

Summary
Understanding the immune cell abundance of cancer and other disease-related tissues has an important role in guiding disease treatments. Computational cell type proportion estimation methods have been previously developed to derive such information from bulk RNA sequencing data. Unfortunately, our results show that the performance of these methods can be seriously plagued by the mismatch between training data and real-world data. To tackle this issue, we propose the DAISM-DNNXMBD (XMBD: Xiamen Big Data, a biomedical open software initiative in the National Institute for Data Science in Health and Medicine, Xiamen University, China.) (denoted as DAISM-DNN) pipeline that trains a deep neural network (DNN) with dataset-specific training data populated from a certain amount of calibrated samples using DAISM, a novel data augmentation method with an in silico mixing strategy. The evaluation results demonstrate that the DAISM-DNN pipeline outperforms other existing methods consistently and substantially for all the cell types under evaluation in real-world datasets.
Keywords: cell type proportion estimation, deconvolution, data augmentation, data simulation, deep learning
Graphical abstract
Highlights
-
•
We propose a data augmentation method (DAISM) for DNN-based cell type deconvolution
-
•
DAISM-DNN enables accurate cell type deconvolution with dataset-specific training data
-
•
DAISM-DNN is robust to random errors in calibration samples
-
•
Trained DAISM-DNN model is reusable across biomedical experiments following same SOP
The bigger picture
Computational cell type deconvolution methods were developed to understand the cellular heterogeneity in disease-related tissues from bulk RNA-seq data. Due to the presence of strong batch effects, the performance of existing methods could fluctuate greatly when applied to different datasets even with the latest development in batch normalization or platform-agnostic signature designs. To tackle this issue, we proposed a DNN-based cell abundance estimation method with dataset-specific training data populated from a certain number of calibrated samples from a target dataset using DAISM, a data augmentation method using an in silico mixing strategy. DAISM-DNN enables accurate cell type proportions prediction and is robust to random errors in the ground truth cell type proportions of calibration samples. Importantly, we showed that with strict SOPs, it is possible to create a “train once, reuse many times” DAISM-DNN model for multiple biomedical experiments without the need for retraining.
Lin et al. propose the DAISM-DNN pipeline for cell type deconvolution from bulk RNA-seq data using deep neural networks trained with dataset-specific training data generated from calibration samples augmented with in silico mixing strategies. DAISM-DNN enables accurate intra- and inter-sample deconvolution and is robust to random errors in ground truth cell type proportions of calibration samples. The trained DAISM-DNN model can also be used across multiple biomedical experiments if these experiments are conducted with a strict SOP to ensure quality consistency.
Introduction
It has been shown that the cellular composition of immune infiltrates in tumors is directly linked to tumor evolution and response to treatments.1,2 A high intratumoral infiltration of lymphocytes and dendritic cells is a favorable prognostic marker for cancer treatment,3,4 while a high stromal content of cancer-associated fibroblasts and M2 macrophages has been shown to be associated with poor outcomes.5,6 Particularly, recent progress in immunotherapy has led to durable clinical benefits, but only in a subpopulation of patients with “hot” tumor immune microenvironments that are characterized by a high infiltration of lymphocytes.7 Therefore, knowledge of the patient-specific immune cell type proportion of solid tumors is invaluable in predicting disease progression or drug response as well as stratifying patients to select the most suitable treatment options.
In the past, fluorescence-activated cell sorting (FACS) and immunohistochemistry (IHC) were used as gold standards to measure the cellular components in a patient sample.8 FACS requires a large number of cells, which limits its clinical applications. On the other hand, IHC only provides information on the cellular composition of a single biopsy slice, which may not represent the full tumor microenvironment (TME) due to its heterogeneity.
With the increasing availability of RNA quantification technologies, such as microarrays, high-throughput RNA-seq, and NanoString, the large-scale expression profiling of clinical samples has become feasible in routine clinical settings.9 However, these methods only measure the average expression of genes from the heterogeneous samples in their entirety but do not provide detailed information on their cellular compositions. To bridge this gap, computational methods have been proposed to estimate individual cell type abundance from the bulk RNA data of heterogeneous tissues (see Table S1). In these methods, the abundance of each cell type from the mixed sample is quantified by aggregating the expression levels of the marker genes into an abundance score (MCP-counter10), by measuring the enrichment level of the marker genes using statistical analysis (xCell11), or by using computational deconvolution methods, such as least-squares regression (quanTIseq,12 EPIC13), support vector regression (SVR) (CIBERSORT,14 CIBERSORTx15), or nonnegative matrix factorization (NMF),16 to derive an optimal dissection of the original sample based on a set of pre-identified cell-type-specific expression signatures.
Undoubtedly, it is very challenging in practice for any of these computational methods to meet the rigid robustness and reliability requirements of biomedical or clinical studies over a broad range of sample types and conditions as well as sequencing technical platforms. For example, in deconvolution-based algorithms, it is expected that the cell-type-specific expression signature should truly represent the expression characteristics of the underlying immune cells from the mixture samples. Unfortunately, the signature gene expression levels employed in existing methods are derived from either FACS-purified and in vitro differentiated or simulated cell subsets or single-cell experiments. The application of antibodies, culture material, or physical disassociation may affect the cell status, resulting in signatures that deviate from those of the actual cells in vivo. Moreover, technical and biological variations between RNA quantification experiments may introduce additional confounding factors that lead to sample or dataset-specific bias in cell type estimation. Similarly, marker gene expression aggregating methods such as MCP-counter require highly specific signatures with genes that are exclusively and stably expressed in certain cell types, which may not be possible for some immune cell lineages.17
Recently, the development of deep neural networks (DNNs) has granted computational power to resolve complex biological problems using data-driven approaches with the vast trove of data available from the biomedical research community powered by high-throughput genomic sequencing technologies.18,19 An application of DNN in cell type proportion estimation was proposed in Scaden,20 where a neural network was trained on bulk RNA-seq data simulated from the scRNA-seq data of different immune cell types to predict cell type proportions from the bulk expression of cell mixtures. A DNN-based model could automatically create optimal features for cell fraction estimation during the training process, thus alleviating the need to generate reliable gene expression profile (GEP) matrices for different cell types. Moreover, it learns the potentially intricate non-linear relationships between the gene expression composition and cell type proportions from training data, which are not possible to be captured by linear models used in other deconvolution algorithms. However, as the performance of DNN is still subject to the same statistical learning principle that test and train conditions must match, it is challenging for a DNN-based algorithm to deliver consistent performance under different experimental conditions unless sufficient ground truth data are available to train a specific predictive model for each distinct experimental condition. As a DNN model usually requires tens of thousands of training samples, the cost of implementing such a method would be prohibitive in practice.
