

Abstract
The morphological plasticity of microglia has fascinated neuroscientists for 100 years. Attempts to classify functional phenotypes are hampered by similarities between endogenous brain microglia and peripheral myeloid cells that can enter the brain under pathological conditions. Recent advances in single cell -omic methodologies have led to an explosion of data regarding gene expression in microglia. Herein, we review the diversity of microglial phenotypes in healthy brain, aging and Alzheimer’s disease, identify knowledge gaps in the body of evidence and suggest areas where new knowledge would be useful. Data from human samples and mouse models are compared and contrasted. Understanding the molecular complexity of the microglial response repertoire will suggest new avenues for therapeutic treatments in Alzheimer’s disease.
Keywords: microarray, microglia, gene expression, sequencing, immunity, neurodegeneration
INTRODUCTION
Identification of Microglia
Discrimination of microglia, the resident immune cells of the brain, from other myeloid cells biochemically has been challenging historically. Initially this was thought to represent common mesodermal embryonic origins. However, fate mapping in mice has shown that microglia originate from erythromyeloid progenitors from the yolk sac in a Pu.1 (transcription factor) and Irf (interferon regulatory factor)-8-dependent and Myb (proto-oncogene transcriptional factor)-independent manner1,2, with Tgfb1 expression essential for microglial development and maintenance. The original identification of microglia was based on morphology after silver stains of brain sections3 and electron microscopy4. Cellular stains based on cell surface markers revealed that microglia and myeloid cells of the monocyte/macrophage lineage share many of the same phenotypic markers. Functional similarities between microglia and other myeloid cells such as phagocytosis may underlie these similarities in marker expression5. Attempts to identify markers to discriminate microglia from myeloid cells have been intense since at least the 1980s. Iba1, F4/80 and Cd-68 (ED-1) were are all markers originally believed to be macrophage specific, but were later shown to stain microglia6,7. By analogy with polarization of T cell or macrophage activation responses, attempts to classify the types of up-regulated proteins and their functions along two general pathways termed pro-inflammatory or M1 vs anti-inflammatory or M2 sparked significant research effort8. Refinement of this schema included subtypes of M2 activation patterns and recognition that multi-dimensional activation occurred, with M1 and M2 representing the most widely divergent responses along a continuum. The utility of the M1/M2 classification has been questioned because it fails to capture the complexity of microglial responses to aging, injury and disease, and because single stimuli can induce both M1 and M2 responses9.
Some success in discriminating brain myeloid populations was obtained using markers with different magnitudes of expression. Staining for Cx3cr1 (Cx3c chemokine receptor 1) is more intense on microglial cells compared with myeloid cells, while Cd45 is more intense on myeloid cells10. Microglia can be identified using flow cytometry as Cx3cr1+/Cd45 low or moderate, while myeloid cells are generally Cx3cr1 low or negative/Cd45 high. However, these low vs high staining intensities are difficult or impossible to discriminate on tissue sections with traditional immunostaining methods. Fluorescent reporter mice were generated using some of these markers, such as Cx3cr1-GFP to label microglia11 and Ccr2 (chemokine receptor 2)-RFP to label peripheral monocyte/myeloid cells12. While these mice have been useful for elucidating roles of different cell populations, it is clear that the markers are expressed in multiple cell populations13. Finally, peripheral monocytes and/or macrophages infiltrating into the brain are reported to adopt expression of microglial selective genes5. These factors complicate interpretation of central vs peripheral contributions to brain myeloid cell populations.
