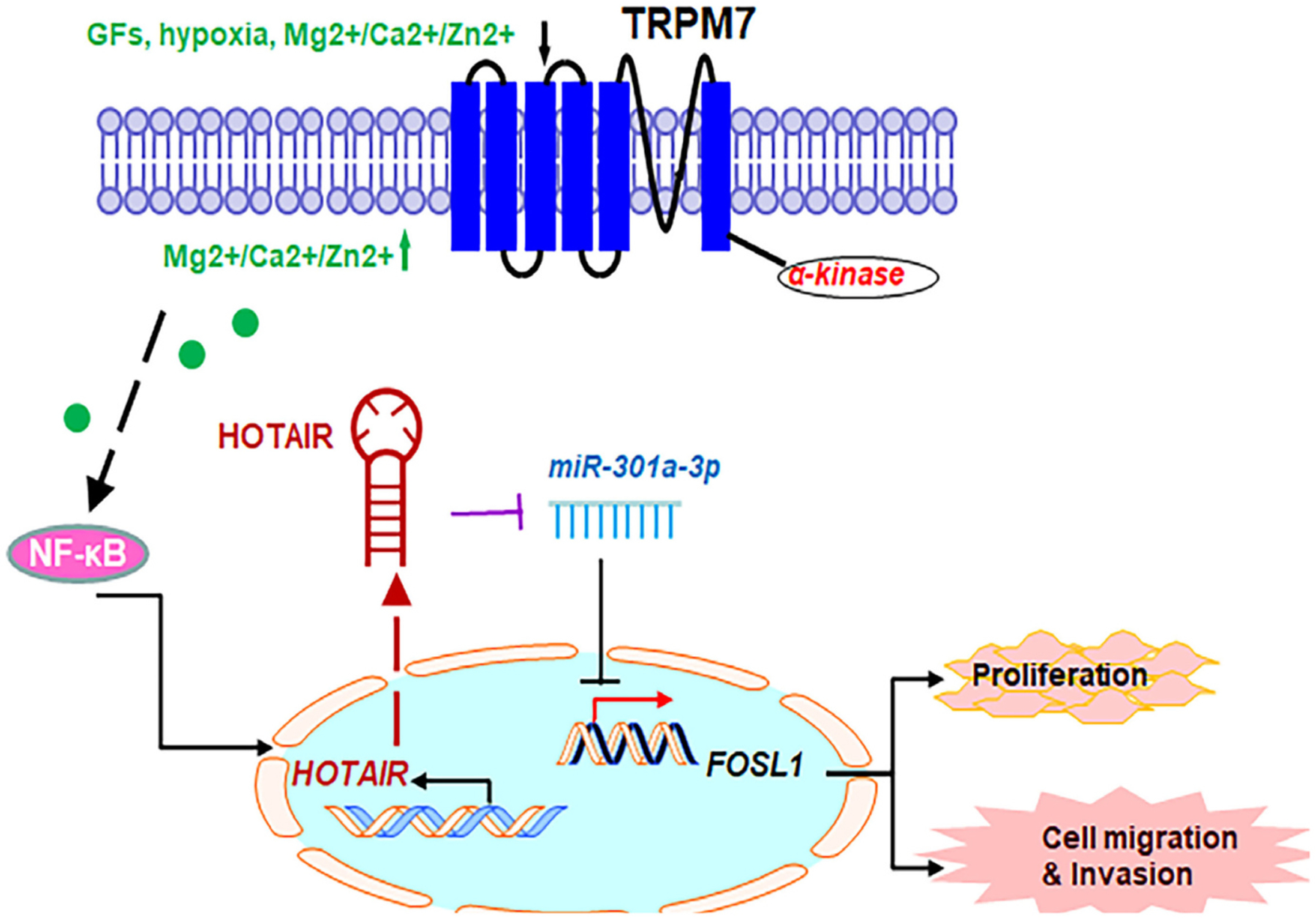
The proposed model for the role of TRPM7-mediated HOTAIR/miR-301a-3p/FOSL1 axis in glioma tumorigenesis. TRPM7 mediates the Ca2+ influx necessary for NF-κB activation, which transcriptionally activates lncHOTAIR. In turn, lncHOTAIR directly inhibits miR-301a-3p expression, which directly functionally impede FOSL1 gene expression activity.
Fig. 2.
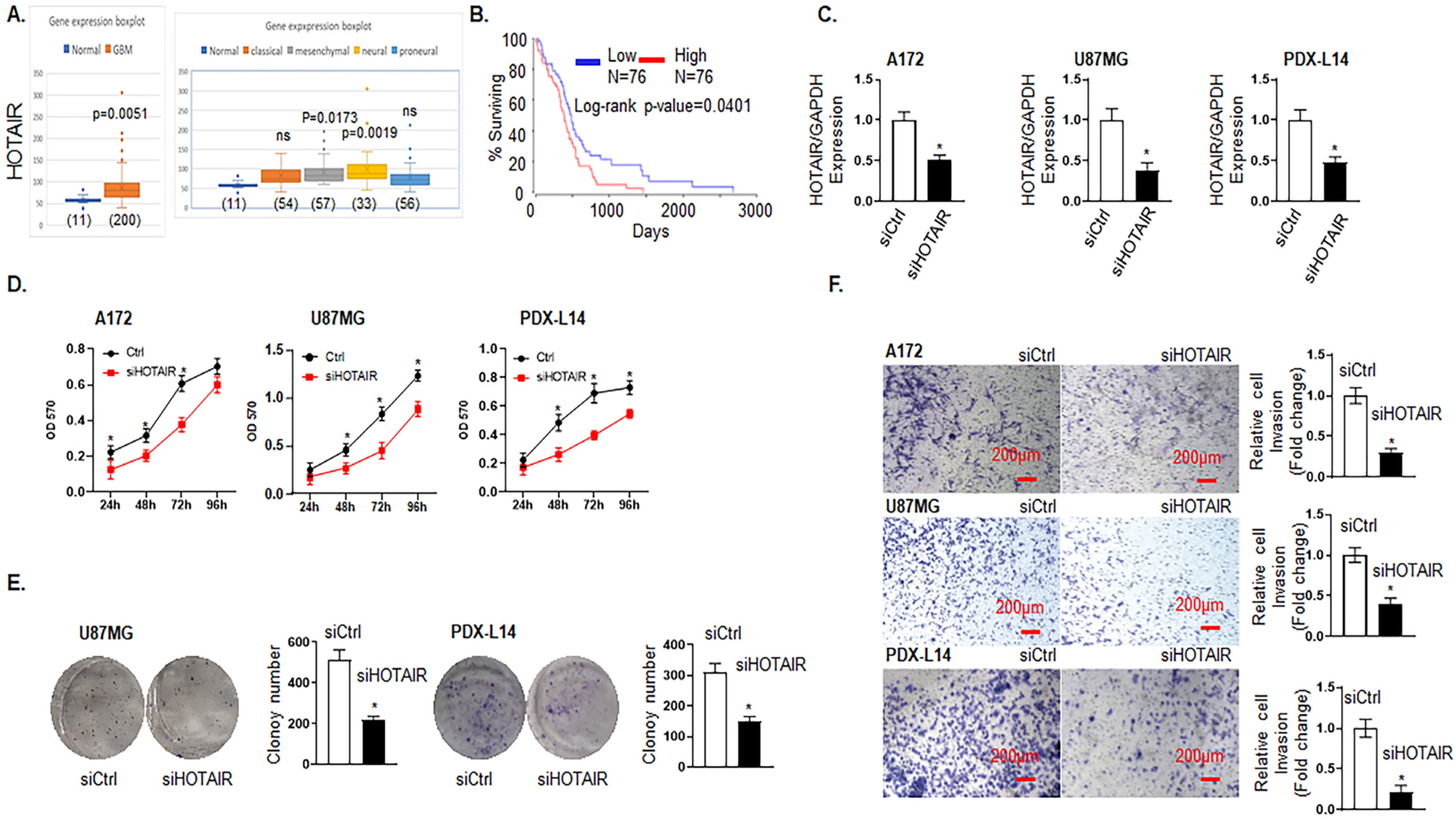
Knockdown of HOTAIR inhibits glioma cell proliferation and migration/invasion. A, HOTAIR expression levels in brain tissues of GBM (left panel) and different molecular subtypes of GBM patients (right panel) were analyzed by Affymetrix Human Exon 1.0 ST of TCGA data. B, Kaplan-Meier survival curve for the 10-year OS rate of glioma patients were analyzed by OncoLnc TCGA data. The cut-off was set at the median. C-F, Different sets of A172, U87MG, and PDX-L14 cells were transfected with siRNA HOTAIR (siHOTAIR) for certain period of times, as indicated, followed by MTT (D), clonogenic (E) and cell invasion (F) assay. C, Transfection efficiency of A172, U87MG, and PDX-L14 was revealed by the expression of HOTAIR at 48 h via qPCR. D, MTT assays were conducted to examine the effects of HOTAIR on cell viability after transfecting A172, U87MG, and PDX-L14 with siHOTAIR targeting HOTAIR mRNA from 24 h to 96 h. E, Clonogenic assays were performed to examine the effects of HOTAIR on long-term cell viability after transfecting A172, U87MG, and PDX-L14 cells with siHOTAIR for two weeks. F, Cell invasion assays were performed to detect the effects of HOTAIR on invasion after transfecting A172, U87MG, and PDX-L14 cells with siHOTAIR targeting HOTAIR mRNA. Images were captured at magnification of x10, *p < 0.05.
