

Abstract
Pancreatic ductal adenocarcinoma (PDA) tumor cells are deprived of oxygen and nutrients and therefore must adapt their metabolism to ensure proliferation. In some physiological states, cells rely on ketone bodies to satisfy their metabolic needs, especially during nutrient stress. Here, we show that PDA cells can activate ketone body metabolism and that β‐hydroxybutyrate (βOHB) is an alternative cell‐intrinsic or systemic fuel that can promote PDA growth and progression. PDA cells activate enzymes required for ketogenesis, utilizing various nutrients as carbon sources for ketone body formation. By assessing metabolic gene expression from spontaneously arising PDA tumors in mice, we find HMG‐CoA lyase (HMGCL), involved in ketogenesis, to be among the most deregulated metabolic enzymes in PDA compared to normal pancreas. In vitro depletion of HMGCL impedes migration, tumor cell invasiveness, and anchorage‐independent tumor sphere compaction. Moreover, disrupting HMGCL drastically decreases PDA tumor growth in vivo, while βOHB stimulates metastatic dissemination to the liver. These findings suggest that βOHB increases PDA aggressiveness and identify HMGCL and ketogenesis as metabolic targets for limiting PDA progression.
Keywords: HMGCL, ketone bodies, metastasis, pancreatic cancer, β‐hydroxybutyrate
Subject Categories: Cancer, Immunology, Metabolism
Systemic and cell‐intrinsic ketone body metabolism supports pancreatic cancer metastasis.

Introduction
Among cancers, pancreatic ductal adenocarcinoma (PDA) is particularly deadly as almost 90% of PDA patients die from their disease in 5 years. PDA is predicted to become the third most common cause of cancer‐related death in the European Union (Ferlay et al, 2016), a ranking already reached in the USA (Siegel et al, 2019). Among PDA patients, 80–85% are not eligible for a potentially curative surgical resection because of an unresectable tumor or presence of metastases, and chemotherapy remains the only therapeutic option with limited impact on overall survival (Mizrahi et al, 2020; O'Kane et al, 2021). This therapeutic void in part reflects an incomplete understanding of disease pathogenesis that limits opportunities to uncover new therapies. PDA tumors contain a dense extracellular matrix derived from non‐cancerous stromal cells that limits vacularization and leads to poor oxygen and nutrient delivery to tumor cells (Guillaumond et al, 2013). Several studies have described the metabolic plasticity of pancreatic tumor cells and how these cells might adapt to the complex PDA microenvironment. For example, PDA cells can scavenge macromolecules from the microenvironment, including catabolism of the extracellular matrix to obtain amino acids and support tumor growth (Commisso et al, 2013; Kamphorst et al, 2015; Davidson et al, 2017; Olivares et al, 2017). PDA cells are also reliant on glucose, lactate, alanine, glutamine, and/or cysteine to sustain bioenergetics, limit ROS‐induced damage, and fuel anabolism (Ying et al, 2012; Guillaumond et al, 2013; Chakrabarti et al, 2015; Daher et al, 2019; Badgley et al, 2020; Parker et al, 2020). This metabolic flexibility provides PDA tumor cells with the ability to adapt to glucose‐ and glutamine‐deprived conditions and use various amino acids as nitrogen donors for glutamine and nucleotide synthesis (Tsai et al, 2021). Indeed, the characteristic metabolic plasticity of PDA can limit the effectiveness of targeting some metabolic dependencies. For example, pancreatic cancer cells are sensitive to inhibitors of glutamine metabolism in vitro, but adapt to glutaminase inhibitors when grown as PDA tumors in mouse models (Biancur et al, 2017). In line with this, recent screens have compared the metabolic dependencies of PDA tumor cells in vivo and in vitro and thereby highlighted the importance of select metabolic pathways in the PDA pathophysiologic context and that could constitute relevant therapeutic metabolic targets (Biancur et al, 2021; Zhu et al, 2021).
Given the high metabolic demand of PDA tumor cells, numerous studies have explored the use of nutrient‐deprived diets as a potential anti‐carcinogenic approach. The ketogenic diet, which is used to treat epileptic patients (Kessler et al, 2011), is a low carbohydrate, low protein, and high fat diet that induces an increase in circulating ketone bodies (KB) (acetoacetate (AcAc) and β‐hydroxybutyrate (βOHB)) that are produced largely by hepatocytes from fatty acids. The rationale for the use of the KD stems from the high glycolytic activity of many tumor cells that, upon KD exposure, may experience a metabolic crisis from diet‐induced reductions in blood glucose and insulin levels. This diet has been proposed a potential cancer therapy, especially for brain, digestive, or breast cancers (Weber et al, 2020). However, given the metabolic heterogeneity of cancers, some tumor cells express ketolytic enzymes that allow them to degrade KB. This metabolic ability was highlighted in hepatocarcinoma cells that adapt to nutrient deprivation and express the ketolytic enzyme SCOT1 (succinyl‐CoA:3‐oxoacid‐CoA transferase encoded by OXCT (3‐oxoacid‐CoA transferase) 1) to support proliferation (Huang et al, 2016). Moreover, in melanoma cells, upregulation of the ketogenic enzyme HMG‐CoA lyase (HMGCL), which converts 3‐hydroxy‐3‐methylglutaryl‐CoA (HMG‐CoA) to AcAc and acetyl‐CoA, induces activation of the MEK1/ERK1 axis by AcAc and thereby promotes the growth of BRAF‐mutated tumors (Kang et al, 2015). However, the metabolic effects of KB on PDA have been less studies, and understanding KB metabolism in tumors with high metabolic flexibility such as PDA is important to understand the consequences of dietary interventions like the KD which may increase the availability of this nutrient to tumors.
Here, we report that βOHB can contribute to PDA cancer cell metabolism, and that pancreatic tumors express high levels of KB metabolic enzymes. In vitro, we show that PDA cells require HMGCL to migrate, invade, and aggregate into tumor spheres. Finally, using PDA murine models, we provide evidence that HMGCL and KB can be required for PDA tumor growth and metastatic invasion.
Results
βOHB contributes to pancreatic tumor aggressiveness and is metabolized by PDA cells
To explore the role of KB on PDA progression, we examined the consequences of intraperitoneal (IP) injection of βOHB in spontaneous PDA bearing Pdx1‐Cre; Ink4a/Arffl/fl; LSL‐KrasG12D (KIC) mice. KIC mice were daily treated with βOHB (100 mg/kg) or vehicle (NaCl 0.9%), starting at 5 weeks of age, and continued for 3 weeks. We observed that βOHB increased pancreatic tumor weight (Fig 1A) and proliferative index (Fig EV1A) without impacting tumor grade (Fig EV1B). We confirmed the growth promoting effects of βOHB on murine pancreatic tumor cells derived from PDA of KIC mice (namely PK4A cells) (Guillaumond et al, 2013) when cultured as tumor spheroids and organoids, and clinically validated these data using human PDA tumor cell‐derived organoids (Fig 1B and C). Interestingly, βOHB treatment increased staining for monocarboxylate transporters 2 (MCT2) and sodium MCT1 (SMCT1) in βOHB‐treated compared to NaCl‐treated tumors (45.8% vs. 20.9% and 42.6% vs. 15.6% staining area for MCT2 and SMCT1, respectively) (Fig 1D and E). MCT2, as part of the MCT 1–4 family, and SMCT1 are both MCTs known to transport KB (Martin et al, 2006; Perez‐Escuredo et al, 2016; Huang et al, 2017; Li et al, 2021), and together with known high expression of MCT1 and MCT4 in PDA (Guillaumond et al, 2013), these data suggest that PDA cells have the capacity to take up KBs in tumors. Importantly, we observed more metastatic lesions in livers from βOHB‐treated compared to NaCl‐treated PDA‐bearing mice (13 vs. 8 mice presented metastatic lesions) suggesting that βOHB promote liver metastases (Fig 1F). In addition, one of 14 βOHB‐mice presented evidence of spleen metastases. Altogether these data demonstrate that βOHB promote PDA aggressiveness and metastasis formation.
Figure 1. βOHB promotes PDA aggressiveness.

-
ATumor weight in KIC mice treated with βOHB (100 mg/kg/day, i.p.) or 0.9% NaCl (i.p.) (n = 13 mice for NaCl and n = 15 mice for βOHB). Data are expressed as mean ± SEM. Significance was defined by Mann‐Whitney test. *P < 0.05.
-
BKinetic of spheroid area from PK4a cells cultured during 12 days in medium alone (untreated) or with βOHB 1 or 10 mM (n = 4, and 5 respectively). Data are expressed as mean ± SEM. Significance compared to untreated cells was defined by two‐way ANOVA followed by a Dunnett’s multiple comparisons test. **P < 0.01, ****P < 0.0001.
-
CRepresentative images of mouse PDA and human PDA primary cells‐derived organoids after 7 days of culture in medium alone (untreated) or supplemented with 1 or 10 mM of βOHB. Scale bar: 1000 µm.
-
D, EImmunostaining of MCT2 and SMCT1 in tumors of KIC mice treated with βOHB (100 mg/kg/day, i.p.) or 0.9% NaCl (i.p.) (n = 5 mice/group). Areas of MCT2 (upper panel) and SMCT1 (lower panel) stainings (D) are expressed as mean of percentage of total tissue area ± SEM. Significance was defined by Mann–Whitney test. *P < 0.05, **P < 0.01. Representative images of MCT2 and SMCT1 stainings (E) in tumors from KIC mice treated with βOHB or NaCl. Scale bar: 100 µm.
-
FHistological characterization of liver and spleen of KIC mice treated with βOHB (100 mg/kg/day, i.p.) or 0.9% NaCl (i.p.) (n = 14 mice/group). Number of mice displaying metastatic liver or spleen in each experimental group is reported.
-
GSchematic showing isotopomer transition from [U‐13C]βOHB to label TCA‐cycle intermediates, glutamate, and proline. Gray filled circles indicate 13C carbon derived from labeled βOHB. Empty circles illustrate unlabeled 12C‐species.
-
H[U‐13C]βOHB tracing into the TCA intermediate: citrate in poorly differentiated PDA explants from KIC mice (n = 5). Data are expressed as mean ± SEM.
-
ISchematic showing ketone metabolism pathway (left panel). Reversible enzymes involved in the production of acetyl‐CoA, acetoacetyl‐CoA, acetoacetate, βOHB, mT, SCOT1/2, and BDH1/2 are indicated in bold case. Immunoblots of ketone metabolic enzymes (BDH1/2, SCOT1/2, mT) (right panel) in pancreatic tissues from 7‐week‐old KI (n = 3 for BDH1/2, SCOT1/2, and n = 4 for mT) and KIC mice (n = 4).
Source data are available online for this figure.
Figure EV1. βOHB is metabolized by PDA cells to fuel their TCA cycle.

- Ki67 immunostaining in tumors of KIC mice treated with βOHB (100 mg/kg/day, i.p.) or 0.9% NaCl (i.p.) (n = 13 mice for NaCl and n = 15 mice for βOHB). Data are expressed as mean of percentage of total tissue area ± SEM. Significance was defined by Mann‐Whitney test. ***P < 0.001 (left panel). Representative images of Ki67 staining in pancreatic tumors from treated KIC mice. Scale bar: 100 µm (right panel).
- Histological characterization of tumors from KIC mice treated with βOHB or 0.9% NaCl. Number of mice with a glandular + undifferentiated (U) or undifferentiated tumors in each experimental group is reported.
- [U‐13C]βOHB tracing into TCA intermediates: citrate, α‐ketoglutarate (α‐KG), succinate, malate and into glutamate and proline in PK4A cells cultured in indicated glucose concentrations. Data are expressed as mean ± SEM (n = 3 technical replicates). Significance was defined by one‐way ANOVA followed by a Bonferroni’s multiple comparisons test. ns: not significant, ***P < 0.001, and mentioned for 0 mM glucose condition compared to other conditions of culture.
- [U‐13C]βOHB tracing into TCA intermediates: αKG, succinate, malate and into glutamate and proline in poorly differentiated PDA explants from KIC mice (n = 5). Data are expressed as mean ± SEM. Source data for this figure can be found in source data Fig 1H.
- Immunochemistry of the ketone metabolic enzymes, BDH1/2 and SCOT1/2, in pancreatic tumors sections from 9‐week‐old KIC mice. Tumor glands (yellow stars) and poorly differentiated cancer cells disseminated into the stroma (blue crosses) are indicated. Representative images and their relative insets from n = 3 KIC mice are illustrated. Scale bar: 100 µm.
Source data are available online for this figure.
To determine how βOHB carbons contribute to PDA cell metabolism, we next traced the metabolic fate of U‐13C‐labeled βOHB (Fig 1G) in PDA cells when cultured under decreasing glucose concentrations. Interestingly, some carbon from βOHB was incorporated into TCA cycle metabolites (citrate, α‐ketoglutarate (αKG), succinate, malate) as well as glutamate and proline even when high levels of glucose were present (Fig EV1C and Appendix Fig S1A and B). Complete glucose limitation was associated with both increased labeling and decreased levels of these metabolites across several metabolites. These data are consistent with previous work showing that pancreatic cancer can catabolize other fuels for cell survival when glucose is limiting (Gouirand & Vasseur, 2018). We next evaluated the metabolism of βOHB in well‐differentiated PDA from 7‐week‐old KIC mice and in late stage 9‐week‐old KIC mouse‐derived poorly differentiated tumors, which exhibit extensive desmoplasia, by tracing U‐13C‐labeled βOHB in the tumors ex vivo. Importantly, and consistent with in vitro data, we observed labeling from βOHB‐derived carbon in TCA intermediates as well as glutamate and proline in later stage PDA tumors (Figs 1H and EV1D and Appendix Fig S1C and D). Interestingly, we observed the same labeling pattern from βOHB in TCA intermediates, glutamate and proline in well‐differentiated tumors, suggesting that KBs can be metabolized by earlier stage tumors as well (Appendix Fig S1E and F). For terminal oxidation of βOHB, PDA cells rely on the expression of several enzymes that are essential for ketolysis (Fig 1I, left panel). We therefore examined levels of a subset of these enzymes in PDA from KIC mice, specifically BDH1/2 (βOHB dehydrogenase 1/2), SCOT1/2, and mT (mitochondrial thiolase encoded by at least ACAT (acetyl‐CoA acetyltransferase) 1). Except for BDH2, we found robust over‐expression of all enzymes examined in tumors relative to healthy pancreatic tissues (Fig 1I, right panel). Importantly, using histological analysis, we observed that all these KB metabolic enzymes are mainly expressed in tumor gland‐like structures (Fig EV1E). Together, these data show that PDA tumor cells express required ketolytic enzymes to metabolize KB.
