

Abstract
Fusion of plasma membranes is essential for skeletal muscle development, regeneration, exercise-induced adaptations, and results in a cell that contains hundreds to thousands of nuclei within a shared cytoplasm. The differentiation process in myocytes culminates in their fusion to form a new myofiber or fusion to an existing myofiber thereby contributing more synthetic material to the syncytium. The choice for two cells to fuse and become one could be a dangerous event if the two cells are not committed to an allied function. Thus, fusion events are highly regulated with positive and negative factors to fine-tune the process, and requires muscle-specific fusogens (Myomaker and Myomerger) as well as general cellular machinery to achieve the union of membranes. While a unified vertebrate myoblast fusion pathway is not yet established, recent discoveries should make this pursuit attainable. Not only does myocyte fusion impact the normal biology of skeletal muscle, but new evidence indicates dysregulation of the process impacts pathologies of skeletal muscle. Here, I will highlight the molecular players and biochemical mechanisms that drive fusion events in muscle, and discuss how this key myogenic process impacts skeletal muscle diseases.
Keywords: Myoblast fusion, Myomaker, Myomerger, Myomixer, Muscle development, Regeneration
1. Introduction
Skeletal muscle cells require hundreds to thousands of nuclei within a shared cytoplasm for proper function. Multinucleation is achieved through cell fusion events that occur during embryogenesis and postnatal muscle growth. These fusion events are not restricted to development, as skeletal myofibers require accretion of additional myonuclei, from satellite cells (muscle stem cells) in the adult for adaptive growth and repair [1]. The fusing cells in skeletal muscle include myocytes, myoblasts that have exited the cell cycle and differentiated, and myofibers that were previously formed. In this review, we use the term ‘myoblast fusion’ because that is convention, even though myocytes are likely the myogenic cell type that fuses, and we also use ‘myocyte fusion’. The general cellular pathway for the fusion of myocytes encompasses cell recognition, signaling, adhesion, actin nucleation, and lipid remodeling [2]. Thus, successful fusion requires multiple cellular processes orchestrated by the interplay between proteins and the lipids that compose membranes. In vertebrates, cell fusion is catalyzed by the muscle-specific fusogens, Myomaker (Mymk) [3,4] and Myomerger/Myomixer/Minion (Mymx) [5–7], and regulated by a host of additional protein and lipid species that ensure a favorable biochemical event [8–10]. In this review I highlight the centrality of cell fusion for the development, regeneration, and maintenance of vertebrate skeletal muscle and give perspectives on the key molecules and pathways that drive the process with a focus on the uniqueness of cell fusion in the skeletal muscle system.
The fusion of membranes occurs in multiple biological settings including between intracellular membranes, between a virus and cell, and between two cells [11]. In addition to skeletal muscle, the mammalian cell types that are regulated by plasma membrane fusion events include zygotes, osteoclasts, placental trophoblasts, and giant cells. Of these cell types, skeletal muscle exhibits characteristics that may suggest the need for uniquely regulated fusion events. First, skeletal myofibers are long-lived and progenitor cells fuse throughout the lifespan of the organism, whereas the other fusogenic cell types are present for a relatively short time. Second, skeletal muscle exhibits a complex morphology of being an extremely large cell packed with sarcomeres that allow contraction and movement, and in comparison, trophoblasts, osteoclasts, and giant cells have less defined shape with fewer structural complexities. Due to these characteristics of skeletal muscle, perhaps it is not surprising that skeletal muscle fusogens are not expressed in other cell types and exhibit unique structural characteristics.
When thinking about the fusion of membranes, one must consider that membranes do not spontaneously fuse and that it is a thermodynamically unfavorable event due to repulsion forces between the lipid head groups in the opposing membranes [12]. Indeed, in biological systems, cells rarely come closer than 30 nm. Dedicated proteins (fusogens) provide catalysts for the reaction by remodeling membranes to generate new structures (a fusion event) [12,13]. A generalized pathway for lipid rearrangements that are necessary for fusion of membranes includes formation of a hemifusion intermediate, where the outer membrane leaflets of the two fusing membranes come together [14]. This intermediate connection is thermodynamically unstable and either quickly dissociates or a fusion pore is formed, completing the fusion reaction. The fusion reactions for intracellular membrane fusion and virus-cell fusion are driven by proteins that have long extracellular domains, which interact between the membranes and undergo a conformational change to bring the membranes close enough to fuse [15–18]. This canonical fusion reaction is unlikely to be sufficient to explain the fusion of myoblast membranes since the muscle fusogens are structurally divergent from the factors (soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) and viral fusogens) that catalyze other membrane fusion reactions.
In addition to membrane events, myocyte fusion requires cellular processes that prepares cells to fuse. Much of our knowledge on these pathways has emanated from studies in Drosophila, which yielded a generalized fusion pathway [19]. In vertebrates, systems to study fusion include in vitro myogenic cells, zebrafish, chick, and mouse development, as well as adult mouse regeneration. These systems have been important tools for the identification of genes that regulate myocyte fusion and revealed some conservation of proteins and pathways with the Drosophila system. However, there is a general lack of understanding for the exact step of the fusion pathway that is regulated by many of the proteins in the vertebrate system. Many of the discrepancies in the vertebrate system can be attributed to redundancy of factors and difficult interpretations as some genetic experiments reveal effects during adult muscle regeneration but not development. Due to the overlap between differentiation and fusion, identifying proteins exclusively involved in fusion has been challenging. Nonetheless, using information from the Drosophila and vertebrate systems, one can distinguish molecules that play a role in the fusion step of myogenesis compared to a process that precedes fusion, and generate cognate fusion pathways based on genetic experiments that determine whether proteins are necessary for fusion.
2. Perspectives
2.1. Recognition and adhesion
The proposed first step of the fusion pathway, during the differentiation of satellite cells, is that cells must recognize and adhere to each other. The need for recognition is to make certain that cells are of the same lineage and a union is beneficial, and adhesion theoretically allows areas of the membrane to be more closely aligned (10–30 nm) and forming a site of contact for fusion. The best evidence that recognition and adhesion are an important step comes from work in Drosophila and zebrafish [19]. In Drosophila, the cell adhesion molecules Dumbfounded (Duf or Kirre) or Roughest (Rst) on founder cells interact in trans with Sticks and stones (Sns) or Hibris (Hbs) on the surface of fusion-competent myoblasts [20–25]. These cell adhesion molecules initiate signals (Dock) to actin nucleating factors (Rac1, Wasp, Arp 2/3) to induce cytoskeletal rearrangements required for fusion [19].
Adhesion and signaling molecules have also been identified in zebrafish. Loss of kirrel, a homolog of Duf in the fly, results in reduced myoblast fusion [26]. In addition to Kirrel, Jamb and Jamc are adhesion molecules that interact in trans and are necessary for fusion of fast muscles in zebrafish [27]. Whether these adhesion proteins in zebrafish also connect to actin nucleating pathways similar to the fly is unknown. While genetic evidence for recognition and adhesion being critical in fly and fish myoblast fusion, similar data is lacking for mammalian myoblast fusion. To date, a cell adhesion molecule genetically necessary in vivo for mouse myoblast fusion has not been discovered. Highlighting potential evolutionary divergence between the fly, zebrafish, and mammalian system is the function of Nephrin, a homolog of fly Sns. Nephrin knockout mice do not show an overt developmental phenotype in muscle, but primary myoblasts exhibit a fusion reduction in vivo [28]. Also, it is not clear if loss of Jamb and Jamc in mouse skeletal muscle impacts fusion events [27]. Cadherin molecules were also proposed as putative adhesion factors for mouse myoblast fusion, but genetic loss-of-function studies do not support a critical role [29]. Beta1 integrins are necessary for myoblast fusion in the mouse although whether the defect is due to adhesion or some other pathway is not known [30]. Secreted factors, such as Interlukin-4 (IL-4) from myoblasts, may be a recognition signal for myoblast-myotube fusion [31].
It is possible that cell adhesion molecules in mammals have gone undiscovered or another potential explanation for the lack of a bona fide recognition or adhesion factor in mammalian myoblast fusion could be that fusing cells interact using a number of cell surface molecules (Fig. 1), thus genetic loss of function studies do not reveal a phenotype when any single molecule is deleted. While cell fusion needs to be regulated to prevent inappropriate mixing of cells, it is possible that adhesion is not a regulatory checkpoint for fusion of mammalian myoblasts. Along those lines, recognition for vertebrate fusion may need redefining and not occur in the classical sense of a receptor-ligand interaction. Recognition would seem like a necessary regulatory checkpoint for fusion, and could be related to the lipid rearrangements induced by the muscle fusogens (discussed below). In this scenario, recognition is not confined to a time stamp prior to adhesion but instead the two cells are ‘recognizing’ each other throughout the process.
