

Abstract
Tumor immunosurveillance requires tumor cell-derived molecules to initiate responses through corresponding receptors on antigen presenting cells (APC) and a specific effector response designed to eliminate the emerging tumor cells. This is supported by evidence from immunodeficient individuals and experimental animals. Recent discoveries suggest that adjuvanticity of tumor-derived heat shock proteins (HSPs) and dsDNA are necessary for tumor-specific immunity. There is also the obligatory early transfer of tumor antigens to APCs. We argue that tumor-derived HSPs deliver sufficient chaperoned antigen for cross-priming within the quantitative limits set by nascent tumors. In contrast to late-stage tumors, we are only just beginning to understand the unique interactions of the immune system with precancerous/nascent neoplastic cells, which is important for improved cancer prevention measures.
Keywords: Adjuvant, Antigen, Cross-priming, CD91/LRP1, Heat shock proteins, cGAS, STING, IgM
Emergence of nascent cancer cells and immune responses
The idea that spontaneous eradication of neoplasms by the immune system can occur prior to any clinical manifestations was put forward over a hundred years ago[1]. The current model of cancer immunosurveillance is based on historical data from over 6 decades and summarized in Box 1. It proposes that T cells and NK cells recognize and eliminate emerging cancer cells. Two major gaps persist in how these immune effector cells are primed and/or activated, related to the source of adjuvanticity, and the cross-presentation mechanism that is sufficiently sensitive to account for the quantitative limits on available tumor antigen. We posit here, first, that tumors harbor molecules, specifically heat hock proteins (HSPs) and double stranded DNA (dsDNA), that are sensed by receptors on antigen presenting cells (APCs), and act as adjuvants. Second, at least one mechanism of cross-priming involving HSP-peptide complexes exists which satisfies the low amount of antigen present in nascent, emerging tumors. As pathway dysfunctions can lead tumorigenesis, understanding the mechanistic details related to immune responses to emerging tumors may allow the development of new experimental models and measures for cancer prevention
Box 1: Historical Perspective on cancer immunosurveillance.
The idea that spontaneous eradication of neoplasms by the immune system could occur prior to any clinical manifestations was first made in 1909[1]. Decades later, several investigators showed that purebred animals could be immunized against a tumor that was originally induced in an animal of the same strain and such immunity was not caused by MHC differences between the host and tumor cells[76,77]; this established the concept that tumors were antigenic and immunogenic. The formal paradigm of cancer immunosurveillance described an evolutionary necessity for a mechanism of elimination or inactivation of potentially dangerous mutant cells and speculated that lymphocytes mediated this process[78,79]. The paradigm suggested that there was a constant liberation of “self-markers”, which in this opinion, we propose include HSPs and dsDNA, for recognition by immune cells. Evidence for this hypothesis comes primarily from immunocompromised individuals and experimental animals bearing a higher incidence of cancer than individuals with intact immune systems. Transplant recipients on immunosuppressive medications and individuals with HIV-1/AIDS develop skin, lung, bladder, prostate, rectum, and colon cancers at rates 10–25 times higher than healthy individuals[80]. Mice deficient in key cells or molecules required for adaptive immunity are generally more susceptible to developing experimentally-induced tumors than wildtype mice[66,81], although some exceptions have been reported[82,83]. Other anecdotal evidence has been derived from observations of increased cancer incidence in the elderly --often ascribed to or associated with immunosenescence -- and in individuals living with chronic stress and associated immune decline[84,85]. So, how do these tumors emerge in the first place? Recent estimates suggest that in an adult human, 50–70 billion nucleated cells turn over each day, or 30–50 million cells per minute[86]. Each time a mammalian cell divides, 1.2 × 1010 nucleotides are polymerized[87]. DNA polymerases δ, which are involved in the bulk of genomic DNA replication, have error rates of 10−4 to 10−5 per nucleotide polymerized[87]. Based on these error rates and accounting for base mismatch repair, on average, it has been estimated that 100,000 to 1,000,000 errors occur per diploid genome during every cell division[87]. Given this net error rate of DNA replication during cell division and the possible occurrence of these mutations in oncogenes or mutational hotspots, the calculated risk of generating a malignant cell is not infrequent [8].
Sensing of nascent, emerging tumors by the immune system
With the exception of virally-induced cancers, tumor cells are antigenically abnormal versions of self; moreover, they do not express Pathogen Associated Molecular Patterns (PAMPs) and therefore typically do not activate innate Pattern Recognition Receptors (PRR) – a necessary step for the generation of innate and adaptive immune responses. With cancer, the mammalian immune system thus relies on sensing aberrantly expressed, non-microbial, self-molecules/markers, or Damage Associated Molecular Patterns (DAMPs)[2]. These are typically intracellular molecules that are abruptly exposed to the extracellular milieu, accumulated in vesicular compartments, or abnormally modified. Similar to the PAMP-PRR axis, DAMPS generally require corresponding Damage Sensing Receptors (DSR) that mediate downstream signaling networks and co-stimulation[3]. Priming of CD8+ and CD4+ T cells, and activation of NK cell responses, implicated in cancer immunosurveillance, are dependent on this co-stimulation (Figure 1)[4]. Below, we discuss recent studies assessing the effects of HSPs and dsDNAs in cancer immunosurveillance; these studies have been conducted with molecules at physiological doses close to those naturally present within mammalian tissues at very early stages of tumorigenesis.