PDA can produce KB from various nutrient sources
As PDA cells are able to use glucose, glutamine, and other carbon sources to support the TCA cycle, we next examined the ability of PDA to produce βOHB from glucose, glutamine, and acetate. We found that a minimal amount of labeled carbon from U‐13C‐glucose, ‐glutamine and ‐acetate can be detected in βOHB in ex vivo PDA tumors (Fig 2A–D). In line with this, we found that PDA expresses the ketogenic enzymes HMGCL/L1 and HMG‐CoA synthase 2 (HMGCS2) in the tumor glands (Fig EV2A–D), in addition to the reversible enzymes SCOT1/2 and mT (Fig 1I). To further explore whether PDA could use other amino acids as carbon sources to produce βOHB, we examined expression of genes coding for enzymes and transporters involved in amino acid metabolism that were differentially expressed between pancreatic tumors at early and late stages of development and age‐matched healthy pancreas. Specifically, we established the transcriptomes of pancreas from control mice (Ink4a/Arffl/fl; LSL‐KrasG12D (KI)) and of pancreas from KIC mice at 6 and 9 weeks of age (Guillaumond et al, 2015; Appendix Fig S2A). Using gene set library from the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database, we identified genes related to amino acid metabolism (469 genes, 2.15% among all genes) and by a Gene Set Enrichment Analysis (GSEA) those with altered expression in tumors from 6‐week‐old or 9 week‐old PKI mice (205 genes, 0.94% among all genes; and 120 genes, 0.55% among all genes, respectively) compared to control pancreas from age‐matched KI mice (Fig EV2E). Among the amino acid metabolic genes deregulated in tumors, we identified, using the percentage of leading‐edge genes, the BCAA pathway gene set as among the most significantly deregulated compared to control pancreas with a progressive enrichment for this pathway when the tumor becomes undifferentiated and anaplastic (43.6% vs. 47.3% in PDA from 6‐week‐old vs. 9‐week‐old PKI mice, respectively; Fig 2E and F, Appendix Table S1). We next examined expression profiles of genes coding for metabolic enzymes or transporters of leucine, isoleucine, and valine and expanded our analysis to KB metabolic genes. Interestingly, we observed a large diversity of genes with altered expression that code for enzymes involved in leucine breakdown towards HMG‐CoA (Bcat1/2, Bckdha/b, Mccc1/2, Ivd, Auh) and that catalyze the common last step of leucine degradation and KB production (Hmgcl/l1), for the reversible enzyme mT (Acat1), and for leucine, branched‐chain ketoacids and KB transporters (Slc7a5, Slc16a1, Slc16a7) (Fig EV2F, Appendix Table S2). Analysis of BCAT1 expression (BCAA transaminase, which catalyzes the first step of BCAAs degradation) revealed increased levels of this enzyme in PDA as well as its localization in the tumoral compartment (Fig EV2G) as observed for other KB metabolic enzymes. To assess whether the ketogenic BCAA leucine may be used by PK4A PDA‐derived tumor cells to support βOHB synthesis, the fate of U‐13C‐labeled leucine was assessed in PK4A cells at various glucose concentrations. This analysis revealed that cells take up leucine similarly regardless of media glucose concentration and that some leucine‐derived carbons can be incorporated into citrate, αKG, succinate, malate, pyruvate, glutamate, and proline with increased labeling under glucose restricted conditions (Fig EV2H and Appendix Fig S2B) and that is associated with a drop in levels of these metabolites (Appendix Fig S2C). Interestingly, we found that leucine carbon also could contribute to the βOHB pool in these cells (Fig EV2I and Appendix Fig S2B). We next cultured these cells in conditions where leucine was absent and found that leucine deprivation drastically reduced PK4A cell proliferation. Moreover, addition of βOHB partly rescued proliferation of leucine‐starved cells, suggesting that PDA cells can use KB to support proliferation for at least the short term when extracellular leucine is absent (Fig EV2J).
Figure 2. PDA can produce KB.

-
ASchematic showing isotopomer transition from [U‐13C]Glucose, [U‐13C]Glutamine, [U‐13C]Acetate, or [U‐13C]Leucine to label TCA‐cycle intermediates, glutamate, and proline. Red, violet, gray, and blue filled circles indicate 13C carbon derived from labeled glucose, glutamine, acetate and leucine, respectively. Empty circles denote 12C‐species.
-
B–D[U‐13C]Glucose (n = 3 mice) (B), [U‐13C]Glutamine (n = 5 mice) (C) and [U‐13C]Acetate (n = 6 mice) (D) tracing into βOHB in poorly differentiated PDA explants from KIC mice. Data are expressed as mean ± SEM.
-
E, FPercentages of leading‐edges amino acid genes in 6 weeks (E) or 9 weeks (F) KIC mice as compared to age‐matched KI mice that contribute to the GSEA enrichment. Red spots represent the P‐value of the deregulated amino acid pathways in KIC mice as compared to KI mice. Red line corresponds to a P‐value of 0.05.
Source data are available online for this figure.
Figure EV2. Increased levels of KB and BCAAs metabolic enzymes are observed in PDA.
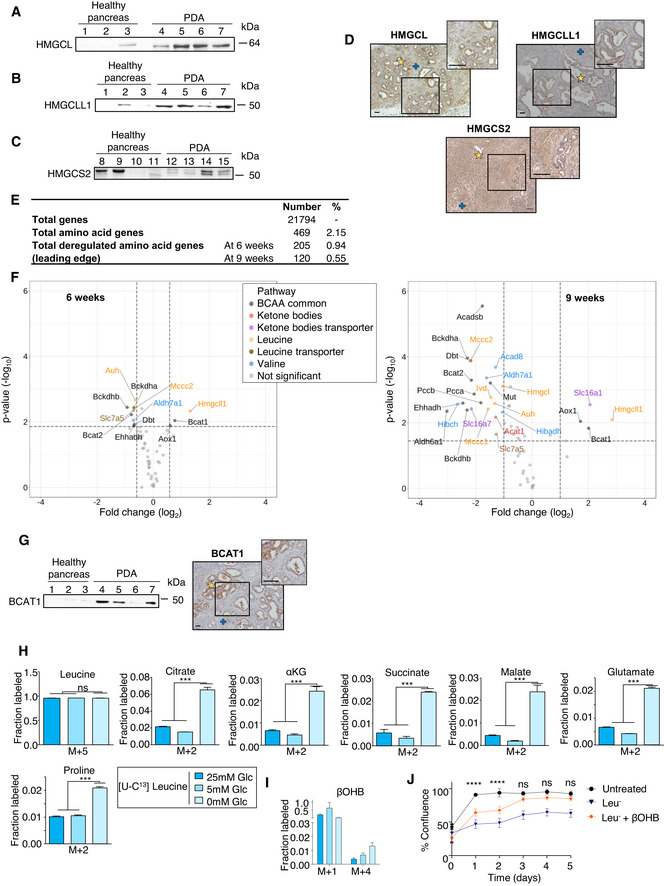
-
A–CImmunoblots of HMGCL (A), HMGCLL1 (B) and HMGCS2 (C) in pancreatic tissues from 7‐week‐old KI (n = 3 for HMGCL, HMGCLL1 and n = 4 for HMGCS2) and KIC mice (n = 4).
-
DImmunochemistry of KB enzymes, HMGCL, HMGCLL1 and HMGCS2 in pancreatic tumors sections from 9‐week‐old KIC mice. Tumor glands (yellow stars) and poorly differentiated cancer cells disseminated into the stroma (blue crosses) are indicated. Representative images and their relative insets from n = 3 KIC mice are illustrated. Scale bar: 100 µm.
-
ENumber and percentage of amino acid genes analyzed by GSEA in KIC tumor bearing mice as well as the number and percentage of amino acid related‐genes involved in significant gene sets (P < 0.05) in PDA from KIC mice at 6 or 9 weeks of age (n = 8 and 3 mice respectively) as compared to control pancreas from age‐matched KI mice (n = 5 and 3 mice respectively).
-
FVolcano plot illustrating BCAA and KB genes differentially and significantly expressed in 9‐week‐old (adjusted P‐value < 0.05; absolute fold change > 2) or 6‐week‐old (adjusted P‐value < 0.05; absolute fold change > 1.5) KIC mice as compared to age‐matched KI mice. The vertical dashed lines represent the fold change cutoff thresholds. The horizontal dashed lines represent the selection cutoff based on the adjusted P‐value cutoff threshold of 0.05.
-
GImmunoblot (left) of BCAT1 in pancreatic tissues from 7‐week‐old KI (n = 3) and KIC mice (n = 4) and immunochemistry (right) of BCAT1 in pancreatic tumors sections from 9‐week‐old KIC mice. Tumor glands (yellow star) and poorly differentiated cancer cells disseminated into the stroma (blue cross) are indicated. Representative images and their relative insets from n = 3 KIC mice are illustrated. Scale bars: 100 µm.
-
H, I[U‐13C]Leucine tracing into TCA intermediates: citrate, αKG, succinate, malate, into glutamate and proline (H) and into βOHB (I) in PK4A cells cultured in indicated glucose concentrations. Data are expressed as mean ± SEM (n = 3 technical replicates). Significance was defined by one‐way ANOVA followed by a Bonferroni’s multiple comparisons test. ns: not significant, ***P < 0.001, and mentioned for 0 mM glucose condition compared to other conditions of culture.
-
JReal time viability of PK4a cells cultured during 5 days in untreated medium or in leucine‐depleted media (Leu‐) alone or supplemented with βOHB (n = 4/condition). Data are expressed as mean ± SEM. Significance was defined by two‐way ANOVA followed by a Dunnett’s multiple comparisons test. ns: not‐significant, ****P < 0.0001, and only mentioned for untreated medium compared to Leu− + βOHB.
Source data are available online for this figure.
Disruption of HMGCL impedes oncogenic properties of pancreatic tumor cells
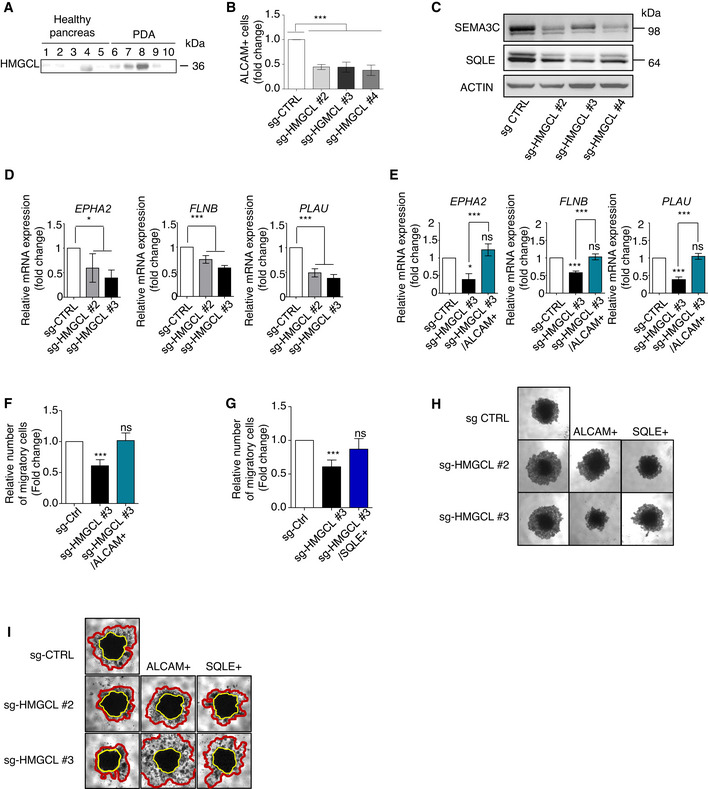
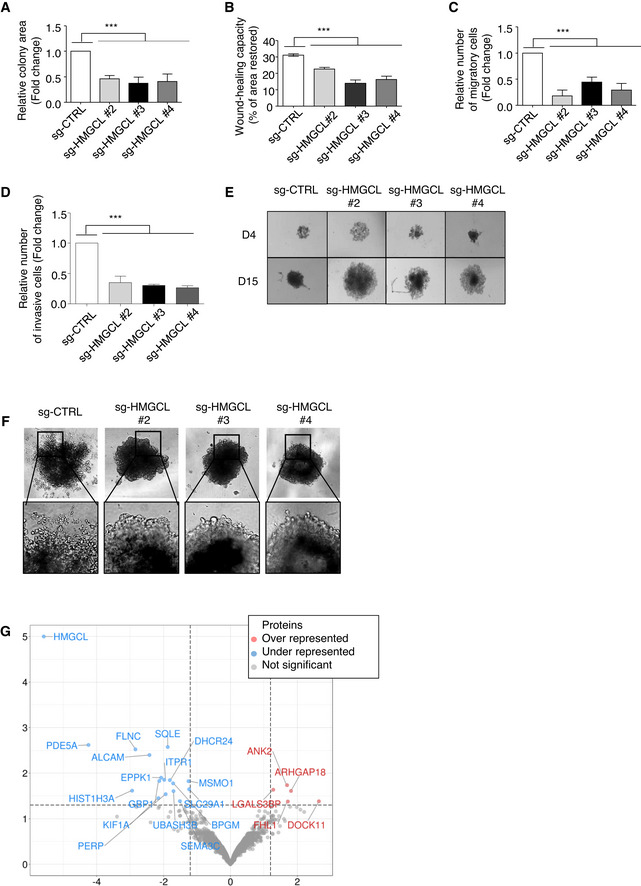
Given that PDA cells actively metabolize KB, and KB can promote tumor growth, we questioned whether inhibiting KB metabolism in PDA cells might affect their oncogenic properties. We specifically investigated the effects of disrupting HMGCL, the key enzyme upstream of βOHB synthesis, which in addition to being highly expressed in murine PDA, was found to be up‐regulated in human PDA (Fig EV3A). The contribution of HMGCL to tumor progression other than PDA has been suggested to be cancer‐type and stage specific (Saraon et al, 2013), (Luo et al, 2017). In PDA, little is known about the role of HMGCL in tumor cell proliferation, migration, and invasion. We generated clonal populations of sg‐HMGCL PANC‐1 cells (sg‐HMGCL) in which HMGCL expression was completely abolished (in clones #2‐6) as compared to sg‐Control PANC‐1 cells (sg‐CTRL) (Appendix Fig S3A). HMGCL disruption did not impact PANC‐1 cell proliferation (P‐value > 0.05; Appendix Fig S3B and C) but prevented the clonogenic capacity of the cells (Fig 3A, Appendix Fig S3D). Using wound healing assays and transwell migration or invasion chambers, we found that HMGCL disruption drastically decreased the migratory potential of PDA cells (Fig 3B and C, Appendix Fig S3E) and also affected their invasive behavior (Fig 3D). We confirmed in MiaPaCa‐2 human PDA cells, which also express significant amounts of HMGCL, that inhibition of HMGCL does not impact proliferation but impedes clonogenic and migratory potential (Appendix Fig S3F–I). In three dimensions, when cultured under anchorage‐independent conditions, although sg‐HMGCL and sg‐CTRL PANC‐1 had the same proliferation index, sg‐HMGCL PANC‐1 spheroids were larger, indicating that HMGCL disruption impeded proper aggregation of PDA cells when cultured as spheroids (Fig 3E, Appendix Fig S3J and K). Interestingly, HMGCL silencing also abolished the invasive abilities of PANC‐1 spheroids (Fig 3F). We further compared the proteome of sg‐CTRL and sg‐HMGCL PANC‐1 cells to identify changes in protein expression associated with loss of 3D‐associated oncogenic features following HMGCL disruption. Among downregulated proteins upon HMGCL loss, we identified ALCAM, SEMA3C, FLNC, and GBP1/3 involved in tumor cell migration, invasion, cell‐to‐cell adhesion, and cytoskeleton homeostasis (Bagci et al, 2009; Li et al, 2017; Honkala et al, 2019; Kamil et al, 2019; Ni et al, 2021), and SQLE, MSMO1, and DHCR24 that belong to sterol metabolism (Fig 3G, Appendix Table S3). In line with this proteomic data, we validated decreased levels of ALCAM and several of its targets (namely EPHA2, FLNB, PLAU), and of SEMA3C and SQLE following HMGCL silencing (Fig EV3B–D). Interestingly, ALCAM positively regulates cell adhesion, migration, and invasion of endometrial cancer cells (Devis et al, 2017), and SQLE, a rate‐limiting enzyme in the cholesterol biosynthesis pathway, promotes invasiveness of hepatocellular carcinoma (Sui et al, 2015) and tumor aggressiveness in breast cancer patients (Kim et al, 2021). Indeed, we observed that increased expression of ALCAM and SQLE in sg‐HMGCL PANC‐1 cells (Appendix Fig S3L and M, and Fig EV3E) does not alter cell proliferation (Appendix Fig S3N) but does restore the ability of these cells to migrate (Fig EV3F and G) and to form well‐aggregated spheroids (Fig EV3H) with invasive capacities (Fig EV3I). Collectively, these data show that HMGCL is dispensable for pancreatic cancer cell proliferation but supports their aggressive, invasive phenotype as well as their migratory abilities in vitro.
