

Abstract
The role of glucose-6-phosphate dehydrogenase (G6PD) in human cancer is incompletely understood. In a metabolite screening, we observed that inhibition of H3K9 methylation suppressed aerobic glycolysis and enhances the PPP in human mesothelioma cells. Genome-wide screening identified G6PD as an H3K9me3 target gene whose expression is correlated with increased tumor cell apoptosis. Inhibition of aerobic glycolysis enzyme LDHA and G6PD had no significant effects on tumor cell survival. Ablation of G6PD had no significant effect on human mesothelioma and colon carcinoma xenograft growth in athymic mice. However, activation of G6PD with the G6PD-selective activator AG1 induced tumor cell death. AG1 increased tumor cell ROS production and the resultant extrinsic and intrinsic death pathway, mitochondrial processes, and unfolded protein response in tumor cells. Consistent with increased tumor cell death in vitro, AG1 suppressed human mesothelioma xenograft growth in a dose-dependent manner in vivo. Furthermore, AG1 treatment significantly increased tumor-bearing mouse survival in an intra-peritoneum xenograft athymic mouse model. Therefore, in human mesothelioma and colon carcinoma, G6PD is not essential for tumor growth. G6PD acts as a metabolic checkpoint to control metabolic flux towards the PPP to promote tumor cell apoptosis, and its expression is repressed by its promotor H3K9me3 deposition.
Keywords: G6PD, Pentose phosphate pathway, aerobic glycolysis, ROS, H3K9me3
Introduction
Glucose is catabolized by two parallel metabolic pathways, glycolysis and the pentose phosphate pathway (PPP). Under physiological conditions, glycolysis converts glucose to pyruvate, which enters the tricarboxylic acid cycle to generate adenosine triphosphate and reduced nicotinamide adenine dinucleotide. Under pathological conditions such as cancer, tumor cells possess a prodigious anabolic and energetic requirement for increased proliferation. To meet this requirement, aerobic glycolysis and preferential conversion of pyruvate to lactate to produce ATP and to regenerate nicotinamide adenine dinucleotide increases in a process termed aerobic glycolysis or the Warburg effect [1, 2]. Thus, glucose metabolic flux primarily goes through aerobic glycolysis in tumor cells, and the function of the aerobic glycolysis metabolic pathway in promotion of tumor growth and progression is a well-documented phenomenon [3].
Glucose-6-phosphate dehydrogenase (G6PD) is the rate-limiting enzyme in the PPP. Since the PPP generates ribose-5-phosphate for nucleic acid synthesis [4], G6PD and the PPP have both been shown to be essential for survival of rapidly dividing tumor cells [5–8]. Indeed, it has been observed that tumor cells often express high levels of G6PD and, thus, the level of expression of G6PD is associated with poor prognosis in multiple human cancers [5, 6, 9, 10]. However, emerging experimental data determined that G6PD deficiency has no effects on the growth and progression of certain types of tumors [11]. Furthermore, G6PD-deficient tumor cells still proliferate via the compensatory malic enzyme 1 and isocitrate dehydrogenase 1 flux [12]. In addition, because the PPP is a parallel and competing pathway of glycolysis for glucose metabolic flux [13], and because aerobic glycolysis is the dominant metabolic pathway in tumor cells [2], it makes sense that G6PD-PPP should be suppressed in tumor cells. Therefore, the role of G6PD in human tumor cells may be cellular context-dependent and remains to be determined.
We report here that H3K9me3 represses G6PD expression to enhance aerobic glycolysis in human mesothelioma. We determined that G6PD functions as a tumor suppressor, not a tumor promoter, in human mesothelioma cells. Activation of G6PD increases ROS production to enhance the ROS-regulated cell death pathways to promote mesothelioma apoptosis. A pharmacological strategy that activates G6PD may represent an effective approach to suppress human mesothelioma growth in vivo.
Materials and Methods
Mice.
Female 7-8 week old Nu/J mice were obtained from Jackson Laboratory (Bar Harbor, ME). Use of mice for this study were approved in advance by Augusta University Institutional Animal Care and Use Committee (Protocol #: 20080162).
Cell lines.
The human epithelioid mesothelioma NCI-H226, biphasic MSTO-211H cell line and human colon carcinoma SW620 cell lines were obtained from American Type Culture Collection (ATCC, Manassas, VA). ATCC characterized these cell lines by morphology, immunology, DNA fingerprint, and cytogenetics. Cell lines were tested bimonthly for mycoplasma contamination and were mycoplasma-free at time of experiments.
Reagents.
Verticillin A (purity >98%) was isolated and characterized from the fungus, Clonostachys rogersoniana (strain MSX59553), as previously described [14, 15]. AG1 was synthesized in LeadGen Labs (Orange, CT) as previously described [16, 17]. G6PDi-1 was also synthesized in LeadGen Labs based on an established procedure [18]. The purity of the synthesized AG1 and G6PDi-1 were both >99% as determined by LC-MS analysis (Figs. S1 and S2).
Chromatin Immunoprecipitation (ChIP).
ChIP was performed as previously described [19]. Anti-H3K9me3 antibody (Cat# 8898, Abcam, Cambridge, MA) and the Chromatin Immunoprecipitation (ChIP) Assay Kit (Cell Signaling, Danvers, MA) were used in ChIP procesures according to the manufacturer’s instructions. The primer sequences are listed in Table S1.
Metabolite Assay.
Tumor cells were treated with verticillin A at 10 nM for 24 hours, washed once with PBS, snap frozen in liquid nitrogen, and stored at −80 °C until analysis. Approximately 1x107 cells/sample were analyzed for each group. Metabolite extraction and metabolomic analysis were carried out in the National Institutes of Health West Coast Metabolomics Center at the University of California Davis (Davis, CA). Data were acquired using ALEX-CIS GCTOF mass spectrometry with a Restek corporation Rtx-5Sil MS column and processed using ChromaTOF software version 2.32 (Leco, St. Joseph, MI) and the BinBase algorithm, as described previously [20, 21]
Western Blotting.
Tumor cells cells were cultured in the presence of various concentrations of verticillin A for 24 hours. Cells were then lysed in total protein lysis buffer [20 mM Hepes, pH 7.4, 20 mM NaCl, 10% Glycerol, 1% Triton X100, proteinase inhibitor cocktails (Cat# 539131, EMD Millipore Corp, Billerca, MA) and phosphotase inhibitor cocktail (Cat# 524628, EMD Millipore Corp)], blotted, and probed with anti-caspase 8, anti-cleaved caspase 9, anti-cleaved caspase 3, anti-cleaved PARP, anti-LDHA, anti-G6PD, and anti-β-actin antibodies (all antibody information is in listed in Table S1).
Analysis of histone methylation.