To examine whether HOTAIR expression could affect the biological activity of glioma cells, siRNA targeting the coding region of HOTAIR (siHOTAIR) was tested for their knockdown efficiency in A172 and U87MG glioma cells as well as in PDX-L14, which represent different molecular subtypes of glioma cells. Briefly, A172 and U87MG both have wild-type TP53, PTEN mutations, and p14ARF/p16 deletion. U87MG cells express high levels of VEGF as compared to A172 express high levels of bFGF [22]. Compared to glioma cell lines, PDX more closely mimic the biological and physiological characteristics of in vivo real cells and tissues. PDX-L14 cells with proneural subtype have wild-type genes including EGFR, PTEN, CDKN2A, NF-κB, CDK4/MDM2, and amplified gene of CSNK2A, deleted TP53, and expression of CD133. The siHOTAIR was very efficient in reducing HOTAIR expression in each glioma cell line (Fig. 2C) by HOTAIR expression reduction at 49.0, 61.0, and 52.0% in A172, U87MG, and PDX-L14 cells, respectively. MTT assay demonstrated that downregulated expression of HOTAIR attenuated the proliferation of A172, U87MG, and PDX-L14 cells (Fig. 2D). In detail, the cell growth viability was reduced from 43.9 to 35.4% with the maximum at 24 h in A172 cell, 46.2 to 28.5% with the maximum at 72 h in U87MG cells, and 45.8 to 25.2% with the maximum at 48 h. The colony formation assay also showed that downregulated HOTAIR expression inhibited the long-term proliferation of U87MG and PDX-L14 glioma cells. The colony numbers were reduced by 57.4 and 51.3% in U87MG and PDX-L14 cells, respectively. However, no colonies were formed by A172 cells (Fig. 2E). The invasion of A172, U87MG, and PDX-L14 cells was significantly reduced following downregulation of HOTAIR expression as shown by transwell assay (Fig. 2F). The number of invasive cells that crossed the Matrigel transwell was dramatically reduced by 70.0, 60.0, and 79.0% in A172, U87MG, and PDX-L14 cells, respectively. These data showed that knockdown of HOTAIR could impede glioma cell growth, invasion, and metastasis.
3.3. HOTAIR acts as a competing endogenous RNA (ceRNA) by sponging miR-301a-3p and indirectly regulates FOSL1 expression
3.3.1. HOTAIR negatively regulates miR-301a-3p, which inhibits glioma cell growth and invasion
Our previous published in-depth data analysis from miRNA microarray data revealed that miR-301a-3p was significantly upregulated by TRPM7 knockdown [Fig. 2A in Frontiers in Oncology, 2019, 9:1413 [20]], while the lncRNA data in Fig. 1 showed that HOTAIR is positively regulated by TRPM7, therefore, we hypothesized that cytoplasmic lncRNA HOTAIR may directly bind to sncRNA, miR-301a-3p, and function as ceRNAs or sponges. To test our hypothesis, we first examined the relationship between HOTAIR and miR-301a-3p. Through the bioinformatics database (LncBase Predicted v.2), we predicted the potential miR-301a-3p binding sites in HOTAIR, which was among the numerous targets of miR-301a-3p (Fig. 3A). Next, by quantification of both miR-301a-3p expression using stem-loop pulsed reverse transcription PCR, and HOTAIR at endogenous levels by qRT-PCR in 14 glioma cell lines, followed by Pearson’s correlation analysis, we found a significant negative correlation between HOTAIR and miR-301a-3p in glioma cells (r = −0.6496, p = 0.0009, Fig. 3B). To further verify the direct binding between HOTAIR and miR-301a-3p, an anti-Ago2 RIP assay was performed in U87MG and PDX-L14 cells that were overexpressing miR-301a-3p by transiently transfected with miR-301a-3p mimic. Compared to the control group, the endogenous HOTAIR was specifically enriched in the miR-301a-3p mimic transfected-U87MG and -PDX-L14 glioma cells (Fig. 3C), indicating that HOTAIR directly targeted miR-301a-3p, and miR-301a-3p was negatively regulated by HOTAIR.
Fig. 3.
