

Abstract
Conjugation of ubiquitin (Ub) to numerous substrate proteins regulates virtually all cellular processes. Eight distinct ubiquitin polymer linkages specifying different functional outcomes are generated in cells. However, the roles of some atypical poly‐ubiquitin topologies, in particular linkages via lysine 27 (K27), remain poorly understood due to a lack of tools for their specific detection and manipulation. Here, we adapted a cell‐based ubiquitin replacement strategy to enable selective and conditional abrogation of K27‐linked ubiquitylation, revealing that this ubiquitin linkage type is essential for proliferation of human cells. We demonstrate that K27‐linked ubiquitylation is predominantly a nuclear modification whose ablation deregulates nuclear ubiquitylation dynamics and impairs cell cycle progression in an epistatic manner with inactivation of the ATPase p97/VCP. Moreover, we show that a p97‐proteasome pathway model substrate (Ub(G76V)‐GFP) is directly modified by K27‐linked ubiquitylation, and that disabling the formation of K27‐linked ubiquitin signals or blocking their decoding via overexpression of the K27 linkage‐specific binder UCHL3 impedes Ub(G76V)‐GFP turnover at the level of p97 function. Our findings suggest a critical role of K27‐linked ubiquitylation in supporting cell fitness by facilitating p97‐dependent processing of ubiquitylated nuclear proteins.
Keywords: cell cycle, K27‐linked ubiquitylation, p97/VCP, ubiquitin, ubiquitin‐proteasome system
Subject Categories: Post-translational Modifications & Proteolysis
Cellular roles of atypical K27‐linked polyubiquitin chains are revealed by disabling their formation via conditional ubiquitin replacement or by blocking their decoding via a K27‐linkage‐specific binder.
Introduction
Protein modification by the polypeptide ubiquitin (Ub) is a central and versatile regulatory mechanism impacting virtually all aspects of eukaryotic cell biology. Ubiquitylation of substrate proteins proceeds via a three‐step enzymatic cascade involving multitudinous combinations of E1 Ub‐activating, E2 Ub‐conjugating and E3 Ub ligase enzymes, targeting a major fraction of the proteome (Swatek & Komander, 2016; Oh et al, 2018). These Ub modifications are reversed by numerous deubiquitinases (DUBs), making ubiquitylation processes reversible and highly dynamic (Clague et al, 2019). In addition to undergoing attachment to lysine residues in cellular target proteins, Ub contains seven internal lysine residues (K6, K11, K27, K29, K33, K48 and K63) that along with its N‐terminal methionine (M1) can serve as acceptor sites for Ub conjugation, enabling the formation of eight distinct Ub linkage types, all of which have been shown to be generated in cells, albeit at remarkably different levels (Dammer et al, 2011; Kim et al, 2011; Swatek & Komander, 2016; Oh et al, 2018; Swatek et al, 2019). Together with a large number of proteins containing Ub‐binding domains that recognize individual Ub‐dependent modifications, the multifaceted architecture of Ub polymers forms the basis of a complex cellular Ub code dictating the functional outcome of Ub‐dependent signaling processes (Swatek & Komander, 2016; Oh et al, 2018). A common fate of ubiquitylated proteins is their degradation via the 26S proteasome, which is largely signalled through K48‐linked poly‐Ub chains but can also be facilitated by other Ub chain types, including K29‐ and K11‐linkages (Matsumoto et al, 2010; Bard et al, 2018; Kaiho‐Soma et al, 2021). Accordingly, K48 linkages represent the most abundant Ub chain type in cells (Dammer et al, 2011; Swatek et al, 2019). In many cases, ubiquitylated proteins require partial unfolding to enable their degradation by the proteasome. This is mediated by the highly abundant hexameric AAA+‐type ATPase p97 (also known as VCP, or Cdc48 in lower eukaryotes), which in conjunction with a broad range of cofactors recognizes and extracts ubiquitylated client proteins from macromolecular complexes or cellular membranes by promoting their Ub‐dependent unfolding, typically accompanied by Ub chain editing (Bodnar & Rapoport, 2017; Twomey et al, 2019; van den Boom & Meyer, 2018). Ub‐binding proteasomal shuttle factors including RAD23 subsequently escort the client protein to the proteasome (Collins & Goldberg, 2017). The central role in the Ub system occupied by p97 implicates it in a plethora of cellular processes and functionally connects it to several diseases; indeed, small molecule inhibitors of p97 activity show promising potential as anticancer agents in preclinical studies (Anderson et al, 2015; Huryn et al, 2020; Le Moigne et al, 2017; van den Boom & Meyer, 2018).
In contrast to K48‐, K29‐ and K11‐linked Ub chains, K63‐linked ubiquitylation is a non‐proteolytic modification with well‐established critical regulatory roles in a range of key cellular processes, including the DNA damage response, innate immunity and vesicular transport (Chen & Sun, 2009; Swatek & Komander, 2016; Oh et al, 2018). However, several less abundant atypical Ub chains have more poorly defined roles in the cellular Ub code. In particular, K27‐linked Ub chains, which represent < 1% of total Ub conjugates in human cells (Dammer et al, 2011; Swatek et al, 2019), have largely eluded functional characterization so far, due to the low cellular abundance and an absence of high‐affinity reagents for their specific detection and isolation. This is compounded by a lack of knowledge on the enzymatic machinery responsible for the formation and removal of K27 linkages. These shortcomings render addressing the cellular function of K27‐linked ubiquitylation highly challenging. Structural studies revealed that K27 is the least solvent‐exposed lysine residue in Ub and thus may not be readily available for enzymatic modification, potentially accounting for the low abundance of K27‐linked Ub chains in cells (Esadze et al, 2011; Castaneda et al, 2016). Correspondingly, the K27 linkage is the least accessible among all di‐Ub conformations, which may explain why most DUBs display poor activity towards K27 linkages (Castaneda et al, 2016; Pan et al, 2016). In cells, recent work implicated K27‐linked ubiquitylation in processes including DNA damage repair, innate immunity and inhibition of the tumor suppressor PTEN (Liu et al, 2014; Gatti et al, 2015; Lee et al, 2019; van Huizen & Kikkert, 2019; Yin et al, 2019), but the precise functional roles of K27‐linked Ub signals in these processes are not fully understood. In yeast, preventing K27‐linked ubiquitylation via a Ub(K27R) mutation only mildly impacts growth (Xu et al, 2009b; Roscoe et al, 2013). However, the relative importance of K27‐linked ubiquitylation for cell fitness in higher eukaryotes has not been addressed.
The paucity of insights into the mechanisms and functions of atypical Ub chains in general, and K27‐linked ubiquitylation in particular, represents a fundamental gap in the understanding of the cellular Ub code and its biological ramifications. Studies of Ub chain linkages for which specific affinity reagents are not available have been typically performed by overexpressing Ub mutants in which one lysine (K) residue is mutated to arginine (R) to selectively abrogate Ub chain formation via this linkage. However, overexpressed Ub may skew endogenous Ub equilibria and lead to artefacts, while the presence of endogenous Ub complicates interpretation of data obtained with Ub K‐R mutants. Accordingly, we reasoned that a previously described Ub replacement strategy enabling the exchange of endogenous Ub with a Ub K‐R mutant expressed at a similar level in a chemically inducible manner (Xu et al, 2009a) could circumvent these caveats and illuminate the cellular phenotypes resulting from targeted disruption of K27‐linked ubiquitylation. Here, by carefully establishing and characterizing such conditional Ub replacement cell lines, we demonstrate that K27‐linked ubiquitylation is instrumental for the proliferation of human cells. We show that K27‐linked Ub polymers are predominantly present in the nucleus and function epistatically with p97 to promote progression through the cell cycle. Abrogating formation of K27‐linked Ub chains or blocking their recognition via overexpression of a K27 linkage‐specific binder impairs the turnover of a ubiquitylated p97‐proteasome pathway model substrate, which is directly modified by K27‐linkages, at the level of p97. Together, these findings provide fundamental insights into the consequences of disabling K27‐linked Ub chain formation in human cells and functionally couple these Ub polymers to the processing of polyubiquitylated proteins via the p97 machinery.
Results
A cellular system for conditional abrogation of K27‐linked ubiquitylation
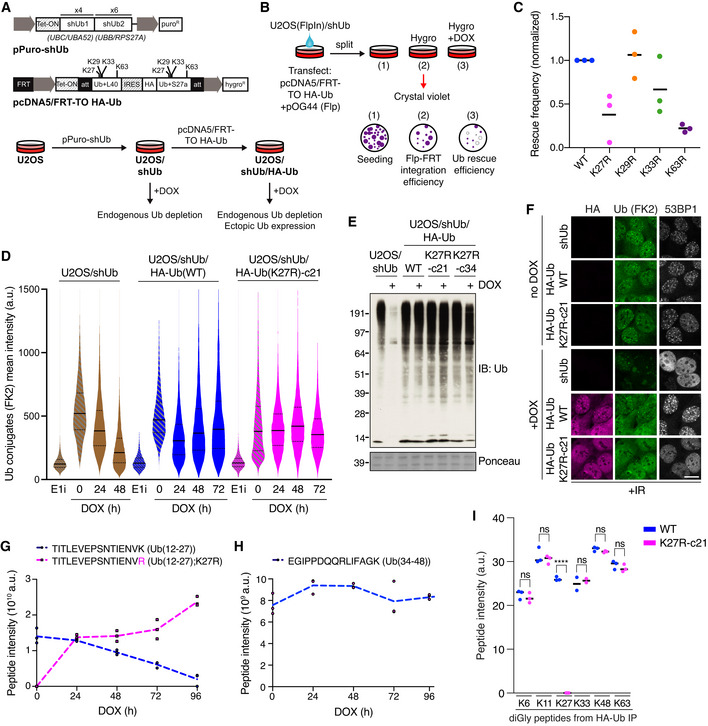
To explore the functions of atypical Ub linkages in human cells, we utilized a previously described Ub replacement strategy enabling targeted linkage‐specific abrogation of Ub chain formation in a Doxycycline (DOX)‐inducible manner (Xu et al, 2009a). This entails the two‐step generation of stable cell lines capable of replacing endogenous Ub with a Ub mutant containing a single lysine to arginine (K‐R) point mutation to selectively abolish ubiquitylation via one specific linkage type (Fig 1A). We first established a human U2OS osteosarcoma derivative cell line conditionally expressing shRNAs targeting all four human Ub‐encoding genes (U2OS/shUb; Figs 1A and EV1A). These cells displayed approximately 90% Ub depletion 48 h after DOX treatment, accompanied by a strong loss of cell viability (Fig EV1B and C). This may in part reflect the co‐depletion of the ribosomal proteins L40 and S27a that are encoded as fusions with Ub by the UBA52 and RPS27A genes, respectively (Fig EV1A). As an initial test for the relative importance of individual atypical Ub linkages for cell fitness, we transiently transfected U2OS/shUb cells with expression constructs encoding the UBA52 and RPS27A genes with Ub in wild‐type (WT) or K‐R mutant configuration (Fig 1A and B; note that the Ub allele encoded by RPS27A contains an N‐terminal HA‐tag). Notably, relative to wild‐type Ub (Ub(WT)), a Ub(K27R) mutant only poorly rescued the colony formation ability of DOX‐treated U2OS/shUb cells, comparable to the expected strong defect imposed by a K63R mutation (Figs 1C and EV1D and E). By contrast, the expression of Ub(K29R) in cells depleted for endogenous Ub restored clonogenic survival almost as efficiently as Ub(WT), while Ub(K33R) had an intermediate effect (Figs 1C and EV1D and E). Prompted by these observations, we set out to further explore the requirement for K27‐linked ubiquitylation in underpinning cellular fitness. To this end, we carefully selected stable cell lines capable of uniformly replacing endogenous Ub with ectopically expressed Ub(WT) or Ub(K27R) alleles at a similar, near‐endogenous level upon DOX treatment (U2OS/shUb/HA‐Ub(WT) and U2OS/shUb/HA‐Ub(K27R) (clones 21 (c21) and 34 (c34)); Figs 1A, D–F and EV1F). Successful DOX‐induced replacement of endogenous Ub with Ub(K27R) in U2OS/shUb/HA‐Ub(K27R) cells was verified by mass spectrometry (MS), showing a progressive exchange of the tryptic peptide spanning Ub‐K27 with the corresponding mutant K27R peptide, while the levels of other Ub‐derived peptides remained essentially unchanged (Figs 1G and H, and EV1G). Importantly, MS‐based quantification of di‐Gly Ub remnants on individual lysines in tryptic Ub peptides revealed a strong and specific loss of K27‐linked ubiquitylation in Ub(K27R)‐replaced cells relative to Ub(WT)‐replaced cells, whereas the abundance of other Ub linkage types was not significantly affected (Fig 1I). Consistently, quantitative image‐based analysis showed no overt impact of Ub(K27R) replacement on total levels of Ub conjugates as well as K48‐ and K63‐linked ubiquitylation, and the subcellular localization of the Ub(WT) and Ub(K27R) transgenes was indistinguishable (Figs 1D–F and EV1H–J). Moreover, whereas depletion of endogenous Ub in U2OS/shUb cells reduced both transcription and translation rates, these defects were corrected by both Ub(WT) and Ub(K27R) replacement (Appendix Fig S1A and B). While the K27R mutation has been recently suggested to negatively impact Ub stability (Kudriaeva et al, 2021), we observed no pronounced difference in the half‐lives of the Ub(WT) and Ub(K27R) proteins upon inhibition of protein synthesis by cycloheximide in our Ub‐replaced cells (Fig EV1K). Further supporting the validity of this cellular system for targeted ablation of K27‐linked ubiquitylation, we found that recruitment of the DNA repair factor 53BP1 to damaged DNA, which has been reported to require K27‐linked ubiquitylation (Gatti et al, 2015), was abolished upon the replacement of endogenous Ub with Ub(K27R) but not Ub(WT) (Figs 1F and EV1L and M). Collectively, these observations demonstrate that our Ub(K27R) replacement cells enable efficient targeted abrogation of K27‐linked ubiquitylation while faithfully preserving overall ubiquitylation patterns, providing a unique opportunity to study the phenotypes associated with selective blockage of K27‐linked Ub chain formation in human cells.