Sensing extracellular Heat Shock Proteins
HSPs were the first cancer cell-derived molecules shown to be immunogenic[5–9]. HSP gp96, hsp70, hsp90, calreticulin, hsp110, and grp170 are abundant intracellular proteins that constitute >5% of the mammalian proteome[10,11]. These six HSPs are the immunogenic HSPs, discussed below. As a consequence of cellular necrosis (also characterized as immunogenic cell death)[12–15], or aberrant secretory pathways, tumor cells can expose HSPs to the extracellular environment where they can be sampled by the immune system[5,13,16]. In mammalian in vitro systems, murine or human APCs, such as dendritic cells (DC) or macrophages cultured with purified mammalian HSPs lead to the upregulation in B7, CD40, and MHC II as well as the release of cytokines and chemokines, including IL-β, IL-6, IL-12p40, TNF-α, CXCL10, and others[13,17,18]. The production of such factors has been reported to be fully dependent on the DC-expressed receptor CD91/LRP1[19–22] – the first DSR to be identified; indeed, CD91 inhibition with antibodies or competitive ligands, or CD91 deletion in genetically modified mice (CD91fl/flCD11cCre) or knockdown via siRNA, renders DCs and macrophages, unresponsive to extracellular immunogenic HSPs[21–23]. Moreover, upon HSP binding to DCs, tyrosines within the intracellular domains of CD91 are synergistically phosphorylated (identified via anti-pTyr antibodies)[17]; this in turn mediates signal transduction events via a network of molecules including p38 MAPK, STAT1, NF-κB, components of the inflammasome (NLRP3, ASC, caspase 1), and representative intermediaries[17,18]. The activation status of these molecules has been monitored through their phosphorylation or catalytic function in murine and human APCs. Aside from CD91, other receptors for HSPs such as scavenger receptor A, TLR2/4, and CD40, have been reported[24–26]; however, conclusive evidence of their in vivo biologic activity in APC activation is lacking[27]. Immunization of mice with microgram quantities of purified HSPs leads to the migration of antigen-bearing DCs from tumor sites to draining lymph nodes/spleen (as observed by flow cytometry of CD11c+ cells)[28], priming of tumor-specific CD8+ and CD4+ T cells (as measured by cytotoxicity of a large number of murine tumors, and cytokine release assays)[5,29,30], and activation of NK cells (as measured by IFN-γ release)[31]. Similar observations have been made in patients with melanoma, renal cell carcinoma, and glioblastoma[32–34]. Given that a typical mammalian cell harbors approximately 2–3pg of gp96, ~20–30pg of hsp90 and ~10–15pg of hsp70, the amount of HSPs released by tens of cells during early tumorigenesis falls well within the effective range that can modulate APCs function[35]. The immunobiology of HSPs and their quantity make them superb candidates as tumor-derived molecules that can generate adjuvanticity for cancer immunosurveillance. Testing this requirement has proven difficult though, given the redundancy of multiple immunogenic HSPs in providing adjuvanticity; generating sequential knock-out mice for all immunogenic HSPs is a necessary but unviable approach. Instead, CD91 has been targeted. Mice lacking CD91 expression in DCs (CD91fl/flCD11cCre) have a significantly higher incidence of methylcholanthrene (MCA)-induced tumors when compared to littermates with wildtype expression of CD91 (CD91fl/fl)[22]. The tumors that arose in CD91fl/flCD11cCre mice were larger and less infiltrated by effector CD4+ and CD8+ T cells and NK cells than in CD91fl/fl mice, with evidence of significantly reduced immunoediting in the tumors, as measured by whole exome sequencing and calculation of Differential Aggretope Index (DAI) of mutated and immunogenic neoepitopes[22]. In addition, transplantable tumors such as D122 carcinoma and SVB6 fibrosarcoma generally grew faster in CD91fl/flCD11cCre compared to CD91fl/fl mice[23]. These observations suggest that loss of CD91 eliminates immunological pressure and allows tumors to grow faster. Moreover, these mouse studies are in strong agreement with data from specifically, patients with CD91 mutations predicted (via Hex Protein Docking simulations) to negatively impact HSP binding and exhibited reduced tumor T cell-infiltration compared with patients with no CD91 mutations [22]. Furthermore, melanoma patients with high expression of CD91 showed better prognoses with slower growing tumors compared to low CD91 expressors, suggesting that high CD91 expression might potentially allow for better functionality of HSPs in providing adjuvanticity for anti-tumor CD8+ and CD4+ T cell responses[22,36].
Of note, responses triggered by HSPs are hormetic and context-dependent; Approximately 1 ug of purified gp96, hsp70 or hsp90 elicits anti-tumor immune responses in mice and humans as evidenced by the rejection of MethA, D122 and CMS5 tumors, to name a few[5,29,37–41]. Ten times this amount however, in skin and lung cancer patients with CD91 defects, primes regulatory T cells (measured by flow cytometry) and pro-tumor responses in mice (enhanced tumor growth) via engagement of CD91 on plasmacytoid dendritic cells, leading to TGF-β production, and upregulation of neuropilins (flow cytometry)[37–40]. Hsp60 has also been shown to instigate biphasic immunoregulatory effects in mice[42]. Therefore, in exceptional situations of excessively high extracellular immunogenic HSPs, as may be the case for late stage bulky/necrotized solid tumors, we propose the HSP-CD91 axis might have a contradictory effect by promoting tumor growth[5,29,37–41].
We posit that collectively, these studies establish a role for the HSP-CD91 axis in tumor immunosurveillance, providing a rationale for further investigating the mechanisms and outcomes of such signaling networks.
Sensing double stranded DNA
Human cytosolic sensor cyclic GMP-AMP (cGAMP) synthase (hcGAS) can be activated and lead to the release of type I IFN when human monocytic THP-1 cells are pulsed in a titrated manner by long fragments of DNA (>45bp) but which remain inactive in the presence of short DNA (≤17bp)[43]. Therefore, there is a direct proportional relationship between DNA length and efficiency of cGAS activation; for DNA of 45 to 70 bp in size, >1 μg/ml is required for cGAS activation, and this is reduced to > 0.167 μg/ml for DNA of 300–500 bp and > 0.017 μg/ml for DNA of 800–2,000 bp in size, respectively[44]. This suggests that long DNA molecules might overcome the need for high DNA concentrations in eliciting effective activation of cGAS, which is relevant as high dsDNA concentrations are typically not present in emerging tumors[43,44]. Of note, cGAS in mouse transdifferentiated BLaER1 cells respond by releasing CXCL10 to both long and short DNA with equal efficiency[45], suggesting species-specific differences in DNA sensing and a mechanism of detecting emerging tumors. Cellular factors such as nucleic acid-stress HMGB proteins and nucleoid-structuring proteins (TFAM, HU) that nucleate and stabilize cGAS dimers by prearranging DNA[45] can also enhance the initial detection of long cytosolic DNA even under conditions of low cGAS concentrations[44], which is pertinent for detecting emerging tumors. Tumor-derived DNA can therefore act as a danger signal when it engages cGAS in a sequence-independent and length-dependent manner at low ligand concentrations[44,46]. DNA binding to cGAS catalyzes the synthesis of cGAMP which activates the ER membrane adaptor stimulator of interferon genes complex (STING) as shown in human embryonic kidney 293 cells. In these cells, activated STING recruits kinases TBK and IKK that phosphorylate transcription factors IRF3 and NF-κB, respectively (as shown by immunoprecipitation and western blotting) [47,48], which induces IL-12, CXCL9, type I IFN production. Type I IFN can mediate cancer immunosurveillance because Ifnar1−/− mice have a higher incidence of tumors compared to WT mice when injected subcutaneously with MCA[49–51]. When the cGAS-deficient colon carcinoma cell line CT26, or Lewis lung carcinoma cells, were injected into mice, the tumor cells were not rejected, indicating cGAS expression by tumor cells, in addition to STING expression by DCs, was required for CD8+ T cell-mediated control of cancer progression[52]. BC2, YAC1, and EμM1 lymphoma cell lines exposed to DNA damaging agents such as aphidicolin in vitro intrinsically activated transcription factor IRF3, leading to the induction and surface expression of RAE1, a ligand for NKG2D on NK cells[53]. These observations have led to experiments that demonstrate the ability of NK cells to mediate rejection of murine transplantable RMA lymphomas[54].
The mechanism of DNA acquisition by DCs is not completely resolved. DCs can phagocytose entire tumor cells[55] or tumor-derived vesicles[56], and intrinsically activate cGAS. However, tumor cGAMP itself, rather than cytoplasmic DNA, can also be transferred to DCs via gap junctions[52,54] which then promotes STING activation and DC-mediated priming of CD8+ T cell responses to B16 tumors[52,54].