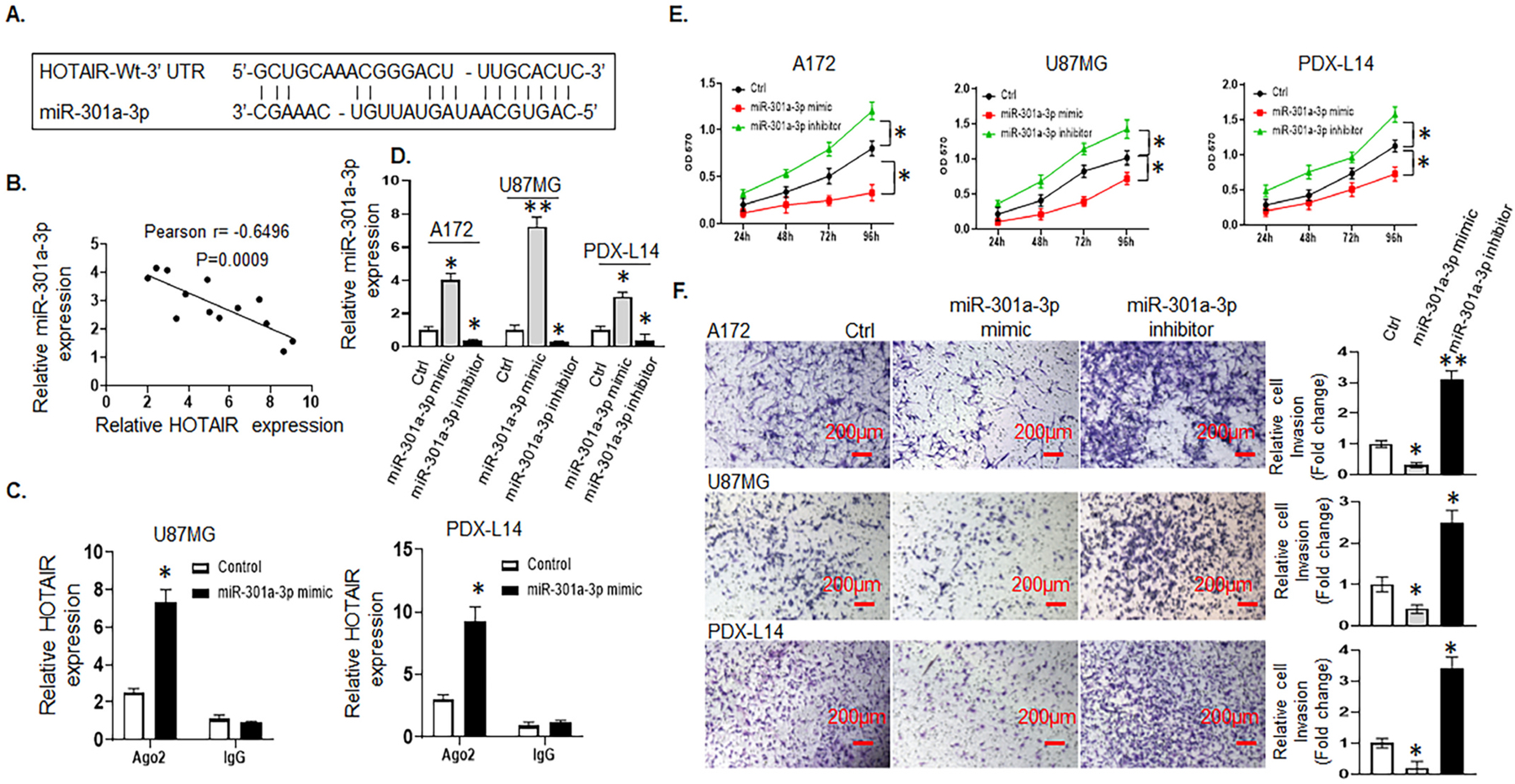
HOTAIR acts as a ceRNA by sponging miR-301a-3p and indirectly regulates FOSL1 expression. HOTAIR negatively regulates miR-301a-3p, which inhibits glioma cell growth and invasion. A, The predicted binding sites of miR-301a-3p to the HOTAIR sequence were shown by analyzing LncBase Predicted v.2 database. B, Pearson’s correlation curve revealed the negative relationship between HOTAIR and miR-301a-3p expression in glioma cells. C, RIP was performed in U87MG and PDX-L14 cells transfected with miR-301a-3p mimics and controls, HOTAIR expression was detected using qRT-PCR. D, The transfection efficiency represented by the differential expression of miR-301a-3p was examined in A172, U87MG, and PDX-L14 cells transfected with miR-301a-3p mimic, inhibitor, or control. E, A172, U87MG, and PDX-L14 cells were transfected with miR-301a-3p mimic, inhibitor, or control for 24 to 96 h followed by an MTT assay. F, Cell invasion assays were performed to examine the effects of miR-301a-3p on invasion after transfecting A172, U87MG, and PDX-L14 with miR-301a-3p mimic, inhibitor, or control. Images were captured at a magnification of x10, *p < 0.05.
To understand the biological effects of miR-301a-3p on glioma cell proliferation, miR-301a-3p expression was manipulated by transfecting A172, U87MG, and PDX-L14 cells with either miR-301a-3p mimics, inhibitors, or controls (Ctrl) for 72 h to increase or decrease miR-301a-3p expression levels. The stem-loop RT-PCR assay confirmed that miR-301a-3p expression levels were dramatically increased in A172, U87MG, and PDX-L14 cells transfected with miR-301a-3p mimics as compared to those in cells transfected with control (Fig 3 D). Similarly, miR-301a-3p was significantly decreased in the above three cell lines transfected with miR-301a-3p inhibitors as compared to that in cells transfected with control (Fig 3 D). As shown in Fig. 3E, the overexpression of miR-301a-3p by transient transfection of miR-301a-3p mimic dramatically inhibited the growth rate of A172, U87MG, and PDX-L14 glioma cells, whereas cell proliferation decreased from 59.4 to 44.2% with the maximum at 96 h in A172, from 52.8 to 29.2% with the maximum at 72 h in U87MG, and from 35.4 to 25.4% with the maximum at 96 h in PDX-L14 cells. In contrast, reduced miR-301a-3p expression by miR-301a-3p inhibitor augmented the growth of the three glioma cell lines above, whereas cell proliferation increased in A172 from 33.3 to 37.9%, in U87MG from 27.8 to 41.2%, and in PDX-L14 from 23.9 to 44.5% for a 96 h period. Next, the Matrigel transwell invasion assays were performed on A172, U87MG, and PDX-L14 cell lines that were transiently transfected with miR-301a-3p mimic, miR-301a-3p inhibitor, or control. The results showed that the ectopic expression of miR-301a-3p inhibited the number of invaded cells by 69.0, 60.0, and 80.0%, respectively (Fig. 3F); on the other hand, miR-301a-3p inhibitors enhanced the number of invaded cells by 67.7, 60.0, and 70.6%, respectively (Fig. 3F). These data showed that miR-301a-3p served as a tumor suppressor inhibiting glioma cell growth and invasion.
3.3.2. HOTAIR positively regulates while miR-301a-3p negatively regulates the target gene FOSL1 in glioma cells
While in the search for miR-301a-3p targeting genes, FOSL1 was initially identified as a potential miR-301a-3p targeted gene using public prediction algorithms (miRbase.org and Starbase v2.0) (Fig. 4A). Experimentally, as shown in Fig. 4B, we found that FOSL1 mRNA and miR-301a-3p had a negative correlation in 14 glioma cell lines by Pearson’s correlation analysis. Then, a dual-luciferase reporter assay was performed to further verify the surmise that FOSL1 was directly targeted by miR-301a-3p in U87MG and PDX-L14 cells. To this end, we constructed FOSL1 3’ UTR sequence into a pGL3 promoter vector to generate a luciferase reporter (wild-type FOSL1 or Wt-FOSL1). Concurrently, we mutated the binding sites of miR-301a-3p to FOSL1 to generate another luciferase reporter, mutant-FOSL1 (Mut-FOSL1) (Fig. 4C, two upper panels). The luciferase activity of U87MG and PDX-L14 cells cotransfected with miR-301a-3p mimic and control, along with luciferase reporters containing Wt-FOSL1 or Mut-FOSL1, were performed. The luciferase activity was discovered to be pronouncedly reduced when Wt-FOSL1 and miR-301a-3p mimic were cotransfected into U87MG and PDX-L14 cells in comparison with the activity in those cells cotransfected with Wt-FOSL1 and control. However, the luciferase activity of Mut-FOSL1 showed no statistical changes. The results of the dual-luciferase reporter assay showed that FOSL1 was indeed the direct target of miR-301a-3p (Fig. 4C, two lower panels). To further explore the association between miR-301a-3p and FOSL1 in glioma cells, we analyzed endogenous FOSL1 expression after transiently transfecting cells with either miR-301a-3p mimics, miR-301a-3p inhibitor, or control in A172, U87MG, and PDX-L14 cells. As shown in Fig. 4D, FOSL1 protein expression levels were significantly reduced by miR-301a-3p mimics and significantly enhanced by miR-301a-3p inhibitor in each of the three glioma cell lines [(Fig. 4D (a)]. Note, in U87MG cells, FOSL1 protein expression changes in response to miR-301a-3p were more obvious at the underexposure setting due to highly expressed FOSL1 in this cell line [(Fig. 4D (b)]. The above results suggested that FOSL1 was the direct target of miR-301a-3p and was negatively regulated by miR-301a-3p.
Fig. 4.