Isolation of Microglia and Profiling Methodology
Two recent advances appear to have solved these difficulties. First, mechanical and/or enzymatic methods to dissociate adult brain into intact cells or nuclei have been developed14. Dissociation of intact cells requires unfrozen, unfixed (fresh) tissue, but intact nuclei can be obtained from frozen tissue. Once isolated, dissociated cells may be selected or sorted based on expression of cell surface molecules. Microglia have been selected or sorted based primarily on Cd11b or Cx3cr1 expression to date15–17. Positive cell selection utilizes cell surface protein immunostaining to couple antigen expression with magnetic beads, retention of cells in a column under a strong magnetic field, followed by release of cells after removing columns from the magnetic field. A wide variety of products for dissociation, positive and negative selection are available from multiple companies. Cell sorting requires immunostaining cell populations with fluorescently tagged antibodies and a sorting instrument. Most universities and research centers operate core facilities with sorting capacity based on multiple fluors and options, but bench-top instruments are available also. Transcriptomic, proteomic or single cell RNA sequencing (RNAseq) techniques may be applied to the sorted or unsorted cells (Table 1). Some caveats about the populations of cells/nuclei that result should be mentioned. First, neurons with large processes and complex morphology, such as pyramidal neurons, are damaged by physical dissociation methods, so recovered populations of neurons probably over-represent smaller neuronal morphologies. Isolation of nuclei circumvents this problem. Comparative cell vs nuclear single cell RNAseq studies report similar transcript identification, although the nuclear RNA includes more pre-processed, unspliced sequences18,19. On the other hand, the small size of microglial nuclei makes it challenging to discriminate them from debris and cell fragments. It is also conceivable that cells or nuclei already damaged by aging or disease are less likely to be physically isolated in these methodologies.
Table 1:
Experimental methodologies for gene expression analyses.
| Platform | Assay Type | Comments |
|---|---|---|
| Transcriptomics Platform | Microarray | Analysis of the transcriptome using hybridization-based methods Probes physically attached to substrate (slide, chip, plate) or beads Limits detection of transcripts to already existing genomic sequence information Limited to reference information available Does not allow discovery of new, unknown genes Limited dynamic range with no detection in the variation of highly or low expressed genes Used on tissue or isolated cells |
| RNA Sequencing (RNASeq) | High throughput sequencing-based technique Allows characterization and quantification of the transcriptome of a sample Detects known RNA, novel RNA and RNA variants, as well as absolute quantification, providing the dynamic landscape of the transcriptome On bulk sample: identify genes and gene modules associated with biologically relevant structure in samples such as treated vs control samples, samples of different cellular states On single cell or single nucleus: identification of the heterogeneity of response, stochasticity of gene expression and inference of gene regulatory networks across the same cell population Compared to microarray platform: Better signal-to noise (hybridization issue) Higher sensitivity and specificity allowing detection of rare and low-abundance transcripts 3-5 times more expensive Single cell analyses widely used in animal models due to requirement for unfrozen, unfixed tissue Single nucleus analyses emerging; limited data from human brain but frozen samples may be used |
|
| Analysis method | Clustering | To explore statistically the similarity between gene expression data, providing a common denominator for a group of genes/proteins Different algorithms provide different cluster models. Therefore understanding these models is essential for the interpretation of the differences between the various algorithms (e.g. connectivity model, distribution-based model) |
| Fold enrichment | Identify classes of genes or proteins that are over-represented in a large set of genes or proteins. Identify coregulated gene networks rather than individual genes that are up- or down-regulated in different cell populations. Use statistical approaches to identify significantly enriched or depleted groups of genes. |
Transcriptomics methodologies using these cell/nuclei separation techniques have been booming in the last 2 years with the ultimate aim to decipher cell behavior using single-cell RNA sequencing and network analyses. Mouse and human data have identified many microglial and inflammatory genes associated with Aβ plaques, but also with tau protein, underscoring the significance of genes associated with innate immunity as risk factors for late-onset AD. By comparing expression changes identified in whole (bulk) homogenates to previously published normative gene or protein expression in specific cell types20,21, gene expression can be attributed (imputed) to specific cell types. In this way, changes in gene or gene set expression may be attributed to microglia if the markers in question are known to arise from microglia. These types of analyses will continue due to the large banks of existing archival Alzheimer tissue. However, single cell/nuclei sequencing technologies are improving rapidly and will see increasing utilization in the foreseeable future. Recently, spatial transcriptomics, cytometry by time of flight (CyTOF) and multiplexed fluorescent in situ hybridization have emerged to correlate changes in gene expression with neuropathology, allowing the identification of different cell populations and brain regions susceptible to neurodegeneration.