Figure 1. A cell‐based system for conditional and selective abrogation of K27‐linked ubiquitylation.
-
AExpression constructs (top) and two‐step procedure (bottom) for generation of Doxycycline (DOX)‐inducible Ub replacement cell lines.
-
BExperimental setup for assessing the ability of exogenous Ub variants to rescue clonogenic survival of U2OS/shUb cells. U2OS/shUb cells were co‐transfected with indicated plasmids to allow for FLP‐FRT‐mediated integration of an ectopic Ub expression cassette (1), which was selected with Hygromycin B (2). Expression of shUb and ectopic Ub was induced by treatment with Doxycycline (DOX) (3), and colony formation was visualized by crystal violet staining.
-
CQuantification of rescue efficiency (3), normalized to FLP‐FRT integration efficiency (2), for individual Ub variants (black bars, mean; n = 3 independent experiments). See also Fig EV1D.
-
DU2OS/shUb and derivative Ub replacement cell lines treated with DOX for the indicated times or Ub E1 inhibitor (E1i) for 1 h were fixed and immunostained with Ub conjugate‐specific antibody (FK2). Levels of Ub conjugates were quantified using quantitative image‐based cytometry (QIBC) (solid lines, median; dashed lines, quartiles (a.u., arbitrary units); > 5,000 cells analyzed per condition).
-
EImmunoblot (IB) analysis of stable Ub replacement cell lines treated or not with DOX for 72 h.
-
FAs in (E), except cells were exposed to ionizing radiation (IR, 2 Gy) 1 h before fixation and co‐immunostained with indicated antibodies. Scale bar, 10 μm.
-
GMass spectrometry‐based quantification of native Ub(12‐27) and Ub(12‐27);K27R) peptides after Trypsin digestion. Data represent triplicate technical replicate samples taken over a 96‐h DOX induction time course of U2OS/shUb/HA‐Ub(K27R)‐c21 cells.
-
HAs in (G), but for Ub(34‐48) peptide.
-
IUbiquitin linkage abundance in Ub(WT)‐ and Ub(K27R)‐replaced cells as indicated by Ub di‐Gly peptide quantification. HA‐Ub conjugates from whole cell extracts of Ub(WT) and Ub(K27R)‐c21 replacement cell lines treated with DOX for 72 h were isolated by HA immunoprecipitation (IP) under denaturing conditions and analyzed by quantitative label‐free mass spectrometry (Figs 3D and EV3C and D) (black bars, median; n = 2–3 technical replicates; ****P < 0.0001, ns: not significant, unpaired t‐test). See Fig EV2C for the impact of Ub(K27R) replacement on K29‐linkage abundance.
Data information: Data are representative of four (D) and three (E,F) independent experiments with similar outcome.
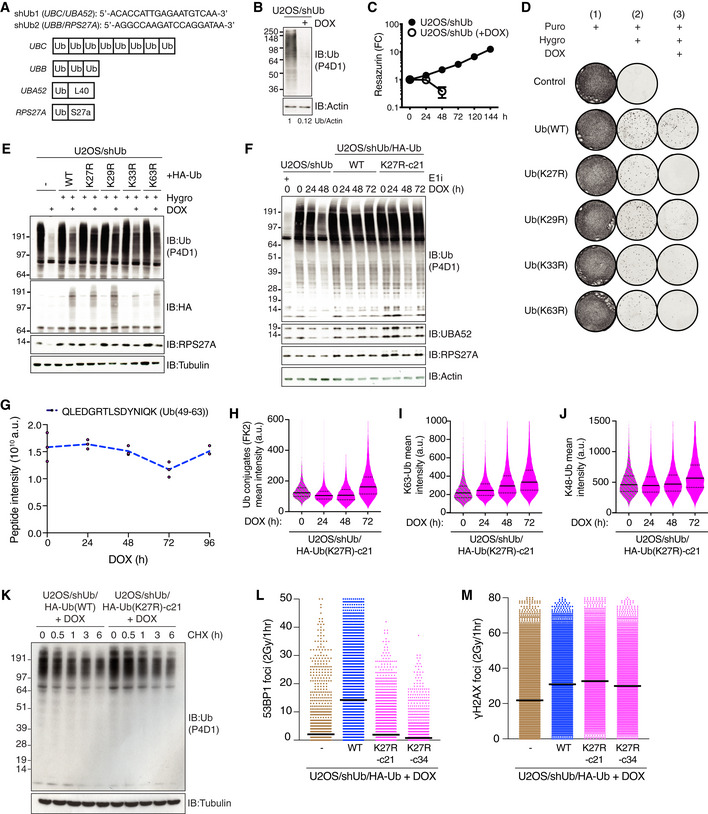
Figure EV1. (related to Fig 1) Generation and characterization of Ub replacement cell lines.
-
AShort hairpin RNA (shRNA) target sequences cloned into pPuro‐shUb (top) and schematic depicting mammalian genes encoding Ub (bottom).
-
BImmunoblot analysis of Ub depletion in U2OS/shUb cells after 48 h of DOX treatment.
-
CViability of U2OS/shUb cells exposed or not to DOX, as determined by resazurin metabolic assay (mean ± SD; n = 3 independent experiments).
-
DRepresentative images of clonogenic rescue assay to restore viability to U2OS/shUb cells after DOX treatment (Fig 1A–C). Where indicated, integration of the Ub replacement cassettes was selected with Hygromycin B, and expression was induced with DOX.
-
EImmunoblot analysis of cells in (D).
-
FImmunoblot analysis of stable U2OS/shUb/HA‐Ub Ub replacement cell lines treated with DOX for the indicated times or Ub E1 inhibitor (E1i) for 1 h.
-
GMass spectrometry‐based quantification of native Ub(49‐63) peptide spanning after Trypsin digestion. Data represent triplicate technical replicate samples taken over a 96‐h DOX induction time course of U2OS/shUb/HA‐Ub(K27R)‐c21 cells.
-
HU2OS/shUb/HA‐Ub(K27R) cells treated with DOX for the indicated times were fixed and immunostained with Ub conjugate‐specific antibody (FK2). Levels of Ub conjugates were quantified using quantitative image‐based cytometry (QIBC) (solid lines, median; dashed lines, quartiles (a.u., arbitrary units); > 5,000 cells analyzed per condition).
-
IAs in (H), but using Ub‐K63 antibody.
-
JAs in (H), but using Ub‐K48 antibody.
-
KImmunoblot analysis of U2OS/shUb/HA‐Ub cell lines treated or not with cycloheximide (CHX) for the indicated times.
-
LNuclear 53BP1 foci in DOX‐treated U2OS/shUb, U2OS/shUb/HA‐Ub(WT) and U2OS/shUb/HA‐Ub(K27R) cell lines exposed to IR (2 Gy), fixed 1 h later and immunostained with 53BP1 antibody was quantified by QIBC (black bars, mean; > 5,000 cells analyzed per condition).
-
MAs in (L), except cells were stained with γ‐H2AX antibody (black bars, mean; > 5,000 cells analyzed per condition).
Data information: Data are representative of three (B,D‐F,H‐J) and two (K‐M) independent experiments with similar outcome.
K27‐linked ubiquitylation is required for cell proliferation
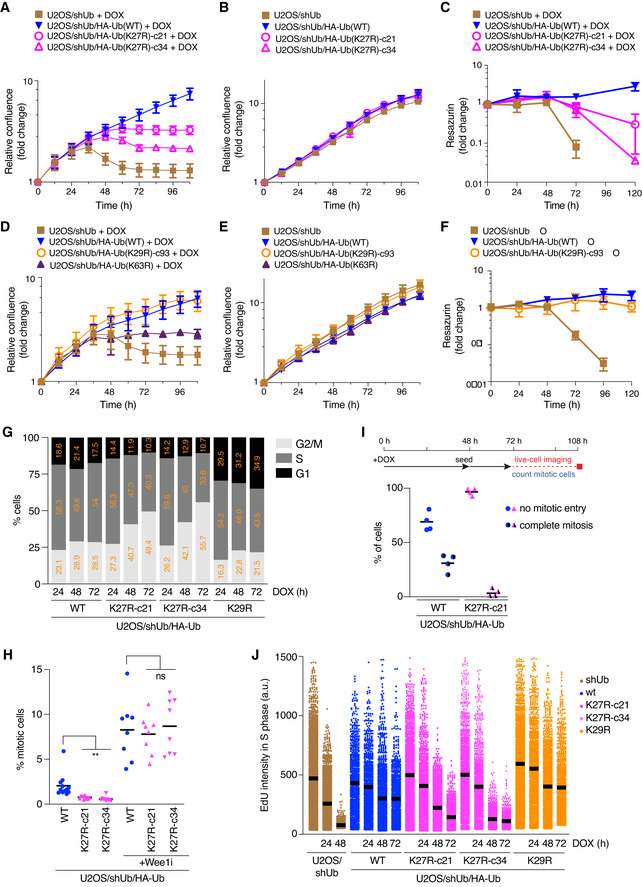
Having established a cellular system for selective and conditional abrogation of K27‐linked ubiquitylation, we first explored the overall impact of Ub(K27R) replacement on cell proliferation and fitness. Notably, whereas exogenous Ub(WT) rescued the cell proliferation defect arising from loss of endogenous Ub, Ub(K27R) expression did not support long‐term proliferation and viability, and only moderately improved the fitness of cells depleted for endogenous Ub (Fig 2A–C). A U2OS/shUb/HA‐Ub(K63R) cell line generated in parallel to conditionally prevent formation of K63‐linked Ub polymers likewise displayed strongly impaired proliferation upon Ub(K63R) replacement (Figs 2D and E, and EV2A), consistent with the key role of K63‐linked ubiquitylation in many aspects of cell physiology (Chen & Sun, 2009; Swatek & Komander, 2016). By contrast, analysis of a corresponding Ub(K29R) replacement cell line we generated showed that abrogating K29‐linked ubiquitylation, which we confirmed using a recently reported specific K29‐linkage binder (Yu et al, 2021), only had a modest impact on cell proliferation (Figs 2D–F and EV2B and C), despite these Ub polymers being considerably more abundant in cells than K27‐linkages (Dammer et al, 2011; Swatek et al, 2019). Unlike the expected impact of Ub(K29R) replacement, K29‐linkage abundance was not affected by abrogation of K27‐linked ubiquitylation (Fig EV2C). In the absence of DOX‐induced Ub replacement, all cell lines displayed identical rates of proliferation (Figs 2B and E, and EV2D and E). Further underscoring the importance of K27‐linked ubiquitylation for cell proliferation, Ub(WT) replacement at a sub‐endogenous level in an independent U2OS/shUb/HA‐Ub(WT) clone was sufficient to rescue the proliferation defect arising from the depletion of endogenous Ub (Fig EV2F and G), implying that the adverse impact on proliferation induced by Ub(K27R) replacement is not simply due to subtle differences in overall ectopic Ub expression levels between Ub(WT) and Ub(K27R) cells.
Figure 2. K27‐linked ubiquitylation is required for proliferation and viability of human cells.
-
ANormalized logarithmic proliferation quantification for U2OS/shUb, U2OS/shUb/HA‐Ub(WT) and U2OS/shUb/HA‐Ub(K27R) cell lines subjected to DOX treatment for the indicated times, as determined by Incucyte image‐based confluence analysis (mean ± SEM; n = 3 independent experiments).
-
BAs in (A), except cells were grown in the absence of DOX (mean ± SEM; n = 3 independent experiments).
-
CViability of U2OS/shUb, U2OS/shUb/HA‐Ub(WT) and U2OS/shUb/HA‐Ub(K27R) cell lines treated or not with DOX, as determined by Resazurin metabolic assay (mean ± SEM; n = 3 independent experiments).
-
DNormalized logarithmic proliferation quantification for U2OS/shUb, U2OS/shUb/HA‐Ub(WT), U2OS/shUb/HA‐Ub(K29R) and U2OS/shUb/HA‐Ub(K63R) cell lines subjected to DOX treatment for the indicated times, as determined by Incucyte image‐based confluence analysis (mean ± SEM; n = 3 independent experiments).
-
EAs in (D), except cells were grown in the absence of DOX (mean ± SEM; n = 3 independent experiments).
-
FViability of U2OS/shUb, U2OS/shUb/HA‐Ub(WT) and U2OS/shUb/HA‐Ub(K27R) cell lines treated or not with DOX, as determined by Resazurin metabolic assay (mean ± SEM; n = 3 independent experiments).
-
GCell cycle profiles of Ub replacement cell lines treated with DOX for the indicated times, pulsed with EdU and stained with HA antibody were determined by QIBC analysis of DAPI and EdU signal intensities (> 2,000 cells analyzed per condition). Cells were gated for Ub replacement based on HA signal positivity.
-
HUb replacement cells treated or not with Wee1i (1 μΜ) for 1.5 h were fixed and immunostained with H3‐pS10 antibody. Mitotic indices were determined by quantifying H3‐pS10‐positive cells using QIBC (black bars, mean; > 4,000 cells analyzed per condition; n = 8 independent experiments; **P < 0.01, ns: not significant, paired t‐test).
-
ILive‐cell imaging‐based quantification of mitotic entry in Ub replacement cell lines (bottom) treated as indicated (top) (black bars, mean; > 30 cells analyzed per condition; n = 4 independent experiments).
-
JDNA synthesis rates of Ub replacement cell lines in (G) were analyzed by quantifying EdU signal intensity in S phase cells using QIBC (black bars, mean (a.u., arbitrary units); > 1,000 S phase cells analyzed per condition).
Data information: Data (G,J) are representative of three independent experiments with similar outcome.