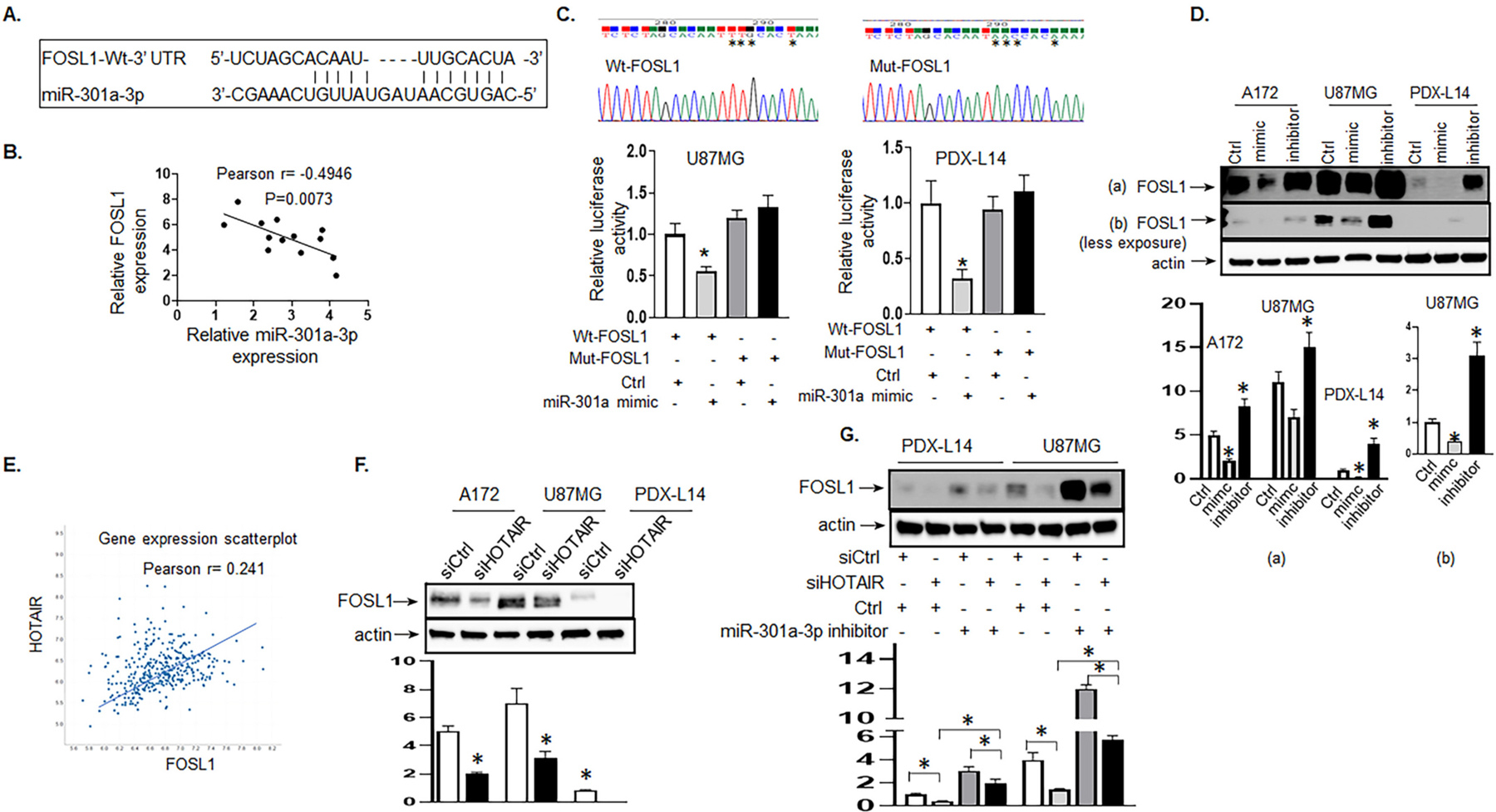
HOTAIR positively regulates while miR-301a-3p negatively regulates the target FOSL1 gene in glioma cells. A, The predicted binding sites of miR-301a-3p to the FOSL1 sequence were shown. B, Pearson’s correlation curve revealed the negative relationship between FOSL1 and miR-301a-3p. C, DNA sequencing of binding sites of miR-301a-3p to FOSL1 3’ UTR (Wt-FOSL1) and to mutated FOSL 3’ UTR (Mut-FOSL1) as indicated by stars were shown in the two upper panels. Luciferase activity of U87MG (left lower panel) and PDX-L14 cells (right lower panel) cotransfected with miR-301a-3p mimic and luciferase reporters containing Wt-FOSL1 or Mut-FOSL1 were performed. D, The endogenous FOSL1 protein expression levels were examined by Western blot after transiently transfecting with miR-301a-3p mimic, inhibitor, or control in A172, U87MG, and PDX-L14 cells. The gel images of both over-and less-exposure were shown. The bar graphs indicate the mean ± SD of three independent experiments. All data represent a representative experiment from three independent experiments, * p < 0.05 (Note, miR-301a-3p mimic was labeled as “mimic,” while miR-301a-3p inhibitor was labeled as “inhibitor” due to space limitations). E, Pearson’s correlation between HOTAIR and FOSL1 in brain tissues of glioma patients was analyzed by Affymetrix Human Exon 1.0 ST. F, FOSL1 protein was examined when the HOTAIR gene was silenced by siHOTAIR in A172, U87MG, and PDX-L14 cells. The bar graphs indicate the mean ± SD of three independent experiments. All data represent a representative experiment from three independent experiments, * p < 0.05. G, The levels of FOSL1 transfected with miR-301a-3p inhibitor along with siHOTAIR in PDX-L14 and U87MG cells were analyzed by Western blot.
As HOTAIR negatively regulated miR-301a-3p by directly targeting it (Fig. 3A–C), FOSL1 was the direct target of miR-301a-3p and was negatively regulated by miR-301a-3p (Fig 4A–D); therefore, it is reasonable for us to conclude that HOTAIR could positively regulate the FOSL1 gene. To determine the relationship between HOTAIR and FOSL1, gene co-expression network was constructed by TCGA (http://www.betastasis.com/glioma/tcga_gbm/) to visually study the relationship between the lncRNA HOTAIR and FOSL1 mRNA expression levels. The HOTAIR and FOSL1 mRNA expression showed a moderate positive correlation in GBM patients by Pearson’s correlation analysis (r = 0.241, Fig. 4E). We further assessed FOSL1 protein expression levels when downregulating HOTAIR using siHOTAIR by Western blot analysis. In A172, U87MG, and PDX-L14 cell lines, the protein level of FOSL1 was significantly decreased in cells transfected with siHOTAIR compared with that in cells transfected with scrambled control (Fig. 4F), further supporting a significant positive correlation between HOTAIR and FOSL1.
To further determine how HOTAIR integrated with miR-301a-3p to target FOSL1 gene, we designed an experiment to understand whether the effects of HOTAIR on FOSL1 expression could be reversed by miR-301a-3p. As shown in Fig. 4G, FOSL1 expression was significantly decreased after transfecting with siHOTAIR in PDX-L14 and U87MG cells. The inhibitory effect of siHOTAIR was notably reversed by co-transfection with siHOTAIR and miR-301a-3p inhibitor in PDX-L14 and U87MG cells. These results indicated that HOTAIR functioned as a ceRNA by sponging miR301a-3p and indirectly regulated FOSL1 expression.
3.4. FOSL1’s high expression is associated with poor survival in GBM patients and elevates glioma cell proliferation and invasion
The TCGA database, which contained information on 454 glioblastoma patients, classified based on WHO classification as GBM, was then analyzed for the clinical significance of FOSL1 mRNA expression. FOSL1 mRNA expression levels in GBM patients were significantly higher than those in normal individuals as detected by Affymetrix HT HG U133A (Fig. 5A left panel); furthermore, classical, mesenchymal, and neural molecular subtypes in glioma tissue were higher than those in the normal brain (p < 0.05; Fig. 5A, right panel). Although a similar tendency was displayed in the proneural subtype, it did not reach a significant difference (Fig. 5A, right panel). To determine whether FOSL1 gene expression is related to patient survival, all GBM patients from The Human Protein Atlas (https://www.proteinatlas.org) were analyzed in a pooled setting, and the median value was selected as the cut-off point, the 7-year OS rate, as revealed by the Kaplan-Meier survival curves (Fig. 5B), was significantly higher (p < 0.001; log-rank test) in those with low FOSL1 transcript levels (blue curve) compared with those with high expression (red curve).