Fig. 1. Adhesion factors for mammalian myoblast fusion.
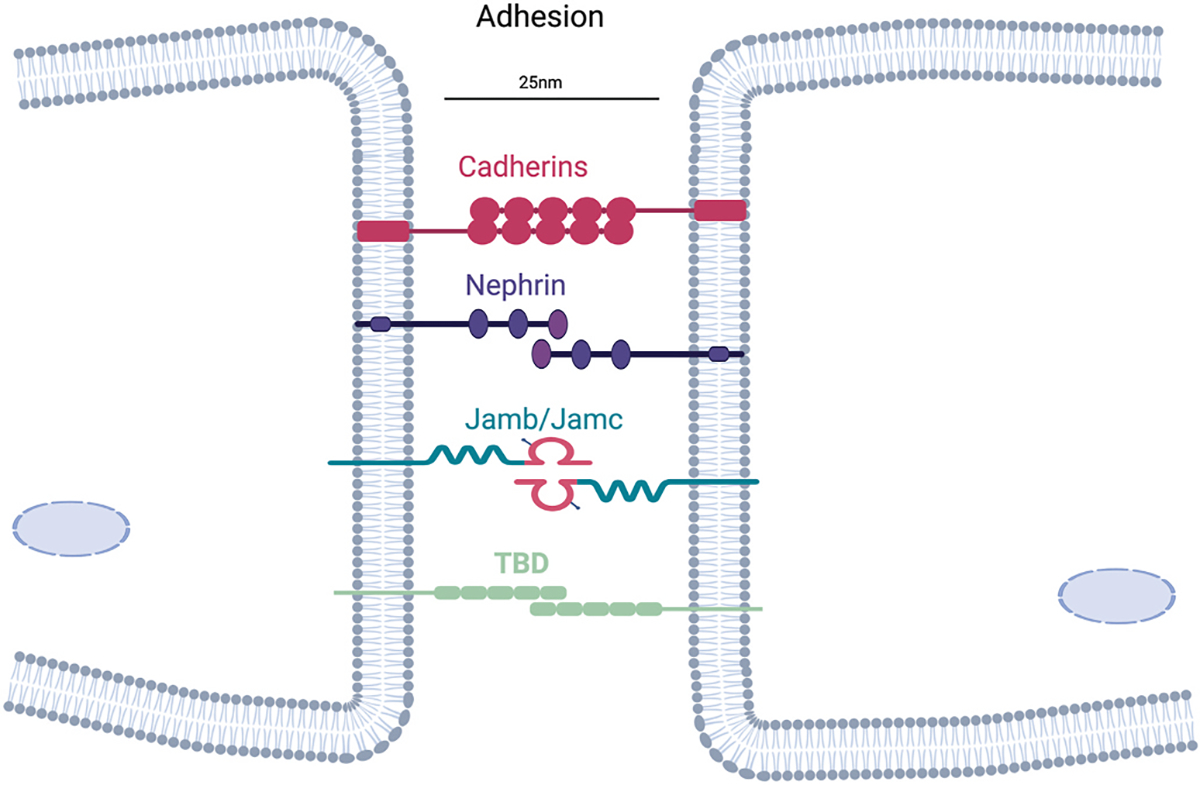
Myocytes can interact through multiple cell surface proteins, which function to bring membranes within 25 nm. Genetic deletion of one of these adhesion factors has not resulted in a myoblast fusion phenotype in vivo, suggesting compensation or that the central adhesion factor remains to be discovered (TBD).
2.2. Actin dynamics
In contrast to recognition and adhesion, cytoskeletal rearrangements are highly conserved between Drosophila and vertebrates, including mammals. An F-actin focus forms in the fusion-competent myoblasts, and these structures resemble a podosome-like structure or invasive protrusions, that suggest this myoblast population is an ‘attacking’ cell [32–34]. In the founder cell, there is strong cortical mechanical tension that resists the attacking cell, and this interaction is thought to promote fusion pore formation [35]. While this level of detail is not available for vertebrate myoblast fusion, an actin focus is present, invasive protrusions have been observed, and many of the actin nucleation pathways are conserved. Signals for cytoskeletal rearrangements converge on the Arp 2/3 complex, which functions to nucleate branched actin. Upstream of the Arp 2/3 complex in mammals are Dock1, Rac, and N-Wasp, similar to the pathways activated in the fly [36–39]. Overall, cytoskeletal remodeling is a critical pathway for myoblast fusion (Fig. 2).
Fig. 2. Signaling to generate F-actin foci is necessary for myoblast fusion.
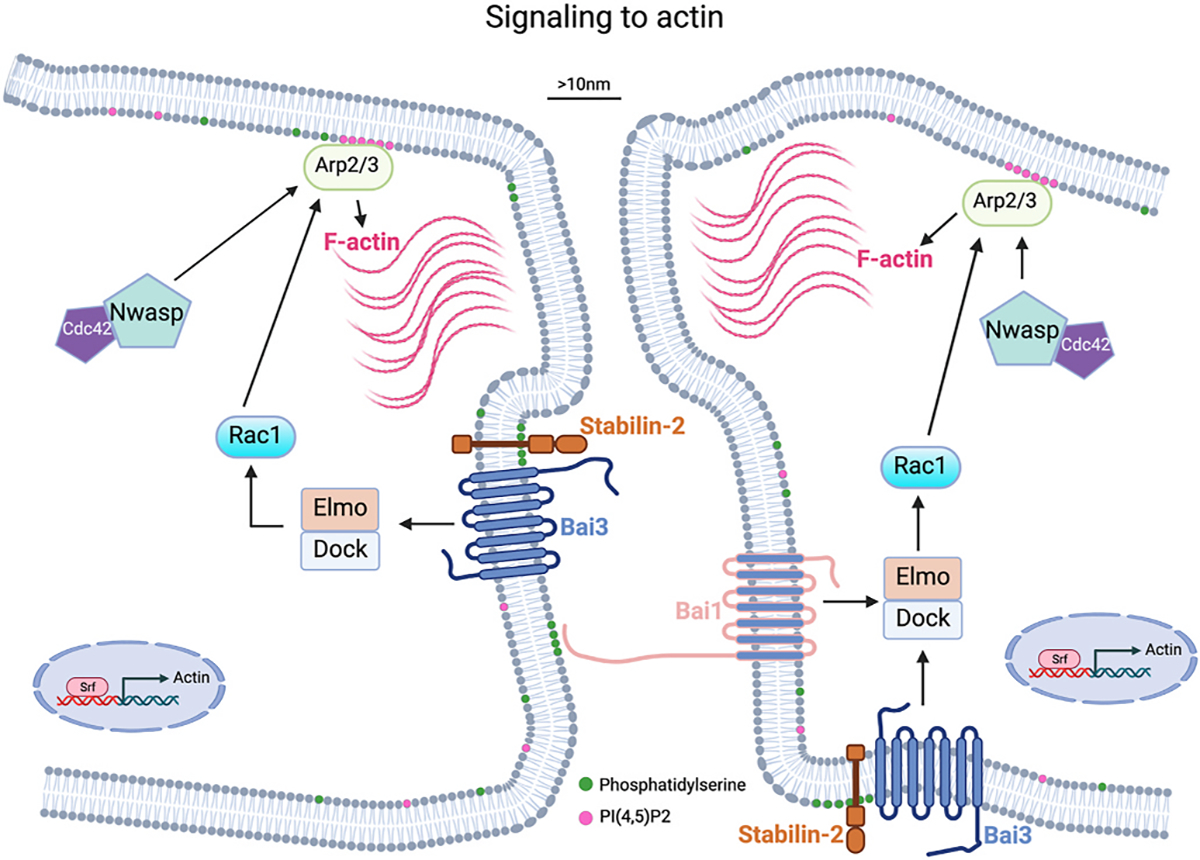
Schematic showing molecular players and cellular pathways that result in a F-actin focus, which push membranes close to 10 nm and begin to establish a fusion synapse. Serum response factor (Srf) increases Actin transcription. Phosphatidylserine (PS) is normally located on the inner leaflet of the plasma membrane. Movement of PS to the outer leaflet provides a signal for PS receptors including Bai1 and Stabilin-2, which activate signaling to generate F-actin through the Arp2/3 complex. Localization of actin polymerization is proposed to be regulated by binding of PI(4,5)P2 to Arp2/3.