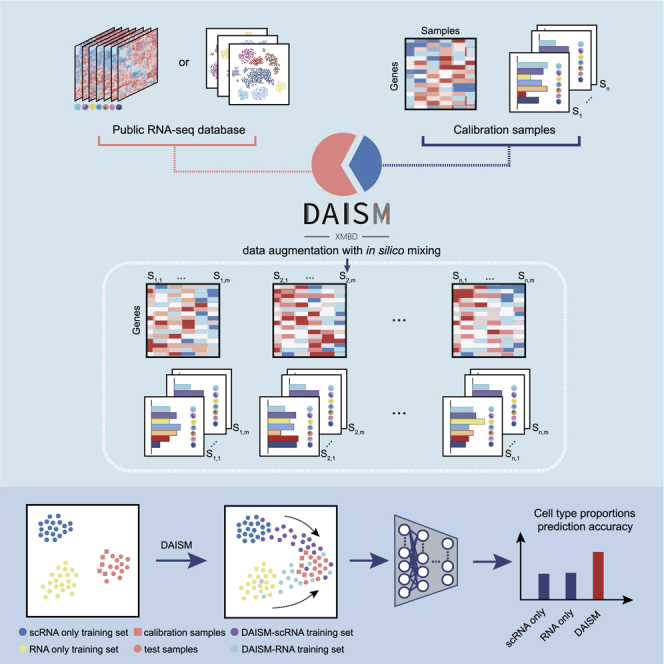
To address these challenges, we developed the DAISM-DNN pipeline (Figure 1) that consists of an in silico data augmentation method in collaboration with a DNN model to achieve robust and highly accurate cell type proportion estimation. The DAISM-DNN pipeline performs model training on a dataset augmented from a calibration dataset comprised of a certain amount of the actual data from the same batch of RNA-seq experiments, of which the ground truth cell type proportions are available for calibration. DAISM-DNN is able to deliver consistent cell type abundance profiling accuracies over different datasets. In addition, it is highly customizable and can be tailored to estimate the abundance of a large variety of cell types including those that are difficult for existing methods to estimate due to the lack of marker genes or GEP signature matrices, and immune cells with overlapping markers or signature genes.21,22
Figure 1.

The DAISM-DNN pipeline
A typical DAISM-DNN workflow involves the following steps to perform cell type proportion estimation. (1) Measure the ground truth proportions of the cell types of interest in a certain portion of calibration samples from the batch of samples to be evaluated. (2) Perform bulk RNA-seq on the calibration and test samples to obtain their expression profiles. (3) Perform data augmentation on the expression profiles of the calibration samples through in silico mixing with the RNA-seq data of purified cells or scRNA-seq data (DAISM). (4) Train a DNN using the augmented data. (5) Use the trained DNN model for cell type proportion estimation in the remaining samples with their bulk expression profiles. Steps (1)–(4) could be optional if the DNN model has already been trained for the given RNA-seq experimental conditions.
Results
There is no one-size-fits-all algorithm for cell type proportion estimation
We first evaluated nine state-of-the-art cell type proportion estimation algorithms, namely, CIBERSORT, CIBERSORTx, EPIC, quanTIseq, MCP-counter, xCell, ABIS,21 MuSiC,23 and Scaden, on 11 independent real-world datasets (n = 685 total samples) acquired using different techniques or platforms (see Table S2) and three simulated datasets generated with scRNA-seq, bulk RNA-seq of purified cells, and microarray, respectively (Experimental procedures). Importantly, the methods under evaluation included the most recent developments to improve the cross-dataset robustness of cell type proportion estimation. For CIBERSORT, we further included four established basis signature matrices, namely, IRIS,24 LM22,14 TIL10,12 and immunoStates25 in our evaluation. ImmunoStates used a basis matrix built using 6,160 samples with different disease states across 42 microarray platforms to mitigate the technical bias from different platforms. In MuSiC, the deconvolution algorithm further included appropriate weighting of genes showing cross-subject and cross-cell consistency to enable the transfer of cell-type-specific expression information from one dataset to another. CIBERSORTx also implemented two batch correction modes (B-mode and S-mode) to reduce the potential bias from batch effects, and we tested with both modes.
Our results show that none of these methods were able to address the estimation bias problem to deliver consistently better results than others across multiple datasets. With regard to the overall prediction accuracy across all cell types and datasets, the DNN-based method (Scaden) achieved the highest average rank in terms of performance in RNA-seq data deconvolution, while ABIS ranked first dealing with microarray data. However, the improvement over most other methods was not significant (Friedman test with post hoc two-tailed Nemenyi test, ; Figures S1, S2A, and S2B). In fact, as the performance of DNN is still subject to the same statistical learning principle that test and train conditions must match, Scaden, similar to other algorithms, showed inconsistent performance on different datasets. A t-distributed stochastic neighbor embedding (t-SNE) analysis of all test datasets demonstrated significant batch effects among those datasets, and the difference among the testing samples was dominated by batch effects rather than cellular composition (Figure S2C). It is understandable that traditional signature-based approaches could suffer from batch effect when the dataset used to derive the signature is very different from the test datasets. Furthermore, as both signature-based and marker-based deconvolution methods assume linear relationships between gene expressions and cell type proportions, their performance could also be influenced by the actual non-linear relationships between them (Figure S3). This result partially explains the inconsistent performance of the existing methods on different datasets and the challenge in developing a one-size-fits-most cell type proportion estimation method that performs uniformly well under different experimental conditions.
DAISM-DNN enables accurate and robust cell type proportion estimation
To overcome the aforementioned limitations, we developed DAISM, a data augmentation method, to produce dataset-specific training data for DNN model training, provided that a certain amount of RNA-seq data with ground truth cell type proportions from the same batch are available for calibration. DAISM generates a large amount of dataset-specific pseudo training data by performing in silico mixing of the calibration data with publicly available scRNA-seq data or RNA-seq data from purified cells at predefined ratios that are known to the training process. A DNN model that predicts the cell type proportions for the remaining samples is then trained using the DAISM-generated pseudo training data (Figure 1).
We evaluated the performance of DNN models trained from DAISM-generated pseudo training data (DAISM-DNN) on the RNA-seq dataset SDY67. A total of 250 samples with ground truth proportions of five cell types (B cells, CD4 T cells, CD8 T cells, monocytes, and NK cells) from SDY67 were used for analysis in this paper. The performance of DAISM-DNN was measured from 30 permutation tests independently. For each permutation test, we used 50 randomly selected samples from SDY67 as testing data, and the remaining 200 samples were served as calibration data, which were augmented with the scRNA-seq data PBMC8k of the five cell types to create the training data (Experimental procedures). DNN was trained on DAISM-generated training data. For comparison, we also employed other cell type proportion estimation algorithms on the same 50 testing samples. Overall, DAISM-DNN outperformed all other algorithms by a significant margin from 30 permutated tests for all the cell types under evaluation (Figures 2 and S4–S6). When evaluated by the average per-cell-type Pearson correlation between the predicted and ground truth cell type proportions, DAISM-DNN achieved the highest correlation, followed by Scaden (Figure 2B). In addition, DAISM-DNN had the lowest root-mean-square error (RMSE) and the highest Lin’s concordance correlation coefficient (CCC), followed by ABIS (Figure 2B). We replaced the scRNA-seq data with the RNA-seq data of purified cells to generate training data with DAISM and did not find a significant difference in performance between the DNN models derived from these two approaches (DAISM-scRNA versus DAISM-RNA; Figure S7).
Figure 2.

Performance of different algorithms on datasets SDY67, GSE59654, and GSE107990
(A) Scatterplots of ground truth fractions (x axis) and predicted cell fractions (y axis) for DAISM-DNN and CIBERSORTx (S-mode). The bar plot shows the Pearson correlation for each cell type in 30 permutation experiments. The value in bar plots indicates the mean value of 30 experiments.
(B) Boxplot of the mean of per-cell-type Pearson correlations for 11 methods, and bar plots of RMSE (right) and CCC (left) for nine methods. All data in bar plots are presented as the mean ± SD. Note that RMSE and CCC are not suitable for evaluating the two marker-based methods, MCP-counter and xCell. Two-sided paired Student’s t tests were used for comparing DAISM-DNN with other methods.