In this paper, we review significant recent findings regarding phenotypic diversity of microglial cells in healthy, aging and Alzheimer’s disease (AD) brain. Attempts to review this field are challenging due the rapid release of large data sets. This article will highlight similarities and differences between human and mouse studies. By understanding the state of knowledge in the field, major gaps will be elucidated, allowing development of a roadmap for directing future work.
“HOMEOSTATIC” MICROGLIA
Differentially Expressed Genes in Microglia: Species and Region Effects
Microarray and single cell RNA sequencing experiments have revealed that microglia are defined by a unique molecular signature, known as their homeostatic signature, which is driven by the expression of the transforming growth factor (Tgf)-β1 cytokine22. Numerous transcriptomic studies on isolated murine cells have identified genes specifically associated with microglia when compared to other cell populations of the brain, myeloid cells or peripheral macrophages consistent with a homeostatic signature of microglia (Table 2). The Immunological Genome (ImmGen) consortium revealed 65 mRNA transcripts increased by five-fold or more in microglia relative to macrophage populations, with an additional of 13 mRNA transcripts encoding transcription factors up-regulated by twofold or more23. This study identified Siglech (sialic acid-binding immunoglobulin-type lectin) and Cx3cr1 as selectively present in microglia. The selectivity of Cx3cr1 was called into question as its expression has been observed on monocytes, macrophages, dendritic cells, T cells, and natural killer (NK) cells24. However, Cx3cr1 is clearly enriched in murine and human microglia25,26. In a whole mouse brain transcriptome study, 29 genes specifically distinguished microglia relative to both CNS cell types and other myeloid populations, with the most specific microglial genes being Olfml3 (olfactomedin-like protein 3), Tmem119 (transmembrane protein 119) and Siglech27. Seven genes, Tmem119, Fcrls (Fc receptor-like molecule), P2ry12 (chemoreceptor for adenosine diphosphate), P2ry13, Gpr34, Gpr84 and Il1a were identified as highly expressed and enriched in microglia, with Tmem119 found to be microglia-specific in both mouse and human28. Interestingly, a study identified specific microglial genes associated with surface molecules “surfaceome” which included ion transporters, molecules involved in lipid metabolism, a potential efflux marker, the co-stimulatory molecule CD40 and non-fully characterised surface markers29, whereas another group highlighted genes that constitute the microglial sensing apparatus known as the “sensome”30. A meta-analysis of 5 datasets revealed 143 genes enriched in microglia relative to macrophages, including P2ry12, Tmem119, Slc2a5 and Fcrls25.
Table 2.
Examples of transcriptomic studies to delineate homeostatic microglial genes in healthy mouse and human brain.