The signals from the membrane to intracellular actin nucleation for vertebrate myoblast fusion are not as clear as the pathways in the fly. The best candidates for the initiation of the signaling cascade is phosphatidylserine (PS) activation of the brain-specific angiogenesis inhibitor (Bai) family of proteins. In normal cells, PS is in the inner leaflet of the plasma membrane but flipping to the outside is proposed as a trigger for fusion of myoblasts and other cell types that fuse [40–42]. Bai3 is a GPCR that promotes myocyte fusion in the chick and mouse and functions to link cell surface signals to actin nucleation pathways by recruiting ELMO/DOCK to the membrane, which activates Rac1 resulting in cytoskeletal rearrangements [43]. Bai3 is required on both fusing cells, but it is unclear if these proteins interact in trans (between two cell membranes). Bai3 is regulated positively by Stabilin-2 and negatively by Complement C1q Like 4 (C1qL4) [44], and such regulation potentially suggests that the pathway is a critical factor for the initiation of fusion. Stabilin-2 is a receptor for phosphatidylserine (PS), which is one of the few lipids shown to function at multiple steps in the fusion pathway. C1qL4 is a secreted molecule that binds to the extracellular domain of Bai3 and inhibits its activity. Bai1 is also involved in myoblast fusion through its PS receptor activity. Apoptotic cells, which have PS on the outer leaflet, could activate Bai1 on myoblasts to initiate the fusion cascade [45]. It should be noted that genetic loss-of-function of Bai1, Bai3, or Stabilin-2 result in a mild myoblast fusion phenotype suggesting that other critical proteins may be involved [43,45,46]. In these models, muscle still forms but with a reduction in myofiber size and myonuclear numbers, and there is a defect in muscle regeneration. Disrupting these genes specifically in muscle satellite cells should clarify their specific requirement for fusion during muscle formation in vivo.
In addition to canonical actin nucleating pathways driven by Rac and N-Wasp, regulators of actin dynamics include PI(4,5)P2, dynamin, and serum response factor (Srf). PI(4,5)P2 helps localize Arp 2/3 complex and results in local actin nucleation and potential marking of the fusion site [47]. Dynamin is typically associated with endocytosis, but also directly bundles actin providing a stronger cytoskeletal network and dynamin is important for myoblast fusion in the fly and mouse myoblasts [22,48]. Srf is a transcription factor and controls expression of cytoskeletal regulators. Deletion of Srf specifically in mouse satellite results in a fusion defect after a hypertrophic stimulus and is attributed to a lack of actin cytoskeleton and actin-based protrusions [49]. The presence of multiple regulatory factors impacting actin nucleation is consistent with that pathway being critical for myoblast fusion across various organisms.
The overall function of cytoskeletal remodeling is that these forces cause close membrane apposition (>10 nm) and thus help assemble membranes for fusogens to act. However, it has been noted that membranes surrounding the invasive protrusions are not closer than membrane areas outside the protrusions [12]. This model also suggests formation of a fusion pore at the end one of these projections. A separate model has been proposed for indirect flight muscles in the fly, which require all of the canonical pathways that initiate actin nucleation but protrusions were detected at a low frequency precluding functional interpretation, and protrusions were more pronounced in mutants where fusion was disrupted. Moreover, through electron microscopy, a relatively flat myoblast membrane is juxtaposed to the myofiber membrane after adhesion, and fusion occurs through multiple pores [50]. It is possible that the two models are both correct depending on the physiological context. During primary fusion, where two myoblasts fuse to form a nascent myofiber invasive protrusions could be needed to form a fusogenic synapse. However, during secondary fusion where a myoblast fuses to an existing myofiber, perhaps multiple fusion pores are needed due to the lack of curvature in the myofiber membrane. The concept that fusion to form new myofibers could be different than fusion with established muscle cells is further discussed below.
2.3. Membrane remodeling by muscle fusogens
While the molecular underpinnings for most of the Drosophila fusion pathway is known, the fusogens that drive the final step of fusion (membrane rearrangements) have not yet been identified. A molecule that acts as a fusogen should be dedicated to the fusion process, expressed when cells are fusing, and be essential and sufficient for fusion. In vertebrates, Myomaker and Myomerger are the only factors currently identified that meet these criteria. Both proteins are spatially and temporally restricted to muscle cells during times of myoblast fusion, and deletion of either gene results in accumulation of mononucleated myoblasts during development and regeneration and thus functional muscle formation is completely absent [4–7,51–57]. Transcriptional regulation of the muscle fusogens is achieved by canonical muscle transcription factors, MyoD and myogenin [51,58]. Ectopic expression of Myomaker in normally non-fusing cells renders them fusion-competent, where they are able to fuse to fusogenic cells, such as myoblasts [4]. Addition of Myomerger to Myomaker-expressing non--fusing cell types, such as fibroblasts, induces fusogenicity and formation of multinucleated cells [6,7,56]. Evidence that Myomaker and Myomerger are key factors for myoblast fusion is demonstrated by their functional conservation in zebrafish, chicken, and trout [52–55,57, 59–61]. In zebrafish, loss of myogenin, a transcription factor needed to activate Myomaker resulted in some residual fusion [58], suggesting that there could be Myomaker-independent fusion pathways although MyoD could compensate and drive Myomaker expression. The complete lack of muscle in Myomaker null mice and lack of fusion in Mymk-deficient human myogenic cells suggests that if Myomaker can be bypassed in fish, those factors are not able to compensate at levels needed to drive fusion in mammals [4,62]. Cells of the fibroblast lineage, derived from the lateral plate mesoderm, that fuse to growing myofibers and help pattern the myotendinous junction have also been reported to not require Myomaker, but more work is needed to define these unique fusion events between cells of different lineages [63]. Interesting work on the evolution of the fusogens showed that Myomaker and Myomerger are present and functional in non-vertebrate chordates such as lamprey [64]. Taken together, Myomaker and Myomerger are critical factors for myoblast fusion and muscle formation, and a unified myoblast fusion pathway should be gleaned from more knowledge about their activities and regulation.
Fusogens act to allow membranes to come closer than distances that can be achieved through strong adhesion and actin protrusions. The proposed membrane remodeling pathway for all fusion events proceeds through hemifusion connections, where the outer membrane leaflets merge, followed by disruption of that intermediate leading to pore formation and cytoplasmic mixing [14]. Given that membrane bilayers are thermodynamically stable, fusogens must function by acting on lipid species to generate energy in the system that would favor rearrangements needed for fusion. Fusogens in intracellular (SNAREs), viral (human immunodeficiency virus (HIV), vesicular stomatitis virus (VSV-G)), and cell fusion (C. Elegans epithelial fusion failure-1 (EFF-1)) act by pulling the membranes close together [11]. However, this mechanism is unlikely to explain fusogen activity in vertebrate myoblasts given that Myomaker and Myomerger are highly embedded in the membrane and lack long extracellular domains typical of classical fusogens [65]. Myomaker is 221 amino acids and spans the bilayer 7 times, with an extracellular N-terminus and intracellular C-terminus [66]. Myomerger contains just 84 amino acids, has a single transmembrane domain, and an ectodomain that harbors two putative helical regions [3, 57].
Currently, there are two models for the activities of Myomaker and Myomerger that lead to fusion of myoblasts. One model poses that the two proteins interact where Myomerger activates the fusogenic activity of Myomaker to establish a fusion pore [5,62]. Interaction between Myomaker and Myomerger is detected through biochemical co-immunoprecipitation studies from cells which overexpress epitope-tagged versions [3,5]. However, it is not clear if these two proteins possess obvious domains that would interact. A separate model to explain fusions induced by Myomaker and Myomerger is that they both function to perturb lipids through different mechanisms and drive distinct steps of the myoblast fusion reaction [3]. Myomaker null myoblasts do not hemifuse indicating Myomaker functions at or before this critical fusion step. Hemifusion is detected in myoblasts lacking Myomerger but cytoplasmic mixing is blocked, indicating that Myomerger functions at the pore formation step. Perhaps the best evidence that Myomaker and Myomerger do not require an interaction for activity is that their individual activities are detected in the absence of the other protein. In human cells, Myomaker is sufficient to induce low level fusions even without the presence of Myomerger [62] and fusion of mouse myoblasts that only have Myomaker (Myomerger KO) can be induced with agents that promote hemifusion-to-fusion transition [3]. These data suggest that Myomaker is functional in the absence of Myomerger. Likewise, Myomerger can induce fusion completion in a fibroblast-Hemagglutinin system and recombinantly purified Myomerger ectodomains are active on synthetic liposomes, even though both systems are devoid of Myomaker [3]. Like all biochemical reactions, the process of membrane rearrangements leading to fusion depends on kinetics and thus it is possible that the independent activities of the muscle fusogens Myomaker and Myomerger are needed for kinetically favorable reactions needed to achieve the high level of fusions to form large multinucleated myofibers.
