

Abstract
Excessive adipose tissue mass underlies much of the metabolic health complications in obesity. Although exercise training is known to improve metabolic health in individuals with obesity, the effects of exercise training without weight loss on adipose tissue structure and metabolic function remain unclear. Thirty-six adults with obesity (BMI = 33±3 kg/m2) were assigned to 12 wks (4d/wk) of either moderate-intensity continuous training (MICT; 70% HR max, 45min; n=17) or high-intensity interval training (HIIT; 90% HR max, 10 X 1min; n=19), maintaining their body weight throughout. Abdominal subcutaneous adipose tissue (aSAT) biopsy samples were collected once before and twice after training (1 day after last exercise and again 4 days later). Exercise training modified aSAT morphology (i.e., reduced fat cell size, increased Collagen type 5a3, both p≤0.05, increased capillary density, p=0.05), and altered protein abundance of factors that regulate aSAT remodeling (i.e., reduced matrix metallopeptidase 9; p=0.02; increased angiopoietin-2; p<0.01). Exercise training also increased protein abundance of factors that regulate lipid metabolism (e.g., hormone sensitive lipase and fatty acid translocase; p≤0.03) and key proteins involved in mitogen-activated protein kinase pathway when measured the day after the last exercise session. However, most of these exercise-mediated changes were no longer significant 4 days after exercise. Importantly, MICT and HIIT induced remarkably similar adaptations in aSAT. Collectively, even in absence of weight loss, 12 weeks of exercise training induced changes in aSAT structure, as well as factors that regulate metabolism and inflammatory signal pathway in adults with obesity.
Keywords: Exercise training, high-intensity interval training, adipose tissue
Graphical abstract
3-month of MICT or HIIT without weight loss induced similar structural and metabolic adaptations in subcutaneous adipose tissue in adults with obesity.
INTRODUCTION
Abdominal subcutaneous adipose tissue (aSAT) in obesity is usually characterized by hypertrophic adipocytes, accumulation of fibrotic collagen proteins in extracellular matrix (ECM), and relatively low capillary density (Summers et al., 1996; Sun et al., 2011; Chun, 2012), all of which are tightly linked with insulin resistance, cell necrosis, and inflammatory macrophage infiltration (Després, 1993; Clément et al., 2004; Cinti et al., 2005). The increased inflammation in aSAT is considered an important contributor to many systemic metabolic health complications (Hotamisligil et al., 1993; Shimomura et al., 1996; Kern et al., 2001). In addition, these aSAT abnormalities in obesity can limit fat storage capacity, resulting in the high rates of fatty acid release into the systemic circulation that underlies metabolically harmful ectopic lipid deposition commonly found in tissues such as the liver and skeletal muscle of many adults with obesity (McQuaid et al., 2011). Therefore, strategies aimed at remodeling aSAT to create more smaller adipocytes (i.e., adipogenesis), modify the content and composition of the ECM, increase capillarization (i.e., angiogenesis), reduce inflammatory pathway activation, and increase lipid storage capacity could lead to improved metabolic health outcomes, even in the absence of weight loss.
Exercise is one of the first-line strategies for treating of obesity-related health complications, but how exercise “works” to improve metabolic health in obesity is still incompletely understood. Exercise training increases skeletal muscle mitochondria and oxidative capacity in adults with obesity (Tremblay et al., 1994; Menshikova et al., 2005; Ryan et al., 2020). But, increasing muscle oxidative capacity, per se, is often found to do rather little to improve insulin sensitivity and other key markers of metabolic health in adults with obesity (Østergård et al., 2006; Hutchison et al., 2012). Alternatively, evidence from our lab and others suggest exercise triggers responses in aSAT that may lead to ECM remodeling, increased angiogenesis, and alterations in the inflammatory profile (Cullberg et al., 2013; Kawanishi et al., 2013; Van Pelt et al., 2017; Ludzki et al., 2018). High-intensity interval training (HIIT) has garnered considerable attention because of its time efficiency and distinct physiological responses compared with more “conventional” moderate-intensity continuous training (MICT) (Little et al., 2014; Weston et al., 2014). However, whether and/or how the intensity of an exercise training program may modify adaptive responses within aSAT remains unclear. Differential effects of HIIT vs. MICT on circulating adipokine levels and adipocyte size in rodent models suggests a possibility that HIIT may induce more robust adaptations in aSAT morphology and metabolism compared with MICT (Shirvani & Arabzadeh, 2020; Sun et al., 2020).
The primary aims of this study were to compare the effects of 12 weeks of MICT vs. HIIT on subcutaneous aSAT structure and whole-body fatty acid mobilization in adults with obesity. This project also examined the effects of MICT and HIIT on the factors that regulate aSAT remodeling (i.e., adipogenesis, ECM remodeling, angiogenesis), metabolism (i.e., lipolysis, esterification, mitochondrial adaptation) and inflammatory signal pathway. Importantly, because even modest changes in body weight and fat mass can induce some aSAT remodeling (Clément et al., 2004; Kos et al., 2009; Magkos et al., 2016), this study was designed to require participants to maintain their body weight throughout the training intervention. We hypothesized that even without weight loss, 12 weeks of exercise training would induce detectable modifications to aSAT structure and metabolic regulation. Moreover, we hypothesized that these training-induced effects would be more pronounced in response to HIIT compared with MICT.
METHODS & MATERIALS
Subjects
Thirty-six adults with obesity (BMI: 30-40 kg/m2) participated in the study. All participants were sedentary, and reported to have had a stable body weight at least 6 months before their first pre-training clinical measurement. Subjects were not taking any medications or supplements known to affect their metabolism except contraceptive medications for some female participants (n=16 out of 24). Eligible participant also had no history of cardiovascular or metabolic disease. All female participants were premenopausal and not pregnant or lactating. All participants completed a detailed medical history survey and resting electrocardiogram, which were reviewed by a physician before any testing. Subjects participating in this study also participated in a related study from our laboratory, focused primarily on the effects of 3 months of exercise training in whole-body insulin sensitivity and muscle lipid metabolism (Ryan et al., 2020). Some of the methods (e.g., exercise training protocols) and results describing the basic responses to training (e.g., anthropometric characteristics, peak oxygen uptake) have been reported elsewhere (Ryan et al., 2020) but are repeated here for convenience. Written informed consent was obtained from all subjects before the study. This study conformed to the standards set by the Declaration of Helsinki, except for registration in a database. This study was approved by the University of Michigan Institutional Review Board (reference no. HUM00106883) and registered at clinicaltrials.gov (NCT02706093).
Study Design
Enrolled subjects were assigned to either moderate-intensity continuous training (MICT; n = 17) or high-intensity interval training (HIIT; n=19) groups in a counter balanced manner to optimize matching of sex and baseline anthropometric characteristics and aerobic fitness between training groups. Before training, subjects underwent one “clinical trial” (i.e., hyperinsulinemic-euglycemic clamp, [1-13C]palmitate tracer infusion to measure whole-body fatty acid mobilization, aSAT biopsies; see details in Clinical trial section, below). After 12-weeks of MICT or HIIT, subjects underwent two clinical trials, one conducted the day after the last exercise session (1d-Post training) and the other conducted after subjects refrained from exercise for 4 days (4d-Post training) to wash out potential influence of the acute exercise effects. Importantly, subjects were required to maintain their body weight throughout the intervention (see details in Training intervention section, below). A schematic of the study design is presented in Figure 1.
Figure 1. Schematic of study design.

Body composition and aerobic fitness testing
Before and after subjects completed the 12-week MICT or HIIT, body composition and aerobic fitness (i.e., peak oxygen uptake; VO2peak) were measured. Fat mass and fat free mass (FFM) were determined by dual-energy x-ray absorptiometry (Lunar DPX DEXA scanner, GE, WI). VO2peak was assessed on a stationary cycle ergometer (Corvival, Lode, Netherlands) using an incremental exercise protocol starting at 40 watts for 4min and was increased 20 watts each minute until volitional exhaustion. The rate of oxygen consumption was measured throughout this test using a metabolic cart (Max-II, Physiodyne Inc, NY) and VO2peak was determined as the highest 30-second average before volitional fatigue. Measurements of respiratory exchange ratio (RER) ≥ 1.1 and maximal heart rate (HRmax) ≥ 90% of age predicted values (i.e., 220-age) were used as secondary indices to help confirm maximal effort during these tests.
Training interventions
Training protocol.
Subjects in both groups exercised 4 days per week for 12 weeks. Each exercise training session for the MICT group consisted of continuous exercise for 45 minutes at 70% of their HRmax. For the HIIT group, each training session involved a 3-minute warm-up at ~65% HRmax, and then 10 x 1-minute intervals at 90% HRmax interspersed with 1 minute low-intensity active recovery. These high intensity intervals were then followed by 3-minute cool-down at 65% HRmax. Participants in both groups were allowed to select their preferred mode of exercise among stationary cycling, treadmill, elliptical or rowing ergometers. Exercise time (MICT; 45 minutes, HIIT; 25 minutes) and energy expenditure (MICT; ~250 kcals, HIIT; ~150 kcals) between groups were considerably different to address the study goal of comparing the low-volume HIIT protocol (Gibala et al., 2012) with a conventional and commonly prescribed steady-state exercise training program.
Training familiarization/ramp-up.
Subjects in both training groups followed a strictly regulated ramp-up protocol to gradually increase their exercise intensity and duration during the first two weeks of training. During the first week, participants in both groups performed 4 sessions of continuous 25 minutes exercise at 65% HRmax. During the second week of the ramp-up, subjects in the MICT group performed 4 sessions of continuous 35-minute exercise at 70% HRmax, while participants in the HIIT group performed 4 sessions of exercise progressively increasing the number of 1-minute intervals at 90% HRmax (i.e., 2 x 1, 4 x 1, 6 x 1, 8 x 1). Both groups began their full training prescription at the beginning of week 3.
Monitoring of training adherence and weight maintenance.
Study staff supervised all exercise sessions for the first 3 weeks of training (total 12 sessions). For the remainder of the 12-week training program, subjects were required to be present for two supervised training sessions each week, with two unsupervised sessions monitored using downloadable telemetry heart rate devices (Polar, Finland) that were provided to all participants, and were required to be worn during all exercise training sessions. Heart rate data during all sessions were reviewed by study staff to ensure subjects’ appropriate completion of all sessions at the assigned intensity and duration. In order to monitor weight stability, participants were weighed several times each week and if body mass deviated by 1-2% from their baseline, our research dietitian provided nutritional guidance to adjust their calorie intake to maintain their weight at their baseline levels.
Clinical trials
Subjects completed three separate clinical trials; Once before training (Pre-training) and twice after training (1d-Post training and 4d-Post training; Figure 1). For each of these three visits, subjects arrived at the Michigan Clinical Research Unit at 1730 h the evening before the clinical trial. For the 1d-Post training visit, participants performed their usual exercise session beginning at 1800 h (before dinner). Immediately after the exercise session, participants ingested a nutritional supplement drink, which was individually determined to replace the calories expended during their exercise session with their dinner (Boost Plus, Nestle, Switzerland; 50% carbohydrates, 35% fat, and 15% protein) to prevent an exercise-induced energy deficit state during the trial. During all trials, subjects were provided a standardized dinner (30% of estimated total daily energy expenditure) and snack (10% of estimated total daily energy expenditure) at 1900 h and 2200 h respectively. The macronutrient composition of the meal and snacks was 55% carbohydrates, 30% fat, and 15% protein. Participants had access to water ad libitum, but after their evening snack they remained fasted until completion of all measurements the next morning. Participants slept in their hospital room overnight.