Tumor cells were centrifuged at 2,000 RPM for 5 min at 4 °C in a microcentifuge. The cell pellets were resuspend in 100 μl of NETN buffer (20 mM Tris-HCl, pH8.0, 150 mM NaCl, 5 mM EDTA, 0.5% Nonidet P-40, 1.5mM phenylmethylsulfonyl fluoride, 1.7 μg/ml aprotinin, 1 mM Na3VO4, and 0.5 mM NaF), incubated on ice for 30 min, and then centrifuged for 5 min at 10,000 RPM. The supernatants were discarded. The detergent-insoluble pellets were resuspend in 90 μl 0.1 N HCl, incubated on ice for 30 min, and then centrifuged for 10 min at 13,000 RPM in a microcentrifuge. The supernatant histone preparations were transferred to a new 1.5 ml microtube and 10 μl 1 M Tris-HCl, pH 9.0 was added to each tube. The microtubes were vortexed and centrifuged briefly. The histone concentration was determined by measuring protein concentration. The purified histones were analyzed by Western blotting using anti-H3K9me3 antibody. The membranes were stripped and reprobed with anti-H3 antibody. Antibody information is listed in Table S1.
RNA Sequencing.
NCI-H226 and MSTO-211H cells were treated with verticillin A at 10 nM for 24 hours and used for RNA isolation. RNA-Seq was performed at Novogene Corp. Reference genome (mm10) and gene model annotation files were downloaded directly from Ensembl. Indices were built and paired-end clean reads were aligned to the reference genome using STARv2.5. Reads were quantified using HTSeqv0.6.1. Rank-log transformed normalized counts from DESeq2 were used as inputs for GSEA, GO and PCA analysis. GSEA was performed using GSEA4.0 (Broad) with gene-set permutation. GO pathway enrichment was performed with clusterprofiler.
OCR and ECAR Measurement.
OCR and ECAR were measured with an XF96 extracellular flux analyzer (Seahorse Bioscience) following protocols recommended by the manufacturer. Briefly, freshly isolated NCI-H226 cells were seeded on XF96 microplates (150,000 cells/well) that had been pre-coated with Cell-Tak adhesive (BD Biosciences) in the absence or presence of verticillin A (5 nM). The plates were centrifuged to immobilize cells. Cells were maintained in a non-buffered assay medium (Seahorse Biosciences) in a non-CO2 incubator for 30 min before assay. Glycolysis was measured with the XF glycolysis stress test kit (Seahorse Biosciences). Initially, cells were incubated in the glycolysis stress test medium without glucose, and ECAR was assessed. Three baseline recordings were made, followed by sequential injection of 10 mM glucose and 1 μM oligomycin that inhibited mitochondrial ATP production and shifted the energy production to glycolysis. The final injection was 100 mM 2-deoxy glucose. The Mito stress test kit (Seahorse Biosciences) was used to test OCR under different conditions. Three baseline recordings were made, followed by sequential injection of 1 μM oligomycin and 0.25 μM FCCP that uncoupled oxygen consumption from ATP production to obtain maximal OCR.
Lactate measurement.
Tumor cells were seeded at 1x105 cells/ml in 24-well plate and treated with 10 nM verticillin A for 24 hours. For intracellular lactate measurement, cells were collected and lysed in total protein lysis buffer. For extracellular lactate measurement, culture supernatants were collected. The flow through fractions were analyzed for lactate concentrations using the Lactate Assay Kit II (Cat# MAK065, Sigma-Aldrich, St Louis, MO) according to the manufacturer’s instructions.
Flow cytometry.
Tumor cells were resuspended in Annexin V-binding buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2, pH 7.4), stained with Annexin V (Biolegend, San Diego, CA) and/or propidium iodide (PI). Stained cells were analyzed in a FACS Calibur flow cytometer (BD Biosciences, San Diego, CA). All flow cytometry data analysis was conducted with the FlowJo program (BD Biosciences).
G6PD knockout tumor cell line generation.
CRISPR was used to knockout G6PD in tumor cells as previously described [22]. HEK293FT cells were co-transfected with pCMV-VSV-G (Addgene #8454), psPAX2 (Addgene #12260) and lentiCRISPRv2 (Genscript, Piscataway, NJ) plasmids containing scramble sgRNA (GGAAGACTTAGTCGAATGAT), G6PD-specific sgRNA1 (GATCCGCGTGCAGCCCAACG), and G6PD-specific sgRNA 2 (ACGGGCATAGCCCACGATGA) using Lipofectamine 2000 (Life Technologies) to produce CRISPR lentivirus. MSTO-211H, NCI-H226 and SW620 cells were transduced with the lentivirus particle. Cells were selected with puromycin to generate stable cell lines.
Cell death pathway analysis.
Tumor cells were cultured in the presence of verticillin A (25 nM) plus Z-VAD (10 μM), necropstatin-1 (10 μM), and ferrostatin-1 (2 μM), either alone or in combinations, for 24 hours. Cell death was determined by PI staining or combined Annexin V and PI staining, followed by flow cytometry analysis.
Total intracellular ROS measurement.
Tumor cells were harvested, washed in PBS, and then stained for ROS using the ROS staining kit (Cat # 88-5930, Life Technologies, Carlsbad, CA) according to the manusfacuturer’s instructions. The stained cells were analyzed by flow cytometry and ROS was quantified using the FlowJo program.
Xenograft mouse models and treatments.
To establish subcutaneous tumors, NCI-H226 cells (4x106/mouse) were injected subcutaneously to the right flank of athymic nude mice. Sample size was based on a previous study [23] and was not estimated by power analysis. Individual mice were not randomized due to concern of fight between mice from different cages. The mouse cages were randomized before treatments. Tumor-bearing mice were treated with AG1 i.p. from day 7 daily for 12 times. To establish intra-peritoneum tumors, NCI-H226 cells (4x106/mouse) were injected intraperitoneally to athymic nude mice. The mice-containing cages were randomized. Tumor-bearing mice were treated with AG1 at day 7 every 2 days for 7 times. Mouse body weights were recorded. Tumor volume was measured and calculated by the formula [(major axis x (minor axis)2)/2].
Immunohistochemistry.
Tumor tissue was fixed in 10% formalin overnight. The fixed tissues were processed into paraffin blocks. The sections were stained with anti-Ki67 antibody (Cat#NB500-170, Novus, Centennial, CO) and in situ TUNEL, respectively, as previously described [24]. De-identified human malignant mesothelioma samples were obtained from the Georgia Cancer Center Biorepository. The tissue was stained with anti-G6PD antibody (Abcam ab210702) using a previously described method [22].
Statistical analysis.
Unless otherwise indicated, statistical analyses were conducted using Prism8 (Graphpad). Data are presented as mean ± SD, and p values were calculated by a two-tailed Student’s t-test. Survival was analyzed using Kaplan-Meier survival plots with the log rank p-value.