We further tested DAISM-DNN on two microarray datasets GSE59654 and GSE107990 with 153 and 164 samples, respectively. Similarly, we randomly selected 50 samples as test data, and the remaining samples were augmented with the scRNA-seq data of the respective cell types to generate the training dataset for DAISM-DNN. Results from 30 permutation tests showed that DAISM-DNN outperformed the other algorithms by a significant margin for all the cell types under evaluation (Figures 2 and S4–S6), except for GSE107990 where the difference between ABIS and DAISM-DNN was not significant.
We extended our evaluation to a fine-grained cell population of 11 cell types: naive B cells, memory B cells, naive CD4 T cells, memory CD4 T cells, regulatory T cells, naive CD8 T cells, memory CD8 T cells, monocytes, NK cells, macrophages, and myeloid dendritic cells (mDCs). The results were compared with those of CIBERSORTx (S-mode and B-mode), ABIS, and xCell, which are also able to produce estimations of fine-grained cell type proportions. Comparisons were only performed on cell types where ground truth cell type proportion information was available for each dataset (Figure S8). The results indicate clear advantages of DAISM-DNN over traditional methods not only in overall performance but also for all individual cell types and datasets in terms of the Pearson correlation (Figure S8B), RMSE, and CCC (Figure S8C).
To understand if the performance gain of DAISM-DNN indeed comes from the data-specific training set generated using DAISM, we generated in silico mixed training data using DAISM with calibration data from SDY67 as well as the direct mixing of RNA-seq data from sorted cells or scRNA-seq data of selected cell types (Experimental procedures). All the in silico mixed data from different mixing strategies followed the same cell type proportions. The t-SNE plot revealed highly distinct clusters of these datasets. Importantly, only the clusters of the DAISM-generated dataset strongly overlapped with SDY67, while the clusters from the remaining datasets showed a clear gap from SDY67, demonstrating strong batch effects between them and the real samples (Figure 3). We also used a combination of simulated training data and real data with a priori cell fraction information as suggested in Menden et al.20 to train DNN models. We integrated five RNA-seq real-world data, including 200 samples excluded for testing of SDY67, with simulated training data generated by scRNA-seq data respectively. The training data size was kept the same in these separate trainings. Obvious performance gains were observed only when calibration samples largely overlapped with the simulated training dataset. Furthermore, DNN models trained with DAISM-generated training datasets achieved significantly better performance than those trained with other in silico training data (Figure 3B), demonstrating the critical role of training data in determining the performance of DNN-based models and the effectiveness of DAISM in creating a training dataset that matches the intrinsic distributions of the real-life data to enable highly accurate cell type proportion estimation.
Figure 3.

Performance of DNN on different training datasets
(A) The t-SNE projection of the SDY67 dataset (n = 50 samples) and training datasets (n = 500 samples per platform) constructed by different strategies and purified samples from different platforms, colored according to training datasets.
(B) The cell type proportion estimation performance on SDY67 was evaluated using Pearson correlation (left), CCC (middle), and RMSE (right). Models trained by nine training datasets generated by different mixing strategies.
In addition, we asked whether DAISM-generated training sets can be similarly leveraged by machine learning models other than DNN to deliver the same performance gain. To this end, we performed experiments on k nearest neighbor regression and SVR. Interestingly, although these two methods were able to achieve better prediction performance when real-life samples were included in the training sets, they were not able to gain further performance improvement when the training sets were augmented with DAISM-generated data, as these non-neural-network models lack the ability to fit a sophisticated model toward better prediction when a large amount of high-quality data are available. As a result, their performances were not as good as DAISM-DNN (Figure S9).
We further asked whether the performance gain of DAISM-DNN is mainly due to the dataset-specific training data generated from DAISM or its DNN design. To this end, we first trained DAISM-DNN and Scaden on the same training set (S4) that combined the four pre-generated peripheral blood mononuclear cells (PBMC) in silico mixtures provided by Menden et al., and we tested their abundance prediction accuracy using 50 randomly selected samples from SDY67. Results from 30 permutation tests showed that there was no significant difference between the two models in Pearson correlations (Figure S10). In terms of CCC and RMSE, the DNN used in DAISM-DNN generated slightly inferior results than the ensemble model in Scaden. On the other hand, when trained on DAISM-generated training datasets with calibration samples from SDY67, both models gained sizable and highly similar improvements in prediction accuracy.
We further evaluated DAISM-DNN with DNNs of different model complexity (layers, number of neurons). In total, we tested 25 hyperparameter settings of different numbers of neurons and layers (Table S3). Results on three datasets (SDY67, GSE59654, and GSE107990) showed that only small variations of prediction performance over different hyperparameter settings were observed for both coarse-grained and fine-grained deconvolution tasks except for some extreme configurations with one hidden layer and small number of hidden neurons (<64) (Figure S11).
Scaling and error sensitivity of DAISM-DNN
To understand how the number of calibration samples would affect the performance of DAISM, we compared the cell type proportion estimation performance of DAISM-DNN when different numbers of calibration samples were used in creating the augmented training data. We found that in general, the estimation accuracy improved with an increasing number of calibration samples used in creating the in silico mixed training data (Figures 4A and S12). When evaluated by CCC or RMSE, which require that the predicted cell fractions follow the real fractions in terms of absolute numbers, the estimation performance improved dramatically when the number of real samples used in in silico mixing increased from zero to 20–40. Beyond that, the rate of improvement slowed down significantly with more calibration samples. Therefore, the actual number of calibration samples have to be decided based on the balance between the desired prediction quality and the costs to create the calibration samples.
Figure 4.

Scaling and error sensitivities of DAISM-DNN
(A) The effect of calibration sample size on the DAISM-DNN pipeline, assessed by the Pearson correlation, CCC, and RMSE of the cell type proportion estimation results (on SDY67). For each calibration sample size, 30 permutation tests were conducted.
(B) The performance of DAISM-DNN when ground truth of calibration samples (n = 30) has different degrees of deviation (5, 10, 15, 20%). ns, not significant; paired one-way ANOVA test.
As cell type proportion measurements on the calibration sample can never be perfect, the performance of DAISM may be affected by measurement errors. To understand how the quality of calibration samples affects the performance of DAISM-DNN, we generated artificial RNA-seq data of different cell type proportions from scRNA-seq data as our test dataset, and then added different levels of random permutations to the ground truth cell type proportions on the selected calibration samples to simulate the noise in the real world (Experimental procedures). Interestingly, regardless of the level of permutations, random measurement errors in the calibration samples did not significantly affect the prediction accuracy of DAISM-DNN when the number of calibration samples is small (n = 30; Figure 4B). For the case where a larger number of calibration samples (n = 200) is available, the prediction accuracy would be affected, and the level of degradation increases with the level of permutation (Figure S13). However, the overall degradation in prediction accuracy is actually very small, suggesting that DAISM-DNN is robust to random errors in the cell type proportions of calibration samples.
Moreover, we investigated at which abundance can DAISM-DNN reliably identify the presence of immune cells using in silico mixed bulk RNA-seq samples that contain an increasing amount of the cell type of interest with a background of other immune or cancer cells (Experimental procedures). We defined the minimal detection fraction as the minimal fraction of spike-in cells needed for the score to be significantly different from zero. For all cell types, the minimal detection fractions of DAISM-DNN were smaller than those of CIBERSORTx and xCell (Figures S14A and S14B), indicating the superior detection sensitivities of DAISM-DNN at low cell abundance.