At ~0700 h the next morning, intravenous catheters were inserted into a hand/forearm vein on each arm. A baseline blood sample was collected at ~0800 h. At ~0830 h we collected aSAT biopsy samples 10-15cm lateral to the umbilicus, as previously described (Ludzki et al., 2020). Briefly, we collected approximately 100mg of a core aSAT sample, which was immediately fixed in 10% formalin for histological analyses of aSAT morphology and structure (see details below), and another ~300mg of aSAT was collected via aspiration. These samples were immediately frozen in liquid nitrogen and stored at −80°C for later quantification of specific proteins using Western blot analyses (see details below). At ~0900 h, we began continuous infusion of [1-13C]palmitate (0.04 μmol/kg/min) to assess whole-body fatty acid mobilization. Beginning at ~0950 h, we collected 3 arterialized (heated hand technique) blood samples separated by 5 minutes each—these plasma samples were used to assess overnight fasted fatty acid mobilization. At ~1000 h, a ~2-hour hyperinsulinemic-euglycemic clamp began to determine anti-lipolytic sensitivity to insulin (DeFronzo et al., 1979). Briefly, a primed, continuous infusion of insulin (40mU/m2/min) was administered; blood glucose was assessed every 5 minutes and 20% dextrose was infused at a variable rate to accommodate changes in blood glucose to maintain blood glucose at the participants’ overnight fasted levels. After blood glucose concentration stabilized without further changes in the rate of D20 infusion for ≥ ~20 minutes (typically 100min after beginning the clamp procedure), five arterialized blood samples were collected every 5 minutes for the determination of steady state plasma fatty acid kinetics.
Analytical procedures
Histological assessment of aSAT morphology and structure
The core aSAT samples that were fixed in 10% formalin at the time of the biopsy were later paraffin embedded (Sakura Tissue Tek TEC, Japan). Embedded samples were sectioned in 10μm thickness by a microtome (#RM2235, Leica, Germany) and placed on a microscopy slide. Histological analysis was performed on samples collected before the 12-week training intervention and those collected 4d-Post training. To keep staining conditions consistent as possible, samples from Pre-training and 4d-Post training visits were placed on the same slide. Sections were prepared for staining by being deparaffinized and dehydrated with xylene and series of ethanol respectively, as described previously (Parlee et al., 2014). To minimize variance between subjects, staining was performed in several batches and all the microscopy conditions (i.e., exposure time) were kept consistent.
Adipocyte size:
Histological sample sections were stained with Harris’ hematoxylin (#HHS16; Sigma Aldrich) and eosin (#318906; Sigma Aldrich) followed by dehydration by ethanol. Xylene based permount was used to mount the slides. Images of the stained sections were obtained with brightfield channel in 10x by Keyence BZ-X700 microscope (Keyence, Japan). Adipocyte size was determined by using Image J (NIH, USA) as described previously (Parlee et al., 2014). Proportion of small adipocytes were measured by calculating the percentage of adipocytes with mean size larger than 1000μm2 and lower than 3000μm2.
Extracellular matrix fibrosis:
Deparaffinized sections were stained with Picro Sirius red (#36554-8; Sigma Aldrich) for 1 hour to determine total fibrosis in aSAT extracellular matrix. Sections were then rinsed with acidified water, followed by dehydration with ethanol and mounted with xylene based permount. To determine the abundance of subtypes of collagen fibers in extracellular matrix, we performed fluorescence immunohistochemistry. Deparaffinized sections were incubated in 0.5mM HCl-Glycine buffer (pH 3.0) at 90°C for 20 min for antigen retrieval. Sections were subsequently blocked with 3% hydrogen peroxide in methanol for 15 minutes followed by 5% normal goat serum in PBS for 1 hour. Sections were then incubated with primary antibodies overnight at 4°C, and then incubated with appropriate secondary antibody in dark. Sections were mounted with antifade mounting medium (Prolong Gold; Thermofisher Scientific). Images of the stained sections were obtained by Keyence BZ-X700 microscope (10x; fluorescence) and analyzed by using Image J. The proportions of each collagen were calculated by dividing the positively stained area by the entire section area. Primary antibodies were Collagen type 4 (Col4, #C1926, Sigma Aldrich), Collagen type 5a3 (Col5a3, #LS-C353420, LifeSpan BioSciences), Collagen type 6 (Col6, # ab6588, Abcam). Secondary antibodies were AlexaFlour 488 (# A11008, A21422, Thermofisher Scientific) and 555 (# A21428, Thermofisher Scientific).
Capillary density:
Sections were incubated with anti-von Willebrand Factor (#AB7356; Abcam) primary antibody followed by incubation with HRP conjugated secondary antibody and DAB substrate for visualization. Hematoxylin was used for counterstaining. Images were obtained in 10x by Keyence BZ-X700 microscope (brightfield). Image J was used to analyze capillary density (i.e., number of capillaries per section area and number of capillaries per adipocytes) and capillary size.
Protein abundance of factors regulating aSAT remodeling, metabolism, and inflammatory pathway.
A portion of each aspirated aSAT biopsy sample (~140mg wet weight) was homogenized in ice-cold 1X RIPA buffer (#9806, Cell Signaling Technology, MA) with freshly added protease and phosphatase inhibitors (P8340, P5726, and P0044; Sigma Aldrich) using two 5 mm steel beads (TissueLyser II, Qiagen, CA). Homogenates were rotated at 50 rpm for 60 min at 4°C and then centrifuged at 4°C for 3 x 15 minutes at 15,000g, disposing the lipid fraction between each centrifugation. Protein concentration was assessed using the bicinchoninic acid method (#23225, Thermofisher Scientific). Samples for Western blotting were prepared in 4x Laemmli buffer, heated for 5 minutes at 95°C, and equal amounts of protein (15μg) were loaded onto handcast gels ranging from 8 to 15%. Following separation by SDS-PAGE, proteins were transferred onto nitrocellulose membranes. Membranes were blocked for 2 hours with 5% bovine serum albumin in tris-buffered saline, 0.1% Tween 20 (TBST) at room temperature and incubated with primary antibodies overnight at 4°C. After primary antibody incubation, membranes were washed and incubated with appropriate secondary antibody (#7074 or #7076, Cell Signaling Technology). All blots were developed using enhanced chemiluminescence (#1705061, Biorad or #34095, Fisher) and imaged (Fluorchem E Imager, ProteinSimple, CA). Primary antibodies were Adipose triglyceride lipase (ATGL, #2138, Cell Signaling Technology), Hormone-sensitive lipase (HSL, #18381, Cell Signaling Technology), Comparative gene identification-58 (CGI-58, ab183739, Abcam), Glycerol-3-phosphate acyltransferase 1 (GPAT1, PA5-20524, Thermofisher Scientific), Diglyceride acyltransferase 1 (DGAT1, NB110-41487, Novus Biologics), CD36 (sc-9154, Santacruz biotechnology), Cytochrome c oxidase subunit IV (COX-IV, #4844, Cell Signaling Technology), Peroxisome Proliferator Activated Receptor Gamma (PPARγ, #2435, Cell Signaling Technology), CCAAT Enhancer Binding Protein Alpha (CEBPα, #8178, Cell Signaling Technology), Fatty acid-binding protein 4 (FABP4, sc-271529, Santacruz Biotechnology), Matrix metalloproteinase 2 (MMP2, #87809, Cell Signaling Technology), Matrix metalloproteinase 9 (MMP9, #13667, Cell Signaling Technology), Tissue inhibitor metalloproteinase 1 (TIMP1, #8926, Cell Signaling Technology), Tissue inhibitor metalloproteinase 2 (TIMP2, #5738, Cell Signaling Technology), Vascular Endothelial Growth Factor A (VEGFα, ab46154, Abcam), Angiopoietin 1 (HPA018816, Sigma Aldrich), Angiopoietin 2 (sc-74403, Santacruz Biotechnology), p38 MAPK (#9212, Cell Signaling Technology), Phospho-p38 MAPK (Thr180/Tyr182) (#9211, Cell Signaling Technology), p44/42 MAPK (Erk1/2, #4695, Cell Signaling Technology), Phospho-p44/42 MAPK (Thr202/Tyr204) (phopho-Erk1/2, # 4376, Cell Signaling Technology), SAPK/JNK (#9252, Cell Signaling Technology), Phospho-SAPK/JNK (Thr183/Tyr185) (#9251, Cell Signaling Technology). To normalize proteins to total protein level, Memcode (#24580, ThermoFisher Scientific) was used to stain total protein in the membranes (Moritz, 2017). To reduce gel-to-gel variability, an internal standard sample (IS; composite aSAT lysate from 8 obese individuals) was also loaded onto each gel for normalization.
Circulating concentrations of adipokines and fatty acids
Plasma concentrations of total adiponectin (DRP300, R&D Systems), high molecular weight (HMW) adiponectin (DHWAD0, R&D Systems) and leptin (EZHL-80SK, Sigma Aldrich) were assessed by ELISA. Plasma concentrations of fatty acids (NC9517309, NC9517311, Fujifilm Medical Systems) were measured using commercially available kits.
Plasma fatty acid kinetics.
Gas chromatography-mass spectroscopy (Agilent 5973 Networks, Mass Selective Detector, Agilent, DE) was used to determine the tracer-to-tracee ratio (TTR) for plasma palmitate and fatty acid rate of appearance (Ra) into plasma was calculated as we have reported previously (Newsom et al., 2010).
Statistical analysis
Non-normally distributed data were log-transformed before statistical analysis. Linear mixed models were applied to examine the main effects of training group (MICT vs. HIIT) and training status (pre training vs. 4d-Post training or pre training vs. 1d-Post training vs. 4d-Post training) and training group x training status interactions (IBM Corp. Released 2019. IBM SPSS Statistics for Windows, Version 26.0. Armonk, NY: IBM Corp). Fisher’s least significant different method was used for post hoc comparisons when significant interaction effects (i.e., training group x training status interaction) were observed. Pairwise comparisons were also conducted between different training status (i.e., Pre-training vs. 1d-Post training vs. 4d-Post training). In the few measurements where missing data were unavoidable due to inadequate tissue yield or tracer availability, data for these measurements from all trials were excluded and specific sample size is reported for these few outcomes. All data are presented as mean ± SD.
RESULTS
Body weight, body composition, VO2peak, and blood parameters in response to training
A total of 36 subjects with obesity completed the exercise training interventions (MICT; n = 17, HIIT; n = 19; Table 1). Both MICT and HIIT significantly increased both absolute (L/min) and relative (ml/kgFFM/min) VO2peak (p < 0.001), and there was a trend for the increase in absolute VO2peak in HIIT (11±5%) to be greater than MICT (6±8%) (p=0.1). As designed, body mass and fat free mass did not change after training (Table 1), but there was a trend for body fat mass to be slightly lower after both HIIT and MICT (p=0.06), which was the result of very small (≤ ~0.5kg), yet the consistent reduction in total body fat mass across subjects (Table 1).
