

Abstract
Multicomponent reactions (MCRs) are powerful synthetic tools in which more than two starting materials couple with each other to form multi-functionalized compounds in a one-pot process, the so-called “tandem”, “domino” or “cascade” reaction, or utilizing an additional step without changing the solvent, the so-called a sequential-addition procedure, to limit the number of synthetic steps, while increasing the complexity and the molecular diversity, which are highly step-economical reactions. The Ugi reaction, one of the most common multicomponent reactions, has recently fascinated chemists with the high diversity brought by its four- or three-component-based isonitrile. The Ugi reaction has been introduced in organic synthesis as a novel, efficient and useful tool for the preparation of libraries of multifunctional peptides, natural products, and heterocyclic compounds with stereochemistry control. In this review, we highlight the recent advances of the Ugi reaction in the last two decades from 2000–2019, mainly in the synthesis of linear or cyclic peptides, heterocyclic compounds with versatile ring sizes, and natural products, as well as the enantioselective Ugi reactions. Meanwhile, the applications of these compounds in pharmaceutical trials are also discussed.
We highlight the recent advances of the Ugi reaction in the last two decades from 2000–2019, mainly in the synthesis of linear or cyclic peptides, heterocyclic compounds with versatile ring sizes, and natural products, as well as the enantioselective Ugi reactions.
1. Introduction
Multicomponent reactions (MCRs) are powerful synthetic tools, in which more than two starting materials react with each other to form multi-functionalized compounds in a one-pot experimental procedure. Nowadays, MCRs are an exceptional strategy to synthesize many pharmaceutical and drug-like structure compounds due to their atom economy, high efficiency, low cost and simple experimental procedure.1,2 Isocyanide-based multicomponent reactions (IMCRs) have attracted the attention of organic chemists since the development of the first one by Mario Passerini 1921,3 which is known as the Passerini-three component reaction (P-3CR), where the coupling between aldehyde, carboxylic acid and isonitrile affords α-acyloxycarboxamide.4,5 In 1959, a new era of isonitrile chemistry started when Ugi discovered that the reaction between primary amine, carbonyl compound (aldehyde or ketone), carboxylic acid and isonitrile formed highly substituted α-aminoacyl amides with high biological activity and structural diversity.6 The Ugi-four component reaction (U-4CR) gives peptide-like structure known as bis-amides or peptomers, which are classified as peptidomimetics and have promising pharmacological properties.7 Furthermore, U-4CR is an economic and environmentally friendly reaction since it can also be performed in water as the solvent.8 Moreover, the optimization of the conditions of the Ugi reaction by stirring the reactants in a concentrated solution of polar protic solvents such as methanol, ethanol or 2,2,2-trifluoroethanol (TFE) has been investigated.9,10 Due to the high flexibility of the Ugi reaction with different functional groups, a wide range of linear bis-amides and pseudo-peptides (linear or cyclic) can be obtained and post-modifications can be achieved to synthesize versatile heterocyclic compounds with unusual structures and a wide range of biological activity.11 Furthermore, U-4CR has become increasingly efficient in the synthesis of many natural products and macro-cyclic molecules.12–14 It has been proven that many important compounds and natural products studied for their biological efficiency can be synthesized simply via U-4CR, which had led to the emergence of many important drugs for the treatment of several diseases. For instance, Crixivan® I (Indinavir™, MK 639) made by Merck is an important HIV protease inhibitor (Fig. 1), where the Ugi reaction contributes to the short synthesis of the piperazine derivative that is the main starting compound in production of Crixivan® I.12,15 Also, philanthotoxin-12 analogues that show potential noncompetitive inhibitory effects on various types of ionotropic receptors in the central nervous system have been synthesized via the Ugi reaction.16
Fig. 1. Structure of Crixivan I, furanomycin II and demethyldysidenin III.

The Ugi reaction is considered a key step in the synthesis of many natural products, for example, the naturally potent α-amino acid antibiotic (+)-furanomycin II isolated from Streptomyces threomyceticus was synthesized using U-4CR.14 Also, the natural product (+)-demethyldysidenin III and its epimer (−)-demethylisodysidenin, which is an antihypertensive, which was demonstrated by Williard and co-workers,17 was synthesized through the condensation of the traditional four components.14
In this review, highlighted the recent advances of the Ugi reaction in the last two decades from 2000–2019, mainly in the organic syntheses of linear and cyclic peptides, heterocyclic compounds, natural products, asymmetric Ugi reaction, and the valuable applications of these compounds in pharmaceuticals trials.
2. The synthetic and pharmaceutical applications of peptide and natural products
2.1. Synthetic and pharmaceutical applications of linear peptides
Recently, in the last two decades, chemists have become more interest in the synthesis of peptidomimetics and naturally occurring peptides via the Ugi reaction due to their special biological characters, great applicability and use in drug discovery.18 U-4CR reactions have proven their efficiency in the synthesis of these structures with the least number of steps and lowest cost, ensuring the synthesis of the target structure using different aliphatic or aromatic isonitriles, with carboxylic acid, carbonyl compound or amine derivatives with structural diversity to synthesize α-aminoacyl amides (Scheme 1).7,18
Scheme 1. Synthesis of α-aminoacyl amides via (U-4CR).

Fentanyl or fentanil is a synthetic opioid analgesic used as a pain medication to relieve pain after different treatments. Novel substituted amino acid-tethered norsufentanil derivatives 2 were synthesized by Ugi reaction utilizing the carboxylic acid-tethered norsufentanil 1 with a variety of aldehydes, amines, and isocyanides to produce a library of the desired norsufentanil derivatives compounds 2. The vivo analgesic activity of the synthesized compounds was evaluated by a tail flick test. Some of the synthesized compounds were found to be more potent than sufentanil, sufentanil citrate, and norsufentanil (Scheme 2).19
Scheme 2. Synthesis of a synthetic opioid analgesic norsufentanil derivative 2.

Alternatively, the convertible isocyanide 2-bromo-6-isocyanopyridine was reported in MCR, where it is good leaving group when it is attacked by the nucleophile of the resulting Ugi bis-amide moiety under both basic and acidic conditions. This reaction sequence was used to prepare the potent opioid carfentanil 4via acidic methanolysis of Ugi product 3 to give carfentanil 4 in near-quantitative yield (98%) (Scheme 3).20 Carfentanil 4 is an opioid analgesic belong to the fentanyl class, which is ∼100 000 times more potent than morphine.20
Scheme 3. Synthesis of carfentanil 4.

(−)-Viridic acid is a tetrapeptide generated by many fungi of the Penicillium group,21 which was first isolated from P. viridicatum.22 Wessjohann and co-workers used the Ugi reaction to prepare the racemate mixture of viridic acid 10 to shorten the pathway of other conventional methods. U-4CR between dipeptide 5, isobutyraldehyde 6, methylamine 7 and the anthranilate-derived isonitrile 8a and b were formulated to give 9a and b, and saponification of 9a and b afforded the racemic (±)-viridic acid 10 in 83% yield (Scheme 4).23 Furthermore, as a result of the unsuccessful separation of the epimers by the classical separation methods, the racemic mixture of viridic acid was investigated as an antibacterial agent against the Gram-negative bacterium Aliivibrio fischeri. Compounds (−)-10 and (±)-10 were the most potent ones with IC50 values of 45.0 ± 4.4 and 38.4 ± 5.8 μM, respectively.23
Scheme 4. Synthesis of (±)-viridic acid 10 using anthranilate-derived isonitriles 8a and b.

The Ugi reaction presents a rapid and simple method to synthesize highly biologically active phosphonic pseudo-peptides using a special type of isonitriles, which is 1-isocyanoalkylphosphonate diaryl ester derivatives.24 The structural features of the synthesized phosphonic pseudo-peptides were designed to act as inhibitors to target human neutrophil elastase, a serine protease, which can be inactivated through exclusive reaction with the aromatic ester and hydroxyl group of serine protease.25,26 This may lead to the development of many pathophysiological states such as cystic fibrosis and rheumatoid arthritis, or tumor growth and invasion.24
Two different types of phosphonic pseudo-peptides were prepared via the Ugi reaction using 1-isocyano-2-methylpropanephosphonic acid di(4-S-methylphenyl)ester (11) with butyl amine 14, Z-Gly 13, and aldehyde 12a and b, or piperazine 16, aliphatic acid 17 with aldehyde 12a and b to afford phosphonic pseudo-peptides 15a and b and 18a and b, respectively. Higher ability to inhibit the proteolytic activity of human neutrophil elastase was observed when 1-isocyano-2-methylpropanephosphonic acid di(4-S-methylphenyl)ester derivative (11) was used to obtain a series of Ugi products with different activity against examined serine protease (Scheme 5).24 According to Sienczyk, the most active Ugi products against the examined serine protease were when R = –CH2Bn (kobs/[I] = 1415 M−1 s−1) and when R = Bn (kobs/[I] = 1360 M−1 s−1). It is worth mentioning that both products showed high potency for the inhibition of human neutrophil elastase.24
Scheme 5. Synthesis of phosphonic pseudo-peptides via Ugi reaction.

Alternatively, a series of Ugi compounds containing a zinc-chelating moiety were found to act as histone deacetylase (HDAC) inhibitors, where HDACs are potential compounds that inhibit the growth of tumor and are in clinical trials as promising antitumor agents.27 The Ugi reaction is the key step in the synthesis of target HDAC inhibitors, giving the Ugi products with an ester function 20a and b, which are subsequently transformed into benzamides 22a and b or hydroxamate 23a and bvia the carboxylic acid intermediate 21a and b (Scheme 6).28
Scheme 6. Synthesis of bis-peptides containing benzamides 22a and b or hydroxamate 23a and bvia Ugi reaction.

Alternatively, another interesting strategy was applied to synthesize Ugi products that could disturb protein–protein interaction. These compounds were designed to be planar, aromatic and functionalized with four points of diversity to easily recognise protein surfaces. The strategy was based on 6-alkyloxy-naphthalene-2-carbaldehyde hydrolysis of Ugi product 24 when (R1 = CH2COOt-Bu), with trifluoroacetic acid to give the corresponding acid 25 (Scheme 7).29
Scheme 7. Synthesis of bis-amides 24 and 25via Ugi reaction.

Furthermore, the Ugi reaction was employed in the synthesis of a targeted library of C-capped dipeptide efflux pump inhibitors, where C-capped dipeptides BU-005 27 were prepared via Ugi four-component reaction via the full deprotection of Boc and DMB of Ugi product 26 using TFA (Scheme 8).30 Inhibitors of drug efflux pumps have great potential as pharmacological agents, which restore the drug susceptibility of multidrug resistant bacterial pathogens. C-capped dipeptides BU-005 27 inhibit two chloramphenicol-specific efflux pumps in Streptomyces coelicolor, a Gram-positive bacterium, which is a relative of the human pathogen Mycobacterium tuberculosis.30
Scheme 8. Synthesis of C-capped dipeptides BU-005 27.

N-Nucleosides have also been synthesized via the U-4CR protocol. A protocol was developed to afford 5-substituted pyrimidine nucleoside N-acylamino acid amide scaffolds 30, which show promising anti-leishmanial activity in the 10−5 M range. The reaction was based on utilizing aniline derivatives 28, 5-formyl-2′-deoxyuridine (29), carboxylic acid and isonitrile to afford the desired 5-position-substituted product 30. In contrast, using trimethylsilylazide 31 instead of an ordinary carboxylic acid gave tetrazolo compounds 32, which show more flexibility than 30 and show striking antiviral and anti-leishmanial activities (Scheme 9).31 It should be noted that pyrimidine derivatives show potential biological activities and wide clinical use in the few last years.32,33
Scheme 9. Synthesis of Ugi nucleosides using 5-formyl-2′-deoxyuridine 29.

Linderman and co-workers prepared convertible isonitrile 35,34 which was subsequently used for the synthesis analogues of uracil polyoxin C (UPOC) methyl ester 38via the Ugi reaction. The polyoxins are group of nucleoside antibiotics that have found use as agricultural fungicides. Because of their antimicrobial activities, polyoxins that play a crucial biological role against chitin synthase (CS) were obtained from Saccharomyces cerevisiae and Candida albicans.35 Chitin synthases are tempting targets for inhibition in fungi and insects.36 The synthesis of the polyoxins and the structurally related nikkomycins was achieved by Ugi reaction using 2′,3′-isopropylidine-protected uridine-5′-aldehyde (33), 2,4-dimethoxybenzylamine (34), isoxazolecarboxylic acid derivative 36, and the convertible isonitrile 35. Subsequently, acidic hydrolysis achieved complete deprotection of the isopropylidene group and DMB group, and conversion of the isonitrile-derived amide into the corresponding uracil polyoxin C (UPOC) methyl ester 38 (Scheme 10).36
Scheme 10. Synthesis of (UPOC) methyl ester 38via Ugi reaction.

Fluorinated peptides have attracted significant interest because the insertion of fluorine into peptides can modulate their acidity, basicity, hydrophobicity, geometry, conformation, reactivity, and biological activity.9,37–39 The Ugi reaction was used to synthesize bis-amide with exceptional structural features such as gem-di-fluorinated pseudo-peptides. The Ugi reaction using 2,2-difluoro-3-(2-hydroxy-1R-phenylethylamino)-3S-phenylpropionic acid (39), isonitrile, aldehyde, and primary amine afforded di-fluorinated pseudo-peptides derivatives 40 in methanol with up to 95% yield (Scheme 11).40
Scheme 11. Synthesis of gem-di-fluorinated pseudo-peptides via Ugi reaction.