Applicability of DAISM-DNN in real-life biomedical experiments
A limitation of DAISM-DNN is that it requires calibration samples to train the dataset-specific models, which may not always be available. On the other hand, it is common that in many scenarios, rigorous standard operation procedure (SOP) and quality control are enforced to derive consistent gene expression quantification results across experiments, such as defined by the gene expression-based biomarker Oncotype DX.26,27 In cases where strict SOPs are enforced, it is possible to pre-train a generic DAISM-DNN model that can be used across different batches without the need for retraining every time. To verify this concept, we generated a validation dataset comprising 36 human PBMCs samples assayed by the same RNA-seq panel in two separate batches (Experimental procedures). The first batch consisted of 30 samples that were used as calibration samples to generate the DAISM-mixed training dataset, while the other batch consisted of six samples for testing. The ground truth cell type proportions of both batches were established using mass cytometry (CyTOF, see Experimental procedures).
To generate the DAISM-mixed data for training in this study, we used CITE-seq28 data, which provide single-cell transcriptome and surface proteins simultaneously, from two public CITE-seq datasets (PBMC5k and PBMC10k) for augmentation. Clusters of different cell types were identified separately for both CyTOF and CITE-seq datasets through meta-clustering on 11 surface marker proteins in common in these two datasets, and manually annotated based on canonical marker expression patterns consistent with known immune cell types. These clusters were further pairwise linked according to Pearson correlation of normalized mean marker expression of each cluster to identify matching populations between them (Figures S15 and S16A; Experimental procedures). It can be seen from the results that with a strict SOP, it is possible to minimize the technical variance of gene expression results between batches (Figure S16B) and enable more stable, robust, and accurate cell type proportion estimation compared with other established methods through a pre-established DAISM-DNN model trained on different batches (Figure S16C).
Discussion
Understanding the cellular heterogeneity in disease-related tissues is essential for the identification of cellular targets for treatments. To this end, computational methods have been developed to quantify cell type compositions from the GEPs of bulk samples, thus allowing the elucidation of cell type contributions to disease from highly available disease-related bulk RNA-seq data. Existing deconvolution methods rely on pre-selected cell-type-specific marker genes or signatures based on cell-type-specific gene expression, which could be derived from existing RNA-seq datasets of single cells or purified cell lines of target cells. The accuracy of these methods is therefore subject to the effectiveness of the selected GEPs to represent different bulk RNA-seq datasets under testing. Unfortunately, due to the presence of strong non-biological cross-platform variations, the performance of such methods could fluctuate greatly when applied to different datasets even with the latest advancements in batch normalization15 or platform-agnostic signature designs.23,25
We developed DAISM-DNN to meet the challenge of accurate cell type proportion quantification for bulk tissues using GEPs derived from disparate sources. One of the key features that differentiates our method from previous works is that we used a DNN-based, data-driven approach that is free from manually curated marker genes or expression signatures. By learning directly from the data, DAISM-DNN not only discovers new features that were previously unrevealed from conventional methods, but also leverages their intricate interactions with target phenotypes to achieve accurate prediction results, which is impossible when shallow models are used.
Another key feature of DAISM-DNN is that instead of relying on normalization or platform-agnostic reference profiles to overcome the cross-platform variation problem, DAISM-DNN builds a dataset-specific prediction model from a certain amount of calibration samples from the testing dataset, thus fundamentally avoiding the problem. This requirement may seem stringent and restrictive as the calibration samples need to be sorted to establish the ground truth cell type proportions. However, our results indicated that only a certain amount of calibration samples are needed to train the prediction model thanks to the DAISM data augmentation strategy. More importantly, we have also shown that with stringent SOPs in the overall experimental procedure, such as those being practiced for GEP-based assays for clinical usage,27 it is possible to create a “train once, reuse many times” assay-specific DAISM-DNN model for data generated under the same or similar experimental conditions. Overall, DAISM-DNN is particularly suitable for large cohort studies or routine clinical applications of which highly reliable and accurate cell fraction information is expected and relatively stable RNA-seq experimental conditions are involved.
Finally, despite the success of deep learning, people find it challenging to apply such models in genomic studies in a supervised learning setup due to the scarcity of training samples. The data augmentation strategy as in DAISM provides a broadly applicable framework to create a large amount of in silico mixed artificial training data from a certain amount of real-life samples with the aid of the increasing availability of scRNA-seq datasets or other datasets that provide comprehensive characteristic maps of different cell types. We hypothesize that the algorithmic principles underlying DAISM could be generalized to the deconvolution of other data modalities, e.g., DNA methylation,29, 30, 31 or other gene expression-based prediction tasks that are currently incompatible with deep learning due to limited availability of training data. Of note, despite the flexibility of DAISM, it still has limitations. For example, as a supervised machine learning algorithm, it requires annotated training data. Therefore, it is not possible to extend DAISM-DNN to learning tasks where such data are not available, e.g., prediction tasks that involve cell types that do not have clear annotations or data for augmentation. Future works are warranted to bring the latest development in unsupervised learning32 or few-shot learning,33 where the model can be trained with little data, to overcome this limitation.
Experimental procedures
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Rongshan Yu (rsyu@xmu.edu.cn).
Materials availability
There are no physical materials associated with this study.
Data augmentation through in silico mixing (DAISM)
Deep learning-based approaches require a large amount of training data. In general, existing data from real tissue samples with known fractions of cell types and gene expression levels could be insufficient to use as a training set. In this regard, we extracted a small number of real-life samples with ground truth cell type proportions to use as a calibration dataset, and we applied the DAISM strategy to create a large number of in silico mixed samples from this calibration dataset.
The expression profile of a DAISM-generated (i.e., in silico mixed) sample is calculated as follows. First, we generate a random variable f with uniform distribution between 0 and 1 to determine the fraction of the calibration sample in the mixed sample, and C random variables with Dirichlet distribution , such that to determine the fractions of the immune cells in the mixed sample, where C is the number of cell types. The expression profile of the final mixed sample is then calculated as follows:
where is the expression profile of a real-life sample randomly selected from the calibration dataset as a seed sample for this in silico mixed sample, and is the aggregated expression of single cell samples or purified samples used for data augmentation. When using scRNA-seq dataset for data augmentation (DAISM-scRNA), we have
where is the number of cells of type k extracted randomly from scRNA-seq datasets for mixing, and denote their expression profiles. Note that is further TPM-normalized before mixing. When using RNA-seq data from purified cells (DAISM-RNA) for augmentation, we have
where is the expression profile of a randomly selected purified sample of cell type k from the respective RNA-seq dataset. Once the expression profile of the in silico sample is created, its “ground truth” cell fractions can be calculated as follows:
where is the fraction of cell type k in the in silico mixed sample, and is the ground truth fraction of cell type k in the calibration samples, which is known a priori through experiments, e.g., flow cytometry analysis.
To decide a suitable range of the fractions of calibration samples in the in silico mixed samples, we tested the performance of DAISM-DNN with different maximum fraction of calibration samples on SDY67, and the result indicated that a wider range of the fractions of calibration samples in the mixed samples would ensure that the final generated training data provide both statistical similarity with the test data and diversity for training the DNN, thus leading to better prediction results (Figures S17A and S17B).