Figure EV3. HMGCL deficiency limits aggressiveness of pancreatic tumor cells.
-
AImmunoblots of HMGCL in healthy pancreatic tissue (n = 5) and PDA (n = 5) from PDA patients.
-
BQuantification by flow‐cytometry of protein levels of ALCAM in sg‐CTRL and sg‐HMGCL #2, #3 and #4 PANC‐1 spheroids. ALCAM positive cells are expressed as mean fold change relative to sg‐CTRL ± SEM (n = 3 independent experiments). Significance was defined by one‐tailed Student’s t‐test. ***P < 0.001.
-
CImmunoblots for SEMA3C, SQLE and ACTIN proteins in sg‐CTRL and sg‐HMGCL #2, #3 and #4 PANC‐1 spheroids (Photos are representative of n = 3 independent experiments).
-
DRelative mRNA expression of ALCAM target transcripts: EPHA2, FLNB, PLAU in sg‐CTRL and sg‐HMGCL (#2 and #3) PANC‐1 cells. Relative expressions are expressed as mean fold change relative to sg‐CTRL ± SEM (n = 3 independent experiments). Significance was defined by one‐tailed Student’s t test. *P < 0.05, ***P < 0.001.
-
ERelative mRNA expression of ALCAM target transcripts: EPHA2, FLNB, PLAU in sg‐CTRL, sg‐HMGCL (#3 clone) and sg‐HMGCL/ALCAM+ (#3 clone) PANC‐1 cells. Relative expressions are expressed as mean fold change relative to sg‐CTRL ± SEM (n = 3 independent experiments). Significance was defined by one‐way ANOVA followed by a Bonferroni’s multiple comparisons test. *P < 0.05, ***P < 0.001, ns: not significant.
-
F, GQuantification of migratory sg‐CTRL, sg‐HMGCL, and sg‐HMGCL overexpressing ALCAM (F) or SQLE (G) PANC‐1 cells. Migratory cell numbers are expressed as mean fold change relative to sg‐CTRL cells ± SEM (n = 3 independent experiments). Significance was defined by one‐tailed Student’s t test. ***P < 0.001, ns: not significant.
-
HRepresentative images at day 15 of sg‐CTRL and sg‐HMGCL #2 and #3 PANC‐1 spheroids stably over‐expressing ALCAM (ALCAM+) or SQLE (SQLE+) protein. (Photos are representative of n = 3 independent experiments).
-
IRepresentative images of spheroid invasion assays of sg‐CTRL and sg‐HMGCL #2 and #3 PANC‐1 cells stably over‐expressing ALCAM (ALCAM+) or SQLE (SQLE+) protein, after 10 days of culture with Matrigel (n = 3 independent experiments). Compact cores of spheroids are surrounded by yellow lines. Invaded areas are surrounded by red lines.
Source data are available online for this figure.
Figure 3. HMGCL targeting limits oncogenic properties of pancreatic tumor cells.
-
AQuantification of colony forming area of sg‐CTRL and sg‐HMGCL (#2, #3 and #4 clones) PANC‐1 cells. Colony area is expressed as mean fold change relative to sg‐CTRL ± SEM (n = 3 independent experiments). Significance was defined by one‐tailed Student’s t‐test. ***P < 0.001.
-
BQuantification of wound healing capacity of sg‐CTRL and sg‐HMGCL (#2, #3 and #4 clones) PANC‐1 cells. Wound healing capacity is expressed as mean of percentage of restored area at 72 h ± SEM (n = 4 independent experiments). Significance was defined by one‐tailed Student’s t test. ***P < 0.001.
-
C, DQuantification of migratory (C) and invasive (D) sg‐CTRL and sg‐HMGCL (#2, #3 and #4 clones) PANC‐1 cells. Migratory or invasive cell numbers are expressed as mean fold change relative to sg‐CTRL cells ± SEM (n = 3 independent experiments). Significance was defined by one‐tailed Student’s t test. ***P < 0.001.
-
ERepresentative images of spheroid formation from sg‐CTRL and sg‐HMGCL (#2, #3 and #4 clones) PANC‐1 cells after 4 or 15 days of culture. Photos are representative of n = 3 independent experiments.
-
FRepresentative images of spheroid invasion assay from sg‐CTRL and sg‐HMGCL (#2, #3 and #4 clones) PANC‐1 cells after 10 days of culture in presence of Matrigel. Photos are representative of n = 3 independent experiments.
-
GVolcano plot illustrating proteins significantly under‐ or over‐represented in sg‐CTRL and sg‐HMGCL PANC‐1 spheroids (n = 3 independent experiments). Significance was defined by one‐tailed Student’s t‐test. Protein levels with a q‐value < 0.05 (horizontal axis) and a fold change < −1.5 or > +1.5 (vertical axis) are considered as significantly down or up‐regulated in sg‐HMGCL PANC‐1 spheroids.
Source data are available online for this figure.
HMGCL and βOHB contribute to pancreatic tumor aggressiveness
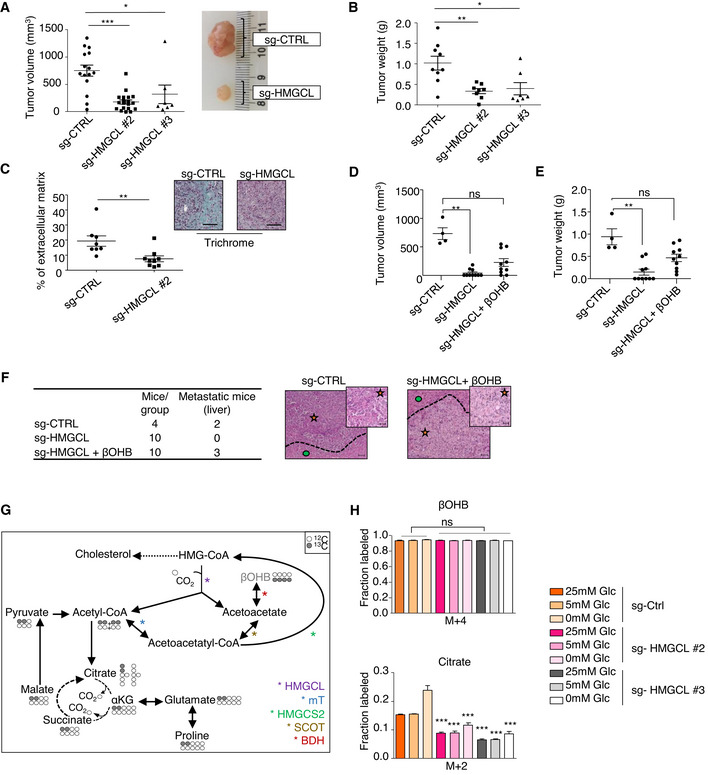
To examined the role of HMGCL on PDA progression, we orthotopically injected sg‐HMGCL PANC‐1 cells or sg‐CTRL cells into nude mice. HMGCL depletion impeded tumor growth as demonstrated by a decrease in size and weight of sg‐HMGCL tumors compared to control tumors (Fig 4A and B). Moreover, in these tumors we observed a reduced stromal content compared to sg‐CTRL tumors (Fig 4C). We next assessed the effect of βOHB administration on tumor growth when given twice a week beginning 7 days after orthotopic injections of sg‐HMGCL PANC‐1 cells. Although providing βOHB did not restore tumor growth to the same levels as sg‐CTRL tumors, βOHB administration significantly increased size and weight of sg‐HMGCL tumors (Fig 4D and E and Appendix Fig S4A) and liver metastasis incidence in sg‐HMGCL tumor‐bearing mice (Fig 4F and Appendix Fig S4B). We next assessed βOHB fate in conditions of HMGCL disruption to examine the consequences of HMGCL deficiency on the ability of PDA cells to metabolize exogenous βOHB. We found that the contribution of βOHB to TCA intermediates, glutamate, and proline was less in these conditions than in control cells (Figs 4G and H, and EV4A and Appendix Fig S4C). While it is unclear how loss of HMGCL affects metabolism of exogenous βOHB, these data may explain why providing βOHB does not fully rescue HMGCL loss in regards with tumor growth. Importantly, there was no change in glucose or glutamine consumption between sg‐CTRL and sg‐HMGCL PANC‐1 cells, and glucose and glutamine consumption is unchanged by βOHB treatment, suggesting that glucose and glutamine uptake is not affected by HMGCL disruption and/or βOHB supplementation in PDA cells (Appendix Fig S4D and E). βOHB is catabolized to acetoacetyl‐CoA which is further converted either by mT to acetyl‐CoA for terminal oxidation into the TCA cycle or is potentially rewired towards HMG‐CoA production by HMGCS2. The decreased catabolism of βOHB to TCA intermediates in sg‐HMGCL cells led us to ask whether βOHB was preferentially converted to HMG‐CoA, which might contribute to cholesterol (Fig 4G). We found that a contribution of βOHB carbon to cholesterol synthesis was decreased in sg‐HMGCL PANC‐1 compared to sg‐CTRL PANC‐1 (Fig EV4B). This inability of HMGCL‐depleted PANC‐1 cells to use βOHB for cholesterol synthesis could be explained by decreased levels of SQLE, MSMO1, and DHCR24, three enzymes of the sterol arm of the cholesterol biosynthesis pathway (Fig 3G). Taken together, these data suggest that HMGCL is important for PDA tumor growth and that exogenous βOHB can partly compensate lack of HMGCL without totally recapitulating growth and metabolism of HMGCL‐expressing PDA cells.
Figure 4. HMGCL and βOHB promote pancreatic tumor aggressiveness.
-
A, BQuantification of volume with representative images (A) and weight (B) of sg‐CTRL (n = 15 for A and n = 9 for B), sg HMGCL #2 (n = 19 for A and n = 8 for B), #3 (n = 7 for A and B) pancreatic tumors. Data are expressed as mean of tumor volume or weight ± SEM. Significance was defined by Mann–Whitney test. *P < 0.05, **P < 0.01, ***P < 0.001.
-
CExtracellular matrix quantification following trichrome staining in sg‐CTRL and sg‐HMGCL #2 pancreatic tumors sections (n = 8 and 9 mice/group respectively, left panel). Data are expressed as mean of percentage of total tissue area ± SEM. Significance was defined by Mann–Whitney test. **P < 0.01. Representative images of trichrome staining in sg‐CTRL and sg‐HMGCL pancreatic tumors. Scale bar: 100 µm (right panel).
-
D, EQuantification of volume (D) and weight (E) of sg‐CTRL pancreatic tumors treated with 0.9% NaCl (i.p.) (n = 4), and pancreatic tumors from two different clones of sg‐HMGCL treated with 0.9% NaCl (i.p.) or βOHB (100 mg/kg/bi‐weekly, i.p.) (n = 10/group). Data are expressed as mean of tumor volume or weight ± SEM. Significance was defined by Mann–Whitney test. ns: not significant, **P < 0.01.
-
FHistological characterization and representative picture of liver from mice orthotopically xenografted with sg‐CTRL or sg‐HMGCL PANC‐1 cells and treated with 0.9% NaCl (i.p.) or βOHB (100 mg/kg/bi‐weekly, i.p.). Number of mice displaying healthy or metastatic liver in each experimental group is reported. Metastatic area (orange star) is separated from liver (green circle) by dotted lines. Scale bar: 100 µm (inset images scale bar: 20 µm).
-
GSchematic showing isotopomer transition from [U‐13C]βOHB to label TCA‐cycle intermediates, glutamate, and proline. Gray filled circles indicate 13C carbon derived from labeled βOHB. Empty circles illustrate unlabeled 12C‐species.
-
H[U‐13C]βOHB tracing into TCA intermediate: citrate in sg‐CTRL and sg‐HMGCL #2 and #3 PANC‐1 cells cultured in indicated glucose concentrations. Data are expressed as mean ± SEM (n = 2 independent experiments). Significance was defined by one‐way ANOVA followed by a Bonferroni’s multiple comparisons test, only significances between sg‐HMGCL #2, #3 PANC‐1 cells and sg‐CTRL PANC‐1 cells under the same culture condition are mentioned. ***P < 0.001.
Source data are available online for this figure.
Figure EV4. βOHB metabolism is affected in PDA cells upon HMGCL deficiency.
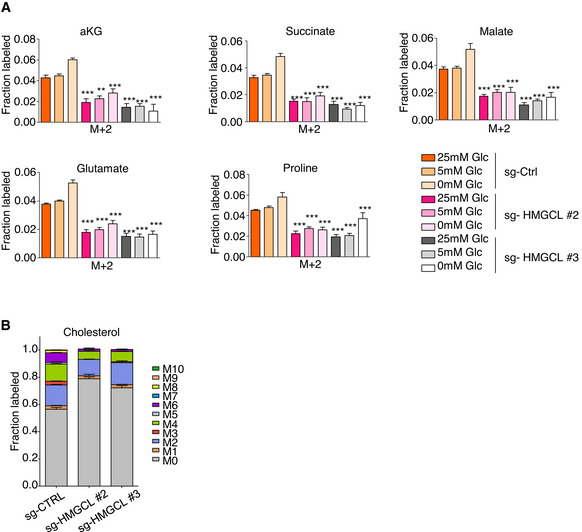
- [U‐13C]βOHB tracing into TCA intermediates: αKG, succinate, malate and into glutamate and proline in sg‐CTRL and sg‐HMGCL #2 and #3 PANC‐1 cells cultured in indicated glucose concentrations. Significance was defined by one‐way ANOVA followed by a Bonferroni’s multiple comparisons test, only significances between sg‐HMGCL #2, #3 PANC‐1 cells and sg‐CTRL PANC‐1 cells under the same culture condition are mentioned (n = 2 independent experiments). **P < 0.01, ***P < 0.001. Source data for this figure can be found in source data Fig 4H.