Alternatively, tertiary glycosyl amide derivatives 45 were prepared via the Ugi reaction using d-β-galactopyranosyl pentaacetate amine (41) with formaldehyde 42, methyl isocyanoacetate (43) and terephthalic acid (44) followed by deacetylation. The applications of tertiary glycosyl amides are as frameworks for the design and preparation of ligands that will facilitate protein–protein or other receptor–receptor interactions. The partiality of limited divalent ligands (or higher order) planned to bind to protein that recognize carbohydrates would encourage clustering and associatively advance protein–protein interactions, which may be essentially higher than monovalent partners or multivalent ligands without these properties. This may be valuable as a modern methodology within the advancement of therapeutics dependent on carbohydrates (Scheme 12).41
Scheme 12. Synthesis of tertiary glycosyl amides 45via Ugi reaction.

The Ugi reaction differs from other traditional peptide coupling techniques, where it represents a short and efficient pathway to design and synthesize linear bis-amides in a one-pot reaction, which can provide pharmaceutical applications.
2.2. Synthetic and pharmaceutical applications of macro and cyclic peptides
Cyclic peptides are widely found in marine organisms and fungi.42 Many of these peptides show particular biological activity, and therefore are highly interesting from a pharmaceutical point of view.42,43 Cyclic peptides show interesting biological activity, putting them on the map of organic chemists to create new methods with minimal steps, which is beneficial for the synthesis of cyclic peptides with exceptional structural features. U-4CR is a promising strategy to synthesize these cyclic peptides through the ring-closing metathesis (RCM) approach.44 It was shown that not only the length or the sequence of the peptide chain is responsible for the success of RCM, but also the synthetic approach. Moreover, the Ugi reaction using allyloxycarbonyl (Alloc)-protected isonitrile 46 and Alloc-protected carboxylic acids 48 and 51, 1-(3-methoxyphenyl)ethanamine (47), and isobutyraldehyde (6) gives Ugi adducts 49 and 52 in excellent yields, respectively. The peptides formed via U-4CR undergo the RCM to form cyclic peptides 50 and 53 in the presence of Ru-catalyst under different reaction conditions, respectively (Scheme 13).44
Scheme 13. Synthesis of cyclic peptide via Ugi reaction.

Another interesting example reported was the synthesis of biaryl ether-containing macrocycle amide via a two-step reaction starting with U-4CR using α-(4′-fluoro-3′-nitro)phenethyl isocyanoacetate (55) with m-hydroxy phenyl alkylcarboxylic derivatives 54a and b followed by intramolecular SNAr-based cycloetherification to give products 57a and b, respectively. The two-step synthesis increased the molecular complexity and points of diversity, where the presence of the nitro group in macrocycles facilitates macrocyclization and provides functional groups in the existing macrocycles, and accordingly increases the molecular diversity (Scheme 14).45
Scheme 14. Two-step synthesis of biaryl ether-containing macrocycles.

Furthermore, a general strategy for the effective synthesis of artificial disulfide-linked macrocycles via U-4CR after oxidative cyclization was introduced. The Ugi reaction using cysteine isocyanide 58 and Fmoc–Cys(Trt)–OH acid 59 with a variety of amine or aldehyde yielded diastereoisomeric combinations 60 in MeOH/THF/DMF (1 : 1 : 0.1, 0.2 M). This protocol was used to generate the novel isolated disulfide-bridged peptides 61 by Ugi cyclization using I2/MeOH/DCM (Scheme 15).46
Scheme 15. Sulfur-switch Ugi reaction for the synthesis of disulfide-bridged peptides 61.

A successive combination between the Ugi reaction and click reaction was utilized for the synthesis of triazole connected to cyclic glycopeptidomimetics. This combination mixed the usual four Ugi reactants, protected glycosyl amine 64, propargyloxycarbonyl Poc-amino alkyl isonitriles derivatives 62, α-azido carboxylic acid 63, and aldehydes in methanol afford Ugi products 65 with a terminal alkyne and azido group. It is worth mentioning that the double nature of the propargyloxycarbonyl (Poc) group was investigated for amine protection and the catalytic cycloaddition with an azide in the presence of CuSO4·5H2O/sodium ascorbate in tBuOH/water, giving 66 as a mixture of two diastereomers (Scheme 16).47
Scheme 16. Synthesis of triazole-linked cyclic glycopeptidomimetics via Ugi reaction.

On the other hand, the macrocyclization of pentapeptides was performed using the Ugi reaction through a three-step protocol to obtain the expected biologically active compounds 70a and b and 74a and b in good yield. This methodology was based on the use of 2,4-dimethoxybenzylamine (34) in MCR of dipeptide acids 68 and 72 and isocyanides 67 and 71 with aldehyde to afford N-alkylated pentapeptides 69 and 73, respectively. The deprotection of both pentapeptides followed by macrolactamization by propanephosphonic acid anhydride (T3P) or benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) afforded cyclic peptides 70a and 74a, respectively. Treatment of 70a and 74a with TFA in the presence of DCM easily removed the DMB N-substituent to afford 70b and 74b in good yield (Scheme 17).48
Scheme 17. Macrocyclization of pentapeptides assisted by a traceless turn-inducing core derived from the Ugi reaction.

Ditopic bifunctional chelating agents 78a and b were synthesized via U-4CR in a single synthetic step utilizing 1,4,7,10-tetraazacyclododecane-N,N′,N′′,N′′′-tetraacetic acid DOTA and DOTA monoamide (DOTAMA) analogues as amine 75a and b and acid 76 input. Different aldehydes and isocyanides afforded Ugi products 77a and b using methanol with ultrasound irradiation. Hydrolysis of the t-butyl esters of 77a and b was successfully carried out by TFA, and the dimeric ligands 78a and b were obtained as analytically pure white solids by precipitation with ether (Scheme 18).49
Scheme 18. General synthesis of ditopic bifunctional chelating agents 78a and bvia U-4CR.

The wide applicability of optical and nuclear properties of yttrium (Y) and gadolinium (Gd) cations can be used in the synthesis of homo- and heterodimetallic systems for diagnostic and therapeutic applications. The Y(iii) complex of DOTAMAEn 79 and the Gd(iii) complex of DOTAMAC6OH 80 were then reacted together with paraformaldehyde and cyclohexyl isocyanide 81 to give heteroditopic Y–Gd complex 82 (Scheme 19). One of the Gd(iii) lipophilic complexes (R1 = H, R2 = C18H37) forms micelles at concentrations <0.1 mM and bind to human serum albumin (HSA) and has been studied by proton relaxometry, and the affinity constant of the complex and the relaxivity of the macromolecular adduct (rb1p = 38.1 mM−1 s−1; 20 MHz and 310 K, respectively) were derived.49
Scheme 19. Synthesis of a heteroditopic Y–Gd complex by U-4CR.

2.3. Synthetic and pharmaceutical application of natural products
Furthermore, the Ugi reaction considered as a key step in the synthesis of many natural products, where not only bacteria contain attractive natural biologically active compounds, but also marine organisms contain interesting natural products.50 Exigurin 89 was isolated from the marine sponge Geodia exigua collected in Oshima Island, and boneratamide A 83 isolated from the marine sponge Axinyssa aplysinoides collected in Indonesia.51 Boneratamide A 83 has an extraordinary structure, which contains terpene and amino acid units connected through an amide linkage. Despirocyclic boneratamide A analogue 86 was simply prepared via the Ugi reaction of terpene isocyanides 84 with α-methyl l-glutamate 85 in yield 43%.52 Moreover, the synthesis of exigurin 89 originated from four building blocks including (−)-10-epi-axisonitrile-3 87, and the simple substances formaldehyde 42, sarcosine 88 and methanol. Similarly, the Ugi reaction between axisonitrile-3 83 acetone 85 and glutamic acid 84 was the key step in the biosynthesis of boneratamide A 83 (Scheme 20).13,53,54
Scheme 20. Total synthesis of a despirocyclic boneratamide A analogue 86 and exigurin 89via the Ugi reaction.

Pacidamycin D 90, a member of a class of uridylpeptide antibiotics, was first isolated from Streptomyces coeruleorubidus AB 1183F-64 in 1989.55 The analogues of pacidamycin D 90 are 3′-hydroxypacidamycin D 91 (Fig. 2), a type of uridylpeptide antibiotics, which are selective antibacterial agents against Pseudomonas aeruginosa, and also act as inhibitors of phospho-MurNAc-pentapeptide transferase (MraY). Their IC50 for MraY is 42 nM and MIC for different types of P. aeruginosa range between 8–32 μg mL−1. MraY is a vital enzyme in bacteria and plays an important role in the peptidoglycan biosynthesis pathway, where it is responsible for the formation of lipid I.56
Fig. 2. Structure of pacidamycin D 90 and its analogue 3′-hydroxypacidamycin D 91.

3′-Hydroxypacidamycin D 91 can be synthesized via U-4CR. The condensation between urea dipeptide 92, 2,4-dimethoxybenzylamine (34), 2-N-methylaminopropionaldehyde derivative 93 and the α,β-unsaturated isonitrile derivative of uridine 94 led to the formation of compound 95a and its diastereomer in acceptable yields, which could be separated by column chromatography. Upon the selective removal of the Boc group of 95a, the liberated secondary amine of 95b was coupled with N-Boc-l-Ala 96 to afford the protected 3′-hydroxyl pacidamycin D 97. This was followed by deprotection steps until the final target products 3′-hydroxypacidamycin D 91 and its epimer racemate (Scheme 21).57
Scheme 21. Total synthesis of 3′-hydroxypacidamycin D 91.

The Ugi adduct 95a was then used to prepare several analogues by reaction with different Boc-protected amino acids to afford compounds with positive biological activity.57
The glutarimide moiety (2,6-piperidinedione) is present in natural product scaffolds that showed diverse biological activities.58,59 This scaffold is present in the structure of julocrotine 105, which has shown antiproliferative effects in vitro against the promastigote and amastigote forms of Leishmania amazonensis. The glutarimide alkaloid julocrotine 105 was prepared through the reaction between (R)-(+)-1-(4-methoxyphenyl)-ethylamine (100) as a chiral auxiliary and the usual Ugi reactants to give 102a, followed by ester basic hydrolysis to give the corresponding carboxylic acid 102b. Subsequently, intermolecular cyclization was carried out with acetic anhydride and sodium acetate to yield the natural julocrotine 105 and its diastereomer 106 (Scheme 22).60
Scheme 22. Synthetic route for julocrotine 105.

3. Synthetic and pharmaceutical applications of heterocyclic compounds
Versatile methods have been reported for the synthesis of different heterocycles, but most of them depend on multi-step systems, which result in high costs and long reaction time.61 On the other hand, isocyanide-based multicomponent reactions (IMCRs) guarantee a one-pot procedure, shorter time and products with high functionality and high yields. U-4CR is one of the most common reactions used in the synthesis of heterocyclic compounds through several routes, including post-modification reactions (from linear adduct to heterocyclic compound) or the UDC strategy (Ugi/deprotect/cyclize), where the final step of ring closing is mediated through amide bond formation.62 Due to the extreme importance of different heterocycles as biologically active compounds,63,64 this review will present the most efficient synthetic routes of U-4CR to synthesize different heterocyclic systems with interesting biological activity, attracting valuable attention from organic and medicinal chemists.
3.1. Synthesis of five-membered heterocyclic compounds
Dihydroxypyrrolidines are nitrogen-based five-membered heterocyclic compounds, which are found in many natural occurring compounds that have promising biological activities. For instance, some alkaloids contain dihydroxypyrrolidine moieties such as swainsonine 107,65 hyacinthacine 108,66 and uniflorine A 109 67 (Fig. 3), which are used as anticancer or antidiabetes compounds.
Fig. 3. Structures of some biologically active dihydroxypyrrolidines.

The synthesis of 1,2-disubstituted-cis-3,4-dihydroxypyrrolidine derivatives 114a was reported using the Ugi reaction protocol. Polindara-García and co-workers synthesized different Ugi products in TFE/InCl3 under microwave irradiation using propargyl amine 110 and glyoxal derivatives 111 to afford Ugi product 112. Subsequently, cesium carbonate and MWI facilitated the cyclization process to obtain 3-pyrroline derivatives 113. Treatment of 113 with osmium tetraoxide and N-methylmorpholine-N-oxide (NMO) was carried out to afford the medicinally dihydroxylated diastereoisomers 114a (cis-isomer) and 114b (trans-isomer) (Scheme 23).68
Scheme 23. Synthesis of 1,2-disubstituted-cis-3,4-dihydroxypyrrolidine derivatives 114a and b.

The reaction between isocyanoalkyl carbonate 115 with the different Ugi components gave the usual Ugi products 116 in moderate to excellent yields. Compounds 116 gave N-acyloxazolidinones 117 in the presence of t-BuOK/THF and molecular sieves 4 Å (MS 4 Å).69 On the other hand, upon treatment of N-acyloxazolidinones 117 with lithium thiolate derived from n-BuLi and n-dodecanethiol 118, the conversion occurred effectively to afford n-dodecanethiol esters 119 (Scheme 24).69
Scheme 24. Synthesis of N-acyloxazolidinones.