Downregulation of cGAS and/or STING expression has been associated with tumorigenesis in humans and poor prognosis in patients with melanoma[57], lung[58], gastric[59] and colorectal cancers[60,61]. Presumably, these observations might support this pathway’s involvement in cancer immunosurveillance. Recently, in intratumoral mouse splenic conventional DCs (cDCs), blocking the inhibitory receptor TIM-3 by using an anti-TIM-3 antibody promoted extracellular DNA uptake by cDCs and activated the cGAS-STING pathway[62]. TIM-3 is frequently upregulated in the tumor microenvironment[63] and can suppress this pathway of cancer immunosurveillance[62]. Of note, studies such as these have led to therapies of advanced carcinomas, and lymphomas, with targeted STING agonists[64].
The synopsis of experimental observations presented here suggest that the HSP-CD91 and dsDNA-cGAS-STING axes are likely involved in cancer immunosurveillance by providing molecular pathways that are important for tumor-associated adjuvanticity, addressing one of the major gaps in current cancer immunosurveillance models. Other tumor-derived molecules with adjuvanticity, but for which direct evidence in cancer immunosurveillance is lacking, are described in Box 2.
Box 2: Other stimuli that may provide adjuvanticity.
There are several other tumor-derived molecules worth mentioning that have been shown to possess adjuvanticity in studies in vitro, but there is no evidence of their role in tumor immunosurveillance. These include nuclear proteins (High mobility box-1; HMGB1, HMGN1), Adenosine triphosphate (ATP), Uric Acid, and IL-33[3]. Several recent studies have also linked certain microbial communities to specific types of cancers, particularly gut-associated cancers[88]. Recently, gut-distal tumors such as lung[89,90], bone[91] and breast tumors[92–94] have been shown to be colonized by bacteria. Bacteria have been localized within both cancer cells and immune cells, with bacterial strain composition varying based on tumor type[91]. Therefore, these are important considerations as such stimuli may influence anti-cancer immunological responses within the microenvironment of established tumors, along with regulatory mechanisms (e.g. the CD24-Siglec G or -Siglec10 pathway in mice and humans, respectively)[95].
Cross-priming of T cells in response to nascent, emerging tumors
Tumor Antigens of nascent, emerging tumors that are recognized by T cells
The innate immune sensing mechanisms described above provide signals such as co-stimulation (signal 2) and cytokines (signal 3) that are necessary for priming adaptive immunity. In addition, another signal that is required is antigen cross-presentation (signal 1) by APCs such as DCs to T cells (Figure 1). CD8+ T cell immunity is essential to achieve effective tumor immunosurveillance; in mice, the obstruction of optimal effector T cell responses through either the loss of T cells (Rag−/−), STAT1 (Stat1−/−), or Th1 effector molecules (e.g. IFNγ, Ifng−/−; perforin, Pfn−/−, granzyme B, Gzmb−/−; and others) can lead to a higher incidence of tumors at tissue sites of induction compared to wildtype mice[65]. Although many tumor antigens have been described, only a few represent tumor rejection antigens. Indeed, tumor antigens can largely be classified as neoantigens (i.e., unique and derived from random somatic mutations and give rise to mutated peptides), shared antigens (i.e., aberrantly or over-expressed unmutated antigens) and (onco) viral antigens. Tumors, induced by intramuscular injection of lentiviral vectors expressing Cre recombinase into genetically engineered KrasLSL-G12D/+;p53flox/flox mice, and lacking neoantigens, are generally not cleared by the immune system, suggesting that these antigens are important for immunosurveillance[66]. Given the random nature of mutation events, antigenic epitopes presented to T cells are also randomly generated. A seminal study showed that tumor immunity was rarely cross-reactive because exposure of mice to one tumor protected the mice against secondary challenge by the same tumor, but rarely against a different tumor[67]. Novel algorithms such as the DAI are improving the predictability of which mutated peptides serve as rejection neoepitopes[68] and we anticipate that these will be tested in the context of cancer immunosurveillance. Since nascent tumors comprise a few cells, one might assume that the net amount of antigen available is minute, and therefore requires efficient cross-presentation of those antigens by APCs to CD8+ T cells. This certainly merits further investigation.
Key Figure, Figure 1. Model of sensing and elimination of emerging tumors by the mammalian immune system.
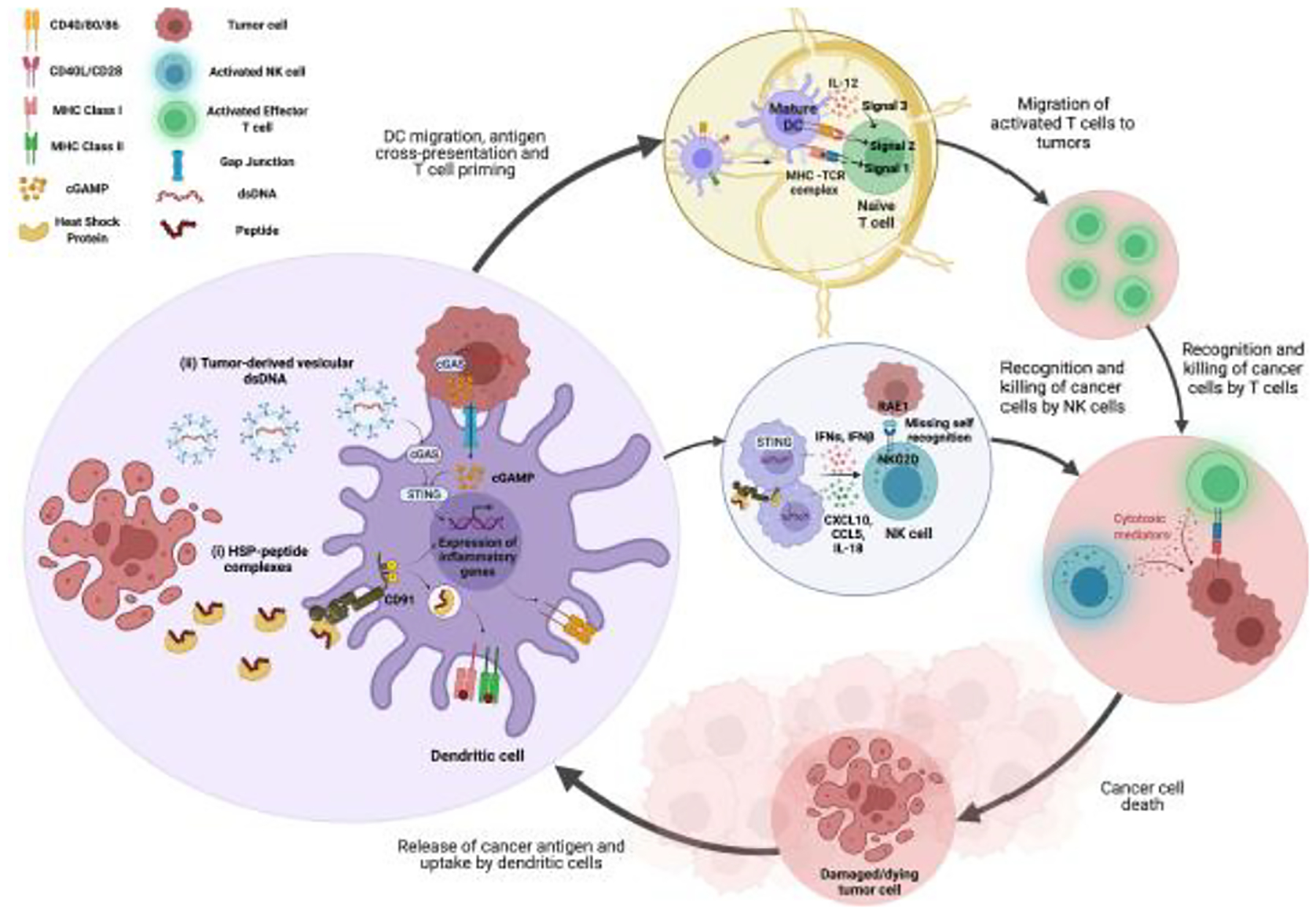
(A) Tumor-derived molecular stimuli can provide adjuvanticity that is necessary for priming and activating anti-tumor T cell and NK cell responses during cancer immunosurveillance. Tumor cell death leads to the release of HSPs[5,13,16] and dsDNA[56]. (i) Tumor-derived HSPs bind to CD91 expressed on DCs and undergo endocytosis along with the chaperoned peptides[21–23]. Peptides are processed and presented by MHC I and MHC II[9,35,72]. Upon binding HSPs, CD91 is phosphorylated and initiates signaling pathways within the DC leading to upregulation of CD86 and CD40, the release of cytokines, and maturation of DCs[17,20–23,75]. (ii) dsDNA binds to cGAS in DCs after internalization[55,56]. dsDNA may bind cGAS in the tumor cell itself and cGAMP can transport into DCs via gap junctions[52,54]. cGAS activates STING in the DC leading to upregulation of CD28, release of cytokines, and maturation of DCs[43–45,51,52]. Mature DCs migrate to lymph nodes where they prime naïve CD8+ and CD4+ T cells to become anti-tumor effector T cells. Mature DCs can also activate NK cells via cytokines[31,53,54]. Effector T cells and activated NK cells mediate cancer cell death.