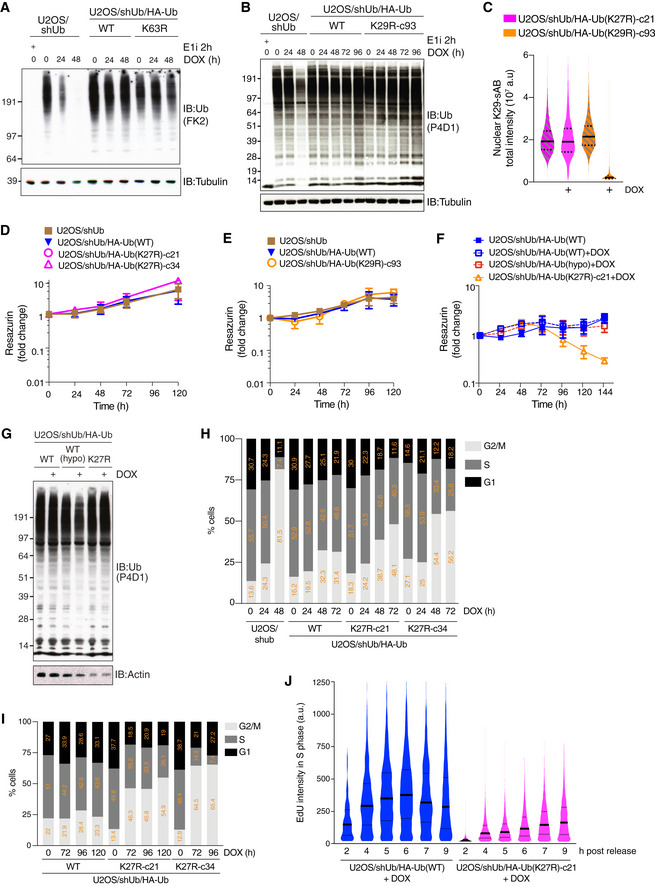
Figure EV2. (related to Fig 2) K27‐linked ubiquitylation is required for cell proliferation and cell cycle progression.
-
A, BImmunoblot analysis of U2OS/shUb and U2OS/shUb/HA‐Ub cell lines treated with DOX for the indicated times or Ub E1 inhibitor (E1i) for 1 h.
-
CU2OS/shUb/HA‐Ub replacement cell lines were treated with DOX for 72 h where indicated, fixed and stained with K29‐Ub specific synthetic antigen binder sAB‐K29 (Yu et al, 2021). Levels of K29‐Ub conjugates were quantified using QIBC (solid lines, median; dashed lines, quartiles (a.u., arbitrary units); > 5,000 cells analyzed per condition).
-
DViability of U2OS/shUb, U2OS/shUb/HA‐Ub(WT) and U2OS/shUb/HA‐Ub(K27R) cell lines without DOX treatment, as determined by Resazurin metabolic assay (mean ± SEM; n = 3 independent experiments).
-
EAs in (D), but U2OS/shUb, U2OS/shUb/HA‐Ub(WT) and U2OS/shUb/HA‐Ub(K29R) cell lines are shown. (mean ± SEM; n = 3 independent experiments).
-
FAs in (D), but U2OS/shUb/HA‐Ub(WT), U2OS/shUb/HA‐Ub(K27R) and U2OS/shUb/HA‐Ub(WT hypomorph) cell lines with or without DOX treatment are shown (mean ± SEM; n = 3 independent experiments).
-
GImmunoblot analysis of U2OS/shUb/HA‐Ub(WT), U2OS/shUb/HA‐Ub(WT hypomorph) and U2OS/shUb/HA‐Ub(K27R) cell lines treated or not with DOX for 72 h.
-
H, ICell cycle profiles of Ub replacement cell lines treated with DOX for the indicated times were determined by QIBC analysis of DAPI and either EdU (H) or PCNA (I) signal intensities (> 2,000 cells analyzed per condition).
-
JQuantification of EdU incorporation in DOX‐treated Ub replacement cell lines at the indicated times after release from a single thymidine block (solid lines, median; dashed lines, quartiles; > 2,000 S phase cells analyzed per condition).
Data information: Data are representative of five (H,I) and three (A‐C,G,J) independent experiments.
We next investigated how abrogating K27‐linked ubiquitylation impairs cell proliferation. Notably, Ub(K27R)‐ but not Ub(WT)‐ or Ub(K29R)‐replaced cells progressively accumulated in the G2/M phase of the cell cycle (Figs 2G and EV2H and I). This was a consequence of cell cycle arrest in G2 phase, as the mitotic index of Ub(K27R)‐replaced cells was markedly lower than that of their Ub(WT) counterparts (Fig 2H). Consistently, live‐cell imaging demonstrated that cells deficient for K27‐linked Ub chain formation exhibited a strong block to mitotic entry (Fig 2I). While Ub(K27R)‐replaced cells did not display any appreciable increase in markers of DNA damage formation including nuclear γH2AX and RPA foci (Appendix Fig S1C and D), they could be forced into mitosis but subsequently failed to complete chromosome segregation upon the inhibition of the Wee1 kinase, which restrains mitotic entry via inhibitory phosphorylation of cyclin‐dependent kinases (Fig 2H; Appendix Fig S1E). This raised the possibility that the G2 cell cycle block imposed by Ub(K27R) replacement might at least in part arise due to defective DNA replication. Indeed, EdU incorporation experiments revealed a precipitous decline in DNA replication efficiency upon the depletion of endogenous Ub that was rescued by re‐expression of both Ub(WT) and Ub(K29R) but not the Ub(K27R) mutant (Fig 2J). Moreover, unlike Ub(WT)‐replaced cells, in which DNA synthesis rates increased and subsequently declined during a 9‐h interval following release from cell cycle synchronization in early S phase as expected, DNA replication in Ub(K27R)‐replaced cells was markedly attenuated and continued to slowly increase throughout this period (Fig EV2J). Together, these findings show that K27‐linked ubiquitylation is required for cell cycle progression and proliferation of human cells.
Loss of K27‐linked ubiquitylation deregulates p97 functionality in the nucleus
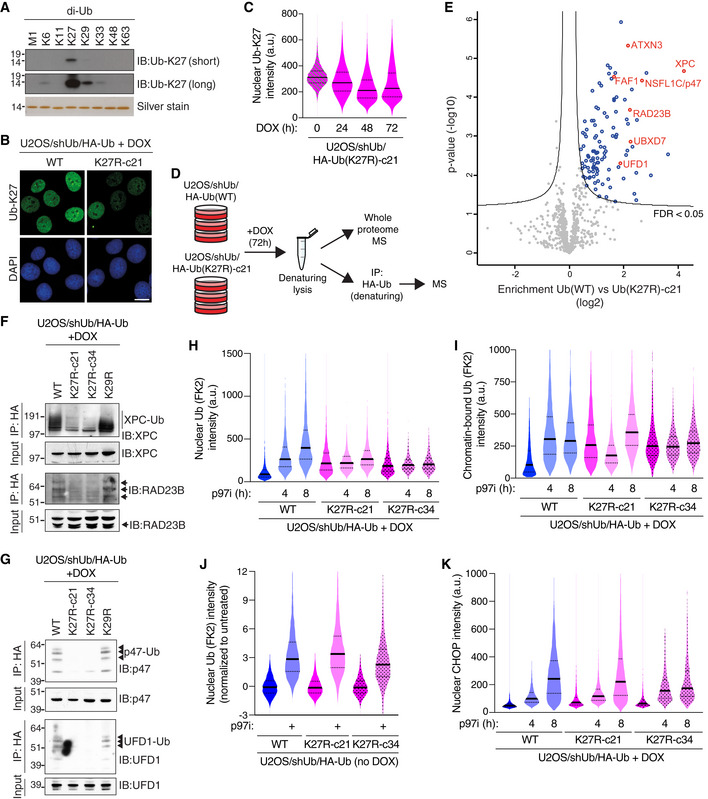
Given the critical role of K27‐linked ubiquitylation in promoting cell proliferation, we utilized the Ub(K27R) replacement approach to further explore the nature and functions of K27‐linked ubiquitylation in cells. We initially probed this using a commercially available antibody to these Ub polymers (Ub‐K27). Although the Ub‐K27 antibody preferentially recognized K27‐linkages among all eight purified di‐Ub chains, it showed weak but detectable reactivity with other atypical Ub linkages, including K29 and K6, and lost specificity for K27 linkages in longer, slow‐migrating Ub chains in cell extracts (Figs 3A and EV3A), limiting its suitability for specific isolation of K27‐linked Ub conjugates from cells. However, in immunofluorescence experiments, the Ub‐K27 antibody exclusively reacted with nuclear epitopes that were substantially reduced upon treatment with a Ub E1 enzyme inhibitor and declined progressively upon DOX‐induced Ub(K27R) replacement unlike total levels of Ub (Figs 3B and C, and EV3B), indicating that this signal partially represents K27 linkages. These observations suggest that in human cells K27‐linked ubiquitylation is predominantly a nuclear modification, in good overall agreement with its observed importance for DNA replication and cell cycle progression.
Figure 3. K27‐linked ubiquitylation predominantly occurs in the nucleus and impacts the integrity of the p97 system.
-
AImmunoblot analysis of Ub‐K27 antibody reactivity with recombinant linkage‐specific di‐Ub proteins.
-
BRepresentative images of U2OS/shUb/HA‐Ub(WT) or U2OS/shUb/HA‐Ub(K27R) cells immunostained with antibody to K27‐linked Ub (Ub‐K27). Scale bar, 10 μm.
-
CIntensity of nuclear signal recognized by Ub‐K27 antibody in U2OS/shUb/HA‐Ub(K27R) cells treated with DOX for the indicated times was quantified using QIBC (solid lines, median; dashed lines, quartiles (a.u., arbitrary units); > 3,000 cells analyzed per condition).
-
DSchematic depicting experimental setup for isolation and quantitative label‐free mass spectrometry analysis of HA‐Ub conjugates isolated from whole cell lysates of DOX‐treated Ub(WT) and Ub(K27R)‐c21 replacement cell lines by HA immunoprecipitation (IP) under denaturing conditions.
-
EGlobal ubiquitylation changes induced by Ub(K27R) replacement, profiled by mass spectrometry analysis of HA‐Ub conjugates isolated as shown in (D) (see also Fig EV3C and D; for full results see Dataset EV1). Volcano plot shows enrichment of individual proteins (HA‐Ub(WT)/HA‐Ub(K27R) ratio) plotted against the P value. Significance thresholds (FDR < 0.05; s0 = 1) are indicated. Nuclear proteins with a known interplay with p97 are highlighted in red. See Fig 1I for Ub di‐Gly peptide quantification.
-
F, GHA‐Ub conjugates isolated as in (D) were immunoblotted with indicated antibodies. See also Fig EV3F.
-
HDOX‐treated Ub replacement cell lines treated with p97i for the indicated times were fixed and immunostained with Ub conjugate‐specific antibody (FK2). Levels of Ub conjugates in the nucleus were quantified using QIBC (solid lines, median; dashed lines, quartiles; > 2,500 cells analyzed per condition).
-
IAs in (H), except cells were subjected to stringent pre‐extraction before fixation (solid lines, median; dashed lines, quartiles; > 5,000 cells analyzed per condition).
-
JUninduced U2OS/shUb/HA‐Ub cell lines treated or not with p97i for 8 h were analyzed as in (H) (solid lines, median; dashed lines, quartiles; > 5,000 cells analyzed per condition).
-
KAs in (H), except cells were immunostained with CHOP antibody (solid lines, median; dashed lines, quartiles (a.u., arbitrary units); > 3,000 cells analyzed per condition).
Data information: Data (A‐C,F‐K) are representative of three independent experiments with similar outcome.
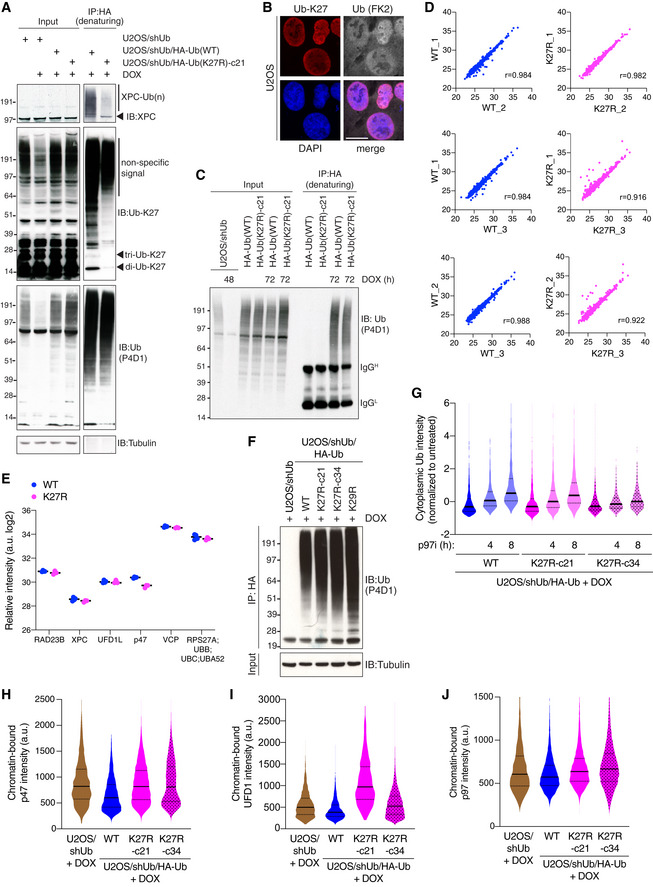
Figure EV3. (related to Fig 3) K27‐linked ubiquitylation predominantly occurs in the nucleus and affects the integrity of the p97 machinery.
-
AU2OS/shUb and DOX‐treated U2OS/shUb/HA‐Ub cell lines were lysed, subjected to HA IP under denaturing conditions and immunoblotted with indicated antibodies.