| Phenotype | Murine genes | Human genes | Methodology and tools |
|---|---|---|---|
| Homeostatic signature of microglia | Tgfbr1, Smad3, C1qa, C1qb, Cst3, Csf1r, Ctsd, Ctss, Cx3cr1, Entpd1, Fcrls, Hexb, Olfml3, P2ry12, Tmem119, Tmsb4x, Sparc, Lgmm, Tppp, Bin1, Rgs10, Gpr34, Sall1 |
P2RY12, P2RY13, C1QA, PROS1, GAS6, GPR34, MERTK, CX3CR1, TMEM119, CSFR1, SELPLG, MARCKS, AIF1, APBB1IP, ABI3, FCER1G |
Isolated cell populations from 2 month old C57BL/6 mice, CX3CR mice and Mr1-deficient mice22; miRNA array analysis and quantitative proteomic analysis of human microglia isolated from patients undergoing surgery22; scRNAseq of isolated microglia from 15 adults that underwent surgery for epilepsy or tumors26; Comparison of 9 published microglial signatures in human79 |
| microglia vs. myeloid populations |
Olfml2b, Cd34, Serpine2, Eya4, Lrrc3, Rtn4rl1, Slc46a1, Ccl12, Ccl4, Socs3, Sparc, Serpinf1, Rapgef5, Rtn1, Sema4d, Hexb, Arhgap22, Spata13, Hn1l, Hn1l, ll7r, Csmd3, Gpr84, Upk1b, St3gal6, H2-Oa, Trem2, Tmem204, Cables1, Cxxc5, Smad7, Ecscr, Ldhb, Ak1, Slc24a3, Slco4a1, Adora3, Fcrls, Fam46c, Olfml3, Slc2a5, Zfp691, Crybb1, Fscn1, Tmem119, Gal3st4, Siglech, Tmc7, Gpr56, Gas6, Sall1, Cx3cr1, Kcnd1, Tspan7, Gpr165 Transcription Factors: Bach2, Erf, Etv5, Junb, Jund, Klf12, Lmo2, Mycl1, Smad7, Sox4, Zfpm1, Zfp691 |
C1QA, C1QB, C1QC, GPR34 CCR2 absent in microglia |
Comparison of gene expression profiles - 6 week old C57BL/6J mice (Immunological Genome Project)23; RNAseq on ex vivo isolated microglia from 39 donors and 10 epilepsy surgery biopsies16; Profiling of isolated microglia from surgical- and autopsy-derived cortical brain samples47 |
| microglia vs. other CNS cells vs. macrophages | Olfml3, Tmem119, Siglech, Slc2a5, Gal3st4, Csmd3, Slco2b1, Gpr84, Lag3, F11r, Adora3, Ccl4, Golm1, P2ry13, Ccl3, Lrrc3, Egr1, Capn3, Tagap, Bco2, IL21r, Cx3cr1, Ccrl2, Grap, Fosb, Gtf2h2, Ptgs1 | CX3CR1, ITGAM, P2RY12, TYROBP | Whole brain transcriptome - C57BL/6 mice27; RNAseq on ex vivo isolated microglia from 39 donors and 10 epilepsy surgery biopsies16 |
| Microglia vs. macrophages | 95 genes in microglia but not macrophages 65 homeostatic genes, including Crybb1, Garnl3, Gpr34, Lag3, Nuak1, Olfml3, Rtn1, Rtn4rl1, Sall1, Sall3, Siglech, Slc1a3, Sparc, Tnfrsf17 |
1063 up- and 832 down-regulated (↑) B2M, MAPK6, ECM1, CSF1, SLC26A11 (↓) GARNL3, TMEM119, TLR10, TLR3, OLFML3, C3, CLEC17A, IGFSF10, BIN1, CX3CR1, P2RY12 |
C57BL/6 mice, 2 months old Cd11b+/Cd45low microglia5; RNAseq on human microglia from parietal cortex tissue and temporal lobe epilepsy surgery biopsies16 |
| microglial surfaceome | Slco4a1 , Slc30a5, Mcoln3, Lrp8, Lpact3, Stab1, Pap2c, Mfsd10, Cd40, Ketcap2, Tmem55b, Tmem48, Cmtm4 | No information available | Transcriptome of FACS-sorted cells – CX3CR1-GFP transgenic mice29 |
| microglial sensome | P2ry12, P2ry13, P2ry6, Gpr34, Adora3, Entpd1, Tmem173, P2yG, Csfr1, Csfr3, Tgfbr1, Tgfbr2, Ifngr1, Il10ra, Il6ra, Il21r, Tnfrsf17, Tnfrsf1b, Cx3cr1, Ccr5, C3ar1, Ptafr, Gpr77, Cmklr1, Cysltr1, Ccrl2, Cmtm6, C5ar1, Fcgr3, Fcer1g, Fcgr2b, Fcgr1, Cmtm7, Fcrl1, Fcgr4, Selplg, Ly86, Cd68, Trem2, Cd180, Tlr2, Cd37, Tlr7, Cd14, Clec4a3, Tlr4, Tlr13, Clec5a, havcr2, Clec7a, Cxcl16, Cd48, Ltf, Cd74, Uk1b, Tlr12, Tlr1, Pilra, Tlr6, Lfitm6, Itgam, Itgb2, Emr1, Ecscr, Lair1, Siglech, Slco2b1, Slc2a5, Lgals9, Gpr183, Tmem37, Cd33, Gpr84, Slc7a7, Cd52, Siglec5, Cd79b, Slc16a3, Icam1, Icam4, Cd94, Lag-3, Cd86, Ptprc, Dap12, Tnfrs13b, Tnfrsf17, Cd22, Tmem119, Cd53, Gi54, Slamf9, Clec4b1, Lilra5, Tmem8c, Gpr160, Cd101 | No information available | RNAseq - 5 month old C57BL/6 mice30 |
In bold, common microglial genes identified from transcriptomic studies.