HSP-peptide complexes are key players in nascent tumor antigen cross-presentation
HSPs are intracellular chaperones of peptides which can include minute quantities of tumor antigens. When highly enriched HSP preparations from human renal cell carcinomas and mouse mastocytoma or RL♂1 were stripped of their chaperoned peptides and analyzed by mass spectrometry, the peptides were shown to essentially represent the antigenic landscape of that respective tumor [27,69,70]. One microgram of total immunogenic HSP will chaperone approximately a nanogram-femtogram of a specific antigenic/mutated peptide[23,71]. Human and mouse cross-presentation systems, set up by incubating HSP-peptide complexes with APCs, show that this quantity of antigen is sufficient for cross-presentation only when chaperoned by the HSP [9,35,72]. Identical amount of soluble antigen (without HSPs) is not sufficient for cross-presentation[71,73]. The readouts in these cross-presentation systems were stimulation, by cytokine release, of antigen-specific CD8+ or CD4+ T cells. There are other mechanisms of cross-presentation in which a 10,000-fold higher amount of antigen than present in nascent tumors have been used and these are not the focus here[74]. For the former cross-priming protocols[9,35,72], HSP-(neo)peptide complexes, upon release by tumor cells in vitro, are taken up via CD91-mediated endocytosis by DCs and macrophages in co-culture experiments and the peptides are cross-presented to CD8+ and CD4+ T cells[19,20,75]. Removal of four HSPs from mouse lymphoma (EL.4) lysates by immunoprecipitation showed that the depleted lysates were incapable of cross-priming when injected intradermally into mice -- as shown by the lack of tumor-specific CD8+ T cell-mediated cytotoxicity ex vivo one week later, despite the presence of soluble tumor antigen in those lysates[71]. Of note, depletion of multiple HSPs in vivo is problematic given that mice and cells are not viable without them. Therefore, despite its caveats, a single HSP receptor, i.e. CD91, has been targeted; in this study, transplantable (D122 lung carcinoma and SVB6 fibrosarcomas) and induced tumors grew faster in CD91flox/floxCD11cCre mice compared to wildtype mice, indicating that they failed to mount anti-tumor-specific immune responses, even though these CD91flox/floxCD11cCre mice could still mount efficient CD8+ T cell responses against CD91-independent immunogens such as CFA/OVA[22]. Moreover, titration of antigen abundance using the D122 tumor further showed that the CD91-HSP pathway was essential for effective antigen cross-presentation to T cells when the target antigen quantity was low, as evidenced by the loss of tumor rejection in mice with loss of CD91 (CD91flox/floxCD11cCre) or in mice in which CD91 was inhibited with the antagonist receptor-associated protein[23]. Once again, this supports the importance of the HSP receptor CD91 in cancer immunosurveillance and suggests that HSP-peptide complexes are the likely ligands, although further assessments are warranted. To support the notion that HSPs constitute CD91 ligands in this context, DCs derived from CD91flox/floxCD11cCre mice were found to not cross-present gp96-chaperoned antigen to T cells when antigen loads were low and could no longer provide signals 2 and 3[23]. As expected, CD91flox/floxCD11cCre mice failed to prime anti-tumor CD8+ and CD4+ T cell responses when immunized with HSPs (as evidenced by the decreased number of tumor-infiltrating T cells), while CD91flox/flox littermates did [23]. We posit that CD91 is dispensable at higher antigen loads, and that when mice are challenged with large doses of tumor cells, presumably, alternative molecular mechanisms might mediate antigen transfer[23]. We argue that the collective evidence supports the idea that tumor antigens which are chaperoned by HSPs, are cross-presented by APCs via CD91 to mount CD8+ and CD4+ T cell responses that can reject nascent, emerging tumors.
Concluding remarks
Newly emerging data support a role for HSP-CD91 and dsDNA-cGAS-STING signaling axes in cancer immunosurveillance but further rigorous work is warranted to confirm this, especially in humans. Individual sensing mechanisms initiating downstream signaling responses might potentially be generalizable among emerging tumors, and we speculate that these sensing mechanisms might overlap and/or be redundant, presumably as a mechanism of host defense to reduce the incidence of tumor escape. This hypothesis certainly raises numerous questions (see Outstanding Questions). Alternative pathways may also exist under specialized conditions; one of these involves natural IgM, and is summarized in Box 3. Presumably, at least one established mechanism of cross-presentation exhibits some degree of redundancy given that multiple HSPs can cross-present their chaperoned antigens. Given the recent advances in whole exome sequencing and proteomics, human inborn errors of immunity are increasingly being discovered, which may provide unique opportunities for molecular discovery and targeting in the context of cancer immunosurveillance. We advocate for pathways involving CD91 and cGAS/STING as important in cancer immunosurveillance, and suggest that they be considered as new candidate targets for enhancing adjuvanticity. In this context, we are also excited about the possibility of studying the underlying molecular mechanisms that support effective immunosurveillance in a unified systems-immunology framework, as dictated by tumor types and histologic origins.