Table 1. Subject characteristics and whole-body clinical biomarker measures before and after training.
Data are presented as mean ± SD. *Significant main effect of training status (p < 0.05). ***Significant main effect of training status (p<0.001). There were no significant effects of training group or significant training group x training status interactions. FFM = Fat Free Mass. HMW = High Molecular Weight.
| MICT (n = 17) | HIIT (n = 19) | |||
|---|---|---|---|---|
| Age (years) | 30 ± 6 | 31 ± 7 | ||
| Sex | 12 Women, 5 Men | 12 Women, 7 Men | ||
| Height (m) | 1.71 ± 0.09 | 1.70 ± 0.08 | ||
| Pre training |
4d-Post Trained |
Pre training |
4d-Post Trained |
|
| Body mass (kg) | 98.0 ± 11.6 | 97.6 ± 11.7 | 96.4 ± 13.7 | 96.6± 13.6 |
| Fat mass (kg) | 40.5 ± 7.1 | 40.1 ± 7.2 | 42.5 ± 7.7 | 42.0 ± 7.7 |
| Fat free mass (kg) | 55.5 ± 9.3 | 55.6 ± 9.6 | 55.9 ± 10.5 | 56.5 ± 10.3 |
| BMI (kg/m2) | 33.7 ± 3.2 | 33.4 ± 3.2 | 33.0 ± 2.9 | 33.1 ± 3.1 |
| Body fat % | 43.4 ± 6 | 43.1 ± 6.4* | 42.1 ± 5.4 | 41.6 ± 5.4* |
| VO2peak (L/min) | 2.3 ± 0.5 | 2.5 ± 0.6*** | 2.5 ± 0.6 | 2.8 ± 0.6*** |
| VO2peak (ml/kgFFM /min) | 41.9 ± 7.7 | 44.9 ± 7.7*** | 43.8 ± 6.1 | 48.5 ± 5.3*** |
| Plasma fatty acids (uM) | 392 ± 159 | 408 ± 118 | 433 ± 156 | 387 ± 122 |
| Plasma total adiponectin (ug/ml) | 4.7 ± 2.6 | 4.0 ± 2.3*** | 4.6 ± 1.9 | 3.9 ± 1.8*** |
| Plasma HMW adiponectin (ug/ml) | 2.6 ± 2.0 | 2.3 ± 1.8* | 2.4 ± 1.3 | 2.0 ± 1.3* |
| Plasma leptin (ng/ml) | 53.2 ± 25.1 | 48.5 ± 22.6* | 49.0 ± 24.7 | 48.0 ± 27.2* |
Three months of exercise training without weight loss did not alter fasting plasma concentrations of fatty acids. Interestingly, fasting plasma concentrations of total adiponectin, HMW adiponectin and leptin were all significantly reduced after training (p=0.001, p=0.03, p=0.03, respectively; Table 1) with no differences between MICT and HIIT.
Morphological and structural aSAT remodeling in response to training
Adipocyte cell size.
Twelve weeks of MICT and HIIT slightly (~10%), yet significantly reduced mean adipocyte size (p=0.002; Figure 2A and 2B) and increased the proportion of small adipocytes (p=0.018; Figure 2C). This was evidenced by a leftward shift in the frequency distribution for 4d-Post training adipocyte size (Figure 2D). There were no distinguishable differences between MICT and HIIT (Figure 2).
Figure 2. Changes in adipocyte cell size in response to training.
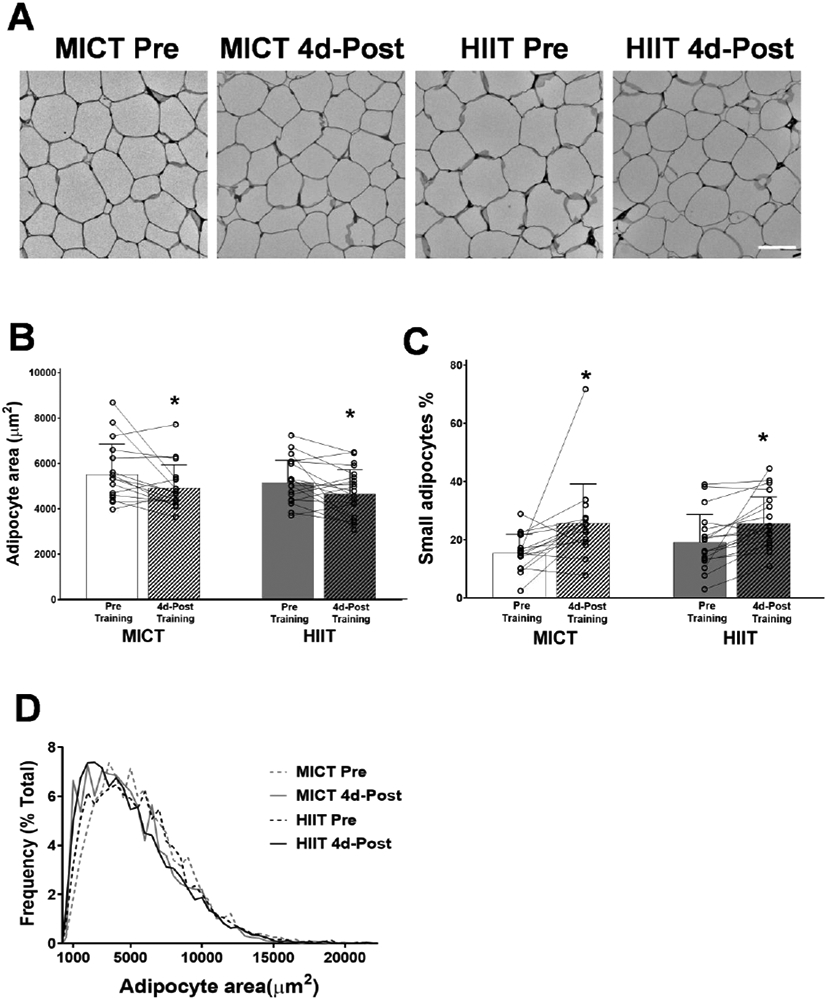
A) Representative images of H&E stained aSAT. White scale bar refers to 100μm. B) Mean adipocyte area. C) Proportion of smaller adipocytes (1000μm2 ≤ area ≤ 3000μm2). C) Frequency distribution of adipocyte area. Sample sizes were MICT n = 16 and HIIT = 18. *Significant main effect of training status (p < 0.05). ** Significant main effect of training status (p < 0.01). There were no significant effects of training group or training group x training status interactions.
aSAT fibrosis and ECM collagen.
aSAT fibrosis (as assessed by Sirius Red staining) was not altered after training in either MICT or HIIT (Figure 3A and 3B). Using immunohistochemistry to assess potential changes in the abundance of some specific aSAT ECM proteins, we found a small, yet significant increase in Col5a3 4 days after training (p=0.04) with no differences between MICT and HIIT (Figure 3A and 3B). Conversely, the abundance of Col4 and 6 were not significantly altered by exercise training (Figure 3A and 3B).
Figure 3. Adaptation of aSAT fibrosis and ECM proteins in response to training.

A) Representative staining images of Sirius Red, Col4, Col5a3, and Col6 in aSAT section. White scale bar refers to 100μm. B) Abundance of ECM proteins quantified from histology images. Sample sizes were MICT n = 14 and HIIT n = 17 for Sirius Red, MICT n = 14 and HIIT n = 16 for Col4 and Col5a3, and MICT n = 16 and HIIT n = 18 for Col6. *Significant main effect of training status (p<0.05). There were no significant effects of training group or training group x training status interactions.
aSAT capillarization.
Exercise training increased capillarization when expressed as the number of capillaries per mm2 (Figure 4A and 4B; p=0.05). Conversely, the change of capillarization when expressed as the number of capillaries per adipocyte (Figures 4A and 4C), did not quite reach statistical significance (p=0.1). There were no differences in capillarization observed between MICT and HIIT. Additionally, capillary cross-sectional area also did not change 4 days after training in either MICT or HIIT (Figure 4D).
Figure 4. aSAT capillarization in response to training.

A) Representative staining images of vWF in aSAT section. Capillaries are visible as dark-brown puncta. Black scale bar refers to 100μm. B) Number of capillaries per field area (mm2). C) Number of capillaries per adipocytes. D) Mean capillary cross-sectional area (μm2). *Significant main effect of training status (p≤0.05). Sample sizes were MICT n = 15 and HIIT = 18. There were no significant effects of training group or training group x training status interactions.
Factors regulating aSAT remodeling
The morphological/structural measurements presented above were all measured in aSAT samples collected 4 days after the last exercise training session. However, because many longer-term adaptations to exercise training like these often stem from relatively short-lived responses to exercise, we measured changes in factors that regulate these structural adaptations both 1 day and 4 days after the last exercise training session.
Markers of adipogenesis regulation
The protein abundance of total PPARγ was not different after training in either group (Figure 5A). However, we found a significant training group x training status interaction for CEBPα, another primary transcription factor for adipogenesis. Post-hoc testing revealed that the protein abundance of total CEBPα was significantly increased in aSAT 1 day and 4 days after the last exercise session in HIIT (p=0.02 and p=0.03 respectively) but not MICT (Figure 5B). The protein abundance of FABP4, a marker for differentiated/mature adipocytes was significantly increased when measured 1 day after exercise (p = 0.01) and tended to remain elevated when measured 4 days after exercise (p=0.09) with no differences between MICT and HIIT (Figure 5C).
Figure 5. Adaptations in aSAT remodeling factors in response to exercise training.