-
BRepresentative images of U2OS cells co‐immunostained with antibodies to K27‐linked Ub (Ub‐K27) and total Ub conjugates (FK2). Scale bar, 10 μm.
- C
-
DCorrelation analysis comparing label‐free quantitation values of protein groups from individual replicate HA‐Ub conjugate samples isolated as in Fig 3D.
-
ERelative intensity of peptide detection for indicated proteins in U2OS/shUb/HA‐Ub(WT) and U2OS/shUb/HA‐Ub(K27R) whole cell extract analyzed by MS (black bars, median; n = 3 technical replicates). Full proteome data are shown in Dataset EV2.
-
FDOX‐treated U2OS/shUb (48 h) and U2OS/shUb/HA‐Ub (72 h) cell lines were lysed, subjected to HA IP under denaturing conditions and immunoblotted with indicated antibodies as in Fig 3F and G.
-
GDOX‐induced U2OS/shUb/HA‐Ub cell lines treated or not with p97i for the indicated times were fixed and immunostained with Ub conjugate‐specific antibody (FK2). Levels of Ub conjugates in the cytoplasm were quantified using QIBC (solid lines, median; dashed lines, quartiles; > 1,000 cells analyzed per condition).
-
HDOX‐induced U2OS/shUb and U2OS/shUb/HA‐Ub cell lines were pre‐extracted, fixed and immunostained with p47 antibody, and analyzed by QIBC (solid lines, median; dashed lines, quartiles; > 3,000 cells analyzed per condition).
-
IAs in (H), except cells were immunostained with UFD1 antibody.
-
JAs in (H), except cells were immunostained with p97 antibody.
Data information: Data (A,B,F‐J) are representative of three independent experiments with similar outcome.
In the absence of a suitable affinity reagent for the specific isolation of K27‐linked Ub conjugates, we utilized our Ub replacement cell lines to globally quantify protein ubiquitylation changes resulting from targeted disruption of K27‐linked ubiquitylation. To this end, proteins modified by HA‐tagged Ub following DOX‐dependent Ub replacement in the U2OS/shUb/HA‐Ub(WT) and U2OS/shUb/HA‐Ub(K27R) cell lines were isolated by HA immunoprecipitation (IP) under denaturing buffer conditions to avoid co‐purification of non‐ubiquitylated proteins and analyzed by label‐free MS‐based proteomics (Figs 3D and EV3C and D). Interestingly, while a large majority of quantified proteins displayed similar ubiquitylation status in Ub(WT)‐ and Ub(K27R)‐replaced cells, further underscoring that the induced Ub(K27R) protein retains full overall functionality in supporting cellular ubiquitylation processes, we identified a small subset of factors whose ubiquitylation level was significantly decreased upon Ub(K27R) replacement (Figs 3E; Dataset EV1). Among these proteins, we noted a striking enrichment of cofactors for the p97 ATPase found in the nuclear p97 interactome (UFD1‐NPL4, p47, UBXD7, FAF1 and ATXN3) (Singh et al, 2019) as well as nuclear proteins with a known interplay with p97, including RAD23B and XPC (Fig 3E). Importantly, whole proteome analysis showed that the altered ubiquitylation status of these factors in Ub(K27R)‐replaced cells was not due to changes in protein abundance (Fig EV3E; Dataset EV2). For several of these proteins, we biochemically verified their reduced ubiquitylation in Ub(K27R)‐ but not Ub(K29R)‐replaced cells (Figs 3F and G, and EV3F). These observations suggested a functional connection between K27‐linked ubiquitylation and p97 complexes in the nucleus. Consistent with this possibility, steady‐state levels of nuclear and chromatin‐associated Ub conjugates were markedly elevated in Ub(K27R)‐replaced cells and largely refractory to the impact of inhibiting p97 activity by the allosteric p97 inhibitor NMS‐873 (p97i), which caused strong accumulation of nuclear and chromatin‐bound Ub conjugates in Ub(WT)‐replaced cells as expected (Fig 3H and I). This was a specific consequence of substituting endogenous Ub with Ub(K27R), as U2OS/shUb/HA‐Ub(WT) and U2OS/shUb/HA‐Ub(K27R) cells showed comparable steady‐state levels of nuclear ubiquitylation and a similar increase upon p97i treatment when Ub replacement was not induced (Fig 3J). Interestingly, disabling K27‐linked Ub chain formation only had a marginal impact on the p97i‐induced accumulation of cytoplasmic Ub conjugates and levels of CHOP, an established marker of endoplasmic reticulum‐associated p97 activity (Figs 3K and EV3G) (Anderson et al, 2015). This suggests that K27‐linked ubiquitylation mainly affects p97 functions in the nucleus, consistent with the nuclear enrichment of K27‐linkages. In line with this, Ub(K27R)‐replaced cells displayed increased chromatin association of p97 and nuclear cofactors including p47 and UFD1 relative to their Ub(WT) counterparts (Fig EV3H–J), further indicating perturbed dynamics of nuclear p97‐mediated processes upon disabling K27‐linked Ub chain formation. Together, these observations suggest that K27‐linked ubiquitylation is functionally coupled to nuclear p97‐regulated processes.
K27‐linked ubiquitylation promotes p97 substrate processing
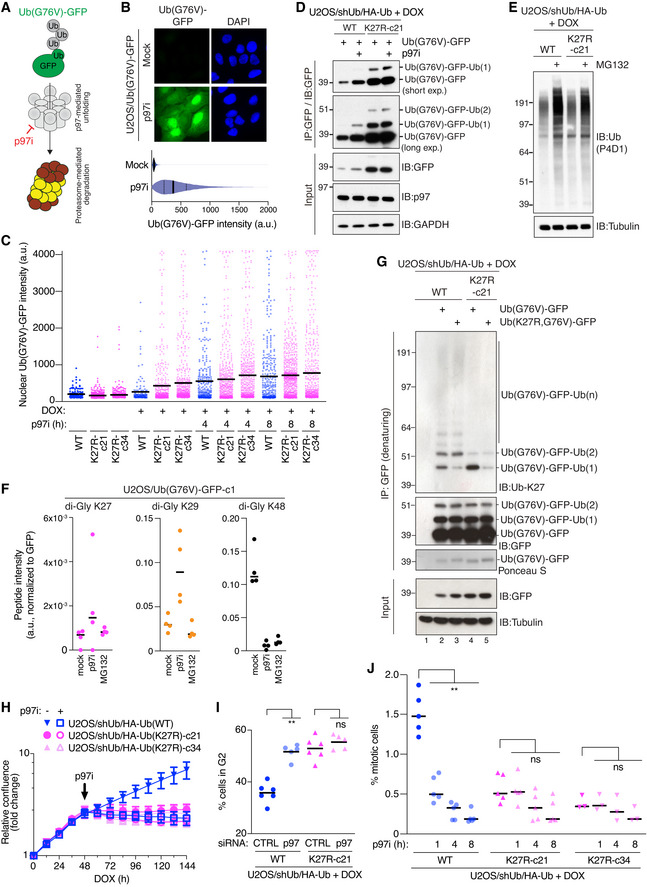
Motivated by the above findings, we set out to address more directly whether abrogating K27‐linked ubiquitylation affects substrate processing by p97. To this aim, we utilized an established fluorescent p97 model substrate, comprising a non‐hydrolyzable Ub(G76V)‐GFP fusion construct that cannot be conjugated to substrate proteins but is highly unstable under steady‐state conditions due to its polyubiquitylation and subsequent proteasomal destruction via the Ub fusion degradation pathway (Dantuma et al, 2000). As degradation of this model substrate via the proteasome requires p97‐dependent unfolding of polyubiquitylated Ub(G76V)‐GFP molecules (Beskow et al, 2009; Chou & Deshaies, 2011), inhibiting p97 strongly stabilizes Ub(G76V)‐GFP, thus providing a robust cell‐based fluorescent readout for p97 activity (Fig 4A and B). We generated derivative Ub(WT) and Ub(K27R) replacement cell lines with a stably integrated shUb‐resistant Ub(G76V)‐GFP reporter and confirmed that in the absence of Ub replacement Ub(G76V)‐GFP expression was tightly repressed but increased substantially upon p97i treatment in both backgrounds (Fig EV4A). Importantly, however, replacing endogenous Ub with Ub(K27R) but not Ub(WT) led to markedly elevated Ub(G76V)‐GFP expression in the absence of p97i (Fig 4C and D). Addition of p97i only modestly increased Ub(G76V)‐GFP abundance in Ub(K27R)‐replaced cells, unlike in Ub(WT) cells (Fig 4C and D). By contrast, the basal expression of Ub(G76V)‐GFP and its p97i‐induced stabilization were virtually identical in Ub(WT)‐ and Ub(K29R)‐replaced cells (Fig EV4B). Although degradation of Ub(G76V)‐GFP requires the activity of both p97 and the proteasome, high‐molecular weight Ub conjugates accumulated to a similar extent in U2OS/shUb/HA‐Ub(WT) and U2OS/shUb/HA‐Ub(K27R) cells upon inhibition of the proteasome (Fig 4E), suggesting that proteasomal activity remains overtly normal in Ub(K27R) cells. Moreover, ubiquitylated forms of Ub(G76V)‐GFP, which accumulate upon inhibition of p97 but not the proteasome (Fig EV4C), possibly due to the action of proteasome‐associated DUBs (Collins & Goldberg, 2017; Bard et al, 2018), were prominent in Ub(K27R)‐ but not Ub(WT)‐ or Ub(K29R)‐replaced cells even in the absence of p97 inhibition (Figs 4D and EV4B). These observations suggest that K27‐linked ubiquitylation promotes the processing of ubiquitylated substrates at the level of p97 but not the proteasome.
Figure 4. K27‐linked ubiquitylation directly targets a p97 model substrate to promote its p97‐dependent turnover and is functionally epistatic with p97 inhibition.
-
ASchematic of p97‐ and proteasome‐dependent degradation of fluorescent Ub(G76V)‐GFP reporter.
-
BRepresentative images (upper) and QIBC analysis (lower) (solid lines, median; dashed lines, quartiles; > 10,000 cells analyzed per condition) of Ub(G76V)‐GFP expression in stable U2OS/Ub(G76V)‐GFP cells treated or not with p97i for 4 h.
-
CU2OS/shUb/HA‐Ub replacement cell lines stably expressing shUb‐resistant Ub(G76V)‐GFP reporter were treated with DOX and p97i as indicated, and nuclear Ub(G76V)‐GFP signal intensity was analyzed by QIBC (black bars, mean; 150–750 GFP‐positive cells analyzed per condition).
-
DDOX‐treated U2OS/shUb/HA‐Ub(WT) and U2OS/shUb/HA‐Ub(K27R) cell lines were transfected with shUb‐resistant Ub(G76V)‐GFP construct and treated or not with p97i for 4 h. Cell extracts were subjected to GFP IP followed by immunoblotting with indicated antibodies.
-
EImmunoblot analysis of DOX‐treated U2OS/shUb/HA‐Ub cell lines treated or not with MG132 for 4 h.
-
FU2OS/Ub(G76V)‐GFP cells left untreated or incubated with p97i or MG132 for 4 h were subjected to denaturing GFP IP and analyzed by quantitative label‐free mass spectrometry. Intensity of indicated Ub di‐Gly peptides was normalized to GFP peptide intensity (black bars, median; n = 4 independent samples (technical replicates)).
-
GDOX‐treated U2OS/shUb/HA‐Ub(WT) and U2OS/shUb/HA‐Ub(K27R) cell lines transfected with indicated Ub(G76V)‐GFP expression constructs were subjected to GFP IP under denaturing conditions followed by immunoblotting with indicated antibodies.
-
HNormalized logarithmic proliferation quantification for U2OS/shUb/HA‐Ub(WT) and U2OS/shUb/HA‐Ub(K27R) cell lines subjected or not to low‐dose (1.5 μM) p97i treatment 48 h after DOX addition, as determined by Incucyte image‐based confluence analysis (mean ± SEM; n = 3 independent experiments).
-
IProportion of DOX‐treated, siRNA‐transfected Ub replacement cells in G2 phase, assayed by EdU incorporation, was determined by QIBC (black bars, mean; n = 5–6 independent experiments; **P < 0.01, ns: not significant, Mann–Whitney test).
-
JProportion of DOX‐treated U2OS/shUb/HA‐Ub(WT) and U2OS/shUb/HA‐Ub(K27R) cell lines, which were subjected to p97i or not for different times, in M phase was determined by staining with a H3‐pS10 antibody using QIBC (black bars, mean; n = 3–5 independent experiments; > 4,000 cells analyzed per condition, **P < 0.01, ns: not significant, Mann–Whitney test).
Data information: Data are representative of four (G) and three (B‐E) independent experiments with similar outcome.
Figure EV4. (related to Fig 4) Disabling K27‐linked ubiquitylation impairs p97‐dependent substrate processing and is functionally epistatic with p97 inhibition.
-
AUninduced U2OS/shUb/HA‐Ub replacement cell lines stably expressing shUb‐resistant Ub(G76V)‐GFP reporter were treated or not with p97i as indicated, and nuclear Ub(G76V)‐GFP signal intensity was analyzed by QIBC (black bars, mean; 150–1,000 GFP‐positive cells analyzed per condition).
-
BDOX‐treated U2OS/shUb/HA‐Ub(WT) and U2OS/shUb/HA‐Ub(K29R) cell lines were transfected with shUb‐resistant Ub(G76V)‐GFP construct and treated or not with p97i for 4 h. Cell extracts were subjected to GFP IP followed by immunoblotting with indicated antibodies.
-
CU2OS/Ub(G76V)‐GFP‐c1 cells treated with p97i (4 h) or MG132 (6 h) where indicated were subjected to GFP IP under denaturing conditions and analyzed by silver staining.
-
DDOX‐treated U2OS/shUb/HA‐Ub(WT) and U2OS/shUb/HA‐Ub(K27R) cell lines were transfected with the indicated Ub(G76V)‐GFP expression constructs for 24 h, and nuclear GFP signal was analyzed by QIBC (black bars, mean; > 1,000 GFP‐positive cells were analyzed per condition).