Outstanding Questions:
Is there functional redundancy or dominance among the HSP-CD91 and dsDNA-cGAS-STING pathways in tumor immunosurveillance? These could be addressed in mouse systems lacking both STING and CD91. Reintroduction of one or the other might establish the necessity or sufficiency of each pathway.
Given the heterogeneity of tissue-resident populations of APCs, what is the identity of APC subset(s) responsible for initiating and orchestrating immunity to emerging tumors and are these tissue-dependent? The availability of many DC knockout mouse models makes this question readily addressable using tissue-specific tumor induction procedures. The reported differences between males and females in tissue-specific density, development and function of DCs might have a bearing on sex dependent differences in tumor incidence.
Are there inborn errors of the HSP-CD91 and dsDNA-cGAS-STING pathway? Although a few mutations in CD91 that affect function have been identified, a comprehensive search for other mutations in these pathways has yet to be undertaken.
Does the microbiome influence the emergence of tumors? The presence of gut microbes and microbial-derived products in established tumors has been reported. However, to our knowledge, it is not known if these may influence the establishment of immune responses to nascent, emerging tumors by shaping the tumor microenvironment. Germ-free mice colonized with specific gut microbiota may be used to address this question.
Box 3: Natural IgM may mediate alternative cancer immunosurveillance mechanisms.
Natural IgM antibodies are germline encoded, do not undergo affinity maturation, and are commonly observed early in polyreactive immune responses to antigenic stimulation[96]. If natural IgM antibodies cross-react with an antigen expressed by an emerging tumor, they might provide an alternate mechanism for cancer immunosurveillance[97,98]. In mice, natural IgM can recognize neoantigens on tumor cells, such as changes in glycoprotein patterns. The resulting antigen-IgM complexes can be acquired by Ly6C+ monocytes via Fc receptors which results in activation of the monocytes[98]. These circulating mouse monocytes present neoantigen-derived peptides by MHC to CD4+ T cells which in turn, facilitate such CD4+ T cell engagement with Batf3+ DCs through CD40-CD40L interactions[98]. The now licensed Batf3+ DCs can cross-prime effector CD8+ T cells against tumor (neo)-antigens (Figure 2)[98]. In this study, recipient transgenic PMEL, Batf3−/−, Cd4−/−, Ccr2−/−, and μMT female mice, were used to demonstrate the necessity for diverse CD8+ T cell repertoires, as well as diverse Batf3+ DCs, CD4+ T cells, tissue-trafficking monocytes, and B cells, respectively, in immune-mediated control of B16F10 tumors[98]. However, this mechanism does not address the cross-priming of antigen- specific T cells by IgM. Relatively large (1–100 mg) doses of ovalbumin and adoptive transfer of OT-1 cells were used, which poorly reflect genuine tumor antigen in amounts, location, and context, in tumor-bearing hosts. Of note, in addition to cellular adaptive responses, anti-tumor IgM antibodies can also mediate direct cytopathic effects and the destruction of nascent tumors by identifying tumor-modified cell surface epitopes, as shown in mice[98]. In humans, natural IgM antibodies reactive against certain tumor-associated antigens have been reported during the asymptomatic stages of cancer and used as an early biomarker for breast cancer[99], indicative of active cancer immunosurveillance, although this warrants further investigation[97,100]. Overall, these studies also highlight the importance of B cells and their associated antibody repertoire in recognizing tumor-associated neoantigens and in facilitating cancer immunosurveillance.
Figure I in Box 3.
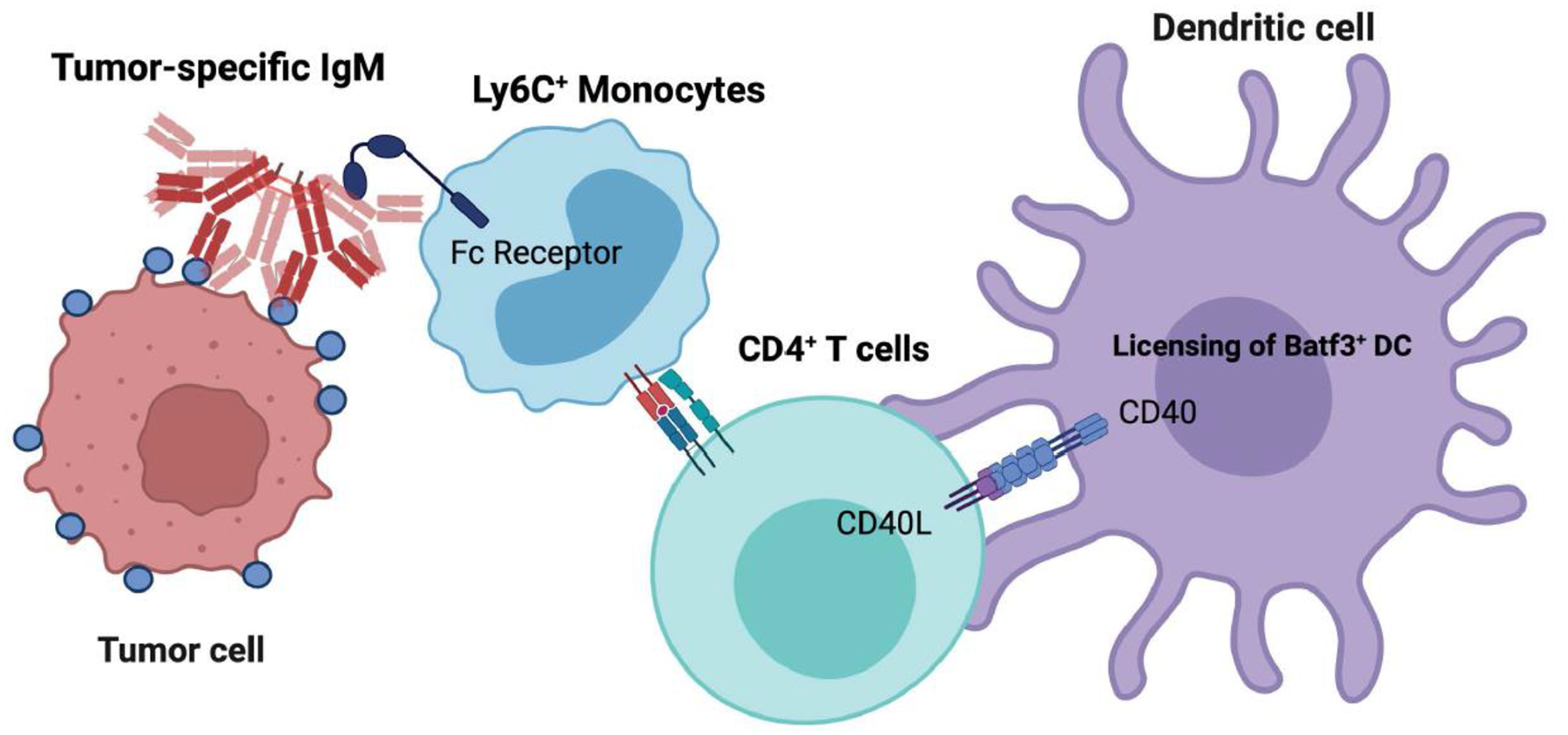
Natural IgM as an alternative pathway for cancer immunosurveillance.