Results
H3K9 methylation regulates human mesothelioma metabolism
Tumor cell metabolism is regulated by epigenetic mechanisms [25]. Specifically, H3K9 methylation has been shown to regulate metabolic pathways and in turn to regulate tumor cell survival [21]. To determine the epigenetic regulation of the global metabolism profile in human mesothelioma cells, we made use of the H3K9 methylation-selective small molecule inhibitor, verticillin A. Verticillin A is a natural product isolated from fungi that selectively inhibits 6 of the 23 known human histone methyltransferases (i.e., SUV39H1, SUV39H2, G9a, GLP, MLL1, and NSD2) [23]. We treated the human mesothelioma cell line NCI-H226 with verticillin A and performed metabolite profiling. Verticillin A increased the PPP metabolites, amino acids, and TCA cycle metabolites in NCI-H226 cells. Lactate, a hallmark of aerobic glycolysis, was decreased by verticillin A treatment in NCI-H226 cells (Fig. 1A). To validate this finding, we treated MSTO-211H and NCI-H226 cells with verticillin A and measured intracellular lactate content using a lactate quantification kit. Verticillin A treatment significantly decreased intracellular lactate levels in both cell lines (Fig. 1B). We also collected tumor cell culture supernatants to determine extracellular lactate levels. Verticillin A treatment also significantly decreased the extracellular lactate levels in both MSTO-211H and NCI-H226 cells (Fig. 1B). However, intracellular glutamine levels increased more than 8-fold in the verticillin A-treated NCI-H226 cells (Fig. 1A), suggesting that verticillin A might suppress glutamine conversion to lactate, thereby inducing its accumulation [26]. In addition, verticillin A also increased the major PPP metabolite, ribulose-5-phosphate, levels in the tumor cells (Fig. 1A). Citric acid is increased in verticillin A-treated cells (Fig. 1A). These observations indicate that inhibition of H3K9 methylation suppresses aerobic glycolysis and increases the PPP and TCA cycle activity.
Figure 1. Epigenetic regulation of metabolism in human mesothelioma cells.

A. NCI-H226 tumor cells were treated with verticillin A (10 nM) for 24h and analyzed for metabolites. Shown are ratios of the indicated metabolites of treated cells/untreated cells. B. The indicated tumor cells were treated with verticillin A (10 nM) for 24h. Cells were harvested and lysed to make total lysates and then measured for intracellular lactate content (left panel). Right panel: Tumor cells were treated with verticillin A (10 nM) for 24h. The culture supernatants were collected and measured for lactate content. Shown are results of one of two experiments. Column: Mean, Bar: SD. C& D. NCI-H226 cells were treated with verticillin A (5 nM). Extracellular acidification rate (ECAR, C) and oxygen consumption rate (OCR, D) were measured with a glycolysis stress test kit (C) or a Mito stress test kit (D). Stressed ECAR and OCR were calculated and shown on the right panel. Shown are results of one of two experiments. Column: Mean, Bar: SD.
To strengthen these finding, extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) measurements were performed to determine the mesothelioma cellular metabolic status. To minimize the cytotoxic effect of verticillin A, a low concentration of verticillin A (i.e., 5 nM) was used. Verticillin A significantly decreased ECAR (Fig. 1C) and increased OCR (Fig. 1D) in human mesothelioma cells. Our data thus suggest that H3K9 methylation promotes aerobic glycolysis and suppresses the PPP and mitochondrial oxidative phosphorylation.
G6PD expression is repressed by its promoter H3K9me3 deposition
Our above findings indicate that H3K9 methylation regulates glucose metabolic flux partitioning between aerobic glycolysis and the PPP. It is known that H3K9 methylation represses gene expression [27]. We then sought to identify H3K9 methylation target genes in human mesothelioma cells by using genome-wide RNA-Seq analysis to screen differentially expressed genes. Verticillin A altered the expression of a broad range of genes in NCI-H226 cells, and a large subset of these genes are involved in metabolic processes (Fig. 2A). G6PD is one of the identified metabolic genes whose expression is up-regulated by verticillin A (Fig. 2B). This is an unexpected observation since G6PD is generally considered to be essential for tumor cell survival and proliferation [6, 8, 10], but verticillin A induced apoptosis and increased G6PD expression in mesothelioma cells. We therefore focused our further studies on G6PD function in mesothelioma cells. Western blotting analysis determined that G6PD protein levels were also increased by verticillin A treatment in human mesothelioma cells (Fig. 2C). Analysis of a human malignant mesothelioma specimen indicates that G6PD is expressed in all tumor cells (Fig. 2D). It was reported that G6PD is active in lymphocytes [18] and plays a critical role in T cell anti-tumor function [28]. As expected, G6PD protein is detected in tumor-infiltrating lymphocytes (Fig. 2D). In contrast, LDHA, the rate-limiting enzyme that converts pyruvate to lactate, was not changed by verticillin A (Fig. 2C). Among the six verticillin A target HMTases, four (i.e., SUV39H1, SUV39H2, G9a, and GLP) catalyze H3K9 methylation with SUV39H1 and SUV39H2 functioning as H3K9me3-selective HMTases [29–31]. We therefore analyzed H3K9me3 levels in verticillin A-treated mesothelioma cells. Consistent with previous finding that verticillin A acts as a H3K9me3-selective inhibitor, verticillin A inhibited H3K9me3 in a dose-dependent manner (Fig. 2E). ChIP analysis revealed that H3K9me3 is enriched in the G6PD promoter region, and verticillin A treatment decreases H3K9me3 deposition at the G6PD promoter in human mesothelioma cells (Fig. 2F). We therefore conclude that G6PD is repressed by its promoter H3K9me3 deposition in human mesothelioma cells.
Figure 2. G6PD is repressed by its promoter H3K9me3 deposition in human mesothelioma cells.

A. RNA-Seq analysis of human mesothelioma cell line NCI-H226 treated with verticillin A. The differentially expressed genes were functionally grouped using the PANTHER program. B. The differentially expressed genes were clustered and presented. C. Western blotting analysis of LDHA and G6PD in human tumor cells treated with verticillin A for 24h. D. A malignant human mesothelioma tissue was stained with G6PD-specific antibody by immunohistochemical method. The brown color indicates G6PD protein staining, and the blue color is nucleus staining. The right panel shows an amplified area from the left panel. The red arrow points to lymphocytes. E. The indicated tumor cells were treated with verticillin A at the indicated concentrations for 24h. Histones were purified from the treated cells and analyzed by Western blotting for H3K9me3 level using anti-H3K9me3 antibody. H3 was used as normalization control. F. Top panel: The human G6PD promoter structure showing ChIP sites. Bottom panel: NCI-H226 cells were treated with verticillin A for 24h and analyzed by ChIP using H3K9me3-specific antibody. Column: Mean, Bar: SD.
Epigenetic regulation of mesothelioma cell survival.
Histone methylation is known to play a key role in regulation of gene expression in tumor cells [21, 32, 33], and H3K9me3 is known to repress expression of genes [34], including those with known functions in apoptosis in tumor cells [23, 35, 36]. Another major group of genes whose expression was changed by verticillin A are those with known functions in cell proliferation (Fig. 2A). As expected, verticillin A induced human mesothelioma cell death in a dose-dependent manner (Fig. 3A). To determine the mechanism of action of these HMTase inhibitors in human mesothelioma cell death, we used cell death pathway inhibitors to determine which cell death pathways are regulated by HMTases. Verticillin A induced mesothelioma cell death through apoptosis, since the caspase inhibitor Z-VAD blocked verticillin A-induced cell death (Fig. 3B). Consistent with this observation, verticillin A induced activation of caspases 3, 8 and 9, and PARP cleavage (Fig. 3C). Verticillin A effectively induced an apoptotic cell death morphology in both NCI-H226 and MSTO-211H cells (Fig. S3). These findings indicate that human mesothelioma survival is regulated by H3K9 methylation.