The DAISM-DNN pipeline
We trained deep feed-forward, fully connected neural networks (multilayer perceptron networks) on DAISM-generated training data to predict the cell fractions from bulk expression data. The network consists of one input layer, three fully connected hidden layers (1024-512-256) and one output layer, implemented with PyTorch (v1.5.1) in Python (v3.7.7). As a DNN can fit a large feature space with a large number of parameters (i.e., connection weights), we did not perform feature selection in advance. Instead, we used all the genes that were present in both the training and testing datasets as input to the neural network. Moreover, the expression profile of each sample was log2-transformed, and scaled to the range of [0,1] through min-max scaling before training:
Here, is the log2-transformed expression level of gene i, is the vector of the log2-transformed expression levels of all genes of a sample, and is the vector of min-max scaled values.
The network was trained using the back-propagation algorithm with randomly initialized network parameters. The mean-square error (MSE) between the ground truth and predicted absolute cell fractions was used as the loss function. The optimization algorithm Adam was used with an initial learning rate of 1 x 10–4. During the training process, the training set was randomly divided into mini-batches with a batch size of 64. When the average MSE of all mini-batches in the current epoch was higher than that of the last epoch, the learning rate was multiplied by an attenuation coefficient until a minimum of 1 x 10–5 was reached to avoid training noise from excessively large learning rates when the network converged to steady state. We randomly split the training set and the validation set at a ratio of 8:2. Early-stopping strategy was adopted to stop training when the validation error did not decrease for 10 epochs, and the model producing the best results on the validation set during training was selected as the final model for prediction.
To evaluate the effect of the size of training data generated from the same number of calibration samples on the deconvolution performance of DAISM-DNN, we tested DAISM-DNN with different training data sizes ranging from 640 up to 32,000 simulated samples on three PBMC datasets (GSE59654, GSE107990, SDY67), respectively. The performance of each model was measured from 30 permutation tests. In each permutation test, 50 randomly selected samples were held out as the test samples, and the remaining samples from the same dataset were used as calibration samples. The mean CCC performance across cell types improved as the training data size increased on all three datasets (Figure S18). However, the increment started to level off when the training data size increased to 3,200 simulated samples for GSE59654 and GSE107990, and about 12,800 simulated samples for SDY67. Based on this result, we used 16,000 simulated samples as the default size of training data in our experiments.
RNA-seq datasets of purified cells
For the RNA-seq data of purified immune cells to serve as augmentation data, we used 1,533 purified cell samples of the eight immune cell types (B cells, CD4 T cells, CD8 T cells, monocytes, NK cells, neutrophils, endothelial cells, and fibroblast) in this study (Table S4). The raw FASTQ reads were downloaded from the NCBI website. Transcription and gene-level expression quantification were performed using Salmon34 (version 0.11.3) with Gencode v29 after quality control of FASTQ reads using fastp.35 All the software tools were used with their default parameters. Transcripts per million (TPM) normalization was then performed on all the samples.
scRNA-seq datasets
For the scRNA-seq data of different immune cell types, two scRNA-seq datasets of PBMCs from patient blood samples were downloaded from the 10× Genomics website (https://support.10xgenomics.com/single-cell-gene-expression/datasets, “8k PBMCs from a healthy donor” and “6k PBMCs from a healthy donor”), denoted as PBMC8k and PBMC6k respectively. PBMC8k was sequenced on Illumina Hiseq4000 with approximately 92,000 reads per cell, and 8,381 cells were detected in total. We used PBMC8k dataset for augmentation in both coarse-grained and fine-grained deconvolution and PBMC6k dataset for generating artificial simulated mixtures to evaluate error sensitivities of DAISM-DNN. The raw scRNA-seq reads were aligned to the GRCh38 reference genome and quantified by Cell Ranger36 (10× Genomics version 2.1.0). The resulting expression matrix was then processed using Seurat37 (v3.1.1). First, cells with less than 500 genes or greater than 10% mitochondrial RNA content and genes expressed in less than five cells were excluded from analysis. Then, cells with abnormally high gene counts were considered as cell doublets and were excluded from further analysis. The raw unique molecular identifier (UMI) counts were log-normalized and the top 2,000 highly variable genes were called based on the average expression (between 0.0125 and 3) and average dispersion (). Principal component analysis was performed on the highly variable genes to further reduce the dimensionality of the data. Finally, clusters were identified using the shared nearest neighbor (SNN)-based clustering algorithm on the basis of the first 20 principal components with an appropriate resolution.
The identified clusters were annotated on the basis of marker genes' expression levels. The marker genes were obtained from the CellMarker database38 for the target cell types in peripheral blood, specifically, CD4 for CD4 T cells, CD8A and CD8B for CD8 T cells, MS4A1 and CD79A for B cells, CD14 and FCGR3A for monocytes, GNLY for NK cells, and FLT3 and FCER1A for dendritic cells. Cell types were identified manually by checking if the respective marker genes were highly differentially expressed in each cluster. The clusters without high expression on the selected marker genes or with high expression on the marker genes of other cell types were grouped into the “unknown” type.
For fine-grained deconvolution, PBMC8k was further clustered into finer groups based on the major cell types and served as augmentation data. B cells were subclustered into naive B cells and memory B cells. CD4 T cells and CD8 T cells were further grouped into naive CD4 T cells, memory CD4 T cells, naive CD8 T cells, and memory CD8 T cells, respectively. Myeloid dendritic cells and monocytes came from monocyte lineages.
CITE-seq datasets
In deconvolving the in-house validation dataset, we used two CITE-seq PBMC datasets that provide read counts of both mRNAs and cell surface proteins as augmentation data. The datasets were downloaded from 10× Genomics website (PBMC5k from https://support.10xgenomics.com/single-cell-gene-expression/datasets/3.0.2/5k_pbmc_protein_v3, PBMC10k from https://support.10xgenomics.com/single-cell-gene-expression/datasets/3.0.0/pbmc_10k_protein_v3). The PBMCs from a healthy donor were stained with TotalSeq-B antibodies (BioLegend) in both datasets for identification of cell surface proteins and were sequenced by Illumina NovaSeq for scRNA-seq. For mRNA profiles in CITE-seq, the same preprocessing steps for scRNA-seq data were applied for quality control. The protein counts were pre-processed with the centered-log-ratio normalization28 prior to clustering.
Datasets for benchmarking
We used 11 public bulk RNA-seq and microarray datasets to evaluate the performance of different cell type proportion estimation methods, of which the expression data and the corresponding ground truth cell fractions were publicly available with reference to the original publications and used accordingly in our benchmarking tests. The cell fractions for SDY67, GSE107990, and GSE59654 were taken from the supplementary materials of the publication.21 Only samples that have the complete ground truth of coarse- or fine-grained cell types were used for analysis. Details on all datasets including references to the original publications can be found in Table S2.
We also generated three datasets of simulated mixtures using single cell samples from PBMC8k, samples from bulk RNA-seq of purified cells, and microarray, respectively, denoted as “simS”, “simR,” and “simM”. Each dataset contains 50 samples.
In-house PBMC validation samples
To further evaluate the validity of our method, we generated an in-house validation dataset of human PBMCs under IRB approval from The First Affiliated Hospital of Xiamen University.
PBMC processing. A total of 36 PBMC samples were isolated from whole blood by Ficoll density gradient centrifugation and stored in liquid nitrogen with frozen solution (90% fetal bovine serum [FBS] and 10% DMSO). Cryopreserved PBMCs were thawed with pre-warm complete RPMI 1640 medium (RPMI1640, 10% FBS) containing 25 U/ml benzonase. Cells from each sample were washed, counted, adjusted to 2 x 106 living cells/stain and transferred to a new 5-mL polystyrene round-bottom tube. Simultaneously, 1 x 105 living cells/sample were washed with PBS and quickly frozen in liquid nitrogen for RNA-seq.