-
EImmunoblot analysis of DOX‐treated U2OS/shUb/HA‐Ub(WT) and U2OS/shUb/HA‐Ub(K27R) cells transfected with non‐targeting control (CTRL) or p97 siRNA.
-
F, GDOX‐treated U2OS/shUb/HA‐Ub cell lines were subjected to local UV irradiation through pore filters in the presence or absence of p97i, fixed at the indicated times and co‐immunostained with XPC and DDB2 antibodies. XPC and DDB2 enrichment at local UV spots was quantified (black bars, mean; > 150 cells with UV spots analyzed per condition; n = 4 independent experiments; **P < 0.01, ***P < 0.001, ns: not significant, unpaired t‐test).
-
HRepresentative images of DOX‐treated Ub replacement cell lines shown in (F,G). Scale bar, 10 μm.
Data information: Data are representative of four (F,G), three (A) and two (B‐E) independent experiments with similar outcome.
Given the importance of K27‐linked ubiquitylation for p97‐dependent turnover of Ub(G76V)‐GFP, we considered the possibility that this model substrate is a direct target of K27‐linked ubiquitylation. Supporting this notion, MS analysis of the Ub(G76V)‐GFP reporter isolated under denaturing buffer conditions confirmed that it was modified by K27‐linked ubiquitylation, albeit to a lesser extent than by K48‐ and K29‐linkages (Fig 4F), both of which have been previously shown to target Ub(G76V)‐GFP (Liu et al, 2017). Consistent with the MS data, the ubiquitylated forms of Ub(G76V)‐GFP were recognized by the partially specific Ub‐K27 antibody (Fig 4G; compare lanes 1 and 2; Fig 3A). Importantly, a K27R mutation within Ub(G76V)‐GFP (Ub(K27R,G76V)‐GFP), which prevents the ubiquitylation of K27 within the fused Ub protein but not overall K27‐linkage formation, selectively reduced the recognition of the band corresponding to Ub(G76V)‐GFP monoubiquitylated at K27 but not slower‐migrating ubiquitylated forms of Ub(G76V)‐GFP (Fig 4G; compare lanes 2 and 3). By contrast, expressing Ub(G76V)‐GFP in Ub(K27R)‐replaced cells, which only allows Ub(G76V)‐GFP ubiquitylation at K27 within the fused Ub but not K27‐linked extensions of Ub modifications on Ub(G76V)‐GFP reduced the level of all but the Ub(G76V)‐GFP monoubiquitylation band recognized by the Ub‐K27 antibody (Fig 4G; compare lanes 2 and 4). In cells expressing Ub(K27R), this band was stronger than in Ub(WT)‐replaced cells, consistent with the impaired processing of ubiquitylated forms of Ub(G76V)‐GFP in Ub(K27R)‐replaced cells and the inability of these cells to extend this initial K27‐linkage on Ub(G76V)‐GFP into longer K27‐linked chains. When Ub(K27R,G76V)‐GFP was expressed in Ub(K27R)‐replaced cells, thereby preventing all K27‐linkage formation on this substrate, little reactivity with the Ub‐K27 antibody was observed (Fig 4G; lane 5). These findings show that the Ub(G76V)‐GFP model p97 substrate is directly modified by K27‐linked ubiquitylation impacting both the K27 residue in Ub(G76V) as well as Ub moieties attached to other lysines in Ub(G76V)‐GFP. Extending these observations, we found that the extent of K27‐linked ubiquitylation on Ub(G76V)‐GFP model substrates inversely correlated with their stability in cells. To this end, Ub(K27R/G76V)‐GFP was expressed at a moderately higher level than Ub(G76V)‐GFP in Ub(WT)‐replaced cells but was further stabilized in Ub(K27R)‐replaced cells (Figs 4G and EV4D), as would be expected if K27‐linkage signals on Ub(G76V)‐GFP stimulate its turnover via the p97‐proteasome pathway. In line with this, our MS analysis showed a trend that both K27‐ and K29‐linked Ub modifications accumulated on Ub(G76V)‐GFP upon inhibition of p97, although in the case of K27 linkages, the increase was modest and varied between individual samples (Fig 4F). Combined, the data suggest that K27‐linked ubiquitylation of the Ub(G76V)‐GFP model substrate facilitates its p97‐dependent proteasomal turnover.
In further support of an important role of K27‐linked ubiquitylation in facilitating p97 functions, phenotypical evidence at several levels suggested an epistatic relationship between Ub(K27R) replacement and p97 inhibition in cells: First, unlike its growth‐suppressive effect in Ub(WT)‐expressing cells, low‐dose p97i treatment did not exacerbate the negative impact on proliferation induced by Ub(K27R) replacement (Fig 4H). Second, whereas knockdown of p97 led to accumulation of cells in G2 phase as reported previously (Magnaghi et al, 2013; Heidelberger et al, 2018), similar to the effect of Ub‐K27R replacement, reduced p97 expression did not further enhance the accumulation of Ub(K27R)‐replaced cells in G2 phase (Figs 4I and EV4E). Likewise, p97i treatment diminished the mitotic index of Ub(WT)‐replaced cells but had no significant effect in Ub(K27R)‐expressing cells (Fig 4J). Third, in line with defective ubiquitylation of the nucleotide excision repair (NER) factor XPC in Ub(K27R)‐replaced cells (Fig 3E and F), we found that its extraction from UV damage sites, which is mediated by p97 (Puumalainen et al, 2014), was impaired to a similar extent by p97i and Ub(K27R)‐replacement whereas no additive impact of these perturbations was observed (Fig EV4F–H). Similar effects were seen for DDB2 (Fig EV4G and H), another DNA damage sensor in NER whose displacement from UV‐damaged chromatin is promoted by p97 activity (Puumalainen et al, 2014). These data are consistent with and further support defective p97‐dependent substrate processing in cells incapable of generating K27‐linked Ub chains.
Blocking K27‐linkage signal decoding impairs p97‐mediated substrate turnover
We sought to ascertain the role of K27‐linked ubiquitylation in promoting the processing of p97 client proteins by means of an orthogonal, Ub replacement‐independent approach for interfering with the functionality of these Ub polymers. To this aim, we took advantage of the recently established ability of the DUB UCHL3 to recognize K27 linkages with high affinity and selectivity (Zhang et al, 2017; Pan et al, 2019; van Tilburg et al, 2020). We reasoned that when expressed at high levels in cells, catalytically inactive UCHL3 (UCHL3 C95S) may efficiently bind K27 linkages and thereby block the biological signals elicited by these modifications. Consistent with this rationale, the overexpression of UCHL3 C95S strongly stabilized the Ub(G76V)‐GFP p97 model substrate, similar to Ub(K27R) replacement (Fig 5A and B). By contrast, a corresponding catalytically inactive version of the structurally most closely related DUB UCHL5 (UCHL5 C88S) that is not known to interact with K27‐linked Ub (Zhang et al, 2017) failed to stabilize Ub(G76V)‐GFP (Fig 5A and B). The expression of UCHL3 C95S also stabilized Ub(G76V)‐GFP in non‐transformed RPE‐1 cells (Fig EV5A). Importantly, the ability of catalytically inactive UCHL3 to stabilize the p97 model substrate was dependent on its ability to recognize K27 linkages. This was evident from a lack of Ub(G76V)‐GFP stabilization by UCHL3 C95S containing a D33A mutation that has been shown to prevent UCHL3 binding to K27 linkages (Pan et al, 2019), a finding we verified by showing that UCHL3 C95S, but neither UCHL3 C95S/D33A nor UCHL5 C88S, interacts with purified K27‐linked di‐Ub and associates with Ub(G76V)‐GFP in cells, consistent with this model substrate being directly modified by K27‐linkages (Figs 5A–C and 4F and G). Moreover, whereas Ub(G76V)‐GFP molecules stabilized by p97i were rapidly degraded upon withdrawal of the inhibitor in control cells, this could be efficiently blocked by expression of UCHL3 C95S but not UCHL3 C95S/D33A or UCHL5 C88S (Fig 5D), further supporting an impairment of p97‐mediated substrate processing upon interference with the functional decoding of K27‐linked Ub signals. To address whether UCHL3 C95S impedes Ub(G76V)‐GFP turnover at the level of p97 or the proteasome, we employed a variant Ub(G76V)‐GFP protein containing a C‐terminal 20‐amino acid extension (Ub(G76V)‐GFP‐20AA), which has been shown to override the requirement for p97‐mediated unfolding to enable its proteasomal degradation (Beskow et al, 2009; Godderz et al, 2015) (Fig 5E). We verified that in human cells the 20AA extension alleviated the need for p97 activity to enable Ub(G76V)‐GFP degradation, whereas Ub(G76V)‐GFP and Ub(G76V)‐GFP‐20AA were stabilized to a similar extent upon inhibition of the proteasome (Fig 5F and G). Importantly, Ub(G76V)‐GFP‐20AA was much less responsive to the stabilizing effect of overexpressed UCHL3 C95S than Ub(G76V)‐GFP (Fig 5H), suggesting that the shielding of K27‐linkages by UCHL3 C95S mainly impairs the turnover of ubiquitylated substrates at the level of p97. Interestingly, in line with its inefficient stabilization by UCHL3 C95S, Ub(G76V)‐GFP‐20AA exhibited greatly reduced interaction with UCHL3 C95S compared to Ub(G76V)‐GFP (Fig 5I). This raises the possibility that K27‐linked ubiquitylation of proteasomal substrates may be positively correlated with their requirement for p97‐mediated processing to enable subsequent degradation.
Figure 5. Blocking the decoding of K27‐linked ubiquitylation signals impairs p97 substrate turnover.
-
AFLAG IPs from U2OS cells transfected with indicated FLAG‐tagged expression constructs were incubated with purified K27 di‐Ub and analyzed by immunoblotting.
-
BU2OS/Ub(G76V)‐GFP cells were transfected with indicated FLAG‐tagged expression constructs, and levels of Ub(G76V)‐GFP in cells gated for FLAG expression were quantified using QIBC (solid lines, median; dashed lines, quartiles; > 2,000 transfected cells analyzed per condition).
-
CCells transfected as in (B) and treated or not with p97i for 4 h were subjected to GFP IP followed by immunoblotting.
-
DCells transfected as in (B) were treated with p97i (4 h) and harvested at the indicated times after removal of the drug. Levels of Ub(G76V)‐GFP in cells gated as in (B) were quantified using QIBC (solid lines, median; dashed lines, quartiles; > 1,000 transfected cells analyzed per condition).
-
ESchematic of fluorescent Ub(G76V)‐GFP and Ub(G76V)‐GFP‐20AA reporters and their dependency on p97 and/or proteasome activity for their turnover.
-
FU2OS/Ub(G76V)‐GFP and U2OS/Ub(G76V)‐GFP‐20AA cell lines were treated or not with p97i (5 μΜ) for the indicated periods of time, and levels of GFP signal were quantified using QIBC (solid lines, median; dashed lines, quartiles; > 10,000 cells analyzed per condition).
-
GAs in (F), except cells were treated with MG132 for the indicated times (solid lines, median; dashed lines, quartiles; > 10,000 cells analyzed per condition).
-
HU2OS/Ub(G76V)‐GFP and U2OS/Ub(G76V)‐20AA‐GFP cell lines were transfected with indicated FLAG‐tagged expression constructs, and GFP levels in cells gated for FLAG expression were quantified using QIBC (solid lines, median; dashed lines, quartiles; > 2,000 transfected cells analyzed per condition).
-
IU2OS/Ub(G76V)‐GFP and U2OS/Ub(G76V)‐20AA‐GFP cell lines were transfected or not with FLAG‐UCHL3 C95S expression construct, treated with MG132 for 2 h and subjected to GFP IP followed by immunoblotting.
Data information: Data are representative of six (B) and three (A,C,D,F‐I) independent experiments with similar outcome.
Figure EV5. (related to Fig 5) Blocking the decoding of K27‐linked ubiquitylation signals impairs p97 substrate turnover.
-
ARPE‐1 cells stably expressing Ub(G76V)‐GFP were transfected with indicated FLAG‐tagged expression constructs, fixed and immunostained with FLAG antibody. Levels of Ub(G76V)‐GFP in FLAG‐positive cells were quantified using QIBC (solid lines, median; dashed lines, quartiles; > 1,000 transfected cells analyzed per condition).
-
BU2OS cells transfected with FLAG‐UCHL3 constructs were subjected to FLAG IP followed by immunoblotting with indicated antibodies.
-
CDOX‐treated Ub replacement cell lines were subjected to p97 IP followed by immunoblotting with the indicated antibodies.
-
DATPase activity of p97 immobilized by IP under denaturing conditions from U2OS/shUb/HA‐Ub(WT) or U2OS/shUb/HA‐Ub(K27R) (mean ± SEM; n = 3 independent experiments).
-
ECoomassie staining of p97 IPs in (D).
-
FCrystal structure of human Ub (PDB ID: 5UJL; Castaneda et al, 2016) (left), modeled structure of a Ub(K27R) mutant (middle) using the PyMol mutagenesis tool, and their overlay (right). Amino acids at the Ub K27 position are highlighted by space‐filling representation.
Data information: Data are representative of three (A,D,E) and two (B,C) independent experiments with similar outcome.
Although nuclear p97 cofactors displayed reduced ubiquitylation status in Ub(K27R)‐replaced cells, they did not show detectable interaction with UCHL3 C95S unlike Ub(G76V)‐GFP (Fig EV5B), indicating that p97 cofactors may not be direct targets of K27‐linked ubiquitylation. Likewise, Ub(K27R) replacement neither affected interactions between p97 and nuclear cofactors nor p97 ATPase activity (Fig EV5C–E). Thus, whereas the p97‐proteasome pathway substrate Ub(G76V)‐GFP is modified by K27‐linked ubiquitylation, we did not obtain supportive evidence for K27‐linkages directly impacting the p97 system per se. Altogether, these findings are consistent with a role of K27‐linkages impinging directly on polyubiquitylated substrates and possibly other factors in facilitating their processing by p97 and ensuing degradation by the proteasome.