On the other hand, N-acylpyrroles building block scaffolds were prepared using 4-isocyanopermethylbutane-1,1,3-triol (IPB) 120 as a convertible isonitrile via the Ugi reaction to afford bis-amide 121. Bis-amide 121 obtained from the Ugi reaction was converted into highly activated N-acylpyrroles 122 using TFA (Scheme 25).70
Scheme 25. Synthesis of N-acylpyrroles 122.

The functionalized N-acyl-2-vinylpyrrolidines derivatives 126 were prepared via the Ugi reaction using unsaturated isocyanide 123 together with a variety of carbonyl compounds, primary amines, and carboxylic acids for the preparation of Ugi derivatives 124, followed by a second transformation involving different Pd catalysts and SN2′ cyclization to give 126, where in most cases the Pd(0)-catalyzed reaction gave the final product in low yield (Scheme 26).71
Scheme 26. Synthesis of N-acyl-2-vinylpyrrolidines derivatives 126.

Also, Ugi–Dieckmann reaction combination was utilized in the synthesis of the tetramic acid derivatives, which are an important class of nitrogen-containing heterocycles and considered as key structures in many natural products with interesting biological activity, including antiviral72 and phytotoxic activities.73 The convertible isonitrile 1,1-dimethyl-2-isocyano-ethyl-methylcarbonate (127) was condensed with other Ugi reactants to give 128, which was deprotonated and then cyclized to 129via intramolecular attack of the enolized anion, allowing 5,5-dimethyloxazolidin-2-one to act as a leaving group and a Dieckmann-like cyclization to tetramic acid derivatives 131a and b occurred (Scheme 27).74
Scheme 27. Synthesis of tetramic acid derivatives 131a and bvia Ugi–Dieckmann reactions.

On the other hand, a series of 2,4,5-trisubstituted oxazoles were synthesized through another Ugi combination via Ugi/Robinson–Gabriel reactions, condensation of 2,4-dimethoxybenzylamine (34) with phenylglyoxal 132 and other Ugi reactants to afford bis-amide 133. The conversion of Ugi product 133 to oxazole 135 was subsequently carried out via a debenzylation/cyclodehydration protocol using (POCl3), which led to the desired Robinson–Gabriel cyclodehydration reaction (Scheme 28).75
Scheme 28. Synthesis of 2,4,5-trisubstituted oxazoles via Ugi/Robinson–Gabriel reactions.

In another interesting synthesis employing the Ugi reaction is the synthesis of aryloxazolone compounds, which are found in many potent non-steroidal drugs and found to be anti-inflammatory and selective COX-2 inhibitors.76 The reaction of different derivatives of arylglyoxals 136 and anilines 138 with cyclohexyl isocyanide (81) and trichloroacetic acid in dichloromethane in the presence of molecular sieves gives aryloxazolones 141 in single synthetic steps, where the formed Ugi bis-amide is not isolated and spontaneously cyclized with elimination of chloroform to irreversibly afford the target product (Scheme 29). The same reaction under the classical Ugi reaction conditions (MeOH, rt) gives bis-amide 140, which is converted to 141 after treatment with Et3N/DCM. Also, the same Ugi reactants can afford deacylated Ugi products 139 in isopropyl ether at room temperature (Scheme 29).77
Scheme 29. Synthesis of aryloxazolone derivatives utilizing trichloroacetic acid via Ugi reaction.

A series of benzene/imidazole systems with more than three connected aromatic rings was synthesized using U-4CR through the condensation of terephthalic acid 44, c-hexylisocyanide 81 with n-butylamine 14, and phenylglyoxal hydrate 142 to afford Ugi adduct 143 followed by condensation with ammonia 144 to give the target compound 145 (Scheme 30).78
Scheme 30. Two-pot synthesis of benzene/imidazole systems.

3.2. Synthesis of fused five-membered heterocyclic compounds
As mentioned before, nitrogen-containing heterocycles are an important class of organic compounds, which represent a wide section of drugs and medicinal chemistry.79,80 A series of fused benzimidazole–isoquinolinones (BIDs) was prepared via Ugi reaction utilizing N-Boc-2-aminophenylisonitrile 147, methyl 2-formylbenzoate 146, amine and carboxylic acid in methanol. The observed Ugi products 148 were deprotected and then cyclized using 10% TFA/DCE, MW to afford the desired biologically active BIDs 150 (Scheme 31). The obtained BIDs were screened against human colorectal cancer (CRC), which showed anti-cancer activity against the SW620 and HT29 CRC lines. It was found that R1 = i-Bu and R2 = 3-pyridyl gave the most potent compound with the lowest growth inhibition (GI50) values among the other observed BIDs for both CRC cell lines (GI50 = 23.78 μM for SW620 and 24.13 μM for HT29). Also, the compound molecular mechanism was explored and the result showed that it suppresses CRC proliferation, growth and survival, and addition, it induces mitochondrial-mediated apoptosis through prohibition of the PI3K/AKT/mTOR signaling cascade (Scheme 31).81
Scheme 31. Synthesis of fused benzimidazole–isoquinolinones 150via Ugi reaction.

Alternatively, the UDC (Ugi/deprotection/cyclization) strategy was used in the synthesis of fused benzimidazole–quinoxalinones scaffolds in good yields using N-Boc-2-aminophenylisonitrile (147) with different 2-fluoroaniline derivatives 151a–c and other Ugi reactants after stirring in methanol at room temperature. After deprotection and cyclization occurred in TFA/DCE to afford 153a–c, a nucleophilic substitution reaction of 153a–c was carried out in DMF and catalyzed by cesium carbonate, affording benzimidazole–quinoxalinones 154a–c (Scheme 32).82
Scheme 32. Synthesis of fused benzimidazole–quinoxalinones 154a–cvia UDC reaction.

Using the same UDC (Ugi/deprotection/cyclization) strategy, the polycyclic heterocycles benzimidazole–quinoxalinones 160a–f were synthesized utilizing a range of tethered ketone acids 155. This strategy will accelerate the preparation of a large number of compounds in high through-put screening in pharmaceutical studies (Scheme 33).82
Scheme 33. Synthesis of fused BIDs polycyclic 160a–fvia UDC reaction.

In another application of the UDC strategy, two series of fused piperazine–benzimidazole compounds were prepared using the same isonitrile 147 under similar reaction conditions, followed by intermolecular nucleophilic substitution reaction to give the target compounds, which have great applicability in medicinal chemistry (Scheme 34).83
Scheme 34. Synthesis of piperazine–benzimidazoles 164via Ugi reaction.

Furthermore, the same strategy was used to synthesize a wide range of more complex piperazine–benzimidazole compounds, where isonitrile 147, bromoacetic acid 161, N-Boc-protected o-phenylenediamine derivatives 165 and aldehydes react with each other to give 166 followed by deprotection to finally give 169 through intermediates 167 and 168 (Scheme 35).83
Scheme 35. Synthesis of piperazine–bis-benzimidazoles 169via Ugi reaction.

Oxindoles are important nitrogen-containing heterocycles that are found in a large number of natural products and pharmaceuticals.84,85 An important two-step synthesis of oxindoles 172 was achieved by Ugi reaction in the first key step using 2-iodobenzaldehyde derivatives 170 followed by efficient intramolecular Pd(ii)-catalyzed Buchwald–Hartwig reaction in the presence of microwave heating to advance the second step reaction (Scheme 36).86
Scheme 36. Synthesis of oxindoles 172via Ugi/Buchwald–Hartwig reaction.

Furthermore, oxoisoindoles 174 are an important class of heterocyclic compounds that represent valuable building blocks for the synthesis of pharmaceutical compounds and natural products.87 They were synthesized via a Ugi four-center, three-component reaction (Ugi-4C-3CR) by condensation between 2-formylbenzoic acid (173), amines and isonitriles. The reaction was promoted by propylphosphonic anhydride (T3P®) to avoid the problem of long reaction time (Scheme 37).88
Scheme 37. Synthesis of oxoisoindoles 174via Ugi reaction.

A series of benzimidazole derivatives were synthesized by Ding et al., where they extended their study on Ugi and aza-Wittig reactions to synthesize more valuable heterocycles. They developed Ugi reaction combined with catalytic aza-Wittig reaction to synthesize multi-substituted benzimidazoles under the classical Ugi reaction conditions in the presence of a catalytic amount of 3-methyl-1-phenyl-2-phospholene 1-oxide (178), 2-aminobenzoyl azide derivatives 175, aldehydes, carboxylic and isonitriles a one-pot reaction proceeds easily to give the target poly-substituted benzimidazoles 179 (Scheme 38).89 On the other hand, benzimidazoles act as antibacterial,90 anti-HIV-1,91 anti-inflammatory,92 and anticancer agents.93
Scheme 38. Synthesis of benzimidazoles derivatives 179via Ugi reaction/catalytic aza-Wittig cyclization.

3.3. Synthesis of pyrazinone or diketopiperazine compounds
On the other hand, the UDC strategy was applied to achieve a series of anti-inflammatory peptidic pyrazinones 184 in a two-step methodology starting with the usual Ugi reaction using N-Boc glycine 180, aromatic aldehyde 181, aminoacetaldehyde dimethyl acetal 182 and isonitrile, followed by microwave-assisted deprotection and oxidative cyclization step (Scheme 39).94 The synthesized pyrazinones were evaluated using in vivo topical anti-inflammatory model using tetradecanoylphorbol acetate (TPA) as an inducer of mouse ear edema, and all the compounds showed interesting results. The most potent compounds were obtained when R1 = c-Hex and R2 = Me or CF3 and their infective dose ID50 values were determined to be 0.45 and 0.46 μmol per ear, respectively, where both compounds were more active than the selective COX-2 inhibitor celecoxib.94
Scheme 39. Synthesis of peptidic pyrazinones 184via Ugi reaction.

Moreover, a methodology based on the Ugi reaction was applied to obtain pyrazin-2(1H)-one analogues, which are precious moieties in drug discovery and many therapeutic agents.95 α-Ketoaldehyde 185, N-Boc-amino acid 186, amine, and isonitrile were utilized to afford Ugi bis-amide 187, followed by deprotection–cyclization via HCl/ether at 0 °C to obtain 3,4-dihydropyrazin-2(1H)-ones (188) or via TFA/DCE at 80 °C to obtain pyrazin-2(1H)-ones (189) (Scheme 40).96
Scheme 40. Synthesis of pyrazin derivatives 188 and 189via Ugi reaction.

Epelsiban 190 is one of the most important orally bioavailable drugs, which is a powerful and selective oxytocin receptor (OXTR) antagonist (Fig. 4).97 The synthesis of epelsiban and its derivatives could be achieved through U-4CR by condensation of N-benzyloxycarbonyl-R-indanylglycine 191, pyridylaldehydes 192, o-benzyloxy phenylisonitrile (193) and R-α-aminoacid esters 194 to give tri-peptide product 195 followed by hydrogenolysis to eliminate the Z and benzyl groups to give 196, and subsequent treatment of 196 with carbonyldiimidazole CDI and a variety of secondary amines gave the desired RRR amides (epelsiban derivatives) 197 (Scheme 41).98 Investigation of the pharmacokinetic profile in rats, dogs, and cynomolgus monkeys of these derivatives showed that 2′,6′-dimethyl-3′-pyridyl R-sec-butyl morpholine amide epelsiban 190 is a significantly powerful oxytocin antagonist (pKi = 9.9) with >31 000-fold selectivity with no significant P450 inhibition.98
Fig. 4. Structure of epelsiban 190.
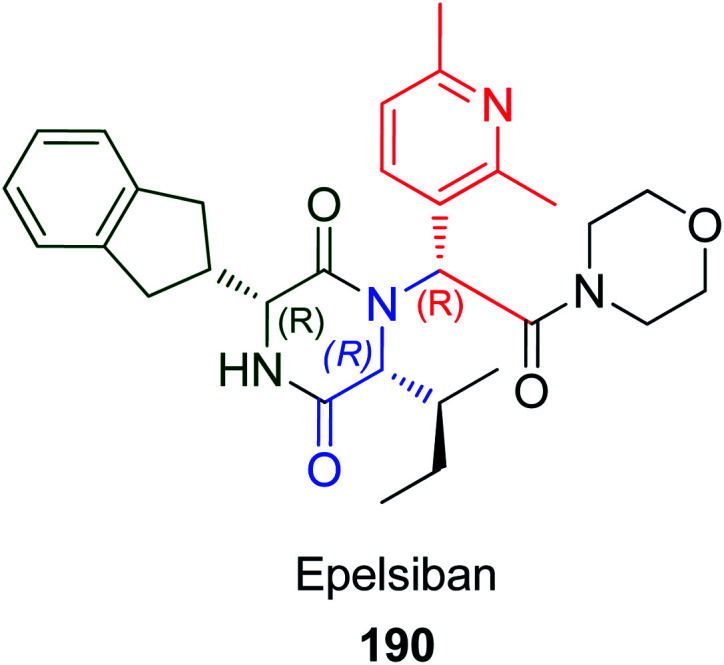
Scheme 41. Synthesis of epelsiban derivatives 197via Ugi reaction.