If one considers that Myomaker and Myomerger function independently but their activities overlap for fusion, then how could Myomaker promote hemifusion and Myomerger pore formation? The precise biochemical activity of Myomaker has not been experimentally validated, but the protein has characteristics similar to ceramidases, such as Alkaline ceramidase 3 (ACER3) and Adiponectin receptor (ADIPOR) [67]. Use of novel software that predicts protein structure (Alphafold, RoseTTAFold) and ChimeraX (Millay lab, unpublished observations) to overlap predicted and known structures demonstrates structural homology to ADIPOR [64]. While there are sequence and structural similarities between Myomaker and ceramidases, there are also differences that could suggest a unique mechanism of action and lead to questions if Myomaker is a ceramidase. ACER3/ADIPOR, and other ceramidases, contain a serine in TM2 and an aspartic acid in TM3, but in Myomaker those residues are valine and serine, respectively. It is possible that the lack of these residues on Myomaker does not allow hydrolase activity, or permits catalysis of a unique substrate. One curious aspect of Myomaker is that structural models indicate that important histidine residues would be closer to the outer leaflet, whereas in ACER3/ADIPOR these residues are juxtaposed to the inner leaflet.
It is interesting that a ceramidase, which breaks down ceramide to sphingosine and a fatty acid, could be the central factor for myoblast fusion. There are different types ceramidases in various compartments in many cell types [68,69], so the need for this type of activity to drive such a specific process like myoblast fusion is not obvious. The answer could be that the trigger to activate the ceramidase activity for Myomaker is unique to the myoblast fusion process, thus achieving specificity. Or it is possible that Myomaker is not a ceramidase and instead binds ceramides through the critical histidine residues, but does not catalyze their breakdown. In this scenario, accumulation of ceramide-rich domains in the outer leaflet (where histidine residues are present on Myomaker) would cause thermodynamic stress requiring lipids to rearrange, and thus promoting hemifusion of closely opposed membranes (Fig. 3). There is only circumstantial evidence for this mechanism including that ceramide facilitates negative curvature of membranes [70], which is predicted to promote formation of the hemifusion stalk [71]. Also, there is a role for ceramide to drive fusion as intracellular membrane fusion is disrupted in sphingomyelinase (synthesizes ceramides) null cells [41]. If Myomaker is a ceramidase, breakdown of ceramide could lead to changes in membrane fluidity, a characteristic known to increase fusion [72,73]. Membrane fluidity is increased prior to fusion in muscle cells [74] and has been proposed to regulate myoblast fusion by very long chain fatty acids (VLCAs) [26], but the key feature for membrane fluidity is that it is not globally changed but instead is altered in local domains of the membrane. Finally, it’s possible that ceramide is not involved in Myomaker activity, but that the muscle fusogen acts on a different lipid substrate to generate a fusogenic lipid with negative spontaneous curvature, or that Myomaker acts as an opportunistic fusogen, where it activates another molecule that normally does not function to regulate fusion. In addition to cell culture and in vivo muscle formation as assays for myoblast fusion, use of cell-free biochemical systems, which were critical to understand mechanistic details in intracellular and viral fusion systems, should be employed to understand biochemical activities of critical fusion factors.
Fig. 3. Membrane remodeling to drive myoblast fusion.
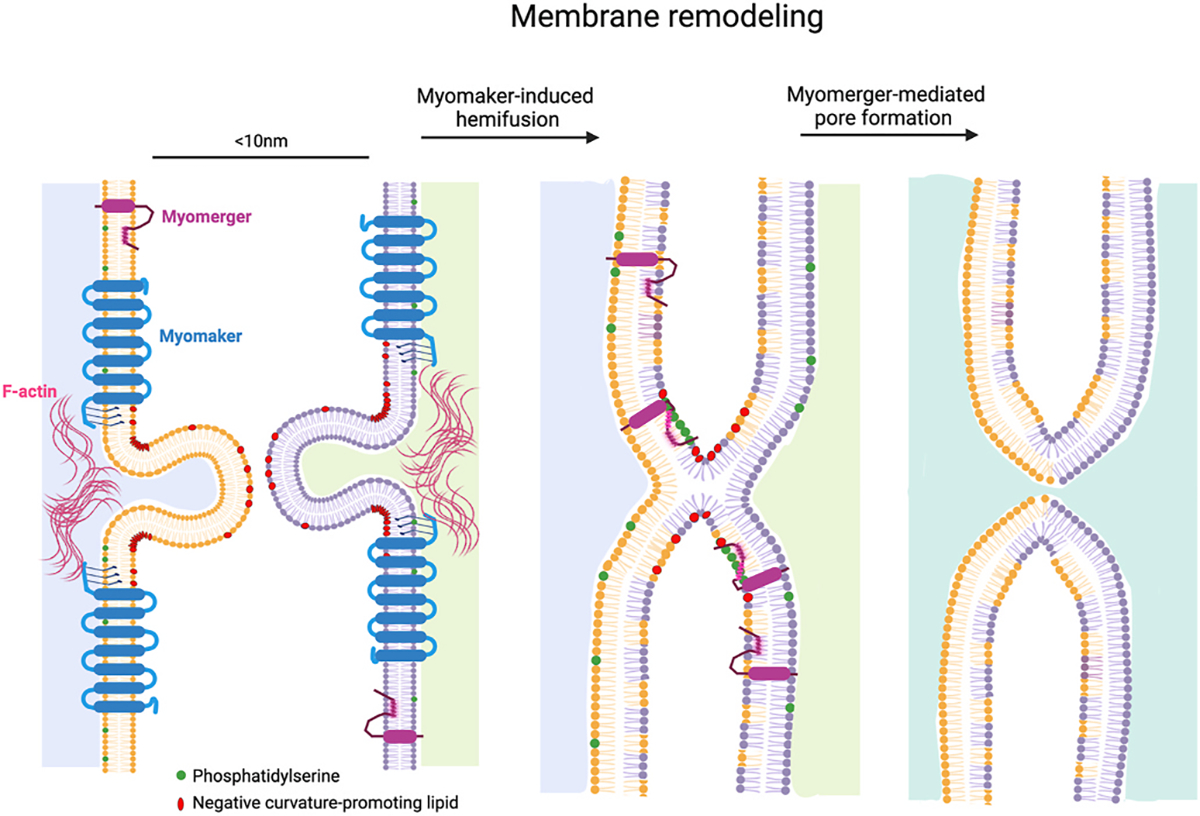
The independent activities of Myomaker and Myomerger culminate in myoblast fusion. Possible function for Myomaker is that it generates domains of negative curvature, promoting closer membrane apposition and outer leaflet membrane mixing (hemifusion). F-actin may provide mechanical support for these domains of negative curvature. Myomerger has two helical regions in its ectodomain. The helix proximal to the transmembrane domain responds to phosphatidylserine exposure to insert in membranes, causing membrane stress and promoting positive curvature, leading to pore formation and completion of the fusion reaction.
Cell and biochemical studies demonstrate that Myomerger possesses a membrane-stressing function. The presence of Myomerger sensitives cells to hypotonic osmotic shock, a stimulus that induces membrane tension and pore formation [3]. Recombinant myomerger causes destabilization of synthetic membranes [3]. Myomerger contains a transmembrane domain followed by two regions in its ectodomain that are predicted to be helical. It is likely that these helical regions, which are present in the recombinant ectodomain, insert in membranes and mediate the membrane-stressing activity of myomerger. One unique characteristic of Myomerger is that its ectodomain functions from outside the cell but that lipids of the hemifusion diaphragm that need remodeling are inside the cell. Theoretical modeling and experimental evidence demonstrate that Myomerger-generated positive curvature in the outer leaflet is propagated to the lipids in the inner diaphragm resulting in pore formation [75].