- [U‐13C]βOHB tracing into cholesterol in sg‐CTRL and sg‐HMGCL #2 and #3 PANC‐1 cells when cultured in 5 mM glucose medium (n = 3 technical replicates).
Data information: in A, B: data are expressed as mean ± SEM.
Source data are available online for this figure.
HMGCL and βOHB favor metastatic dissemination of PDA cells
To assess whether HMGCL supports the metastatic potential of cancer cells in vivo, we performed an in ovo assay by inoculating sg‐CTRL and sg‐HMGCL PANC‐1 cells onto chick‐embryo chorioallantoic membranes (CAMs). CAM is a supportive environment to evaluate the PDA metastatic potential (Hagedorn et al, 2005) (Dumartin et al, 2010) (Mikaelian et al, 2013). After 10 days of PANC‐1 cell engraftment on the upper CAM, we monitored metastasis formation by evaluating the presence of tumor cells at the lower CAM that had migrated through the vicinity of preexisting vessels and disseminated to distant sites. We observed that in the absence of HMGCL the metastatic potential of PANC‐1 cells was reduced as compared to HMGCL expressing PANC‐1 cells (Fig 5A). In PDA patients, primary pancreatic tumor cells preferentially disseminate to the liver (76–94%), compared to peritoneum (41–56%), abdominal lymph nodes (41%), and lungs (45–48%) (Makohon‐Moore et al, 2017). To determine whether ketone metabolism was associated with metastasis in PDA patients, we examined expression of 15 KB metabolism genes in 728 primary pancreatic carcinoma samples and 76 metastatic samples (mainly liver and peritoneal sites) extracted from 15 publicly available whole‐genome mRNA expression data sets (Appendix Table S4). We defined a global measurement of the 15 KB metabolism genes using a metagene approach in which the KB‐related metagene was computed as the first component of a principal component analysis (PCA). The comparison between metastatic vs primary tumor samples showed higher expression of the KB metagene score in metastases than in primary tumors (Fig 5B, Appendix Table S5). Interestingly, KB genes expression is robustly increased in liver metastases compared to PDA as illustrated for Hmgcl, Hmgcs2, Bdh1, and Slc16a1 (Fig EV5). Moreover, immunostaining of KB transporters in liver metastasis of PDA‐bearing KIC mice revealed the presence of SMCT1 and MCT2 in tumor cells within liver metastasis (Fig 5C, and Appendix Fig S5A). These data, combined with the increased liver metastatic index of βOHB‐treated PDA‐bearing mice (Figs 1F and 4F), suggested that PDA cells, when established in liver, have the ability to use KB. To test this hypothesis, we developed a mouse model of induced‐liver metastasis (I‐LM) and implanted PK4A cells into the liver of syngenic healthy KI mice. The same number of tumor cells was implanted into two separate liver lobes for all mice. Three days after cell engraftment, mice were treated with βOHB for 10 days and then sacrificed for examination of liver metastasis. Although macroscopic liver metastases were observed in both vehicle control‐ and βOHB‐treated groups (Appendix Fig S5B), histological analysis of livers revealed more numerous tumors in βOHB‐treated as compared to NaCl‐treated mice (Fig 5D). PK4A cells implanted in livers of vehicle‐treated mice formed fewer tumor foci that were localized at the injection site with a negligible number of small disseminated lesions, whereas βOHB treatment promoted colonization of liver lobes by PK4A cells and the appearance of high numbers of small‐ and medium‐sized tumors (Fig 5E). Moreover, the mice injected with βOHB developed a higher percentage of well‐differentiated tumors in liver than that of the mice in the control group (Fig 5F). When I‐LM mouse models were submitted to leucine‐enriched diet, the examination of liver metastases 10 days after grafts revealed that leucine‐enriched diet also promoted metastatic dissemination with an increased number of small and medium metastasis (Appendix Fig S5C–E). Consistent with these in vivo data, treatment of mouse or human liver metastatic tumor cell derived‐organoids with βOHB promotes their growth (Fig 5G). Collectively, these data support a role for KB as a promoter of PDA metastasis.
Figure 5. HMGCL and βOHB favor metastatic dissemination of PDA cells.

-
AHuman ALU sequences quantification in lower chorioallantoic membrane (CAM) of chicken embryo injected with sg‐CTRL or sg‐HMGCL #2 PANC‐1 cells. Relative amount of metastasis is expressed as mean fold change relative to sg‐CTRL ± SEM (n = 8/group). Significance was defined by one‐tailed Student’s t‐test. *P < 0.05.
-
BBox‐and‐whisker plots of ketone bodies (KB) metagene score defined as first component of PCA of genes in primary tumors (n = 728), in all metastases (n = 76), and in liver metastases specifically (n = 35). Box‐and‐whisker plot were defined with default parameters by median value (central band at the 50th percentile), interquartile ranges (IQR, box limited by 25th and 75th percentile) and whisker boundaries defined at 1.5× IQR. Significance was defined by Student’s t‐test.
-
CRepresentative immunostaining of SMCT1 and MCT2 in metastatic livers of KIC mice (n = 3 mice). Metastatic area (orange star) is separated from liver (green circle) by dotted lines. Scale bar: 100 µm.
-
D–FEffect of βOHB treatment on liver metastatic incidence, size and status. Representative HPS staining of liver lobes from metastatic mice treated with βOHB (100 mg/kg/day, i.p.) or 0.9% NaCl (i.p.) (n = 8 mice/group). Metastatic areas are separated from liver by yellow dotted lines. Scale bar: 1 mm and 500 µm for insets (D). Number and size of metastasis per lobe and classified in small, medium, and large size (E). Pathological status of metastasis from mice treated with βOHB or NaCl (n = 13 or 5 lobes/group, respectively). Data are expressed as percentage of total metastasis in all liver lobes presenting metastasis (F).
-
GRepresentative images of mouse and human liver metastatic cells‐derived organoids after 7 days of culture in medium alone (untreated) or supplemented with 1 or 10 mM of βOHB. Scale bar: 1,000 µm.
-
HWorking model: HMGCL and βOHB promote pancreatic tumor expansion and dissemination. PDA tumor cells expressed HMGCL (left panel). In this setting, PDA can use βOHB from direct source as tumor surrounding βOHB, mimicked in our study by direct injection of βOHB, and indirect source from degradation of various nutrients. βOHB and HMGCL contribute to tumor growth and metastases formation. In metastases, ketone bodies genes are upregulated. When HMGCL is knock‐out (right panel), contribution of βOHB in TCA cycle is reduced and tumor growth as well as dissemination capacity of PDA tumor cells are strongly disturbed.
Source data are available online for this figure.
Figure EV5. KB metabolism genes are highly expressed in metastasis of PDA patients.
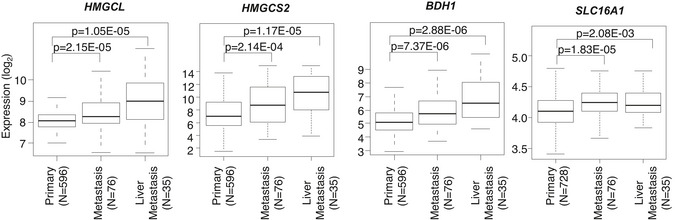
Box‐and‐whisker plots representing differential expression of four KB genes in human primary tumors, all metastases and in liver metastases specifically. Box‐and‐whisker plot were defined with default parameters by median value (central band at the 50th percentile), interquartile ranges (IQR, box limited by 25th and 75th percentile) and whisker boundaries defined at 1.5× IQR. Significance was defined by Student’s t‐test.
Source data are available online for this figure.
In summary, our data demonstrate that βOHB strongly participates in the acquisition of the metastatic phenotype of PDA and that PDA cells can activate KB metabolism. In this context, we show that HMGCL, upstream of βOHB production, supports tumor expansion and PDA cells dissemination (Fig 5H).
Discussion
Pancreatic ductal adenocarcinoma evolution is characterized by a silent, asymptomatic progression phase, followed by a tumor burden that almost inevitably gives rise to a symptomatic aggressive metastatic cancer. Moreover, PDA progression is associated with a massive metabolic reprogramming which supports tumor expansion (Gouirand et al, 2018; Vaziri‐Gohar et al, 2018). Indeed, one of the hallmarks of PDA is its metabolic plasticity that makes this tumor able to grow despite a dense microenvironment as well as an inefficient vascular network which encloses and deprives tumor cells of nutrients and oxygen. This led us to investigate if PDA’s ability to activate KB metabolism represents a new facet of PDA metabolic flexibility, as KB represent an alternative fuel in mammals that is particularly prominent in conditions of nutrient stress. Interestingly, concomitant ketogenesis versus ketolysis has been described in the context of breast cancer in cancer‐associated‐fibroblasts versus epithelial tumor cells respectively (Martinez‐Outschoorn et al, 2012). Ketone oxidation also has been reported in several solid cancers such as brain and liver tumors in which BDH and SCOT enzymes are upregulated (De Feyter et al, 2016) (Huang et al, 2016; Zhang & Xie, 2017). Hence, it appears that tumor cells derived from multiple organs are able to activate KB metabolism, which may be relevant to whether the use of the KD is effective as an anti‐cancer therapy. In the context of PDA, while the KD has been shown to reduce pancreatic tumor growth in vivo in some models (Shukla et al, 2014), other studies revealed no effect of KD alone on PDA tumor growth or animal survival (Hopkins et al, 2018; Lien et al, 2021). Moreover, the limited number of PDA patients studied with this dietary intervention and the lack of long‐term follow‐up make the role of the KD in treating PDA patients unclear (Zhang et al, 2021).
In order to directly evaluate the impact of KB on PDA evolution, we examined the effect of βOHB on PDA and found evidence for a direct benefit of ketones on PDA expansion. These findings are consistent with data from a mouse model of spontaneous mammary tumors (Rodrigues et al, 2017). In addition, using in vitro tracing of βOHB fate, we found that βOHB can be metabolized by PDA cells, particularly when glucose is limiting and that this ketolysis is preserved even in PDA tissue slices containing multiple cell types. Accordingly, expression of the ketolytic enzymes BDH, SCOT, and mTH in PDA enables SCOT‐dependent oxidation of KB, as for liver, breast, prostate, and colon tumors (Martinez‐Outschoorn et al, 2012; Saraon et al, 2013; Huang et al, 2016; Lee et al, 2016), and is in accordance with its active consumption of KB.
The reversible activity of mT and SCOT as well as their high levels in PDA led us to hypothesize that PDA cells have also the ability to produce their own pool of KB from acetyl‐CoA (Fig 4G). A second mechanism of KB production is the HMGCS2‐dependent ketogenesis, which is favored in ketogenic organs, such as liver, and has been suggested to occur in other tissues and cells including tumor cells (Grabacka et al, 2016). Interestingly, we found that PDA cells express HMGCS2 and HMGCL, which are both required for de novo ketogenesis, suggesting that both HMGCS2‐ and SCOT‐dependent ketogenesis may be active in PDA. Ex vivo, we showed that some nutrients can contribute carbon to KB production, even though labeling of KB is relatively low from any carbon source. Hence, the dependence of PDA cells on HMGCL suggests that production of KB plays some role. Importantly, loss of HMGCL drastically impacts on PDA growth and aggressiveness and reduces βOHB metabolism via a mechanism still to be determined, which probably explains the partial rescue of HMGCL loss by exogenous KB. This may suggest that a link between ketogenesis and ketolysis that needs to be further explored exists in these cells and underlies how ketone metabolism affects PDA progression. Another HMGCS2‐independent ketogenic pathway relies on catabolism of BCAAs. Previous studies have demonstrated that BCAAs are increased in the circulation early during PDA development, with the BCAAs being derived from the increased breakdown of tissue protein (Mayers et al, 2014). Although the uptake of BCAAs appears to be low in PDA tumors and the tumors do not require BCAT to grow (Mayers et al, 2016), it has been reported that leucine supplementation can still promote pancreatic tumor burden, suggesting that leucine metabolism may still contribute in some way to PDA growth (Liu et al, 2014). In mammals under nutrient starvation, catabolism of leucine towards KB is rapid and accounts for approximately 4% of KB supply (Thomas et al, 1982). By transcriptomic analysis of PDA, we revealed that enzymes of BCAAs catabolism together with KB metabolic enzymes are commonly up‐regulated during tumor progression and tracing of leucine revealed that carbon from this amino acid can also contribute to the ΒOHB pool. Hence, a contribution of leucine to de novo ketogenesis, either in cancer cells or in tissues outside of the cancer, may be one way that this amino acid can promote PDA growth.
A role for HMGCL in cancer is controversial, with both pro‐ and anti‐tumoral effect of this enzyme reported in various cancers (Saraon et al, 2013; Kang et al, 2015; Luo et al, 2017). We find that HMGCL can promote PDA progression and whether HMGCL represents a good cancer target will depend on the cancer context. Ketogenesis as a metabolic process is crucial in specific physiological contexts associated with fasting and starvation periods. Indeed, loss of function of HMGCL is associated with hypoglycemia, acidosis, hyperammonemia, and severe neurological symptoms (Grunert et al, 2017). However, the pathogenesis associated with the metabolic crisis caused by congenital HMGCL deficiency is not fully understood, and the enzyme may not be required in all cases in otherwise normal adults. As PDA cells are submitted to nutritional stress, one might suspect that HMGCL plays a more important role in this context. In addition, the proteomic profile of HMGCL depleted PDA cells revealed alteration of several, so far unsuspected, proteins involved in sterols synthesis such as SQLE, MSOM1, and DHCR24. Hence, HMGCL depletion might slow cholesterol biosynthesis, thereby increasing levels of squalene, a toxic product when accumulated in cells with low levels of SQLE. Such accumulation might provoke alterations in oncogenic functions of PDA cells, independently of sterol synthesis inhibition, as previously shown for lung neuroendocrine tumors (Mahoney et al, 2019). In light of these results, it appears important to further explore the molecular mechanisms regulated by HMGCL that lead to these unsuspected protein alterations in order to validate HMGCL as a druggable candidate to target PDA aggressiveness.