Another protocol was used to prepare derivatives of heterocycles with a pyrazine moiety utilizing a two-step synthesis including U-4CR followed by [5 + 1] cyclization to obtain the target 1,6-dihydro-6-oxopyrazine-2-carboxylic acid derivatives 201. Product 201 was prepared via the reaction between arylglyoxals 198, benzoylformic acid 199, amines and isonitrile to afford the usual Ugi adduct 200a and its tautomer 200b, which cyclized in the next step in the presence of ammonium acetate in acetic acid to obtain 201 in good yields (Scheme 42).99
Scheme 42. Synthesis of 6-oxopyrazine-2-carboxamide derivatives 201via Ugi reaction.

Moreover, a wide range of heterocyclic compounds are commercially available drugs to treat many infections. Praziquantel (PZQ) 206 100 is a drug containing a pyrazino isoquinoline ring system as its core structure, which can be synthesized via the Ugi reaction under mild conditions (MeOH, rt) (Scheme 43).101,102 PZQ 206 is widely used in the treatment of schistosomiasis, which is disease also known as bilharzia or silent pandemic.103,104 Indeed, PZQ 206 showed activity against trematode and cestode infections in humans and animals.105
Scheme 43. Synthesis of anti-schistosomal praziquantel drug via Ugi reaction.

Additionally, the Ugi strategy can be applied to synthesize many complex spin-labeled structures for EPR analysis for screening methods in biomedically significant ligand binding investigations. Labelled diketopiperazines (DKPs) and peptide–peptoid chimera were synthesized through the reaction between 4-amino-2,2,6,6-tetramethylpiperidinyloxy (4-amino TEMPO) (207) and the other Ugi reactants to form Ugi products 208a–d. After the formation of Ugi products 208a–d, the corresponding acylpyrroles 209 were obtained in the presence of camphorsulfonic acid (CSA) under reflux. Similarly, the basic cleavage of the Fmoc group by DBU led to the formation of the desired spin-labeled DKPs 210a–i (Scheme 44).106
Scheme 44. Synthesis of DKPs 210a–i with spin-labels attached.

3.4. Synthesis of seven-membered heterocyclic compounds
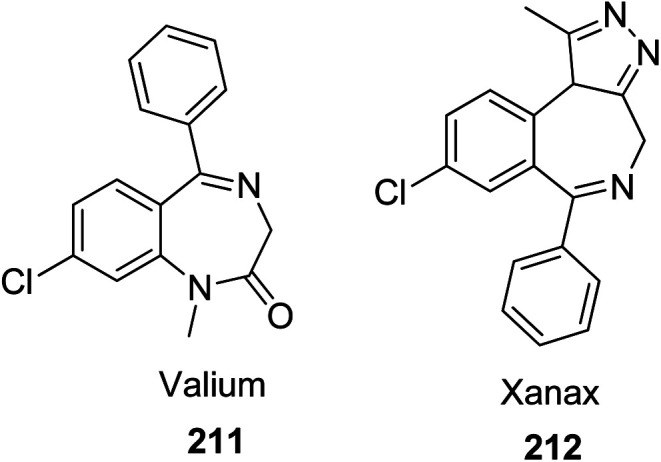
Benzodiazepines (BDZs) show a wide range of applicability as biologically active structures, where they are used as anti-anxiety drugs due to their ability to slow the central nervous system. Benzodiazepines enhance the effect of the neurotransmitter gamma-aminobutyric acid (GABA) at the GABAA receptor, resulting in sedative, hypnotic (sleep-inducing), anxiolytic (anti-anxiety), anticonvulsant, and muscle relaxant properties107 or antitumor drugs.108 Both Valium 211 and Xanax 212 are the best known biologically active BDZs (Fig. 5).109
Fig. 5. Structure of Valium 211 and Xanax 212.
Also, several 1,4-benzodiazepine (BDZ) scaffolds 217a and b and 218a and b were synthesized using anthranilate 213a and b as an amine component for Ugi-4CR together with the isocyanide Boc-glycinal 214, and a carboxylic acid with the UDC protocol followed by deprotection of the Boc group by TFA in 1,2-dichloroethane (DCE), respectively. Subsequently, the free amine group is condensed with the orthogonal ester group to form the 1,4-diazepine ring in the third step (cyclization) (Scheme 45).110 Alternatively, using trimethysilylazide instead of carboxylic acid affords 2-tetrazolo substituted 1,4-benzodiazepines 218a and b.110
Scheme 45. Synthesis of 1,4-benzodiazepines derivatives 217a and 218b using Boc-glycinal 214.

Subsequently, a series of N-Boc-amino acids 219 were subjected to the same protocol for the synthesis of 1,4-benzodiazepines-2-one derivatives 221a and b, which are abundant in the protein–protein interaction interface (Scheme 46).110
Scheme 46. Synthesis of 1,4-benzodiazepine-2-one derivatives 221a and b using N-Boc-amino acids 219.

On the other hand, another protocol was reported to synthesize pyridodiazepinediones by utilizing the Ugi reaction as a key step using 2-isocyanophenyl benzoate 223 and nicotinic acid derivatives 222. The Ugi bis-amide was obtained in poor yield using this method, and thus the pyridodiazepinediones 224 and 225 were formed after treatment of the crude mixture of Ugi reaction with KOt-Bu in dry THF. When R1 is a butyl or benzyl group, another step including Suzuki cross-coupling reaction is needed to afford product 227. In the case of 224, methylation of the NH– group was necessary to obtain 226, which in turn may engage to Suzuki reaction to obtain the tetra-substituted pyridodiazepinedione scaffolds 227 (Scheme 47).109
Scheme 47. Synthesis of tetra-substituted pyridodiazepinedione 224–227.

A different series of novel bicyclic and tricyclic macrolactams were synthesized using a methodology similar to the UDC method known as Ugi-deprotection–carbonylation/intramolecular amidation. This strategy based on the use of bi-functional acids 219 or bi-functional amines 229 with o-iodobenzaldehyde 228 and different isonitriles to afford Ugi-protected bis-amides 230 and 232, which are transformed into macrolactams 231 or 233via Boc deprotection followed by carbonylation/intramolecular amidation sequential reactions (Scheme 48).111
Scheme 48. Synthesis of macrolactams 231 and 233.

Further, the interesting synthesis of BDZs was applied by combining the Ugi reaction and SNAr chemistry. In the U-4CR of N-Boc-α-aminoaldehydes 234, 2-fluoro-5-nitrobenzoic acid (235) as an electrophilic component with amines and isonitriles were used to give Ugi product 236. Notably, the scavenging resins PS-tosylhydrazine (PS-TsNHNH2) and PS-diisopropylethylamine (PS-DIEA) were utilized for purification, where they could remove excess aldehyde or unreacted acid, while PS-morpholine acted a as proton scavenger. Indeed, this protocol represents an additional instance of the UDC strategy, and hence, after Boc group removal by TFA, with aid of PS-morpholine, cyclization occurs to give desired benzodiazepines 237 by elimination of HF (Scheme 49).112
Scheme 49. Synthesis of BDZs 237via UDC/SNAr strategy.

On the other hand, transition metal catalysis plays an important role in the Ugi reaction, affording different strategies to obtain an increasing number of novel structures.113,114 Zhu and co-workers reported the palladium- and copper-catalyzed synthesis of 1,4-benzodiazepine-2,5-diones 241 and ring-fused dihydroazaphenanthrenes 242via U-4CR as a key step using o-iodophenylisocyanide 238 and o-iodobenzoic acid 239 with other Ugi starting materials followed by catalytic treatment of the bis-amide 240 with Cu(i)-catalyst to give 1,4-benzodiazepine-2,5-dione 241 or using Pd(ii)-catalyst to give ring-fused dihydroazaphenanthrenes 242. Also, the conversion of 241 to 242 could be achieved easily through Pd(ii) catalysis (Scheme 50).115
Scheme 50. Synthesis of 1,4-benzodiazepine-2,5-diones 241 and ring-fused dihydroazaphenanthrenes 242via U-4CR.

In another strategy to synthesize the biologically active BDZ derivatives, a series of 1,2,4,5-tetrahydro-1,4-benzodiazepin-3-ones 244 or 245 using the Ugi reaction in the presence of the two-electron reducing agent Fe(0)/NH4Cl with E-α,β-unsaturated carboxylic acid 243 and microwave irradiation afforded the target products 244 or 245 without obtaining the Ugi linear bis-amide (Scheme 51).116 However, in the absence of microwave irradiation or any heat sources in the one-pot protocol, only the linear bis-amide Ugi product was obtained.116
Scheme 51. Synthesis of 1,2,4,5-tetrahydro-1,4-benzodiazepin-3-one 244 or 245.

A post-Ugi reductive aza-Michael reaction was performed using isopropyl aldehyde 6, o-nitrobenzylamine 246, benzyl isocyanide 247 and (E)-fumaric acid monoethyl ester 248. It is worth mentioning that at high microwave intensity, a one-pot 6-exo aza-Michael cyclization occurred to afford 2,5-diketopiperazines 249 and 250, while in the case of a decreasing microwave intensity, only BDZ 251 was formed, as illustrated in Scheme 52.116 Moreover, Ugi reaction using (E)-fumaric acid monoethyl ester 248 gives a series of 2,5-diketopiperazines or 2-azaspiro[4.5]deca-6,9-diene-3,8-diones derivatives in a single synthetic operation under the influence of microwaves with complete control of pathway selectivity.117
Scheme 52. Microwave intensity controls the pathway selectivity of aza-Michael cyclization.

Some resins such as Wang resin and hydroxymethyl resin have been utilized in the two-step solid phase synthesis of the biologically active ketopiperazines and BDZs through the UDC (Ugi/Boc-deprotect/cyclize) strategy. The reaction of N-Boc-anthranilic acids or N-Boc-α-amino aldehydes with resin-bound α-amino acids 252 for the synthesis of 1,4-benzodiazepine-2,5-diones 255 and ketopiperazines 257via both Wang resin and hydroxymethyl resin, respectively, as illustrated in Scheme 53,118 was performed via the deprotection/cyclization of resin-bound Ugi products 253 and 254.118
Scheme 53. Synthesis of BDZs 255 and ketopiperazines 257 in the solid phase via UDC.

Furthermore, the UDC strategy dependent on the use of N-Boc-protected amino acid 259 with 2-aminobenzoyl derivatives 213a to obtain linear Ugi bis-amide 260 in methanol, and subsequent deprotection–cyclization process to obtain functionalized BDZ scaffolds 261 was reported (Scheme 54).119
Scheme 54. Synthesis of 1,4-benzodiazepin-2-one derivatives 261 using N-Boc amino acid 259via UDC.

The one-pot synthesis of substituted 2,3-dihydro-1H-2-benzazepin-1-ones 265via U-4CR/Wittig reaction was developed, where phosphonium salt 262 was introduced into U-4CR as the acidic partner with p-substituted arylglyoxals 263, amine and isonitrile to afford 264, which were converted to 2,3-dihydro-1H-2-benzazepin-1-ones (265) via the intramolecular Wittig reaction in the presence of triethyl amine Et3N (Scheme 55).120
Scheme 55. One-pot synthesis of 2,3-dihydro-1H-2-benzazepin-1-ones 265.

The combination of the Ugi reaction, SN2′ cyclization and intramolecular Heck reaction gave natural product-like compounds of polycyclic heterocyclic-containing benzo[d]pyrrolo[1,2-a]azepin-5(6H)-one analogues 269 in excellent yields. The protocol was achieved using unsaturated isonitrile 123, which carries a methyl carbonate that acts as a leaving group in the cyclization, and several o-halobenzaldehydes 266 suitable for the Heck step were obtained. It is important to note that the Ugi conditions were different from the classical conditions, where the solvent mixture of trifluoroethanol (TFE) and ethanol was used in an equivalent ratio at 45 °C, while both the SN2′ cyclization and intramolecular Heck steps were carried out using the optimized Pd(0)-catalyzed conditions in the presence of 1,2-diphenylphosphinoethane (DPPE) as the ligand (Scheme 56).121
Scheme 56. Synthesis of benzo[d]pyrrolo[1,2-a]azepin-5(6H)-ones 269via Ugi–Heck reactions.

In the last decades, several methods have been reported for the synthesis of amino-triazoloazepinone (Ata) analogues, including the use of ruthenium catalyst followed by lactimization process or heating in the presence of DMF as a solvent.122 It was investigated if amino-triazoloazepinone could be synthesized via U-4CR followed by [3 + 2] Huisgen thermal cycloaddition. This procedure has the advantages of the minimum number of synthetic steps, no additives or catalysts, high yield and low-cost solvents.123 Condensation between azidoamino acid 270, aldehydes, propargylamine 110 and several aliphatic and aromatic isonitriles gave the desired (Ata) 272 (Scheme 57).123
Scheme 57. Synthesis of amino-triazoloazepinone (Ata) 272via Ugi/Huisgen cycloaddition.

3.5. Synthesis of fused six-membered heterocyclic compounds
A series of coumarin-based structures was synthesized by coupling the Ugi and Knoevenagel reactions to report a powerful pathway to one-pot five-component Knoevenagel–Ugi adducts. Coumarin derivatives have been reported to exhibit valuable biological behaviors, for instance, anticancer,124 antiviral,125 anti-Alzheimer's,126 and anti-HIV activities.127 The synthesis of coumarin-based α-acyl amino amides 277 in a one-pot five-component Knoevenagel–Ugi approach was achieved by the reaction among 2,2-dimethyl-1,3-dioxane-4,6-dione (273), which known as Meldrum's acid, salicylaldehyde, 274, aniline 275, benzaldehyde derivatives 276 and isonitrile in ethanol (Scheme 58).2
Scheme 58. Synthesis of coumarin-based α-acyl amino amides 277via Knoevenagel–Ugi strategy.