To note: The discrimination between microglia, macrophages and infiltrating monocytes in human remains challenging partly due to the lack of reliable markers to distinguish these populations during the FACS isolation procedure.
Abbreviations: FACS, fluorescent cell sorting; sc, single cell.
Despite differences in methodology and source of microglia between these experimental studies, 17 genes were repetitively identified associated with microglia, with the transcripts classified into purinergic receptors (P2ry12, P2ry13, Adora3, Gpr34, Entpd1); cytokines and chemokines or their receptors (Tgfbr1, Cx3cr1, Ccrl2); Fc receptors (Fcrls); endogenous ligands, receptors and transporters (Siglech, Gpr84, Slco4a1, Hexb); potential sensome proteins (Tmem119), developmental proteins (Olfml3, Sall1) and some with unknown function (Csmd3). In contrast, genes associated with non-microglial myeloid cells in brain include CD14, Fcgrl, Mertk, Ctsd and Fert231, while genes selective for peripheral macrophages include Emilin2, Gda, Hp and Sell25 or Fn1, Cxcl13 and Ednrb32. The markers typically used for immunohistochemical identification of microglia, including Aif1 (Iba1) and Cd68 (Cd68), are generally poor at discriminating microglia from myeloid cells23,25. Proteomic confirmation of microglial specific markers includes P2ry12, Tmem119, Fcrls and Slc2a525. These proteins are predicted to be expressed on the microglial cell surface, so future studies could select or sort microglia based on markers more specific than Cd11b or Cx3cr1 to identify novel patterns of co-regulated gene expression. Similarly, a number of putative microglial genes identified in mouse models were confirmed at the gene and/or protein level in humans such as TMEM119, P2RY12, CX3CR1, CCR2 and Fcγ receptors15,16,22,28,33–38. Although similarities in homeostatic gene constituents were noted above, different groups find different numbers of genes in the homeostatic panel.
Microglia in different brain regions vary in gene expression, especially in genes associated with bioenergetic pathways and immunoregulatory pathways. Microglia from cerebellum and to a lesser extent hippocampus appear to exist in a more “immune vigilant” state compared with microglia from cerebral cortex or striatum39. Similarly, sex differences have been reported in both mouse and human40–43. Consequently, the homeostatic signature may be modulated by many biological variables. Additional confirmation and harmonization of a panel of genes accepted by multiple investigators would help to define this population in more detail.