Materials and Methods
Reagents and Tools table
| Reagents or Tools | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Rabbit Polyclonal Anti‐ACAT1 (WB) | Sigma | Cat# AV54278, RRID: AB_1844483 |
| Mouse Monoclonal Anti‐ALCAM/CD166 (WB) | Santa Cruz Biotechnolgy | Cat# sc‐74558, RRID : AB_2289495 |
| Mouse Monoclonal Anti‐ALCAM/CD166 (FACS) | BD Biosciences | Cat# 559260, RRID:AB_397209 |
| Mouse Monoclonal Anti‐β‐Actin | Sigma‐Aldrich | Cat# A5316, RRID:AB_476743 |
| Rabbit Polyclonal Anti‐BCAT1 | Aviva Systems Biology | Cat# ARP46132_P050, RRID:AB_10640125 |
| Rabbit Polyclonal Anti‐BDH1 | Novus Biologicals | Cat# NBP1‐88673, RRID:AB_11009202 |
| Rabbit Polyclonal Anti‐BDH2 | Sigma | Cat# HPA036028, RRID:AB_10670674 |
| Rabbit Polyclonal Anti‐Hmgcl | Abcam | Cat# ab197022, RRID:AB_2797140 |
| Rabbit Polyclonal Anti‐Hmgcll1 | Abcam | Cat# ab101576, RRID:AB_10712203 |
| Rabbit monoclonal Anti‐HMGCS2 (IHC) | Abcam | Cat# ab137043, RRID: AB_2749817 |
| Rabbit Polyclonal Anti‐HMGCS2 (WB) | Sigma | Cat# AV41562, RRID: AB_1850800 |
| Rat Monoclonal Anti‐Ki67 | Biolegend | Cat# 652402, RRID:AB_11204254 |
| Rabbit Polyclonal Anti‐SLC16A7/MCT2 | Bioss | Cat# bs‐3995R, RRID:AB_10855300 |
| Rabbit Polyclonal Anti‐OXCT1 (SCOT1) | Novus Biologicals | Cat# NBP1‐82462, RRID:AB_11027140 |
| Rabbit Polyclonal Anti‐OXCT2 (SCOT2) | Thermo Fisher Scientific | Cat# PA5‐49312, RRID:AB_2634766 |
| Rat Polyclonal Human/Mouse anti‐Semaphorin 3C | R&D Systems | Cat# MAB1728, RRID:AB_2301533 |
| Rabbit Polyclonal Anti‐SQLE | Proteintech | Cat# 12544‐1‐AP, RRID:AB_2195888 |
| Rabbit Polyclonal Anti‐SLC5A8/SMCT1 | Bioss | Cat# bs‐6106R, RRID:AB_11108966 |
| Goat Polyclonal Anti‐Rat IgG ‐ HRP | Santa Cruz Biotechnolgy | Cat# sc‐2006, RRID:AB_1125219 |
| Goat Polyclonal Anti‐Mouse IgG Human ads ‐ HRP | Southern Biotechnolgy | Cat# 1030‐05, RRID:AB_2619742 |
| Goat Polyclonal Anti‐Rabbit IgG – HRP | Southern Biotechnolgy | Cat# 4030‐05, RRID:AB_2687483 |
| Rabbit Polyclonal Anti‐Goat IgG (H + L) – HRP | Southern Biotechnolgy | Cat# 6160‐05, RRID:AB_2796231 |
| Goat Polyclonal Anti‐Mouse IgG (H + L) ‐ Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A‐11029, RRID:AB_138404 |
| Biological Samples | ||
| Healthy pancreas and PDA protein extract from PDA patients | Leca et al (2016) | https://doi.org/10.1172/JCI87734 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lipofectamine 3000 | Thermo Fisher Scientific | Cat# L3000015 |
| G418 | Thermo Fisher Scientific | Cat# 10131027 |
| Mitomycin C | Sigma Aldrich | Cat# 10107409001 |
| Collagenase type V | Sigma Aldrich | Cat# C9263 |
| Critical Commercial Assays | ||
| CellTrace™ CFSE Cell Proliferation Kit | Thermo Fisher Scientific | Cat# C34554 |
| VECTASTAIN ABC kit | Vector | Cat# PK‐6100 |
| Liquid DAB and substrate chromogen system | DAKO | Cat# K3468 |
| Corning BioCoat™ Matrigel Invasion Chamber | Corning | Cat# 11553570 |
| Trichrome Masson kit | RAL DIAGNOSTICS | Cat# 361350‐0000 |
| Deposited Data | ||
| 9‐week RMA data | Guillaumond et al (2015) | GSE61412 |
| 6‐week RMA data | GSE127891 | |
| Experimental Models: Cell Lines | ||
| PK4a mouse cells | Guillaumond et al (2013) | RRID:CVCL_WB21 |
| PANC‐1 human cells | ATCC | Cat# CRL‐1469, RRID:CVCL_0480 |
| MiaPaCa‐2 human cells | ATCC |
Cat# CRL‐1420 RRID:CVCL_0428 |
| Experimental Models: Organisms/Strains | ||
| Pdx1‐Cre; Ink4a/Arffl/fl;LSL‐KrasG12Dmice | Aguirre et al (2003) | https://doi.org/10.1101/gad.1158703 |
| HsdCpb:NMRI‐Foxn1nu | Envigo | Cat# 5652667, RRID:MGI:5652667 |
| Oligonucleotides | ||
| 36B4 primer Forward AATCCCTGACGCACCGCCGTGATG | Guillaumond et al (2015) | https://doi.org/10.1073/pnas.1421601112 |
| 36B4 primer Reverse TGGGTTGTTTTCCAGGTGCCCTCG | Guillaumond et al (2015) | https://doi.org/10.1073/pnas.1421601112 |
| Filamin B primer Forward AGTTAACCAGCCAGCATCCT | Devis et al (2017) | https://doi.org/10.1002/path.4851 |
| Filamin B primer Reverse TGACATCGATGGTGTGGACA | Devis et al (2017) | https://doi.org/10.1002/path.4851 |
| Epha2 primer Forward GGCTTCTTTATCCACCGCAG | Devis et al (2017) | https://doi.org/10.1002/path.4851 |
| Epha2 primer Reverse CGAGGATGTCTTCAGCATGC | Devis et al (2017) | https://doi.org/10.1002/path.4851 |
| Plau primer Forward TTCACCACCATCGAGAACCA | Devis et al (2017) | https://doi.org/10.1002/path.4851 |
| Plau primer Reverse TTGCGTGTTGGAGTTAAGCC | Devis et al (2017) | https://doi.org/10.1002/path.4851 |
| Recombinant DNA | ||
| HMGCL Crispr/Cas9 KO plasmids | Santa Cruz Biotechnologies | Cat# sc‐422425 |
| HMGCL HDR plasmids | Santa Cruz Biotechnologies | Cat# sc‐403841‐HDR |
| Control CRISPR/Cas9Plasmid | Santa Cruz Biotechnologies | Cat# sc‐418922 |
| pcDNA3.1(+)‐N‐DYK | GeneScript | / |
| ALCAM cDNA (pcDNA3.1(+)‐N‐DYK) | GeneScript | / |
| SQLE cDNA (pcDNA3.1(+)‐N‐DYK) | GeneScript | / |
| Software and Algorithms | ||
| R studio 3.4.4 | RStudio, Inc. | RRID:SCR_000432 |
| Gene Set Enrichment Analysis with the fgsea package | Bioconductor | RRID:SCR_003199 |
| Image J | National Institutes of Health | RRID:SCR_003070 |
| Prism Version 5.03 | Graph Pad | RRID:SCR_002798 |
| Ingenuity Pathway Analysis | IPA® | RRID:SCR_008653 |
| FlowJo Version 10 | FlowJo LLC | RRID:SCR_008520 |
| Other | ||
| DMEM medium without glutamine (Gln‐) | Thermo Fisher Scientific | Cat# 11960‐044 |
| DMEM medium without glutamine nor branched chain amino acid (BCAA‐) | Thermo Fisher Scientific | Cat# ME15212L1 |
| DMEM medium without glutamine nor glucose (Glc‐) | Thermo Fisher Scientific | Cat# A14430‐01 |
| Filtered foetal bovine serum | Hyclone | Cat# SH30070.02 |
| Dialyzed foetal bovine serum | Hyclone | Cat# SH30079.02 |
| Foetal bovine serum | BioSera | Cat# FB‐1280 |
| Antibiotic/antimycotic solution | Thermo Fisher Scientific | Cat# 15240062 |
| Leucine | Sigma Aldrich | Cat# L8912 |
| Isoleucine | Sigma Aldrich | Cat# I7403 |
| Valine | Sigma Aldrich | Cat# V0513 |
| Sodium 3‐hydroxybutyrate | Sigma Aldrich | Cat# 54965 |
| Acetoacetate | Sigma Aldrich | Cat# A8509 |
| l‐Leucine (13C6) | Cambridge Isotope Laboratories | Cat# CLM‐2262‐H‐PK |
| Sodium D‐3‐Hydroxybutyrate (13C4) | Cambridge Isotope Laboratories | Cat# CLM‐3853‐PK |
| l‐glutamine (200 mM) | Thermo Fisher Scientific | Cat# 25030024 |
| TransIT®‐LT1 | Mirus | Cat# MIR2300 |
Methods and Protocols
Spontaneous PDA models
Male PDA‐bearing mice Pdx1‐Cre; LSL‐KrasG12D; Ink4a/Arffl/fl (KIC), and control LSL‐KrasG12D; Ink4a/Arffl/fl (KI) littermates were obtained as previously described (Guillaumond et al, 2013). After sacrifice at 6 weeks (n = 5 KI mice and n = 8 KIC mice) or 9 weeks of age (n = 3 KI mice and n = 3 KIC mice), pieces of tumor or control pancreas were either fixed in 4% (wt/vol) formaldehyde, snap‐frozen in cold isopentane for further analysis, or directly homogenized in 4 M guanidinium isothiocyanate lysis buffer for efficient pancreatic RNA extraction, according to previously published protocols (Guillaumond et al, 2013). All animal care and experimental procedures were performed in agreement with the Animal Ethics Committee of Marseille under reference (APAFIS#19183‐2019021414576818). To study the impact of βOHB on tumors, 5‐week‐old KIC mice were daily injected for 3 weeks either with 100 mg/kg of βOHB or with an equivalent volume of 0.9% NaCl. After sacrifice, mice and tumors were weighed. Tumors, livers, and spleens were fixed in 4% (wt/vol) formaldehyde for histologic scoring and hematoxylin‐phloxine‐saffron (HPS) staining.
PDA xenograft models
Under isoflurane anesthesia [induction: 4% (vol/vol) and maintenance: 1.5% (vol/vol)], mice were injected subcutaneously with 0.2 mg/kg of buprenorphine (Vetergesic; Sogeval) and lidocaine (Xylovet; Ceva) was administered at 3.5 mg/kg by infiltration in the abdominal cavity. Five‐week‐old male HsdCpb:NMRI‐Foxn1nu mice were orthotopically implanted with 1 × 106 of sg‐CTRL PANC‐1 cells (n = 15) or sg‐HMGCL #2 clone (n = 19) or sg‐HMGCL #3 clone (n = 7) PANC‐1 cells. For βOHB treatment, mice were injected with 1 × 106 of sg‐CTRL PANC‐1 cells (n = 4) or sg‐HMGCL #2,3 clones (n = 20). Seven days after engraftment, mice received twice a week 0.9% NaCl (n = 4 and n = 10 for sg‐CTRL group and sg‐HMGCL group, respectively) or 100 mg/kg of βOHB (n = 10 for sg‐HMGCL group) for 2 months and sacrificed. At the time of sacrifice, tumors and livers were removed and weighed, and tumor length (L) and width (W) were determined with caliper. Tumor volume was determined with the following formula: π/6 × [(L + W)/2]. Tumors and livers were fixed in 4% (wt/vol) formaldehyde. For collagen staining, Masson’s trichrome coloration of formalin‐fixed paraffin‐embedded (FFPE) tumor sections was performed using an Artisan Link Special Staining System (Dako), according to manufacturer’s instructions.
In ovo assay
Fertilized white Leghorn eggs were incubated at 37.5°C and 50% relative humidity for 9 days. At this time (E9), the chicken embryo chorioallantoic membrane (CAM) was dropped by drilling a small hole through the eggshell into the air sac and a 1 cm² window was cut in the eggshell above the CAM. 3 × 106 sg‐CTRL and sg‐HMGCL#2 clone PANC‐1 cells were inoculated onto the CAM of each egg (n = 8 eggs/group). At embryonic day 18 (E18), a 1 cm2 portion of the lower CAM was collected to evaluate the number of metastatic cells. Genomic DNA was extracted from the lower CAM and analyzed by qPCR with specific primers for Alu sequences (Zijlstra et al, 2002). Data were analyzed with CFX Manager 3.1 software (Bio‐Rad).
Assessment of βOHB injection or leucine‐enriched diet on liver metastasis
As an induced metastatic model, KI mice were injected in two distinct liver lobes with 1 × 105 PK4A cells. Three days post‐engraftment, mice were daily treated with 0.9% NaCl (n = 8) or 100 mg/kg of βOHB (n = 8) for 10 days. To evaluate the effect of leucine‐enriched diet on liver metastasis, control amino acid defined diet (l‐leucine 12 g/kg, TD110839, Envigo) and leucine‐enriched diet (l‐leucine 24 g/kg, TD110842 produced as isocaloric diet based on control diet TD110839, Envigo) were used. Mice were fed these diets one week before cell engraftment, for treatment of 18 days total. At the end of the experiments, livers were removed, washed with PBS 1×, and then fixed in 4% (wt/vol) formaldehyde. For each mouse, 1, 2, or 3 liver lobes with macroscopic metastasis were paraffin‐embedded. 5 μm tissue sections were stained by HPS method. HPS‐stained slides were scanned at 10× and 20× magnification. Liver metastasis characterization was performed by counting the number of metastasis per slide divided by the number of lobes present on the slide, and then classified into three different subtypes: small‐, medium‐, and large‐size metastasis. In addition, anatomopathological status of metastasis was determined for each metastasis. Three different statuses were defined: undifferentiated, differentiated, and glandular metastasis.
Cell culture conditions
PK4A cell line was isolated from KIC murine PDA as previously described (Olivares et al, 2017), and used from passages 18–28. PANC‐1 and MiaPaCa‐2 cells were obtained from the American Type Culture Collection. Cells were maintained in 25 mM glucose, glutamine free Dulbecco’s modified Eagle’s medium (DMEM) without pyruvate, supplemented with 2 mM of l‐glutamine, 10% (vol/vol) fetal bovine serum (FBS). Human PDA and liver metastasis‐derived primary cell cultures were obtained as previously described (Nicolle et al, 2021). Briefly, PDA‐ and liver metastasis‐PDX (patient‐derived‐xenograft) samples were split into several small pieces (1 mm3) and processed in a biosafety chamber. After a fine mincing, they were treated with collagenase type V (C9263; Sigma‐Aldrich, Inc., St. Louis, Missouri, USA) and trypsin/EDTA and suspended in DMEM supplemented with 1% w/w penicillin/streptomycin and 10% of FBS. After centrifugation, cells were re‐suspended in Serum Free Ductal Media (SFDM) adapted from Schreiber et al (2004) and conserved at 37°C in a 5% CO2 incubator. The three PDA patients from which the PDA‐ and liver metastasis‐primary cell cultures are originated are part of the PaCaOmics cohort registered under the Paoli‐Calmettes Institute clinical trial number 2011‐A01439‐32. For survival assays under leucine‐deprived conditions, PK4A cells were cultured in 25 mM glucose, glutamine, and branched‐chain amino acids (BCAAs) free Dulbecco’s modified Eagle’s medium DMEM without pyruvate, supplemented with 2 mM of l‐glutamine, with 10% filtered FBS (SH30070, Hyclone), 1 mM of valine, isoleucine, and leucine (untreated medium), or with dialyzed FBS (SH30079, Hyclone), 1 mM of valine, isoleucine (Leu‐ medium) with or without 10 mM of βOHB. For nutrient uptake experiments, glucose free‐media (glc‐) consists of glutamine‐free DMEM without glucose nor pyruvate and supplemented with 2 mM of l‐glutamine, 10% dialyzed FBS, and complete media consists of media used for cell maintenance, both with and without 1 mM of βOHB. Cells were cultured with 1% antibiotic/antimycotic solution at 37°C, 21% oxygen in a 5% CO2 incubator and were mycoplasma free.