As mentioned before, the Ugi reaction is an efficient route to synthesize several heterocyclic compounds. Another fruitful strategy represents the synthesis of benzoxazinone analogues via U-4CR/Mitsunobu cyclization starting from o-amino phenols 278, α-hydroxy acids 279, aldehydes and isonitriles to give Ugi product 280 followed by Mitsunobu cyclization using DEAD/PPh3 protocol to afford benzo[b][1,4]oxazin-3-ones 281 in two high yielding steps (Scheme 59).128 It is worth mentioning that benzo[b][1,4]oxazin-3-ones 281 represent a drug-like structure involved in many pharmaceutical compounds.129,130
Scheme 59. Synthesis of benzoxazinones 281via U-4CR/Mitsunobu cyclization.

Moreover, the preparation of 3-triazolyl-quinolin-2-(1H)-one analogues was carried out via the combination of Ugi, click and Knoevenagel reactions. The Ugi reaction between o-acylanilines 213a, 2-azidoacetic acid 282, and different aldehydes and isocyanides affords azido-decorated Ugi products 283, which are subsequently reacted with terminal alkyne derivatives under the influence of Cu(i)-catalysis via [3 + 2] cycloaddition to give 1,2,3-triazolo derivatives 284, followed by intramolecular Knoevenagel fashion to afford the 1,2,3-triazolo-quinolinone scaffold 285 (Scheme 60).131
Scheme 60. Synthesis of 1,2,3-triazolo-quinolinone 285via Ugi “click”–Knoevenagel condensations.

The first synthesis of quinoxaline derivatives via the Ugi reaction was reported by Ayaz and co-workers utilizing aryl glyoxal derivatives 286, N-Boc-o-phenylenediamine derivatives 287, and formic acid 288, where the reaction proceeds smoothly in methanol at room temperature to obtain the linear Ugi product, which is easily cyclized in the presence of TFA/DCE solution and a catalytic amount of water with microwave irradiation at 140 °C for 20 min to afford quinoxalines 289 (Scheme 61).132
Scheme 61. Synthesis of quinoxaline derivatives 289via Ugi reaction.

Searching for expansion of the structural diversity under the same reaction conditions, it was reported that the use of o-N-Boc-phenylisonitrile (147) facilitates access to 2-benzimidazolylquinoxalines 292 and 293, in which the combination between the benzimidazole and quinoxaline ring can give an opportunity to improve their biological activity. Also 2-benzimidazolylquinoxalines 292 can be obtained during this reaction when ortho-substituted aromatic carboxylic acids are used (Scheme 62).132
Scheme 62. Synthesis of 2-benzimidazolylquinoxalines derivatives 292 and 293via Ugi reaction.

Through the same one-pot protocol, Yan, Ding and co-workers successfully synthesized multi-substituted quinoxalin-2(1H)-ones via a U-4CR/catalytic aza-Wittig reaction. They developed a new strategy to synthesize their target compounds through the Ugi reaction between 2-aminobenzoyl azides 175, aldehydes, ketoacids 294, and isonitriles in the presence of a catalytic amount of the same phosphorus catalyst they used before, 3-methyl-1-phenyl-2-phospholene 1-oxide (178), to produce quinoxalin-2(1H)-ones 297 as the target product (Scheme 63).133 With regard to other heterocycles, quinoxalin-2(1H)-one derivatives are classified as crucial pharmacologically active compounds,133 where they act as antidiabetic agents,134 anti-inflammatory agents135 and antibacterial agents.136 It has been noted that Ding's protocol guarantees high atom efficiency, mild reaction conditions, ready availability of starting materials and yield of up to 90%.89,133
Scheme 63. Synthesis of quinoxalin-2(1H)-ones 297via a U-4CR/catalytic aza-Wittig reaction.

Several bioactive natural products contain quinazolinone moieties, which have promising biological properties such as antibacterial,137 antileishmanial,138 anti-HIV,139 anti-inflammatory,140 and antifungal141 activities. Balalaie and co-workers successfully synthesized pseudo-peptides containing a quinazolinone moiety via U-4CR142 using 3-amino-1,2,3,4-tetrahydro-4-oxoquinazoline-2-carboxylic acid derivatives 298.142 The Ugi reaction using 298 with different aromatic aldehydes, primary amines and isonitriles gave the desired pseudo-peptides, which contain quinazolinone moiety 299 (Scheme 64).142 Notably, the major diastereoisomer was observed in crystalline form and the minor one remained dissolved in ethanol.
Scheme 64. Synthesis of pseudo-peptides containing quinazolinone moiety 299via U-4CR.

A two-step solution phase protocol for the synthesis of dihydroquinazoline scaffolds was reported, where the method is based on the trans-modification of the Ugi products with acid treatment to obtain new highly functionalized drug-like dihydroquinazolines.129,143 The Ugi reaction with isonitrile, carboxylic acid, aldehyde and mono-Boc protected 2-aminobenzylamines afford Ugi product 302, which can be treated by acid to obtain the target products 304 (Scheme 65, Path A).144 Notably, changing the position of the Boc protecting group in the used amine can modify the resulting product from 1,4-dihydroquinazoline 304 to 3,4-dihydroquinazoline 305 (Scheme 65, Path B).144
Scheme 65. Synthesis of dihydroquinazolines 304 and 305via Ugi reaction.

3.6. Synthesis of polycyclic heterocyclic compounds
In the same context, the aza-Wittig reactions have received considerable attention as a powerful tool to synthesize heterocycles.145,146 Recently, the Ugi reaction followed by a post-condensation Staudinger–aza-Wittig reaction was proven to be beneficial in the synthesis of several biologically active heterocycles.147,148 Ding and co-workers reported the first cyclization of the Ugi product to afford an indolo[1,2-c]quinazoline ring system via the nucleophilic addition of the methine group to the carbonyl group or what it is known as the Staudinger–aza-Wittig–nucleophilic addition reaction,149 and the Ugi reaction using o-acylaniline 213a, o-azidobenzaldehyde 306, carboxylic acid and isonitrile was carried out in methanol at room temperature to give Ugi products 307 in fair yield. Ugi products 307 were further reacted with triphenyl phosphine followed by heating in toluene to give the desired indolo[1,2-c]quinazolines 310 (Scheme 66).149
Scheme 66. Synthesis of indolo[1,2-c]quinazolines 310via the sequential (U-4CR)–Staudinger–aza-Wittig–nucleophilic addition reaction.

Furthermore, the synthesis of fused dihydroquinazoline–benzodiazepine tetracycles was reported by a similar route but with different conditions utilizing propyl aldehyde 311, butyl isonitrile 312, N-Boc-protected anthranilic acid 313 and 2-amino-N-Boc-protected benzylamines 300 to afford the Ugi product 314, which was treated by acid to get the target products (Scheme 67, Path A).144 The imine was pre-formed in DCM in the presence of magnesium sulphate followed by microwave irradiation at 120 °C for 10 min to get the desired Ugi product, which was deprotected by an acid to form tetracyclic adduct 316, and with the same steps and reactants 317 was afforded by utilizing amine 301 instead of 300 (Scheme 67, Path B).144
Scheme 67. Synthesis of dihydroquinazoline–benzodiazepines 316 and 317.

In continuation of our study on the Ugi reaction and its post-condensations, Ugi reaction followed by the Pictet–Spengler reaction was used to synthesize useful heterocycles such as 1,2,3,4-tetrahydro-β-carboline (THBC), which are based on tetracyclic peptidomimetics and considered tryptophan analogues. These analogues (e.g. THBC) are known to bind with various receptor sites such as serotonin to cause inhibition of monoamine oxidase A enzyme.150 Moreover, some of the tetracyclic β-carbolines were found to have antimalarial characters151 or act as selective inhibitors in the anticancer field.152 The use of 2-aminoacetaldehyde 320 as a carbonyl partner in the Ugi reaction was investigated for the first time by Lesma and co-workers. Their target was to condense N-protected tryptophan-derived isonitrile 318, N-protected-2-aminoacetaldehyde 320, aminoacetaldehyde diethylacetal 319 and acetic acid 321 as a carboxylate nucleophile to obtain the desired Ugi products 322 and 323. The cyclization of both 322 and 323 was carried out smoothly in the presence of formic acid at 60 °C to afford 324 and 325a and b, respectively (Scheme 68).153
Scheme 68. Synthesis of 1,2,3,4-tetrahydro-β-carboline 324 and 325a and bvia U-4CR/Pictet–Spengler reaction.

Furthermore, an unusual protocol was developed utilizing the Ugi reaction to synthesize azaspiro compounds, which are natural product-like compounds. The combination of the Ugi and Michael reaction afforded a unique protocol known as the Ugi/Michael/aza-Michael reaction (UMAM), which was used to synthesize fused azaspiro tricycles and azaspiro tetracycles in good yield using γ-keto-α,β-unsaturated carboxylic acid and different types of acids amines, aldehydes and isonitriles under microwave irradiation, in the presence of water as the solvent (Scheme 69).154
Scheme 69. Synthesis of azaspiro compounds 327–329via Ugi reaction.

Paulvannan illustrated Ugi MCR reaction followed by intramolecular Diels–Alder reaction of furan (IMDAF)155 to build natural product-like libraries. Ugi condensation of 2-furaldehyde 330, acetylenic acids 331 as dienophiles, isonitriles and amines generated acetylenic amides 332 (Scheme 70).155 Transformation of 332 to the oxabicyclo[2.2.1]heptadiene derivatives 333 was accomplished under thermal conditions. However, it was observed that when the Ugi products were allowed to stand at room temperature, they were partially converted to the corresponding cycloadduct 333. In contrast, exposure of 332 to catalytic Yb(OTf)3 in 1,4-dioxane at high temperatures cleanly converted it to isoindolinone 334a in tautomerism with 334b.156
Scheme 70. Ugi MCR reaction followed by the intramolecular Diels–Alder reaction of furan (IMDAF).

4. Enantioselective Ugi reaction
Enantioselective Ugi-4CR or Ugi-3CR with stereochemical control is appropriate for molecular diversity of α-acylaminoamide with applicable synthesis in drug discovery since it facilitates rapid access to diverse libraries of biologically important molecules and natural products, which demand multistep synthetic pathways.157–159
A series of novel chiral morpholin-2-one-3-carboxamide derivatives or ketopiperazine-2-carboxamide compounds 336 was prepared via the asymmetric Ugi three-component reaction using chiral cyclic aldimines or ketoimines 335. The chiral imines showed promising stereo induction for the new chiral center of the Ugi products, and predominantly trans-isomers were obtained in most cases with good diastereoselectivity of the new stereocenter at C-3 in the products (Scheme 71).160
Scheme 71. Synthesis of chiral morpholin-2-one-3-carboxamide derivatives 4 and ketopiperazine-2-carboxamide.

The catalyzed enantioselective Ugi-4CR or Ugi-3CR with high optical purity was accomplished recently using chiral phosphoric acids (CPAs), which are diesters derived from the 1,1′-bi-2-naphthol (BINOL) or (S)-1,1′-spirobiindane-7,7′-diol (SPINOL) motifs possessing chiral backbones. CPA are used in asymmetric catalysis due to their enhanced acidity compared to carboxylic acids, which is perceived to accelerate the kinetics of the enantioselective Ugi reaction. Consequently, the chiral phosphoric acid CPA 338-catalyzed enantioselective Ugi four-center, three-component reaction of isocyanides, anilines, and 2-formylbenzoic acids 337 was employed for the synthesis of isoindolinones 339 in excellent yields. Chiral isoindolinone derivatives are considered a key structural motif in many bioactive natural products such as (S)-(+)-lennoxamine (340) and medicinally relevant compounds such as (R)-pazinaclone (341), a sedative and anxiolytic drug (Scheme 72).161,162
Scheme 72. Synthesis of chiral isoindolinones 339.

Alternatively, lacosamide is a medication used in the adjunctive treatment of partial-onset seizures and diabetic neuropathic pain. It is administered orally or intravenously and sold under the brand name Vimpat.163 α-Amino amide derivatives were prepared via a catalytic enantioselective three-component Ugi reaction utilizing SPINOL-derived phosphoric acid with bulky 2,4,6-tricyclohexylphenyl groups at the 6,6′ positions 344 in good to excellent yields (62% to 99%) and enantiocontrol (81% to >99% enantiomeric excess). This method was used to prepare the anticonvulsive drug (R)-lacosamide 347 using 2-methoxy acetaldehyde (342), 4-nitroaniline (343), and benzyl isocyanide 247, affording the Ugi product 345 in 90% yield and 94% ee, which was transformed into 346 in 95% yield after dearylation via sequential reduction and oxidation. Acylation of the chiral amine 346 afforded (R)-lacosamide 347 in 88% yield and 96% ee (Scheme 73).164
Scheme 73. Enantioselective synthesis of (R)-lacosamide (347) via Ugi-3CR.

