

Abstract
Peptides are gaining substantial attention as therapeutics for human diseases. However, they have limitations such as low bioavailability and poor pharmacokinetics. Periostin, a matricellular protein, can stimulate the repair of ischemic tissues by promoting angiogenesis. We have previously reported that a novel angiogenic peptide (amino acids 142-151) is responsible for the pro-angiogenic activity of periostin. To improve the in vivo delivery efficiency of periostin peptide (PP), we used proteins self-assembled into a hollow cage-like structure as a drug delivery nanoplatform in the present study. The periostin peptide was genetically inserted into lumazine synthase (isolated from Aquifex aeolicus) consisting of 60 identical subunits with an icosahedral capsid architecture. The periostin peptide-bearing lumazine synthase protein cage nanoparticle with 60 periostin peptides multivalently displayed was expressed in Escherichia coli and purified to homogeneity. Next, we examined angiogenic activities of this periostin peptide-bearing lumazine synthase protein cage nanoparticle. AaLS-periostin peptide (AaLS-PP), but not AaLS, promoted migration, proliferation, and tube formation of human endothelial colony-forming cells in vitro. Intramuscular injection of PP and AaLS-PP increased blood perfusion and attenuated severe limb loss in the ischemic hindlimb. However, AaLS did not increase blood perfusion or alleviate tissue necrosis. Moreover, in vivo administration of AaLS-PP, but not AaLS, stimulated angiogenesis in the ischemic hindlimb. These results suggest that AaLS is a highly useful nanoplatform for delivering pro-angiogenic peptides such as PP.
Keywords: Angiogenesis, Angiogenic peptide, Periostin, Peripheral artery disease, Protein cage nanoparticle
INTRODUCTION
Peripheral artery disease (PAD) is a common circulatory problem in which narrowed arteries show reduced blood flow to limbs. PAD can progress into critical limb ischemia in the severe disease stage, which is associated with a higher risk of limb amputation. Therefore, it is essential to enhance blood perfusion and angiogenesis to treat PAD (1, 2). Therapeutic angiogenesis using growth factors and peptides is regarded as a promising strategy for the treatment of PAD (3, 4). Peptides can be efficiently delivered to target tissues because of their small sizes, making them attractive therapeutics for ischemic diseases. However, there are disadvantages when using of peptide drugs because peptides cannot fully retain the bioactivity of their parental protein, needing higher concentrations than their parental protein to achieve similar effects (5). Moreover, peptides are highly susceptible to protease degradation and rapid clearance by kidney, leading to poor pharmacokinetics when delivered systematical-ly (6-8). Therefore, in vivo delivery efficacy of peptide drugs must be improved to develop pro-angiogenic peptide therapeutics.
Protein cage nanoparticles are formed by self-assembly of protein subunits to produce a cage-like structure (9). Lumazine synthase originating from hyperthermophile Aquifex aeolicus (AaLS) consists of 60 identical subunits with spherical and hollow icosahedral capsid structures having exterior and interior diameters of 15.4 nm and 9 nm, respectively (10). Accumulating evidence suggests that AaLS can display various types of ligands such as target-binding ligands, contrast agents, and large functional proteins on its surface through genetic or chemical modifications (11-14). Furthermore, AaLS can uniformly display peptides on the surface without losing essential properties of protein cage architectures (13, 15). Protein cage nanoparticles have been used to deliver drugs and antigens (14, 16, 17). However, the delivery of therapeutic peptides has not been reported yet.
Periostin, also known as osteoblast-specific factor 2 (OSF2), is a 93-kDa matricellular protein which was identified as a cell adhesion protein (18). It was recently classified as a novel matricellular protein related to the extracellular matrix, sharing homology with insect cell adhesion molecule fasciclin 1 (18, 19). Periostin can promote migration, invasion, and angiogenesis of human umbilical endothelial cells (20-22). It also can stimulate angiogenesis in tumors (23, 24). Previously, we have identified a pro-angiogenic peptide consisting of 10 amino acids (amino acids 142-151, WDNLDSDIRR) of periostin (21). The periostin peptide (PP) can stimulate the migration, adhesion, proliferation, and tube-forming abilities of human umbilical cord blood-derived endothelial colony-forming cells (ECFCs).