RNA extraction and library preparation. Total RNA from PBMC samples was extracted using RNeasy Mini Kit (QIAGEN) according to the manufacturer’s instructions. Quantification of RNA concentration was performed by Quantus fluorometer and Quantus RNA HS Assay Kit (Promega). Fragment length was assessed using an Agilent 2100 Bioanalyzer and RNA HS Kit (Agilent). During library preparation, RNA was first fragmented at 95°C for 0–15min according to the DV200 value estimated by Agilent 2100 Bioanalyzer System, then it underwent reverse-transcript, cDNA synthesis, and strand-specific library preparation using NEBNext Ultra II Directional RNA Library Prep Kit for Illumina(NEB).
Hybrid capture and sequencing. RNA libraries were captured separately by AmoyDx Master Panel, which contains 2,396 genes for RNA expression detection and 44 genes for fusion detection. Captured products were amplified and quantified by Quantus fluorometer. Library size was assessed using Agilent 2100 Bioanalyzer. After pooling, libraries were then sequenced on Illumina NovaSeq 6000 instrument (Illumina) with PE150 strategy. Sequencing data were analyzed and annotated with an in-house developed pipeline. A set of experimental and data quality control parameters were set up. For processing RNA-seq data, quality assessment was carried out using in-house script FormatFastQ (v2.4.0). Alignment to targeted genes from Gencode hg37 was performed using STAR (v2.7.2b) and gene counts were quantified using RSEM (v1.3.3).
Mass cytometry of validation samples. Isotope-labeled antibodies were purchased from PLT Tech (Hangzhou, China), where antibodies conjugation and testing were performed. After thawing and preprocessing, PBMC samples were stained by surface antibodies (see Table S5) to 2 x 106 cells for 30 min at room temperature (RT). All samples were then washed with Maxpar Cell Staining Buffer and incubated in Nuclear Antigen Staining Buffer for 30 min at RT. Then, samples were washed and stained by intracellular antibody cocktail for 30–45 min at RT. Subsequently, cells were washed and incubated in Ir intercalation solution overnight at 4°C. Immediately prior to data acquisition, cells were washed, and Maxpar water with EQ beads was added to adjust cell concentration to 2.5–5 x 104/ml. All samples were acquired on a CyTOF2 mass cytometer (Fluidigm, Helios) at an event rate of 200–500 cells per second.
CyTOF data analysis. CyTOF data were normalized by EQ-bead normalization in the CyTOF2 equipment and uploaded to Cytobank (https://community.cytobank.org/) for data cleaning, doublets, and dead cell removal. We removed EQ beads, used channel DNA2 and Event_length to exclude aggregated cells, and used channels DNA2 and Rh103 to select alive cells. The results were then exported as .fcs files for analysis. Data were scaled with arcsinh-transformation and further analyzed in R (v3.6.3). Single alive cells were first clustered using FlowSOM39 (R package, v.1.18.0), which used self-organizing maps for high-dimensional data reduction. Subsequently, we used Phenograph (R package, v0.99.1), a graph-based community detection method using the Louvain algorithm, for second clustering on the groups from FlowSOM. After every single cell was assigned to a cluster, we manually annotated each cluster based on its marker expression pattern compared with patterns of known immune cell types.
Mapping between CyTOF and CITE-seq cell clusters
To find proper augmentation data for deconvolving the in-house validation dataset, we first performed clustering on normalized protein profiles of CyTOF and CITE-seq, respectively, using both FlowSOM and Phenograph. Eleven surface markers in common were used for clustering: CD3, CD4, CD8a, CD14, CD16, CD56, CD19, CD25, CD45RA, CD45RO, and CD127. Then the Pearson correlation between each cluster of CyTOF and CITE-seq data was calculated based on the mean values of marker expressions. For each CyTOF cluster, we identified the best-matching cluster of CITE-seq according to the correlation between two clusters. We allowed one-to-many mapping in pairwise linking. After building these pairwise constraints, we further manually annotated these clusters based on similar typical marker expression patterns compared with patterns of known immune cell types. Only the clusters that can be clearly annotated were used for further experiments. Finally, we selected eight cell types (B cells, CD14 monocytes, NK cells, Treg, naive CD4 T cells, naive CD8 T cells, CD4 T effector memory, and CD8 T effector memory) to perform deconvolution validation.
Performance of DNN on different training datasets
We trained DNNs in DAISM-DNN on training datasets generated from expression data of purified cells to compare the performance of DNN with and without using real-life calibration samples. To this end, we first generated two training datasets using DAISM with calibration data from SDY67 and further augmented with scRNA-seq or RNA-seq data of purified samples. These two training sets were denoted as “DAISM-scRNA” and “DAISM-RNA,” respectively. For each training set, 30 permutation tests were conducted. In each permutation test, 50 samples were randomly drawn as hold-out test samples, and the remaining samples were candidates for calibration samples to create augmented training data with DAISM mixing strategy.
In addition, we also generated two training datasets using only RNA-seq expression profiles of sorted cells or scRNA-seq data and denoted them as “RNAonly” and “scRNAonly,” respectively. The generation of these training datasets followed the same linear mathematical operation as defined previously, with the only exception that real-life samples with ground truth cell fractions were not used in the mixing process. Briefly, for RNA-seq datasets, the expression of a simulated sample was calculated as
where C is the number of cell types involved in mixing, are random variables with Dirichlet distribution that determined the fractions of different cells in the in silico mixed sample, and is the expression profile of a randomly selected purified sample of cell type k from the respective RNA-seq dataset. For scRNA-seq dataset, is given by
where is the number of cells of type k extracted randomly from scRNA-seq datasets for mixing, and denote their expression profiles. Note that were further TPM-normalized before being used for training.
As suggested in Scaden, we also generated five training datasets that directly combined five different real-world bulk RNA-seq samples, namely SDY67, GSE107011, GSE127813, GSE107572, and GSE130824, with simulated mixtures respectively. For fair comparison, the same number of training samples (16,000) were used in all training sets.
For t-SNE analysis, all 50 test samples of SDY67 and 500 randomly selected artificial mixtures from each training dataset were plotted based on the common genes from SDY67 and the five training datasets. The parameter perplexity was set to 30 and the other parameters were set to their default values.
Scaling and error sensitivity
To identify the impact of the number of calibration samples on the performance of DAISM, we tested the cell type proportion estimation performance of DAISM-DNN with respect to the number of calibration samples used in creating the augmented training data on both RNA-seq dataset SDY67 and microarray dataset GSE59654. The experiments were started at zero calibration samples, and we gradually increased the number of calibration samples to the maximum available calibration samples at a step size of 10 samples. At each step, 30 permutation tests were conducted. In each permutation test, we randomly drew 50 samples as hold-out test samples, and the remaining samples were candidates for calibration samples to create augmented training data with the DAISM mixing strategy. Pearson correlation coefficients, CCC coefficients, and RMSE were used to measure the performance of the estimation of each cell type.
To test the sensitivity of DAISM-DNN to errors in ground truth cell type fraction, we generated artificial simulated mixtures with permutations in ground truth cell proportions to mimic potential measurement errors in real-life calibration samples. We used PBMC6k single cell reference data to generate 250 artificial bulk RNA-seq data with known cell type proportions as our test set. Thirty permutation tests were conducted in each experiment condition. In each permutation test, we randomly selected 50 samples as hold-out test samples, and the remaining samples were used as candidates for calibration samples. During the training, the ground truth proportions of all cell types of calibration samples were further perturbed with random noises between % to d% of the original proportions. After that, the perturbed calibration samples were augmented by using DAISM with PBMC8k single cell dataset as described previously to generate the training data.