Alternatively, the enantioselective synthesis of 2-(1-aminoalkyl)-5-aminooxazoles 350 from α-isocyanoacetamides (348), aldehydes, and anilines was accomplished using U-3CR (Scheme 74) and CPA catalyst by stirring a toluene solution of aldehydes, anilines, and α-isocyanoacetamides (348) (c = 0.05 M, −20 °C) in the presence of CPA 349 (20 mol%), affording the target oxazoles 350 in excellent yields with enantioselectivities of up to 90% ee.162
Scheme 74. CPA 349-catalyzed enantioselective reaction of aldehydes, amines, and α-isocyanoacetamides.

Recently, Houk, Tan and coworkers constructed α-acylaminoamide compounds by combining the traditional starting materials in a one-pot reaction. Stereochemistry control of the enantioselective four-component Ugi reaction was achieved using a variety of chiral phosphoric acid derivative catalysts CPA 351 and 344 and the type of aldehyde (aryl or alkyl), providing more than 80 examples of α-acylaminoamides in good to excellent enantiomeric excess, as illustrated in Fig. 6.165,166
Fig. 6. Stereochemistry control of enantioselective four-component Ugi reaction.

5. Conclusion
In this review, we summarized the preliminary applications of the Ugi reaction in organic synthesis and pharmaceutical research during the last 20 years from 2000 to 2019 due to the amazing properties of the Ugi reaction. We have highlighted the use of the Ugi reaction to prepare a variety of linear or cyclic peptides with multifunctional structures, which have promising biological activities. Also, the application of the Ugi reaction was extended to the synthesis of fused or unfused heterocyclic compounds with 5, 6, and 7-membered rings. Furthermore, the Ugi reaction is considered as a magic tool to prepare natural products with complex structures or stereochemistry with high enantioselectivity. However, despite the successful application of the Ugi reaction in the preparation of functional peptides, heterocyclic compounds, and natural products in the last two decades, there are still some limitations of the Ugi reaction that must be illustrated. The intolerable odor, difficulty in synthesis, relatively high price and sensitivity in acidic medium of isonitriles are the major disadvantages, which limit its application for large-scale synthesis. Additionally, the functional modification of isocyanide is difficult, which also limits the modification of functional groups of peptides, heterocyclic or natural products. Accordingly, other MCRs may be used to overcome the limitations of Ugi reactions, such as the Mannich reactions, Povarov MCRs, Petasis reaction, and Biginelli reaction, for applications with different purposes. A future challenge is to complete the isocyanide MCR by summarizing the applications of the Passerini reaction in a separate perspective.
Conflicts of interest
There are no conflicts of interest to declare.
Supplementary Material
Acknowledgments
M. A. F., H. A. and M. S. A. express sincere gratitude to Dr Qingzhi Zhang (University of St Andrews, DOH group, UK) for her support, also we are grateful to Faculty of Science, Alexandria University, Egypt and Academy of Scientific Research and Technology.
References
- Janežič D. Hodošček M. Ugi I. Internet Electron. J. Mol. Des. 2002;1:293–299. [Google Scholar]
- Kumar S. Mukesh K. Harjai K. Singh V. Tetrahedron Lett. 2019;60:8–12. doi: 10.1016/j.tetlet.2018.11.030. [DOI] [Google Scholar]
- Passerini M. Simone L. Gazz. Chim. Ital. 1921;51:126–129. [Google Scholar]
- Chandgude A. L. Dömling A. Green Chem. 2016;18:3718–3721. doi: 10.1039/C6GC00910G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoup M. S. Wahby Y. Abdel-Hamid H. Ramadan E. S. Teleb M. Abu-Serie M. M. Noby A. Eur. J. Med. Chem. 2019;168:340–356. doi: 10.1016/j.ejmech.2019.02.051. [DOI] [PubMed] [Google Scholar]
- Ugi I. Werner B. Dömling A. Molecules. 2003;8:53–66. doi: 10.3390/80100053. [DOI] [Google Scholar]
- Barreto A. d. F. S. Santos V. A. d. Andrade C. K. Z. Beilstein J. Org. Chem. 2016;12:2865–2872. doi: 10.3762/bjoc.12.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dömling A. Chem. Rev. 2006;106:17–89. doi: 10.1021/cr0505728. [DOI] [PubMed] [Google Scholar]
- Ayoup M. S. Fouad M. A. Abdel-Hamid H. Ramadan E. S. Abu-Serie M. M. Noby A. Teleb M. Eur. J. Med. Chem. 2020;186:111875. doi: 10.1016/j.ejmech.2019.111875. [DOI] [PubMed] [Google Scholar]
- Kreye O. Türünç O. Sehlinger A. Rackwitz J. Meier M. A. R. Chem.–Eur. J. 2012;18:5767–5776. doi: 10.1002/chem.201103341. [DOI] [PubMed] [Google Scholar]
- Váradi A. Palmer T. C. Notis Dardashti R. Majumdar S. Molecules. 2016;21:1–22. doi: 10.3390/molecules21010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dömling A. Wang W. Wang K. Chem. Rev. 2012;112:3083–3135. doi: 10.1021/cr100233r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa S. Nakanishi K. Udagawa Y. Maeda M. Sato S. Nakano K. Masuda T. Ichikawa Y. Org. Biomol. Chem. 2020;18:687–693. doi: 10.1039/C9OB02249J. [DOI] [PubMed] [Google Scholar]
- Touré B. B. Hall D. G. Chem. Rev. 2009;109:4439–4486. doi: 10.1021/cr800296p. [DOI] [PubMed] [Google Scholar]
- Rossen K. Pye P. J. DiMichele L. M. Volante R. P. Reider P. J. Tetrahedron Lett. 1998;39:6823–6826. doi: 10.1016/S0040-4039(98)01484-1. [DOI] [Google Scholar]
- Liu N. Cao S. Wu J. Yu J. Shen L. Feng X. Qian X. Tetrahedron. 2008;64:3966–3974. doi: 10.1016/j.tet.2008.02.051. [DOI] [Google Scholar]
- De Laszlo S. E. Williard P. G. J. Am. Chem. Soc. 1985;107:199–203. doi: 10.1021/ja00287a036. [DOI] [Google Scholar]
- Fathi V. Ramezanpour S. Balalaie S. Rominger F. Reza Bijanzadeh H. Helv. Chim. Acta. 2014;97:1630–1637. doi: 10.1002/hlca.201400061. [DOI] [Google Scholar]
- Nami M. Salehi P. Dabiri M. Bararjanian M. Gharaghani S. Khoramjouy M. Al-Harrasi A. Faizi M. Chem. Biol. Drug Des. 2018;91:902–914. doi: 10.1111/cbdd.13157. [DOI] [PubMed] [Google Scholar]
- van der Heijden G. Jong J. A. W. Ruijter E. Orru R. V. A. Org. Lett. 2016;18:984–987. doi: 10.1021/acs.orglett.6b00091. [DOI] [PubMed] [Google Scholar]
- Bräse S. Encinas A. Keck J. Nising C. F. Chem. Rev. 2009;109:3903–3990. doi: 10.1021/cr050001f. [DOI] [PubMed] [Google Scholar]
- Ziarani G. M. Moradi R. Mahammadkhani L. Arkivoc. 2019;(part i):18–40. [Google Scholar]
- Neves Filho R. A. W. Stark S. Westermann B. Wessjohann L. A. Beilstein J. Org. Chem. 2012;8:2085–2090. doi: 10.3762/bjoc.8.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieńczyk M. Podgórski D. Błażejewska A. Kulbacka J. Saczko J. Oleksyszyn J. Bioorg. Med. Chem. 2011;19:1277–1284. doi: 10.1016/j.bmc.2010.12.008. [DOI] [PubMed] [Google Scholar]
- Sabidó E. Tarragó T. Giralt E. Bioorg. Med. Chem. 2010;18:8350–8355. doi: 10.1016/j.bmc.2010.09.066. [DOI] [PubMed] [Google Scholar]
- Sienczyk M. Oleksyszyn J. Curr. Med. Chem. 2009;16:1673–1687. doi: 10.2174/092986709788186246. [DOI] [PubMed] [Google Scholar]
- Paris M. Porcelloni M. Binaschi M. Fattori D. J. Med. Chem. 2008;51:1505–1529. doi: 10.1021/jm7011408. [DOI] [PubMed] [Google Scholar]
- Grolla A. A. Podestà V. Chini M. G. Di Micco S. Vallario A. Genazzani A. A. Canonico P. L. Bifulco G. Tron G. C. Sorba G. Pirali T. J. Med. Chem. 2009;52:2776–2785. doi: 10.1021/jm801529c. [DOI] [PubMed] [Google Scholar]
- Xu Y. Shi J. Yamamoto N. Moss J. A. Vogt P. K. Janda K. D. Bioorg. Med. Chem. 2006;14:2660–2673. doi: 10.1016/j.bmc.2005.11.052. [DOI] [PubMed] [Google Scholar]
- Okandeji B. O. Greenwald D. M. Wroten J. Sello J. K. Bioorg. Med. Chem. 2011;19:7679–7689. doi: 10.1016/j.bmc.2011.10.011. [DOI] [PubMed] [Google Scholar]
- Fan X. Zhang X. Bories C. Loiseau P. M. Torrence P. F. Bioorg. Chem. 2007;35:121–136. doi: 10.1016/j.bioorg.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Fahmy H. Kuppast B. Teleb Ismail M. Curr. Mol. Pharmacol. 2017;10:270–281. doi: 10.2174/1874467209666161101144155. [DOI] [PubMed] [Google Scholar]
- Teleb M. Kuppast B. Spyridaki K. Liapakis G. Fahmy H. Eur. J. Med. Chem. 2017;138:900–908. doi: 10.1016/j.ejmech.2017.07.016. [DOI] [PubMed] [Google Scholar]
- Linderman R. J. Binet S. Petrich S. R. J. Org. Chem. 1999;64:336–337. doi: 10.1021/jo982186e. [DOI] [Google Scholar]
- Cohen E. Annu. Rev. Entomol. 1987;32:71–93. doi: 10.1146/annurev.en.32.010187.000443. [DOI] [Google Scholar]
- Plant A. Thompson P. Williams D. M. J. Org. Chem. 2009;74:4870–4873. doi: 10.1021/jo900244m. [DOI] [PubMed] [Google Scholar]
- Awad L. F. Ayoup M. S. Beilstein J. Org. Chem. 2020;16:1022–1050. doi: 10.3762/bjoc.16.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoup M. S. Cordes D. B. Slawin A. M. Z. O'Hagan D. Org. Biomol. Chem. 2015;13:5621–5624. doi: 10.1039/C5OB00650C. [DOI] [PubMed] [Google Scholar]
- Ayoup M. S. Cordes D. B. Slawin A. M. Z. O'Hagan D. Beilstein J. Org. Chem. 2015;11:2671–2676. doi: 10.3762/bjoc.11.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouge V. Jubault P. Quirion J.-C. Tetrahedron Lett. 2004;45:773–776. doi: 10.1016/j.tetlet.2003.11.019. [DOI] [Google Scholar]
- Murphy P. V. Bradley H. Tosin M. Pitt N. Fitzpatrick G. M. Glass W. K. J. Org. Chem. 2003;68:5692–5704. doi: 10.1021/jo034336d. [DOI] [PubMed] [Google Scholar]
- Lee Y. Phat C. Hong S.-C. Peptides. 2017;95:94–105. doi: 10.1016/j.peptides.2017.06.002. [DOI] [PubMed] [Google Scholar]
- Whelan A. N. Elaridi J. Harte M. Smith S. V. Jackson W. R. Robinson A. J. Tetrahedron Lett. 2004;45:9545–9547. doi: 10.1016/j.tetlet.2004.10.165. [DOI] [Google Scholar]
- Kazmaier U. Hebach C. Watzke A. Maier S. Mues H. Huch V. Org. Biomol. Chem. 2005;3:136–145. doi: 10.1039/B411228H. [DOI] [PubMed] [Google Scholar]