In the present study, we utilized a lumazine synthase as a template protein cage nanoparticle for multivalent display of PP. Angiogenic and therapeutic capacities of PP-expressing AaLS (AaLS-PP) were explored using in vitro ECFCs and in vivo hindlimb ischemia models.
RESULTS
Engineering of a PP in lumazine synthase protein cage nanoparticles
We utilized a previously developed lumazine synthase protein cage nanoparticle (AaLS) (13) as a template and genetically fused 10 amino acids of PP (WDNLDSDIRR), amino acid sequences 142-151 of periostin (21), with an extra linker (GSGG) to form AaLS-PP (Fig. 1A). We have previously reported that the C-termini of AaLS subunits are fully exposed to the surface of the AaLS and that they can tolerate peptide insertion of various lengths (25). In addition, a short extra linker is sufficient for target recognition of fused peptides (25). AaLS-PP was over-expressed in E. coli and purified using immobilized metal affinity chromatography (IMAC). We obtained a highly pure (> 98%) protein that migrated as a single protein band between apparent molecular masses of 35 kDa and 28 kDa in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (Fig. 1B). The actual molecular mass of the AaLS-PP subunit was determined to be 28,970.0 Da using a mass spectrometer, which was almost identical to the calculated mass value (28,970.2 Da) based on its amino acid sequences (Fig. 1C). AaLS-PP was eluted as a single peak. It was eluted much earlier than that the 29 kDa monomeric protein in size exclusion chromatography (Fig. 1D). It exhibited a hydrodynamic diameter of 28 nm during dynamic light scattering analysis (Fig. 1E). A spherical and hollow structure of AaLS-PP was confirmed by transmission electron microscopy (TEM; Fig. 1F). These results indicated that PPs were successfully fused to AaLS and that AaLS-PP formed a spherical cage nanoparticle architecture with a uniform size. The concentration of AaLS-PP used in this study was calculated based on the molar concentration of the AaLS protein cage nanoparticle.
Fig. 1.

Construction and characterization of AaLS-PP. (A) Schematic illustration of AaLS-PP construction consisting of 60 AaLS-PP subunits. (B) SDS-PAGE analysis of purified AaLS-PP subunits. (C) Mass spectrometric analyses of AaLS-PP subunits. (D) Size exclusion chromatography elution profile of AaLS-PP. (E) Dynamic light scattering analysis of AaLS-PP. (F) Transmission electron microscopic image of AaLS-PP stained with 1% uranyl acetate.
AaLS-PP stimulates angiogenic activities of ECFCs
To explore pro-angiogenic effects of ECFCs, cell migration ability was measured using a transwell cell migration assay. In previous studies, we have identified the PP sequence from the first fasciclin domain (D1), which is responsible for periostin-mediated angiogenesis (20, 21). VEGF, recombinant periostin proteins (full length and D1 domain), and PP all stimulated the migration of ECFCs (Fig. 2A). To examine effects of AaLS-PP on ECFC migration, we compared AaLS-PP and its negative control, AaLS, at different concentrations. As shown in Fig 2A, AaLS-PP dose-dependently promoted the migration of ECFCs with maximal stimulation at 5 nM, whereas AaLS had no significant effect on cell migration, suggesting that AaLS-PP specifically stimulated the migration of ECFCs through a PP-mediated mechanism. In the following experiments, we used 5 nM of recombinant protein cage nanoparticles to compare pro-angiogenic activities of AaLS-PP and AaLS in vitro.
Fig. 2.