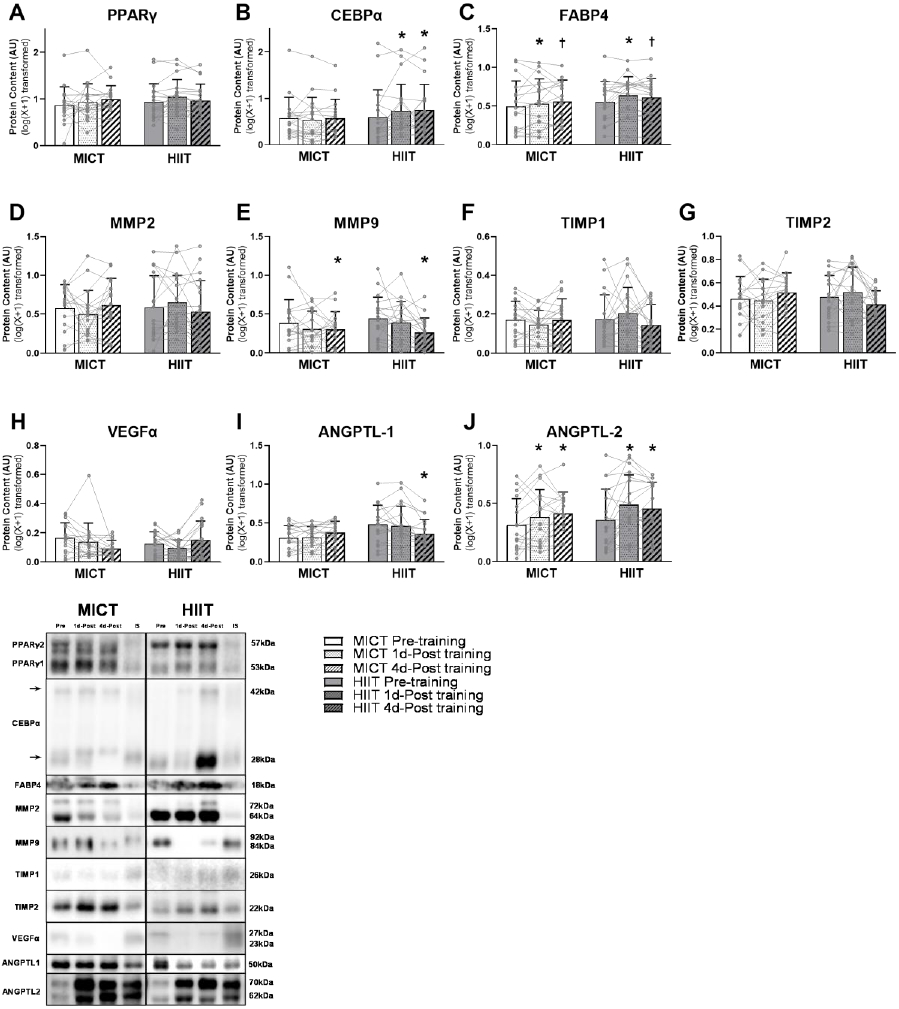
(A~C) Protein expression of factors/markers involved in adipogenesis. (D~G) Protein expression of ECM regulators. (H~J) Protein expression of angiogenic regulators. *Significant main effect of training status, with post-hoc analysis identifying a significant difference compared with Pre-training (p<0.05). † Trend of main effect of training status, with post-hoc analysis identifying a trend of difference compared with Pre-training (0.05<p<0.1). There were significant training group x training status interactions in the abundance of CEBPα (p=0.046) and Angiopoetin-1 (p=0.002).
Extracellular matrix regulators
Although we did not find an effect of exercise training on total aSAT fibrosis (as assessed by Sirius Red staining; Figure 3), we did find that abundance of MMP9, one of the key proteins involved in ECM remodeling was reduced 4 days after exercise training (p=0.02; Figure 5E). There was no significant difference in MMP9 abundance between MICT and HIIT. The protein abundance of MMP2, TIMP1, and TIMP2 were not altered either 1 day or 4 days after exercise training (Figure 5D, F, and G).
Angiogenesis regulators
Aligning with the observed increase in aSAT capillarization after both MICT and HIIT, protein abundance of the angiogenic regulator, Angiopoietin-2 was increased both 1 day and 4 days after exercise training (p<0.01) with no differences between groups (Figure 5J). In contrast, we did not observe an increased abundance of VEGFα, which is widely considered a “master regulator” of angiogenic activity (Figure 5H). Additionally, we found a significant training group x training status interaction for Angiopoietin-1, another angiogenic regulator. Post-hoc testing revealed that Angiopoietin-1 was significantly reduced below pre-training levels when measured 4 days after training after HIIT (p=0.006) but not MICT (Figure 5I).
Whole-body fatty acid mobilization
After an overnight fast, fatty acid release from adipose tissue into the systemic circulation (fatty acid Ra) was not affected by either MICT or HIIT when measured either 1 day or 4 days after exercise training (Figure 6). During the insulin infusion, the potent antilipolytic effects of insulin reduced fatty acid Ra to levels ~70% below basal levels in both training groups (p<0.001), but there was no effect of exercise training on the antilipolytic effects of insulin when measured after 1 day and 4 days of exercise training (Figure 6).
Figure 6. Whole body fatty acid mobilization rate in response to exercise training.
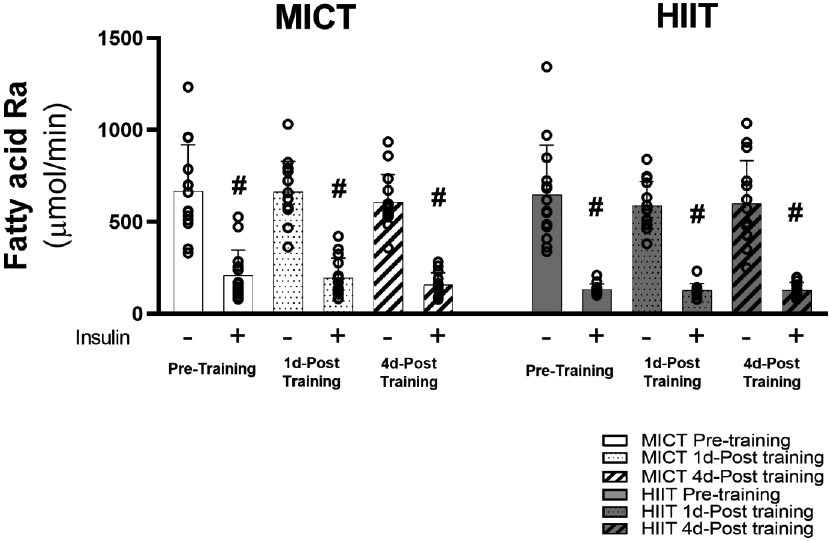
Sample sizes were MICT n = 14 and HIIT = 14. Fatty acid rate of appearance in the systemic circulation after over-night fasting and insulin-stimulated conditions. #Significant main effect of insulin (p<0.001). There were no significant main effects of training group, training status, or training group x training status interactions.
Proteins regulating aSAT metabolism
In agreement with our observation that training did not affect fatty acid release from adipose tissue, protein abundance of the main triacylglycerol lipase protein, ATGL and its primary activator, CGI-58 also did not change after training (Figure 7A and B). Interestingly, however, abundance of HSL, which hydrolyzes diacylglycerol to monoacylglycerol, was significantly elevated above pre-training levels when measured 1 day after exercise (p = 0.01) and tended to remain elevated 4 days later (p=0.06; Figure 7C). The protein abundance of the fatty acid trafficking protein, FAT/CD36 was significantly increased 1 day after exercise (p=0.03), which then returned to pre-training level 4 days later (Figure 7D). Neither of the primary fatty acid esterification proteins, GPAT1 and DGAT1 were altered by training (Figure 7E and F). Exercise training significantly increased the protein abundance of COX-IV, a classical marker of mitochondrial density (p<0.01; Figure 7G). However, the protein abundance of SDHA, an enzyme involved in citric acid cycle and electron transport chain, was not affected after exercise training (Figure 7H). Importantly, we did not observe any differences between HIIT and MICT for any of these proteins that regulate aSAT metabolism (Figure 7).
Figure 7. Adaptations in aSAT metabolic factors in response to exercise training.
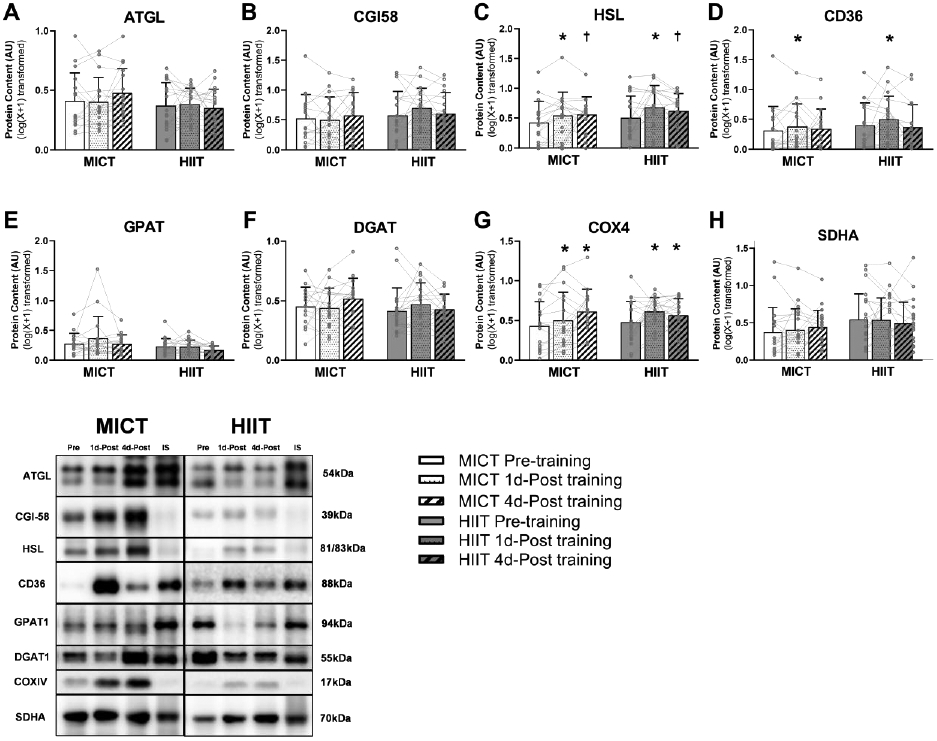
(A~C) Protein expression of lipolytic enzymes. D). Protein expression of fatty acid trafficking protein. (E and F) Protein expression of esterific enzymes. (G and H) Protein expression of mitochondrial markers. *Significant main effect of training status, with post-hoc analysis identifying a significant difference compared with Pre-Training (p<0.05). † Trend of main effect of training status, with post-hoc analysis identifying a trend of difference compared with Pre-Training (0.05<p<0.1). There were no significant main effects of training group or training group x training status interactions
Key signal proteins involved in MAPK pathway
The protein abundance of phosphorylated P38 MAPK (Thr180 and Tyr182) was significantly increased 1 day after exercise training (p=0.02) with no differences between MICT and HIIT (Figure 8A). However, this exercise-induced increase in phosphorylated P38 was no longer evident when expressed relative to total P38 MAPK (Figure 8C). We also found the total protein abundances of JNK and ERK1/2 to be significantly elevated 1 day after last exercise session (both p=0.02) with no difference between MICT and HIIT (Figure 8D and G). Importantly, when measured 4 days after the last exercise training session, we could no longer detect significant changes in the abundance of any of these proteins (total or phosphorylated forms) compared with pre-training levels.
Figure 8. Adaptations in aSAT MAPK proteins in response to exercise training.