Homeostatic Microglial Subpopulations
Identification of a set of genes exclusively expressed in microglia has allowed unprecedented discrimination of cellular responses of peripheral myeloid cells and central microglia that was not possible a decade ago. However, emerging evidence suggests considerable diversity even within homeostatic microglia that remains to be elucidated and harmonized. T-distributed stochastic neighbor embedding (t-SNE) analyses allow clustering of cells based on gene expression similarities among individual cells of a population. Phenotypic diversity of microglia revealed by these recent analyses suggests the presence of several microglial populations within the same brain in physiological conditions in both rodents and humans. However, naming standards have not developed, so clusters are given random numbers or color discriminations that vary between groups, publications and analysis packages. Therefore, a consensus has not formed regarding the classification of microglial subtypes nor the gene sets that would unambiguously define these subpopulations. Clusters range from 444 to 1526 microglial clusters using different models and analyses. Nine microglial subtypes, based on their gene expression profiles, were identified from microglia sourced from 15 donors who underwent surgery for tumor or epilepsy26. Single-cell RNAseq and mass cytometry revealed differences across the transcriptional spectrum of microglia with a core set of homeostatic genes such as TMEM119, CX3CR1, CSFR1, P2RY12, P2RY13, SELPLG (P-selectin glycoprotein ligand-1) and MARCKS (myristoylated alanine-rich C-kinase substrate) expressed by all microglial populations. These different clusters reflected microglial populations with subtypes characterised by: major histocompatibility (MHC) class II and antiviral immunity genes HLA-DRA (human leucocyte antigen), CD74 and IFI44L (interferon induced protein 44 like); integrin receptor binding protein and metabolism genes, SPP1 (secreted phosphoprotein 1, also known as osteopontin), APOE (apolipoprotein E) and LPL (lipoprotein lipase); and chemokines and pro-inflammatory cytokines genes such as CCL2 (chemokine C-C motif ligand 2) and IL1B (interleukin 1β)26. Interestingly, using several antibodies against myeloid markers, distinction between grey vs white matter microglia was observed. Despite the presence of a common core signature for grey and white matter microglia comprising P2RY12, TMEM119, ADGRG1, P2RY13, SLC2A5 and GRP3417, grey matter microglia expressed higher levels of homeostatic proteins, while microglia isolated from the white matter tended to be more involved in antigen presentation (MHCII genes), inflammation and lipid metabolism (APOE)26. Another study combined antibody detection followed by mass spectrometry analysis on isolated microglia from post-mortem tissue. This methodology allows identification of microglial phenotypes based on protein rather than gene expression. This confirmed the phenotypic homeostatic signature of microglia as previously reported in mice and human transcriptomic studies with P2RY12 and TMEM119 markers45. The findings were also consistent with both proteins specific to microglia, distinguishing these cells from other myeloid cells. Of note, the authors identified four subsets of microglial phenotype distributed differently between brain areas. Subset 1, characterised by higher expression of activation markers CD11C, CCR5, CD45, FCGRI (CD64), CD68, CX3CR1, EMR1 (EGF-like module-containing mucin-like hormone receptor-like 1) and HLA-DR, was mainly present in the subventricular zone and thalamus. High levels of proliferation markers (cyclin A, cyclin B1 and KI67) were associated with subset 1, implying a more activated, potentially primed, microglial phenotype. Subsets 2 and 3 were mainly detected in frontal and temporal cortical regions, with remarkably, both phenotypes associated with expression of the mannose receptor CD206, a known perivascular macrophage marker44. Subset 4 was the most challenging to detect, being less abundant than the other subsets, but was more prominent in temporal than in frontal cortex45. Similarly, another group identified regional phenotypic signatures of microglia within the human brain, in line with mouse and human studies34,39. Single cell RNAseq performed on CD45+ FACS-sorted cells revealed the presence of 4 major microglial clusters in healthy human brain from resected tissue without evidence of pathology46, with some of their findings overlapping the gene expression the same group identified in adult mouse microglia. Clusters 1 and 2 were characterised by the expression of CST3 (cystatin C protein) and the purinergic receptor P2RY13, while cluster 4 displayed expression of chemokines (CCL4, CCL2), zinc transcription factors (EGR2, EGR3) and a marker of mature dendritic cells (CD83). Interestingly these studies support evidence for four microglial populations co-existing in the healthy brain, but their differential gene expression muddles our understanding of microglia. To add to the complexity, another group identified 14 clusters of microglia assumed to represent distinct states of the cells, emphasizing the complement components as important effectors of microglia, (e.g. C1QA, C1QB, C1QC and GPR34)47. Interestingly in this paper, cluster 1 was present in all brains and thus was considered as the homeostatic microglial cluster, while clusters detected only in the older individuals were associated with an interferon response, in accordance with the expression of more inflammatory genes with aging (see below). At the present time, it is not known whether these microglia subtypes represent true subpopulations or phenotypic diversity. Clustering analyses effectively discriminate populations that are highly divergent, but appreciating gradual transitions is more challenging.