Regulation of Myomaker and Myomerger, beyond transcriptional activation in the myogenic lineage, is likely to represent an important aspect of the myoblast fusion pathway. Myoblasts presumably need a mechanism to spatially and temporally control these membrane destabilizing activities to maintain the integrity of the membrane and regulate fusion. Myomaker and Myomerger are not uniformly present on myoblast membranes suggesting one component of the regulation is trafficking [76]; another potential regulatory mechanism is movement of the proteins to actin rich areas where fusion may occur [77]. Lipids could also help control these membrane-bound muscle fusogens. PS has been proposed as a trigger for myoblast fusion [41] and in addition to its function in activation of cell surface molecules that lead to actin nucleation, PS also functions during the membrane remodeling stage of fusion [42]. Myomerger contains positively-charged arginine residues in the first helix (amphipathic) of its ectodomain, which theoretically would promote association with negatively charged PS in membrane bilayers. While robust protein-protein interaction studies are not yet available, there are likely additional factors that cooperate with Myomaker to drive fusion [62].
In addition to Myomaker and Myomerger, other factors may perturb the myoblast membrane to achieve sufficient fusion. Indeed, GRAF1 and myoferlin, have not been shown to interact with the muscle fusogens, but function in myoblast fusion events [78,79]. GRAF1 is a curvature-generating protein and increases pore formation induced by viral fusogens [80], while myoferlin regulates endocytic trafficking potentially affecting fusion through movement of proteins and lipids. Syncytins are encoded by endogenous elements likely captured from retroviruses that are structurally similar to viral fusogens, and essential for fusion of placental trophoblasts [81]. Intriguingly, male Syncytin-B null mice exhibit reduced muscle size with a concomitant decrease in the number of myonuclei [82]. While loss-of-function experiments specifically in satellite cells should be performed to test the relative importance of these factors, it is likely that a multitude of molecules synergize for the complex process of myoblast fusion.
3. Regulation of fusion in myofibers
A key aspect of muscle development and regeneration is to control fusions at the proper time and place, which prevents formation of aberrant syncytia. Fusion occurs during stages where two myoblasts fuse to form a nascent myofiber, then secondary fusion follows between a myoblast and a myofiber. On the surface, it would appear that myoblast-myoblast fusions and myoblast-myofiber fusions are similarly regulated given that the mergers transpire between cells of the same lineage. However, the myofiber is not only undergoing fusion but also maturing into a functional, contractile muscle cell, so perhaps it is not surprising that recent evidence suggests differential regulation of primary and secondary fusion events. Activation of CaMKII in growing myotubes is proposed to be a specific pathway that increases fusion on the myofiber side [83]. Molecular fusion brakes are present in the myofiber compartment, where both the membrane and cortical actin cytoskeleton need to be restored to a non-fusogenic state after fusion. Additionally, there are questions if the muscle fusogens are required on the myofiber for fusion. The lack of knowledge related to the myofiber fusion pathway could be attributed to the models and tools typically used to study fusion. In vitro myogenic differentiation and in vivo development and regeneration require primary fusion events to initiate the process followed by secondary fusion to growing myofibers, thus it is difficult to parse apart regulators that function within both or one of the fusion processes. Postnatal muscle development and exercise models, when there are already established myofibers, are likely better models to study secondary fusion. Genetic tools to delete genes in myofibers are also problematic because even promoters of genes expressed in the myofiber can also be turned on in differentiated myoblasts prior to fusion.
The phospholipid flippase complex of ATP11A (member of the type IV subfamily of P-type adenosine triphosphatases (P4-ATPases) and CDC50A function to move PS from the outer to inner leaflet of muscle cells in vitro [84]. Reduction of either of these factors results in excessive fusion, suggesting that clearance of PS from the outer leaflet is necessary to control myoblast fusion. I presume the PS removal must occur in myotubes, since PS on the outer leaflet is important in myoblasts to activate the fusion pathway, although this was not directly tested. ATP11A- and CDC50A-deficient cells exhibit reduced F-actin and non-muscle myosin IIA (NMIIA) in myotubes, which is proposed to prevent uncontrolled fusions. PIEZO1 is a mechanosensitive Ca2+ channel, and depletion of this protein also results in excessive cell fusion and no accumulation of cortical F-actin or NMIIA in the multinucleated cells. Overall, the data indicate that movement of PS to the inner leaflet of the membrane in myotubes activates the Ca2+ channel activity of PIEZO1 resulting in formation of cortical actomyosin assembly to limit uncontrolled cell fusion [84].
TGF-β signaling is also a negative regulator of the fusion pathway in chick and mice [85,86]. Two independent studies showed that for myoblast fusion to occur, TGF-β signaling needs to be reduced. In chick myoblast fusion, this regulation occurs in the myofiber compartment, but it is less clear if that is also true during mouse myoblast fusion. The activation of TGF-β signaling in the chick system is achieved through a receptor complementation mechanism, where the myofiber expresses TGF-βR1 and the myoblast contributes TGF-βR2 through fusion, causing heterodimerization of TGF-β receptors and initiation of the SMAD2/3 signaling cascade. In mouse myoblasts, TGF-β signaling prevents or breaks down actin architecture, which suggests that this pathway would reduce fusion of myoblasts since actin is needed in that compartment. It is difficult to interpret if a TGF-β signaling-induced reduction of actin in the myofiber after fusion would also limit fusion. The PIEZO1 study suggests formation of cortical actin will limit fusion, although different actin structures could be regulated by these different pathways. More defined studies where the fusion regulators are manipulated specifically in the myoblast or myofiber compartments in vivo could yield a more definitive myofiber fusion pathway.
The functional requirement of the skeletal muscle fusogens on myoblasts and myofibers also points to differential regulation in the two compartments. In vitro cell-based experiments show that during primary fusion Myomaker is needed on both fusing cells, and Myomerger is required on one fusing cell [6,7]. In contrast, expression and genetic evidence suggests that Myomaker expression is not needed on the myofiber for fusion. Myomaker mRNA is not detected in muscle after fusion has ceased (postnatal day 21 in the mouse) until there is an injury [51]. In the mdx model, Myomaker in the myofiber is contributed exclusively through fusion of myoblasts [87]. Thus, Myomaker mRNA is quickly dowregulated after fusion, and existing myonuclei possess limited ability to upregulate Myomaker, although the kinetics of Myomaker protein are not yet understood. Deletion of Myomaker using an inducible human skeletal actin-Cre (HSA-CreER), which is expressed in myofibers and during late-stage differentiation of myoblasts, does not impact fusion dynamics in the mdx dystrophic mouse model [87] or during load-induced muscle hypertrophy (Millay lab, unpublished observations). Caveats to these genetic experiments include that one cannot be sure that Myomaker was deleted from all myonuclei, and there is the possibility that Myomaker protein could be placed on the myofiber membrane through extracellular vesicles emanating from myoblasts. Overall, there is an emerging paradigm that the fusion pathway in the myofiber is distinct from myoblasts, even for the muscle fusogens, although the need for Myomerger in the myofiber is not known.
3.1. Myoblast fusion and muscle diseases
In addition to the role of myoblast fusion for muscle development and maintenance, dysregulation of critical fusion factors impact genetic diseases of skeletal muscle through direct and indirect mechanisms. Mutations in Myomaker in humans has been proposed to cause Carey Fineman Ziter syndrome (CFZS) [88], characterized by facial abnormalities, generalized muscle hypoplasia with hypotonia and relatively mild proximal weakness, failure to thrive, delayed motor milestones, scoliosis, and normal intelligence. Currently, there are 13 reported patients (age 7–69) who exhibit CFZS associated with autosomal recessive mutations in Myomaker [88–91], and the onset of this heterogenous phenotype occurs at birth for facial abnormalities or in the early childhood to juvenile period for muscle hypotonia and weakness. Histological analysis of the vastus lateralis muscle from one patient at 15 years of age indicates hypotrophy of type I myofibers and hypertrophy of type II myofibers, but minimal central nucleation or activation of the myogenic program, both hallmarks of regeneration [88]. A previous biopsy at a younger age apparently showed a small reduction in myofiber size and increase in connective tissue, suggesting as the patient aged, the disease progressed potentially due to limitations in regenerative capacity. A biopsy of an independent patient revealed hypertrophy of both type I and type II myofibers [91]. Levels of muscle creatine kinase in the serum was only mildly elevated in a few patients indicating minimal damage to muscle cell membranes, at least at the time of analysis. Taken together, CFZS associated with Myomaker mutations are phenotypically heterogenous due to currently unclear reasons.