Figure 3. Inhibition of histone methylation induces human mesothelioma cell apoptosis.

A. Tumor cells were treated by verticillin A at indicated concentrations for 24h, analyzed by flow cytometry for Annexin V and PI. Percentage of Annexin V+ and Annexin V+PI+ cells were quantified and presented. B. MSTO-211H and NCI-H226 were treated by verticillin A and cell death pathway inhibitors, analyzed by flow cytometry for Annexin V and PI staining. The percentage of Annexin V+PI+ cells were quantified. Column: Mean, Bar: SD. C. MSTO-211H and NCI-H226 were treated by verticillin A at indicated doses for 24h and analyzed by Western Blotting for apoptosis-associated proteins.
Inhibition of LDHA and G6PD does not affect human mesothelioma survival
To determine whether aerobic glycolysis maintains human mesothelioma cell survival, tumor cells were treated with GSK2837808A, a specific inhibition of LDHA, the key enzyme that converts pyruvate to lactate, and analyzed for cell death. Inhibition of LDHA did not cause significant cell death in vitro (Fig. 4A). Next, the tumor cells were treated with the G6PD-selective inhibitor G6PDi-1 [18]. Inhibition of G6PD also does not cause significant tumor cell death (Fig. 4B). These observations indicate that aerobic glycolysis and G6PD are not essential for human mesothelioma cell survival.
Figure 4. Aerobic glycolysis and G6PD do not maintain human mesothelioma cell survival in vitro.

A. Tumor cells were cultured in the presence of the indicated concentrations of GSK2837808A for 24h. Cells were stained with PI and analyzed by flow cytometry. % cell death is calculated as % PI+ cells and presented at the bottom panels. Shown are results of one of two experiments. Cycle: Mean, Bar: SD. B. Tumor cells were cultured in the presence of the indicated concentrations of G6PDi-1 for 24h. Cells were stained with PI and analyzed by flow cytometry. % cell death is calculated as % PI+ cells and presented at the right panels. Shown are results of one of two experiments. Cycle: Mean, Bar: SD.
Activation of G6PD promotes tumor cell apoptosis in vitro
Our above findings indicate that the G6PD-PPP metabolic flux promotes tumor cell apoptosis, and tumor cells use H3K9me3 deposition at the G6PD promoter to down-regulate G6PD expression to decrease tumor cell apoptosis. To further strengthen this notion, we took a complementary approach and made use of the recently developed G6PD enzymatic activity activator AG1 [17]. The rationale is that if down-regulating G6PD protects tumor cells from apoptosis, pharmacological activation of G6PD with AG1 should induce tumor cell apoptosis. Although H3K9me3 repressed G6PD expression in the tumor cells, G6PD protein is detectable in the tumor cells (Fig. 2C). Therefore, increasing its enzymatic activity at the protein level should enhance the PPP. To test this hypothesis, we treated tumor cells with AG1 and analyzed tumor cell death. Indeed, AG1 treatment induced human mesothelioma cell death in a dose-dependent manner (Fig. 5A). AG1 treatment induced cell death with shrinkage of the cell and the nucleus (Fig. S4), which are morphology changes typical of apoptotic cells.
Figure 5. G6PD increases ROS promotion to regulate ROS-regulated cell death pathway to promote human mesothelioma cell death in vitro.

A. Tumor cells were cultured in the presence of the indicated concentrations of AG1 for 24h. Cells were stained with PI and analyzed by flow cytometry. % cell death is calculated as % PI+ cells and presented at the right panels. Shown are results of one of two experiments. Cycle: Mean, Bar: SD. B. NCI-H226 cells were treated with AG1 at 10 μM for 24h and analyzed by RNA-Seq. Differentially expressed genes induced by AG1 in the intrinsic and extrinsic apoptosis, mitochondria, and unfolded protein response pathways are shown. C. Tumor cells were labeled by ROS assay stain, treated with AG1 at 10μM for 24h, and analyzed by flow cytometry. The MFI of ROS were quantified and presented at the bottom panel. Column: Mean, Bar: SD.
To elucidate the molecular mechanism underlying G6PD activation-induced tumor cell death, we treated tumor cells with AG1 and performed genome-wide gene expression analysis by RNA-Seq. Pathway analysis of the differentially expressed genes revealed that AG1 treatment altered the expression of gene sets in three pathways: the extrinsic and intrinsic cell death pathway, mitochondria process, and unfolded protein response (Fig. 5B). Several genes with increased expression are TNF family genes, including TNFRSF9, TNFRSF21, TNFRSF10A/b, with known functions in apoptosis [37]. In addition, pro-apoptotic genes BID and BAXBNIP3L [38] were also increased, whereas the anti-apoptotic gene BRCA1 is decreased. Furthermore, ATF4 is known to regulate glucose metabolism-dependent cell apoptosis [39], and its expression is increased (Fig. 5B). These three cellular pathways are known to bridge ROS to apoptosis [40]. ROS is a key product of the mitochondrial oxidative phosphorylation process [41], and activation of the PPP has been shown to increase ROS production [42–44]. Thus, we hypothesized that AG1 increases ROS production to induce tumor cell apoptosis. To test this hypothesis, we treated tumor cells with AG1 and then measured intracellular ROS production levels. Indeed, AG1 significantly increased ROS production in human mesothelioma cells (Fig 5C). Taken together, our data indicate that G6PD increases ROS production to activate ROS-regulated cell death pathways to induce human mesothelioma cell apoptosis in vitro.
G6PD is not essential for human mesothelioma and colon carcinoma survival in vivo.
To determine whether G6PD is essential for tumor cell survival, we ablated G6PD in human mesothelioma and colon carcinoma cell lines. The CRISPR technique almost completely knocked out G6PD in both MSTO-211H and NCI-H226 cells (Fig. 6A) and, to a lesser degree, in SW620 cells (Fig. S5A). Ablating G6PD has no significant effect on MSTO-211H and NCI-H226 cell viability in vitro (Fig. 6B). Furthermore, ablating G6PD did not affect MSTO-211H and NCI-H226 xenografts grown in athymic nude mice (Fig. 6C). Ablating G6PD also did not significantly affect human colon carcinoma SW620 xenografts (Fig. S5B). To validate that AG1 induces mesothelioma cell apoptosis through G6PD, the Scramble and the G6PD KO cells were treated with AG1 and analyzed for apoptosis. Indeed, ablating G6PD dramatically decreased MSTO-211H and NCI-H226 cell sensitivity to AG1-induced apoptosis (Fig. 6D).
Figure 6. G6PD is not essential for human mesothelioma survival in vitro and in vivo.