Discussion
Although K27‐linked Ub chains have been implicated in several cellular processes, including the DNA damage response and innate immunity (Gatti et al, 2015; Swatek & Komander, 2016; van Huizen & Kikkert, 2019), deciphering the precise function of K27‐linked ubiquitylation and its relative importance for cell growth and survival has remained a major challenge due to a lack of approaches for specifically isolating and manipulating this Ub chain type. To remedy this fundamental knowledge gap, we leveraged a validated Ub replacement strategy to characterize the cellular phenotypes arising from targeted disruption of K27‐linked Ub chain formation, revealing for the first time that K27‐linked ubiquitylation has a critical role in supporting proliferation and viability of human cells. This is notable considering the low abundance of K27‐linkages in mammalian cells and is further reinforced by our observation that preventing the formation of K29‐linked Ub chains, which make up a considerably larger proportion of cellular Ub conjugates (Dammer et al, 2011; Swatek et al, 2019), only had a modest impact on cell cycle progression and proliferation. We consider it unlikely for several reasons that the phenotypes resulting from induction of the Ub(K27R) allele are an indirect consequence of overtly perturbed Ub functionality rather than a block to K27‐linked Ub chain formation per se. First, Ub(K27R)‐replaced cells show unchanged levels of most other Ub linkage topologies and ubiquitylated proteins, implying that the ectopic Ub(K27R) protein retains general functionality in cells. Second, a Ub mutant‐independent approach for blocking recognition of K27 linkages in cells via overexpression of a specific high‐affinity binder of this modification (UCHL3 C95S) to outcompete the binding of endogenous decoders of K27‐linkage signals phenocopies impaired p97 substrate processing seen in Ub(K27R)‐replaced cells. Third, modeling analysis indicates that a K27R substitution has no significant impact on the structure and folding of Ub (Fig EV5F), and yeast strains carrying a Ub(K27R) mutation only show mildly reduced growth (Xu et al, 2009b; Roscoe et al, 2013). We note in this context that while the K27R mutation has been suggested to decrease the stability of Ub (Kudriaeva et al, 2021), no pronounced difference in the half‐lives of the ectopic Ub(WT) and Ub(K27R) proteins was apparent in our Ub‐replaced cells.
While the observed impact of Ub(K27R) replacement in undermining cell cycle progression and proliferation may represent a composite effect of modulating different cellular processes regulated by K27‐linked ubiquitylation, several lines of evidence presented here functionally couple this Ub chain type to the p97 machinery and suggest impaired processing of p97 substrates as one key determinant of the reduced fitness of Ub(K27R)‐replaced cells. First, unbiased proteomic profiling of ubiquitylation changes resulting from abrogation of K27‐linked ubiquitylation revealed a signature of nuclear p97 cofactors and proteins with a known interplay with p97 among the small number of regulated proteins. Although these factors may not be direct targets of K27‐linked ubiquitylation and it remains to be established why their overall ubiquitylation is reduced in Ub(K27R)‐replaced cells, such global insights offer important clues to the identity of cellular proteins and processes affected by manipulating K27‐linked Ub chain formation. Second, abrogating K27‐linked ubiquitylation and inhibiting p97 activity not only had an equivalent but also epistatic impact on processes including cell proliferation, cell cycle progression and protein recruitment to damaged DNA, in which key roles of p97 are well established (van den Boom & Meyer, 2018). Third, using fluorescent reporters of p97 and proteasome activity we obtained direct evidence for impaired p97‐ but not proteasome‐dependent substrate processing upon Ub(K27R) replacement or K27‐linkage binder (UCHL3 C95S) overexpression. Moreover, abolishing K27‐linked Ub chain formation selectively deregulated nuclear p97 activity, in agreement with the enrichment of these Ub linkages in the nucleus. Thus, despite the limitations imposed by the current lack of specific tools for identifying direct targets of K27‐linked ubiquitylation, a consistent theme emanating from our study is the importance of these Ub polymers in human cells for promoting the processing of ubiquitylated nuclear substrates by the p97 system, which is crucial for proteostasis and cell fitness (van den Boom & Meyer, 2018). This may help to explain why in our replacement cell lines K27‐linked Ub chains appear to be more critical than K29‐linked ubiquitylation for proliferation, despite their lower abundance.
Precisely how K27‐linked ubiquitylation facilitates the turnover of ubiquitylated substrates via p97 remains to be established, but our data suggest that this is at least partially mediated by K27‐linkage modifications targeting p97 substrates directly. To this end, our proteomic analysis and ubiquitylation assays demonstrated that the p97‐proteasome pathway model substrate Ub(G76V)‐GFP is directly modified by K27‐linked ubiquitylation, albeit at a lower level than K48‐ and K29‐linkages, which are well‐established signals for proteasomal degradation and have previously been implicated in Ub(G76V)‐GFP turnover (Liu et al, 2017). Our complementary findings from experiments with the K27‐selective binder UCHL3 C95S provide further evidence in support of a role for K27‐linked ubiquitylation in impacting p97‐mediated processing at the level of the ubiquitylated substrate. Specifically, we found that UCHL3 C95S interacted with and strongly stabilized Ub(G76V)‐GFP in a manner that was dependent on its previously reported ability to bind K27‐linkages with high selectivity (Zhang et al, 2017; Pan et al, 2019; van Tilburg et al, 2020). By contrast, an extended version of the Ub(G76V)‐GFP substrate that is largely refractory to the requirement of p97‐mediated unfolding for proteasomal degradation neither bound to nor showed pronounced stabilization by UCHL3 C95S. This suggests that the direct binding of UCHL3 C95S to K27‐ubiquitylated Ub(G76V)‐GFP impairs its processing by p97 that enables subsequent proteasomal destruction. A role for K27‐linked Ub polymers in facilitating proteasomal degradation of ubiquitylated substrates via p97 is consistent with previous findings showing that total K27‐linkage abundance increases upon proteasome inhibition, similar to K48‐ and K29‐linkages (Kim et al, 2011; Heidelberger et al, 2018). It is worth noting in this context that the Ub‐binding UBA2 domain in the predominantly nuclear proteasomal shuttle factor RAD23A not only binds K48‐linked Ub chains but has also been shown to recognize K27‐linkages with comparable affinity (Castaneda et al, 2016). Thus, K27‐linkages residing on p97‐processed ubiquitylated substrates might potentially help to promote RAD23‐mediated escorting of the substrate to the proteasome, a possibility that awaits experimental clarification. Unlike the p97‐proteasome pathway model substrate Ub(G76V)‐GFP, we found no evidence for K27‐linked ubiquitylation being directly targeted to the p97 machinery per se. Moreover, the function of the proteasome appeared overtly normal in Ub(K27R)‐replaced cells. However, it remains possible that in addition to targeting p97 substrates directly, K27‐linked Ub polymers may impact p97‐dependent processing of ubiquitylated proteins in additional ways that were not picked up in our assays, and further studies will be required to delineate the precise mechanistic underpinnings of the emerging role of K27‐linked Ub chains in supporting cell proliferation and fitness via the p97 system and other pathways. The findings and experimental models reported here provide an entry point for further targeted exploration of the hitherto concealed biological functions of atypical ubiquitylation via K27‐linkages.
Materials and Methods
Cell culture
All mammalian cells were cultured under standard conditions at 37°C and 5% CO2 in DMEM (Thermo) supplemented with 10% FBS (v/v) and penicillin‐streptomycin (Thermo). Recombination cloning‐compatible U2OS cells were generated using the Flp‐In T‐Rex Core Kit (Thermo) according to manufacturer instructions and maintained under selection using Blasticidin (Invivogen) and Zeocin (Thermo). To generate U2OS/shUb cells, U2OS(FlpIn) cells were transfected with pPuro‐shUb and selected with puromycin (Sigma). Derivative cell lines stably expressing the Ub replacement expression cassette (U2OS/shUb/HA‐Ub) were generated by co‐transfection of U2OS/shUb with pcDNA5/FRT/TO‐UBA52/HA‐RPS27A and pOG44 (Thermo) plasmids, followed by selection with Hygromycin B (Thermo). Stable clones were carefully screened for uniform expression of ectopic Ub at levels matching those of endogenous Ub and maintained under selection with blasticidin, puromycin and hgromycin B. Ub replacement was initiated by treatment with Doxycycline (DOX) (0.5 µg/ml) for 72 h unless otherwise indicated. U2OS cells and derivative cell lines stably expressing shUb‐resistant Ub(G76V)‐GFP or Ub(G76V)‐GFP‐20AA were generated by transfection and selection with G418 (Invivogen). U2OS/Ub(G76V)‐GFP and U2OS/Ub(G76V)‐GFP‐20AA clones were visually screened for low basal GFP expression. All cell lines used in this study were regularly tested negative for mycoplasma infection but were not authenticated. Unless otherwise indicated, the following drug concentrations were used: MLN‐7243 (E1i; 5 μM, Active Biochem), NMS‐873 (p97i; 5 μM, Sigma), MG132 (20 μM; Sigma), MK‐1775 (Wee1i; 1 μM, Selleckchem), RO‐3306 (CDK1i; 10 μM, Sigma), Actinomycin D (ActD; 2 μg/ml, Sigma).
Plasmids and siRNAs
Inducible silencing of endogenous Ub expression was performed using pPuro‐shUb (kind gift from Zhijian Chen, University of Texas Southwestern Medical Center), as previously described (Xu et al, 2009a). In brief, shUb encodes tandem sequences of 5′‐ACACCATTGAGAATGTCAA‐3′ targeting UBC and UBA52, and 5′‐AGGCCAAGATCCAGGATAA‐3′ targeting UBB and RPS27A (Fig EV1A). An inducible expression cassette encoding shRNA‐resistant UBA52 and RPS27A (kind gift from Zhijian Chen, University of Texas Southwestern Medical Center) was cloned into pcDNA5/FRT/TO (Thermo). Ub mutagenesis was performed using the Q5 Site‐Directed Mutagenesis kit (New England Biolabs) and the following primer sets: UBA52 K27R: 5′‐GAAAACGTCAGAGCCAAAATTC‐3′ and 5′‐GATTGTATCACTGGGCTC‐3′; RPS27A K27R: 5′‐GAAAATGTAAGAGCTAAAATTCAGGAC‐3′ and 5′‐TATCGTATCCGAGGGTTC‐3′; UBA52 K29R: 5′‐GTCAAAGCCAGAATTCAAGAC‐3′ and 5′‐GTTTTCGATTGTATCACTG‐3′; RPS27A K29R: 5′‐ GTAAAAGCTAGAATTCAGGACAAG‐3′ and 5′‐ATTTTCTATCGTATCCGAG‐3′; UBA52 K33R: 5′‐ATTCAAGACAGGGAGGGTATC‐3′ and 5′‐TTTGGCTTTGACGTTTTC‐3′; RPS27A K33R: 5′‐ATTCAGGACAGGGAAGGAATTC‐3′ and 5′‐TTTAGCTTTTACATTTTCTATCG‐3′. Mammalian expression construct encoding Ub(G76V)‐GFP (Dantuma et al, 2000) was a kind gift from Nico Dantuma (Karolinska Institute, Stockholm, Sweden). An shUb‐resistant version of Ub(G76V)‐GFP was generated using the Q5 Site‐Directed Mutagenesis kit with primer set 5′‐AAAACGTAAAGGCAAAGATCCAAGATAAGG‐3′ and 5′‐CGATAGTATCACTGGGCTCAACCTCG‐3′ according to manufacturer’s instructions. Ub(K27R/G76V)‐GFP was derived from shUb‐resistant Ub(G76V)‐GFP using the Q5 Site‐directed mutagenesis kit and primers 5′‐GAAAACGTAAGGGCAAAGATC‐3′ and 5′‐GATAGTATCACTGGGCTC‐3′. Ub(G76V)‐GFP‐20AA (Godderz et al, 2015) was derived from Ub(G76V)‐GFP using the Q5 Site‐Directed Mutagenesis kit with primer set 5′‐TTCGAGGGCAAGCCCATCCCCAACCCCCTGTAAAGCGGCCGCGACTCT‐3′ and 5′‐GGGGCCCCTGCTCTCCAGGGGCCTGCAGCTCTTGTACAGCTCGTCCATGC‐3′. Inactivating mutations (FLAG‐UCHL3 C95S, FLAG‐UCHL3 C95S/D33A and FLAG‐UCHL5 C88S) were introduced into DUB expression plasmids described previously (Mosbech et al, 2013) using the Q5 Site‐Directed Mutagenesis kit.
All siRNAs were used at 20 nM and transfected with Lipofectamine RNAiMAX reagent (Thermo) according to manufacturer’s instructions. The following siRNA oligonucleotides were used: Non‐targeting control (CTRL): 5′‐GGGAUACCUAGACGUUCUA‐3′; p97: Dharmacon SmartPool (D‐008727).
Cell lysis and immunoprecipitation
Immunoprecipitation (IP) of HA‐tagged proteins was performed using Anti‐HA Affinity matrix (Roche) under denaturing conditions, unless otherwise stated. GFP‐trap (Chromotek) was used according to manufacturer’s instructions. Cell lysis was performed using RIPA buffer (140 mM NaCl; 10 mM Tris‐HCl (pH 8.0); 0.1% sodium deoxycholate (w/v); 1% Triton X‐100 (v/v); 0.1% SDS (w/v); 1 mM EDTA; 0.5 mM EGTA). IPs were washed in non‐denaturing buffer (150 mM NaCl; 50 mM Tris‐HCl; 0.5mM EDTA). All lysis and IPs indicated as under denaturing conditions were performed in denaturing buffer (50 mM NaCl; 20 mM Tris‐HCL (pH 7.5); 0.5% sodium deoxycholate (w/v); 0.5% igepal (v/v); 0.5% SDS (w/v); 1 mM EDTA). All lysis and wash buffers were supplemented with 1 mM fresh PMSF Protease Inhibitor (Thermo), complete EDTA‐free protease inhibitor Cocktail Tablets (Roche), 1.25 mM N‐ethylmaleimide (Sigma) and 50 µM PR‐619 (Calbiochem).
