

Abstract
Regulation of intracellular pH (pHi) in cardiomyocytes is crucial for cardiac function; however, currently known mechanisms for direct or indirect extrusion of acid from cardiomyocytes seem insufficient for energetically-efficient extrusion of the massive H+ loads generated under in vivo conditions. In cardiomyocytes, voltage-sensitive H+ channel activity mediated by the HVCN1 proton channel would be a highly efficient means of disposing of H+, while avoiding Na+-loading, as occurs during direct acid extrusion via Na+/H+ exchange or indirect acid extrusion via Na+-HCO3− cotransport. PCR and immunoblotting demonstrated expression of HVCN1 mRNA and protein in canine heart. Patch clamp analysis of canine ventricular myocytes revealed a voltage-gated H+ current that was highly H+-selective. The current was blocked by external Zn2+ and the HVCN1 blocker 5-chloro-2-guanidinobenzimidazole (ClGBI). Both the gating and Zn2+ blockade of the current were strongly influenced by the pH gradient across the membrane. All characteristics of the observed current were consistent with the known hallmarks of HVCN1-mediated H+ current. Inhibition of HVCN1 and the NHE1 Na+/H+ exchanger, singly and in combination, showed that either mechanism is largely sufficient to maintain pHi in beating cardiomyocytes, but that inhibition of both activities causes rapid acidification. These results show that HVCN1 is expressed in canine ventricular myocytes and provides a major H+-extrusion activity, with a capacity similar to that of NHE1. In the beating heart in vivo, this activity would allow Na+-independent extrusion of H+ during each action potential and, when functionally coupled with anion transport mechanisms, could facilitate transport-mediated CO2 disposal.
Graphical Abstract
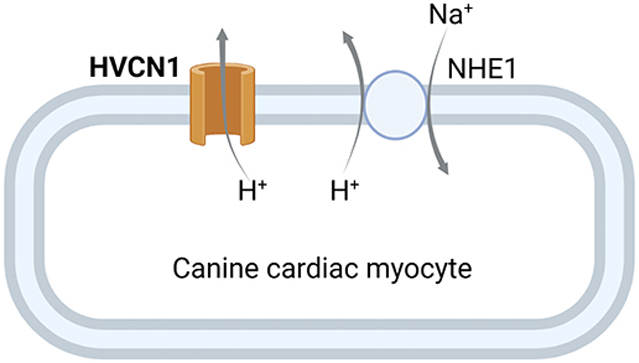
The HVCN1 proton channel is expressed in canine ventricular myocytes and contributes to H+ extrusion.
INTRODUCTION
Cardiac myocytes are metabolically highly active, and H+ and HCO3− are continuously generated by hydration of CO2 as it exits the mitochondria (Schroeder et al., 2013). H+-extrusion and maintenance of intracellular pH (pHi) in cardiomyocytes are essential for cardiac function. A decrease in pHi leads to depression of cardiac contractility through both inhibition of excitation-contraction coupling and a reduction in myofilament Ca2+ sensitivity (Fabiato & Fabiato, 1978; Orchard & Kentish, 1990). Acidification also affects myocyte electrical properties and Ca2+ handling, promoting the development of cardiac arrhythmias (Orchard & Cingolani, 1994).
Na+/H+ exchanger isoform 1 (NHE1) is generally viewed as the major mechanism for H+-extrusion in cardiomyocytes (Wakabayashi et al., 2013), but an additional mechanism seems likely. Despite the clear importance of H+ extrusion, loss or inhibition of NHE1 does not impair cardiac performance (Prasad et al., 2013b) and can be cardioprotective due to a reduction in Na+- and Ca2+-loading (Wang et al., 2003). Further, use of Na+/H+ exchange as the only major H+-extrusion mechanism would cause an equivalent amount of Na+-loading, which is detrimental under some conditions (Wakabayashi et al., 2013), and would require expenditure of energy to extrude excess Na+. HCO3−-uptake via Na+-HCO3− cotransport has been proposed as an alternative mechanism of acid-disposal (Garciarena et al., 2013a), but it also would cause Na+-loading. In addition, use of HCO3−-uptake to dispose of H+ would require that CO2 be reformed via dehydration of the imported HCO3−, followed by its diffusion from the cell.
An alternative mechanism of H+-extrusion that would avoid both the Na+-loading problem and formation of additional CO2 is voltage-gated H+ channel activity (Capasso et al., 2011; DeCoursey, 2013). Voltage-gated H+ channels (or Hv1 channels) are a family of H+-selective channels that are gated by both transmembrane voltage and the pH gradient (Cherny et al., 1995; DeCoursey, 2013). In mammals the channel is encoded by the Hvcn1 gene (HVCN1 in human) (Ramsey et al., 2006). The HVCN1 channel has been identified in many tissues, where its H+-extrusion activity contributes to diverse physiological functions (Capasso et al., 2011; DeCoursey, 2013). Our previous RNA sequencing study noted that HVCN1 is expressed in heart tissues of mouse, human and other mammalian species (Vairamani et al., 2017). More recently, a study using quantitative 3D confocal microscopy suggested that HVCN1 is expressed in a variety of cardiac cells, including human cardiomyocytes (Bkaily & Jacques, 2017). Although HVCN1 has not yet been functionally identified in cardiac myocytes, its voltage and pH sensitivities would make it ideally suited for direct extrusion of H+ during each action potential. This would allow efficient dissipation of acid on a beat-to-beat basis while avoiding an equivalent uptake of Na+ and stimulation of Ca2+-loading, as occurs when Na+/H+ exchange is activated (Wakabayashi et al., 2013). In addition to providing a mechanism for Na+-independent acid-extrusion, the presence of HVCN1 in cardiac myocytes would support the recent proposal that H+ channel activity, operating in a functionally coupled system with Cl−/HCO3− exchange, carbonic anhydrase, and Cl− channel activity, could serve as a mechanism for energetically-efficient transport-mediated CO2 disposal (Vairamani et al., 2017). Here, we demonstrate that the HVCN1 H+ channel is expressed in canine heart and plays a major role in pHi regulation in beating ventricular cardiomyocytes.
METHODS
Ethical approval.
Handling and usage of animals were in accordance with protocols approved by the University of Cincinnati Institutional Animal Care and Use Committee (protocol number 18-11-26-01), and in compliance with The Journal’s ethical policies.
Animals and tissue preparation.
Adult mongrel dogs, weighing 15-20 kg, were obtained from a USDA approved vendor. The sex of the dogs was randomly selected by the vendor, with a ratio of about 1:1. The animals had free access to water during transport and upon arrival. The animals were not housed onsite and were euthanized upon arrival. For euthanasia, the front leg was shaved and prepped with betadine or nolvasan scrub and alcohol rinsed 3 times. A 18-20 gauge intravenous (IV) catheter was placed in the cephalic vein and secured with porous tape. Euthasol was administered IV (80 mg/kg body weight) and death was confirmed by negative heart auscultation as determined by stethoscope exanimation by a Laboratory Animal Medical Services veterinarian staff. Hearts were excised and washed, and tissue was collected from the hearts and either frozen in liquid nitrogen for preparation of protein or RNA or used to isolate ventricular myocytes.
Western blotting.
Frozen tissue was homogenized with 1X cell lysis buffer (Cell Signaling Technology) containing complete protease inhibitor cocktail (Roche Applied Science) and phosphatase inhibitor cocktail sets I and II (EMD Millipore Chemicals). After centrifugation, the supernatants were collected. Protein samples were separated by 12% SDS-PAGE and transferred to nitrocellulose (Bio-Rad). Blots were blocked with 5% nonfat milk (0.1% Tween in PBS) and incubated overnight with rabbit polyclonal primary antibodies anti-HVCN1 (1:100 dilution, Santa Cruz, sc-136712) and anti-β actin (1:1000 dilution, Cell Signaling Technology) at 4°C. Membranes were then probed with HRP-conjugated anti-rabbit IgG secondary antibodies (1:5000 dilution, Cell Signaling Technology) for 1 hour at room temperature. In preliminary experiments using homogenates from dog and mouse left and right ventricles and spleen, which expresses very high levels of HVCN1, the antibody identified a 32 kD band (the size of HVCN1) that was blocked by the blocking peptide (data not shown). The protein bands were detected by the ECL Western blotting detection system (GE Healthcare). Quantification of the band intensities were performed by AlphaEaseFC software (Alpha Innotech). Experiments were repeated in tissues from 3 hearts.
Total RNA extraction and real-time RT-PCR.
Total RNA was extracted from frozen canine cardiac tissues using TRIzol Reagent (Life Technologies) according to manufacturer’s instructions. After DNase I (DNA-free™, Ambition) digestion, 1 μg total RNA was reverse transcribed using oligo dT primers random primers and SuperScript™ III (Invitrogen). Real-time PCR was performed using a StepOnePlus™ system (Applied Biosystems) with the following conditions: 2 min hold at 50°C (uracil DNA glycosylase (UDG) incubation) and 10 min hold at 95°C (UDG inactivation and DNA polymerase activation), followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. The following primer sets were used: HVCN1 forward: 5’-CTCCCACAGGTTTCAGGTTATC-3’, reverse: 5’-GAACACCTTGGTGGCATAGT-3’; GADPH forward: 5’-ATTCTACCCACGGCAAATTCC-3’, reverse: 5’-TTCTCCATGGTGGTGAAGACC-3’. These primer sets generated products of 127 and 172 bp for HVCN1 and GADPH, respectively. Specificity of the PCR product was confirmed by analysis of melting curve. All experiments were done in triplicate. The relative expression levels of HVCN1 were normalized to GADPH and were quantified by the △△Ct method. StepOne Software version 2.3 (Applied Biosystems) was used for data analysis.