The initial report on CFZS caused by Myomaker identified five different mutations. The genetics indicate that disease in patients is caused by the combination of a hypomorphic allele with a null allele [88]. Three mutations (Met1-mutation of initiating methionine, Gly100Ser, Cys185Arg) may be null mutations as it was difficult to detect protein after transfection in Hela cells. Pro91Thr and Ile154Thr were classified as hypomorphic based on some ability to induce fusion of fibroblasts with myoblasts, a classical function of wild-type (WT) Myomaker. A more recent analysis of a Swedish family identifies a new mutation, Trp79Arg, along with Pro91Thr. Trp79Arg is likely a null allele (Millay lab, unpublished observations). Overall, data suggest that CFZS is associated with one hypomorphic allele and one null allele of Myomaker, which results in a lower level of Myomaker activity.
Mutations in Myomaker are clearly associated with CFZS, although more work remains to fully understand how a Myomaker hypomorphic protein causes muscle disease. Molecularly, Myomaker Pro91Thr functioned in fibroblast heterologous fusion is similar to WT, and it was also able to rescue fusion in Mymomaker null zebrafish but not to WT levels [88]. While these data are consistent with a hypomorphic activity, whether Myomaker Pro91Thr was expressed at similar levels as WT was not tested and the mechanistic underpinning for reduced function is not known. The link between Myomaker mutations and the muscle pathology is also unclear. The simplest explanation is that muscle pathology is directly caused by a small fusion deficit resulting in reduced number of myofibers or reduced number of nuclei in myofibers. It is not clear if CFZS patients exhibit developmental defects that progressively lead to muscle pathology or if they exhibit issues with the maintenance of muscle once formed. Reductions of myonuclear number in the mouse results in an adaptive response that allows normal muscle development, albeit with myofibers that have reduced size [92], which would suggest some level of flexibility for the minimum level of myonuclei needed for development. The effect of Myomaker mutations on myonuclear number or myofiber number is very difficult to study in patients, necessitating the generation of new models. Answers to these questions should improve knowledge about the disease pathology in humans and generally aid in the delineation of a myoblast fusion pathway.
Skeletal muscle diseases can also be impacted indirectly by dysregulation of the skeletal muscle fusogens. Typically, fusion events in muscle occur during development and after a stimulus in the adult, but this is not the case during dystrophic disease pathology where continuous fusion takes place. Myomaker is down-regulated after fusion in WT mice but remains elevated in dystrophic myofibers [87]. Genetic deletion of Myomaker in mdx myofibers does not impact fusion but results in reduced membrane damage, indicating that the fusion process itself is not a contributing factor to dystrophic membrane instability. These data suggest that ectopic expression of Myomaker in myofibers and its potential membrane-remodeling activities has deleterious consequences [87]. Similarly, ablation of satellite cells early in the dystrophic disease progression results in larger myofibers that exhibit healthier membranes [93]. This situation may be similar to the hypertrophy observed in CFZS where new myofiber formation is disrupted leading to hypertrophy of existing myofibers. Hence, during chronic muscle regeneration there are unintended consequences of chronic myoblast fusion, such as sustained expression of Myomaker in the myofiber, that negatively impact disease etiology.
4. Conclusions
While the molecules and mechanisms of the myoblast fusion pathway have become clearer over the last few years, there are still gaps in our understanding. The requirement of actin nucleation and the muscle fusogens are the major components of the pathway, based on their absolute requirement for myoblast fusion in vivo. The majority of other factors have more modest effects on the pathway, and often defects are only observed during regeneration, nonetheless it takes many proteins and lipids to drive efficient myoblast fusion. Critical unanswered questions in the field include if biochemical evidence will support the proposed function of Myomaker based on structural homology, the fusion pathway in myofibers compared to myoblasts, the identification of key adhesion and recognition factors for mammalian myoblast fusion, the role of lipids and membrane homeostasis during different steps of fusion, and precise mechanisms of how impaired fusogen function contribute to muscle pathology. Productive answers to these questions has the potential to improve strategies for the treatment of muscle diseases and augment adaptations of muscle after exercise.
Acknowledgements
I thank members of the Millay laboratory for critical reading of the review. I apologize to those whose work I am unable to cite due to space limitations. The work in the Millay laboratory is funded by grants to D.P. M. from the Cincinnati Children’s Hospital Research Foundation, United States, Pew Charitable Trusts, National Institutes of Health (R01AR068286, R01AG059605, R61AR076771), and a sponsored research agreement with Sana Biotechnology.
Footnotes
Credit author statement
D.P.M. wrote the manuscript.
References
- [1].Prasad V, Millay DP, Skeletal muscle fibers count on nuclear numbers for growth, Semin. Cell Dev. Biol. (2021), 10.1016/j.semcdb.2021.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kim JH, Chen EH, The fusogenic synapse at a glance, J. Cell Sci. 132 (2019), 10.1242/jcs.213124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Leikina E, et al. , Myomaker and myomerger work independently to control distinct steps of membrane remodeling during myoblast fusion, Dev. Cell 46 (2018) 767–780 e767, 10.1016/j.devcel.2018.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Millay DP, et al. , Myomaker is a membrane activator of myoblast fusion and muscle formation, Nature 499 (2013) 301–305, 10.1038/nature12343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bi P, et al. , Control of muscle formation by the fusogenic micropeptide myomixer, Science 356 (2017) 323–327, 10.1126/science.aam9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Quinn ME, et al. , Myomerger induces fusion of non-fusogenic cells and is required for skeletal muscle development, Nat. Commun. 8 (2017) 15665, 10.1038/ncomms15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhang Q, et al. , The microprotein Minion controls cell fusion and muscle formation, Nat. Commun. 8 (2017) 15664, 10.1038/ncomms15664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kim JH, Jin P, Duan R, Chen EH, Mechanisms of myoblast fusion during muscle development, Curr. Opin. Genet. Dev. 32 (2015) 162–170, 10.1016/j.gde.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sampath SC, Sampath SC, Millay DP, Myoblast fusion confusion: the resolution begins, Skeletal Muscle 8 (2018) 3, 10.1186/s13395-017-0149-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Deng S, Azevedo M, Baylies M, Acting on identity: myoblast fusion and the formation of the syncytial muscle fiber, Semin. Cell Dev. Biol. 72 (2017) 45–55, 10.1016/j.semcdb.2017.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Brukman NG, Uygur B, Podbilewicz B, Chernomordik LV, How cells fuse J Cell Biol. 218 (2019) 1436–1451, 10.1083/jcb.201901017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hernandez JM, Podbilewicz B, The hallmarks of cell-cell fusion, Development 144 (2017) 4481–4495, 10.1242/dev.155523. [DOI] [PubMed] [Google Scholar]
- [13].Segev N, Avinoam O, Podbilewicz B, Fusogens. Curr Biol 28 (2018) R378–R380, 10.1016/j.cub.2018.01.024. [DOI] [PubMed] [Google Scholar]
- [14].Chernomordik LV, Kozlov MM, Membrane hemifusion: crossing a chasm in two leaps, Cell 123 (2005) 375–382, 10.1016/j.cell.2005.10.015. [DOI] [PubMed] [Google Scholar]
- [15].Martens S, McMahon HT, Mechanisms of membrane fusion: disparate players and common principles, Nat. Rev. Mol. Cell Biol. 9 (2008) 543–556, 10.1038/nrm2417. [DOI] [PubMed] [Google Scholar]
- [16].Podbilewicz B, Virus and cell fusion mechanisms, Annu. Rev. Cell Dev. Biol. 30 (2014) 111–139, 10.1146/annurev-cellbio-101512-122422. [DOI] [PubMed] [Google Scholar]