Chromatin fractionation
Fractionation of cell pellets was performed as described (Borgermann et al, 2019). In brief, cell pellets were washed and resuspended in cytoplasmic extraction buffer (10 mM Tris; 10 mM KCl; 1.5 mM MgCl2; 0.34 M sucrose; 10% glycerol (v/v); 0.1% Triton (v/v)) and incubated for 15 min on ice. Cells were pelleted and supernatant (cytoplasmic fraction) was removed to a separate tube. Cells were washed in cytoplasmic extraction buffer and pellets resuspended in RIPA buffer and incubated for 15 min at 37°C. Cells were pelleted and supernatant (chromatin fraction) was removed to a separate tube.
Immunofluorescence
Mammalian cells grown on coverslips were fixed in formalin buffer (VWR) for 15 min at room temperature (RT). Cells were permeabilized with PBS containing 0.5% Triton‐X for 5 min and blocked with 5% BSA (Sigma) for 1 h prior to staining with indicated primary antibodies overnight at 4°C. After 3 washes cells were stained with a combination of Alexa Fluor secondary antibodies and 4′,6‐Diamidino‐2‐Phenylindole (DAPI; Molecular Probes) for 1 h at RT in the dark. Finally, after 4 further rounds of washing, coverslips were dried and mounted on glass slides using Mowiol (Sigma). Where indicated, cells were pre‐extracted prior to fixation using either CSK buffer (100 mM NaCl; 10 mM HEPES; 3 mM MgCl2; 300 mM sucrose; 0.25% Triton‐X; 1 mM PMSF) or a stringent pre‐extraction buffer (10 mM Tris‐HCl, pH 7.4; 2.5 mM MgCl2; 0.5% igepal; 1 mM PMSF). K29‐linked Ub conjugates were visualized using a K29‐Ub‐specific synthetic antigen binder (sAB‐K29; kind gift from Minglei Zhao (University of Chicago)) (Yu et al, 2021) and Alexa Fluor 488 AffiniPure F(ab′)2 Fragment Donkey Anti‐Human IgG (Jackson Immunoresearch). Local UV damage was induced by irradiating cells on coverslips with 100 J/m2 through a PBS‐equilibrated membrane of 8 μm pores and fixing at the indicated timepoints. To determine cell cycle distribution, nascent DNA synthesis was estimated by assessing 5‐ethynyl‐2′‐deoxyuridine (EdU; Thermo) incorporation. Cells were incubated with 10 μΜ EdU for 1 h prior to labelling with Click‐iT Plus EdU Alexa Fluor 647 Imaging Kit (Thermo) according to manufacturer’s instructions. To assess transcription efficiency, nascent RNA production was detected using the Click‐iT RNA Alexa Fluor 594 Imaging Kit (Thermo) according to manufacturer’s instructions. Finally, to assess translation efficiency, nascent protein production was detected by incubating cells in methionine‐free DMEM (Thermo) supplemented with 50 μM of the methionine analog L‐Azidohomoalanine (AHA; Thermo) for 3 h, prior to labeling with Alexa Fluor 488 Alkyne (Thermo). Quantitative image‐based cytometry (QIBC) was performed as described (Toledo et al, 2013). In brief, cells were fixed, permeabilized and stained as described above. Images were acquired with a ScanR high‐content screening microscope (Olympus). Automated and unbiased image analysis was carried out with the ScanR analysis software (version 2.8.1). Data were exported and processed using Spotfire software (version 10.5.0; Tibco).
Antibodies
Antibodies used for immunoblotting included: Actin (MAB1501, Millipore (1:50,000 dilution), RRID: AB_2223041), GAPDH (sc‐20357, Santa Cruz (1:5,000),RRID: AB_641107), GFP (11814460001, Sigma (1:1,000), RRID: AB_390913), HA (11867423001, Roche (1:1,000), RRID: AB_390918), FLAG (F1804, Sigma (1:5,000), RRID: AB_262044), K27‐linked Ub (ab181537, Abcam (1:5,000), RRID: AB_2713902), NPL4 (13489, Cell Signaling (1:1,000), RRID: AB_2798232), p47 (NBP2‐13677, Novus (1:1,000)), p97 (ab11433, Abcam (1:1,000), RRID: AB_298039), PCNA (sc‐56, Santa Cruz (1:1,000), RRID: AB_628110), RAD23B (A302‐306A, Bethyl (1:1,000), AB_1850216), RPS27A (ab111598, Abcam (1:1,000), RRID: AB_10863285), Tubulin (T9026, Sigma (1:50,000)), UBA52 (PA5‐23685, Thermo (1:1,000), RRID: AB_2541185), Ub (BML‐PW8810, Enzo (1:1,000) and sc‐8017 (P4D1), Santa Cruz (1:1,000), RRID: AB_2762364), UFD1 (13789, Cell Signaling (1:1,000), RRID: AB_2798313), XPC (A301‐122A, Bethyl (1:1,000), RRID: AB_2288476). Antibodies used for immunofluorescence included: 53BP1 (sc‐22760, Santa Cruz (1:1,000), RRID: AB_2256326 and sc‐10911, Santa Cruz (1:500), RRID: AB_633428), DDB2 (ab51017, Abcam (1:100), RRID: AB_869426), DDIT3/CHOP (ab11419 (1:100)), HA (11867423001, Roche (1:500), RRID: AB_390918), K27‐linked Ub (ab181537, Abcam (1:1,000), RRID: AB_2713902), K48‐linked Ub (05‐1307, Sigma (1:200), RRID: AB_1587578), K63‐linked Ub (BML‐PW0600‐0025, Enzo (1:100), RRID: AB_2052278), p47 (NBP2‐13677, Novus (1:200)), p97 (ab11433, Abcam (1:400), RRID: AB_298039), Ub (BML‐PW8810, Enzo (1:100)), UFD1 (13789, Cell Signaling (1:200), RRID: AB_2798313), XPC (X1129, Sigma (1: 500), RRID: AB_796182), γ‐H2AX (05‐636, Sigma (1:1,000), RRID: AB_309864).
Di‐Ub binding assays
The reactivity of K27‐Ub antibody with individual recombinant linkage‐specific di‐Ub proteins (Boston Biochem) was analyzed using standard immunoblot procedure. Loading was determined on a duplicate di‐Ub gel stained using Silver stain kit (Pierce) according to manufacturer’s instructions. For analysis of DUB binding to K27 di‐Ub, U2OS cells were transfected with DUB plasmids overnight and lysed in 150 mM NaCl; 50 mM Tris, pH 7.5; 1 mM EDTA; 0.5% Igepal supplemented with 1 mM fresh PMSF protease inhibitor (Thermo) and complete EDTA‐free protease inhibitor cocktail tablets (Roche). Lysates were cleared prior to 5‐fold dilution in high salt wash buffer (500 mM NaCl; 20 mM Tris, pH 8.0; 2 mM EDTA; 1% Triton‐X). Recombinant DUBs were then enriched with Anti‐FLAG M2 Affinity Gel (Sigma). Beads were washed with high salt wash buffer prior to equilibration with di‐Ub binding buffer (150 mM NaCl; 50 mM Tris, pH 8.0; 10% glycerol; 0.5% Igepal). K27‐linked di‐Ub (Boston Biochem) was incubated with beads for 3 h at room temperature prior to di‐Ub binding buffer washes and elution by boiling in the presence of loading buffer.
ATPase activity assay
Endogenous p97 was enriched from whole cell lysates under denaturing conditions and ATPase activity determined using the ATPase/GTPase Activity Assay Kit (Sigma), according to manufacturer’s instructions.
Clonogenic assay
U2OS/shUb cells were seeded at low density and co‐transfected with pcDNA5/FRT/TO‐UBA52/HA‐RPS27A (WT or K‐R mutants) and pOG44 plasmids to induce recombination. Transfection pools were split into two plates and colonies were established for 48 h under Hygromycin B selection. Doxycycline was added to one of the dishes and colonies expanded for 120 h, prior to staining with crystal violet solution (Sigma). DOX‐treated plates were seeded at 5 times density to Hygromycin B only plates. Colonies on the Hygromycin B plate and the Hygromycin B/Doxycycline plate were counted for recombination efficiency and Ub viability rescue efficiency, respectively. Colony counts performed using GelCount (Oxford Optronix) imaging system and software (version 1.2.4.2).
Proliferation and viability assays
Relative cell proliferation was measured using an Incucyte S3 Live‐Cell Analysis System. Cells (1 × 104) were seeded in triplicate into a 24‐well plate 24 h prior to Doxycycline addition. Cells were imaged at 3 h intervals after doxycycline addition with a mean confluency determined from 4 images per well. Relative cell viability was determined by metabolic conversion of resazurin sodium salt (Sigma) into fluorescent resofurin. In brief, cells (1 × 104) were seeded into a 96‐well plate and incubated overnight. Resazurin solution was diluted to 10 µg/ml in base medium and 200 µl was added to each well and incubated for 2 h. Fluorescence was measured at 544 nm excitation and 590–10 nm emission using a POLARstar OMEGA plate reader (BMG Labtech). After addition of doxycycline, resazurin conversion was measured in 24 h intervals.
Live‐cell imaging
Cells were seeded into a µ‐slide 8 well (Ibidi) imaging dish 24 h prior to imaging. Imaging was performed for 36 h using a DeltaVision Elite microscope (GE Healthcare) equipped with a 40x oil objective lens (numerical aperture 1.35) and CoolSNAP HQ2 camera (Photometrics). Cells were maintained at 37°C in Leibovitz L‐15 medium (Gibco) supplemented with 10% FCS. Image acquisition and analysis was performed using SoftWoRx (GE Healthcare) software. Mitotic cells were counted as percentage of cells entering mitosis from those imaged throughout the time course.
Whole proteome sample preparation
Snap‐frozen sample pellets were lysed in 20% [v/v] 2,2,2‐trifluoroethanol in 100 mM Tris‐HCl, pH 8.0 supplemented with 5 mM DTT and heated for 10 min at 95°C. Lysates were sonicated in a Biorupter (Diagenode) for 15 min prior to the addition of 25 mM chloroacetamide and incubated for 20 min. Samples were eluted in 100 mM Tris‐HCl, pH 8.0 and digested with trypsin (1:50) overnight at 37°C at 1,500 rpm. Peptides were desalted and purified using styrenedivinylbenzene‐reversed phase sulfonate (SDB‐RPS) StageTips prepared in 0.2% trifluoroacetic acid (TFA). Peptides were washed and eluted with 80% acetonitrile (ACN); 1% ammonia prior to vacuum‐drying. Dried peptides were reconstituted in 2% ACN and 0.1% TFA.
Affinity purification and mass spectrometry (AP‐MS) sample preparation
HA‐tag affinity purification was performed as described above. Partial on‐bead digest was performed using 2 M urea; 2 mM DTT; 20 µg/ml Trypsin; 50 mM Tris, pH 7.5 incubated at 37°C for 30 min at 1,400 rpm. Supernatants were transferred to new tubes, alkylated with 25 mM chloroacetamide and further digested overnight at RT. Digestion was terminated with 1% TFA. Peptides were purified and desalted using SDB‐RPS StageTips and eluted as for whole proteome samples.
Liquid chromatography‐mass spectrometry (LC‐MS) analysis
Nanoflow LC‐MS analysis of tryptic peptides was performed on a quadrupole Orbitrap mass spectrometer (Q Exactive HF‐X, Thermo) coupled to an EASY‐nLC1200 system (Thermo) via a nano‐electrospray ion source. Approx. 0.25 µg of sample was loaded on a 50‐cm HPLC‐column (75‐μm inner diameter, New Objective, USA; in‐house packed using ReproSil‐Pur C18‐AQ 1.9‐µm silica beads; Dr Maisch GmbH, Germany). Peptides were separated using a linear gradient from 5 to 60% solvent B (80% CAN; 0.1% formic acid) in solvent A (0.1% formic acid). Run duration was 100 min (whole proteome) or 60 min (AP‐MS). Column temperature was maintained at 60°C. Peptide detection occurred using “top‐15” (whole proteome) or “top‐10” (AP‐MS) data‐dependent mode, collecting MS spectra in the Orbitrap mass analyzer (60,000 resolution, 300–1,650 m/z range) with an automatic gain control (AGC) target of 3 × 106 and a maximum ion injection time of 25 ms. The most intense ions from the full scan were isolated with an isolation width of 1.4 m/z. Following higher‐energy collisional dissociation (HCD) with a normalized collision energy (NCE) of 27%, MS/MS spectra were collected in the Orbitrap (15,000 resolution) with an AGC target of 1 × 105 and a maximum ion injection time of 28 ms (whole proteome) or 50 ms (AP‐MS). Precursor dynamic exclusion was enabled with a duration of 30 s.
Raw file processing and bioinformatic analyses
MS raw files were processed with MaxQuant software (Cox & Mann, 2008) (version 1.5.0.38). The integrated Andromeda search engine (Cox et al, 2011) was used for peptide and protein identification at an FDR of < 1%. The human UniProtKB database (October 2017) was used as forward database and the automatically generated reverse database for the decoy search. A modified ubiquitin sequence substituting lysine at position 27 with arginine (K27R substitution) was included. A minimum threshold of 7 amino acids was used for peptide identification. Proteins that could not be discriminated by unique peptides were assigned to the same protein group (Cox & Mann, 2008). Label‐free protein quantification was performed using the MaxLFQ algorithm (Cox et al, 2014). Protein ratios were determined based on median peptide ratios and only common peptides were used for pair‐wise ratio calculations. The “match‐between‐runs” feature of MaxQuant was enabled to transfer peptide identifications across runs based on high mass accuracy and normalized retention times. Prior to data analysis, proteins found as reverse hits or only identified by site‐modification, were filtered out. All statistical and bioinformatic analyses were performed using Perseus (Tyanova et al, 2016) or the R framework (https://www.r‐project.org/).