Canine cardiomyocyte isolation.
Dogs were euthanized and hearts excised. Wedge-shaped left ventricular free wall was dissected and cannulated via a descending branch of the left circumflex artery. Ventricular myocytes were dissociated by perfusion with a Tyrode’s solution containing 85 unit ml−1 collagenase (type II, Worthington) at 37°C, as we have previously described (Dong et al., 2011). Isolated myocytes were harvested and stored in a standard Tyrode’s solution containing 0.1 mM Ca2+ at room temperature or 4°C, for recordings on the same day or the following day.
Electrophysiological recordings.
Isolated canine left ventricular myocytes were perfused with Tyrode’s solution containing (in mM): NaCl 140, KCl 5.4, MgCl2 1, CaCl2 1.8, HEPES 5, and glucose 10 (pH = 7.4). Whole-cell patch clamp recordings were performed with an Axopatch-200B amplifier. Glass pipettes were filled with solution containing (in mM): tetramethylammonium (TMA) methanesulfonate (MeSO3) 80, bis(2-hydroxyethyl)amino-tris(hydroxymethyl)methane (Bis-Tris) 100, MgCl2 2, EGTA 1, pH adjusted to 6.5 with TMA hydroxide (TMAOH), and had a resistance of 1.5 – 2 MΩ. After the membrane was ruptured, the perfusion solution was switched to external solution containing (in mM): TMAMeSO3 80, MgCl2 2, and EGTA 1, buffered with Bis-Tris 100 (for pHo = 6.5), HEPES 100 (for pHo = 7.0 and 7.5) or N-[Tris(hydroxymethyl)methyl] glycine (Tricine) 100 (for pHo = 8.0). pH of all external solutions was adjusted with TMAOH. The pipette and external solutions followed previously described recipes for H+ channel recording (Cherny et al., 1995; Schilling et al., 2002; Ramsey et al., 2006). When studying Zn2+ blockade of HVCN1 currents, EGTA was omitted from the external solutions. Washing in of the H+ channel recording solution eliminated contaminating ionic and exchanger currents, and revealed a time- and voltage-dependent voltage gated H+ current. Recording of the H+ current commenced once the current became relatively stable. The H+ current continued to increase in amplitude and activation rate, although at a slow rate, as reported in other tissues (Byerly et al., 1984; Cherny et al., 1995). All recordings were performed at room temperature (24 - 25°C) unless noted otherwise. Data collection and analysis were performed using pCLAMP software (Axon Instruments, Foster City, CA). Activation of the HVCN1 current was sigmoidal at low voltages. To determine the activation rate, following previously described methods, the current traces were fitted with a single exponential after a brief delay (DeCoursey & Cherny, 1995).
Myocyte pHi measurement.
Myocyte pHi was measured using the pH sensitive fluorescent dye 2’, 7’-bis-(2-carboxyethyl)-5-(and-6)-carboxy-fluorescein - acetoxymethyl ester (BCECF-AM, Molecular Probes). Isolated canine ventricular myocytes were incubated in 5 μM BCECF-AM in Tyrode’s solution containing (in mM): NaCl 118, KCl 5.4, HEPES 10, NaH2PO4 0.33, glucose 10, CaCl2 1.8 and MgCl2 2 (pH 7.4) at room temperature for 20 min, then washed to remove the extracellular dye. Fluorescence images were recorded using a Zeiss LSM 710 inverted confocal microscope (Carl Zeiss Microscopy, LLC, Thornwood, NY, USA), measured at excitation wavelengths of 435 nm and 488 nm and emission at 535 nm. The collected images were analyzed and processed with ImageJ software (National Institute of Health). The fluorescence emission ratio (488 nm/435 nm) vs. pHi relationship was determined using solutions buffered at designated pH values and containing the H+ ionophore nigericin (Thomas et al., 1979). Myocytes were perfused with solutions containing: 140 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 5 mM Glucose, 10 μM nigericin and 20 mM buffer of HEPES, Bis-Tris or MES, pH = 7.5, 7, 6.5, 6 and 5.5. The emission ratio was measured at each pH value to determine the emission ratio vs. pH relationship. To determine the effects of HVCN1 and/or NHE1 blockers on myocyte pHi, myocytes were placed in a chamber containing Tyrode’s solution containing (in mM): NaCl 118, KCl 5.4, HEPES 10, NaH2PO4 0.33, glucose 10, CaCl2 1.8 and MgCl2 2 (pH 7.4) at 37°C, and paced continuously with field stimulation (Grass S48 stimulator, Grass Instruments) at 1 Hz. Field stimulations were briefly paused for ~2 seconds during the fluorescence measurement to stop myocyte contraction and allow more stable sampling. After establishing a stable baseline measurement for at least 10 min, treatment drug as described in specific experiments was added. After background subtraction, the fluorescence ratio at each time point was calculated and converted to pHi values based on the emission ratio vs. pH relationship. All measurements of myocyte pHi were performed at 37°C. All reagents used in the study were from Sigma-Aldrich unless otherwise stated.
Estimation of Acid Production and Corresponding H+ Currents in Myocytes.
Estimates of acid production and H+ currents require pHi measurements, along with estimates of buffering power, total myocyte volume (from which H+ are extruded), and the volume of intracellular free water in which acid detected by BCECF occurs. Under conditions similar to those of the current study (nominally CO2/HCO3−-free HEPES buffer), the increase in intracellular buffered acid in the pHi range 7.2-6.2, determined previously for guinea-pig (Lagadic-Gossmann et al., 1992) and mouse (Nakamura et al., 2008) myocytes, was ~26 mM. On the basis of morphometric analyses of myocytes for various mammalian species (Bensley et al., 2016), the volume of canine left ventricular myocytes was estimated to be ~27 pL. On the basis of data in Haworth et al (Haworth et al., 1983), Poole-Wilson & Cameron (Poole-Wilson & Cameron, 1975), and Persson & Halle (Persson & Halle, 2008), intracellular free water was estimated to be ~60% of cell volume (~16 pL) if mitochondrial volume (~30% of cell volume) (Tsushima et al., 2018) was included. With mitochondrial volume excluded, the relevant water volume would be ~11 pL. HVCN1-mediated H+ currents that would be needed to extrude this quantity of acid were estimated using the formula: I = (mM H+/sec) x (cytosolic free water volume) x F, where F is the Faraday constant 96500 Coulombs/Mole.
Data analysis.
Statistical analyses were performed using Student’s t-test or one-way ANOVA. Differences were considered statistically significant at a value of P < 0.001. Comparison of EC50 values for Zn2+ blockade dose responses under different pHo was performed using GraphPad Prism 9.2.0. Linear regression was performed with GraphPad Prism 9.2.0 or SigmaPlot 11.0. Data was analyzed with Microsoft Excel and are expressed as mean ± standard deviation (SD). N values indicate the number of myocytes. If n ≤ 30, all data points are plotted in the figures; if n > 30, data points are plotted as mean ± SD and the raw datasets are provided.
RESULTS
HVCN1 is present in canine ventricular myocytes.
HVCN1 protein (Fig. 1A) and mRNA (Fig. 1B) were detected in canine left and right ventricles and atria, and appeared to be expressed at higher levels in atria than in ventricles. Transmural gradients across the left and right ventricles were observed at the mRNA level, but not at the protein level. Whole-cell patch clamp recordings under conditions that eliminate other ionic currents revealed a voltage-gated outward H+ current in isolated canine left ventricular myocytes (Fig. 1C). In response to depolarizing voltage steps, the current had slow activation, no detectable inactivation, and relatively rapid deactivation. In addition to its voltage-dependence, the gating of the current depended strongly on the pH gradient across the membrane (ΔpH, extracellular pH (pHo) – pHi). Increases in ΔpH, equivalent to relative intracellular acidification, markedly shifted both the current-voltage curve and activation threshold (Vthr) to more negative voltages (Fig. 1D and E). Average Vthr was 14.3 mV under symmetrical pH, and was shifted to −6.7, −28.9 and −57.1 mV at ΔpH of 0.5, 1.0 and 1.5, respectively (P < 0.0001). The Vthr vs ΔpH relationship had a slope of −47.1, agreeing with the 40 mV shift in activation/ΔpH reported for HVCN1 and related proton channels (Cherny et al., 1995). With increased ΔpH, the activation rate-voltage relationship was also shifted to the left (Fig. 1F), indicating faster activation at higher pHo. These properties closely resemble those of HVCN1 currents in other cell types (Cherny et al., 1995; Capasso et al., 2011).
Fig. 1. HVCN1 is expressed and active in canine ventricular myocytes.