Highlights:
Tumor-derived heat shock proteins (HSPs) and dsDNA are self-derived mammalian molecules that can provide adjuvanticity necessary to initiate adaptive immune responses.
We propose that the receptors for HSPs and dsDNA, which are CD91 and STING, respectively, may play a crucial role in cancer immunosurveillance; their loss, dysfunction, or inhibition allows for an increased incidence of cancer in mice and humans.
Tumor antigens chaperoned by HSPs are cross-presented by dendritic cells via CD91 to prime T cell responses. Estimates of the amount of chaperoned antigen and HSPs satisfies the quantitative limits available in nascent, emerging tumors, and we argue that this strongly supports such a mechanism as a likely important cross-presenting pathway in cancer immunosurveillance.
Acknowledgements
We would like to thank Professor Walter Storkus, Department of Dermatology, University of Pittsburgh for his critical review and evaluation of this manuscript. Figures in this manuscript were created with BioRender (BioRender.com). This work was funded in part by National Institute of Health Grants (NIH) CA233803 (RJB).
Glossary:
- Adjuvant
molecules that can trigger activation and maturation of APCs, e.g. DCs, that prime T cell responses by triggering co-stimulation and cytokine production
- Adjuvanticity
the condition of a molecule to confer innate immune stimulation
- Antigen presenting cells
immune cells that can take up and present antigen to induce an adaptive immune response
- Cellular Necrosis
type of cell death where plasma membranes are disrupted and cellular contents are leaked into the extracellular space
- CD91/LRP1
cell surface receptor on APCs ; involved in endocytosis of HSPs and signaling in response to HSPs
- Cross-priming
The ability of certain APCs to acquire exogenous antigen and present derivative peptides on the cell surface by MHC I to initiate a CD8+ T cell response
- Differential Aggretope Index (DAI)
algorithm index that represents differences in MHC binding affinity between wildtype and corresponding mutated peptides; it is a strong predictor of which mutated peptides will generate a T cell response
- Damage Sensing Receptor (DSR)
Receptors of the innate immune system that recognize molecules released by damaged or dying host cells
- Effector T cell
subset of T cells that execute effector functions by producing cytokines (CD4+) and/or cytotoxic mediators (CD8+) to target specific antigens
- Gap Junctions
intercellular channels that allow the exchange of small molecules and ions
- Heat shock protein (HSP)
Family of intracellular proteins that respond to cellular stress. A subset of HSPs, called the immunogenic HSPs, chaperone antigens and initiate immune responses when released from cells by binding to CD91 on APCs
- Immunoediting/Tumor editing
During the clonal expansion of tumors, tumors are modified by the immune system as they adapt. These modifications include downregulation or elimination of tumor antigens that are targeted by T cells
- Inflammasome
multiprotein oligomers in the cytosol of innate cells responsible for the activation of inflammatory responses following their activation and assembly
- Neoantigen
antigens expressed uniquely by tumors; typically resulting from mutated proteins processed into peptides and presented by MHC to T cells
- Neuropilins
Transmembrane glycoproteins expressed in neurons and DCs. On DCs, neuropilins can engage and promote Treg function
- Pathogen Associated Molecular Patterns
broad molecular epitopes expressed by pathogens and recognized by receptors on the innate immune system
- Pattern Recognition Receptors (PRR)
Receptors of the innate immune system that recognize broad patterns expressed by pathogens
- Regulatory T cells
subset of CD4+ T cells involved in immunoregulation or suppression of other immune cells
- Stimulator of interferon genes (STING)
Adaptor protein downstream of cGAS that recruits kinases TBK1 and IRF3 to induce type 1 IFNs and pro-inflammatory cytokine expression
- Shared antigen
antigens expressed across cancers and tissues of the same type
- Th1 effector molecules
Proteins of the immune system that are necessary for the development of CD4+ helper T cell type 1 immune responses responsible for the clearance of tumors and pathogens. These proteins include signaling and cytolytic proteins
- Tumor rejection antigens
Tumor-derived molecules that when targeted by the immune system, lead to elimination of the tumor
- Viral antigen
molecules expressed by viruses processed and presented by infected cells to the immune system
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ehrlich P (1909) Über den jetzigen Stand der Chemotherapie. Ber. Dtsch. Chem. Ges 42, 17–47 [Google Scholar]
- 2.Matzinger P (1994) Tolerance, danger, and the extended family. Annu. Rev. Immunol 12, 991–1045 [DOI] [PubMed] [Google Scholar]
- 3.Hernandez C et al. (2016) Damage-associated molecular patterns in cancer: a double-edged sword. Oncogene 35, 5931–5941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Acuto O and Michel F (2003) CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat. Rev. Immunol 3, 939–951 [DOI] [PubMed] [Google Scholar]
- 5.Srivastava PK et al. (1986) Tumor rejection antigens of chemically induced sarcomas of inbred mice. Proc. Natl. Acad. Sci. USA 83, 3407–3411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Z and Srivastava PK (1993) Tumor rejection antigen gp96/grp94 is an ATPase: implications for protein folding and antigen presentation. EMBO J. 12, 3143–3151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Udono H and Srivastava PK (1993) Heat shock protein 70-associated peptides elicit specific cancer immunity. J. Exp. Med 178, 1391–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Srivastava PK (1993) Peptide-binding heat shock proteins in the endoplasmic reticulum: role in immune response to cancer and in antigen presentation. Adv. Cancer Res 62, 153–177 [DOI] [PubMed] [Google Scholar]
- 9.Blachere NE et al. (1997) Heat shock protein-peptide complexes, reconstituted in vitro, elicit peptide-specific cytotoxic T lymphocyte response and tumor immunity. J. Exp. Med 186, 1315–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Binder RJ (2019) Immunosurveillance of cancer and the heat shock protein-CD91 pathway. Cell Immunol. 343, 103814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindquist S and Craig EA (1988) The heat-shock proteins. Annu. Rev. Genet 22, 631–677 [DOI] [PubMed] [Google Scholar]
- 12.Vabulas RM et al. (2002) The endoplasmic reticulum-resident heat shock protein Gp96 activates dendritic cells via the Toll-like receptor 2/4 pathway. J. Biol. Chem 277, 20847–20853 [DOI] [PubMed] [Google Scholar]
- 13.Basu S et al. (2000) Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int. Immunol 12, 1539–1546 [DOI] [PubMed] [Google Scholar]
- 14.Kroemer G et al. (2013) Immunogenic cell death in cancer therapy. Annu. Rev. Immunol 31, 51–72 [DOI] [PubMed] [Google Scholar]
- 15.Galluzzi L et al. (2017) Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol 17, 97–111 [DOI] [PubMed] [Google Scholar]
- 16.Ménoret A et al. (1999) Association of peptides with heat shock protein gp96 occurs in vivo and not after cell lysis. Biochem. Biophys. Res. Commun 262, 813–818 [DOI] [PubMed] [Google Scholar]