Comparison of Mouse and Human Data
Regulation of microglial homeostasis remains incompletely understood, but knowledge starts to emerge, mainly from mouse studies. Overall, gene co-expression analysis confirmed that microglia transmit “resting” signals to neurons via Cx3Cr1, Trem2 and Tyrobp initiate phagocytosis, purinergic receptors P2RYx signal neuronal injury, and Csfr1 induces cell survival or proliferation48. This remains to be determined in humans at the protein level for some of the genes49, as methodology used to isolate microglia can impact expression of their transcriptome50. Although many homeostatic genes are conserved across species, as many as 50% of the genes may vary in mice vs humans, with human specific homeostatic genes including APOC1, MP2L1, SORL1, CD58, ERAP2, GNLY and S100A1251. Discrepancies could be due to the source of the human tissue (resected tissue from surgery vs. autopsy brain), methodology applied to isolate and analyse microglia (mixed populations composed of microglia, perivascular macrophages, meningeal macrophages, monocytes), brain region investigated (cortex, hippocampus, grey/white matter) and/or clinical information not always reported such as the age of the patients, post-mortem delay, cause of death, and the presence of comorbidities or treatment. All these different elements add to the challenges in getting a clear consensus of the microglia landscape in healthy conditions.
Nonetheless, a consensus is emerging to acknowledge regional microglial heterogeneity and phenotypes. It is recognised that microglia adapt to their environment8, potentially providing an explanation for the different microglial populations detected. However, the environmental culprit behind the regional phenotypes, and as a consequence, their significance remains unclear. Could they be explained by highly specialised functions performed by the neurons in selected brain regions? Could these populations reflect different functions of microglia within the same brain? Indeed, microglia express receptors for most of the neurotransmitters, and thus responses to a specific neurotransmitter might direct microglial function and transcriptomic expression, adding to their heterogeneity52. Do the grey vs white microglia originate from the same pool? It remains controversial whether myeloid cells that are recruited to brain in response to injury or disease can acquire expression of microglial selective genes5,22. Nevertheless, these disparities could have implications regarding brain vulnerability in the context of neurodegenerative diseases in brain areas prone to pathology development and/or neurodegeneration.
“AGED” MICROGLIA
Homeostatic Genes
Reduced expression of homeostatic genes during aging is reported by multiple, but not all, authors (Table 3). Note that this reduction is not accompanied by increased gene expression of macrophage selective genes39. Genes identified as belonging to the TGFβ signaling pathway were down-regulated in aged human microglia, highlighting perturbation of microglial homeostasis in response to aging36. Similarly, genes involved in early microglial development (RUNX1, IRF8, and PU1) were also identified as master regulators for an age-dependent microglia module, implying a role for them in microglial homeostasis during aging48.
Table 3.
Examples of gene expression changes in microglia with aging.