Effects of periostin peptide-bearing protein cage nanoparticle on endothelial colony-forming cells. (A) The migration ability of endothelial colony-forming cells (ECFCs) for AaLS was evaluated after treatment with VEGF (10 ng/ml), full-length recombinant periostin (10 μg/ml), periostin domain 1 (10 μg/ml), PP (0.5 μM), AaLS, or AaLS-PP at indicated concentrations. Data are presented as mean ± S.D. (n = 7). (B) Representative images of tube formation of ECFCs after treatment with VEGF (10 ng/ml), full-length recombinant periostin (10 μg/ml), periostin D1 (10 μg/ml), PP (0.5 μM), AaLS (5 nM), and AaLS-PP (5 nM). Scale bar = 100 μm. (C) Lengths of tubes were quantified in four random fields from each well and values were normalized relative to those of the corresponding control. Data are presented as mean ± S.D. (n = 7). ECFCs were incubated with PP (0.5 μM) or AaLS-PP (5 nM) for 24 h. (D) Representative images of ECFCs stained with Ki67 (green) and DAPI (blue). Scale bar = 50 μm. (E) The number of Ki67-positive cells was quantified per DAPI-positive cells. Data are presented as mean ± S.D. (n = 9). *P < 0.05 vs. PBS; #P < 0.05 vs. PP.
We performed a tube-forming assay using ECFCs to confirm the pro-angiogenic effect of AaLS-PP. The endothelial tube-form-ing ability of ECFCs was increased by treatment with 5 nM of AaLS-PP, which was as potent as treatment with VEGF, FL, D1, or PP (Fig. 2B, C), suggesting that AaLS-PP could specifically activate the endothelial tube-forming ability of ECFCs. Moreover, we have reported that PP can promote the migration and tube formation of ECFCs and increase their proliferation (21). To determine whether AaLS-PP also promote their proliferation, ECFCs were treated with PP or AaLS-PP for 24 h and then immunostained with an antibody against Ki67, a proliferation marker, with their nuclei labeled with DAPI. The number of Ki67-positive ECFCs was increased by treatment with PP or AaLS-PP (Fig. 2D). The percentage of Ki67-positive cells per DAPI-positive total cells was increased by treatment with AaLS-PP (Fig. 2E). These results suggest that AaLS-PP can activate the migration, tube-forming ability, and proliferation of ECFCs in vitro.
AaLS-PP promotes blood perfusion and alleviates tissue necrosis in a murine hindlimb ischemia model
Previously, we have demonstrated that periostin and D1 can increase blood perfusion in ischemic limbs and alleviate tissue necrosis (20). However, effects of PP and AaLS-PP on the repair of ischemic tissues have not been reported yet. To investigate effects of PP and AaLS-PP on vascular regeneration in vivo, we examined therapeutic effects of PP, AaLS, and AaLS-PP in an ischemic hindlimb animal model. PP, AaLS, and AaLS-PP were administered by intramuscular injection into ischemic hindlimbs three times per week for four weeks. Blood flow was then measured by laser Doppler imaging analysis. As shown in Fig. 3A, intramuscular injection of AaLS-PP, but not AaLS, improved blood perfusion in ischemic limbs. Intramuscular administration of PP also increased blood perfusion in ischemic limbs (Fig. 3B). Notably, on days 7 and 14, blood flow was improved more rapidly in the PP group than in the AaLS-PP group. However, on days 21 and 28, PP and AaLS-PP groups exhibited similar blood flow levels. Tissue necrosis score was measured before sacrifice on day 28. Administration of either PP or AaLS-PP reduced tissue necrosis compared to PBS. However, there was no significant difference between PP and AaLS-PP groups (Fig. 3C). These results suggest that intramuscular injection of either PP or AaLS-PP could augment blood perfusion and alleviate tissue necrosis in ischemic hindlimbs.
Fig. 3.