- Cristau P. Vors J.-P. Zhu J. Org. Lett. 2001;3:4079–4082. doi: 10.1021/ol0168420. [DOI] [PubMed] [Google Scholar]
- Vishwanatha T. M. Bergamaschi E. Dömling A. Org. Lett. 2017;19:3195–3198. doi: 10.1021/acs.orglett.7b01324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samarasimhareddy M. Hemantha H. P. Sureshbabu V. V. Tetrahedron Lett. 2012;53:3104–3107. doi: 10.1016/j.tetlet.2012.04.034. [DOI] [Google Scholar]
- Puentes A. R. Morejón M. C. Rivera D. G. Wessjohann L. A. Org. Lett. 2017;19:4022–4025. doi: 10.1021/acs.orglett.7b01761. [DOI] [PubMed] [Google Scholar]
- Tei L. Gugliotta G. Avedano S. Giovenzana G. B. Botta M. Org. Biomol. Chem. 2009;7:4406–4414. doi: 10.1039/B907932G. [DOI] [PubMed] [Google Scholar]
- Ichikawa Y. Yamasaki T. Nakanishi K. Udagawa Y. Hosokawa S. Masuda T. Synthesis. 2019;51:2305–2310. doi: 10.1055/s-0037-1610867. [DOI] [Google Scholar]
- Williams D. E. Patrick B. O. Tahir A. Van Soest R. Roberge M. Andersen R. J. J. Nat. Prod. 2004;67:1752–1754. doi: 10.1021/np049798g. [DOI] [PubMed] [Google Scholar]
- Saito K. Nishimori A. Kotsuki H. Nakano K. Ichikawa Y. Synlett. 2013;24:757–761. doi: 10.1055/s-0032-1318305. [DOI] [Google Scholar]
- Ichikawa Y. Saito K. Mimura R. Kitamori A. Matsukawa A. Ikeda A. Masuda T. Kotsuki H. Nakano K. Heterocycles. 2016;92:1040–1053. doi: 10.3987/COM-16-13442. [DOI] [Google Scholar]
- Pelliccia S. Alfano I. A. Galli U. Novellino E. Giustiniano M. Tron G. C. Symmetry. 2019;11:798–837. doi: 10.3390/sym11060798. [DOI] [Google Scholar]
- Fernandes P. B. Swanson R. N. Hardy D. J. Hanson C. W. Coen L. Rasmussen R. R. Chen R. H. J. Antibiot. Res. 1989;42:521–526. doi: 10.7164/antibiotics.42.521. [DOI] [PubMed] [Google Scholar]
- Winn M. Goss R. J. M. Kimura K.-i. Bugg T. D. H. Nat. Prod. Rep. 2010;27:279–304. doi: 10.1039/B816215H. [DOI] [PubMed] [Google Scholar]
- Okamoto K. Sakagami M. Feng F. Takahashi F. Uotani K. Togame H. Takemoto H. Ichikawa S. Matsuda A. Bioorg. Med. Chem. Lett. 2012;22:4810–4815. doi: 10.1016/j.bmcl.2012.05.050. [DOI] [PubMed] [Google Scholar]
- Guimarães L. R. C. Rodrigues A. P. D. Marinho P. S. B. Muller A. H. Guilhon G. M. S. Santos L. S. do Nascimento J. L. M. Silva E. O. Parasitol. Res. 2010;107:1075–1081. doi: 10.1007/s00436-010-1973-0. [DOI] [PubMed] [Google Scholar]
- Peixoto R. N. S. Guilhon G. M. S. P. Das Graças M. Zoghbi B. Araújo I. S. Uetanabaro A. P. T. Santos L. S. Brasil D. D. S. B. Molecules. 2013;18:3195–3205. doi: 10.3390/molecules18033195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidou M. Kurpiewska K. Kalinowska-Tłuscik J. Dömling A. Eur. J. Org. Chem. 2018:6714–6719. doi: 10.1002/ejoc.201801276. [DOI] [Google Scholar]
- Zhu J. Eur. J. Org. Chem. 2003:1133–1144. doi: 10.1002/ejoc.200390167. [DOI] [Google Scholar]
- Xu Z. Ayaz M. Cappelli A. A. Hulme C. ACS Comb. Sci. 2012;14:460–464. doi: 10.1021/co300046r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teleb M. Rizk O. H. Zhang F.-X. Fronczek F. R. Zamponi G. W. Fahmy H. Bioorg. Chem. 2019;88:102915. doi: 10.1016/j.bioorg.2019.04.009. [DOI] [PubMed] [Google Scholar]
- Rizk O. H. Teleb M. Abu-Serie M. M. Shaaban O. G. Bioorg. Chem. 2019;92:103189. doi: 10.1016/j.bioorg.2019.103189. [DOI] [PubMed] [Google Scholar]
- Qian B.-C. Kamori A. Kinami K. Kato A. Li Y.-X. Fleet G. W. J. Yu C.-Y. Org. Biomol. Chem. 2016;14:4488–4498. doi: 10.1039/C6OB00531D. [DOI] [PubMed] [Google Scholar]
- Bergeron-Brlek M. Meanwell M. Britton R. Nat. Commun. 2015;6:6903–6908. doi: 10.1038/ncomms7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritthiwigrom T. Willis A. C. Pyne S. G. J. Org. Chem. 2010;75:815–824. doi: 10.1021/jo902355p. [DOI] [PubMed] [Google Scholar]
- Flores-Constante G. Sánchez-Chávez A. C. Polindara-García L. A. Eur. J. Org. Chem. 2018:4586–4591. doi: 10.1002/ejoc.201800756. [DOI] [Google Scholar]
- Rikimaru K. Yanagisawa A. Kan T. Fukuyama T. Synlett. 2004:41–44. [Google Scholar]
- Neves Filho R. A. W. Stark S. Morejon M. C. Westermann B. Wessjohann L. A. Tetrahedron Lett. 2012;53:5360–5363. doi: 10.1016/j.tetlet.2012.07.064. [DOI] [Google Scholar]
- Banfi L. Basso A. Cerulli V. Guanti G. Riva R. J. Org. Chem. 2008;73:1608–1611. doi: 10.1021/jo702087x. [DOI] [PubMed] [Google Scholar]
- Schlegel B. Schmidtke M. Dörfelt H. Kleinwächter P. Gräfe U. J. Basic Microbiol. 2001;41:179–183. doi: 10.1002/1521-4028(200107)41:3/4<179::AID-JOBM179>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Marfori E. C. Kajiyama S. i. Fukusaki E.-i. Kobayashi A. Phytochemistry. 2003;62:715–721. doi: 10.1016/S0031-9422(02)00629-5. [DOI] [PubMed] [Google Scholar]
- Spatz J. H. Welsch S. J. Duhaut D.-E. Jäger N. Boursier T. Fredrich M. Allmendinger L. Ross G. Kolb J. Burdack C. Umkehrer M. Tetrahedron Lett. 2009;50:1705–1707. doi: 10.1016/j.tetlet.2009.01.124. [DOI] [Google Scholar]
- Shaw A. Y. Xu Z. Hulme C. Tetrahedron Lett. 2012;53:1998–2000. doi: 10.1016/j.tetlet.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelidou A. S. Hadjipavlou-Litina D. Chem. Rev. 2005;105:3235–3271. doi: 10.1021/cr040708m. [DOI] [PubMed] [Google Scholar]
- García-Valverde M. Macho S. Marcaccini S. Rodríguez T. Rojo J. Torroba T. Synlett. 2008:33–36. [Google Scholar]
- Sung K. Wu S.-H. Chen P.-I. Tetrahedron. 2002;58:5599–5602. doi: 10.1016/S0040-4020(02)00550-1. [DOI] [Google Scholar]
- George Rosenker K. M. Paquette W. D. Johnston P. A. Sharlow E. R. Vogt A. Bakan A. Lazo J. S. Wipf P. Bioorg. Med. Chem. 2015;23:2810–2818. doi: 10.1016/j.bmc.2015.01.043. [DOI] [PubMed] [Google Scholar]
- Wen L.-R. Sun Q.-C. Zhang H.-L. Li M. Org. Biomol. Chem. 2013;11:781–786. doi: 10.1039/C2OB27137K. [DOI] [PubMed] [Google Scholar]
- He L.-J. Yang D.-L. Li S.-Q. Zhang Y.-J. Tang Y. Lei J. Frett B. Lin H.-k. Li H.-y. Chen Z.-Z. Xu Z.-G. Bioorg. Med. Chem. 2018;26:3899–3908. doi: 10.1016/j.bmc.2018.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.-Z. Zhang J. Tang D.-Y. Xu Z.-G. Tetrahedron Lett. 2014;55:2742–2744. doi: 10.1016/j.tetlet.2014.03.063. [DOI] [Google Scholar]
- Song G.-T. Li S.-Q. Yang Z.-W. Yuan J.-H. Wang M.-S. Zhu J. Chen Z.-Z. Xu Z.-G. Tetrahedron Lett. 2015;56:4616–4618. doi: 10.1016/j.tetlet.2015.06.035. [DOI] [Google Scholar]
- Dounay A. B. Overman L. E. Chem. Rev. 2003;103:2945–2964. doi: 10.1021/cr020039h. [DOI] [PubMed] [Google Scholar]
- Somei M. Yamada F. Nat. Prod. Rep. 2005;22:73–103. doi: 10.1039/B316241A. [DOI] [PubMed] [Google Scholar]
- Bonnaterre F. Bois-Choussy M. Zhu J. Org. Lett. 2006;8:4351–4354. doi: 10.1021/ol061755z. [DOI] [PubMed] [Google Scholar]
- Speck K. Magauer T. Beilstein J. Org. Chem. 2013;9:2048–2078. doi: 10.3762/bjoc.9.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga V. Milen M. Ábrányi-Balogh P. Tetrahedron Lett. 2018;59:3683–3689. doi: 10.1016/j.tetlet.2018.08.063. [DOI] [Google Scholar]
- Yan Y.-M. Rao Y. Ding M.-W. J. Org. Chem. 2016;81:1263–1268. doi: 10.1021/acs.joc.5b02575. [DOI] [PubMed] [Google Scholar]
- Mavrova A. T. Yancheva D. Anastassova N. Anichina K. Zvezdanovic J. Djordjevic A. Markovic D. Smelcerovic A. Bioorg. Med. Chem. 2015;23:6317–6326. doi: 10.1016/j.bmc.2015.08.029. [DOI] [PubMed] [Google Scholar]
- Pan T. He X. Chen B. Chen H. Geng G. Luo H. Zhang H. Bai C. Eur. J. Med. Chem. 2015;95:500–513. doi: 10.1016/j.ejmech.2015.03.050. [DOI] [PubMed] [Google Scholar]
- Vasantha K. Basavarajaswamy G. Vaishali Rai M. Boja P. Pai V. R. Shruthi N. Bhat M. Bioorg. Med. Chem. Lett. 2015;25:1420–1426. doi: 10.1016/j.bmcl.2015.02.043. [DOI] [PubMed] [Google Scholar]
- Reddy T. S. Kulhari H. Reddy V. G. Bansal V. Kamal A. Shukla R. Eur. J. Med. Chem. 2015;101:790–805. doi: 10.1016/j.ejmech.2015.07.031. [DOI] [PubMed] [Google Scholar]
- Hernández-Vázquez E. Chávez-Riveros A. Nieto-Camacho A. Miranda L. D. ChemMedChem. 2019;14:132–146. doi: 10.1002/cmdc.201800634. [DOI] [PubMed] [Google Scholar]
- Hughes R. O. Rogier D. J. Jacobsen E. J. Walker J. K. MacInnes A. Bond B. R. Zhang L. L. Yu Y. Zheng Y. Rumsey J. M. Walgren J. L. Curtiss S. W. Fobian Y. M. Heasley S. E. Cubbage J. W. Moon J. B. Brown D. L. Acker B. A. Maddux T. M. Tollefson M. B. Mischke B. V. Owen D. R. Freskos J. N. Molyneaux J. M. Benson A. G. Blevis-Bal R. M. J. Med. Chem. 2010;53:2656–2660. doi: 10.1021/jm901781q. [DOI] [PubMed] [Google Scholar]
- Azuaje J. El Maatougui A. Pérez-Rubio J. M. Coelho A. Fernández F. Sotelo E. J. Org. Chem. 2013;78:4402–4409. doi: 10.1021/jo4003163. [DOI] [PubMed] [Google Scholar]
- Mahar K. M. Enslin M. B. Gress A. Amrine-Madsen H. Cooper M. Clin. Pharmacol. Drug Dev. 2018;7:33–43. doi: 10.1002/cpdd.363. [DOI] [PubMed] [Google Scholar]
- Borthwick A. D. Liddle J. Davies D. E. Exall A. M. Hamlett C. Hickey D. M. Mason A. M. Smith I. E. D. Nerozzi F. Peace S. Pollard D. Sollis S. L. Allen M. J. Woollard P. M. Pullen M. A. Westfall T. D. Stanislaus D. J. J. Med. Chem. 2012;55:783–796. doi: 10.1021/jm201287w. [DOI] [PubMed] [Google Scholar]
- Faggi C. García-Valverde M. Marcaccini S. Pepino R. Pozo M. C. Synthesis. 2003;2003:1553–1558. [Google Scholar]
- Fenwick A. Trans. R. Soc. Trop. Med. Hyg. 2006;100:200–207. doi: 10.1016/j.trstmh.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Cioc R. C. Ruijter E. Orru R. V. A. Green Chem. 2014;16:2958–2975. doi: 10.1039/C4GC00013G. [DOI] [Google Scholar]
- Cioli D. Pica-Mattoccia L. Parasitol. Res. 2003;90:S3–S9. doi: 10.1007/s00436-002-0751-z. [DOI] [PubMed] [Google Scholar]
- Doenhoff M. J. Cioli D. Utzinger J. Curr. Opin. Infect. Dis. 2008;21(6):659–667. doi: 10.1097/QCO.0b013e328318978f. [DOI] [PubMed] [Google Scholar]