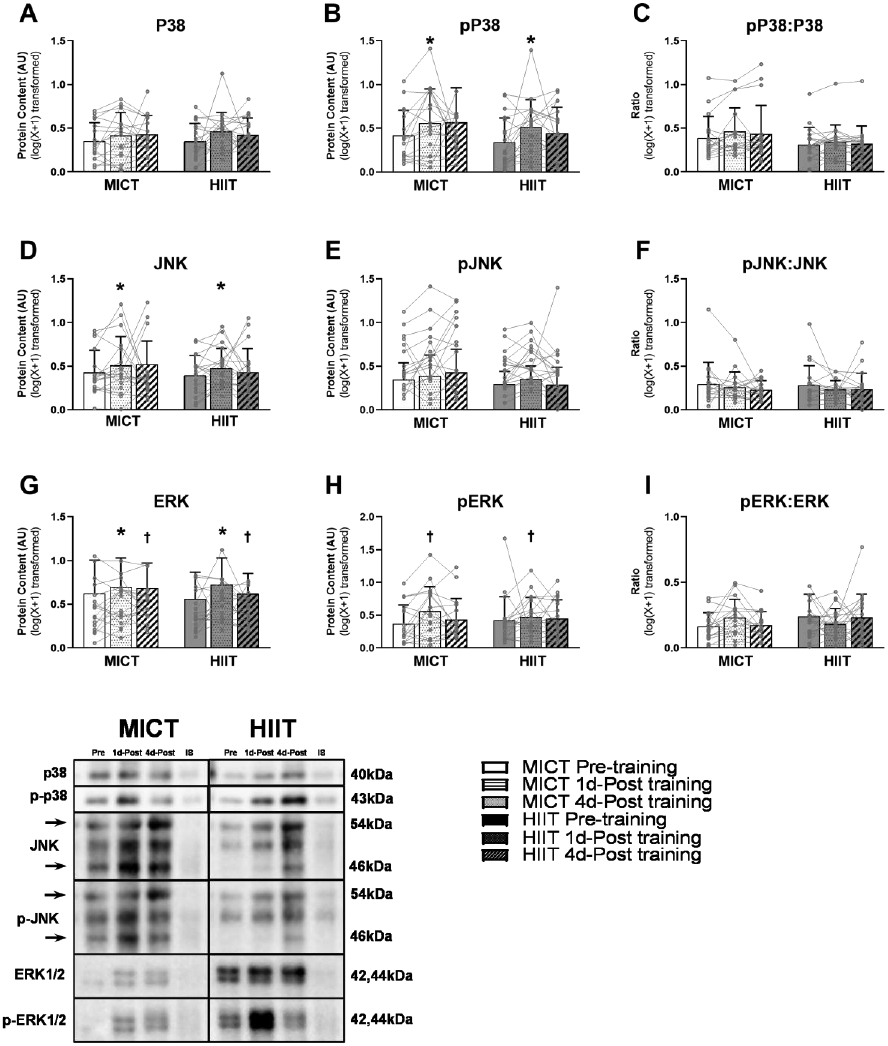
Protein expression of A) total P38 and B) phosphorylated P38. C) The activity of P38 was measured by normalizing the abundance of phosphorylated P38 by total P38. Protein expression of D) total JNK and E) phosphorylated JNK. F) The activity of JNK was measured by normalizing the abundance of phosphorylated JNK by total JNK. Protein expression of G) total ERK and H) phosphorylated ERK. I) The activity of ERK was measured by normalizing the abundance of phosphorylated ERK by total ERK. *Significant main effect of training status, with post-hoc analysis identifying a significant difference compared with Pre-Training (p<0.05). † Trend of main effect of training status, with post-hoc analysis identifying a trend of difference compared with Pre-Training (0.05<p<0.1). There were no significant main effects of training group or training group x training status interactions.
DISCUSSION
The major findings of this study were that 12 weeks of exercise training, in absence of changes in body weight and fat mass, reduced adipocyte size, modified the composition of ECM and increased capillarization in aSAT. Accompanying the changes in ECM and capillarization, we found some key factors that regulate extracellular matrix remodeling (MMP9), and angiogenesis (ANGPTL2) were altered in response to exercise training. Additionally, many of the modest exercise-induced changes in factors that regulate aSAT metabolism (i.e., HSL and CD36) and inflammatory pathway activation (i.e., phosphorylated P38 MAPK, JNK, and ERK1/2) were evident the day after the last session of exercise but were reversed 4 days later. Importantly, despite the robust difference in training intensity and volume, metabolic and aSAT adaptations in response to MICT and HIIT were remarkably similar.
Adipose tissue with a greater proportion of smaller adipocytes is often considered to be more metabolically favorable in terms of lower pro-inflammatory macrophage infiltration (Weisberg et al., 2003), lower lipolytic rates (Laurencikiene et al., 2011), and enhanced sensitivity to insulin (Olefsky, 1976, 1977). Some studies have reported adipocyte size to be smaller after exercise training (Despres et al., 1991; Gollisch et al., 2009; Américo et al., 2019), perhaps contributing to the exercise-induced improvement in metabolic health. However, most of these reductions in adipocyte size after exercise training were also accompanied by a meaningful amount of weight loss, thereby confounding the interpretation of direct effects of exercise on adipocyte size. In contrast, by intentionally requiring our participants to maintain body weight throughout the 12-week training program, we were largely able to remove this confounding influence of weight loss on the effects of exercise training on adipocyte size. We must acknowledge that despite weight maintenance in our study, the very small, non-significant reduction in whole body fat mass (~0.5kg) may have contributed to a portion of the ~10% reduction of adipocyte size we observed after training. However, it seems unlikely that we would be able to detect this very small change in whole body fat mass at the adipocyte-level. This is supported by our observation that the reduction in adipocyte size in our study was not related to the reduction in fat mass (R2 = 0.005, P = 0.7), suggesting factors other than loss of body fat (e.g., formation of more adipocytes) may also underlie the reduction of adipocyte size we observed. Our findings that some of adipogenic markers were increased after training (i.e., trend of increase in protein abundance of FABP4 and significant increase in protein abundance of CEBPα in HIIT) provides some support for this notion. It is unclear why CEBPα was only increased in HIIT, but perhaps, expression of adipogenic transcription factors may be differentially regulated by exercise intensity.
The potential role of exercise on aSAT ECM is important because ECM’s fibrotic content is tightly associated with obesity-induced insulin resistance (Divoux et al., 2010; Spencer et al., 2011). Because changes in body fat mass are known to induce remodeling of adipose ECM (Divoux et al., 2010; Spencer et al., 2011), to accurately assess the effects of exercise on aSAT ECM, once again it is critical to tightly control of body weight/body fat mass during exercise interventions. To our knowledge, only one previous study has examined the effects of exercise training on ECM remodeling independent of weight loss, and it was reported that in high fat diet (HFD)-fed mice, four-months of exercise training markedly attenuated the increase in epidydimal adipose tissue fibrosis despite identical gains in body weight and body fat mass between exercised and non-exercised animals (Kawanishi et al., 2013). In contrast, our findings suggest that three-months of exercise training without weight loss may not induce meaningful modifications in aSAT fibrosis in human subjects with obesity. We recognize that our three-month exercise interventions may have been too short to induce meaningful changes in aSAT fibrotic deposition in humans. An alternative possibility to explain the apparent discrepancy between our findings and those of Kawanishi, et al (Kawanishi et al., 2013) is that exercise may be effective in attenuating the increased fibrotic formation that often occurs in adipose tissue with weight gain but may not be as effective in reducing existing fibrosis. Importantly, however, our observation that exercise training modestly modified the composition of the collagens in aSAT samples may have important clinical implications. Although the increase in Col5a3 abundance may seem to conflict with the notion that increased extracellular matrix deposition is negatively associated with metabolic health, evidence suggest Col5a3 is a crucial signaling component in glucose homeostasis (Huang et al., 2011). Huang et al reported that isolated adipocytes from epididymal fat pads in Col5a3−/− mice exhibited impaired glucose uptake compared with that in wild type, suggesting the critical role of Col5a3 in adipose tissue glucose metabolism. Therefore, increased abundance of Col5a3 in response to exercise training may still reflect a metabolically favorable adaptation.
High capillary density in adipose tissue enhances capacity for oxygen and nutrient delivery (and metabolic by-product removal), possibly preventing local hypoxia, which can lead to increased fibrosis and inflammation (Halberg et al., 2009; Krishnan et al., 2012). Although we found an increase in aSAT capillarization after exercise training (i.e., when expressed as number of capillaries per mm2) – this effect was rather modest and the increase in the number of capillaries per adipocyte did not quite reach statistical significance. One previous study reported that 12 weeks of endurance exercise training significantly increased the number of capillaries per adipocyte in human aSAT(Walton et al., 2015). However, this exercise-induced increase in aSAT capillarization was only found in their non-obese, insulin-sensitive subjects (BMI: 26 ± 0.72 kg/m2) while aSAT capillarization did not increase significantly in their insulin resistant subjects with obesity (BMI: 35.1 ± 0.9 kg/m2) (Walton et al., 2015). This may help explain why we did not find a more robust increase in aSAT capillarization in our subjects. It is possible that insulin resistance may blunt angiogenesis by suppressing the expression of VEGFα (Pasarica et al., 2009). Additionally, our finding that Angiopoietin-2 protein abundance increased after both MICT and HIIT, suggests that exercise training may upregulate components of the angiogenic pathway - but perhaps 12 weeks of exercise training was not long enough to induce a robust increase in capillarization of the hypertrophied adipocytes in adults with obesity. While our finding that Angiopoietin-1 protein abundance in aSAT did not change after MICT is consistent with the previous exercise training study in humans (Walton et al., 2015), the reduction in HIIT suggests that aSAT Angiopoietin-1 may be differentially regulated by exercise intensity, although the underlying mechanisms for this phenomenon are unclear. Accompanying our observed trend for an increase in the number of capillaries per adipocyte in our study, the modest reduction in adipocyte diameter we observed after training may represent a favorable adaptation in the context of oxygen and nutrient delivery, by reducing diffusion distance in adipocytes.
Excessive fatty acid release/mobilization from aSAT into the systemic circulation that is common in obesity, often results in ectopic fat deposition in other tissues (i.e., liver, skeletal muscle, heart, etc.) which underlies many obesity-related cardiometabolic complications (Ravussin & Smith, 2002; McQuaid et al., 2011). Our findings that 12 weeks of exercise training without weight loss did not alter whole-body fatty acid mobilization rate is consistent with previous reports (Horowitz et al., 1999; Horowitz et al., 2000). Our current findings extend on these previous studies by demonstrating that the anti-lipolytic effects of insulin were also not affected by 3 months of training. We acknowledge the possibility that our dose of insulin during the clamp (40 mU/m2/min) may have been high enough to mask potential improvements in anti-lipolytic sensitivity to insulin after training. But based on previous work (Shojaee-Moradie et al., 2007), we do not believe this to be the case. Our observations in this study are also specific to the conditions when exercise training is not accompanied by weight loss. We previously reported that when exercise training is accompanied by weight loss, systemic fatty acid mobilization declines (Schenk et al., 2009). In fact, the reduction in lipolytic rate and systemic fatty acid mobilization was identical when adults with obesity lost 12% body weight with or without exercise training (Schenk et al., 2009). Therefore, it appears that changes in systemic fatty acid mobilization are particularly responsive to weight loss but not exercise training, per se. Our finding that the protein expression of ATGL in aSAT was unaltered by training aligns with the lack of change in fatty acid Ra. Although the trend of increase in aSAT HSL we found in response to training may indicate increased lipolytic capacity, this may be only relevant under conditions when energy expenditure is high (e.g., during exercise) because fatty acid mobilization rate was not different while our subjects were at rest. Interestingly, we found a transient increase in the abundance of the fatty acid transporter, CD36 in aSAT 1 day after last training session, which supports the notion of an adaptive response to increase the capacity of lipid handling in aSAT that the subjects experience during their regular exercise sessions. However, our observation that aSAT CD36 protein abundance returned to pre-training levels 4 days after the last session of exercise suggests this adaptation is transient.