Effects of AaLS-PP on blood perfusion and limb salvage in an ischemic hindlimb animal model. After modeling mouse hindlimb ischemia, PP (5 μM), AaLS (50 nM), and AaLS-PP (50 nM) were intramuscular injected three times a week and observations were made until day 28. (A) Representative images of hindlimbs and blood flow measured by LDPI on days 0 and 28 from eight different mice. (B) Quantitative analysis of blood flow measured by LDPI. LDPI ratio was calculated as the ratio of ischemic to contralateral hindlimb blood perfusion over the observation period. Data are presented as mean ± S.D. (n = 8 per group). *P < 0.05 vs. PBS. (C) Statistical analysis of the necrosis score on day 28. Data are presented as mean ± S.D. (n = 8 per group). *P < 0.05 vs. PBS.
AaLS-PP promotes angiogenesis in ischemic limbs
To explore whether PP and AaLS-PP could promote angiogenesis in ischemic hindlimbs, expression levels of CD31, a capillary marker, and α-smooth muscle actin (α-SMA), an artery vessel marker, were investigated by immunostaining analysis (Fig. 4A). The density of CD31-positive capillaries in PP-injected limbs was significantly higher than that in PBS-injected control limbs. Furthermore, the number of CD31-positive capillaries in AaLS-PP-injected limbs was higher than that in AaLS-injected limbs, with values similar to those of PP-injected limbs (Fig. 4B). Furthermore, the density of α-SMA-positive blood vessels was higher in PP- and AaLS-PP-injected groups than in PBS- and AaLS-injected groups (Fig. 4C). There was no significant difference in the density of α-SMA-positive blood vessels between PP and AaLS-PP groups. These results suggest that AaLS-PP could stimulate blood perfusion in ischemic limbs by augmenting angiogenesis.
Fig. 4.
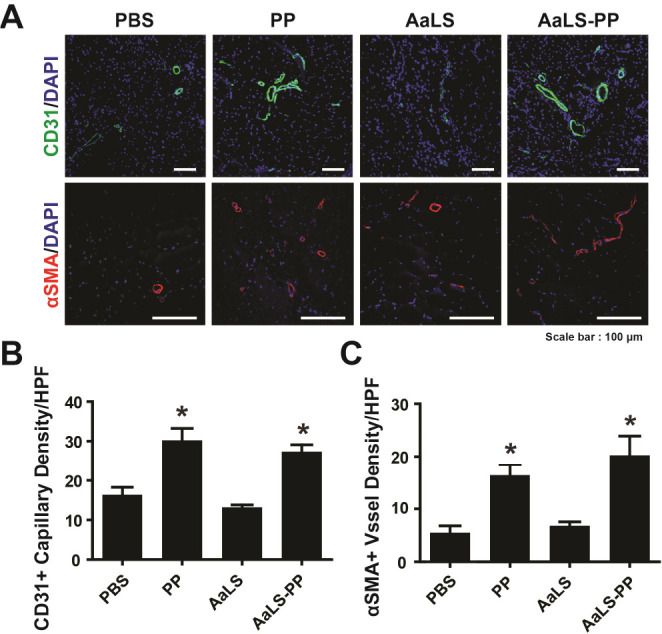
Effects of AaLS-PP on neovascularization in ischemic hindlimbs. Staining capillaries and blood vessels in ischemic limbs 28 days after intramuscular injection of PBS, PP, AaLS, or AaLS-PP. (A) Representative images immunostained with CD31 (green) or α-SMA (red). Nuclei (blue) were counterstained with DAPI. Overlaid images are shown. (B) Quantitative analysis of capillary density expressed by the number of CD31-positive capillaries per high power field (HPF). (C) Quantitative analysis of α-SMA-positive vessels per HPF. Data are presented as mean ± S.D. (n = 24). *P < 0.05 vs. PBS.