- Woelfle M. Seerden J.-P. de Gooijer J. Pouwer K. Olliaro P. Todd M. H. PLoS Neglected Trop. Dis. 2011;5:e1260. doi: 10.1371/journal.pntd.0001260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai J.-Y. Infect. Chemother. 2013;45:32–43. doi: 10.3947/ic.2013.45.1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultani H. N. Haeri H. H. Hinderberger D. Westermann B. Org. Biomol. Chem. 2016;14:11336–11341. doi: 10.1039/C6OB02194H. [DOI] [PubMed] [Google Scholar]
- Ettari R. Micale N. Schirmeister T. Gelhaus C. Leippe M. Nizi E. Di Francesco M. E. Grasso S. Zappalà M. J. Med. Chem. 2009;52:2157–2160. doi: 10.1021/jm900047j. [DOI] [PubMed] [Google Scholar]
- Spencer J. Rathnam R. P. Chowdhry B. Z. Future Med. Chem. 2010;2:1441–1449. doi: 10.4155/fmc.10.226. [DOI] [PubMed] [Google Scholar]
- Van den Bogaert A. M. Nelissen J. Ovaere M. Van Meervelt L. Compernolle F. De Borggraeve W. M. Eur. J. Org. Chem. 2010:5397–5401. doi: 10.1002/ejoc.201000549. [DOI] [Google Scholar]
- Huang Y. Khoury K. Chanas T. Dömling A. Org. Lett. 2012;14:5916–5919. doi: 10.1021/ol302837h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan A. Verzal M. K. Tetrahedron Lett. 2005;46:1697–1701. doi: 10.1016/j.tetlet.2005.01.063. [DOI] [Google Scholar]
- Tempest P. Pettus L. Gore V. Hulme C. Tetrahedron Lett. 2003;44:1947–1950. doi: 10.1016/S0040-4039(03)00084-4. [DOI] [Google Scholar]
- Omar-Amrani R. Thomas A. Brenner E. Schneider R. Fort Y. Org. Lett. 2003;5:2311–2314. doi: 10.1021/ol034659w. [DOI] [PubMed] [Google Scholar]
- Yamada K. Kubo T. Tokuyama H. Fukuyama T. Synlett. 2002:0231–0234. doi: 10.1055/s-2002-19782. [DOI] [Google Scholar]
- Cuny G. Bois-Choussy M. Zhu J. J. Am. Chem. Soc. 2004;126:14475–14484. doi: 10.1021/ja047472o. [DOI] [PubMed] [Google Scholar]
- De Silva R. A. Santra S. Andreana P. R. Org. Lett. 2008;10:4541–4544. doi: 10.1021/ol801841m. [DOI] [PubMed] [Google Scholar]
- Santra S. Andreana P. R. Org. Lett. 2007;9:5035–5038. doi: 10.1021/ol702256t. [DOI] [PubMed] [Google Scholar]
- Hulme C. Ma L. Kumar N. V. Krolikowski P. H. Allen A. C. Labaudiniere R. Tetrahedron Lett. 2000;41:1509–1514. doi: 10.1016/S0040-4039(99)02326-6. [DOI] [Google Scholar]
- Azuaje J. Pérez-Rubio J. M. Yaziji V. El Maatougui A. González-Gomez J. C. Sánchez-Pedregal V. c. M. Navarro-Vázquez A. Masaguer C. F. Teijeira M. Sotelo E. J. Org. Chem. 2015;80:1533–1549. doi: 10.1021/jo502382q. [DOI] [PubMed] [Google Scholar]
- Wang L. Ren Z.-L. Ding M.-W. J. Org. Chem. 2015;80:641–646. doi: 10.1021/jo502275f. [DOI] [PubMed] [Google Scholar]
- Riva R. Banfi L. Basso A. Cerulli V. Guanti G. Pani M. J. Org. Chem. 2010;75:5134–5143. doi: 10.1021/jo100859y. [DOI] [PubMed] [Google Scholar]
- Buysse K. Farard J. Nikolaou A. Vanderheyden P. Vauquelin G. Sejer Pedersen D. Tourwé D. Ballet S. Org. Lett. 2011;13:6468–6471. doi: 10.1021/ol202767k. [DOI] [PubMed] [Google Scholar]
- Barlow T. M. A. Jida M. Tourwé D. Ballet S. Org. Biomol. Chem. 2014;12:6986–6989. doi: 10.1039/C4OB01381F. [DOI] [PubMed] [Google Scholar]
- Devji T. Reddy C. Woo C. Awale S. Kadota S. Carrico-Moniz D. Bioorg. Med. Chem. Lett. 2011;21:5770–5773. doi: 10.1016/j.bmcl.2011.08.005. [DOI] [PubMed] [Google Scholar]
- Curini M. Epifano F. Maltese F. Marcotullio M. C. Gonzales S. P. Rodriguez J. C. Aust. J. Chem. 2003;56:59–60. doi: 10.1071/CH02177. [DOI] [Google Scholar]
- Anand P. Singh B. Singh N. Bioorg. Med. Chem. 2012;20:1175–1180. doi: 10.1016/j.bmc.2011.12.042. [DOI] [PubMed] [Google Scholar]
- Xue H. Lu X. Zheng P. Liu L. Han C. Hu J. Liu Z. Ma T. Li Y. Wang L. Chen Z. Liu G. J. Med. Chem. 2010;53:1397–1401. doi: 10.1021/jm901653e. [DOI] [PubMed] [Google Scholar]
- Banfi L. Basso A. Giardini L. Riva R. Rocca V. Guanti G. Eur. J. Org. Chem. 2011:100–109. doi: 10.1002/ejoc.201001077. [DOI] [Google Scholar]
- Hulme C. Dietrich J. Mol. Diversity. 2009;13:195–207. doi: 10.1007/s11030-009-9111-6. [DOI] [PubMed] [Google Scholar]
- McAtee J. J. Dodson J. W. Dowdell S. E. Erhard K. Girard G. R. Goodman K. B. Hilfiker M. A. Jin J. Sehon C. A. Sha D. Shi D. Wang F. Wang G. Z. Wang N. Wang Y. Viet A. Q. Yuan C. C. K. Zhang D. Aiyar N. V. Behm D. J. Carballo L. H. Evans C. A. Fries H. E. Nagilla R. Roethke T. J. Xu X. Douglas S. A. Neeb M. J. Bioorg. Med. Chem. Lett. 2008;18:3716–3719. doi: 10.1016/j.bmcl.2008.05.058. [DOI] [PubMed] [Google Scholar]
- Qian W. Wang D. Wang H. Yu P. Liu S. Chen S. Tetrahedron Lett. 2018;59:2167–2169. doi: 10.1016/j.tetlet.2018.04.050. [DOI] [Google Scholar]
- Ayaz M. Xu Z. Hulme C. Tetrahedron Lett. 2014;55:3406–3409. doi: 10.1016/j.tetlet.2014.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y.-M. Li H.-Y. Ren J. Wang S. Ding M.-W. Synlett. 2018;29:1447–1450. doi: 10.1055/s-0037-1609846. [DOI] [Google Scholar]
- Gupta D. Ghosh N. N. Chandra R. Bioorg. Med. Chem. Lett. 2005;15:1019–1022. doi: 10.1016/j.bmcl.2004.12.041. [DOI] [PubMed] [Google Scholar]
- El-Sabbagh O. I. El-Sadek M. E. Lashine S. M. Yassin S. H. El-Nabtity S. M. Med. Chem. Res. 2009;18:782. doi: 10.1007/s00044-009-9203-y. [DOI] [Google Scholar]
- Ajani O. O. Obafemi C. A. Ikpo C. O. Ogunniran K. O. Nwinyi O. C. Chem. Heterocycl. Compd. 2009;45:1370–1378. doi: 10.1007/s10593-010-0435-z. [DOI] [Google Scholar]
- Patel N. B. Patel J. C. Med. Chem. Res. 2011;20:511–521. doi: 10.1007/s00044-010-9345-y. [DOI] [Google Scholar]
- Sharma M. Chauhan K. Shivahare R. Vishwakarma P. Suthar M. K. Sharma A. Gupta S. Saxena J. K. Lal J. Chandra P. Kumar B. Chauhan P. M. S. J. Med. Chem. 2013;56:4374–4392. doi: 10.1021/jm400053v. [DOI] [PubMed] [Google Scholar]
- Geitmann M. Unge T. Danielson U. H. J. Med. Chem. 2006;49:2375–2387. doi: 10.1021/jm0504050. [DOI] [PubMed] [Google Scholar]
- Kumar N. Shalini K. Drabu S. Biointerface Res. Appl. Chem. 2011;1:203–208. [Google Scholar]
- Vashi R. Shelat C. Synthesis. 2011;3:911–916. [Google Scholar]
- Balalaie S. Saeedi S. Ramezanpour S. Helv. Chim. Acta. 2016;99:138–142. doi: 10.1002/hlca.201500187. [DOI] [Google Scholar]
- Hulme C. Chappeta S. Dietrich J. Tetrahedron Lett. 2009;50:4054–4057. doi: 10.1016/j.tetlet.2009.04.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich J. Kaiser C. Meurice N. Hulme C. Tetrahedron Lett. 2010;51:3951–3955. doi: 10.1016/j.tetlet.2010.05.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.-Y. Riedrich M. Mikyna M. Arndt H.-D. Angew. Chem., Int. Ed. 2009;48:8137–8140. doi: 10.1002/anie.200903477. [DOI] [PubMed] [Google Scholar]
- Marsden S. P. McGonagle A. E. McKeever-Abbas B. Org. Lett. 2008;10:2589–2591. doi: 10.1021/ol800921n. [DOI] [PubMed] [Google Scholar]
- De Moliner F. Crosignani S. Banfi L. Riva R. Basso A. J. Comb. Chem. 2010;12:613–616. doi: 10.1021/cc100122n. [DOI] [PubMed] [Google Scholar]
- Lecinska P. Corres N. Moreno D. García-Valverde M. Marcaccini S. Torroba T. Tetrahedron. 2010;66:6783–6788. doi: 10.1016/j.tet.2010.06.062. [DOI] [Google Scholar]
- He P. Nie Y.-B. Wu J. Ding M.-W. Org. Biomol. Chem. 2011;9:1429–1436. doi: 10.1039/C0OB00855A. [DOI] [PubMed] [Google Scholar]
- Khorana N. Smith C. Herrick-Davis K. Purohit A. Teitler M. Grella B. Dukat M. Glennon R. A. J. Med. Chem. 2003;46:3930–3937. doi: 10.1021/jm030080s. [DOI] [PubMed] [Google Scholar]
- Gupta L. Srivastava K. Singh S. Puri S. K. Chauhan P. M. S. Bioorg. Med. Chem. Lett. 2008;18:3306–3309. doi: 10.1016/j.bmcl.2008.04.030. [DOI] [PubMed] [Google Scholar]
- Jenkins P. R. Wilson J. Emmerson D. Garcia M. D. Smith M. R. Gray S. J. Britton R. G. Mahale S. Chaudhuri B. Bioorg. Med. Chem. 2008;16:7728–7739. doi: 10.1016/j.bmc.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Lesma G. Cecchi R. Crippa S. Giovanelli P. Meneghetti F. Musolino M. Sacchetti A. Silvani A. Org. Biomol. Chem. 2012;10:9004–9012. doi: 10.1039/C2OB26301G. [DOI] [PubMed] [Google Scholar]
- Santra S. Andreana P. R. Angew. Chem. 2011;123:9590–9594. doi: 10.1002/ange.201103567. [DOI] [PubMed] [Google Scholar]
- Paulvannan K. J. Org. Chem. 2004;69:1207–1214. doi: 10.1021/jo030231z. [DOI] [PubMed] [Google Scholar]
- Wright D. L. Robotham C. V. Aboud K. Tetrahedron Lett. 2002;43:943–946. doi: 10.1016/S0040-4039(01)02299-7. [DOI] [Google Scholar]
- Nunes P. S. G. Vidal H. D. A. Corrêa A. G. Org. Biomol. Chem. 2020;18(39):7751–7773. doi: 10.1039/D0OB01631D. [DOI] [PubMed] [Google Scholar]
- Lesma G. Meneghetti F. Sacchetti A. Stucchi M. Silvani A. Beilstein J. Org. Chem. 2014;10:1383–1389. doi: 10.3762/bjoc.10.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riva R. Science. 2018;361:1072–1073. doi: 10.1126/science.aau7497. [DOI] [PubMed] [Google Scholar]
- Zhu D. Xia L. Pan L. Li S. Chen R. Mou Y. Chen X. J. Org. Chem. 2012;77:1386–1395. doi: 10.1021/jo2021967. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Ao Y.-F. Huang Z.-T. Wang D.-X. Wang M.-X. Zhu J. Angew. Chem., Int. Ed. 2016;55:5282–5285. doi: 10.1002/anie.201600751. [DOI] [PubMed] [Google Scholar]
- Wang Q. Wang D.-X. Wang M.-X. Zhu J. Acc. Chem. Res. 2018;51:1290–1300. doi: 10.1021/acs.accounts.8b00105. [DOI] [PubMed] [Google Scholar]
- Beyreuther B. K. Freitag J. Heers C. Krebsfänger N. Scharfenecker U. Stöhr T. CNS Drug Rev. 2007;13:21–42. doi: 10.1111/j.1527-3458.2007.00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. Wang Y.-Y. Sun H. Li S.-Y. Xiang S.-H. Tan B. Sci. China: Chem. 2020;63:47–54. doi: 10.1007/s11426-019-9606-2. [DOI] [Google Scholar]
- Zhang J. Yu P. Li S.-Y. Sun H. Xiang S.-H. Wang J. Houk K. N. Tan B. Science. 2018;361:eaas8707. doi: 10.1126/science.aas8707. [DOI] [PubMed] [Google Scholar]
- Shaabani S. Dömling A. Angew. Chem., Int. Ed. 2018;57:16266–16268. doi: 10.1002/anie.201811129. [DOI] [PMC free article] [PubMed] [Google Scholar]