Murine PDA transcriptome analysis
All expression data from samples from 6‐week (n = 5 control/healthy pancreas vs. 8 PDA) and 9‐week (n = 3 control/healthy pancreas vs. n = 3 PDA), old mice were normalized with the Robust Multichip Average (RMA; Irizarry et al, 2003) from the package oligo available on Bioconductor (R 3.4.4 software). Gene Set Enrichment Analysis (GSEA) based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database was carried out with the R package fGSEA. The expression fold changes have been used as input of the function fgsea. Leading‐edge genes correspond to the core genes that account for the gene set’s enrichment signal (Subramanian et al, 2005). Hence, the leading‐edge gene number is expressed as the percentage of deregulated leading‐edge genes on total genes that compose each metabolic pathway (Appendix Table S1).
For differential BCAA and KB gene expression analysis, we only considered genes that are exclusively involved in BCAA catabolism and we added genes coding for BCAA and KB transporters to obtain a full representation of BCAA and KB metabolism in PDA. Genes differentially expressed between PDA and control pancreas from 6‐ or 9‐week‐old PKI and KI mice (Appendix Table S2) were determined by a t‐test applied on the normalized RMA expression. The P‐values were adjusted for multiple testing with the Benjamini‐Hochberg method. Only genes with absolute fold change > 1.5 or > 2 and adjusted P‐value < 0.05 are considered as significantly deregulated in 6‐ or 9‐weeks PDA, respectively (Appendix Table S2) and are highlighted in volcano plots. For these illustrations, fold‐changes and raw P‐values are transformed into log2 and –log10 values, respectively. The dashed horizontal lines represent the selection cutoff based on the adjusted P‐value cutoff threshold of 0.05. This cutoff does not match the same raw P‐value (y‐axis) for the two datasets since the calculated adjusted P‐values depend on the input raw P‐values.
KB gene expression in human PDA samples
We collected gene expression data of 728 pancreatic carcinomas and 76 metastatic samples (metastasis sites coming from liver (n = 35, 46%), peritoneal (n = 12, 16%), lymph node (n = 9, 12%), lung (n = 8, 11%), abdominal wall (n = 3, 4%), diaphragm (n = 3, 4%), colon (n = 2, 3%), duodenum (n = 2, 3%), and fat (n = 2, 3%)) from 15 public data sets from the National Center for Biotechnology Information (NCBI)/Genbank GEO, ArrayExpress, European Genome‐phenome Archive (EGA), and TCGA databases (Appendix Table S4). Data analysis required pre‐analytic processing and normalization as previously described (Birnbaum et al, 2017). KB gene expression levels were extracted from each data set and were standardized within each data set using the primary cancer population of the largest set as a reference to be comparable across data sets and to exclude bias from population heterogeneity. All steps were done in R using Bioconductor and associated packages.
Human samples
Freshly frozen tissue samples of PDA (n = 5) and corresponding healthy/adjacent pancreas (n = 5) located at distant site from the tumor were obtained from patients who underwent surgery. Histologic examination confirmed diagnosis of PDA and of healthy pancreases. Tumor staging was performed according to the International Union Against Cancer TNM system. All patients underwent surgery at the Department of Digestive Surgery, North Hospital, Marseille, France between 2009 and 2010. Before surgery, all patients had signed an informed consent form that had been approved by the local ethics committee (agreement reference of CRO2 tissue collection: DC‐2013‐1857; Marseille, France).
Protein extracts and western blots
Pancreatic ductal adenocarcinoma, healthy pancreas samples from PKI and KI mice and cell pellets were lysed in SDS 0,1%, 1% Triton X‐100, Tris pH8 0.01 M and NaCl 0.14 M supplemented with 1 mM Phenylmethylsulfonyl fluoride, 100 µM orthovanadate, 40 mM β‐glycerophosphate, 1 mM Sodium Fluoride, protease inhibitor cocktail (Sigma Aldrich), and sodium deoxycholate 1 or 10% for tissue or cells respectively, prior to centrifugation (14,000 rpm, 10 min, 4°C). Supernatants were collected, and protein concentration was evaluated using BioRad Protein assay. Whole cell protein extracts (75–100 µg/lane) were resolved by SDS/PAGE using a 10% (vol/vol) acrylamide gel and transferred onto 0.2 µm nitrocellulose membranes (Bio‐Rad). After a blocking step (5% (wt/vol) non‐fat milk in Tris‐Buffered Saline (TBS)), primary antibody was incubated overnight (O/N) at 4°C (in 5% (wt/vol) non‐fat milk in TBS 0.1% Tween 20). ECL protein detection (Millipore) was performed with a Fusion Fx7 chemiluminescent imager and the quantification of the amounts of proteins of interest was determined by densitometry using Image J software (NIH) and normalized to ACTIN or to total loaded protein stained with Amido black or Ponceau Red.
Immuno‐histochemistry
Formalin‐fixed paraffin‐embedded sections of mouse PDA (5 µm‐thick) were deparaffinized in xylene and rehydrated through a series of graded ethanol concentrations. Antigen retrieval was performed in citrate buffer pH6 (Diapath)/ 0.05% Tween 20 or in target retrieval solution at pH 6 or 9 (Dako), before the quenching of endogenous peroxidase activity (3% (vol/vol) H2O2). Tissue sections were then incubated with blocking solution prior to primary antibody. Immunoreactivity was visualized using the Vectastain ABC kit (PK‐4001, Vector Laboratories) or with a biotinylated goat anti‐rabbit antibody and peroxidase‐conjugated streptavidin from Dako (France). Peroxidase activity was revealed using liquid DAB+ substrate chromogen system (Dako). Counter‐staining in Mayer’s Haematoxilin was followed by a bluing step in sodium bicarbonate buffer (0.1% in water) before final dehydration and mounting of the sections. Quantifications of staining were performed on seven pictures at 10× or 20× magnification using ImageJ software (NIH).
Establishment of sg‐HMGCL cell lines
PANC‐1 and MiaPaCa‐2 cells were seeded on 6‐well plates 24 h before being transfected with a pool of 3 HMGCL Crispr/Cas9 knock‐out plasmids or with a Crispr/Cas9 control plasmid. Each plasmid codes for the Cas9 nuclease, a Green Fluorescent Protein (GFP), and a target‐specific 20 nt guide RNA (gRNA). PANC‐1 and MiaPaCa‐2 cells were co‐transfected with a Homology‐Directed Repair (HDR) plasmid coding for a Red fluorescent protein (RFP) and containing a puromycin resistance gene. Forty‐eight hour after transfection performed with Lipofectamine 3000, PANC‐1‐RFP cells were cloned by cell sorting using fluorescence‐activated cell sorting (FACS) (ArialII, BD Biosciences) and only PANC‐1 clones depleted in HMGCL were selected and maintained upon puromycin treatment. For sg‐HMGCL MiaPaCa‐2 cells, clones were isolated using limiting dilution and maintained upon puromycin treatment.
Establishment of ALCAM+ and SQLE+ PANC‐1 cell lines
sg‐HMGCL PANC‐1 cells were seeded on 6‐well plates with 2 × 105 cells per well 24 h before transfection. Using TransIT®‐LT1, cells were transfected with either an empty pcDNA3.1(+)‐N‐DYK plasmid, or a pcDNA3.1(+)‐N‐DYK plasmid containing SQLE or ALCAM cDNA. All plasmids contain both a flag on the N‐terminus and a neomycin resistance gene. Forty‐eight hour post‐transfection, stable transfected cells were selected and maintained upon neomycin treatment.
Survival assays
7 × 104 PK4A cells were plated in 48‐well plates using experimental media as described above in Methods and Protocols. Media were changed at day 3. Percentage of confluence was assessed every day for 5 days using IncuCyte™ S3 Live‐Cell analysis system (Sartorius).
Proliferation assays
sg‐CTRL or sg‐HMGCL PANC‐1 cells or MiaPaCa‐2 cells (3 × 104) were plated in 48‐well plates, in triplicates, using media for their maintenance as described above in Methods and Protocols. After the indicated times, triplicates were pooled, and viability was assessed via Trypan blue exclusion using a cell viability analyzer (Vi‐cell XR, Beckman Coulter).
Clonogenic assays
1,000 sg‐CTRL or sg‐HMGCL PANC‐1 and MiaPaCa‐2 cells were seeded in a 6‐well plate and cultured in the same medium as for their maintenance. Fifteen days later, colonies were fixed in 10% (wt/vol) formalin and stained with 0.5% Crystal violet. Pictures were captured using an Evos microscope (Thermo Fisher Scientific) at 4× magnification, and areas of stained colonies were quantified by ImageJ software (NIH). Representative pictures for illustration were obtained using a camera.
Migratory assays
Three 104 sg‐CTRL and sg‐HMGCL PANC‐1 cells were seeded in the same medium as for their maintenance without FBS in a 24‐well insert, pre‐coated with 1% of gelatin and 0.1% of fibronectin, and placed in a 24‐well plate containing the same media with 10% FBS. Four hour after seeding, inserts were fixed in 10% (wt/vol) methanol, stained with Coomassie bleu. Seven pictures of each well were captured using an Evos microscope at 4× magnification and quantified by ImageJ software.
Wound‐healing assays
7.5 × 105 sg‐CTRL and sg‐HMGCL PANC‐1 cells and MiaPaCa‐2 cells were seeded in 6‐well plates in the same medium as for their maintenance. To inhibit proliferation, cells were rendered quiescent using mitomycin C (0.5 µg/ml) 2 h before scratch. After scratch, wells were washed with PBS 1×, and fresh medium containing mitomycin C (0.5 µg/ml) was added. Images were captured at 0 h and 72 h after scratch, at 2× magnification. Scratched area recovered by cells was measured using ImageJ software and expressed as percentage of measured initial scratched area at day 0.
Invasion assays
4.5 × 104 sg‐CTRL and sg‐HMGCL PANC‐1 cells were seeded in the same medium as for their maintenance without FBS into 24‐well invasion chambers already pre‐coated with matrigel and placed in a 24‐well plate containing the same medium as for their maintenance. Twenty‐four hour after invasion, inserts were fixed in 10% (wt/vol) methanol, stained with Coomassie bleu. Images were obtained using an Evos microscope and the number of invasive cells was quantified by ImageJ software. For spheroid invasion assays, 1,500 sg‐CTRL and sg‐HMGCL PANC‐1 cells were seeded in the same medium as for their maintenance in 96‐well ultra‐low attachment plate (corning). After 3 days in culture, 100 µl of medium was replaced by 100 µl of cold matrigel (Corning). Plates were centrifuged 5 min at 4°C to allow the dilution of matrigel in medium and placed for 1 h in a 37°C incubator for matrigel polymerization before adding 100 µl of medium. Pictures were captured at day 10 or 15 at 2×, 4×, 10×, or 20× magnifications to observe invasive cells.
Spheroid/organoid assays
1,500 cells were seeded in 96‐well ultra‐low attachment plate (corning) and cultured as spheroids in the same medium as for their maintenance. For PK4a spheroids, media containing 1 or 10 mM of βOHB, or not (untreated), were replenished at day 3, 7 and 10. Spheroids area was assessed every day for 12 days using IncuCyte™ S3 Live‐Cell analysis system (Sartorius). For sg‐CTRL and sg‐HMGCL PANC‐1 spheroids, media was replenished twice a week. Pictures of PANC‐1 spheroids were captured using Evos microscope at 2, 4, or 10× magnification. After 15 days, PANC‐1 spheroids were dissociated using trypsin, and cells were counted via Trypan blue exclusion. For mouse and human PDA‐ or liver metastasis‐derived organoids, we cultured 5 × 104 cells in Pancreatic Organoid Feeding Media (POFM) medium after seeding in 48‐well plate pre‐coated with Matrigel as previously described (Hoare et al, 2021). After 2 days, once the organoids were formed, media were replaced by the same medium as for their maintenance alone or with 1 or 10 mM of βOHB. Pictures of organoids were captured using Evos microscope at 4× magnification.
Quantification of cell fluorescent dye
To analyze proliferation of cells, CellTraceTM Carboxyfluorescein Succinimidyl Ester (CFSE) Cell proliferation Kit was used according to the manufacturer’s protocol. Briefly, before seeding, sg‐CTRL and sg‐HMGCL PANC‐1 cells were incubated for 20 min with 5 µM of CFSE in a 37°C incubator, then washed twice with PBS 1× containing 2% FBS. 3.5 × 105 cells were seeded in 2 ml of medium in 6‐well plates and maintained for 3 days. For spheroid assays, 1,500 cells were seeded in 200 µl of medium in a 96‐well ultra‐low attachment plate (corning) and maintained for 7 days. At the indicated times, cells were detached and washed with PBS 1×. Mean fluorescence intensity (MFI) of each cell line was measured with the MACSQuant‐VYB cytometer and then reported as the percentage of MFI as indicated in figure legends, using FlowJo software, in order to evaluate the proliferation index.
Tracing experiments
For in vitro tracing of leucine or βOHB, 7.5 × 104 of PK4A, sg‐CTRL or sg‐HMGCL PANC‐1 cells were plated in triplicate in 6‐well plates in medium used for their maintenance. The following day, cells were washed with PBS 1× and refreshed with DMEM without glucose, nor pyruvate, nor glutamine, and supplemented or not with 5 mM or 25 mM of glucose and with 2 mM l‐glutamine, 10% dialyzed FBS, and 0.8 mM U‐13C‐Leucine or 1 mM U‐13C‐βOHB for 24 h at 37°C. Then, cells were harvested, extracted, and analyzed as described below. For ex vivo tracing, tumors were collected from KIC mice at 6‐ or 9‐weeks of age. After collection, thin slices (around 500 µm) were performed from a piece of tumor and then washed twice with PBS 1× and cultured in DMEM with 10% dialyzed FBS without glucose, nor pyruvate, nor glutamine supplemented with 2 mM l‐glutamine and 25 mM U‐13C‐Glucose, or without glutamine and supplemented with 2 mM U‐13C‐Glutamine or supplemented with 2 mM l‐glutamine, 1 mM U‐13C‐βOHB, or 0.2 mM U‐13C‐Acetate, for 24 h. After that, tumor slices were harvested, washed, grounded to powder, and metabolites were extracted and analyzed as described below. A piece of the initial tumor was fixed in 4% (wt/vol) formaldehyde for further histological analysis.
Extraction of metabolites
At the conclusion of the tracing experiments, ex vivo tumors, PK4A, sg‐CTRL, or sg‐HMGCL PANC‐1 cells were rinsed once in blood bank saline, and metabolites were extracted with a methanol:chloroform:water (5:5:3) mix containing norvaline as an internal standard, as previously described (Olivares et al, 2017). After vortexing and centrifugation, the aqueous phase containing polar metabolites was removed, dried under nitrogen gas, and stored at −80˚C for subsequent derivatization. Samples were then run on the Gas Chromatography/Mass Spectrometry (GC/MS) as described in the following section.