A. CRISPR technique was used to knockout G6PD in NCI-H226 and MSTO-211H cells. Cells were selected with puromycin to generate stable cell lines and analyzed by Western blotting for G6PD. KO1 and KO2 are the cell lines transduced with the G6PD sgRNA1 and G6PD sgRNA2, respectively, as described in the method section. B. Scramble and G6PD-knockout human mesothelioma cells were cultured for 24h and analyzed by MTS for proliferation. C. Scramble and G6PD-knockout human mesothelioma cells were injected into athymic mice to establish xenografts. The tumor volume kinetics were calculated and are presented at the bottom panel. Cycle: Mean, Bar: SD. D. Scramble and G6PD-knockout human mesothelioma cells were treated by AG1 and analyzed by flow cytometry for Annexin V and PI staining. The percentage of Annexin V+PI+ cells are quantified and presented at the right. Shown are results of one of two experiments. Column: Mean, Bar: SD.
G6PD functions as a tumor suppressor in vivo.
To determine whether the above finding that G6PD activation induces apoptosis in vitro can be translated to tumor growth suppression in vivo, human mesothelioma cells were injected into athymic nude mice to establish xenografts. The tumor-bearing mice were then treated with AG1, which exhibited a dose-dependent efficacy in suppression of human mesothelioma xenograft growth (Fig. 7A). Consistent with such growth inhibition, AG1 significantly decreased tumor cell proliferation as determined by Ki67 intensity of tumor cells (Fig. 7B) and increased tumor cell apoptosis (Fig. 7C) in the tumor xenografts in vivo. To validate the tumor suppression function of G6PD, NCI-H226 cells were injected intraperitoneally in athymic mice. The intraperitoneal tumor grew aggressively, and eight of the ten tumor-bearing mice in the solvent control group had enlarged abdomens, likely due to ascites, whereas none of the AG1-treated mice developed enlarged abdomens. Median time to death in the solvent control group is 21.5 days (95% CI=20d-22d). The AG1-treated mice had significantly better survival than the control (p=0.0122). The median time to death in AG1-treated group was 23 days (95% CI:20d-25d). However, AG1 treatment caused approximately a 20% body weight loss at the efficacious dose (Fig. 7E). Taken together, our data indicate that pharmacological activation of G6PD with AG1 is an effective approach to suppress human mesothelioma growth in vivo.
Figure 7. Pharmacological activation of G6PD enzymatic activity suppresses human mesothelioma growth in vivo.
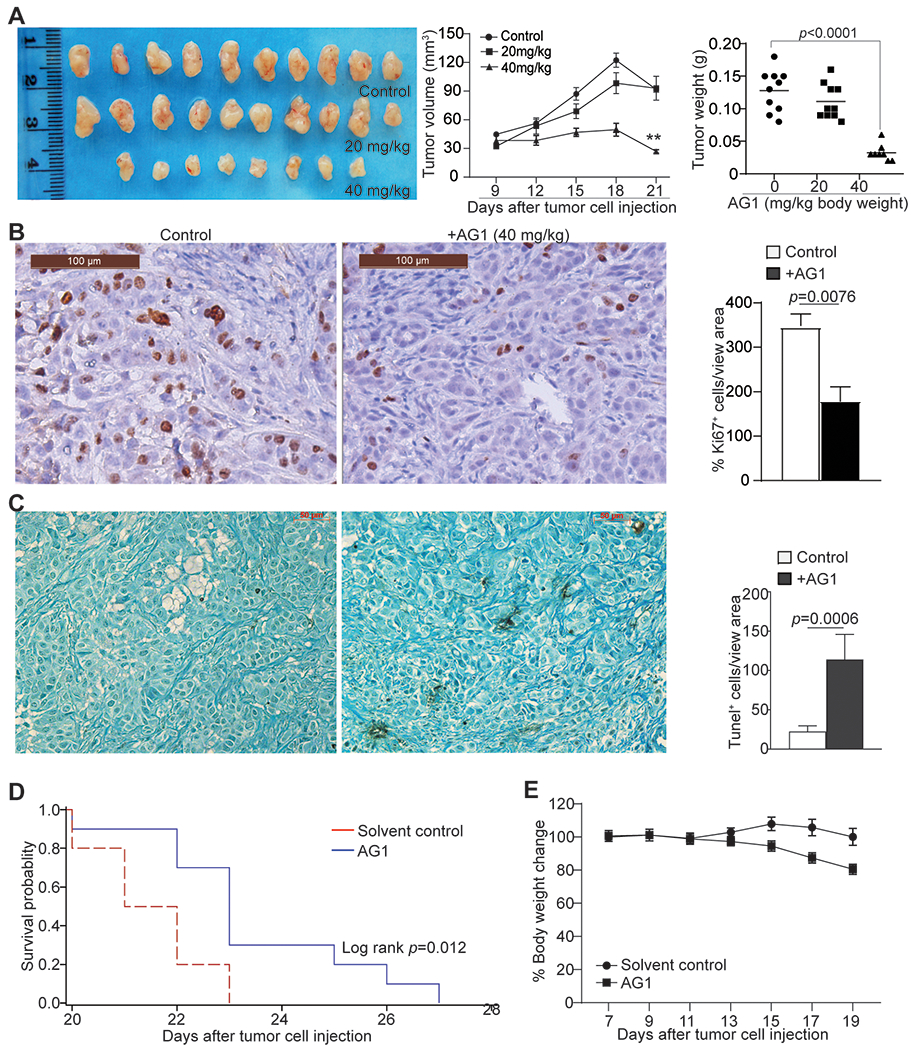
A. NCI-H226 cells were injected to athymic mice to establish xenografts. The tumor-bearing mice were treated with solvent (n=10), AG1 at 20 mg/kg body weight (n=10), and 40 mg/kg body weight (n=8) daily. The tumor growth kinetics and tumor weight at the end of the experiment were calculated and presented at the right. B. The tumor tissues as shown in A were analyzed by immunohistochemical staining with Ki67-specific antibody. The Ki-67 positive cells/view area were quantified and presented at the right. Column: Mean, Bar: SD. C. The tumor tissues as shown in A were analyzed by in situ TUNEL. The TUNEL positive cells/view area were quantified and presented at the right. Column: Mean, Bar: SD. D & E. NCI-H226 cells were injected intraperitoneally to athymic nude mice. Mouse cages were randomized. Tumor-bearing mice were then treated with solvent (n=10) or AG1 (40 mg/kg body weight, n=10) at day 7 every 2 days for 7 times. Survival was recorded. Shown is Kaplan-Meier survival plot with the log rank p-value (D). The tumor-bearing mouse body weight were recorded and presented in E. Cycle/square symbol: Mean, Bar: SD.