Minimal detection fraction and background prediction levels
We followed the method proposed in Sturm et al.22 to evaluate the minimal detection fraction and background prediction levels of DAISM-DNN. We used a single cell dataset GSE115978 that contains cancer and immune cells to create simulated bulk RNA-seq samples of different spike-in levels of cell types of interest. For each cell type of interest, we generated five independent samples of different spike-in levels. For each sample, we randomly drew i cells of cell type of interest and 1,000 background cells containing all cell types except for the cell type of interest or only cancer cell types from the single cell dataset. The ratio of the cell types of interest is then . This results in five batches of 147 samples each (21 spike-in levels × 7 cell types). For calibration samples, we created 100 simulated samples that contained all cell types with random cell type proportions from GSE115978, where the cell type proportions followed Dirichlet distribution. Then we used PBMC8k as augmentation data to create a 16,000 DAISM-simulated training data for training DNN. We defined the minimal detection fraction as the minimal i at which the predicted score of cell type of interest is significantly different from the background prediction level (one-sided Student’s t test, alpha = 0.05) and background prediction level of cell type of interest as the predicted fraction of cell type of interest with i = 0.
Performance benchmarking
Since cell type abundances were resolved at different granularities in different deconvolution methods, regularizing the cell types of all methods to the same granularities had to be performed to facilitate a fair comparison. In this study, we only tested the performance of the benchmarked methods on six specific coarse-grained cell types (B cells, CD4 T cells, CD8 T cells, NK cells, monocytes, neutrophils) for comparison. The fine-grained cell type results of some methods were mapped to coarse-grained cell types according to the hierarchy of cell types defined in Strum et al.22
CIBERSORT (CS) is a signature-based deconvolution algorithm that uses ν-SVR to estimate cell abundance. We obtained R code from the CIBERSORT website (https://cibersort.stanford.edu/). We used CIBERSORT with different signature matrices (LM22, IRIS, immunoStates, and TIL10) and denoted them as four methods. The input data for CIBERSORT was in linear domain and all parameters were set to their default values. CIBERSORTx (CSx) is an extended version of CIBERSORT that generates a signature matrix from scRNA-seq data and provides two batch correction strategies (B-mode and S-mode) for cross-platform deconvolution. The B-mode was designed to remove technical differences between bulk profiles and signature matrices derived from bulk sorted reference profiles, while S-mode was used for signature matrices derived from droplet-based or UMI-based scRNA-seq data. We experimented with both B-mode and S-mode. For B-mode, LM22 was used as the signature matrix. For S-mode, the scRNA-seq dataset PBMC8k was used to generate the signature matrix, which was applied in further deconvolution. While testing the in-house PBMC dataset, PBMC10k was used to create the signature matrix. We ran CIBERSORTx from its website (https://cibersortx.stanford.edu/). Quantile normalization was disabled when input was RNA-seq or scRNA-seq simulated mixture data.
We used the R package MuSiC (https://github.com/xuranw/MuSiC) for MuSiC. MuSiC takes scRNA-seq data with cell type labels as reference. Deconvolution using MuSiC was performed with five coarse-grained cell types (B cells, CD4 T cells, CD8 T cells, NK cells, and monocytes). The single cell PBMC dataset PBMC8k was used as reference data.
ABIS enables absolute estimation of cell abundance from both bulk RNA-seq and microarray data. Deconvolution was performed through an R/Shiny app (https://github.com/giannimonaco/ABIS). The results output from ABIS were absolute cell frequencies and were divided by 100 in our study for comparison with other methods on RMSE.
Scaden is a DNN-based deconvolution algorithm. We used the training datasets provided by Scaden (https://github.com/KevinMenden/scaden), which contain 32,000 artificial mixtures from four scRNA-seq PBMC datasets, denoted as S4. Training was performed for 5,000 steps per model on each dataset as recommended in the original paper.
We ran quanTIseq, MCP-counter, EPIC, and xCell through R package immunedeconv22, which provided an integrated inference to benchmark on six deconvolution methods. The parameter tumor was set to FALSE when performing deconvolution on all PBMC datasets. As recommended in the original paper, EPIC was run with BRef as the signature set on PBMC samples.
Statistical analysis
We used three evaluation criteria to compare the performance of DAISM-DNN methods. Pearson correlation r was used to measure the linear concordance between predicted cell type proportions and the FACS ground truth. Lin’s CCC and the RMSE were further used to evaluate the performance for methods that enable absolute cell type proportion estimation.
Differences in continuous measurements were tested using the two-tailed Student’s t test. Two-sided p values were used unless otherwise specified, and a p value less than 0.05 was considered significant. For comparisons between multiple groups, we used one-way ANOVA using the built-in function aov in R. Ranking of the algorithms over multiple testing sets was determined using Friedman test with post hoc two-tailed Nemenyi test.40 PRISM was used for basic statistical analysis and plotting (http://www.graphpad.com), and the Python or R language and programming environment were used for the remainder of the statistical analysis.
Acknowledgments
We thank Chenxin Li and Jiajing Xie for helping to perform some simulation tests in this paper. This work was supported by the National Natural Science Foundation of China (81788101 to J.H.).
Author contributions
Y.L., H.L., X.X., and M.W. performed the computational analysis, analyzed data, and generated figures. W.Y., J.H., and R.Y. conceived the project and designed the methodology. L.Z., K.W., and J.Z. performed all experiments. F.Z. and M.Z. contributed to discussion and reviewed and edited the manuscript. All authors assisted to write the manuscript.
Declaration of interests
R.Y. and W.Y. are shareholders of Aginome Scientific. J.Z. and F.Z. are employees of Amoy Diagnostics. The authors declare no other competing interests.
Published: February 3, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.patter.2022.100440.
Contributor Information
Wenxian Yang, Email: wx@aginome.com.
Jiahuai Han, Email: jhan@xmu.edu.cn.
Rongshan Yu, Email: rsyu@xmu.edu.cn.
Supplemental information
Data and code availability
All expression datasets analyzed in this work, including accession codes and web links (if available), are listed in Table S2. The source code for DAISM-DNN is available at https://github.com/xmuyulab/DAISM-XMBD, https://doi.org/10.5281/zenodo.5723561. In-house validation data are available from the corresponding authors upon reasonable request.