- 17.Pawaria S and Binder RJ (2011) CD91-dependent programming of T-helper cell responses following heat shock protein immunization. Nat. Commun 2, 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y et al. (2018) Cutting Edge: The Heat Shock Protein gp96 Activates Inflammasome-Signaling Platforms in APCs. J. Immunol 201, 2209–2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Binder RJ et al. (2000) CD91: a receptor for heat shock protein gp96. Nat. Immunol 1, 151–155 [DOI] [PubMed] [Google Scholar]
- 20.Basu S et al. (2001) CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity 14, 303–313 [DOI] [PubMed] [Google Scholar]
- 21.Binder RJ and Srivastava PK (2004) Essential role of CD91 in re-presentation of gp96-chaperoned peptides. Proc. Natl. Acad. Sci. USA 101, 6128–6133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sedlacek AL et al. (2019) CD91 on dendritic cells governs immunosurveillance of nascent, emerging tumors. JCI Insight 4, [Google Scholar]
- 23.Zhou YJ et al. (2014) Establishment of tumor-associated immunity requires interaction of heat shock proteins with CD91. Cancer Immunol Res 2, 217–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berwin B et al. (2003) Scavenger receptor-A mediates gp96/GRP94 and calreticulin internalization by antigen-presenting cells. EMBO J. 22, 6127–6136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Warger T et al. (2006) Interaction of TLR2 and TLR4 ligands with the N-terminal domain of Gp96 amplifies innate and adaptive immune responses. J. Biol. Chem 281, 22545–22553 [DOI] [PubMed] [Google Scholar]
- 26.Becker T et al. (2002) CD40, an extracellular receptor for binding and uptake of Hsp70-peptide complexes. J. Cell Biol 158, 1277–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Binder RJ et al. (2004) The heat-shock protein receptors: some answers and more questions. Tissue Antigens 64, 442–451 [DOI] [PubMed] [Google Scholar]
- 28.Binder RJ et al. (2000) Cutting edge: heat shock protein gp96 induces maturation and migration of CD11c+ cells in vivo. J. Immunol 165, 6029–6035 [DOI] [PubMed] [Google Scholar]
- 29.Basu S and Srivastava PK (1999) Calreticulin, a peptide-binding chaperone of the endoplasmic reticulum, elicits tumor- and peptide-specific immunity. J. Exp. Med 189, 797–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsutake T et al. (2010) High efficiency CD91- and LOX-1-mediated re-presentation of gp96-chaperoned peptides by MHC II molecules. Cancer Immun 10, 7. [PMC free article] [PubMed] [Google Scholar]
- 31.Sedlacek AL et al. (2016) Phenotypically distinct helper NK cells are required for gp96-mediated anti-tumor immunity. Sci. Rep 6, 29889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wood C et al. (2008) An adjuvant autologous therapeutic vaccine (HSPPC-96; vitespen) versus observation alone for patients at high risk of recurrence after nephrectomy for renal cell carcinoma: a multicentre, open-label, randomised phase III trial. Lancet 372, 145–154 [DOI] [PubMed] [Google Scholar]
- 33.Testori A et al. (2008) Phase III comparison of vitespen, an autologous tumor-derived heat shock protein gp96 peptide complex vaccine, with physician’s choice of treatment for stage IV melanoma: the C-100–21 Study Group. J. Clin. Oncol 26, 955–962 [DOI] [PubMed] [Google Scholar]
- 34.Crane CA et al. (2013) Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin. Cancer Res 19, 205–214 [DOI] [PubMed] [Google Scholar]
- 35.Kropp LE et al. (2010) Ovalbumin-derived precursor peptides are transferred sequentially from gp96 and calreticulin to MHC class I in the endoplasmic reticulum. J. Immunol 184, 5619–5627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stebbing J et al. (2004) The common heat shock protein receptor CD91 is up-regulated on monocytes of advanced melanoma slow progressors. Clin. Exp. Immunol 138, 312–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chandawarkar RY et al. (1999) The dual nature of specific immunological activity of tumor-derived gp96 preparations. J. Exp. Med 189, 1437–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chandawarkar RY et al. (2004) Immune modulation with high-dose heat-shock protein gp96: therapy of murine autoimmune diabetes and encephalomyelitis. Int. Immunol 16, 615–624 [DOI] [PubMed] [Google Scholar]
- 39.Kovalchin JT et al. (2006) In vivo treatment of mice with heat shock protein, gp 96, improves survival of skin grafts with minor and major antigenic disparity. Transpl. Immunol 15, 179–185 [DOI] [PubMed] [Google Scholar]
- 40.Kinner-Bibeau LB et al. (2017) HSPs drive dichotomous T-cell immune responses via DNA methylome remodelling in antigen presenting cells. Nat. Commun 8, 15648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Janetzki S et al. (2000) Immunization of cancer patients with autologous cancer-derived heat shock protein gp96 preparations: a pilot study. Int. J. Cancer 88, 232–238 [DOI] [PubMed] [Google Scholar]
- 42.Habich C and Burkart V (2007) Heat shock protein 60: regulatory role on innate immune cells. Cell Mol. Life Sci 64, 742–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou W et al. (2018) Structure of the Human cGAS-DNA Complex Reveals Enhanced Control of Immune Surveillance. Cell 174, 300–311.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luecke S et al. (2017) cGAS is activated by DNA in a length-dependent manner. EMBO Rep. 18, 1707–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andreeva L et al. (2017) cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature 549, 394–398 [DOI] [PubMed] [Google Scholar]
- 46.Sun L et al. (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang C et al. (2019) Structural basis of STING binding with and phosphorylation by TBK1. Nature 567, 394–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ishikawa H and Barber GN (2008) STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Diamond MS et al. (2011) Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med 208, 1989–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fuertes MB et al. (2011) Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J. Exp. Med 208, 2005–2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Woo S-R et al. (2014) STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schadt L et al. (2019) Cancer-Cell-Intrinsic cGAS Expression Mediates Tumor Immunogenicity. Cell Rep. 29, 1236–1248.e7 [DOI] [PubMed] [Google Scholar]
- 53.Lam AR et al. (2014) RAE1 ligands for the NKG2D receptor are regulated by STING-dependent DNA sensor pathways in lymphoma. Cancer Res. 74, 2193–2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marcus A et al. (2018) Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity 49, 754–763.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dhodapkar MV et al. (2008) Interactions of tumor cells with dendritic cells: balancing immunity and tolerance. Cell Death Differ. 15, 39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Diamond JM et al. (2018) Exosomes Shuttle TREX1-Sensitive IFN-Stimulatory dsDNA from Irradiated Cancer Cells to DCs. Cancer Immunol Res 6, 910–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xia T et al. (2016) Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res. 76, 6747–6759 [DOI] [PubMed] [Google Scholar]