Fig. 5.
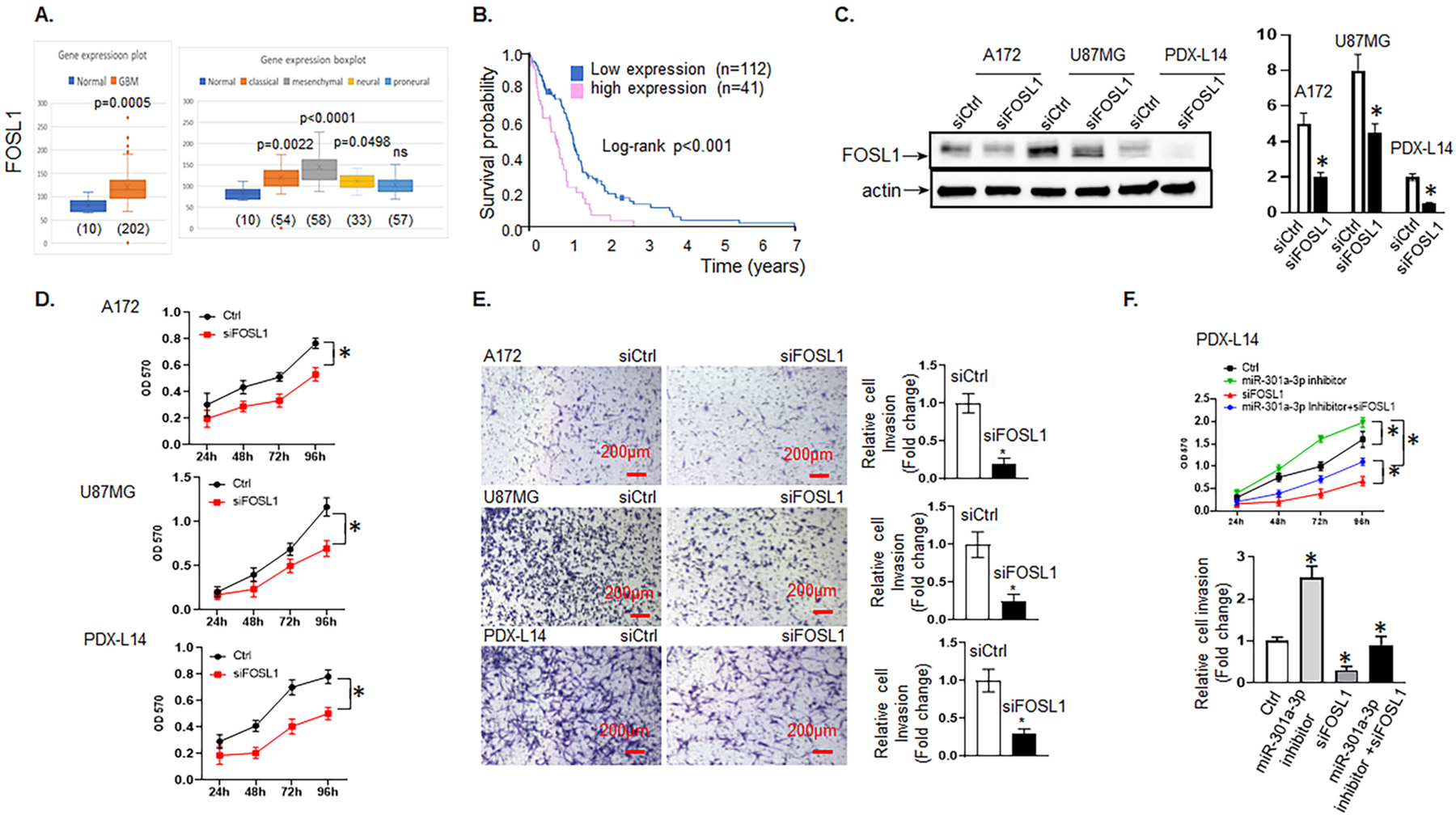
FOSL1’s high expression is associated with poor survival in GBM patients and increases glioma cell proliferation and invasion. A, FOSL1 mRNA expression levels in brain tissues of glioma patients (left panel) and different molecular subtypes (right panel) were analyzed by Affymetrix Human Exon 1.0 ST of TCGA data, respectively. B, Kaplan-Meier survival curve for the 7-year OS rate of glioma patients. The cut-off was set at the median. C, The level of FOSL1 was analyzed in A172, U87MG, and PDX-L14 cells transfected with siFOSL1 and control by Western blot. D, A172, U87MG, and PDX-L14 cells were transfected with siFOSL1 for 24 to 96 h followed by an MTT assay. E, Cell invasion assays were conducted to examine the effects of FOSL1 on invasion after transfecting A172, U87MG, and PDX-L14 with siFOSL1 targeting FOSL1 mRNA. Images were captured at a magnification of x10, *p < 0.05. F, Functional assays were performed to identify the phenomenon of FOSL1 inhibition partially reverses the promoting effects of the miR-301a-3p inhibitor on the growth (upper panel) and metastasis (lower panel) of PDX-L14 cells.
We then determined the biological functions of FOSL1 on glioma by transiently transfecting A172, U87MG, and PDX-L14 glioma cells with siFOSL1 and corresponding controls. The transient transfection efficiency was determined by Western blot. As shown in Fig. 5C, FOSL1 protein expression levels were significantly reduced by siFOSL1 treatment in each of the glioma cell lines. As expected, the reduction of FOSL1 expression by siFOSL1 effectively reduced glioma cell proliferation during 24 to 96 h post-transfection from 30.6 to 35.0% in A172 cells, 15.8 to 41.4% in U87MG cells, and 35.7 to 50.2% in PDX-L14 cells (Fig. 5D). Similarly, the number of invasive cells that crossed the Matrigel transwells was reduced by 80.0, 75.0, and 70.0% in A172, U87MG, and PDX-L14 cells at 72 h post-transfection of siFOSL1, respectively (Fig. 5E). These data suggested that FOSL1 upregulation promoted glioma cell growth and invasion.
As knockdown of the highly endogenously expressed FOSL1 significantly weakened proliferation and invasion of PDX-L14 cells (Fig. 5D–E), while FOSL1 was the direct target of miR-301a-3p and was negatively regulated by miR-301a-3p (Fig. 4A–D), we then further hypothesized that miR-301a-3p exerted its function through regulation of FOSL1 in glioma cells. To test this hypothesis, rescue experiments were conducted. Our results showed that FOSL1 suppression blocked the effect induced by miR-301a-3p reduction on glioma cell growth and invasion (Fig. 5F). In detail, siFOSL1 effectively reduced glioma cell proliferation induced by miR-301a-3p inhibitor during 24 to 96 h post-transfection from 23.8 to 45.6% for PDX-L14. Similarly, siFOSL1 markedly reduced miR301a-3p inhibitor-induced invasion by 66.7%. These data suggested that FOSL1 partially reversed the promoting effect of miR-301a-3p inhibition on the growth and metastasis of glioma cells. These results further support our findings in Fig. 3E–F, that reduced miR-301a-3p expression alone dramatically enhanced the proliferative and invasive ability of PDX-L14. We use the proposed model to summarize the role of the TRPM7-mediated HOTAIR/miR-301a-3p/FOSL1 axis in glioma tumorigenesis (Fig. 6). TRPM7 mediates the Ca2+ influx necessary for NF-κB activation, which transcriptionally activates lncHOTAIR. In turn, lncHOTAIR directly inhibits miR-301a-3p expression, which directly functionally impede FOSL1 gene activity.