- [17].Wang Y, et al. , SNARE-mediated membrane fusion in autophagy, Semin. Cell Dev. Biol. 60 (2016) 97–104, 10.1016/j.semcdb.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rizo J, Sudhof TC, The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices–guilty as charged? Annu. Rev. Cell Dev. Biol. 28 (2012) 279–308, 10.1146/annurev-cellbio-101011-155818. [DOI] [PubMed] [Google Scholar]
- [19].Lee DM, Chen EH, Drosophila myoblast fusion: invasion and resistance for the ultimate union, Annu. Rev. Genet. 53 (2019) 67–91, 10.1146/annurev-genet-120116-024603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ruiz-Gomez M, Coutts N, Price A, Taylor MV, Bate M, Drosophila dumbfounded: a myoblast attractant essential for fusion, Cell 102 (2000) 189–198, 10.1016/s0092-8674(00)00024-6. [DOI] [PubMed] [Google Scholar]
- [21].Strunkelnberg M, et al. , rst and its paralogue kirre act redundantly during embryonic muscle development in Drosophila, Development 128 (2001) 4229–4239. [DOI] [PubMed] [Google Scholar]
- [22].Zhang R, et al. , Dynamin regulates the dynamics and mechanical strength of the actin cytoskeleton as a multifilament actin-bundling protein, Nat. Cell Biol. 22 (2020) 674–688, 10.1038/s41556-020-0519-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rushton E, Drysdale R, Abmayr SM, Michelson AM, Bate M, Mutations in a novel gene, myoblast city, provide evidence in support of the founder cell hypothesis for Drosophila muscle development, Development 121 (1995) 1979–1988. [DOI] [PubMed] [Google Scholar]
- [24].Artero RD, Castanon I, Baylies MK, The immunoglobulin-like protein Hibris functions as a dose-dependent regulator of myoblast fusion and is differentially controlled by Ras and Notch signaling, Development 128 (2001) 4251–4264. [DOI] [PubMed] [Google Scholar]
- [25].Dworak HA, Charles MA, Pellerano LB, Sink H, Characterization of Drosophila hibris, a gene related to human nephrin, Development 128 (2001) 4265–4276. [DOI] [PubMed] [Google Scholar]
- [26].Srinivas BP, Woo J, Leong WY, Roy S, A conserved molecular pathway mediates myoblast fusion in insects and vertebrates, Nat. Genet. 39 (2007) 781–786, 10.1038/ng2055. [DOI] [PubMed] [Google Scholar]
- [27].Powell GT, Wright GJ, Jamb and jamc are essential for vertebrate myocyte fusion, PLoS Biol. 9 (2011), e1001216, 10.1371/journal.pbio.1001216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sohn RL, et al. , A role for nephrin, a renal protein, in vertebrate skeletal muscle cell fusion, Proc. Natl. Acad. Sci. U. S. A. 106 (2009) 9274–9279, 10.1073/pnas.0904398106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Krauss RS, Joseph GA, Goel AJ, Keep your friends close: cell-cell contact and skeletal myogenesis, Cold Spring Harbor Perspect. Biol 9 (2017), 10.1101/cshperspect.a029298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Schwander M, et al. , Beta1 integrins regulate myoblast fusion and sarcomere assembly, Dev. Cell 4 (2003) 673–685, 10.1016/s1534-5807(03)00118-7. [DOI] [PubMed] [Google Scholar]
- [31].Horsley V, Jansen KM, Mills ST, Pavlath GK, IL-4 acts as a myoblast recruitment factor during mammalian muscle growth, Cell 113 (2003) 483–494, 10.1016/s0092-8674(03)00319-2. [DOI] [PubMed] [Google Scholar]
- [32].Sens KL, et al. , An invasive podosome-like structure promotes fusion pore formation during myoblast fusion, J. Cell Biol. 191 (2010) 1013–1027, 10.1083/jcb.201006006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chen EH, Invasive podosomes and myoblast fusion, Curr. Top. Membr. 68 (2011) 235–258, 10.1016/B978-0-12-385891-7.00010-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Shilagardi K, et al. , Actin-propelled invasive membrane protrusions promote fusogenic protein engagement during cell-cell fusion, Science 340 (2013) 359–363, 10.1126/science.1234781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kim JH, et al. , Mechanical tension drives cell membrane fusion, Dev. Cell 32 (2015) 561–573, 10.1016/j.devcel.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Moore CA, Parkin CA, Bidet Y, Ingham PW, A role for the Myoblast city homologues Dock1 and Dock5 and the adaptor proteins Crk and Crk-like in zebrafish myoblast fusion, Development 134 (2007) 3145–3153, 10.1242/dev.001214. [DOI] [PubMed] [Google Scholar]
- [37].Vasyutina E, Martarelli B, Brakebusch C, Wende H, Birchmeier C, The small G-proteins Rac1 and Cdc42 are essential for myoblast fusion in the mouse, Proc. Natl. Acad. Sci. U. S. A. 106 (2009) 8935–8940, 10.1073/pnas.0902501106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Laurin M, et al. , The atypical Rac activator Dock180 (Dock1) regulates myoblast fusion in vivo, Proc. Natl. Acad. Sci. U. S. A. 105 (2008) 15446–15451, 10.1073/pnas.0805546105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gruenbaum-Cohen Y, et al. , The actin regulator N-WASp is required for muscle-cell fusion in mice, Proc. Natl. Acad. Sci. U. S. A. 109 (2012) 11211–11216, 10.1073/pnas.1116065109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jeong J, Conboy IM, Phosphatidylserine directly and positively regulates fusion of myoblasts into myotubes, Biochem. Biophys. Res. Commun. 414 (2011) 9–13, 10.1016/j.bbrc.2011.08.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Whitlock JM, Chernomordik LV, Flagging fusion: phosphatidylserine signaling in cell-cell fusion, J. Biol. Chem. 296 (2021) 100411, 10.1016/j.jbc.2021.100411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Leikina E, et al. , Extracellular annexins and dynamin are important for sequential steps in myoblast fusion, J. Cell Biol. 200 (2013) 109–123, 10.1083/jcb.201207012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hamoud N, Tran V, Croteau LP, Kania A, Cote JF, G-protein coupled receptor BAI3 promotes myoblast fusion in vertebrates, Proc. Natl. Acad. Sci. U. S. A. 111 (2014) 3745–3750, 10.1073/pnas.1313886111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hamoud N, et al. , Spatiotemporal regulation of the GPCR activity of BAI3 by C1qL4 and Stabilin-2 controls myoblast fusion, Nat. Commun. 9 (2018) 4470, 10.1038/s41467-018-06897-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hochreiter-Hufford AE, et al. , Phosphatidylserine receptor BAI1 and apoptotic cells as new promoters of myoblast fusion, Nature 497 (2013) 263–267, 10.1038/nature12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Park SY, et al. , Stabilin-2 modulates the efficiency of myoblast fusion during myogenic differentiation and muscle regeneration, Nat. Commun. 7 (2016) 10871, 10.1038/ncomms10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Bothe I, Deng S, Baylies M, PI(4,5)P2 regulates myoblast fusion through Arp2/3 regulator localization at the fusion site, Development 141 (2014) 2289–2301, 10.1242/dev.100743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chuang MC, et al. , Tks5 and Dynamin-2 enhance actin bundle rigidity in invadosomes to promote myoblast fusion, J. Cell Biol. 218 (2019) 1670–1685, 10.1083/jcb.201809161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Randrianarison-Huetz V, et al. , Srf controls satellite cell fusion through the maintenance of actin architecture, J. Cell Biol. 217 (2018) 685–700, 10.1083/jcb.201705130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Dhanyasi N, et al. , Surface apposition and multiple cell contacts promote myoblast fusion in Drosophila flight muscles, J. Cell Biol. 211 (2015) 191–203, 10.1083/jcb.201503005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Millay DP, Sutherland LB, Bassel-Duby R, Olson EN, Myomaker is essential for muscle regeneration, Genes Dev. 28 (2014) 1641–1646, 10.1101/gad.247205.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Landemaine A, Rescan PY, Gabillard JC, Myomaker mediates fusion of fast myocytes in zebrafish embryos, Biochem. Biophys. Res. Commun. 451 (2014) 480–484, 10.1016/j.bbrc.2014.07.093. [DOI] [PubMed] [Google Scholar]
- [53].Zhang W, Roy S, Myomaker is required for the fusion of fast-twitch myocytes in the zebrafish embryo, Dev. Biol. 423 (2017) 24–33, 10.1016/j.ydbio.2017.01.019. [DOI] [PubMed] [Google Scholar]
- [54].Shi J, Cai M, Si Y, Zhang J, Du S, Knockout of myomaker results in defective myoblast fusion, reduced muscle growth and increased adipocyte infiltration in zebrafish skeletal muscle, Hum. Mol. Genet 27 (2018) 3542–3554, 10.1093/hmg/ddy268. [DOI] [PubMed] [Google Scholar]