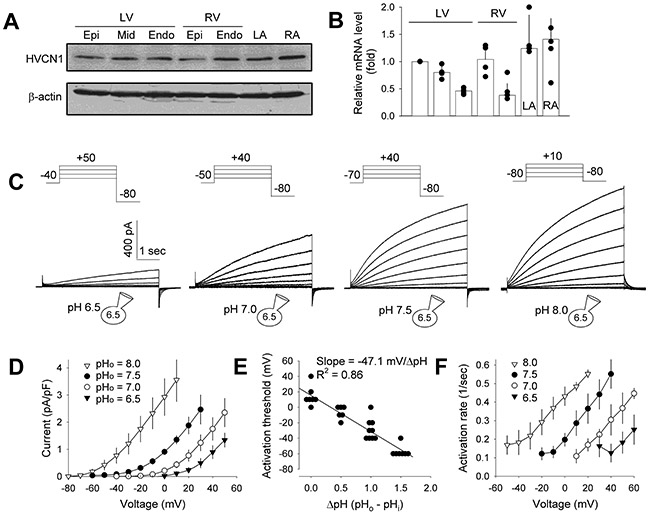
(A and B) HVCN1 protein and mRNA expression, respectively, in canine left and right ventricular (LV and RV) and left and right atrial (LA and RA) tissues, detected using immunoblot (representative of 3 hearts) and quantitative real-time PCR (n = 3 hearts). Epi: epicardium; Mid: midmyocardium; Endo: endocardium. (C) HVCN1 current recorded from canine LV myocytes under pHi = 6.5 and indicated pHo. Insets: voltage clamp protocols; depolarizing voltage step increment = 10 mV. (D) Current-voltage relationships of HVCN1. Current amplitude was measured at the end of 5 s depolarizing steps (n = 5 to 11 myocytes). (E) Activation threshold-ΔpH relationship (n = 6 to 9). Data were fitted with a line with a slope of −47.1 mV/unit change in ΔpH. (F) Activation rate-voltage relationship (n = 4 to 5 myocytes). Data are plotted as individual data points and/or mean ± SD.
Canine ventricular HVCN1 is H+-selective.
The reversal potential of the apparent HVCN1-mediated H+ current was recorded under conditions of varying pHo, using standard voltage-clamp protocols. An increase in ΔpH markedly shifted the reversal of the tail current to negative voltages (Fig. 2A and B). A plot of the average reversal potential (Vrev) vs ΔpH was fitted by a line with a slope of −56.7 mV/unit increase in ΔpH, agreeing well with the Nernst equation. These results show that the HVCN1 channel in canine ventricular myocytes is highly H+ selective. A hallmark of the voltage-gated H+ channel is a linear correlation between Vthr and Vrev that can be described by Vthr = 0.76Vrev + 18 mV (DeCoursey & Cherny, 1997; DeCoursey, 2013). The canine ventricular HVCN1 current also had this feature, with a Vthr vs Vrev relationship of Vthr = 0.85Vrev + 17.6 mV (Fig 2C).
Fig. 2. Canine ventricular HVCN1 is H+-selective.
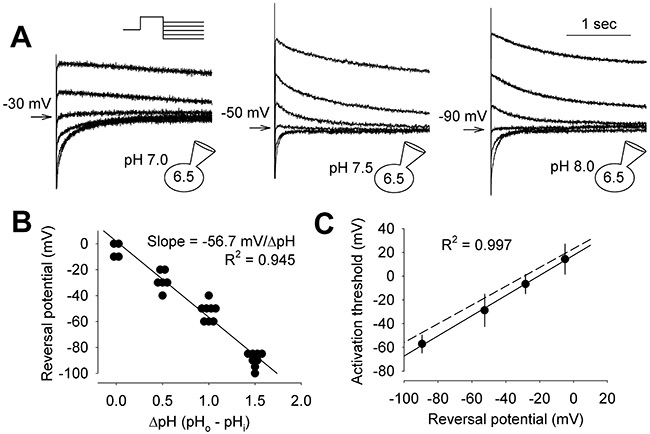
(A) Representative tail currents at indicated intra and extracellular pH. Inset: voltage clamp protocol. Currents were activated by 3 s depolarizing steps, followed by hyperpolarizing steps in −10 mV increments, until clear reversal of the tail current was observed. Holding potential was −70, −70 and −80 mV, and activation step was +40, +40 and +30 mV for the left, middle and right panel, respectively. Arrows: tail currents closest to reversal and the corresponding voltages. (B) Vrev-ΔpH relationship (n = 4 to 8 myocytes). Fitted line had a slope of −56.7 mV/unit change in ΔpH. C, Vthr-Vrev relationship. Data were fitted with Vthr = 0.85Vrev + 17.5 mV (solid line). Dash line: Vthr = 0.76Vrev + 18 mV reported for HVCN1 channel (DeCoursey & Cherny, 1997). Data are plotted as individual data points or mean ± SD.
Dependence of canine ventricular HVCN1 on temperature and ΔpH.
A signature property of the HVCN1 channel is its high Q10, a measure of its temperature sensitivity (DeCoursey & Cherny, 1998; Decoursey, 2012). Increase in temperature substantially increased the amplitude of the HVCN1 current in canine ventricular myocytes and the activation rate (Fig. 3A). The average Q10 was 2.4, which is within the range reported for HVCN1 channel (Decoursey, 2012) and higher than most other ion channels.
Fig. 3. Dependence of canine ventricular HVCN1 on temperature and internal pH.
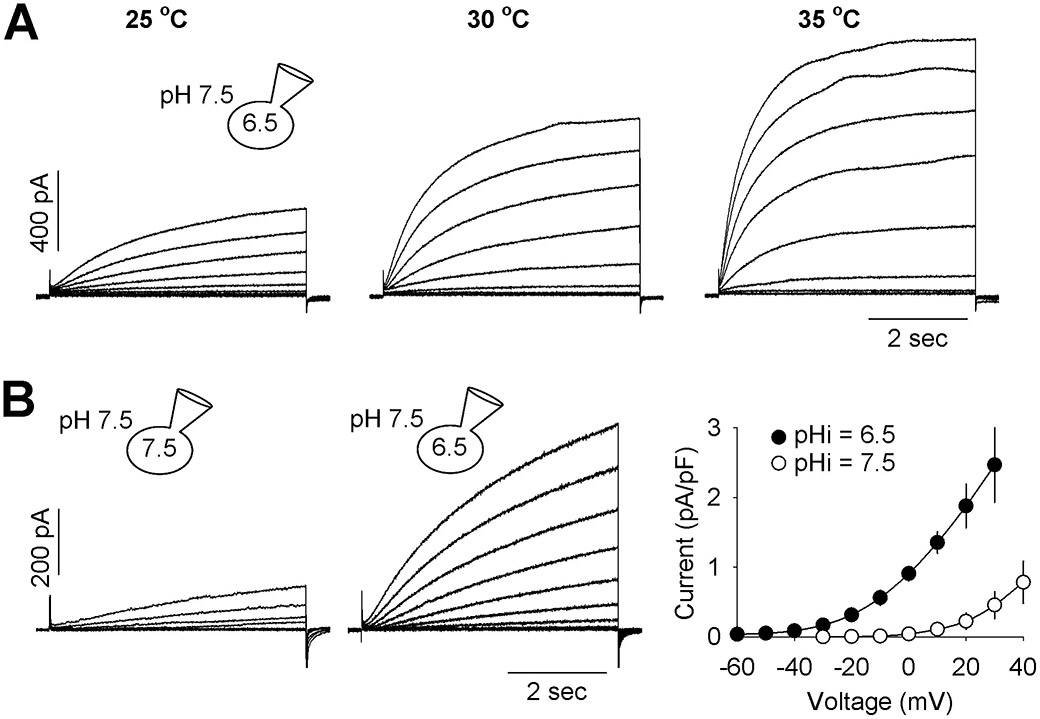
(A) HVCN1 current recorded at 25°C, 30°C and 35°C, from the same myocyte. Holding potentials were −70 mV and activation steps were −60 to +30 mV, in 10 mV increment. Similar results were found in 3 cells with an average fold increase in current amplitude per 10 °C (Q10) of 2.4. (B) HVCN1 current recorded under pHi = 7.5 and 6.5 and pHo = 7.5. (C) Current-voltage relationships of HVCN1 under pHi = 7.5 and 6.5 and pHo = 7.5. Current amplitude was measured at the end of 5 s depolarizing steps (n = 4 and 11 myocytes). Data are expressed as mean ± SD.
To confirm that the gating of canine ventricular HVCN1 was dependent on ΔpH, not just the absolute values of extracellular pH, the current was recorded under pHi = 7.5 and compared to pHi = 6.5, both under pHo = 7.5 (Fig. 3B). Increasing pHi (reducing ΔpH) had a similar effect on the activation kinetics as reducing pHo as shown in Fig. 1.
Canine ventricular HVCN1 H+ channel is blocked by extracellular Zn2+ and the HVCN1 inhibitor ClGBI.
Most known voltage-gated H+ channels are blocked by external Zn2+ in a pHo-dependent manner (Cherny & DeCoursey, 1999; DeCoursey, 2013). Inhibition by Zn2+ is eliminated by mutation of His140 and His193 (Ramsey et al., 2006). Canine HVCN1 has both histidines at the equivalent positions. The HVCN1 current in canine ventricular myocytes was sensitive to Zn2+, with the sensitivity markedly increased at higher pHo (Fig. 4A and B). The IC50 for Zn2+ blockade of HVCN1 was 57.4 μM at pHo = 6.5, and was shifted to 10.2 μM at pHo = 7.0, and 1.4 μM at pHo = 7.5 (Fig. 4B). The IC50 values were statistically different among three pHo (P < 0.0001). Zn2+ also reduced the rate of activation of the HVCN1 current, as shown in Fig. 4C. These properties closely match those reported for Zn2+ blockade of mammalian HVCN1 channels (Cherny & DeCoursey, 1999; DeCoursey, 2013).
Fig. 4. pHo-dependent blockade of canine ventricular HVCN1 by external Zn2+ and HVCN1 blockade by ClGBI.