4. Discussion
Major findings from the present study include: 1) TRPM7 functions as a positive regulator of lncRNA HOTAIR and a negative regulator of miR-301a-3p; 2) HOTAIR acts as a ceRNA by sponging miR-301a-3p and indirectly regulates target gene FOSL1 expression; 3) Both HOTAIR and FOSL1 play oncogenic roles in glioma tumorigenesis, which promote glioma cell growth/proliferation and migration/invasion; increased expression of HOTAIR and FOSL1 are associated with unfavorable survival in GBM patients; 4) miR-301a-3p inhibits glioma cell proliferation and invasion by directly targeting FOSL1. Taken together, our study elucidated the role of TRPM7 mediated HOTAIR as a miR-301a-3p sponge to target the FOSL1 gene in glioma.
TRPM7 is a bifunctional protein fusing a divalent cations channel to a C-terminal serine/threonine kinase. Since cloned two decades ago, TRPM7 has emerged as new therapeutic target and is associated with tumor progression, anoxic neuronal death, tissue fibrosis, giant platelet disorders, neutrophil recruitment, and production of reactive oxygen species [30,31]. Our group has long been focusing on the study of TRPM7’s potential as a therapeutic target in glioblastoma. Our previous work uncovered that aberrant TRPM7 expression and activity is essential for glioma malignancy not only through modulating protein-coding genes such as Notch, STAT3-ALDH1, and CD133 [21,22] but also managing sncRNAs/miRNAs, which play key regulatory roles in shaping glioma cellular functions. Continuously, our current study identified the lncRNA, HOTAIR mediated by TRPM7 competing with its downstream miRNA, miR-301a-3p, positively regulate oncogenic gene FOSL1, which contributes to glioblastoma growth.
The majority of human RNA transcripts do not encode for proteins, instead, non-coding RNAs are crucial to the regulation of cell physiology and shape cellular activity. The aberrant expression of many non-coding RNAs has been documented and associated with human malignancies including GBM [23,32]. Previously, our in-depth data analysis from miRNA microarray data revealed that hsa-miR-301a-3p is statistically significantly upregulated by TRPM7 knockdown [Fig. 2 in Frontiers in Oncology 2019,9:1413 [20]]. The signaling pathways involving lncRNA-miRNA-mRNA axis to fine tune their effects on the gene regulation [33] prompted us to investigate the lncRNAs changes based upon TRPM7 deletion in glioma cells. When A172 glioma cells with TRPM7 depletion were subjected to lncRNA PCR array assay, we did find a panel of 17 significantly altered lncRNAs, by which HOTAIR is the most upregulated one when TRPM7 was deleted. TRPM7 encodes a permeable nonselective cation channel fused with serine/threonine kinase at its carboxyl terminus. Although the channel function is dissociated from intrinsic kinase activity [34] and the kinase activity is not channel dependent, the kinase domain may modulate channel activity [13,35,36]. Our previous studies showed that TRPM7’s channel activity is required for glioma cell growth, while the kinase domain is required for cell migration/invasion [20]. Therefore, we surmise that both the TRPM7 channel and kinase domain are involved in the regulation of TRPM7 on HOTAIR, which is worth further investigation.
HOTAIR, an oncogenic lncRNA located on chromosome 12q13.13, is transcribed from the antisense strand by RNA polymerase II and is 2.2 kb long, capped, spliced, and polyadenylated [37]. HOTAIR is overexpressed in glioma [7], breast [38], lung [39], colorectal [40], and hepatocellular carcinoma [41], and is associated with the promotion of oncogenesis and metastasis [42]. HOTAIR silences target genes via multiple mechanisms. One of the well characterized and well-established mechanisms is HOTAIR’s interaction with chromatin-modifying enzymes polycomb repressive complex 2 (PRC2) and lysin specific demethylase 1 (LSD1) [43]. Beyond that, HOTAIR plays a role in protein degradation [44,45], microRNA sponging [46], and immune modulation [47,48]. However, most of the research into HOTAIR’s role in GBMs has been documented in biomarkers [49,50], with relative few studies on mechanisms, especially the lackluster of studies on relationships with another type of non-coding RNA, miRNA. This leaves a crucial gap in our understanding of glioma pathogenesis. Our current study discovered that HOTAIR acted as an oncogene, mediated by TRPM7, and directly bound to its downstream miR-301a-3p, which ultimately led to the positively regulated oncogene FOSL1 in glioma cells.
FOSL1, encoding FRA-1, is an AP-1 transcription factor with prognostic value in human solid tumors such as breast, lung, pancreatic, and colon cancer, where its overexpression correlated with tumor progression or worse patient survival [51–54]. FOSL1 controls cancer cell proliferation and survival, acts as a master switch of epithelial-to-mesenchymal transition (EMT) [55,56]. In agreement with other studies, our results showed that FOSL1 is responsible for sustaining glioma cell growth and glioma cell invasion in vitro and is an unfavorable prognostic factor for GBM patients. Although previously, Verde’s group postulated that FOSL1 expression depends on the balance between the p53-mediated transactivation and miR-34a-mediated posttranscriptional inhibition of FOSL1 in MCF7 cells [55], we are the first group to report that miR-301a-3p directly bind to FOSL1 and mediate posttranscriptional inhibition of FOSL1 in support of gliomagenesis, as represented by the luciferase reporter assay (Fig. 4C). FOSL1’s role in tumor microenvironment (TME) has also been documented. In colorectal cancer (CRC), FOS-L1 is both a transactivator of IL-6 in tumor associated macrophages (TAMs) and a transcriptional target of the IL-6/Stat3 pathway in cancer cells. FOSL1 induces the IL-6 transcription in TAMs, the IL-6 secreted by TAMs is a major inducer of EMT in CRC cancer cells, in which the IL-6-Stat3-FOSL1 axis critically contribute to FOSL1 accumulation, invasion and metastasis [55,57]. As we previously found that TRPM7 channels regulate glioma stemness through Stat3 [21], in the future, it is worth further investigation into the mutual regulation between FOSL1 and Stat3 in remodeling glioma TME in support of glioma cell invasion and progression. Of note, inhibition of FOSL1 represents “a double-edged sword,” the opposite data from Tian’s group in cervical cancer showed that FOSL1 favors apoptosis, decreases in MDM2, and piles up p53, in which, FOSL1 expression is reduced in tumor with respect to normal tissue [58]. In addition, the treatment (vemurafenib)-induced FOSL1 downregulation, unexpectedly, critically contributes to the increase of growth factors, such as EGF, and decrease of growth inhibitors, such as IGFBP3 in melanoma [59]. Therefore, context-and therapy-dependent mechanisms of FOSL1 should be taken into account when considering any approach to target FOSL1.
miRNAs are thought to regulate the expression of various coding genes simultaneously. miR-301a-3p was reported to promote oxidative stress, inflammation and apoptosis in oxidized low-density lipoprotein (ox-LDL)-induced human umbilical vein endothelial cells (HUVECs) [60]. Previously, we reported that miR-28–5p is negatively regulated by TRPM7 and inhibits glioma cell growth and invasion by targeting Rap1b gene [20]. In addition, we also found that miR-301a-3p showed the same trend as miR-28–5p when TRPM7 is silenced in glioma cells [20]. Thus, we speculated that miR-301a-3p might have a similar effect as miR-28–5p on glioma cell phenotype by targeting different target genes. Several other targets of miR-301a-3p, such as FOSL1, KLF7 [60] and MYBL1, are shown based on TargetScanHuman, which is a tool for microRNA targets prediction (targetscan.org). In the current study, we focused on FOSL1, which was associated with HOTAIR (Fig. 4E). We observed an inverse correlation between HOTAIR and miR-301a-3p (Fig. 3B). Importantly, we found that HOTAIR binds directly to miR-301a-3p, as shown by the endogenous interaction between HOTAIR and miR-301a-3p by coimmunoprecipitation with the Ago2 protein in glioma cells (Fig. 3C). HOTAIR could play a role of endogenous sponge or ceRNA to control miR-301a-3p availability for its target gene FOSL1 (Fig. 4G).