Mitochondrial biogenesis and function are often compromised in aSAT in obesity, which has been associated with metabolic disturbances (Hammarstedt et al., 2003; Semple et al., 2004; Heinonen et al., 2015). These metabolic abnormalities linked to low mitochondria density in aSAT may attributed to increased endoplasmic reticulum stress, oxidative stress, and inflammation in response to nutritional overload (Chen et al., 2010; Wang et al., 2010; Marycz et al., 2018). Our finding that both MICT and HIIT increased the protein abundance of COX-IV in our obese subjects expands on previous works that indicate that exercise training may promote mitochondrial function and biogenesis in adipose tissue in both lean rodents (Sutherland et al., 2009; Trevellin et al., 2014) and non-obese humans (Rönn et al., 2014; Riis et al., 2019). Unlike many of the other regulatory proteins we measured in our aSAT samples, the increase in COX-IV persisted at least 4 days after the last exercise training session, suggesting this was more than an acute/transient adaptation. It is also notable that the increase in aSAT COX-IV abundance was similar in both MICT and HIIT despite the large difference in exercise stimulus. Although COX-IV protein abundance is commonly used as a marker of mitochondrial content, we recognize that additional studies are needed to more comprehensively determine the effects of exercise training on aSAT mitochondrial adaptations in adults with obesity.
MAPK signaling in adipose tissue has been reported to be involved in adipogenesis, inflammation, and lipolysis, raising the possibility of an active role of MAPK signaling in aSAT remodeling (Hu et al., 1996; Camp & Tafuri, 1997; Greenberg et al., 2001; Trujillo et al., 2006). The only previous study to our knowledge that examined the effects of exercise training on MAPK signaling in human aSAT reported a reduced expression of genes involved in the MAPK pathway after 6-month of exercise training healthy adults (Rönn et al., 2014). In contrast, we found no change in our markers of MAPK activity (i.e., abundance of phosphorylated forms of P38, ERK1/2, and JNK relative to the total abundance of each protein) in response to training. Moreover, the transient increase in the total protein abundance of ERK1/2 and JNK we observed the day after the last exercise session suggests a possibility of an increase in the capacity for MAPK activity in aSAT. But this potential for increased MAPK capacity was transient since the abundance of JNK and ERK all returned to pre-training levels after 4 days without exercise.
Leptin and adiponectin are two mostly well-studied peptides released from adipose tissue (“adipokines”). Obesity is often associated with high plasma leptin concentrations and low plasma adiponectin concentrations, and while the effects of weight loss on leptin (decrease) and adiponectin (increase) concentrations are quite consistent, evidence regarding the effects of exercise training on these adipokines is equivocal. Some studies report systemic leptin or adiponectin levels increase (Shadid et al., 2006; Markofski et al., 2014), decrease (Yatagai et al., 2003; Ozcelik et al., 2004; Polak et al., 2006), while others report no change after training (Hulver et al., 2002; Ligibel et al., 2009). An important confounder to the interpretation of many of these exercise studies is the change in body fat mass that often occurred during these training interventions. Our finding that 12 weeks of exercise training without weight loss significantly reduced both systemic adiponectin and leptin level suggests that these adipokines may be regulated by training independent of weight loss. However, the underlying mechanisms that explains the reduction in both adiponectin and leptin after training are unclear.
In contrast to our hypothesis, the metabolic and structural adaptations in aSAT were remarkably similar between HIIT and MICT, despite marked differences in the exercise stimulus. This finding may suggest that even though our MICT and HIIT protocols represent two rather distinct exercise training regimens – the difference in the exercise stimulus between MICT and HIIT was not large enough to evoke meaningful differences in adaptive responses to aSAT we measured in our study. Alternatively, it is possible that adaptations stemming from the more “conventional” 45 min of continuous exercise in our MICT protocol was closely matched by the brief high intensity stimulus during HIIT, despite nearly 70% greater energy expenditure and 50% longer exercise time in MICT vs. HIIT. Regardless, the similar adaptations in aSAT structure and factors that regulate metabolic functions between HIIT and MICT were rather modest compared with previous findings that reported metabolic improvements (i.e., increased insulin sensitivity and reduced whole body inflammation) and functional modifications in aSAT (Marcell et al., 2005; Fabre et al., 2018; Riis et al., 2019) after exercise training. We postulate the modest changes in aSAT structure and markers of metabolic function in our study compared with these others were largely a consequence of our study design to intentionally maintain body weight during our training program to assess the effects of exercise training independent of weight loss.
Although this was a well-controlled study to compare the effects of HIIT vs. MICT on lipid metabolism and aSAT structure and function independent of weight loss in obese adults, there are some important limitations. Despite the modest modifications in aSAT structure in response to exercise training even in the absence of weight loss, we acknowledge that 12 weeks of training intervention may be too short to induce major structural and morphological modifications given that aSAT remodeling may be a slow process (Arner et al., 2010). However, our findings suggest that even 3 months of exercise training may increase the capacity of tissue remodeling (i.e., adipogenesis, angiogenesis, ECM remodeling) and it is possible that training-induced tissue remodeling may be even more robust after longer-term training. We did not tightly control dietary composition during the 12-week training intervention and therefore cannot completely rule out the possibility that the structural and metabolic adaptations in aSAT after training may be influenced by daily variations in nutrient content and/or timing of intake. Although we note that diet composition and meal timing could have some impact on aSAT metabolism (Shostak et al., 2013; Hernández et al., 2017), we closely monitored body weight fluctuations in our participants enough to know that they didn’t experience wide variations in diet composition or meal timing. Also, we successfully managed participants’ body weight to keep it similar to their pre-training levels during the intervention period, which we contend to be the most impactful on our outcomes (Sjöström & Björntorp, 1974; Smith et al., 2001; Eriksson-Hogling et al., 2015). As discussed in our previous report (Ryan et al., 2020), we intentionally did not match total energy expenditure or exercise time between MICT and HIIT to compare two distinct exercise prescriptions that are commonly implemented in real-life environment. This allowed us to conclude that HIIT, even with lower energy expenditure and time commitment compared with MICT induced similar adaptations in aSAT.
In summary, our findings indicate that 12 weeks of exercise training, without weight loss, induced some remodeling within aSAT, as evidenced by a reduction in adipocyte cell size, altered ECM composition, and an increase in capillarization. However, these morphological/structural adaptations in aSAT were quite modest and did not culminate in measurable changes in whole body fatty acid mobilization, which is a major factor underlying insulin resistance in obesity (Roden et al., 1996; Schenk et al., 2009). In line with this, in previously published work from this same overall project (Ryan et al., 2020), we reported neither MICT nor HIIT induced a persistent increase in insulin sensitivity after 12 weeks of training. Therefore, perhaps adaptations to aSAT must be robust enough to lower systemic fatty acid mobilization to manifest into a measurable improvement in whole body insulin sensitivity. We acknowledge that changes to aSAT structure and function may require an exercise intervention longer than 3 months. Alternatively, perhaps the independent effects of exercise training on aSAT structure and metabolic function are relatively subtle when not accompanied by weight loss, which is known to have a robust impact on many aspects of aSAT metabolism and morphology (Rupnick et al., 2002; Clément et al., 2004; Larson-Meyer et al., 2006; Kos et al., 2009). The transient nature in the expression of some key factors that regulate aSAT fatty acid metabolism and inflammatory pathway activation (i.e., increased the day after the last exercise training session – but returned to pre-training levels 4 days later) supports the notion that the effects of exercise on aSAT metabolic function and inflammation are largely affected by the most recent session of exercise, rather than by longer-term adaptations to exercise training. Importantly, our findings also indicate that the effects of MICT and HIIT on aSAT structure, metabolism and inflammatory pathway were remarkably similar despite marked differences in the energy expenditure, duration, and intensity of the exercise stimulus.
Supplementary Material
KEY POINTS.
Exercise training is well-known to improve metabolic health in obesity - but how exercise modifies the structure and metabolic function of adipose tissue, in absence of weight loss, remains unclear.
We report that 12 weeks of moderate-intensity continuous training (MICT) and 12 weeks of high-intensity interval training (HIIT) both induced modifications in adipose tissue structure and factors that regulate adipose tissue remodeling, metabolism, and inflammatory signal pathway in adults with obesity, even without weight loss (with no meaningful differences between MICT and HIIT).
The modest modifications in adipose tissue structure in response to 12 weeks of MICT or HIIT did not lead to changes in the rate of fatty acid release from adipose tissue.
These results expand our understanding about the effects of two commonly used exercise training prescriptions (MICT and HIIT) on adipose tissue remodeling that may lead to advanced strategies for improving metabolic health outcomes in adults with obesity.
Acknowledgment:
The authors gratefully thank the contribution of the study participants. We acknowledge excellent technical assistance provided by exercise training coordinators and supervisors; Dr. Benjamin Carr, Dr. Jacob Haus, Jeffrey Wysocki, RN; the staff at the Michigan Clinical Research Unit; and all the members of the Substrate Metabolism Laboratory.
Fundings:
This study was supported by The National Institutes of Health (R01DK077966, P30DK089503, U24DK097153, T32DK007245, F32DK117522) and the Canadian Institutes of Health Research (338735 and 146190).
Biography

Cheehoon Ahn received his BS in Physical Education with interests in exercise physiology and adipocyte biology from Seoul National University, Korea. He joined Jeff Horowitz’s Substrate Metabolism Laboratory at the University of Michigan where he received his MS and is a current PhD candidate. He is currently exploring the role of exercise in adipose tissue remodeling. His long-term interests include understanding how muscle-adipose crosstalk is regulated by obesity and exercise.

Ben Ryan completed doctoral training in Integrative Physiology under the supervision of Bill Byrnes in the Applied Exercise Science Laboratory at the University of Colorado Boulder. He then completed a postdoctoral research fellowship with Jeff Horowitz in the Substrate Metabolism Laboratory at the University of Michigan. He is currently a research physiologist in the Thermal and Mountain Medicine Division at the U.S. Army Research Institute of Environmental Medicine.
Footnotes
Clinical trials registration: Registered at www.clinicaltrials.gov as NCT02706093.
Disclosure Summary: The authors have no disclosures to report.
Data availability:
All datasets generated and analyzed in this study are available from the corresponding author on reasonable request.