GC/MS analyses
Dried polar metabolites were dissolved in 20 μl of pyridine containing 2% of methoxamine hydrochloride (Thermo Fisher Scientific) and held at 37°C for 1 h. After dissolution and reaction, tert‐butyl‐di‐methyl‐silyl derivatization was initiated by adding 25 μl N‐methyl‐N‐(tert‐butyl‐di‐methyl‐silyl) trifluoroacetamide + 1% tert‐butyl‐dimethyl‐chlorosilane (Sigma Aldrich) and by incubating at 37°C for 30 min. GC/MS analysis was performed using an Agilent 7890 GC equipped with a 30 m DB‐35MS capillary column connected to an Agilent 5975B MS operating under electron impact ionization at 70 eV. One microliter of sample was injected in splitless mode at 270°C, using helium as the carrier gas at a flow rate of 1.2 ml/min. For measurement of polar metabolites, the GC oven temperature was held at 100°C for 1 min, increased to 105°C at 2.5°C/min and held at 105°C for 2 min, increased to 250°C at 3.5°C/min, and increased to 320°C at 20°C/min. The MS source and quadrupole were held at 230°C and 150°C, respectively, and the detector was run in scanning mode, recording ion abundance in the range of 100–605 m/z. MIDs were determined by integrating the appropriate ion fragments.
Nutrient uptake analysis
Glucose and glutamine consumption were assessed using the YSI 2950 BioAnalyser (System‐C‐Industry). Briefly, triplicates of sg‐CTRL or sg‐HMGCL PANC‐1 cells were seeded (3 × 104 cells) in 48‐well plates in experimental media as described above. After 72 h, supernatants were collected and metabolite concentrations of glutamine and glucose were measured. Viable cells numbers were evaluated through Trypan blue exclusion using a cell viability analyzer (Vi‐cell XR, Beckman Coulter) and used for normalization. For every metabolite measurement, fresh maintenance or glc free media were used as starting point for baseline concentrations.
Mass spectrometry proteomic analysis
Extraction of proteins from sg‐CTRL and sg‐HMGCL (#2 and #3) PANC‐1 spheroids was performed as previously described. 15 µg of each protein sample of each cell line was loaded on a NuPAGE gel 4–12% (Life Technologies) and run as a single band for a 6 min electrophoresis at 80V in MOPS buffer (Thermo Fisher Scientific). Gels were incubated with Imperial Protein Stain (Thermo Fisher Scientific) and after extensive washes, stacked protein bands were cut and submitted to classical in gel protein digestion (reduction, ioacetamide alkylation, and trypsin digestion). Extracted peptides were concentrated under speed vacuum and solubilized in 2% acetonitrile, 0.05% Trifluoracetic acid. For mass spectrometry acquisition, 20% of each sample was injected in triplicates on a liquid nanochromatography Ultimate 3000RSLC system (Thermo Fisher Scientific) online in front of an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific). Peptides were first concentrated and purified on a pre‐column from Dionex (C18 PepMap100, 2 cm × 100 µm I.D, 100 Å pore size, 5 µm particle size) in solvent A (0.1% formic acid in 2% acetonitrile). In the second step, peptides were separated on a reverse phase LC EASY‐Spray C18 column from Dionex (PepMap RSLC C18, 50 cm × 75 µm I.D, 100 Å pore size, 2 µm particle size) at 300 nl/min flow rate and 40°C. After column equilibration using 4% of solvent B (20% water–80% acetonitrile–0.1% formic acid), peptides were eluted from the analytical column by a two‐step linear gradient (4‐22% acetonitrile/H2O; 0.1% formic acid for 220 min and 22–32% acetonitrile/H2O; 0.1% formic acid for 20 min). For peptide ionization in the EASY‐Spray nanosource, spray voltage was set at 2.2 kV and the capillary temperature at 275°C. The mass spectrometer was used in data‐dependent mode to switch consistently between MS and MS/MS. Time between Masters Scans was set to 3 s. MS spectra were acquired with the Orbitrap in the range of m/z 400–1,600 at a FWHM resolution of 120,000 measured at 200 m/z. AGC target was set at 4.0e5 with a 50 ms Maximum Injection Time. The more abundant precursor ions were selected, and collision‐induced dissociation fragmentation at 35% was performed and analyzed in the ion trap using the “Inject Ions for All Available Parallelizable time” option with a maximum injection time of 300 ms and an AGC target of 4000. Charge state screening was enabled to include precursors with 2 and 7 charge states. Dynamic exclusion was enabled with a repeat count of 1 and a duration of 60 s. These chromatographic conditions were previously optimized with a protein pool from all the samples.
Quantitative proteomics processing
For data processing, we used the free suite MaxQuant version 15.3.8 (Cox et al, 2011). The relative intensities based on label‐free quantification (LFQ) were calculated using the MaxLFQ algorithm (Cox et al, 2014). The 27 LC‐MS raw acquisitions were processed by the Andromeda search engine integrated into MaxQuant (Cox et al, 2011). The identification of the precursor ions present in the mass spectra was performed by comparison with the protein database of Human extracted from Uniprot on the 31st of May 2017 and containing 20,200 entries. This database was supplemented with a set of 245 proteins that are commonly found as contaminants. The following parameters were used for this search: (i) trypsin cleavage authorization before prolines; (ii) authorization of two failed cleavages; (iii) fixed modification of cysteines by carbamidomethylation (+57.02146 Da) and variable modification of methionines by oxidation (+15.99491) and N‐terminal proteins by acetylation (+42.0116); (iv) authorization of five modifications per peptide; and (v) minimum peptides length of seven amino acids and a maximum mass of 4,600 Da. Spectra alignment was performed in two dimensions; the elution time of the precursor ions (min) and the mass range. The “Match between runs” option has been enabled to allow the transfer of identifications between LC‐MS/MS based on the mass and the retention time using the default settings. The false positive rate on identification was set at 1%. The statistical analysis was carried out with the Perseus program (version 1.6.0.7) of the MaxQuant environment. The normalized intensity LFQ was transformed by a base logarithm 2 to obtain a normal distribution. Differential proteins were evidenced by the application of a multiple ANOVA t‐test or a Student t‐test performed by controlling the false positive rate at 1% using 250 permutations. Proteins differently expressed between samples were analyzed based on the log2 difference of the LFQ intensity and log10 of the associated q value. For visualization of proteomic data sets, volcano plots were created using a cutoff of q‐value< 0.05 and a cutoff of difference of LFQ intensity of < −2.3 or > 2.3 between compared groups. The differential proteomics analysis was carried out on identified proteins after removal of proteins only identified with a modified peptide, peptides shared with other proteins, proteins from contaminant database and proteins which are only represented in two replicates of six of the same condition. Common proteins with deregulated expression levels in sg‐HMGCL #2 and #3 compared to sg‐CTRL PANC‐1 cells were then further analyzed.
ALCAM staining
4 × 105 sg‐CTRL or sg‐HMGCL PANC‐1 cells were detached from plate with accutase, transferred to a 96 round bottom well plate, and were incubated with anti‐ALCAM antibody diluted in PBS 1× containing 2% FBS and 5% EDTA during 30 min at 37°C. Then, cells were washed three times before being incubated with secondary antibody at 37°C for 30 min. MFI of 10,000 cells for each cell line was measured with the MACSQuant –VYB cytometer and analyzed with FlowJo software, in order to evaluate ALCAM quantity.
Reverse transcription
5 × 105 cells were plated in 6 cm dish at day 0 in maintenance medium. The day after, cells were washed twice with PBS 1× and RNA was extracted by Trizol reagent. 5 μg of total RNA were used to synthetize cDNA with the PrimeScript RT reagent kit (Promega) and provided oligo‐dT primers, according to the manufacturer’s instructions.
Quantitative real‐time polymerase chain reaction (qPCR)
Quantitative PCR reactions were performed with EPHA2, FLNB, and PLAU specific primers and the GoTaq qPCR master mix kit (Promega) using the Mx3005P Stratagene system. Differential expression of transcripts of interest was normalized with the respective 36B4 housekeeping transcript expression.
Statistical analysis
One‐tailed unpaired Student’s t‐test was used for comparing means of two experimental groups in the case of one independent factor; for in vitro experiments comparing sg‐CTRL vs. sg‐HMGCL samples, we compared the overall changes between the two groups (i.e. mean of all sg‐HMGCL samples vs. sg‐CTRL samples). For all in vivo experiments, we used a Mann‐Whitney non‐parametric test to compare two experimental groups when data do not follow a normal distribution. One way or two‐way ANOVA, followed by the appropriated multiple comparison test, was used when comparing more than two experimental groups. In all figures, data are presented as a mean ± standard error of the mean (SEM) and exact n values used to calculate the statistics are indicated. All statistical analyses were performed using GraphPad Prism 5. P‐values< 0.05 were considered statistically significant.
Author contributions
Victoire Gouirand: Conceptualization; Data curation; Validation; Methodology; Writing—original draft; Writing—review and editing. Tristan Gicquel: Conceptualization; Data curation; Methodology. Evan C Lien: Conceptualization; Data curation; Formal analysis; Writing—original draft; Writing—review and editing. Emilie Jaune‐Pons: Data curation; Methodology. Quentin Da Costa: Conceptualization; Methodology; Writing—original draft. Pascal Finetti: Data curation; Writing—original draft. Elodie Metay: Data curation; Methodology. Camille Duluc: Methodology. Jared R Mayers: Methodology. Stephane Audebert: Methodology; Writing—original draft. Luc Camoin: Conceptualization; Methodology. Laurence Borge: Methodology. Marion Rubis: Methodology. Julie Leca: Resources; Methodology. Jeremy Nigri: Methodology. François Bertucci: Conceptualization; Methodology; Writing—original draft. Nelson Dusetti: Resources; Methodology. Juan Lucio Iovanna: Resources; Methodology. Richard Tomasini: Conceptualization; Resources; Methodology. Ghislain Bidaut: Conceptualization; Data curation; Methodology. Fabienne Guillaumond: Conceptualization; Methodology; Writing—review and editing. Matthew G Vander Heiden: Conceptualization; Funding acquisition; Methodology; Writing—original draft; Writing—review and editing. Sophie Vasseur: Conceptualization; Resources; Data curation; Formal analysis; Supervision; Funding acquisition; Validation; Investigation; Methodology; Writing—original draft; Project administration; Writing—review and editing.
In addition to the CRediT author contributions listed above, the contributions in detail are
VG, TG, ECL, EJ‐P, CD, and EM performed the experiments. VG, ECL, JRM, and MGVH performed and assisted tracing experiments and GC/MS analysis. QDC and GB analyzed the mouse transcriptomic datasets. PF and FB provided and analyzed human transcriptomic datasets. LC and SA performed proteomics and data analysis. LB and JN assisted and provided advice for cell culture. MR assisted immunochemistry experiments. ND and JLI provided human PDA and liver metastatic derived primary cells. RT and JL provided protein extracts from human samples. SV, VG, MGVH, FG, and RT conceptualized the study. FG and RT provided advice regarding experiments and writing of the manuscript. SV directed the study. SV, MGVH, VG, ECL, PF, QDC, and FG wrote the manuscript. Funding acquisition, SV, MGVH, and FG.
Disclosure and competing interests statement
MGVH discloses that he is on the scientific advisory board of Agios Pharmaceuticals, iTeos Therapeutics, Drioa Ventures, Sage Therapeutics, and Auron Therapeutics. Other authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank the cell culture platform (PCC, TPR2, CRCM, Marseille, France), the mice colony facility (PSEA, TPR2, CRCM, Marseille, France), the bioinformatic platform (Cibi, CRCM, Marseille, France), proteomics platform (CRCM, Marseille, France) for technical assistance. V.G. was supported by the “Ligue contre le cancer” and the “Association pour la Recherche sur le Cancer, ARC.” T.G. was supported by the National Institute of Cancer (INCaPLBIO15‐217). E.J.P. was supported by the Foundation for Medical Research (FRM), C.D. was supported by the National Institute of Cancer (INCa Pair Pancreas 2018‐082), E.M. was supported by the French Ministry for Research and Education (MENRT). E.C.L. is a Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation (DRG‐2299‐17). J.R.M. was supported by F30CA183474 from the NCI. MGVH acknowledges support from the Lustgarten Foundation, the MIT Center for Precision Cancer Medicine, SU2C, the Ludwig Center at MIT, the NCI (R35‐CA242379, R01CA168653, R01CA201276, P30CA1405141), the Emerald Foundation, and is a faculty scholar award from the Howard Hughes Medical Institute. SV acknowledges supports from the National Institute of Cancer (INCa PLBIO15‐217, INCa Pair Pancreas 2018‐082), the Plan Cancer 2009–2013 (C13056AS), the Foundation for Medical Research (FRM SPF2018096968), the association for cancer research (ARC, PJA‐20181208127), and the Cancéropôle PACA (2019‐00100).
The EMBO Journal (2022) 41: e110466.
Data availability
For expression data from samples from 6‐week (n = 5 control pancreas vs 8 PDA) and 9‐week (n = 3 control pancreas vs n = 3 PDA) old mice, the DNA microarray raw data have been deposited in Gene Expression Omnibus under: GSE127891 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE127891) (6‐week RMA data) and GSE61412 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE61412) (9‐week RMA data).