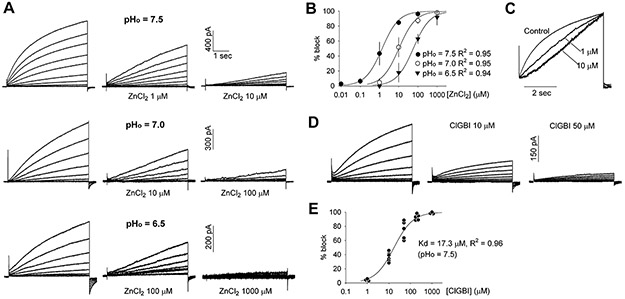
(A) Representative current at pHi = 6.5 and indicated pHo, and under control and indicated concentrations of extracellular Zn2+. Data in each row were from the same myocyte and recorded using identical voltage clamp protocol. (B) Dose-response curves for Zn2+ blockade of HVCN1 expressed as the ratio of current with to without Zn2+ at the end of 5 s pulses (n= 3 to 6 myocytes). Data are mean ± SD and were fitted with a standard Hill equation with Hill coefficient of 1.0 and IC50 = 1.37, 10.25 and 57.36 μM at pHo = 7.5, 7.0 and 6.5, respectively. (C) Current traces at +30 mV from the same families of currents shown in (A) at pHo = 7.5, scaled to the same amplitude and overlaid. (D) Representative current at pHi = 6.5 and pHo= 7.5, and under control and indicated concentrations of extracellular ClGBI. The currents were from the same myocyte and recorded using identical voltage clamp protocol. (E) Dose-response curves for ClGBI blockade of HVCN1 expressed as the ratio of current with to without ClGBI at the end of 5 s pulses (n= 3 to 6 myocytes). Data points were fitted with a standard Hill equation with Hill coefficient of 1.0 and IC50 = 17.3 μM at pHi = 6.5 and pHo= 7.5.
A guanidine derivative, 5-chloro-2-guanidinobenzimidazole (ClGBI), was recently identified as a selective blocker of HVCN1 (Hong et al., 2014). It blocks HVCN1 at micromolar concentrations by binding to the channel’s voltage-sensing domain from the extracellular side. Extracellular ClGBI blocked the HVCN1 current in canine ventricular myocytes in a dose-dependent manner with an IC50 of 17.3 μM (Fig. 4D and E). This is consistent with the IC50 of 26.3 μM for ClGBI blockade of heterologously expressed human HVCN1 (Hong et al., 2014).
HVCN1 contributes to acid extrusion in beating canine ventricular myocytes.
Currently, NHE1 is thought to be the only transport mechanism responsible for direct H+-extrusion in cardiac myocytes (Wakabayashi et al., 2013). A potential role for HVCN1 in pHi regulation in canine ventricular myocytes was examined using the pH-sensitive dye BCECF (Fig. 5). While fluorescence excitation at ~490 nm (λ1, 488 nm used in our experiments) was pH-dependent (shown in green, Fig. 5A), the excitation isosbestic point at ~435 nm (λ2, shown in red) gave an indication for any dye bleaching or changes in myocyte condition. Ventricular myocytes were paced at 1 Hz at 37°C, and a stable baseline was established for at least 10 min in the presence of NHE1 blockade using 1 μM ethyl-isopropyl amiloride (EIPA). Addition of 300 μM Zn2+ to the extracellular solution caused a significant drop in pHi (Fig. 5A and D); average pHi decreased from 7.1 in control to 6.13 at 5 minutes. This intracellular acidification was clearly visible in the merged images (Fig. 5A). Addition of 100 μM ClGBI in the presence of EIPA caused a similar drop in pHi, from 7.16 in control to 5.84 at 5 minutes (Fig. 5B and E). Conversely, when the experiment was performed to examine the effect of NHE1 blockade in the presence of HVCN1 blockade, pHi dropped from 7.15 in control to 6.13 at 5 minutes (Fig. 5C and F). In contrast, HVCN1 blockade alone with Zn2+ (Fig. 5G) or ClGBI (Fig. 5H) in the absence of EIPA, or NHE1 blockade alone in the absence of HVCN1 blockade (Fig. 5I) did not cause significant changes in pHi.
Fig. 5. HVCN1 contributes to acid extrusion in beating canine ventricular myocytes.

(A) Representative fluorescent images at excitations of 488 and 435 nm, and merged, recorded in the presence of EIPA (open bar) at steady state and subsequently with Zn2+ (closed bar) added. (B) Representative fluorescent images merged from recordings at excitations of 488 and 435 nm, in the presence of EIPA at steady state (at least 10 min after EIPA was added) and subsequently with ClGBI added. (C) Representative fluorescent images merged from recordings at excitations of 488 and 435 nm, in the presence of ClGBI at steady state (at least 10 min after ClGBI was added) and subsequently with EIPA added. (D to I) Average pHi under indicated combined or single blockers (n = 12 to 16 myocytes). Drug concentrations were 1 μM for EIPA, 100 μM for ClGBI, and 300 μM for Zn2+. Data are mean ± SD. *: P < 0.001 vs the last point in control in a unpaired t-test.
DISCUSSION
Our data provide unambiguous evidence that HVCN1 is expressed and active in canine cardiac myocytes and that it plays a major role in the regulation of pHi. Whole cell patch clamp analysis revealed a voltage- and pH-sensitive current that has all the hallmark features of the HVCN1 channel (Byerly et al., 1984; Cherny et al., 1995; DeCoursey & Cherny, 1995; Cherny & DeCoursey, 1999; Schilling et al., 2002; Ramsey et al., 2006; Capasso et al., 2011; DeCoursey, 2013). H+-selectivity of the current was demonstrated by the agreement of its reversal potential with the Nernst potential, and the gating kinetics and their pH-dependence closely matched results reported for HVCN1 (Cherny et al., 1995). The current had the pharmacologic fingerprint of HVCN1, including blockade by two structurally unrelated HVCN1 blockers, Zn2+ and ClGBI (Cherny & DeCoursey, 1999; Hong et al., 2014; Asuaje et al., 2017), and the strongly enhanced Zn2+ blockade at increased pHo. In addition, the H+ current exhibited a high Q10, as demonstrated previously for HVCN1-mediated currents (DeCoursey & Cherny, 1998; Decoursey, 2012). Inhibition of Na+/H+ exchange or HVCN1 separately in beating canine cardiomyocytes caused only a minor perturbation of pHi but inhibition of both together caused a rapid and marked drop in pHi, thus showing that both mechanisms mediate direct extrusion of H+. These results are important because they demonstrate, for the first time, a major Na+-independent acid-extrusion mechanism in cardiac myocytes.
The properties of HVCN1 make it well suited as a major mechanism for steady-state pHi regulation in cardiac myocytes in vivo. Because H+ extrusion via HVCN1 is driven by the H+ electrochemical gradient, it would appear to be an ideal mechanism for dissipation of H+ in beating myocytes. With an action potential amplitude of ~120 mV and peak membrane potential of >20 mV in mammalian ventricular myocytes (Jost et al., 2005; Sun & Wang, 2005; Rosati et al., 2008; Feldman et al., 2016), the rhythmic depolarizations occurring with each beat would provide an efficient energy source for extrusion of H+ that accumulate during the interbeat interval. Because HVCN1 gating is strongly regulated by the transmembrane pH gradient, any intracellular acidification would shift the voltage-dependent activation negatively and increase the activation rate, thereby increasing channel activity. Thus, HVCN1 is a powerful mechanism for pHi regulation in rhythmically depolarizing cardiac myocytes, with an activity that would be scalable as H+-generation increases at elevated heart rates. An important feature of HVCN1-mediated H+-extrusion is that it is not coupled to the transport of other ions. Unlike Na+/H+ exchange and Na+-HCO3− cotransport, it does not cause Na+-loading, which has effects on cardiac function that are unrelated to regulation of pHi and would require utilization of energy for extrusion of Na+ via the Na+,K+-ATPase. The existence of two distinct mechanisms for direct extrusion of H+ would allow flexibility in myocardial regulatory responses that involve varying degrees of Na+-loading, which can affect both contractility and energy-utilization.
The NHE1 Na+/H+ exchanger is a major Na+-uptake system in cardiac myocytes, and its activity is regulated by a variety of signaling mechanisms that transmit signals to downstream targets (Lazdunski et al., 1985; Garciarena et al., 2013b; Wakabayashi et al., 2013; Shimada-Shimizu et al., 2014; Yeves et al., 2014; Richards et al., 2019). Na+-loading causes Ca2+-loading, which can be detrimental under pathophysiological conditions (Karmazyn et al., 2008) but enhances contractility under normal conditions (Ennis et al., 2013). Despite its clear role in pHi regulation and Na+-loading, inhibition or genetic loss of NHE1 does not impair normal cardiac function (Prasad et al., 2013b; Wakabayashi et al., 2013), suggesting that other mechanisms can compensate in part for the loss of its activity. Na+-HCO3− cotransporter isoform 1 (NBCe1) is the most abundant Na+-HCO3− cotransporter in rodent heart (Table 1), and there is evidence that it contributes to Na+-loading (Garciarena et al., 2013a; Garciarena et al., 2013b). NBCe1 is expressed in t-tubules and sarcolemma, where its Na+-influx activity causes Ca2+-loading via effects on the Na+/Ca2+ exchanger (Garciarena et al., 2013b). Inhibition or genetic ablation of NHE1 or NBCe1 has cardioprotective effects, including protection against ischemia-reperfusion injury in isolated hearts (Wang et al., 2003; Fantinelli et al., 2014) and reduced apoptosis in response to cardiac hypertrophy (Garciarena et al., 2009) or coronary artery ligation (Vairamani et al., 2018). Conversely, transgenic overexpression of either NHE1 (Nakamura et al., 2008) or NBCe1 (Chen et al., 2020) in mice causes cardiac remodeling and disease, which has been attributed primarily to effects on Na+- and Ca2+-loading. It has been noted that functional coupling of the AE3 Cl−/HCO3− exchanger with NHE1 (Alvarez et al., 2007b) and/or NBCe1 (Prasad et al., 2013a) could facilitate pH-neutral Na+-loading.