REFERENCE
- Américo ALV, Muller CR, Vecchiatto B, Martucci LF, Fonseca-Alaniz MH & Evangelista FS. (2019). Aerobic exercise training prevents obesity and insulin resistance independent of the renin angiotensin system modulation in the subcutaneous white adipose tissue. PloS one 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arner E, Westermark PO, Spalding KL, Britton T, Rydén M, Frisén J, Bernard S & Arner P. (2010). Adipocyte turnover: relevance to human adipose tissue morphology. Diabetes 59, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp HS & Tafuri SR. (1997). Regulation of Peroxisome Proliferator-activated Receptor γ Activity by Mitogen-activated Protein Kinase*. Journal of Biological Chemistry 272, 10811–10816. [DOI] [PubMed] [Google Scholar]
- Chen X-H, Zhao Y-P, Xue M, Ji C-B, Gao C-L, Zhu J-G, Qin D-N, Kou C-Z, Qin X-H & Tong M-L. (2010). TNF-α induces mitochondrial dysfunction in 3T3-L1 adipocytes. Molecular and cellular endocrinology 328, 63–69. [DOI] [PubMed] [Google Scholar]
- Chun T-H. (2012). Peri-adipocyte ECM remodeling in obesity and adipose tissue fibrosis. Adipocyte 1, 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, Wang S, Fortier M, Greenberg AS & Obin MS. (2005). Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. Journal of lipid research 46, 2347–2355. [DOI] [PubMed] [Google Scholar]
- Clément K, Viguerie N, Poitou C, Carette C, Pelloux V, Curat CA, Sicard A, Rome S, Benis A & Zucker JD. (2004). Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. The FASEB Journal 18, 1657–1669. [DOI] [PubMed] [Google Scholar]
- Cullberg KB, Christiansen T, Paulsen SK, Bruun JM, Pedersen SB & Richelsen B. (2013). Effect of weight loss and exercise on angiogenic factors in the circulation and in adipose tissue in obese subjects. Obesity 21, 454–460. [DOI] [PubMed] [Google Scholar]
- DeFronzo RA, Tobin JD & Andres R. (1979). Glucose clamp technique: a method for quantifying insulin secretion and resistance. American Journal of Physiology-Endocrinology And Metabolism 237, E214. [DOI] [PubMed] [Google Scholar]
- Després J-P. (1993). Abdominal obesity as important component of insulin-resistance syndrome. Nutrition (Burbank, Los Angeles County, Calif) 9, 452–459. [PubMed] [Google Scholar]
- Despres JP, Pouliot MC, Moorjani S, Nadeau A, Tremblay A, Lupien PJ, Theriault G & Bouchard C. (1991). Loss of abdominal fat and metabolic response to exercise training in obese women. American Journal of Physiology-Endocrinology And Metabolism 261, E159–E167. [DOI] [PubMed] [Google Scholar]
- Divoux A, Tordjman J, Lacasa D, Veyrie N, Hugol D, Aissat A, Basdevant A, Guerre-Millo M, Poitou C & Zucker J-D. (2010). Fibrosis in human adipose tissue: composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes 59, 2817–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson-Hogling D, Andersson DP, Bäckdahl J, Hoffstedt J, Rössner S, Thorell A, Arner E, Arner P & Rydén M. (2015). Adipose tissue morphology predicts improved insulin sensitivity following moderate or pronounced weight loss. International journal of obesity 39, 893–898. [DOI] [PubMed] [Google Scholar]
- Fabre O, Ingerslev LR, Garde C, Donkin I, Simar D & Barres R. (2018). Exercise training alters the genomic response to acute exercise in human adipose tissue. Epigenomics 10, 1033–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibala MJ, Little JP, MacDonald MJ & Hawley JA. (2012). Physiological adaptations to low-volume, high-intensity interval training in health and disease. The Journal of physiology 590, 1077–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollisch KSC, Brandauer J, Jessen N, Toyoda T, Nayer A, Hirshman MF & Goodyear LJ. (2009). Effects of exercise training on subcutaneous and visceral adipose tissue in normal-and high-fat diet-fed rats. American Journal of Physiology-Endocrinology and Metabolism 297, E495–E504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg AS, Shen W-J, Muliro K, Patel S, Souza SC, Roth RA & Kraemer FB. (2001). Stimulation of lipolysis and hormone-sensitive lipase via the extracellular signal-regulated kinase pathway. Journal of Biological Chemistry 276, 45456–45461. [DOI] [PubMed] [Google Scholar]
- Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, Wang ZV, Landskroner-Eiger S, Dineen S & Magalang UJ. (2009). Hypoxia-inducible factor 1α induces fibrosis and insulin resistance in white adipose tissue. Molecular and cellular biology 29, 4467–4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarstedt A, Jansson PA, Wesslau C, Yang X & Smith U. (2003). Reduced expression of PGC-1 and insulin-signaling molecules in adipose tissue is associated with insulin resistance. Biochemical and biophysical research communications 301, 578–582. [DOI] [PubMed] [Google Scholar]
- Heinonen S, Buzkova J, Muniandy M, Kaksonen R, Ollikainen M, Ismail K, Hakkarainen A, Lundbom J, Lundbom N & Vuolteenaho K. (2015). Impaired mitochondrial biogenesis in adipose tissue in acquired obesity. Diabetes 64, 3135–3145. [DOI] [PubMed] [Google Scholar]
- Hernández EÁ, Kahl S, Seelig A, Begovatz P, Irmler M, Kupriyanova Y, Nowotny B, Nowotny P, Herder C & Barosa C. (2017). Acute dietary fat intake initiates alterations in energy metabolism and insulin resistance. The Journal of clinical investigation 127, 695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz JF, Braudy RJ, Martin Iii WH & Klein S. (1999). Endurance exercise training does not alter lipolytic or adipose tissue blood flow sensitivity to epinephrine. American Journal of Physiology-Endocrinology And Metabolism. [DOI] [PubMed] [Google Scholar]
- Horowitz JF, Leone TC, Feng W, Kelly DP & Klein S. (2000). Effect of endurance training on lipid metabolism in women: a potential role for PPARα in the metabolic response to training. American Journal of Physiology-Endocrinology And Metabolism 279, E348–E355. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS & Spiegelman BM. (1993). Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259, 87–91. [DOI] [PubMed] [Google Scholar]
- Hu E, Kim JB, Sarraf P & Spiegelman BM. (1996). Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARγ. Science 274, 2100–2103. [DOI] [PubMed] [Google Scholar]
- Huang G, Ge G, Wang D, Gopalakrishnan B, Butz DH, Colman RJ, Nagy A & Greenspan DS. (2011). α3 (V) collagen is critical for glucose homeostasis in mice due to effects in pancreatic islets and peripheral tissues. The Journal of clinical investigation 121, 769–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulver MW, Zheng D, Tanner CJ, Houmard JA, Kraus WE, Slentz CA, Sinha MK, Pories WJ, MacDonald KG & Dohm GL. (2002). Adiponectin is not altered with exercise training despite enhanced insulin action. American Journal of Physiology-Endocrinology And Metabolism 283, E861–E865. [DOI] [PubMed] [Google Scholar]
- Hutchison SK, Teede HJ, Rachoń D, Harrison CL, Strauss BJ & Stepto NK. (2012). Effect of exercise training on insulin sensitivity, mitochondria and computed tomography muscle attenuation in overweight women with and without polycystic ovary syndrome. Diabetologia 55, 1424–1434. [DOI] [PubMed] [Google Scholar]
- Kawanishi N, Niihara H, Mizokami T, Yano H & Suzuki K. (2013). Exercise training attenuates adipose tissue fibrosis in diet-induced obese mice. Biochemical and biophysical research communications 440, 774–779. [DOI] [PubMed] [Google Scholar]
- Kern PA, Ranganathan S, Li C, Wood L & Ranganathan G. (2001). Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. American Journal of Physiology-Endocrinology And Metabolism 280, E745–E751. [DOI] [PubMed] [Google Scholar]
- Kos K, Wong S, Tan B, Gummesson A, Jernas M, Franck N, Kerrigan D, Nystrom FH, Carlsson LMS & Randeva HS. (2009). Regulation of the fibrosis and angiogenesis promoter SPARC/osteonectin in human adipose tissue by weight change, leptin, insulin, and glucose. Diabetes 58, 1780–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan J, Danzer C, Simka T, Ukropec J, Walter KM, Kumpf S, Mirtschink P, Ukropcova B, Gasperikova D & Pedrazzini T. (2012). Dietary obesity-associated Hif1α activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes & development 26, 259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson-Meyer DE, Heilbronn LK, Redman LM, Newcomer BR, Frisard MI, Anton S, Smith SR, Alfonso A & Ravussin E. (2006). Effect of calorie restriction with or without exercise on insulin sensitivity, β-cell function, fat cell size, and ectopic lipid in overweight subjects. Diabetes care 29, 1337–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurencikiene J, Skurk T, Kulyté A, Hedén P, Åström G, Sjölin E, Rydén M, Hauner H & Arner P. (2011). Regulation of lipolysis in small and large fat cells of the same subject. The Journal of Clinical Endocrinology & Metabolism 96, E2045–E2049. [DOI] [PubMed] [Google Scholar]
- Ligibel JA, Giobbie-Hurder A, Olenczuk D, Campbell N, Salinardi T, Winer EP & Mantzoros CS. (2009). Impact of a mixed strength and endurance exercise intervention on levels of adiponectin, high molecular weight adiponectin and leptin in breast cancer survivors. Cancer Causes & Control 20, 1523–1528. [DOI] [PubMed] [Google Scholar]
- Little JP, Jung ME, Wright AE, Wright W & Manders RJF. (2014). Effects of high-intensity interval exercise versus continuous moderate-intensity exercise on postprandial glycemic control assessed by continuous glucose monitoring in obese adults. Applied physiology, nutrition, and metabolism 39, 835–841. [DOI] [PubMed] [Google Scholar]
- Ludzki AC, Krueger EM, Baldwin TC, Schleh MW, Porsche CE, Ryan BJ, Muir LA, Singer K, Lumeng CN & Horowitz JF. (2020). Acute aerobic exercise remodels the adipose tissue progenitor cell phenotype in obese adults. Frontiers in physiology 11, 903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludzki AC, Pataky MW, Cartee GD & Horowitz JF. (2018). Acute endurance exercise increases Vegfa mRNA expression in adipose tissue of rats during the early stages of weight gain. Applied Physiology, Nutrition, and Metabolism 43, 751–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magkos F, Fraterrigo G, Yoshino J, Luecking C, Kirbach K, Kelly SC, de Las Fuentes L, He S, Okunade AL & Patterson BW. (2016). Effects of moderate and subsequent progressive weight loss on metabolic function and adipose tissue biology in humans with obesity. Cell metabolism 23, 591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcell TJ, McAuley KA, Traustadóttir T & Reaven PD. (2005). Exercise training is not associated with improved levels of C-reactive protein or adiponectin. Metabolism 54, 533–541. [DOI] [PubMed] [Google Scholar]