References
- Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, DePinho RA (2003) Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev 17: 3112–3126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee H‐J, Purohit V, Sagalovskiy IR, Ma A, Kapilian J, Firl CEM et al (2020) Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368: 85–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagci T, Wu JK, Pfannl R, Ilag LL, Jay DG (2009) Autocrine semaphorin 3A signaling promotes glioblastoma dispersal. Oncogene 28: 3537–3550 [DOI] [PubMed] [Google Scholar]
- Biancur DE, Kapner KS, Yamamoto K, Banh RS, Neggers JE, Sohn ASW, Wu W, Manguso RT, Brown A, Root DE et al (2021) Functional genomics identifies metabolic vulnerabilities in pancreatic cancer. Cell Metab 33: 199–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancur DE, Paulo JA, Małachowska B, Quiles Del Rey M, Sousa CM, Wang X, Sohn ASW, Chu GC, Gygi SP, Harper JW et al (2017) Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat Commun 8: 15965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum DJ, Finetti P, Lopresti A, Gilabert M, Poizat F, Raoul J‐L, Delpero J‐R, Moutardier V, Birnbaum D, Mamessier E et al (2017) A 25‐gene classifier predicts overall survival in resectable pancreatic cancer. BMC Med 15: 170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti G, Moore ZR, Luo X, Ilcheva M, Ali A, Padanad M, Zhou Y, Xie Y, Burma S, Scaglioni PP et al (2015) Targeting glutamine metabolism sensitizes pancreatic cancer to PARP‐driven metabolic catastrophe induced by ss‐lapachone. Cancer Metab 3: 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commisso C, Davidson SM, Soydaner‐Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB et al (2013) Macropinocytosis of protein is an amino acid supply route in Ras‐transformed cells. Nature 497: 633–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M (2014) Accurate proteome‐wide label‐free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13: 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10: 1794–1805 [DOI] [PubMed] [Google Scholar]
- Daher B, Parks SK, Durivault J, Cormerais Y, Baidarjad H, Tambutte E, Pouyssegur J, Vucetic M (2019) Genetic ablation of the cystine transporter xCT in PDAC cells inhibits mTORC1, growth, survival, and tumor formation via nutrient and oxidative stresses. Cancer Res 79: 3877–3890 [DOI] [PubMed] [Google Scholar]
- Davidson SM, Jonas O, Keibler MA, Hou HW, Luengo A, Mayers JR, Wyckoff J, Del Rosario AM, Whitman M, Chin CR et al (2017) Direct evidence for cancer‐cell‐autonomous extracellular protein catabolism in pancreatic tumors. Nat Med 23: 235–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Feyter HM, Behar KL, Rao JU, Madden‐Hennessey K, Ip KL, Hyder F, Drewes LR, Geschwind JF, de Graaf RA, Rothman DL (2016) A ketogenic diet increases transport and oxidation of ketone bodies in RG2 and 9L gliomas without affecting tumor growth. Neuro‐oncology 18: 1079–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devis L, Moiola CP, Masia N, Martinez‐Garcia E, Santacana M, Stirbat TV, Brochard‐Wyart F, García Á, Alameda F, Cabrera S et al (2017) Activated leukocyte cell adhesion molecule (ALCAM) is a marker of recurrence and promotes cell migration, invasion, and metastasis in early‐stage endometrioid endometrial cancer. J Pathol 241: 475–487 [DOI] [PubMed] [Google Scholar]
- Dumartin L, Quemener C, Laklai H, Herbert J, Bicknell R, Bousquet C, Pyronnet S, Castronovo V, Schilling MK, Bikfalvi A et al (2010) Netrin‐1 mediates early events in pancreatic adenocarcinoma progression, acting on tumor and endothelial cells. Gastroenterology 138: 1595–1606 [DOI] [PubMed] [Google Scholar]
- Ferlay J, Partensky C, Bray F (2016) More deaths from pancreatic cancer than breast cancer in the EU by 2017. Acta Oncol 55: 1158–1160 [DOI] [PubMed] [Google Scholar]
- Gouirand V, Guillaumond F, Vasseur S (2018) Influence of the tumor microenvironment on cancer cells metabolic reprogramming. Front Oncol 8: 117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouirand V, Vasseur S (2018) Fountain of youth of pancreatic cancer cells: the extracellular matrix. Cell Death Discov 4: 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabacka MM, Wilk A, Antonczyk A, Banks P, Walczyk‐Tytko E, Dean M, Pierzchalska M, Reiss K (2016) Fenofibrate induces ketone body production in melanoma and glioblastoma cells. Front Endocrinol 7: 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünert SC, Schlatter SM, Schmitt RN, Gemperle‐Britschgi C, Mrázová L, Balcı MC, Bischof F, Çoker M, Das AM, Demirkol M et al (2017) 3‐Hydroxy‐3‐methylglutaryl‐coenzyme A lyase deficiency: clinical presentation and outcome in a series of 37 patients. Mol Genet Metab 121: 206–215 [DOI] [PubMed] [Google Scholar]
- Guillaumond F, Bidaut G, Ouaissi M, Servais S, Gouirand Vi, Olivares O, Lac S, Borge L, Roques J, Gayet O et al (2015) Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc Natl Acad Sci 112: 2473–2478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillaumond F, Leca J, Olivares O, Lavaut M‐N, Vidal N, Berthezène P, Dusetti NJ, Loncle C, Calvo E, Turrini O et al (2013) Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc Natl Acad Sci USA 110: 3919–3924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagedorn M, Javerzat S, Gilges D, Meyre A, de Lafarge B, Eichmann A, Bikfalvi A (2005) Accessing key steps of human tumor progression in vivo by using an avian embryo model. Proc Natl Acad Sci USA 102: 1643–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoare O, Fraunhoffer N, Elkaoutari A, Gayet O, Bigonnet M, Roques J, Nicolle R, McGuckin C, Forraz N, Sohier E et al (2021) Exploring the complementarity of pancreatic ductal adenocarcinoma preclinical models. Cancers 13: 2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honkala AT, Tailor D, Malhotra SV (2019) Guanylate‐binding protein 1: an emerging target in inflammation and cancer. Front Immunol 10: 3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins BD, Pauli C, Du X, Wang DG, Li X, Wu D, Amadiume SC, Goncalves MD, Hodakoski C, Lundquist MR et al (2018) Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560: 499–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CK, Chang PH, Kuo WH, Chen CL, Jeng YM, Chang KJ, Shew JY, Hu CM, Lee WH (2017) Adipocytes promote malignant growth of breast tumours with monocarboxylate transporter 2 expression via beta‐hydroxybutyrate. Nat Commun 8: 14706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DE, Li T, Wang L, Zhang L, Yan R, Li K, Xing S, Wu G, Hu L, Jia W et al (2016) Hepatocellular carcinoma redirects to ketolysis for progression under nutrition deprivation stress. Cell Res 26: 1112–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer‐Barclay YD, Antonellis KJ, Scherf U, Speed TP (2003) Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4: 249–264 [DOI] [PubMed] [Google Scholar]
- Kamil M, Shinsato Y, Higa N, Hirano T, Idogawa M, Takajo T, Minami K, Shimokawa M, Yamamoto M, Kawahara K et al (2019) High filamin‐C expression predicts enhanced invasiveness and poor outcome in glioblastoma multiforme. Br J Cancer 120: 819–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, Vander Heiden MG, Miller G, Drebin JA, Bar‐Sagi D et al (2015) Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res 75: 544–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H‐B, Fan J, Lin R, Elf S, Ji Q, Zhao L, Jin L, Seo J, Shan C, Arbiser J et al (2015) Metabolic rewiring by oncogenic BRAF V600E links ketogenesis pathway to BRAF‐MEK1 signaling. Mol Cell 59: 345–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler SK, Neal EG, Camfield CS, Kossoff EH (2011) Dietary therapies for epilepsy: future research. Epilepsy Behav 22: 17–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NI, Park MH, Kweon SS, Cho N, Lee JS (2021) Squalene epoxidase expression is associated with breast tumor progression and with a poor prognosis in breast cancer. Oncol Lett 21: 259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leca J, Martinez S, Lac S, Nigri J, Secq V, Rubis M, Bressy C, Sergé A, Lavaut M‐N, Dusetti N et al (2016) Cancer‐associated fibroblast‐derived annexin A6+ extracellular vesicles support pancreatic cancer aggressiveness. J Clin Invest 126: 4140–4156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CL, Huang CJ, Yang SH, Chang CC, Huang CC, Chien CC, Yang RN (2016) Discovery of genes from feces correlated with colorectal cancer progression. Oncol Lett 12: 3378–3384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Chen Q, Yin D, Shi S, Yu L, Zhou S, Chen E, Zhou Z, Shi Y, Fan J et al (2017) Novel role of semaphorin 3A in the growth and progression of hepatocellular carcinoma. Oncol Rep 37: 3313–3320 [DOI] [PubMed] [Google Scholar]
- Li Y, Zhang X, Ma A, Kang Y (2021) Rational application of beta‐hydroxybutyrate attenuates ischemic stroke by suppressing oxidative stress and mitochondrial‐dependent apoptosis via activation of the Erk/CREB/eNOS pathway. ACS Chem Neurosci 12: 1219–1227 [DOI] [PubMed] [Google Scholar]
- Lien EC, Westermark AM, Zhang Y, Yuan C, Li Z, Lau AN, Sapp KM, Wolpin BM, Vander Heiden MG (2021) Low glycaemic diets alter lipid metabolism to influence tumour growth. Nature 599: 302–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu KA, Lashinger LM, Rasmussen AJ, Hursting SD (2014) Leucine supplementation differentially enhances pancreatic cancer growth in lean and overweight mice. Cancer Metab 2: 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Qin L, Li BO, Liao Z, Liang J, Xiao X, Xiao X, Mo Y, Huang G, Zhang Z et al (2017) Inactivation of HMGCL promotes proliferation and metastasis of nasopharyngeal carcinoma by suppressing oxidative stress. Sci Rep 7: 11954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney CE, Pirman D, Chubukov V, Sleger T, Hayes S, Fan ZP, Allen EL, Chen Y, Huang L, Liu M et al (2019) A chemical biology screen identifies a vulnerability of neuroendocrine cancer cells to SQLE inhibition. Nat Commun 10: 96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makohon‐Moore AP, Zhang M, Reiter JG, Bozic I, Allen B, Kundu D, Chatterjee K, Wong F, Jiao Y, Kohutek ZA et al (2017) Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat Genet 49: 358–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PM, Gopal E, Ananth S, Zhuang L, Itagaki S, Prasad BM, Smith SB, Prasad PD, Ganapathy V (2006) Identity of SMCT1 (SLC5A8) as a neuron‐specific Na+‐coupled transporter for active uptake of L‐lactate and ketone bodies in the brain. J Neurochem 98: 279–288 [DOI] [PubMed] [Google Scholar]
- Martinez‐Outschoorn UE, Lin Z, Whitaker‐Menezes D, Howell A, Lisanti MP, Sotgia F (2012) Ketone bodies and two‐compartment tumor metabolism: stromal ketone production fuels mitochondrial biogenesis in epithelial cancer cells. Cell Cycle 11: 3956–3963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayers JR, Torrence ME, Danai LV, Papagiannakopoulos T, Davidson SM, Bauer MR, Lau AN, Ji BW, Dixit PD, Hosios AM et al (2016) Tissue of origin dictates branched‐chain amino acid metabolism in mutant Kras‐driven cancers. Science 353: 1161–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayers JR, Wu C, Clish CB, Kraft P, Torrence ME, Fiske BP, Yuan C, Bao Y, Townsend MK, Tworoger SS et al (2014) Elevation of circulating branched‐chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat Med 20: 1193–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikaelian I, Malek M, Gadet R, Viallet J, Garcia A, Girard‐Gagnepain A, Hesling C, Gillet G, Gonzalo P, Rimokh R et al (2013) Genetic and pharmacologic inhibition of mTORC1 promotes EMT by a TGF‐beta‐independent mechanism. Cancer Res 73: 6621–6631 [DOI] [PubMed] [Google Scholar]
- Mizrahi JD, Surana R, Valle JW, Shroff RT (2020) Pancreatic cancer. Lancet 395: 2008–2020 [DOI] [PubMed] [Google Scholar]
- Ni T, Wang H, Zhan D, Tao L, Lv M, Wang W, Chu Z, Zhou Z, Sunagawa M, Liu Y (2021) CD133+/CD166+ human gastric adenocarcinoma cells present the properties of neoplastic stem cells and emerge more malignant features. Life Sci 269: 119021 [DOI] [PubMed] [Google Scholar]
- Nicolle R, Gayet O, Duconseil P, Vanbrugghe C, Roques J, Bigonnet M, Blum Y, Elarouci N, Armenoult L, Ayadi M et al (2021) A transcriptomic signature to predict adjuvant gemcitabine sensitivity in pancreatic adenocarcinoma. Ann Oncol 32: 250–260 [DOI] [PubMed] [Google Scholar]
- O'Kane GM, Ladak F, Gallinger S (2021) Advances in the management of pancreatic ductal adenocarcinoma. Can Med Assoc J 193: E844–E851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares O, Mayers JR, Gouirand V, Torrence ME, Gicquel T, Borge L, Lac S, Roques J, Lavaut M‐N, Berthezène P et al (2017) Collagen‐derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat Commun 8: 16031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker SJ, Amendola CR, Hollinshead KER, Yu Q, Yamamoto K, Encarnación‐Rosado J, Rose RE, LaRue MM, Sohn ASW, Biancur DE et al (2020) Selective alanine transporter utilization creates a targetable metabolic niche in pancreatic cancer. Cancer Discov 10: 1018–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Escuredo J, Van Hee VF, Sboarina M, Falces J, Payen VL, Pellerin L, Sonveaux P (2016) Monocarboxylate transporters in the brain and in cancer. Biochem Biophys Acta 1863: 2481–2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues LM, Uribe‐Lewis S, Madhu B, Honess DJ, Stubbs M, Griffiths JR (2017) The action of beta‐hydroxybutyrate on the growth, metabolism and global histone H3 acetylation of spontaneous mouse mammary tumours: evidence of a beta‐hydroxybutyrate paradox. Cancer Metab 5: 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraon P, Cretu D, Musrap N, Karagiannis GS, Batruch I, Drabovich AP, van der Kwast T, Mizokami A, Morrissey C, Jarvi K et al (2013) Quantitative proteomics reveals that enzymes of the ketogenic pathway are associated with prostate cancer progression. Mol Cell Proteom 12: 1589–1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber FS, Deramaudt TB, Brunner TB, Boretti MI, Gooch KJ, Stoffers DA, Bernhard EJ, Rustgi AK (2004) Successful growth and characterization of mouse pancreatic ductal cells: functional properties of the Ki‐RAS(G12V) oncogene. Gastroenterology 127: 250–260 [DOI] [PubMed] [Google Scholar]
- Shukla SK, Gebregiworgis T, Purohit V, Chaika NV, Gunda V, Radhakrishnan P, Mehla K, Pipinos II, Powers R, Yu F et al (2014) Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab 2: 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A (2019) Cancer statistics, 2019. CA Cancer J Clin 69: 7–34. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Z, Zhou J, Cheng Z, Lu P (2015) Squalene epoxidase (SQLE) promotes the growth and migration of the hepatocellular carcinoma cells. Tumour Biol 36: 6173–6179 [DOI] [PubMed] [Google Scholar]
- Thomas LK, Ittmann M, Cooper C (1982) The role of leucine in ketogenesis in starved rats. Biochem J 204: 399–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PY, Lee MS, Jadhav U, Naqvi I, Madha S, Adler A, Mistry M, Naumenko S, Lewis CA, Hitchcock DS et al (2021) Adaptation of pancreatic cancer cells to nutrient deprivation is reversible and requires glutamine synthetase stabilization by mTORC1. Proc Natl Acad Sci 118: e2003014118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaziri‐Gohar A, Zarei M, Brody JR, Winter JM (2018) Metabolic dependencies in pancreatic cancer. Front Oncol 8: 617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber DD, Aminzadeh‐Gohari S, Tulipan J, Catalano L, Feichtinger RG, Kofler B (2020) Ketogenic diet in the treatment of cancer – Where do we stand? Mol Metab 33: 102–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Kimmelman A, Lyssiotis C, Hua S, Chu G, Fletcher‐Sananikone E, Locasale J, Son J, Zhang H, Coloff J et al (2012) Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149: 656–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Xie C (2017) The role of OXCT1 in the pathogenesis of cancer as a rate‐limiting enzyme of ketone body metabolism. Life Sci 183: 110–115 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhang T, Yang W, Chen H, Geng X, Li G, Chen H, Wang Y, Li L, Sun B (2021) Beneficial diets and pancreatic cancer: molecular mechanisms and clinical practice. Front Oncol 11: 630972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XG, Chudnovskiy A, Baudrier L, Prizer B, Liu Y, Ostendorf BN, Yamaguchi N, Arab A, Tavora B, Timson R et al (2021) Functional genomics in vivo reveal metabolic dependencies of pancreatic cancer cells. Cell Metab 33: 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zijlstra A, Mellor R, Panzarella G, Aimes RT, Hooper JD, Marchenko ND, Quigley JP (2002) A quantitative analysis of rate‐limiting steps in the metastatic cascade using human‐specific real‐time polymerase chain reaction. Cancer Res 62: 7083–7092 [PubMed] [Google Scholar]