Table 1.
mRNA expression for major H+ and HCO3− transporters in heart.
| Gene | Protein | Human | Rhesus Monkey |
C57BI6 Mouse |
Opossum | Rat |
|---|---|---|---|---|---|---|
| Hvcn1 | HVCN1 | 9 | 5 | 1 | 1 | 1 |
| Slc9a1 | NHE1 | 5 | 17 | 8 | 7 | 2.5 |
| Slc9a8 | NHE8 | 2 | 4 | 4 | 3 | 6.5 |
| Slc4a1 | AE1 | 0.3 | 0.5 | 0.1 | 2 | 2 |
| Slc4a2 | AE2 | 7 | 23 | 7 | 16 | 3.5 |
| Slc4a3 | AE3 | 178 | 77 | 47 | 45 | 25 |
| Slc4a4 | NBCe1 | 0.5 | 0.5 | 4 | 4 | 5.5 |
| Slc4a5 | NBCe2 | 0 | 0.1 | 0 | 2 | 0 |
| Slc4a7 | NBCn1 | 1 | 0.4 | 2 | 2 | 3.5 |
| Slc26a6 | PAT1 | 3 | 6 | 4 | 0 | 3.5 |
RPKM (Reads Per Kilobase of transcript per Million mapped reads) values for mRNA expression in Human, Rhesus monkey, Mouse and Opossum heart is from Brawand et al. (Brawand et al., 2011). Data were normalized in this study, allowing approximate cross-species comparisons of individual transporters. Data for rat heart are an average of values for male and female Fischer rats (Yu et al., 2014). These data indicate that HVCN1 is expressed in heart of all species listed and that AE3 is the most abundantly expressed Cl−/HCO3− exchanger.
The differences in mRNA expression levels for NHE1 and HVCN1 in hearts of various species (Table 1) suggest major species-differences in the relative importance of Na+-dependent and Na+-independent H+-extrusion mechanisms. In mouse heart, NHE1 mRNA expression was much greater than that of HVCN1, whereas HVCN1 mRNA expression in human heart was greater than that of NHE1. Mice have very high heart rates and much less cardiac reserve than humans and other large animals; mice exhibit only minimal force-frequency relationships (Georgakopoulos & Kass, 2001) and their heart rates can only increase ~50% vs about 200-300% in humans (Milani-Nejad & Janssen, 2014). Given the effects of Na+ on contractility and force-frequency relationships (Endoh, 2004), an increase in Na+-loading could play a role in the utilization of cardiac reserve. If so, then reciprocal alterations of HVCN1 activity and Na+/H+ exchange could modulate the degree of Na+-loading while maintaining H+-extrusion activity.
There are a number of transporters besides NHE1 and HVCN1 that could contribute to acid removal in cardiac myocytes, although none of them would allow Na+-independent regulation of pHi. RNA Seq data (Brawand et al., 2011; Yu et al., 2014) have revealed cardiac expression of mRNAs for Na+-HCO3− cotransporters and for an additional Na+/H+ exchanger (Table 1). mRNA for NHE8, which is expressed in endosomes and plasma membranes (Zhang et al., 2007; Lawrence et al., 2010), was present in all species examined and was expressed at higher levels than NHE1 in rat heart (Table 1) and in FVBN mouse heart (Vairamani et al., 2017). Single cell RNA Seq analysis of mouse heart showed that NHE8 is expressed in myocytes (Han et al., 2018), raising the possibility that it could contribute to acid extrusion. Although it was not a consideration during the design of our experiments, the EIPA concentrations used to inhibit NHE1 would also inhibit NHE8 (Zhang et al., 2007).
The rate of acidification that occurred when both Na+/H+ exchange and HVCN1 were inhibited was quite high, indicating that under normal circumstances, large amounts of acid are being produced and extruded from the cell. On the basis of buffering power, myocyte volume, and the volume of accessible cell water, the accumulation of acid on a whole cell basis was estimated to be ~2.2-3.1 mM/min, which would correspond to whole cell currents of ~95-135 pA if HVCN1 alone mediated the extrusion of all of this acid. Potential sources of acid include a modest contribution from NADPH oxidase activity, for which H+-extrusion via HVCN1 can provide charge balance (DeCoursey et al., 2000; Musset et al., 2010), and hydrolysis of ATP, which should be a factor only at more acidic pHi levels that impair mitochondrial function (Gursahani & Schaefer, 2004). However, the source of most of the acid generated, particularly at higher pHi levels, is CO2. With glucose as the sole energy source and efficient glucose oxidation in cardiac myocytes (Pascual & Coleman, 2016), O2 utilization would equal CO2 production and CO2 hydration would produce both H+ and HCO3−.
The estimated level of acid production when both Na+/H+ exchange and HVCN1 were inhibited is well within the range of expected values based on earlier studies of O2 utilization by isolated myocytes cultured under similar conditions (Piper et al., 1982). However, the amount of acid (and HCO3−) produced would also depend on the rate of CO2 hydration. A study using 13C-labeled pyruvate and magnetic resonance spectroscopy showed that most of the CO2 generated by mitochondrial respiration in the isolated rat heart (Schroeder et al., 2010) is hydrated by carbonic anhydrase, and that newly formed 13C-labeled CO2 and HCO3− are equilibrated in less than 10 seconds. Furthermore, the labeled CO2 and HCO3− declined rapidly, suggesting that newly formed HCO3− and residual CO2 are quickly eliminated. Similar studies of the rat heart in vivo demonstrated that carbonic anhydrase-mediated conversion of CO2 to HCO3− and H+ occurs as it exits the mitochondria, with an 11.4-fold increase in CO2 hydration relative to the spontaneous rate (Schroeder et al., 2013). The investigators also showed that inhibition of intracellular carbonic anhydrase led to reduced phosphocreatine/ATP ratios and increased acid in the mitochondrial matrix. Thus, hydration of CO2 to H+ + HCO3− as it exits the mitochondria is necessary for efficient energy metabolism; however, a corollary to this finding is that extrusion of both hydration products would be needed.
While it is clear that HVCN1, in the absence of Na+/H+ exchange, can extrude the quantities of acid generated from CO2 hydration in beating cardiac myocytes in vitro, it remains to be determined whether it is capable of extruding the much larger quantities of acid that occur in vivo (Endeward et al., 2010). A point to consider is that the experiments in Figs. 1-3 were carried out using quiescent cells and highly non-physiological buffers needed to suppress other ion currents and to isolate and characterize H+ currents. While these conditions were critical for documenting HVCN1 activity in ventricular myocytes, the observed currents are unlikely to represent the full magnitude of H+ currents that would occur in beating cells under physiological conditions in vivo. For example, the H+ currents in non-beating myocytes likely represent only the activity in surface sarcolemmal membranes, but not activity that might be present in t-tubules. In addition, the biophysical properties of HVCN1 and some of its known regulatory mechanisms suggest that its activity under physiological conditions in vivo would be substantially greater than that occurring in cultured cells.
DeCoursey & Cherny (1998) have documented a high Q10 for proton conductance and an exceptionally high Q10 of 6-9 for gating, consistent with the experiments shown in Fig. 3. In addition, phosphorylation of Thr29 converts the channel to an enhanced gating mode, in which it opens faster and closes more slowly, and causes a 40-mV negative shift in its H+ conductance-voltage relationship (DeCoursey et al., 2000; Musset et al., 2010). Pathak et al. (Pathak et al., 2016) reported that membrane stretch increases the rate of activation and shifts the activation to less depolarized potentials. They also suggested that when primed by other stimuli, membrane stretch can render the channel hyperactive. The unique regulation of HVCN1 by voltage, ΔpH, and mechanical stretch enables it to respond dynamically as these properties change, ensuring that acid extrusion occurs only when the electrochemical gradient is outwards, and that H+ efflux is adjusted according to the acid load and to the electrical and mechanical conditions at each moment. Additional studies will be needed to determine the specific properties and regulatory mechanisms that affect HVCN1 activity in cardiac myocytes and to assess the relative roles of Na+/H+ exchange and HVCN1 in regulating pHi homeostasis in cardiac muscle in vivo.
The hypothesis that HVCN1 is expressed and active in cardiac myocytes was based on data indicating that the AE3 Cl−/HCO3− exchanger is involved in transport-mediated CO2 disposal (Vairamani et al., 2017). A role for AE3-mediated HCO3−-extrusion in CO2 disposal had been proposed earlier for both retina and cardiac myocytes (Alvarez et al., 2007a; Vargas & Alvarez, 2012), but a H+-extrusion mechanism that could operate in a functionally-coupled and energetically-efficient system was not identified. Because most of the HCO3− in a cell is generated by CO2 hydration, which also produces H+, an effective means of CO2 disposal would be extrusion of HCO3− via Cl−/HCO3− exchange, with Cl−-recycling via Cl− channel activity, and H+-extrusion via HVCN1 (Vairamani et al., 2017). Na+/H+ exchange can function in such a system but causes obligatory Na+-loading, which is beneficial in some circumstances, but also increases energy utilization (Kandilci et al., 2020). Thus, if functionally-coupled ion transport, in concert with carbonic anhydrase activities, were to be used for CO2 disposal by cardiac myocytes, it would be useful to have H+-extrusion mechanisms that either contribute to Na+-loading, when the benefits outweigh the energy costs, or that are energetically-efficient and do not cause Na+-loading. HVCN1 is the only known mechanism that would allow energetically-efficient H+-extrusion.