- Markofski MM, Carrillo AE, Timmerman KL, Jennings K, Coen PM, Pence BD & Flynn MG. (2014). Exercise training modifies ghrelin and adiponectin concentrations and is related to inflammation in older adults. Journals of Gerontology Series A: Biomedical Sciences and Medical Sciences 69, 675–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marycz K, Kornicka K, Szlapka-Kosarzewska J & Weiss C. (2018). Excessive endoplasmic reticulum stress correlates with impaired mitochondrial dynamics, mitophagy and apoptosis, in liver and adipose tissue, but not in muscles in EMS horses. International Journal of Molecular Sciences 19, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuaid SE, Hodson L, Neville MJ, Dennis AL, Cheeseman J, Humphreys SM, Ruge T, Gilbert M, Fielding BA & Frayn KN. (2011). Downregulation of adipose tissue fatty acid trafficking in obesity: a driver for ectopic fat deposition? Diabetes 60, 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menshikova EV, Ritov VB, Toledo FGS, Ferrell RE, Goodpaster BH & Kelley DE. (2005). Effects of weight loss and physical activity on skeletal muscle mitochondrial function in obesity. American journal of physiology-endocrinology and metabolism 288, E818–E825. [DOI] [PubMed] [Google Scholar]
- Moritz CP. (2017). Tubulin or not tubulin: heading toward total protein staining as loading control in western blots. Proteomics 17, 1600189. [DOI] [PubMed] [Google Scholar]
- Newsom SA, Schenk S, Thomas KM, Harber MP, Knuth ND, Goldenberg N & Horowitz JF. (2010). Energy deficit after exercise augments lipid mobilization but does not contribute to the exercise-induced increase in insulin sensitivity. Journal of applied physiology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olefsky JM. (1976). The effects of spontaneous obesity on insulin binding, glucose transport, and glucose oxidation of isolated rat adipocytes. The Journal of clinical investigation 57, 842–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olefsky JM. (1977). Insensitivity of large rat adipocytes to the antilipolytic effects of insulin. Journal of lipid research 18, 459–464. [PubMed] [Google Scholar]
- Østergård T, Andersen JL, Nyholm B, Lund S, Nair KS, Saltin B & Schmitz O. (2006). Impact of exercise training on insulin sensitivity, physical fitness, and muscle oxidative capacity in first-degree relatives of type 2 diabetic patients. American Journal of Physiology-Endocrinology and Metabolism 290, E998–E1005. [DOI] [PubMed] [Google Scholar]
- Ozcelik O, Celik H, Ayar A, Serhatlioglu S & Kelestimur H. (2004). Investigation of the influence of training status on the relationship between the acute exercise and serum leptin levels in obese females. Neuroendocrinology Letters 25, 381–385. [PubMed] [Google Scholar]
- Parlee SD, Lentz SI, Mori H & MacDougald OA. (2014). Quantifying size and number of adipocytes in adipose tissue. Methods in enzymology 537, 93–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasarica M, Sereda OR, Redman LM, Albarado DC, Hymel DT, Roan LE, Rood JC, Burk DH & Smith SR. (2009). Reduced adipose tissue oxygenation in human obesity: evidence for rarefaction, macrophage chemotaxis, and inflammation without an angiogenic response. Diabetes 58, 718–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak J, Klimcakova E, Moro C, Viguerie N, Berlan M, Hejnova J, Richterova B, Kraus I, Langin D & Stich V. (2006). Effect of aerobic training on plasma levels and subcutaneous abdominal adipose tissue gene expression of adiponectin, leptin, interleukin 6, and tumor necrosis factor α in obese women. Metabolism 55, 1375–1381. [DOI] [PubMed] [Google Scholar]
- Ravussin E & Smith SR. (2002). Increased fat intake, impaired fat oxidation, and failure of fat cell proliferation result in ectopic fat storage, insulin resistance, and type 2 diabetes mellitus. Annals of the New York Academy of Sciences 967, 363–378. [DOI] [PubMed] [Google Scholar]
- Riis S, Christensen B, Nellemann B, Møller AB, Husted AS, Pedersen SB, Schwartz TW, Jørgensen JOL & Jessen N. (2019). Molecular adaptations in human subcutaneous adipose tissue after ten weeks of endurance exercise training in healthy males. Journal of Applied Physiology 126, 569–577. [DOI] [PubMed] [Google Scholar]
- Roden M, Price TB, Perseghin G, Petersen KF, Rothman DL, Cline GW & Shulman GI. (1996). Mechanism of free fatty acid-induced insulin resistance in humans. The Journal of clinical investigation 97, 2859–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rönn T, Volkov P, Tornberg Å, Elgzyri T, Hansson O, Eriksson KF, Groop L & Ling C. (2014). Extensive changes in the transcriptional profile of human adipose tissue including genes involved in oxidative phosphorylation after a 6-month exercise intervention. Acta physiologica 211, 188–200. [DOI] [PubMed] [Google Scholar]
- Rupnick MA, Panigrahy D, Zhang C-Y, Dallabrida SM, Lowell BB, Langer R & Folkman MJ. (2002). Adipose tissue mass can be regulated through the vasculature. Proceedings of the National Academy of Sciences 99, 10730–10735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan BJ, Schleh MW, Ahn C, Ludzki AC, Gillen JB, Varshney P, Van Pelt DW, Pitchford LM, Chenevert TL & Gioscia-Ryan RA. (2020). Moderate-intensity exercise and high-intensity interval training affect insulin sensitivity similarly in obese adults. The Journal of Clinical Endocrinology & Metabolism 105, e2941–e2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk S, Harber MP, Shrivastava CR, Burant CF & Horowitz JF. (2009). Improved insulin sensitivity after weight loss and exercise training is mediated by a reduction in plasma fatty acid mobilization, not enhanced oxidative capacity. The Journal of physiology 587, 4949–4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple RK, Crowley VC, Sewter CP, Laudes M, Christodoulides C, Considine RV, Vidal-Puig A & O'Rahilly S. (2004). Expression of the thermogenic nuclear hormone receptor coactivator PGC-1 α is reduced in the adipose tissue of morbidly obese subjects. International journal of obesity 28, 176–179. [DOI] [PubMed] [Google Scholar]
- Shadid S, Stehouwer CDA & Jensen MD. (2006). Diet/Exercise versus pioglitazone: effects of insulin sensitization with decreasing or increasing fat mass on adipokines and inflammatory markers. The Journal of Clinical Endocrinology & Metabolism 91, 3418–3425. [DOI] [PubMed] [Google Scholar]
- Shimomura I, Funahasm T, Takahashi M, Maeda K, Kotani K, Nakamura T, Yamashita S, Miura M, Fukuda Y & Takemura K. (1996). Enhanced expression of PAI–1 in visceral fat: Possible contributor to vascular disease in obeisty. Nature medicine 2, 800–803. [DOI] [PubMed] [Google Scholar]
- Shirvani H & Arabzadeh E. (2020). Metabolic cross-talk between skeletal muscle and adipose tissue in high-intensity interval training vs. moderate-intensity continuous training by regulation of PGC-1α. Eating and Weight Disorders-Studies on Anorexia, Bulimia and Obesity 25, 17–24. [DOI] [PubMed] [Google Scholar]
- Shojaee-Moradie F, Baynes K, Pentecost C, Bell J, Thomas E, Jackson N, Stolinski M, Whyte M, Lovell D & Bowes S. (2007). Exercise training reduces fatty acid availability and improves the insulin sensitivity of glucose metabolism. Diabetologia 50, 404–413. [DOI] [PubMed] [Google Scholar]
- Shostak A, Meyer-Kovac J & Oster H. (2013). Circadian regulation of lipid mobilization in white adipose tissues. Diabetes 62, 2195–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöström L & Björntorp P. (1974). Body composition and adipose tissue cellularity in human obesity. Acta Medica Scandinavica 195, 201–211. [DOI] [PubMed] [Google Scholar]
- Smith SR, Lovejoy JC, Greenway F, Ryan D, deJonge L, de la Bretonne J, Volafova J & Bray GA. (2001). Contributions of total body fat, abdominal subcutaneous adipose tissue compartments, and visceral adipose tissue to the metabolic complications of obesity. Metabolism-Clinical and Experimental 50, 425–435. [DOI] [PubMed] [Google Scholar]
- Spencer M, Unal R, Zhu B, Rasouli N, McGehee RE Jr, Peterson CA & Kern PA. (2011). Adipose tissue extracellular matrix and vascular abnormalities in obesity and insulin resistance. The Journal of Clinical Endocrinology & Metabolism 96, E1990–E1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers LKM, Samra JS, Humphreys SM, Morris RJ & Frayn KN. (1996). Subcutaneous abdominal adipose tissue blood flow: variation within and between subjects and relationship to obesity. Clinical science 91, 679–683. [DOI] [PubMed] [Google Scholar]
- Sun K, Kusminski CM & Scherer PE. (2011). Adipose tissue remodeling and obesity. The Journal of clinical investigation 121, 2094–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Li F-H, Li T, Min Z, Yang L-D, Gao H-E, Wu D-S & Xie T. (2020). Effects of high-intensity interval training on adipose tissue lipolysis, inflammation, and metabolomics in aged rats. Pflügers Archiv-European Journal of Physiology 472, 245–258. [DOI] [PubMed] [Google Scholar]
- Sutherland LN, Bomhof MR, Capozzi LC, Basaraba SAU & Wright DC. (2009). Exercise and adrenaline increase PGC-1α mRNA expression in rat adipose tissue. The Journal of physiology 587, 1607–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay A, Simoneau J-A & Bouchard C. (1994). Impact of exercise intensity on body fatness and skeletal muscle metabolism. Metabolism 43, 814–818. [DOI] [PubMed] [Google Scholar]
- Trevellin E, Scorzeto M, Olivieri M, Granzotto M, Valerio A, Tedesco L, Fabris R, Serra R, Quarta M & Reggiani C. (2014). Exercise training induces mitochondrial biogenesis and glucose uptake in subcutaneous adipose tissue through eNOS-dependent mechanisms. Diabetes 63, 2800–2811. [DOI] [PubMed] [Google Scholar]
- Trujillo ME, Lee M-J, Sullivan S, Feng J, Schneider SH, Greenberg AS & Fried SK. (2006). Tumor Necrosis Factor α and Glucocorticoid Synergistically Increase Leptin Production in Human Adipose Tissue: Role for p38 Mitogen-Activated Protein Kinase. The Journal of Clinical Endocrinology & Metabolism 91, 1484–1490. [DOI] [PubMed] [Google Scholar]
- Van Pelt DW, Guth LM & Horowitz JF. (2017). Aerobic exercise elevates markers of angiogenesis and macrophage IL-6 gene expression in the subcutaneous adipose tissue of overweight-to-obese adults. Journal of Applied Physiology 123, 1150–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton RG, Finlin BS, Mula J, Long DE, Zhu B, Fry CS, Westgate PM, Lee JD, Bennett T & Kern PA. (2015). Insulin-resistant subjects have normal angiogenic response to aerobic exercise training in skeletal muscle, but not in adipose tissue. Physiological reports 3, e12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Si Y, Shirihai OS, Si H, Schultz V, Corkey RF, Hu L, Deeney JT, Guo W & Corkey BE. (2010). Respiration in adipocytes is inhibited by reactive oxygen species. Obesity 18, 1493–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL & Ferrante AW. (2003). Obesity is associated with macrophage accumulation in adipose tissue. The Journal of clinical investigation 112, 1796–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston KS, Wisløff U & Coombes JS. (2014). High-intensity interval training in patients with lifestyle-induced cardiometabolic disease: a systematic review and meta-analysis. British journal of sports medicine 48, 1227–1234. [DOI] [PubMed] [Google Scholar]
- Yatagai T, Nishida Y, Nagasaka S, Nakamura T, Tokuyama K, Shindo M, Tanaka H & Ishibashi S. (2003). Relationship between exercise training-induced increase in insulin sensitivity and adiponectinemia in healthy men. Endocrine journal 50, 233–238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets generated and analyzed in this study are available from the corresponding author on reasonable request.