References
- 1.Fridman W.H., Pagès F., Sautes-Fridman C., Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat. Rev. Cancer. 2012;12:298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 2.Palucka A.K., Coussens L.M. The basis of oncoimmunology. Cell. 2016;164:1233–1247. doi: 10.1016/j.cell.2016.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang A.C., Postow M.A., Orlowski R.J., Mick R., Bengsch B., Manne S., Xu W., Harmon S., Giles J.R., Wenz B., et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545:60–65. doi: 10.1038/nature22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fridman W.H., Zitvogel L., Sautès-Fridman C., Kroemer G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017;14:717. doi: 10.1038/nrclinonc.2017.101. [DOI] [PubMed] [Google Scholar]
- 5.Kalluri R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer. 2016;16:582. doi: 10.1038/nrc.2016.73. [DOI] [PubMed] [Google Scholar]
- 6.DeNardo D.G., Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019;19:369–382. doi: 10.1038/s41577-019-0127-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galluzzi L., Chan T.A., Kroemer G., Wolchok J.D., López-Soto A. The hallmarks of successful anticancer immunotherapy. Sci. Translational Med. 2018;10 doi: 10.1126/scitranslmed.aat7807. [DOI] [PubMed] [Google Scholar]
- 8.Petitprez F., Sun C.-M., Lacroix L., Sautès-Fridaman C., de Reyniès A., Fridman W.H. Quantitative analyses of the tumor microenvironment composition and orientation in the era of precision medicine. Front. Oncol. 2018;8:390. doi: 10.3389/fonc.2018.00390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hrdlickova R., Toloue M., Tian B. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA. 2017;8:e1364. doi: 10.1002/wrna.1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Becht E., Giraldo N.A., Lacroix L., Buttard B., Elarouci N., Petitprez F., Selves J., Laurentpuig P., Sautesfridman C., Fridman W.H., et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17:218. doi: 10.1186/s13059-016-1070-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aran D., Hu Z., Butte A.J. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18:220. doi: 10.1186/s13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finotello F., Mayer C., Plattner C., Laschober G., Rieder D., Hackl H., Krogsdam A., Loncova Z., Posch W., Wilflingseder D., et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019;11:1–20. doi: 10.1186/s13073-019-0638-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Racle J., De Jonge K., Baumgaertner P., Speiser D.E., Gfeller D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. eLife. 2017;6:1–25. doi: 10.7554/eLife.26476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Newman A.M., Liu C.L., Green M.R., Gentles A.J., Feng W., Xu Y., Hoang C.D., Diehn M., Alizadeh A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newman A.M., Steen C.B., Liu C.L., Gentles A.J., Chaudhuri A.A., Scherer F., Khodadoust M.S., Esfahani M.S., Luca B.A., Steiner D., et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019;37:773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang W., Wan C., Lu X., Tu S.-w., Sun Y., Zhang X., Zang Y., Zhang A., Huang K., Liu Y., et al. ICTD: a semi-supervised cell type identification and deconvolution method for multi-omics data. bioRxiv. 2019:426593. doi: 10.1101/426593. [DOI] [Google Scholar]
- 17.Danaher P., Warren S., Dennis L., D’Amico L., White A., Disis M.L., Geller M.A., Odunsi K., Beechem J., Fling S.P. Gene expression markers of tumor infiltrating leukocytes. J. Immunother. Cancer. 2017;5:18. doi: 10.1186/s40425-017-0215-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eraslan G., Avsec Ž., Gagneur J., Theis F.J. Deep learning: new computational modelling techniques for genomics. Nat. Rev. Genet. 2019;20:389–403. doi: 10.1038/s41576-019-0122-6. [DOI] [PubMed] [Google Scholar]
- 19.Ching T., Himmelstein D.S., Beaulieu-Jones B.K., Kalinin A.A., Do B.T., Way G.P., Ferrero E., Agapow P.-M., Zietz M., Hoffman M.M., et al. Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interf. 2018;15:20170387. doi: 10.1098/rsif.2017.0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Menden K., Marouf M., Oller S., Dalmia A., Magruder D.S., Kloiber K., Heutink P., Bonn S. Deep learning–based cell composition analysis from tissue expression profiles. Sci. Adv. 2020;6:eaba2619. doi: 10.1126/sciadv.aba2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Monaco G., Lee B., Xu W., Mustafah S., Hwang Y.Y., Carre C., Burdin N., Visan L., Ceccarelli M., Poidinger M., et al. RNA-seq signatures normalized by mRNA abundance allow absolute deconvolution of human immune cell types. Cell Rep. 2019;26:1627. doi: 10.1016/j.celrep.2019.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sturm G., Finotello F., Petitprez F., Zhang J.D., Baumbach J., Fridman W.H., List M., Aneichyk T. Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics. 2019;35:i436. doi: 10.1093/bioinformatics/btz363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X., Park J., Susztak K., Zhang N.R., Li M. Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat. Commun. 2019;10:380. doi: 10.1038/s41467-018-08023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abbas A.R., Baldwin D.T., Ma Y., Ouyang W., Gurney A.L.B., Martin F., Fong S., Campagne M.V.L., Godowski P.J., Williams P., et al. Immune response in silico (IRIS): immune-specific genes identified from a compendium of microarray expression data. Genes Immun. 2005;6:319–331. doi: 10.1038/sj.gene.6364173. [DOI] [PubMed] [Google Scholar]
- 25.Vallania F., Tam A., Lofgren S., Schaffert S., Azad T.D., Bongen E., Haynes W.A., Alsup M., Alonso M.N., Davis M.M., et al. Leveraging heterogeneity across multiple datasets increases cell-mixture deconvolution accuracy and reduces biological and technical biases. Nat. Commun. 2018;9:4735. doi: 10.1038/s41467-018-07242-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melisko M. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. Women’s Oncol. Rev. 2005;5:45–47. doi: 10.1080/14733400500093379. [DOI] [PubMed] [Google Scholar]
- 27.Baehner F.L. The analytical validation of the Oncotype DX Recurrence Score assay. Ecancermedicalscience. 2016;10:675. doi: 10.3332/ecancer.2016.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stoeckius M., Hafemeister C., Stephenson W., Houck-Loomis B., Chattopadhyay P.K., Swerdlow H., Satija R., Smibert P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods. 2017;14:865–868. doi: 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chakravarthy A., Furness A., Joshi K., Ghorani E., Ford K., Ward M.J., King E.V., Lechner M., Marafioti T., Quezada S.A., et al. Pan-cancer deconvolution of tumour composition using DNA methylation. Nat. Commun. 2018;9:3220. doi: 10.1038/s41467-018-05570-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levy J.J., Titus A.J., Petersen C.L., Chen Y., Salas L.A., Christensen B.C. MethylNet: an automated and modular deep learning approach for DNA methylation analysis. BMC Bioinformatics. 2020;21:1. doi: 10.1186/s12859-020-3443-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang H., Cai R., Dai J., Sun W. EMeth: an EM algorithm for cell type decomposition based on DNA methylation data. Scientific Rep. 2021;11:1. doi: 10.1038/s41598-021-84864-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bengio Y., Courville A.C., Vincent P. Unsupervised feature learning and deep learning: a review and new perspectives. arXiv. 2012 1206.5538. [Google Scholar]
- 33.Wang Y., Yao Q. Few-shot learning: a survey. arXiv. 2019 1904.05046. [Google Scholar]
- 34.Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen S., Zhou Y., Chen Y., Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng G.X.Y., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., Mcdermott G., Zhu J., et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8:14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Butler A., Hoffman P.J., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang X., Lan Y., Xu J., Quan F., Zhao E., Deng C., Luo T., Xu L., Liao G., Yan M., et al. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019;47:D721. doi: 10.1093/nar/gky900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Gassen S., Callebaut B., Van Helden M.J., Lambrecht B.N., Demeester P., Dhaene T., Saeys Y. FlowSOM: using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A. 2015;87:636–645. doi: 10.1002/cyto.a.22625. [DOI] [PubMed] [Google Scholar]
- 40.Demšar J. Statistical comparisons of classifiers over multiple data sets. J. Machine Learn. Res. 2006;7:1–30. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All expression datasets analyzed in this work, including accession codes and web links (if available), are listed in Table S2. The source code for DAISM-DNN is available at https://github.com/xmuyulab/DAISM-XMBD, https://doi.org/10.5281/zenodo.5723561. In-house validation data are available from the corresponding authors upon reasonable request.