A possible objection to the idea of transport-mediated CO2 disposal is that diffusion of CO2 through the cytoplasm and across cell membranes, either directly or via gas channels, is very rapid (Endeward et al., 2014; Arias-Hidalgo et al., 2017) and might be no impediment to CO2 disposal (Cooper et al., 2015; Hulikova et al., 2015). However, a high level of carbonic anhydrase activity has been reported in cardiac myocytes (Arias-Hidalgo et al., 2017) and, as discussed above, CO2-venting from mitochondria is facilitated by carbonic anhydrase-catalyzed conversion of CO2 → HCO3− + H+ as it exits the mitochondria (Schroeder et al., 2013). Buffering of H+ by high concentrations of histidyl dipeptides and other components (Swietach et al., 2013) helps to maintain pHi and, by sequestering one of the hydration products, also maintains a strong driving force for CO2 hydration. Thus, with continuous O2 consumption and CO2 hydration, a functionally coupled ion-transport system for extrusion of both H+ and HCO3− would be needed. The AE3 Cl−/HCO3− exchanger is a powerful mechanism for HCO3−-extrusion and Cl−-uptake, one or more of the Cl− channels in myocytes (Vairamani et al., 2017) could allow Cl−-exit, and either Na+/H+ exchange or HVCN1 activity could mediate H+-extrusion. HVCN1 activity is energetically-efficient and would provide charge balance to the process, whereas Na+/H+ exchange would require additional transport mechanisms and energy utilization to deal with the charge imbalance and increased Na+. After H+ and HCO3− are extruded into the extracellular or t-tubular fluid containing high concentrations of HCO3−, carbonic anhydrase activity would complete the process by regenerating CO2. Notably, carbonic anhydrases IV and XIV both enhance the activity of AE3 (Svichar et al., 2009). Also, Casey and colleagues ((Casey et al., 2009; Vargas & Alvarez, 2012) showed that AE3 associates physically with carbonic anhydrase XIV and suggested that this interaction is part of a system to dispose of CO2.
The sites at which transport-mediated CO2 disposal might occur have not been determined but may include the sarcolemma, t-tubules, and intercalated discs. The membrane location of HVCN1 is not known, but the fact that H+ currents could be identified in quiescent cells indicates that at least some HVCN1 is in the sarcolemma. AE3 (Alvarez et al., 2007b) and NHE1 (Petrecca et al., 1999; Lawrence et al., 2010) have been localized to sarcolemmal, t-tubule, and intercalated disc membranes. However, a later study reported that NHE1 was heavily concentrated in intercalated discs, with no evidence of activity in t-tubules (Garciarena et al., 2013a). Whether Na+/H+ exchangers and/or HVCN1 are active in t-tubules is an important issue, as the t-tubule system provides the shortest route for extrusion of H+ from the cytosol (Gadeberg et al., 2016). Also, mechanical forces occurring in beating cardiac myocytes cause substantial changes in t-tubular volume (McNary et al., 2012) that could enhance HVCN1 activity via mechanical stretch (Pathak et al. 2016) and cause rapid exchange of t-tubular and extracellular fluids. Thus, when t-tubular volume is reduced, a portion of its somewhat acidified fluid with newly generated CO2 would be expelled, and when t-tubular volume expands, fluid with normal extracellular pH and high O2 would be drawn in. Furthermore, t-tubule membranes have high levels of cholesterol, which is reported to make membranes relatively impermeable to CO2, which should prevent significant leakage of CO2 back into the cytosol. Interestingly, recent studies show that membrane permeability of O2 increases as cholesterol increases (Dotson et al., 2017; Al-Samir et al., 2021). Thus, one can speculate that the exchange of t-tubular and extracellular fluids, driven by mechanical forces (McNary et al., 2012 PMID: 22884710), may have the potential to contribute not only to CO2 disposal but also to O2 delivery.
In addition to functions in pHi regulation and transport-mediated CO2 disposal, activation of HVCN1 generates an outward current, and likely would also contribute to repolarization of the membrane and influence the action potential morphology. This effect would be more pronounced in the hearts of large animals that have a spike-and-dome cardiac action potential morphology, where the net current flow is small during the plateau phase and even a small current can have a notable effect on action potential duration. The time course and size of HVCN1 current during the action potential would be influenced by pHi, and the electrogenic extrusion of H+ could have a small K+-sparing effect during repolarization. In addition, by sharing the H+ extrusion burden, the presence of HVCN1 could influence Na+/H+ exchange activity, which has its own downstream electrical impacts through its effects on Na+ and Ca2+ loading, and consequently, Na+-Ca2+ exchanger (NCX) activity. The contribution of NCX to cardiac action potential is complex and can either lengthen or shorten the action potential depending on intracellular Na+ concentration (Armoundas et al., 2003). Therefore, the net influence of HVCN1 on action potential morphology likely is complex, would depend on the physiological state, and remains to be defined.
In conclusion, HVCN1, which uses the membrane potential and pH gradient rather than the Na+ gradient as a driving force, serves as a major H+-extrusion mechanism in canine ventricular myocytes. Although additional studies will be needed to further dissect its physiological functions in heart and to determine its relative importance in humans and other species, HVCN1 appears to be an ideal mechanism for disposal of H+, generated predominantly by CO2 hydration (Schroeder et al., 2013), which far exceeds metabolic acid generated from non-carbonic sources (Rose, 1994). HVCN1 activity would allow pHi homeostasis to be regulated independently of Na+-loading, thus allowing greater flexibility in myocardial regulatory responses. In addition, when functionally coupled with Cl−/HCO3− exchange, carbonic anhydrase activity, and Cl− channel activity, HVCN1 could contribute to transport-mediated CO2 disposal.
Supplementary Material
KEY POINTS SUMMARY.
Intracellular pH (pHi) regulation is crucial for cardiac function, as acidification depresses contractility and causes arrhythmias. H+ ions are generated in cardiomyocytes from metabolic processes and particularly from CO2 hydration, which has been shown to facilitate CO2-venting from mitochondria.
Currently, the NHE1 Na+/H+ exchanger is viewed as the dominant H+-extrusion mechanism in cardiac muscle.
We show that the HVCN1 voltage-gated proton channel is present and functional in canine ventricular myocytes, and that HVCN1 and NHE1 both contribute to pHi regulation.
HVCN1 provides an energetically-efficient mechanism of H+-extrusion that would not cause Na+-loading, which can cause pathology, and that could contribute to transport-mediated CO2 disposal.
These results provide a major advance in our understanding of pHi regulation in cardiac muscle.
Funding
This work was supported by NIH grants ES017263, ES027855 (HSW), funds from the University of Cincinnati College of Medicine (GES and HSW), and NIH grant R35-GM126902 (TED).
Biography

Dr. Jianyong Ma obtained his M.D. and Ph.D. degrees from the Nanchang University in China, and was a clinical cardiologist prior to joining The University of Cincinnati in 2009. He is currently a Research Scientist. His research interests include the physiological and electrophysiological properties of the heart, and the impact of environmental chemicals on the cardiovascular system. Discovering HVCN1 in canine heart is a highlight of his career, as the finding is expected to shed important light on cardiac pH regulation. He plans to use his basic research expertise and clinical cardiology experience and pursue a clinical research career.
Footnotes
Competing interests
The authors have no conflict of interest.
Data availability statement
The data that support the findings of this study are available from HSW upon reasonable request.