DISCUSSION
It has been reported that full-length recombinant and the first FAS-I domain of periostin can stimulate angiogenic activities of ECFCs by activating integrins β3 and β5 (20). Moreover, PP can stimulate the migration of ECFCs through an integrin β5-dependent mechanism (21), indicating that PP can act as a ligand for integrin receptors. In the present study, we demonstrated that periostin peptide-decorated protein cage nanoparticles could promote migration, tube formation, and proliferation of ECFCs for the first time. AaLS-PP dose-dependently increased angiogenic activities of ECFCs with maximal stimulation at 5 nM, which corresponded to angiogenic activities of ECFCs treated with 0.5 μM PP, indicating that the pro-angiogenic activity of AaLS-PP was 100-times more potent than that of PP. Because AaLS-PP contains 60 PPs multivalently expressed in the exterior of the AaLS protein cage, this result suggests that PPs displayed on the exterior of AaLS can act as a ligand similar to soluble PP. Therefore, AaLS is likely to be a good carrier for the delivery of peptides.
Renal filtration affects pharmacokinetics of peptides and protein drugs. It is believed that proteins with a molecular weight of less than 15 kDa, but not plasma proteins larger than 60 kDa, can freely filter through the glomeruli of kidneys (26). The half-life of peptide drugs can be extended by several technologies, including genetic fusion to the Fc domain, a long inert polypeptide, PEGylation, and conjugation to serum albumin or IgG (27). The molecular weight of the AaLS-PP subunit was estimated to be approximately 29 kDa. Because AaLS-PP protein cages were composed of 60 subunits, the total molecular weight of AaLS-PP protein cages was 1,740 kDa. Using in vivo near infrared imaging and magnetic resonance imaging analyses, we have previously reported that AaLS variants show prolonged blood circulation time (remaining in mouse for longer than 12 h) (28). This suggests that AaLS-PP protein cages cannot filter through glomeruli and that AaLS-PP protein cages might have a longer half-life than PP. We demonstrated that intramuscular injection of PP and AaLS-PP increased blood perfusion and alleviated tissue necrosis in ischemic hindlimbs. However, AaLS did not affect blood perfusion or angiogenesis in vivo. Intriguingly, blood flow was improved more rapidly in the PP group than in the AaLS-PP group. However, both PP and AaLS-PP groups exhibited similar blood flow levels on day 28. Although the molecular mechanism associated with the delayed blood perfusion in the AaLS-PP group remains unclear, these results suggest that AaLS-PP can be used for the treatment of peripheral artery diseases. Our findings suggest that protein cage nanoplatform is a promising tool for clinical and biotechnological applications of peptides, antibodies, chemicals, and other molecules.
MATERIALS AND METHODS
Materials
The EGM-2MV bullet kit (CC-3202) and EBM-2 (CC3156) medium were purchased from Lonza (Basel, Switzerland). PBS (LB001) was purchased from Welgene (Gyeongsan, South Korea). Fetal bovine serum (FBS) and 0.05% trypsin-EDTA were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Bovine serum albumin (BSA) and gelatin were purchased from Sigma-Aldrich (St. Louis, MO, USA). Recombinant human VEGF and periostin were purchased from R&D Systems (Minneapolis, MN, USA). Anti-CD31 antibody (MEC 13.3) and growth factor-reduced Matrigel (GFR-Matrigel) were purchased from BD Bio-sciences (Bedford, MA, USA). Rabbit anti-α-SMA antibody (ab5694) was purchased from Abcam (Cambridge, United Kingdom). Alexa 488, Alexa 568 goat anti-rabbit, and Alexa 488 goat anti-mouse antibodies were purchased from Life Technologies (Carlsbad, CA, USA).
Protein expression and purification
The AaLS-PP gene was cloned into the pETDuet-1 vector (AmpR) and transformed into the competent E. coli strain BL21 (DE3). Cells were grown in Luria-Bertani (LB) medium (1.0 L) containing 50 mg/L ampicillin at 37°C. For protein expression, cells were induced with 500 μM IPTG at an optical density of 0.5-0.6 at 600 nm and further cultured overnight at 30°C. The cells were harvested and lysed with a lysis buffer containing lysozyme, followed by sonication, and cellular debris was removed by centrifugation (15,000 × g, 1 h). Cell lysates were heated (65°C, 10 min), centrifuged (15,000 × g, 30 min), and subsequently purified by fast protein liquid chromatography (FPLC) using immobilized metal affinity chromatography (IMAC). The endotoxin was removed using Triton X-114 (Sigma) until the resulting level was less than 0.25 EU/ml and quantified using a Limulus Amebocyte Lysate assay (Genescript).