Discussion
Aerobic glycolysis is a hallmark of human cancer [3], and lactate dehydrogenase A (LDHA) is the key enzyme in aerobic glycolysis that converts pyruvate to lactate [45]. Consistent with this phenomenon, inhibition of LDHA redirects pyruvate to mitochondrial oxidative phosphorylation, leading to increased apoptosis and suppression of tumor growth and progression [45–47] and this is likely due, at least in part, to increased ROS production and the resultant ROS-induced apoptosis [48, 49]. However, the pyruvate-lactate metabolic pathway in tumor cell survival is apparently tumor type and cellular context-dependent, since LDHA inhibition alone does not impact human melanoma cell survival in vitro and tumor growth in vivo [2]. Similarly, we observed that inhibition of LDHA does not affect human mesothelioma cell survival. Strikingly, we observed that G6PD expression is down-regulated by its promoter H3K9me3 deposition in human mesothelioma, and activation of G6PD significantly increased human mesothelioma cell death in vitro. Consistent with G6PD-mediated tumor cell death in vitro, pharmacological activation of G6PD suppressed human mesothelioma xenograft growth in a dose-dependent manner in vivo, resulting in improved survival of tumor-bearing mice. Taken together, our data suggest that the GD-PPP pathway, and not the pyruvate-lactate pathway, plays an essential role in human mesothelioma survival.
G6PD and the PPP are considered essential for promoting tumor cell proliferation and survival [8], but we observed that activation of G6PD increased ROS production to activate ROS-regulated cell death pathways, thereby inducing human mesothelioma cell death. It is generally believed that tumor cells increase reduction of NADP+ to NADPH via activating G6PD and thereby PPP to accommodate excess ROS to maintain survival [50]. NADPH also inhibits G6PD through a feedback inhibition mechanism to direct glucose metabolic flux to glycolysis [51]. AG1 is an activator of G6PD and may constantly activate G6PD and, therefore, it overcomes NADPH-mediated negative regulation of G6PD and the PPP to increase NADPH. It is therefore expected that increased NADPH would detoxify ROS to suppress ROS-induced cell death. However, this is not the case in human mesothelioma cells in vitro. A recent study has shown that NADPH also plays a metabolism-independent role and directly binds to HDAC3 to regulate histone acetylation in tumor cells [52]. It has also been shown that lactate inhibits histone deacetylase SIRIT1 expression in renal cell carcinoma cells to increase global H3 and H3K9 acetylation to promote tumor cell aggressiveness [53]. Inhibition of the lactate transporter reverses SIRIT1 inhibition and tumor promotion [53]. It is thus possible that verticillin A may repress lactate transporter expression to decrease lactate secretion, and that the NADPH-HDACs/sirtuins pathway might mediate crosstalk between lactate/aerobic glycolysis and the PPP to control tumor cell survival and proliferation; this remains to be elucidated. Emerging experimental data indicate that G6PD may not be essential for the survival and progression of certain tumors, as previously reported [11, 12]. In this study, we determined that G6PD is not essential for mesothelioma and colon carcinoma survival in vivo. It is therefore possible that NADPH may not play an essential role in anti-oxidative stress, as previously postulated, due to its metabolism-independent role. It is also possible that dysregulation of the NADPH pathway by AG1-mediated constitutive G6PD activation may diminish NADPH function in blocking ROS function in induction of apoptosis in human mesothelioma cells, and this also requires further study.
Epigenetic alteration of tumor-regulatory gene expression is a known mechanism of tumor growth control [54]. The histone methyltransferase inhibitor, verticillin A, has G9A inhibitory activity [23]. G9A is known to regulate PRC2-mediated gene silencing [55], and inactivation of G9A depletes serine and its downstream metabolites, triggering cell death with autophagy in cancer cells [21]. The G9A-PRC2 complex inhibition may thus also contribute to verticillin A-induced mesothelioma cell death. Verticillin A also inhibits SUV39H1 and SUV39H2 enzymatic activity to inhibit H3K9me3 [23]. H3K9me3 likely regulates genome-wide gene expression involved in various cellular pathways in tumor cells. It is known that H3K9me3 regulates apoptosis, cell cycle, and oxidative stress response pathways in colon and ovarian tumor cells [23, 36, 56, 57]. In this study, we determined H3K9me3 represses G6PD expression in human mesothelioma cells to increase mesothelioma cell survival. Our findings thus extend the H3K9me3 function to metabolic regulation of mesothelioma tumor cell survival.
Emerging experimental data indicate that epigenetic reprogramming of metabolism, including both histone methylation and sirtuin-mediated aerobic glycolysis, plays a critical role in tumor growth and progression [53, 58–60]. It is well known that mesothelioma pathogenesis is regulated by the EZH2-H3K27me3 epigenetic axis [61, 62]. Although targeting the EZH2-H3K27me3 epigenetic pathway results in variable outcomes in human clinical trials [61, 63], these epigenetic targets may be targeted to boost the efficacy of immunotherapy in mesothelioma. Specifically, the H3K9me3-G6PD axis may be a target in mesothelioma immunotherapy. Immune check point inhibitor-based immunotherapy has shown potent efficacy in certain mesothelioma patients [64, 65]. Immune checkpoint inhibitors suppress tumors through activating tumor-reactive T cells to induce tumor cell apoptosis. If the target tumor cells are not sensitive to apoptosis, then immune checkpoint inhibitor immunotherapy and T cell-based immunotherapy are not going to be effective [66]. Targeting the H3K9me3-G6PD epigenetic axis or G6PD activity with a H3K9me3 inhibitor or G6PD activator such as AG1 thus has the potential to sensitize human mesothelioma to apoptosis induction to enhance the efficacy of immune checkpoint inhibitor immunotherapy in suppression of human mesothelioma.
A limitation of this study is that only mesothelioma and colon carcinoma cell lines were used for analysis of G6PD function. The function of G6PD in other types of human cancer remains to be determined. G6PD is a target of verticillin A, but verticillin A likely also regulates other genes that play a role in the tumor cell apoptosis. Furthermore, the off-target effect and toxicity of AG1 as a potential anti-cancer agent remain to be determined.
Supplementary Material
Acknowledgements
We thank Dr. Roni Bollag at the Georgia Cancer Center Biorepository for providing the human mesothelioma specimen and for pathological analysis. We also than Dr. Natasha Savage for tumor specimen analysis and Dr. Kimya Jones for immunohistochemical analysis of tumor specimen. This work was supported by the National Cancer Institute grants R01 CA227433 (to M.W.G, Y.L.C., N.H.O., K.L.), R01CA133085 (to K.L), P01 CA125066 (to N.H.O), R01CA190429 and R01CA236890 (to H.-F.D.), F30CA236436 (to J.D.K), and the US Department of Veterans Affairs Award CX001364 (to K. L).