References
- Al-Samir S, Itel F, Hegermann J, Gros G, Tsiavaliaris G & Endeward V. (2021). O permeability of lipid bilayers is low, but increases with membrane cholesterol. Cellular and molecular life sciences : CMLS 78, 7649–7662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez BV, Gilmour GS, Mema SC, Martin BT, Shull GE, Casey JR & Sauve Y. (2007a). Blindness caused by deficiency in AE3 chloride/bicarbonate exchanger. PLoS One 2, e839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez BV, Johnson DE, Sowah D, Soliman D, Light PE, Xia Y, Karmazyn M & Casey JR. (2007b). Carbonic anhydrase inhibition prevents and reverts cardiomyocyte hypertrophy. The Journal of physiology 579, 127–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias-Hidalgo M, Al-Samir S, Weber N, Geers-Knörr C, Gros G & Endeward V. (2017). CO 2 permeability and carbonic anhydrase activity of rat cardiomyocytes. Acta Physiol (Oxf) 221, 115–128. [DOI] [PubMed] [Google Scholar]
- Armoundas AA, Hobai IA, Tomaselli GF, Winslow RL & O'Rourke B. (2003). Role of sodium-calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circulation research 93, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asuaje A, Smaldini P, Martin P, Enrique N, Orlowski A, Aiello EA, Gonzalez Leon C, Docena G & Milesi V. (2017). The inhibition of voltage-gated H(+) channel (HVCN1) induces acidification of leukemic Jurkat T cells promoting cell death by apoptosis. Pflugers Arch 469, 251–261. [DOI] [PubMed] [Google Scholar]
- Bensley JG, De Matteo R, Harding R & Black MJ. (2016). Three-dimensional direct measurement of cardiomyocyte volume, nuclearity, and ploidy in thick histological sections. Sci Rep 6, 23756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bkaily G & Jacques D. (2017). Na(+)-H(+) exchanger and proton channel in heart failure associated with Becker and Duchenne muscular dystrophies. Can J Physiol Pharmacol 95, 1213–1223. [DOI] [PubMed] [Google Scholar]
- Brawand D, Soumillon M, Necsulea A, Julien P, Csárdi G, Harrigan P, Weier M, Liechti A, Aximu-Petri A, Kircher M, Albert FW, Zeller U, Khaitovich P, Grützner F, Bergmann S, Nielsen R, Pääbo S & Kaessmann H. (2011). The evolution of gene expression levels in mammalian organs. Nature 478, 343–348. [DOI] [PubMed] [Google Scholar]
- Byerly L, Meech R & Moody W Jr., (1984). Rapidly activating hydrogen ion currents in perfused neurones of the snail, Lymnaea stagnalis. The Journal of physiology 351, 199–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capasso M, DeCoursey TE & Dyer MJ. (2011). pH regulation and beyond: unanticipated functions for the voltage-gated proton channel, HVCN1. Trends Cell Biol 21, 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey JR, Sly WS, Shah GN & Alvarez BV. (2009). Bicarbonate homeostasis in excitable tissues: role of AE3 Cl-/HCO3- exchanger and carbonic anhydrase XIV interaction. Am J Physiol Cell Physiol 297, C1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Chen L, Chen K, Lin H, Shen M, Chen L, Zhu H, Zhu Y, Wang Q, Xi F, Huang X, Wang Y, Liao W, Bin J, Asakura M, Liu J, Kitakaze M & Liao Y. (2020). Overexpression of Na(+)-HCO3(−) cotransporter contributes to the exacerbation of cardiac remodeling in mice with myocardial infarction by increasing intracellular calcium overload. Biochim Biophys Acta Mol Basis Dis 1866, 165623. [DOI] [PubMed] [Google Scholar]
- Cherny VV & DeCoursey TE. (1999). pH-dependent inhibition of voltage-gated H(+) currents in rat alveolar epithelial cells by Zn(2+) and other divalent cations. J Gen Physiol 114, 819–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherny VV, Markin VS & DeCoursey TE. (1995). The voltage-activated hydrogen ion conductance in rat alveolar epithelial cells is determined by the pH gradient. J Gen Physiol 105, 861–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GJ, Occhipinti R & Boron WF. (2015). CrossTalk proposal: Physiological CO2 exchange can depend on membrane channels. The Journal of physiology 593, 5025–5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decoursey TE. (2012). Voltage-gated proton channels. Compr Physiol 2, 1355–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey TE. (2013). Voltage-gated proton channels: molecular biology, physiology, and pathophysiology of the H(V) family. Physiol Rev 93, 599–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey TE & Cherny VV. (1995). Voltage-activated proton currents in membrane patches of rat alveolar epithelial cells. The Journal of physiology 489 ( Pt 2), 299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey TE & Cherny VV. (1997). Deuterium isotope effects on permeation and gating of proton channels in rat alveolar epithelium. J Gen Physiol 109, 415–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey TE & Cherny VV. (1998). Temperature dependence of voltage-gated H+ currents in human neutrophils, rat alveolar epithelial cells, and mammalian phagocytes. J Gen Physiol 112, 503–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey TE, Cherny VV, Zhou W & Thomas LL. (2000). Simultaneous activation of NADPH oxidase-related proton and electron currents in human neutrophils. Proc Natl Acad Sci U S A 97, 6885–6889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong M, Niklewski PJ & Wang HS. (2011). Ionic mechanisms of cellular electrical and mechanical abnormalities in Brugada syndrome. Am J Physiol Heart Circ Physiol 300, H279–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotson RJ, Smith CR, Bueche K, Angles G & Pias SC. (2017). Influence of Cholesterol on the Oxygen Permeability of Membranes: Insight from Atomistic Simulations. Biophys J 112, 2336–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endeward V, Al-Samir S, Itel F & Gros G. (2014). How does carbon dioxide permeate cell membranes? A discussion of concepts, results and methods. Front Physiol 4, 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endeward V, Gros G & Jürgens KD. (2010). Significance of myoglobin as an oxygen store and oxygen transporter in the intermittently perfused human heart: a model study. Cardiovasc Res 87, 22–29. [DOI] [PubMed] [Google Scholar]
- Endoh M (2004). Force-frequency relationship in intact mammalian ventricular myocardium: physiological and pathophysiological relevance. Eur J Pharmacol 500, 73–86. [DOI] [PubMed] [Google Scholar]
- Ennis IL, Aiello EA, Cingolani HE & Perez NG. (2013). The autocrine/paracrine loop after myocardial stretch: mineralocorticoid receptor activation. Curr Cardiol Rev 9, 230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiato A & Fabiato F. (1978). Effects of pH on the myofilaments and the sarcoplasmic reticulum of skinned cells from cardiace and skeletal muscles. The Journal of physiology 276, 233–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantinelli JC, Orlowski A, Aiello EA & Mosca SM. (2014). The electrogenic cardiac sodium bicarbonate co-transporter (NBCe1) contributes to the reperfusion injury. Cardiovasc Pathol 23, 224–230. [DOI] [PubMed] [Google Scholar]
- Feldman AM, Gordon J, Wang J, Song J, Zhang XQ, Myers VD, Tilley DG, Gao E, Hoffman NE, Tomar D, Madesh M, Rabinowitz J, Koch WJ, Su F, Khalili K & Cheung JY. (2016). BAG3 regulates contractility and Ca(2+) homeostasis in adult mouse ventricular myocytes. Journal of molecular and cellular cardiology 92, 10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadeberg HC, Bond RC, Kong CHT, Chanoit GP, Ascione R, Cannell MB & James AF. (2016). Heterogeneity of T-Tubules in Pig Hearts. PLoS One 11, e0156862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garciarena CD, Caldiz CI, Portiansky EL, Chiappe de Cingolani GE & Ennis IL. (2009). Chronic NHE-1 blockade induces an antiapoptotic effect in the hypertrophied heart. J Appl Physiol (1985) 106, 1325–1331. [DOI] [PubMed] [Google Scholar]
- Garciarena CD, Ma YL, Swietach P, Huc L & Vaughan-Jones RD. (2013a). Sarcolemmal localisation of Na+/H+ exchange and Na+-HCO3− co-transport influences the spatial regulation of intracellular pH in rat ventricular myocytes. The Journal of physiology 591, 2287–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garciarena CD, Youm JB, Swietach P & Vaughan-Jones RD. (2013b). H(+)-activated Na(+) influx in the ventricular myocyte couples Ca(2)(+)-signalling to intracellular pH. Journal of molecular and cellular cardiology 61, 51–59. [DOI] [PubMed] [Google Scholar]
- Georgakopoulos D & Kass D. (2001). Minimal force-frequency modulation of inotropy and relaxation of in situ murine heart. The Journal of physiology 534, 535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gursahani HI & Schaefer S. (2004). Acidification reduces mitochondrial calcium uptake in rat cardiac mitochondria. Am J Physiol Heart Circ Physiol 287, H2659–2665. [DOI] [PubMed] [Google Scholar]
- Han X, Wang R, Zhou Y, Fei L, Sun H, Lai S, Saadatpour A, Zhou Z, Chen H, Ye F, Huang D, Xu Y, Huang W, Jiang M, Jiang X, Mao J, Chen Y, Lu C, Xie J, Fang Q, Wang Y, Yue R, Li T, Huang H, Orkin SH, Yuan G-C, Chen M & Guo G. (2018). Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 172, 1091–1107. [DOI] [PubMed] [Google Scholar]
- Haworth RA, Hunter DR, Berkoff HA & Moss RL. (1983). Metabolic cost of the stimulated beating of isolated adult rat heart cells in suspension. Circ Res 52, 342–351. [DOI] [PubMed] [Google Scholar]
- Hong L, Kim IH & Tombola F. (2014). Molecular determinants of Hv1 proton channel inhibition by guanidine derivatives. Proc Natl Acad Sci U S A 111, 9971–9976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulikova A, Vaughan-Jones RD, Niederer SA & Swietach P. (2015). CrossTalk opposing view: Physiological CO2 exchange does not normally depend on membrane channels. The Journal of physiology 593, 5029–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost N, Virag L, Bitay M, Takacs J, Lengyel C, Biliczki P, Nagy Z, Bogats G, Lathrop DA, Papp JG & Varro A. (2005). Restricting excessive cardiac action potential and QT prolongation: a vital role for IKs in human ventricular muscle. Circulation 112, 1392–1399. [DOI] [PubMed] [Google Scholar]