Cell culture
Human ECFCs were isolated from human umbilical cord blood and characterized as previously described (20, 29, 30). ECFCs were cultured on a 0.1% gelatin-coated dish with EGM-2MV medium at 37°C in a 5% CO2 incubator. When the ECFC density reached 70-80% of the dish area, the cells were subcultured at a ratio of 1:3. To harvest the ECFCs, cells were washed with PBS two times and treated with 0.05% trypsin-EDTA in a 5% CO2 incubator at 37°C for 3 min. The trypsin-treated cells were neutralized with FBS, collected in a conical tube, and centrifuged at 200 × g for 4 min. ECFCs from passage numbers 6-8 were used in this study.
Hindlimb ischemia model and blood flow measurement
Animal experiments were performed using protocols approved by the Pusan National University Institutional Animal Use and Care Committee (PNU-2016-1381). The hind limb ischemia model was established by ligating and burning the artery at the hindlimb, as described previously (29). BALB/C (male, age 6-9 weeks, weighing 22-24 g) mice were anesthetized with an intraperitoneal injection of 400 mg/kg 2,2,2-tribromoethanol (Sigma-Aldrich, St. Louis, MO) and shaved for operative resection of one femoral artery and laser Doppler perfusion imaging (LDPI). The femoral artery was excised from its proximal origin as a branch of the external iliac artery to the distal point where it bifurcates into the saphenous and popliteal arteries. After arterial ligation, ischemic hindlimbs were injected with PBS, PP, AaLS, and AaLS-PP into three sites (20 μl per site, total 60 μl) in the muscle of the medial thigh three times per week. The extent of necrosis in the ischemic hindlimb was recorded on day 28 after surgery.
Measurements of blood flow and tissue necrosis
Blood flow in ischemic and normal limbs was measured on days 0, 7, 14, 21, and 28 after surgery using an LDPI analyzer (Moor Instruments, Ltd., Devon, UK). The contralateral hind limbs served as internal controls. Perfusions in ischemic and contralateral limbs were calculated by counting red (high perfusion) and blue (low perfusion) colored histogram pixels. Blood perfusion was expressed as the LDPI index, which represented the ischemic versus non-ischemic limb blood flow ratio. A ratio of 1 before surgery indicated equal blood perfusion in both legs. The extent of necrosis in the ischemic hindlimb was recorded on day 28 after surgery. Scores for necrosis were assessed as follows: 0, limb salvage; 1, toe amputation; 2, foot amputation; and 3, limb amputation.
Statistical analysis
Results of multiple observations are presented as the mean ± SD. For multivariate data analysis, group differences were assessed using a one-way or two-way analysis of variance, followed by post hoc comparisons using Scheffe’s method.
Supplementary methods
Mass spectrometry analysis and characterization of the protein cage nanoparticles, immunocytochemistry analysis, and assays for migration, tube formation, and proliferation of ECFCs are described in the supplementary information.
SUPPLEMENTARY MATERIALS
ACKNOWLEDGEMENTS
This research was supported by the MRC programs (NRF-2015 R1A5A2009656) of the National Research Foundation of Korea funded by the Ministry of Education, Science, and Technology (NRF-2020R1A2C2011654 and 2019R1A2C2002749).
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicting interests.