- 58.Yang H et al. (2017) cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 114, E4612–E4620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song S et al. (2017) Decreased expression of STING predicts poor prognosis in patients with gastric cancer. Sci. Rep 7, 39858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xia T et al. (2016) Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 14, 282–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang C-A et al. (2017) DNA-Sensing and Nuclease Gene Expressions as Markers for Colorectal Cancer Progression. Oncology 92, 115–124 [DOI] [PubMed] [Google Scholar]
- 62.de Mingo Pulido Á et al. (2021) The inhibitory receptor TIM-3 limits activation of the cGAS-STING pathway in intra-tumoral dendritic cells by suppressing extracellular DNA uptake. Immunity 54, 1154–1167.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sakuishi K et al. (2010) Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med 207, 2187–2194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Amouzegar A et al. (2021) STING agonists as cancer therapeutics. Cancers (Basel) 13, [Google Scholar]
- 65.Dunn GP et al. (2004) The three Es of cancer immunoediting. Annu. Rev. Immunol 22, 329–360 [DOI] [PubMed] [Google Scholar]
- 66.DuPage M et al. (2012) Expression of tumour-specific antigens underlies cancer immunoediting. Nature 482, 405–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Prehn RT and Main JM (1957) Immunity to methylcholanthrene-induced sarcomas. J. Natl. Cancer Inst 18, 769–778 [PubMed] [Google Scholar]
- 68.Duan F et al. (2014) Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J. Exp. Med 211, 2231–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ishii T et al. (1999) Isolation of MHC class I-restricted tumor antigen peptide and its precursors associated with heat shock proteins hsp70, hsp90, and gp96. J. Immunol 162, 1303–1309 [PubMed] [Google Scholar]
- 70.Li HZ et al. (2013) Isolation and identification of renal cell carcinoma-derived peptides associated with GP96. Technol Cancer Res Treat 12, 285–293 [DOI] [PubMed] [Google Scholar]
- 71.Binder RJ and Srivastava PK (2005) Peptides chaperoned by heat-shock proteins are a necessary and sufficient source of antigen in the cross-priming of CD8+ T cells. Nat. Immunol 6, 593–599 [DOI] [PubMed] [Google Scholar]
- 72.Suto R and Srivastava PK (1995) A mechanism for the specific immunogenicity of heat shock protein-chaperoned peptides. Science 269, 1585–1588 [DOI] [PubMed] [Google Scholar]
- 73.Li M et al. (2001) Cell-associated ovalbumin is cross-presented much more efficiently than soluble ovalbumin in vivo. J. Immunol 166, 6099–6103 [DOI] [PubMed] [Google Scholar]
- 74.Blum JS et al. (2013) Pathways of antigen processing. Annu. Rev. Immunol 31, 443–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Srivastava P (2002) Interaction of heat shock proteins with peptides and antigen presenting cells: chaperoning of the innate and adaptive immune responses. Annu. Rev. Immunol 20, 395–425 [DOI] [PubMed] [Google Scholar]
- 76.Basombrío MA and Prehn RT (1972) Immune status of autochthonous and adoptively protected mice toward spontaneous and chemically induced tumors. Cancer Res. 32, 2545–2550 [PubMed] [Google Scholar]
- 77.Gross L (1943) Intradermal immunization of C3H mice against a sarcoma that originated in an animal of the same line. Cancer Res. 3, 326–333 [Google Scholar]
- 78.Burnet M (1957) Cancer: a biological approach. III. Viruses associated with neoplastic conditions. IV. Practical applications. Br. Med. J 1, 841–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Burnet FM (1971) Immunological surveillance in neoplasia. Transplant. Rev 7, 3–25 [DOI] [PubMed] [Google Scholar]
- 80.Peto J (2001) Cancer epidemiology in the last century and the next decade. Nature 411, 390–395 [DOI] [PubMed] [Google Scholar]
- 81.Svane IM et al. (1996) Chemically induced sarcomas from nude mice are more immunogenic than similar sarcomas from congenic normal mice. Eur. J. Immunol 26, 1844–1850 [DOI] [PubMed] [Google Scholar]
- 82.Qin Z and Blankenstein T (2004) A cancer immunosurveillance controversy. Nat. Immunol 5, 3–4; author reply 4 [DOI] [PubMed] [Google Scholar]
- 83.Schreiber RD et al. (2004) Response to “A cancer immunosurveillance controversy.” Nat. Immunol 5, 4–5 [Google Scholar]
- 84.Zhang L et al. (2020) Chronic stress-induced immune dysregulation in cancer: implications for initiation, progression, metastasis, and treatment. Am. J. Cancer Res 10, 1294–1307 [PMC free article] [PubMed] [Google Scholar]
- 85.Zinger A et al. (2017) Cancer and Aging - the Inflammatory Connection. Aging Dis 8, 611–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sender R and Milo R (2021) The distribution of cellular turnover in the human body. Nat. Med 27, 45–48 [DOI] [PubMed] [Google Scholar]
- 87.Preston BD et al. (2010) DNA replication fidelity and cancer. Semin. Cancer Biol 20, 281–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Goodman B and Gardner H (2018) The microbiome and cancer. J. Pathol 244, 667–676 [DOI] [PubMed] [Google Scholar]
- 89.Greathouse KL et al. (2018) Interaction between the microbiome and TP53 in human lung cancer. Genome Biol. 19, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peters BA et al. (2019) The Microbiome in Lung Cancer Tissue and Recurrence-Free Survival. Cancer Epidemiol. Biomarkers Prev 28, 731–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nejman D et al. (2020) The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 368, 973–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Thompson KJ et al. (2017) A comprehensive analysis of breast cancer microbiota and host gene expression. PLoS One 12, e0188873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Banerjee S et al. (2018) Distinct microbial signatures associated with different breast cancer types. Front. Microbiol 9, 951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Costantini L et al. (2018) Characterization of human breast tissue microbiota from core needle biopsies through the analysis of multi hypervariable 16S-rRNA gene regions. Sci. Rep 8, 16893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu Y et al. (2009) CD24-Siglec G/10 discriminates danger- from pathogen-associated molecular patterns. Trends Immunol. 30, 557–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhou Z-H et al. (2007) The broad antibacterial activity of the natural antibody repertoire is due to polyreactive antibodies. Cell Host Microbe 1, 51–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Brändlein S et al. (2003) Natural IgM antibodies and immunosurveillance mechanisms against epithelial cancer cells in humans. Cancer Res. 63, 7995–8005 [PubMed] [Google Scholar]
- 98.Atif SM et al. (2018) Immune surveillance by natural igm is required for early neoantigen recognition and initiation of adaptive immunity. Am. J. Respir. Cell Mol. Biol 59, 580–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fernández Madrid F (2005) Autoantibodies in breast cancer sera: candidate biomarkers and reporters of tumorigenesis. Cancer Lett. 230, 187–198 [DOI] [PubMed] [Google Scholar]
- 100.Vollmers HP and Brändlein S (2005) The “early birds”: natural IgM antibodies and immune surveillance. Histol Histopathol 20, 927–937 [DOI] [PubMed] [Google Scholar]