In summary, overexpressed HOTAIR mediated by TRPM7 promoted glioma cell proliferation and invasion, suggesting that HOTAIR exhibits oncogenic properties in glioma progression. HOTAIR exerted the oncogenic effects partially through TRPM7/HOTAIR/miR301a-3p/FOSL1 axis. These findings provide novel molecular mechanisms and useful information to find new biomarkers for diagnosis and therapeutic applications in glioma progression. To be noted, transcriptional regulation of FOSL1 has been documented through several transcription factors binding to the FOSL1 promoter such as AP-1, SNAIL1, TWIST1, SMADS, and Stat3 [55]. As FOSL1 is a key glioma regulator, it is expected that other TRPM7-mediated FOSL1 activation could contribute to glioma pathogenesis other than HOTAIR and miR-301a-3p. As mentioned above, the mechanism by which Stat3 activated by TRPM7 modulates FOSL1 transcription and translation in glioma will be worth investigating in the future.
Significance:
Our study elucidated the role of TRPM7-mediated HOTAIR as a miRNA sponge to target downstream FOSL1 oncogene and therefore consequently contribute to gliomagenesis, which shed new light on TRPM7/lncRNA-directed diagnostic and therapeutic approach in glioma.
Acknowledgments
We are grateful to Dr. Yancey G. Gillespie at the UAB for providing us with the PDX lines, and Dr. Erwin van Meir at UAB for providing T98G and LN18 glioma cell lines.
Financial support
This study was supported by the SC3 grant from NIH NIGMS GM121230 (ML). The funding agencies had no role in the design, collection, analysis, and interpretation of the study’s data and in writing the manuscript.
Footnotes
Declaration of Competing Interest
The authors declare no potential conflicts of interest.
References
- [1].Prager BC, Bhargava S, Mahadev V, Hubert CG, Rich JN, Glioblastoma Stem Cells: Driving Resilience through Chaos, Trends Cancer 6 (2020) 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Li C, Cho HJ, Yamashita D, Abdelrashid M, Chen Q, Bastola S, et al. , Tumor edge-to-core transition promotes malignancy in primary-to-recurrent glioblastoma progression in a PLAGL1/CD109-mediated mechanism, Neuro-oncology Adv 2 (2020) vdaa163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Taylor JT, Ellison S, Pandele A, Wood S, Nathan E, Forte G, et al. , Actinomycin D downregulates Sox2 and improves survival in preclinical models of recurrent glioblastoma, Neuro-oncology 22 (2020) 1289–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bausart M, Préat V, Malfanti A, Immunotherapy for glioblastoma: the promise of combination strategies, J. Exp. Clin. Cancer Res 41 (2022) 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gritsch S, Batchelor TT, Gonzalez Castro LN, Diagnostic, therapeutic, and prognostic implications of the 2021 World Health Organization classification of tumors of the central nervous system, Cancer 128 (2022) 47–58. [DOI] [PubMed] [Google Scholar]
- [6].Galanis E, Wen PY, de Groot JF, Weller M, Isocitrate dehydrogenase wild-type glial tumors, including glioblastoma, Hematol. Oncol. Clin. North Am 36 (2022) 113–132. [DOI] [PubMed] [Google Scholar]
- [7].Pastori C, Kapranov P, Penas C, Peschansky V, Volmar CH, Sarkaria JN, et al. , The Bromodomain protein BRD4 controls HOTAIR, a long noncoding RNA essential for glioblastoma proliferation, Proc. Natl. Acad. Sci. U. S. A 112 (2015) 8326–8331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Momtazmanesh S, Rezaei N, Long non-coding RNAs in diagnosis, treatment, prognosis, and progression of glioma: a state-of-the-art review, Front. Oncol 11 (2021), 712786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rynkeviciene R, Simiene J, Strainiene E, Stankevicius V, Usinskiene J, Miseikyte Kaubriene E, et al. , Non-coding RNAs in glioma, Cancers 11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ryazanova LV, Dorovkov MV, Ansari A, Ryazanov AG, Characterization of the protein kinase activity of TRPM7/ChaK1, a protein kinase fused to the transient receptor potential ion channel, J. Biol. Chem 279 (2004) 3708–3716. [DOI] [PubMed] [Google Scholar]
- [11].Ryazanova LV, Rondon LJ, Zierler S, Hu Z, Galli J, Yamaguchi TP, et al., TRPM7 is essential for Mg(2+) homeostasis in mammals, Nat. Commun 1 (2010) 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, Stokes AJ, et al. , LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability, Nature 411 (2001) 590–595. [DOI] [PubMed] [Google Scholar]
- [13].Schmitz C, Perraud AL, Johnson CO, Inabe K, Smith MK, Penner R, et al., Regulation of vertebrate cellular Mg2 homeostasis by TRPM7, Cell 114 (2003) 191–200. [DOI] [PubMed] [Google Scholar]
- [14].Matsushita M, Kozak JA, Shimizu Y, McLachlin DT, Yamaguchi H, Wei FY, et al. , Channel function is dissociated from the intrinsic kinase activity and autophosphorylation of TRPM7/ChaK1, J. Biol. Chem 280 (2005) 20793–20803. [DOI] [PubMed] [Google Scholar]
- [15].Mittermeier L, Demirkhanyan L, Stadlbauer B, Breit A, Recordati C, Hilgendorff A, et al. , TRPM7 is the central gatekeeper of intestinal mineral absorption essential for postnatal survival, Proc. Natl. Acad. Sci. U. S. A 116 (2019) 4706–4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yee NS, Role of TRPM7 in cancer: potential as molecular biomarker and therapeutic target, Pharmaceuticals (Basel, Switzerland) 10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Prevarskaya N, Skryma R, Shuba Y, Ion channels and the hallmarks of cancer, Trends Mol. Med 16 (2010) 107–121. [DOI] [PubMed] [Google Scholar]
- [18].Nadolni W, Zierler S, The channel-kinase TRPM7 as novel regulator of immune system homeostasis, Cells 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Leng TD, Li MH, Shen JF, Liu ML, Li XB, Sun HW, et al. , Suppression of TRPM7 inhibits proliferation, migration, and invasion of malignant human glioma cells, CNS Neurosci. Ther 21 (2015) 252–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wan J, Guo AA, Chowdhury I, Guo S, Hibbert J, Wang G, et al. , TRPM7 induces mechanistic target of Rap1b through the downregulation of miR-28–5p in glioma proliferation and invasion, Front. Oncol 9 (2019) 1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu M, Inoue K, Leng T, Guo S, Xiong ZG, TRPM7 channels regulate glioma stem cell through STAT3 and Notch signaling pathways, Cell. Signal 26 (2014) 2773–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wan J, King P, Guo S, Saafir T, Jing Y, Liu M, TRPM7 induces tumorigenesis and stemness through notch activation in glioma, Front. Pharmacol 11 (2020) 590723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Panni S, Lovering RC, Porras P, Orchard S, Non-coding RNA regulatory networks, Biochim. Biophys. Acta 1863 (2020), 194417. [DOI] [PubMed] [Google Scholar]