Footnotes
Conflict of interests
AHC, MWG and NHO have ownership interest in Ionic Pharmaceuticals, LLC. KL has ownership interest in CheMedImmune Inc. NHO is a member of the Scientific Advisory Board of Mycosynthetix, Inc. CJP has an ownership interest in Mycosynthetix, Inc. Other authors have declared that no conflict of interest exists
References
- 1.Warburg O On the origin of cancer cells. Science 1956; 123: 309–314. [DOI] [PubMed] [Google Scholar]
- 2.Pathria G, Scott DA, Feng Y, Sang Lee J, Fujita Y, Zhang G et al. Targeting the Warburg effect via LDHA inhibition engages ATF4 signaling for cancer cell survival. EMBO J 2018; 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324: 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ge T, Yang J, Zhou S, Wang Y, Li Y, Tong X. The Role of the Pentose Phosphate Pathway in Diabetes and Cancer. Front Endocrinol (Lausanne) 2020; 11: 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu S, Wang H, Li Y, Xie Y, Huang C, Zhao H et al. Transcription Factor YY1 Promotes Cell Proliferation by Directly Activating the Pentose Phosphate Pathway. Cancer Res 2018; 78: 4549–4562. [DOI] [PubMed] [Google Scholar]
- 6.Zhang X, Zhang X, Li Y, Shao Y, Xiao J, Zhu G et al. PAK4 regulates G6PD activity by p53 degradation involving colon cancer cell growth. Cell Death Dis 2017; 8: e2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakamura M, Nagase K, Yoshimitsu M, Magara T, Nojiri Y, Kato H et al. Glucose-6-phosphate dehydrogenase correlates with tumor immune activity and programmed death ligand-1 expression in Merkel cell carcinoma. J Immunother Cancer 2020; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding H, Chen Z, Wu K, Huang SM, Wu WL, LeBoeuf SE et al. Activation of the NRF2 antioxidant program sensitizes tumors to G6PD inhibition. Sci Adv 2021; 7: eabk1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kowalik MA, Columbano A, Perra A. Emerging Role of the Pentose Phosphate Pathway in Hepatocellular Carcinoma. Front Oncol 2017; 7: 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagashio R, Oikawa S, Yanagita K, Hagiuda D, Kuchitsu Y, Igawa S et al. Prognostic significance of G6PD expression and localization in lung adenocarcinoma. Biochim Biophys Acta Proteins Proteom 2019; 1867: 38–46. [DOI] [PubMed] [Google Scholar]
- 11.Ghergurovich JM, Esposito M, Chen Z, Wang JZ, Bhatt V, Lan T et al. Glucose-6-Phosphate Dehydrogenase Is Not Essential for K-Ras-Driven Tumor Growth or Metastasis. Cancer Res 2020; 80: 3820–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen L, Zhang Z, Hoshino A, Zheng HD, Morley M, Arany Z et al. NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nat Metab 2019; 1: 404–415. [PMC free article] [PubMed] [Google Scholar]
- 13.Liu B, Bai W, Ou G, Zhang J. Cdh1-Mediated Metabolic Switch from Pentose Phosphate Pathway to Glycolysis Contributes to Sevoflurane-Induced Neuronal Apoptosis in Developing Brain. ACS Chem Neurosci 2019; 10: 2332–2344. [DOI] [PubMed] [Google Scholar]
- 14.Figueroa M, Graf TN, Ayers S, Adcock AF, Kroll DJ, Yang J et al. Cytotoxic epipolythiodioxopiperazine alkaloids from filamentous fungi of the Bionectriaceae. J Antibiot 2012; 65: 559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amrine CSM, Raja HA, Darveaux BA, Pearce CJ, Oberlies NH. Media studies to enhance the production of verticillins facilitated by in situ chemical analysis. J Ind Microbiol Biotechnol 2018; 45: 1053–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raub AG, Hwang S, Horikoshi N, Cunningham AD, Rahighi S, Wakatsuki S et al. Small-Molecule Activators of Glucose-6-phosphate Dehydrogenase (G6PD) Bridging the Dimer Interface. ChemMedChem 2019; 14: 1321–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hwang S, Mruk K, Rahighi S, Raub AG, Chen CH, Dorn LE et al. Correcting glucose-6-phosphate dehydrogenase deficiency with a small-molecule activator. Nat Commun 2018; 9: 4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghergurovich JM, Garcia-Canaveras JC, Wang J, Schmidt E, Zhang Z, TeSlaa T et al. A small molecule G6PD inhibitor reveals immune dependence on pentose phosphate pathway. Nat Chem Biol 2020; 16: 731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu C, Klement JD, Smith AD, Yang D, Waller JL, Browning DD et al. p50 suppresses cytotoxic T lymphocyte effector function to regulate tumor immune escape and response to immunotherapy. J Immunother Cancer 2020; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fiehn O, Wohlgemuth G, Scholz M, Kind T, Lee DY, Lu Y et al. Quality control for plant metabolomics: reporting MSI-compliant studies. Plant J 2008; 53: 691–704. [DOI] [PubMed] [Google Scholar]
- 21.Ding J, Li T, Wang X, Zhao E, Choi JH, Yang L et al. The histone H3 methyltransferase G9A epigenetically activates the serine-glycine synthesis pathway to sustain cancer cell survival and proliferation. Cell Metab 2013; 18: 896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu C, Liu Z, Klement JD, Yang D, Merting AD, Poschel D et al. WDR5-H3K4me3 epigenetic axis regulates OPN expression to compensate PD-L1 function to promote pancreatic cancer immune escape. J Immunother Cancer 2021; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paschall AV, Yang D, Lu C, Choi JH, Li X, Liu F et al. H3K9 Trimethylation Silences Fas Expression To Confer Colon Carcinoma Immune Escape and 5-Fluorouracil Chemoresistance. J Immunol 2015. 195:1868–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu C, Paschall AV, Shi H, Savage N, Waller JL, Sabbatini ME et al. The MLL1-H3K4me3 Axis-Mediated PD-L1 Expression and Pancreatic Cancer Immune Evasion. J Natl Cancer Inst 2017; 109:djw283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ravindran Menon D, Hammerlindl H, Torrano J, Schaider H, Fujita M. Epigenetics and metabolism at the crossroads of stress-induced plasticity, stemness and therapeutic resistance in cancer. Theranostics 2020; 10: 6261–6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A 2007; 104: 19345–19350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lachner M, O’Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 2001; 410: 116–120. [DOI] [PubMed] [Google Scholar]
- 28.Lu C, Yang D, Klement JD, Colson YL, Oberlies NH, Pearce CJ et al. G6PD functions as a metabolic checkpoint to regulate granzyme B expression in tumor-specific cytotoxic T lymphocytes. J Immunother Cancer 2022; 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fritsch L, Robin P, Mathieu JR, Souidi M, Hinaux H, Rougeulle C et al. A subset of the histone H3 lysine 9 methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Mol Cell 2010; 37: 46–56. [DOI] [PubMed] [Google Scholar]
- 30.Rice JC, Briggs SD, Ueberheide B, Barber CM, Shabanowitz J, Hunt DF et al. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol Cell 2003; 12: 1591–1598. [DOI] [PubMed] [Google Scholar]
- 31.Muller MM, Fierz B, Bittova L, Liszczak G, Muir TW. A two-state activation mechanism controls the histone methyltransferase Suv39h1. Nat Chem Biol 2016; 12: 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abbosh PH, Montgomery JS, Starkey JA, Novotny M, Zuhowski EG, Egorin MJ et al. Dominant-negative histone H3 lysine 27 mutant derepresses silenced tumor suppressor genes and reverses the drug-resistant phenotype in cancer cells. Cancer Res 2006; 66: 5582–5591. [DOI] [PubMed] [Google Scholar]