- Kandilci HB, Richards MA, Fournier M, Şimşek G, Chung YJ, Lakhal-Littleton S & Swietach P. (2020). Cardiomyocyte Na/H Exchanger-1 Activity Is Reduced in Hypoxia. Front Cardiovasc Med 7, 617038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmazyn M, Kilic A & Javadov S. (2008). The role of NHE-1 in myocardial hypertrophy and remodelling. Journal of molecular and cellular cardiology 44, 647–653. [DOI] [PubMed] [Google Scholar]
- Lagadic-Gossmann D, Buckler KJ & Vaughan-Jones RD. (1992). Role of bicarbonate in pH recovery from intracellular acidosis in the guinea-pig ventricular myocyte. The Journal of physiology 458, 361–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence SP, Bright NA, Luzio JP & Bowers K. (2010). The sodium/proton exchanger NHE8 regulates late endosomal morphology and function. Mol Biol Cell 21, 3540–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazdunski M, Frelin C & Vigne P. (1985). The sodium/hydrogen exchange system in cardiac cells: its biochemical and pharmacological properties and its role in regulating internal concentrations of sodium and internal pH. Journal of molecular and cellular cardiology 17, 1029–1042. [DOI] [PubMed] [Google Scholar]
- McNary TG, Spitzer KW, Holloway H, Bridge JHB, Kohl P & Sachse FB. (2012). Mechanical modulation of the transverse tubular system of ventricular cardiomyocytes. Prog Biophys Mol Biol 110, 218–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani-Nejad N & Janssen PM. (2014). Small and large animal models in cardiac contraction research: advantages and disadvantages. Pharmacol Ther 141, 235–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musset B, Capasso M, Cherny VV, Morgan D, Bhamrah M, Dyer MJS & DeCoursey TE. (2010). Identification of Thr29 as a critical phosphorylation site that activates the human proton channel Hvcn1 in leukocytes. The Journal of biological chemistry 285, 5117–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura TY, Iwata Y, Arai Y, Komamura K & Wakabayashi S. (2008). Activation of Na+/H+ exchanger 1 is sufficient to generate Ca2+ signals that induce cardiac hypertrophy and heart failure. Circ Res 103, 891–899. [DOI] [PubMed] [Google Scholar]
- Orchard CH & Cingolani HE. (1994). Acidosis and arrhythmias in cardiac muscle. Cardiovasc Res 28, 1312–1319. [DOI] [PubMed] [Google Scholar]
- Orchard CH & Kentish JC. (1990). Effects of changes of pH on the contractile function of cardiac muscle. Am J Physiol 258, C967–981. [DOI] [PubMed] [Google Scholar]
- Pascual F & Coleman RA. (2016). Fuel availability and fate in cardiac metabolism: A tale of two substrates. Biochim Biophys Acta 1861, 1425–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak MM, Tran T, Hong L, Joós B, Morris CE & Tombola F. (2016). The Hv1 proton channel responds to mechanical stimuli. The Journal of general physiology 148, 405–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson E & Halle B. (2008). Cell water dynamics on multiple time scales. Proc Natl Acad Sci U S A 105, 6266–6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrecca K, Atanasiu R, Grinstein S, Orlowski J & Shrier A. (1999). Subcellular localization of the Na+/H+ exchanger NHE1 in rat myocardium. Am J Physiol 276, H709–H717. [DOI] [PubMed] [Google Scholar]
- Piper HM, Probst I, Schwartz P, Hütter FJ & Spieckermann PG. (1982). Culturing of calcium stable adult cardiac myocytes. Journal of molecular and cellular cardiology 14, 397–412. [DOI] [PubMed] [Google Scholar]
- Poole-Wilson PA & Cameron IR. (1975). ECS, intracellular pH, and electrolytes of cardiac and skeletal muscle. Am J Physiol 229, 1299–1304. [DOI] [PubMed] [Google Scholar]
- Prasad V, Lorenz JN, Lasko VM, Nieman ML, Al Moamen NJ & Shull GE. (2013a). Loss of the AE3 Cl(−)/HCO(−) 3 exchanger in mice affects rate-dependent inotropy and stress-related AKT signaling in heart. Front Physiol 4, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad V, Lorenz JN, Miller ML, Vairamani K, Nieman ML, Wang Y & Shull GE. (2013b). Loss of NHE1 activity leads to reduced oxidative stress in heart and mitigates high-fat diet-induced myocardial stress. Journal of molecular and cellular cardiology 65, 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey IS, Moran MM, Chong JA & Clapham DE. (2006). A voltage-gated proton-selective channel lacking the pore domain. Nature 440, 1213–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards MA, Simon JN, Ma R, Loonat AA, Crabtree MJ, Paterson DJ, Fahlman RP, Casadei B, Fliegel L & Swietach P. (2019). Nitric oxide modulates cardiomyocyte pH control through a biphasic effect on sodium/hydrogen exchanger-1. Cardiovasc Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosati B, Dong M, Cheng L, Liou SR, Yan Q, Park JY, Shiang E, Sanguinetti M, Wang HS & McKinnon D. (2008). Evolution of ventricular myocyte electrophysiology. Physiol Genomics 35, 262–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose BD. (1994). Clinical Physiology of Acid-Base and Electrolyte Disorders; 4th Edition. Chapter 11, page 301. McGraw-Hill, Inc. [Google Scholar]
- Schilling T, Gratopp A, DeCoursey TE & Eder C. (2002). Voltage-activated proton currents in human lymphocytes. The Journal of physiology 545, 93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder MA, Ali MA, Hulikova A, Supuran CT, Clarke K, Vaughan-Jones RD, Tyler DJ & Swietach P. (2013). Extramitochondrial domain rich in carbonic anhydrase activity improves myocardial energetics. Proc Natl Acad Sci U S A 110, E958–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder MA, Swietach P, Atherton HJ, Gallagher FA, Lee P, Radda GK, Clarke K & Tyler DJ. (2010). Measuring intracellular pH in the heart using hyperpolarized carbon dioxide and bicarbonate: a 13C and 31P magnetic resonance spectroscopy study. Cardiovasc Res 86, 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada-Shimizu N, Hisamitsu T, Nakamura TY, Hirayama N & Wakabayashi S. (2014). Na+/H+ exchanger 1 is regulated via its lipid-interacting domain, which functions as a molecular switch: a pharmacological approach using indolocarbazole compounds. Mol Pharmacol 85, 18–28. [DOI] [PubMed] [Google Scholar]
- Sun X & Wang HS. (2005). Role of the transient outward current (Ito) in shaping canine ventricular action potential--a dynamic clamp study. The Journal of physiology 564, 411–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svichar N, Waheed A, Sly WS, Hennings JC, Hübner CA & Chesler M. (2009). Carbonic anhydrases CA4 and CA14 both enhance AE3-mediated Cl--HCO3- exchange in hippocampal neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 29, 3252–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swietach P, Youm JB, Saegusa N, Leem CH, Spitzer KW & Vaughan-Jones RD. (2013). Coupled Ca2+/H+ transport by cytoplasmic buffers regulates local Ca2+ and H+ ion signaling. Proc Natl Acad Sci U S A 110, E2064–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JA, Buchsbaum RN, Zimniak A & Racker E. (1979). Intracellular pH measurements in Ehrlich ascites tumor cells utilizing spectroscopic probes generated in situ. Biochemistry 18, 2210–2218. [DOI] [PubMed] [Google Scholar]
- Tsushima K, Bugger H, Wende AR, Soto J, Jenson GA, Tor AR, McGlauflin R, Kenny HC, Zhang Y, Souvenir R, Hu XX, Sloan CL, Pereira RO, Lira VA, Spitzer KW, Sharp TL, Shoghi KI, Sparagna GC, Rog-Zielinska EA, Kohl P, Khalimonchuk O, Schaffer JE & Abel ED. (2018). Mitochondrial Reactive Oxygen Species in Lipotoxic Hearts Induce Post-Translational Modifications of AKAP121, DRP1, and OPA1 That Promote Mitochondrial Fission. Circ Res 122, 58–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vairamani K, Prasad V, Wang Y, Huang W, Chen Y, Medvedovic M, Lorenz JN & Shull GE. (2018). NBCe1 Na(+)-HCO3(−) cotransporter ablation causes reduced apoptosis following cardiac ischemia-reperfusion injury in vivo. World J Cardiol 10, 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vairamani K, Wang HS, Medvedovic M, Lorenz JN & Shull GE. (2017). RNA SEQ Analysis Indicates that the AE3 Cl(−)/HCO3(−) Exchanger Contributes to Active Transport-Mediated CO2 Disposal in Heart. Sci Rep 7, 7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas LA & Alvarez BV. (2012). Carbonic anhydrase XIV in the normal and hypertrophic myocardium. Journal of molecular and cellular cardiology 52, 741–752. [DOI] [PubMed] [Google Scholar]
- Wakabayashi S, Hisamitsu T & Nakamura TY. (2013). Regulation of the cardiac Na(+)/H(+) exchanger in health and disease. J Mol Cell Cardiol 61, 68–76. [DOI] [PubMed] [Google Scholar]
- Wang Y, Meyer JW, Ashraf M & Shull GE. (2003). Mice with a null mutation in the NHE1 Na+-H+ exchanger are resistant to cardiac ischemia-reperfusion injury. Circulation research 93, 776–782. [DOI] [PubMed] [Google Scholar]
- Yeves AM, Villa-Abrille MC, Perez NG, Medina AJ, Escudero EM & Ennis IL. (2014). Physiological cardiac hypertrophy: critical role of AKT in the prevention of NHE-1 hyperactivity. Journal of molecular and cellular cardiology 76, 186–195. [DOI] [PubMed] [Google Scholar]
- Yu Y, Fuscoe JC, Zhao C, Guo C, Jia M, Qing T, Bannon DI, Lancashire L, Bao W, Du T, Luo H, Su Z, Jones WD, Moland CL, Branham WS, Qian F, Ning B, Li Y, Hong H, Guo L, Mei N, Shi T, Wang KY, Wolfinger RD, Nikolsky Y, Walker SJ, Duerksen-Hughes P, Mason CE, Tong W, Thierry-Mieg J, Thierry-Mieg D, Shi L & Wang C. (2014). A rat RNA-Seq transcriptomic BodyMap across 11 organs and 4 developmental stages. Nat Commun 5, 3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Bobulescu IA, Goyal S, Aronson PS, Baum MG & Moe OW. (2007). Characterization of Na+/H+ exchanger NHE8 in cultured renal epithelial cells. Am J Physiol Renal Physiol 293, F761–F766. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from HSW upon reasonable request.