- [55].Hromowyk KJ, Talbot JC, Martin BL, Janssen PML, Amacher SL, Cell fusion is differentially regulated in zebrafish post-embryonic slow and fast muscle, Dev. Biol. 462 (2020) 85–100, 10.1016/j.ydbio.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bi P, et al. , Fusogenic micropeptide Myomixer is essential for satellite cell fusion and muscle regeneration, Proc. Natl. Acad. Sci. U. S. A. 115 (2018) 3864–3869, 10.1073/pnas.1800052115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Shi J, et al. , Requirement of the fusogenic micropeptide myomixer for muscle formation in zebrafish, Proc. Natl. Acad. Sci. U. S. A. 114 (2017) 11950–11955, 10.1073/pnas.1715229114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ganassi M, et al. , Myogenin promotes myocyte fusion to balance fibre number and size, Nat. Commun. 9 (2018) 4232, 10.1038/s41467-018-06583-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Luo W, Li E, Nie Q, Zhang X, Myomaker, regulated by MYOD, MYOG and miR-140-3p, promotes chicken myoblast fusion, Int. J. Mol. Sci. 16 (2015) 26186–26201, 10.3390/ijms161125946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Perello-Amoros M, Ralliere C, Gutierrez J, Gabillard JC, Myomixer is expressed during embryonic and post-larval hyperplasia, muscle regeneration and differentiation of myoblats in rainbow trout (Oncorhynchus mykiss), Gene 790 (2021) 145688, 10.1016/j.gene.2021.145688. [DOI] [PubMed] [Google Scholar]
- [61].Landemaine A, et al. , Trout myomaker contains 14 minisatellites and two sequence extensions but retains fusogenic function, J. Biol. Chem. 294 (2019) 6364–6374, 10.1074/jbc.RA118.006047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Zhang H, et al. , Human myotube formation is determined by MyoD-Myomixer/Myomaker axis, Sci. Adv. 6 (2020), 10.1126/sciadv.abc4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Yaseen W, et al. , Fibroblast fusion to the muscle fiber regulates myotendinous junction formation, Nat. Commun. 12 (2021) 3852, 10.1038/s41467-021-24159-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Zhang H, et al. , Evolution of a chordate-specific mechanism for myoblast fusion, bioRxiv 2021 (2021), 10.1101/2021.07.24.453587, 2007.2024.453587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Petrany MJ, Millay DP, Cell fusion: merging membranes and making muscle, Trends Cell Biol. 29 (2019) 964–973, 10.1016/j.tcb.2019.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Millay DP, et al. , Structure-function analysis of myomaker domains required for myoblast fusion, Proc. Natl. Acad. Sci. U. S. A. 113 (2016) 2116–2121, 10.1073/pnas.1600101113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Pei J, Millay DP, Olson EN, Grishin NV, CREST–a large and diverse superfamily of putative transmembrane hydrolases, Biol. Direct 6 (2011) 37, 10.1186/1745-6150-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Coant N, Sakamoto W, Mao C, Hannun YA, Ceramidases, roles in sphingolipid metabolism and in health and disease, Adv Biol Regul 63 (2017) 122–131, 10.1016/j.jbior.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Vasiliauskaite-Brooks I, Healey RD, Granier S, 7TM proteins are not necessarily GPCRs, Mol. Cell. Endocrinol. 491 (2019) 110397, 10.1016/j.mce.2019.02.009. [DOI] [PubMed] [Google Scholar]
- [70].Alonso A, Goni FM, The physical properties of ceramides in membranes, Annu. Rev. Biophys. 47 (2018) 633–654, 10.1146/annurev-biophys-070317-033309. [DOI] [PubMed] [Google Scholar]
- [71].Chernomordik L, Kozlov MM, Zimmerberg J, Lipids in biological membrane fusion, J. Membr. Biol. 146 (1995) 1–14, 10.1007/BF00232676. [DOI] [PubMed] [Google Scholar]
- [72].Saez R, Goni FM, Alonso A, The effect of bilayer order and fluidity on detergent-induced liposome fusion, FEBS Lett. 179 (1985) 311–315, 10.1016/0014-5793(85)80541-x. [DOI] [PubMed] [Google Scholar]
- [73].Wilschut J, Duzgunes N, Hoekstra D, Papahadjopoulos D, Modulation of membrane fusion by membrane fluidity: temperature dependence of divalent cation induced fusion of phosphatidylserine vesicles, Biochemistry 24 (1985) 8–14, 10.1021/bi00322a002. [DOI] [PubMed] [Google Scholar]
- [74].Prives J, Shinitzky M, Increased membrane fluidity precedes fusion of muscle cells, Nature 268 (1977) 761–763, 10.1038/268761a0. [DOI] [PubMed] [Google Scholar]
- [75].Golani G, et al. , Myomerger promotes fusion pore by elastic coupling between proximal membrane leaflets and hemifusion diaphragm, Nat. Commun. 12 (2021) 495, 10.1038/s41467-020-20804-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Gamage DG, et al. , Insights into the localization and function of myomaker during myoblast fusion, J. Biol. Chem. 292 (2017) 17272–17289, 10.1074/jbc.M117.811372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Hammers DW, et al. , Filopodia powered by class X myosin promote fusion of mammalian myoblasts, Elife 10 (2021), 10.7554/eLife.72419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Lenhart KC, et al. , GRAF1 promotes ferlin-dependent myoblast fusion, Dev. Biol. 393 (2014) 298–311, 10.1016/j.ydbio.2014.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Posey AD Jr., et al. , Endocytic recycling proteins EHD1 and EHD2 interact with fer-1-like-5 (Fer1L5) and mediate myoblast fusion, J. Biol. Chem. 286 (2011) 7379–7388, 10.1074/jbc.M110.157222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Richard JP, et al. , Intracellular curvature-generating proteins in cell-to-cell fusion, Biochem. J. 440 (2011) 185–193, 10.1042/BJ20111243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Dupressoir A, et al. , A pair of co-opted retroviral envelope syncytin genes is required for formation of the two-layered murine placental syncytiotrophoblast, Proc. Natl. Acad. Sci. U. S. A. 108 (2011) E1164–E1173, 10.1073/pnas.1112304108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Redelsperger F, et al. , Genetic evidence that captured retroviral envelope syncytins contribute to myoblast fusion and muscle sexual dimorphism in mice, PLoS Genet. 12 (2016), e1006289, 10.1371/journal.pgen.1006289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Eigler T, et al. , ERK1/2 inhibition promotes robust myotube growth via CaMKII activation resulting in myoblast-to-myotube fusion, Dev. Cell 56 (2021), 10.1016/j.devcel.2021.11.022, 3349-3363 e3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Tsuchiya M, et al. , Cell surface flip-flop of phosphatidylserine is critical for PIEZO1-mediated myotube formation, Nat. Commun. 9 (2018) 2049, 10.1038/s41467-018-04436-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Girardi F, et al. , TGFbeta signaling curbs cell fusion and muscle regeneration, Nat. Commun. 12 (2021) 750, 10.1038/s41467-020-20289-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Melendez J, et al. , TGFbeta signalling acts as a molecular brake of myoblast fusion, Nat. Commun. 12 (2021) 749, 10.1038/s41467-020-20290-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Petrany MJ, Song T, Sadayappan S, Millay DP, Myocyte-derived Myomaker expression is required for regenerative fusion but exacerbates membrane instability in dystrophic myofibers, JCI Insight 5 (2020), 10.1172/jci.insight.136095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Di Gioia SA, et al. , A defect in myoblast fusion underlies Carey-Fineman-Ziter syndrome, Nat. Commun. 8 (2017) 16077, 10.1038/ncomms16077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Alrohaif H, et al. , Whole-exome sequencing identifies mutations in MYMK in a mild form of Carey-Fineman-Ziter syndrome, Neurol Genet 4 (2018) e226, 10.1212/NXG.0000000000000226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Camacho A, et al. , Carey-fineman-ziter syndrome: a MYMK-related myopathy mimicking brainstem dysgenesis, J. Neuromuscul. Dis. 7 (2020) 309–313, 10.3233/JND-200477. [DOI] [PubMed] [Google Scholar]
- [91].Hedberg-Oldfors C, Lindberg C, Oldfors A, Carey-Fineman-Ziter syndrome with mutations in the myomaker gene and muscle fiber hypertrophy, Neurol Genet 4 (2018) e254, 10.1212/NXG.0000000000000254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Cramer AAW, et al. , Nuclear numbers in syncytial muscle fibers promote size but limit the development of larger myonuclear domains, Nat. Commun. 11 (2020) 6287, 10.1038/s41467-020-20058-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Boyer JG, et al. , Satellite cell depletion in early adulthood attenuates muscular dystrophy pathogenesis, bioRxiv (2019) 857433, 10.1101/857433. [DOI] [Google Scholar]