REFERENCES
- 1.Rubanyi GM. Angiogenesis in health and disease : basic mechanisms and clinical applications. Dekker; New York: 2000. [Google Scholar]
- 2.Tongers J, Roncalli JG, Losordo DW. Therapeutic angiogenesis for critical limb ischemia: microvascular thera-pies coming of age. Circulation. 2008;118:9–16. doi: 10.1161/CIRCULATIONAHA.108.784371. [DOI] [PubMed] [Google Scholar]
- 3.Kumar VA, Liu Q, Wickremasinghe NC, et al. Treatment of hind limb ischemia using angiogenic peptide nanofibers. Biomaterials. 2016;98:113–119. doi: 10.1016/j.biomaterials.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sarkar B, Nguyen PK, Gao W, Dondapati A, Siddiqui Z, Kumar VA. Angiogenic self-assembling peptide scaffolds for functional tissue regeneration. Biomacromolecules. 2018;19:3597–3611. doi: 10.1021/acs.biomac.8b01137. [DOI] [PubMed] [Google Scholar]
- 5.Licht T, Tsirulnikov L, Reuveni H, Yarnitzky T, Ben-Sasson SA. Induction of pro-angiogenic signaling by a synthetic peptide derived from the second intracellular loop of S1P3 (EDG3) Blood. 2003;102:2099–2107. doi: 10.1182/blood-2002-12-3634. [DOI] [PubMed] [Google Scholar]
- 6.Craik DJ, Fairlie DP, Liras S, Price D. The future of peptide-based drugs. Chem Biol Drug Des. 2013;81:136–147. doi: 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]
- 7.Frackenpohl J, Arvidsson PI, Schreiber JV, Seebach D. The outstanding biological stability of beta- and gamma-peptides toward proteolytic enzymes: an in vitro investigation with fifteen peptidases. Chembiochem. 2001;2:445–455. doi: 10.1002/1439-7633(20010601)2:6<445::AID-CBIC445>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 8.Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Synthetic therapeutic peptides: science and market. Drug Discov Today. 2010;15:40–56. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 9.Bhaskar S, Lim S. Engineering protein nanocages as carriers for biomedical applications. NPG Asia Mater. 2017;9:e371. doi: 10.1038/am.2016.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X, Meining W, Fischer M, Bacher A, Ladenstein R. X-ray structure analysis and crystallographic refinement of lumazine synthase from the hyperthermophile Aquifex aeolicus at 1.6 A resolution: determinants of thermostability revealed from structural comparisons. J Mol Biol. 2001;306:1099–1114. doi: 10.1006/jmbi.2000.4435. [DOI] [PubMed] [Google Scholar]
- 11.Azuma Y, Edwardson TGW, Hilvert D. Tailoring lumazine synthase assemblies for bionanotechnology. Chem Soc Rev. 2018;47:3543–3557. doi: 10.1039/C8CS00154E. [DOI] [PubMed] [Google Scholar]
- 12.Choi H, Choi B, Kim GJ, et al. Fabrication of nanoreaction clusters with dual-functionalized protein cage nanobuilding blocks. Small. 2018;14:e1801488. doi: 10.1002/smll.201801488. [DOI] [PubMed] [Google Scholar]
- 13.Kim H, Kang YJ, Min J, Choi H, Kang S. Development of an antibody-binding modular nanoplat-form for antibody-guided targeted cell imaging and delivery. RSC Advances. 2016;6:19208–19213. doi: 10.1039/C6RA00233A. [DOI] [Google Scholar]
- 14.Min J, Kim S, Lee J, Kang S. Lumazine synthase protein cage nanoparticles as modular delivery platforms for targeted drug delivery. RSC Adv. 2014;4:48596–48600. doi: 10.1039/C4RA10187A. [DOI] [Google Scholar]
- 15.Kang HJ, Kang YJ, Lee YM, Shin HH, Chung SJ, Kang S. Developing an antibody-binding protein cage as a molecular recognition drug modular nanoplatform. Biomaterials. 2012;33:5423–5430. doi: 10.1016/j.biomaterials.2012.03.055. [DOI] [PubMed] [Google Scholar]