- 33.Matei D, Nephew KP. Epigenetic Attire in Ovarian Cancer: The Emperor’s New Clothes. Cancer Res 2020; 80: 3775–3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nicetto D, Donahue G, Jain T, Peng T, Sidoli S, Sheng L et al. H3K9me3-heterochromatin loss at protein-coding genes enables developmental lineage specification. Science 2019; 363: 294–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olcina MM, Leszczynska KB, Senra JM, Isa NF, Harada H, Hammond EM. H3K9me3 facilitates hypoxia-induced p53-dependent apoptosis through repression of APAK. Oncogene 2016; 35: 793–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salvi A, Amrine CSM, Austin JR, Kilpatrick K, Russo A, Lantvit D et al. Verticillin A Causes Apoptosis and Reduces Tumor Burden in High-Grade Serous Ovarian Cancer by Inducing DNA Damage. Mol Cancer Ther 2020; 19: 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aggarwal BB, Gupta SC, Kim JH. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood 2012; 119: 651–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gu Y, Bouwman P, Greco D, Saarela J, Yadav B, Jonkers J et al. Suppression of BRCA1 sensitizes cells to proteasome inhibitors. Cell Death Dis 2014; 5: e1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iurlaro R, Puschel F, Leon-Annicchiarico CL, O’Connor H, Martin SJ, Palou-Gramon D et al. Glucose Deprivation Induces ATF4-Mediated Apoptosis through TRAIL Death Receptors. Mol Cell Biol 2017; 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta 2016; 1863: 2977–2992. [DOI] [PubMed] [Google Scholar]
- 41.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell 2005; 120: 483–495. [DOI] [PubMed] [Google Scholar]
- 42.Koo SJ, Szczesny B, Wan X, Putluri N, Garg NJ. Pentose Phosphate Shunt Modulates Reactive Oxygen Species and Nitric Oxide Production Controlling Trypanosoma cruzi in Macrophages. Front Immunol 2018; 9: 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tu D, Gao Y, Yang R, Guan T, Hong JS, Gao HM. The pentose phosphate pathway regulates chronic neuroinflammation and dopaminergic neurodegeneration. J Neuroinflammation 2019; 16: 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hayes JD, Dinkova-Kostova AT, Tew KD. Oxidative Stress in Cancer. Cancer Cell 2020; 38: 167–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oshima N, Ishida R, Kishimoto S, Beebe K, Brender JR, Yamamoto K et al. Dynamic Imaging of LDH Inhibition in Tumors Reveals Rapid In Vivo Metabolic Rewiring and Vulnerability to Combination Therapy. Cell Rep 2020; 30: 1798–1810 e1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A 2010; 107: 2037–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yeung C, Gibson AE, Issaq SH, Oshima N, Baumgart JT, Edessa LD et al. Targeting Glycolysis through Inhibition of Lactate Dehydrogenase Impairs Tumor Growth in Preclinical Models of Ewing Sarcoma. Cancer Res 2019; 79: 5060–5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell 2012; 48: 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jian SL, Chen WW, Su YC, Su YW, Chuang TH, Hsu SC et al. Glycolysis regulates the expansion of myeloid-derived suppressor cells in tumor-bearing hosts through prevention of ROS-mediated apoptosis. Cell Death Dis 2017; 8: e2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuehne A, Emmert H, Soehle J, Winnefeld M, Fischer F, Wenck H et al. Acute Activation of Oxidative Pentose Phosphate Pathway as First-Line Response to Oxidative Stress in Human Skin Cells. Mol Cell 2015; 59: 359–371. [DOI] [PubMed] [Google Scholar]
- 51.Dick TP, Ralser M. Metabolic Remodeling in Times of Stress: Who Shoots Faster than His Shadow? Mol Cell 2015; 59: 519–521. [DOI] [PubMed] [Google Scholar]
- 52.Li W, Kou J, Qin J, Li L, Zhang Z, Pan Y et al. NADPH levels affect cellular epigenetic state by inhibiting HDAC3-Ncor complex. Nat Metab 2021; 3: 75–89. [DOI] [PubMed] [Google Scholar]
- 53.Monteiro-Reis S, Lameirinhas A, Miranda-Goncalves V, Felizardo D, Dias PC, Oliveira J et al. Sirtuins’ Deregulation in Bladder Cancer: SIRT7 Is Implicated in Tumor Progression through Epithelial to Mesenchymal Transition Promotion. Cancers (Basel) 2020; 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stomper J, Meier R, Ma T, Pfeifer D, Ihorst G, Blagitko-Dorfs N et al. Integrative study of EZH2 mutational status, copy number, protein expression and H3K27 trimethylation in AML/MDS patients. Clin Epigenetics 2021; 13: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mozzetta C, Pontis J, Fritsch L, Robin P, Portoso M, Proux C et al. The histone H3 lysine 9 methyltransferases G9a and GLP regulate polycomb repressive complex 2-mediated gene silencing. Mol Cell 2014; 53: 277–289. [DOI] [PubMed] [Google Scholar]
- 56.Paschall AV, Liu K. Epigenetic Regulation of Apoptosis and Cell Cycle Regulatory Genes in Human Colon Carcinoma Cells. Genom Data 2015; 5: 189–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lu C, Klement JD, Yang D, Albers T, Lebedyeva IO, Waller JL et al. SUV39H1 regulates human colon carcinoma apoptosis and cell cycle to promote tumor growth. Cancer Lett 2020; 476: 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Williams D, Fingleton B. Non-canonical roles for metabolic enzymes and intermediates in malignant progression and metastasis. Clin Exp Metastasis 2019; 36: 211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wong CC, Qian Y, Yu J. Interplay between epigenetics and metabolism in oncogenesis: mechanisms and therapeutic approaches. Oncogene 2017; 36: 3359–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McDonald OG, Li X, Saunders T, Tryggvadottir R, Mentch SJ, Warmoes MO et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet 2017; 49: 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cui W, Popat S. Pleural mesothelioma (PM) - The status of systemic therapy. Cancer Treat Rev 2021; 100: 102265. [DOI] [PubMed] [Google Scholar]
- 62.Pinton G, Wang Z, Balzano C, Missaglia S, Tavian D, Boldorini R et al. CDKN2A Determines Mesothelioma Cell Fate to EZH2 Inhibition. Front Oncol 2021; 11: 678447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McCambridge AJ, Napolitano A, Mansfield AS, Fennell DA, Sekido Y, Nowak AK et al. Progress in the Management of Malignant Pleural Mesothelioma in 2017. J Thorac Oncol 2018; 13: 606–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hotta K, Fujimoto N. Current evidence and future perspectives of immune-checkpoint inhibitors in unresectable malignant pleural mesothelioma. J Immunother Cancer 2020; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nakajima EC, Vellanki PJ, Larkins E, Chatterjee S, Mishra-Kalyani PS, Bi Y et al. FDA Approval Summary: Nivolumab in Combination with Ipilimumab for the Treatment of Unresectable Malignant Pleural Mesothelioma. Clin Cancer Res 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lemoine J, Ruella M, Houot R. Overcoming Intrinsic Resistance of Cancer Cells to CAR T-Cell Killing. Clin Cancer Res 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.