- [24].Zhang J, Chen G, Gao Y, Liang H, HOTAIR/miR-125 axis-mediated Hexokinase 2 expression promotes chemoresistance in human glioblastoma, J. Cell. Mol. Med 24 (2020) 5707–5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hong Q, Li O, Zheng W, Xiao WZ, Zhang L, Wu D, et al. , LncRNA HOTAIR regulates HIF-1α/AXL signaling through inhibition of miR-217 in renal cell carcinoma, Cell Death Dis 8 (2017), e2772. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [26].Sa L, Li Y, Zhao L, Liu Y, Wang P, Liu L, et al. , The role of HOTAIR/miR-148b-3p/USF1 on regulating the permeability of BTB, Front. Mol. Neurosci 10 (2017) 194. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [27].Varkonyi-Gasic E, Wu R, Wood M, Walton EF, Hellens RP, Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs, Plant Methods 3 (2007) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Schappe MS, Szteyn K, Stremska ME, Mendu SK, Downs TK, Seegren PV, et al., Chanzyme TRPM7 mediates the Ca(2+) influx essential for lipopolysaccharide-induced toll-like receptor 4 endocytosis and macrophage activation, Immunity 48 (2018) 59–74.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Özeş AR, Miller DF, Özeş ON, Fang F, Liu Y, Matei D, et al. , NF-κB-HOTAIR axis links DNA damage response, chemoresistance and cellular senescence in ovarian cancer, Oncogene 35 (2016) 5350–5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nadolni W, Immler R, Hoelting K, Fraticelli M, Ripphahn M, Rothmiller S, et al. , TRPM7 Kinase Is Essential for Neutrophil Recruitment and Function via Regulation of Akt/mTOR Signaling, Front. Immunol 11 (2020), 606893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chubanov V, Gudermann T, Mapping TRPM7 Function by NS8593, Int. J. Mol. Sci 21 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mousavi SM, Derakhshan M, Baharloii F, Dashti F, Mirazimi SMA, Mahjoubin-Tehran M, et al. , Non-coding RNAs and glioblastoma: insight into their roles in metastasis, Mol. Ther. Oncol 24 (2022) 262–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].López-Urrutia E, Bustamante Montes LP, Ladrón de Guevara Cervantes D, Pérez-Plasencia C, Campos-Parra AD, Crosstalk between long non-coding RNAs, micro-RNAs and mRNAs: deciphering molecular mechanisms of master regulators in cancer, Front. Oncol 9 (2019) 669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Clark K, Langeslag M, van Leeuwen B, Ran L, Ryazanov AG, Figdor CG, et al. , TRPM7, a novel regulator of actomyosin contractility and cell adhesion, EMBO J 25 (2006) 290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Demeuse P, Penner R, Fleig A, TRPM7 channel is regulated by magnesium nucleotides via its kinase domain, J. Gen. Physiol 127 (2006) 421–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Desai BN, Krapivinsky G, Navarro B, Krapivinsky L, Carter BC, Febvay S, et al. , Cleavage of TRPM7 releases the kinase domain from the ion channel and regulates its participation in Fas-induced apoptosis, Dev. Cell 22 (2012) 1149–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Price RL, Bhan A, Mandal SS, HOTAIR beyond repression: In protein degradation, inflammation, DNA damage response, and cell signaling, DNA Repair 105 (2021), 103141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bhan A, Hussain I, Ansari KI, Kasiri S, Bashyal A, Mandal SS, Antisense transcript long noncoding RNA (lncRNA) HOTAIR is transcriptionally induced by estradiol, J. Mol. Biol 425 (2013) 3707–3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhao W, An Y, Liang Y, Xie XW, Role of HOTAIR long noncoding RNA in metastatic progression of lung cancer, Eur. Rev. Med. Pharmacol. Sci 18 (2014) 1930–1936. [PubMed] [Google Scholar]
- [40].Peng CL, Zhao XJ, Wei CC, Wu JW, LncRNA HOTAIR promotes colon cancer development by down-regulating miRNA-34a, Eur. Rev. Med. Pharmacol. Sci 23 (2019) 5752–5761. [DOI] [PubMed] [Google Scholar]
- [41].Yang T, He X, Chen A, Tan K, Du X, LncRNA HOTAIR contributes to the malignancy of hepatocellular carcinoma by enhancing epithelial-mesenchymal transition via sponging miR-23b-3p from ZEB1, Gene 670 (2018) 114–122. [DOI] [PubMed] [Google Scholar]
- [42].Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, et al. , Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis, Nature 464 (2010) 1071–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, et al. , Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs, Cell 129 (2007) 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhang H, Diab A, Fan H, Mani SK, Hullinger R, Merle P, et al. , PLK1 and HOTAIR accelerate proteasomal degradation of SUZ12 and ZNF198 during hepatitis B virus-induced liver carcinogenesis, Cancer Res 75 (2015) 2363–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Yoon JH, Abdelmohsen K, Kim J, Yang X, Martindale JL, Tominaga-Yamanaka K, et al. , Scaffold function of long non-coding RNA HOTAIR in protein ubiquitination, Nat. Commun 4 (2013) 2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhang Z, Cheng J, Wu Y, Qiu J, Sun Y, Tong X, LncRNA HOTAIR controls the expression of Rab22a by sponging miR-373 in ovarian cancer, Mol. Med. Rep 14 (2016) 2465–2472. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [47].Fujisaka Y, Iwata T, Tamai K, Nakamura M, Mochizuki M, Shibuya R, et al. , Long non-coding RNA HOTAIR up-regulates chemokine (C-C motif) ligand 2 and promotes proliferation of macrophages and myeloid-derived suppressor cells in hepatocellular carcinoma cell lines, Oncol. Lett 15 (2018) 509–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sun J, Chu H, Ji J, Huo G, Song Q, Zhang X, Long non-coding RNA HOTAIR modulates HLA-G expression by absorbing miR-148a in human cervical cancer, Int. J. Oncol 49 (2016) 943–952. [DOI] [PubMed] [Google Scholar]
- [49].Tan SK, Pastori C, Penas C, Komotar RJ, Ivan ME, Wahlestedt C, et al. , Serum long noncoding RNA HOTAIR as a novel diagnostic and prognostic biomarker in glioblastoma multiforme, Mol. Cancer 17 (2018) 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Elsayed ET, Salem PE, Darwish AM, Fayed HM, Plasma long non-coding RNA HOTAIR as a potential biomarker for gastric cancer, Int. J. Biol. Markers (2018) 528–533. [DOI] [PubMed] [Google Scholar]
- [51].Vallejo A, Perurena N, Guruceaga E, Mazur PK, Martinez-Canarias S, Zandueta C, et al. , An integrative approach unveils FOSL1 as an oncogene vulnerability in KRAS-driven lung and pancreatic cancer, Nat. Commun 8 (2017) 14294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Xu H, Jin X, Yuan Y, Deng P, Jiang L, Zeng X, et al. , Prognostic value from integrative analysis of transcription factors c-Jun and Fra-1 in oral squamous cell carcinoma: a multicenter cohort study, Sci. Rep 7 (2017) 7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Iskit S, Schlicker A, Wessels L, Peeper DS, Fra-1 is a key driver of colon cancer metastasis and a Fra-1 classifier predicts disease-free survival, Oncotarget 6 (2015) 43146–43161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gallenne T, Ross KN, Visser NL, Salony Desmet C.J., Wittner BS, et al. , Systematic functional perturbations uncover a prognostic genetic network driving human breast cancer, Oncotarget 8 (2017) 20572–20587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Talotta F, Casalino L, Verde P, The nuclear oncoprotein Fra-1: a transcription factor knocking on therapeutic applications’ door, Oncogene 39 (2020) 4491–4506. [DOI] [PubMed] [Google Scholar]
- [56].Marques C, Unterkircher T, Kroon P, Oldrini B, Izzo A, Dramaretska Y, et al. , NF1 regulates mesenchymal glioblastoma plasticity and aggressiveness through the AP-1 transcription factor FOSL1, eLife 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Liu H, Ren G, Wang T, Chen Y, Gong C, Bai Y, et al. , Aberrantly expressed Fra-1 by IL-6/STAT3 transactivation promotes colorectal cancer aggressiveness through epithelial-mesenchymal transition, Carcinogenesis 36 (2015) 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Xiao S, Zhou Y, Yi W, Luo G, Jiang B, Tian Q, et al. , Fra-1 is downregulated in cervical cancer tissues and promotes cervical cancer cell apoptosis by p53 signaling pathway in vitro, Int. J. Oncol 46 (2015) 1677–1684. [DOI] [PubMed] [Google Scholar]
- [59].Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, et al. , Therapy-induced tumour secretomes promote resistance and tumour progression, Nature 520 (2015) 368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Jiang H, Lv J, MicroRNA-301a-3p increases oxidative stress, inflammation and apoptosis in ox-LDL-induced HUVECs by targeting KLF7, Exp. Ther. Med 21 (2021) 569. [DOI] [PMC free article] [PubMed] [Google Scholar]