- 16.Ra JS, Shin HH, Kang S, Do Y. Lumazine synthase protein cage nanoparticles as antigen delivery nanoplatforms for dendritic cell-based vaccine development. Clin Exp Vaccine Res. 2014;3:227–234. doi: 10.7774/cevr.2014.3.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rother M, Nussbaumer MG, Renggli K, Bruns N. Protein cages and synthetic polymers: a fruitful symbiosis for drug delivery applications, bionanotechnology and materials science. Chem Soc Rev. 2016;45:6213–6249. doi: 10.1039/C6CS00177G. [DOI] [PubMed] [Google Scholar]
- 18.Horiuchi K, Amizuka N, Takeshita S, et al. Identifi-cation and characterization of a novel protein, periostin, with restricted expression to periosteum and periodontal ligament and increased expression by transforming growth factor beta. J Bone Miner Res. 1999;14:1239–1249. doi: 10.1359/jbmr.1999.14.7.1239. [DOI] [PubMed] [Google Scholar]
- 19.Takeshita S, Kikuno R, Tezuka K, Amann E. Osteoblast-specific factor 2: cloning of a putative bone adhesion protein with homology with the insect protein fasciclin I. Biochem J. 1993;294(Pt 1):271–278. doi: 10.1042/bj2940271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim BR, Jang IH, Shin SH, et al. Therapeutic angiogenesis in a murine model of limb ischemia by recombinant periostin and its fasciclin I domain. Biochim Biophys Acta. 2014;1842:1324–1332. doi: 10.1016/j.bbadis.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 21.Kim BR, Kwon YW, Park GT, et al. Identification of a novel angiogenic peptide from periostin. PLoS One. 2017;12:e0187464. doi: 10.1371/journal.pone.0187464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z, Nie F, Chen X, et al. Upregulated periostin promotes angiogenesis in keloids through activation of the ERK 1/2 and focal adhesion kinase pathways, as well as the upregulated expression of VEGF and angiopoietin1. Mol Med Rep. 2015;11:857–864. doi: 10.3892/mmr.2014.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gillan L, Matei D, Fishman DA, Gerbin CS, Karlan BY, Chang DD. Periostin secreted by epithelial ovarian carcinoma is a ligand for alpha(v)/beta(3) and alpha(v)beta(5) integrins and promotes cell motility. Cancer Res. 2002;62:5358–5364. [PubMed] [Google Scholar]
- 24.Gonzalez-Gonzalez L, Alonso J. Periostin: a matricellular protein with multiple functions in cancer development and progression. Front Oncol. 2018;8:225. doi: 10.3389/fonc.2018.00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Min J, Kim S, Lee J, Kang S. Lumazine synthase protein cage nanoparticles as modular delivery platforms for targeted drug delivery. Rsc Advances. 2014;4:48596–48600. doi: 10.1039/C4RA10187A. [DOI] [Google Scholar]
- 26.Schenk S, Schoenhals GJ, de Souza G, Mann M. A high confidence, manually validated human blood plasma protein reference set. BMC Med Genomics. 2008;1:41. doi: 10.1186/1755-8794-1-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strohl WR. Fusion proteins for half-life extension of biologics as a strategy to make biobetters. BioDrugs. 2015;29:215–239. doi: 10.1007/s40259-015-0133-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim H, Jin S, Choi H, et al. Target-switchable Gd(III)- DOTA/protein cage nanoparticle conjugates with multiple targeting affibody molecules as target selective T-1 contrast agents for high-field MRI. J Control Release. 2021;335:269–280. doi: 10.1016/j.jconrel.2021.05.029. [DOI] [PubMed] [Google Scholar]
- 29.Heo SC, Kwon YW, Jang IH, et al. WKYMVm-induced activation of formyl peptide receptor 2 stimulates ischemic neovasculogenesis by promoting homing of endothelial colony-forming cells. Stem Cells. 2014;32:779–790. doi: 10.1002/stem.1578. [DOI] [PubMed] [Google Scholar]
- 30.Kwon YW, Lee SJ, Heo SC, et al. Role of CXCR2 in the Ac-PGP-induced mobilization of circulating angiogenic cells and its therapeutic implications. Stem Cells Transl Med. 2019;8:236–246. doi: 10.1002/sctm.18-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.