Quantification and statistical analysis
All statistical analyses were performed using Prism 9.0.0 (GraphPad Software). Statistical details including number of independent experiments (n), definition of significance and measurements are defined in figure legends. No statistical method was used to predetermine sample size and no data were excluded from the analyses. Samples were not randomized and investigators were not blinded to group allocation during data collection and analysis.
Author contributions
Robert F Shearer: Conceptualization; Investigation; Methodology; Writing—review & editing. Dimitris Typas: Conceptualization; Funding acquisition; Investigation; Methodology; Writing—review & editing. Fabian Coscia: Funding acquisition; Investigation; Methodology; Writing—review & editing. Sofie Schovsbo: Investigation; Writing—review & editing. Thomas Kruse: Investigation; Writing—review & editing. Andreas Mund: Investigation; Methodology; Writing—review & editing. Niels Mailand: Conceptualization; Supervision; Funding acquisition; Methodology; Writing—original draft; Project administration; Writing—review & editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Acknowledgements
We thank Zhijian Chen, Minglei Zhao and Nico Dantuma for providing reagents, Satya Pentakota for help with structure modeling analysis, Katrine Weischenfeldt for technical assistance and members of the Mailand laboratory for helpful discussions. This work was supported by grants from the Novo Nordisk Foundation (grants no. NNF14CC0001 and NNF18OC0030752), Independent Research Fund Denmark (grant no. 9040‐00038B), Lundbeck Foundation (grant no. R223‐2016‐281), Danish National Research Foundation (grant no. DNRF‐115) and the European Commission’s Horizon 2020 Research and Innovation Programme (Marie Skłodowska‐Curie Individual Fellowship grant agreements no. 744866 (D.T.) and 846795 (F.C.)).
The EMBO Journal (2022) 41: e110145.
Contributor Information
Dimitris Typas, Email: dimitrios.typas@cpr.ku.dk.
Niels Mailand, Email: niels.mailand@cpr.ku.dk.
Data availability
The mass spectrometry proteomics data (Dataset EV1; Dataset EV2) have been deposited to the ProteomeXchange Consortium via the Proteomics Identifications (PRIDE) partner repository (http://www.ebi.ac.uk/pride) (Perez‐Riverol et al, 2019) with the dataset identifier PXD023579. All other data supporting the findings of this study are available within the article and supplementary information. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Anderson D, Le Moigne R, Djakovic S, Kumar B, Rice J, Wong S, Wang J, Yao B, Valle E, Kiss von Soly S et al (2015) Targeting the AAA ATPase p97 as an approach to treat cancer through disruption of protein homeostasis. Cancer Cell 28: 653–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard JAM, Goodall EA, Greene ER, Jonsson E, Dong KC, Martin A (2018) Structure and function of the 26S proteasome. Annu Rev Biochem 87: 697–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beskow A, Grimberg KB, Bott LC, Salomons FA, Dantuma NP, Young P (2009) A conserved unfoldase activity for the p97 AAA‐ATPase in proteasomal degradation. J Mol Biol 394: 732–746 [DOI] [PubMed] [Google Scholar]
- Bodnar N, Rapoport T (2017) Toward an understanding of the Cdc48/p97 ATPase. F1000Res 6: 1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boom J, Meyer H (2018) VCP/p97‐mediated unfolding as a principle in protein homeostasis and signaling. Mol Cell 69: 182–194 [DOI] [PubMed] [Google Scholar]
- Borgermann N, Ackermann L, Schwertman P, Hendriks IA, Thijssen K, Liu JC, Lans H, Nielsen ML, Mailand N (2019) SUMOylation promotes protective responses to DNA‐protein crosslinks. EMBO J 38: e101496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaneda CA, Dixon EK, Walker O, Chaturvedi A, Nakasone MA, Curtis JE, Reed MR, Krueger S, Cropp TA, Fushman D (2016) Linkage via K27 bestows ubiquitin chains with unique properties among polyubiquitins. Structure 24: 423–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ, Sun LJ (2009) Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell 33: 275–286 [DOI] [PubMed] [Google Scholar]
- Chou TF, Deshaies RJ (2011) Quantitative cell‐based protein degradation assays to identify and classify drugs that target the ubiquitin‐proteasome system. J Biol Chem 286: 16546–16554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clague MJ, Urbe S, Komander D (2019) Breaking the chains: deubiquitylating enzyme specificity begets function. Nat Rev Mol Cell Biol 20: 338–352 [DOI] [PubMed] [Google Scholar]
- Collins GA, Goldberg AL (2017) The Logic of the 26S proteasome. Cell 169: 792–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M (2014) Accurate proteome‐wide label‐free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13: 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10: 1794–1805 [DOI] [PubMed] [Google Scholar]
- Dammer EB, Na CH, Xu P, Seyfried NT, Duong DM, Cheng D, Gearing M, Rees H, Lah JJ, Levey AI et al (2011) Polyubiquitin linkage profiles in three models of proteolytic stress suggest the etiology of Alzheimer disease. J Biol Chem 286: 10457–10465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantuma NP, Lindsten K, Glas R, Jellne M, Masucci MG (2000) Short‐lived green fluorescent proteins for quantifying ubiquitin/proteasome‐dependent proteolysis in living cells. Nat Biotechnol 18: 538–543 [DOI] [PubMed] [Google Scholar]
- Esadze A, Li DW, Wang T, Bruschweiler R, Iwahara J (2011) Dynamics of lysine side‐chain amino groups in a protein studied by heteronuclear 1H–15N NMR spectroscopy. J Am Chem Soc 133: 909–919 [DOI] [PubMed] [Google Scholar]
- Gatti M, Pinato S, Maiolica A, Rocchio F, Prato MG, Aebersold R, Penengo L (2015) RNF168 promotes noncanonical K27 ubiquitination to signal DNA damage. Cell Rep 10: 226–238 [DOI] [PubMed] [Google Scholar]
- Godderz D, Heinen C, Marchese FP, Kurz T, Acs K, Dantuma NP (2015) Cdc48‐independent proteasomal degradation coincides with a reduced need for ubiquitylation. Sci Rep 5: 7615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberger JB, Voigt A, Borisova ME, Petrosino G, Ruf S, Wagner SA, Beli P (2018) Proteomic profiling of VCP substrates links VCP to K6‐linked ubiquitylation and c‐Myc function. EMBO Rep 19: e44754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Huizen M, Kikkert M (2019) The role of atypical ubiquitin chains in the regulation of the antiviral innate immune response. Front Cell Dev Biol 7: 392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huryn DM, Kornfilt DJP, Wipf P (2020) p97: an emerging target for cancer, neurodegenerative diseases, and viral infections. J Med Chem 63: 1892–1907 [DOI] [PubMed] [Google Scholar]
- Kaiho‐Soma AI, Akizuki Y, Igarashi K, Endo A, Shoda T, Kawase Y, Demizu Y, Naito M, Saeki Y, Tanaka K et al (2021) TRIP12 promotes small‐molecule‐induced degradation through K29/K48‐branched ubiquitin chains. Mol Cell 81: 1411–1424.e7 [DOI] [PubMed] [Google Scholar]
- Kim W, Bennett E, Huttlin E, Guo A, Li J, Possemato A, Sowa M, Rad R, Rush J, Comb M et al (2011) Systematic and quantitative assessment of the ubiquitin‐modified proteome. Mol Cell 44: 325–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudriaeva AA, Livneh I, Baranov MS, Ziganshin RH, Tupikin AE, Zaitseva SO, Kabilov MR, Ciechanover A, Belogurov AA Jr (2021) In‐depth characterization of ubiquitin turnover in mammalian cells by fluorescence tracking. Cell Chem Biol 28: 1192–1205.e9 [DOI] [PubMed] [Google Scholar]
- Le Moigne R, Aftab BT, Djakovic S, Dhimolea E, Valle E, Murnane M, King EM, Soriano F, Menon M‐K, Wu ZY et al (2017) The p97 inhibitor CB‐5083 is a unique disrupter of protein homeostasis in models of multiple myeloma. Mol Cancer Ther 16: 2375–2386 [DOI] [PubMed] [Google Scholar]
- Lee Y‐R, Chen M, Lee JD, Zhang J, Lin S‐Y, Fu T‐M, Chen H, Ishikawa T, Chiang S‐Y, Katon J et al (2019) Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC‐WWP1 inhibitory pathway. Science 364: aau0159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Liu W, Ye Y, Li W (2017) Ufd2p synthesizes branched ubiquitin chains to promote the degradation of substrates modified with atypical chains. Nat Commun 8: 14274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Han C, Xie B, Wu Y, Liu S, Chen K, Xia M, Zhang Y, Song L, Li Z et al (2014) Rhbdd3 controls autoimmunity by suppressing the production of IL‐6 by dendritic cells via K27‐linked ubiquitination of the regulator NEMO. Nat Immunol 15: 612–622 [DOI] [PubMed] [Google Scholar]
- Magnaghi P, D'Alessio R, Valsasina B, Avanzi N, Rizzi S, Asa D, Gasparri F, Cozzi L, Cucchi U, Orrenius C et al (2013) Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat Chem Biol 9: 548–556 [DOI] [PubMed] [Google Scholar]
- Matsumoto ML, Wickliffe KE, Dong KC, Yu C, Bosanac I, Bustos D, Phu L, Kirkpatrick DS, Hymowitz SG, Rape M et al (2010) K11‐linked polyubiquitination in cell cycle control revealed by a K11 linkage‐specific antibody. Mol Cell 39: 477–484 [DOI] [PubMed] [Google Scholar]
- Mosbech A, Lukas C, Bekker‐Jensen S, Mailand N (2013) The deubiquitylating enzyme USP44 counteracts the DNA double‐strand break response mediated by the RNF8 and RNF168 ubiquitin ligases. J Biol Chem 288: 16579–16587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh E, Akopian D, Rape M (2018) Principles of ubiquitin‐dependent signaling. Annu Rev Cell Dev Biol 34: 137–162 [DOI] [PubMed] [Google Scholar]
- Pan M, Gao S, Zheng Y, Tan X, Lan H, Tan X, Sun D, Lu L, Wang T, Zheng Q et al (2016) Quasi‐racemic X‐ray structures of K27‐linked ubiquitin chains prepared by total chemical synthesis. J Am Chem Soc 138: 7429–7435 [DOI] [PubMed] [Google Scholar]
- Pan M, Zheng Q, Ding S, Zhang L, Qu Q, Wang T, Hong D, Ren Y, Liang L, Chen C et al (2019) Chemical protein synthesis enabled mechanistic studies on the molecular recognition of K27‐linked ubiquitin chains. Angew Chem Int Ed Engl 58: 2627–2631 [DOI] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47: D442–D450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puumalainen MR, Lessel D, Ruthemann P, Kaczmarek N, Bachmann K, Ramadan K, Naegeli H (2014) Chromatin retention of DNA damage sensors DDB2 and XPC through loss of p97 segregase causes genotoxicity. Nat Commun 5: 3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscoe BP, Thayer KM, Zeldovich KB, Fushman D, Bolon DN (2013) Analyses of the effects of all ubiquitin point mutants on yeast growth rate. J Mol Biol 425: 1363–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AN, Oehler J, Torrecilla I, Kilgas S, Li S, Vaz B, Guerillon C, Fielden J, Hernandez‐Carralero E, Cabrera E et al (2019) The p97‐Ataxin 3 complex regulates homeostasis of the DNA damage response E3 ubiquitin ligase RNF8. EMBO J 38: e102361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swatek KN, Komander D (2016) Ubiquitin modifications. Cell Res 26: 399–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swatek KN, Usher JL, Kueck AF, Gladkova C, Mevissen TET, Pruneda JN, Skern T, Komander D (2019) Insights into ubiquitin chain architecture using Ub‐clipping. Nature 572: 533–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker‐Jensen S, Mailand N, Bartek J, Lukas J (2013) ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155: 1088–1103 [DOI] [PubMed] [Google Scholar]
- Twomey EC, Ji Z, Wales TE, Bodnar NO, Ficarro SB, Marto JA, Engen JR, Rapoport TA (2019) Substrate processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science 365: eaax1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740 [DOI] [PubMed] [Google Scholar]
- van Tilburg GBA, Murachelli AG, Fish A, van der Heden van Noort GJ, Ovaa H, Sixma TK (2020) K27‐linked diubiquitin inhibits UCHL3 via an unusual kinetic trap. Cell Chem Biol 28: 191–201.e8 [DOI] [PubMed] [Google Scholar]
- Xu M, Skaug B, Zeng W, Chen ZJ (2009a) A ubiquitin replacement strategy in human cells reveals distinct mechanisms of IKK activation by TNFalpha and IL‐1beta. Mol Cell 36: 302–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, Rush J, Hochstrasser M, Finley D, Peng J (2009b) Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137: 133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Q, Han T, Fang B, Zhang G, Zhang C, Roberts ER, Izumi V, Zheng M, Jiang S, Yin X et al (2019) K27‐linked ubiquitination of BRAF by ITCH engages cytokine response to maintain MEK‐ERK signaling. Nat Commun 10: 1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Zheng Q, Erramilli SK, Pan M, Park S, Xie Y, Li J, Fei J, Kossiakoff AA, Liu L et al (2021) K29‐linked ubiquitin signaling regulates proteotoxic stress response and cell cycle. Nat Chem Biol 17: 896–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Smits AH, van Tilburg GB, Jansen PW, Makowski MM, Ovaa H, Vermeulen M (2017) An interaction landscape of ubiquitin signaling. Mol Cell 65: 941–955.e8 [DOI] [PubMed] [Google Scholar]