| Reference | Age comparison | O>Y # genes | O>Y gene examples | O>Y pathways | O<Y # genes | O<Y gene examples | O<Y pathways | No change with aging | Methodology and Tools |
|---|---|---|---|---|---|---|---|---|---|
| 53 | Mouse 2 vs 15 months | 29 | C4; C1qb; Cats; Gas5 | Complement; lysosome; iron homeostasis | 6 | Acecs2 | Acetyl-CoA biosynthesis | C57BL/6NIA; bulk HPC; microarray | |
| 30 | Mouse 5 vs 24 months | 1831 | Tnf; Cxcl10; Nampt; Birc3; Cxcl9; Spp1; Arg1 | Stat3; neuroregulin-1; 5/12 classic activation markers; 24/37 alternate activation markers | 1672 | Trem2; P2ry12; Dap12; Siglech; Nlrp3 | Oxidative phosphorylation | Itgam (Cd11b); Cd14; Cd68; Icam | C57BL/6NIA; Cd11b/Cd45+; FACS sort; RNAseq |
| 86 | Mouse 2.5 vs 15-18 months | 482 | Ccl3; Pik3cd; Lyz1; Wbscr22; Rdh12; P2ry12 | Cytoplasmic membrane-bounded vesicle; regulation of proliferation; regulation of lymphocyte activation | 169 | Ccr6; Slpi; Clec4d; Ifitm1; Plbd1 | Inflammatory response; glycosamino-glycan binding; cell motility | C57BL/6J; Cd11b/Cd45+; FACS sort; microarray | |
| 39 | Mouse 4, 12, 22 months | 200-500 (varied with brain region) | Immune amplification | 10-100 (varied with brain region) | Tmem119; P2ry12; P2ry13; Fcrls | Homeostatic genes; TGFβ receptor genes; Cell adhesion/migration/motility | Bioenergetic pathways | C57BL/6J; Cd11b+; MACS selection; microarray | |
| 40 | Human 20-99 year | 50-200 depending on region | CLU; S100A8; CD14; CASP1 | Complement; TLRs | CX3CL1 | CX3CR1 | 4 brain regions; Bulk RNA; microarray | ||
| 48 | Human 13-95 year | CX3CR1; CSF1R; P2RY12; P2RY13; TREM2; TYROBP; some TLRs | Cell surface receptors for microglia-neuron crosstalk; M2a genes | M1 marker genes | Frontal Cortex; bulk RNA imputed to microglia; microarray | ||||
| 16 | Human 34-102 year | 212 | cell adhesion; axonal guidance; cell surface receptor expression | 360 | P2RY12; IL6R; TLR10 | Actin dynamics | Parietal cortex; percoll gradient separation; CD11B+/CD45int FACS sort; RNAseq | ||
| 36 | Human 50 (archived data set) vs 95 year | 1060 | C1QA; CD14; GRN; IRF7; TSPO | Amyloid fiber formation | 1174 | CD83; IL1B; NFKB1; TLR4 | TGFβ signaling | Dorsolateral prefrontal cortex (BA 9/46); CD11B+ MACS selection; RNAseq | |
| 26 | Human <30, 30-50, >50 (year) | SPP1 | Percoll gradient separation; CD45+ FACs cell sorting; single cell RNAseq |
Abbreviations: BA, Brodmann area; FACS, fluorescent cell sorting; HPC, hippocampus; MACS, positive cell selection using magnetic beads; O, old; Y, young
Genes Associated with Primed Microglia
Cognitive performance of aged relative to younger mice is impaired in association with pro-inflammatory transcriptomic and microglial changes, and studies on isolated microglia indicate an exacerbated pro-inflammatory state53,54. Experimental evidence suggests that microglia undergo priming during aging, defined as an exacerbated microglial response induced by an acute inflammatory stimulus on microglia already in an activated status caused by repetitive inflammatory stimuli55,56. Importantly, the priming stimulus is critical; microglia in aged mice responded to the cytokines Tnfα + Il1β+ Il12 with larger gene inductions than did young mice, but responses to the anti-inflammatory cytokines Il4+ Il13 were lost with aging57. In a study of microglial depletion and repopulation with new and unprimed microglia in aged mice (16-18 months old), expression of 127 genes normally modified with age were reversed following microglial repopulation, with no difference from the adult control mice58. These included the age-associated increased genes A2m, Apoe, Bmp6, Olr1, Sorl1, and Tgfb2i, or decreased genes Cdkn1a, Dennd2c, and Socs3. Interestingly, the age-associated inflammatory profile of microglia (C3, Clec7a, Ifi44l, Il1b, Il1rn, Mrc1, Tlr8) was not affected by the microglial depletion and repopulation. Gene expression changes in this category were closer to the adult control mice but not fully restored. Indeed, the response by the new microglia to inflammatory challenge was still higher than compared to adult mice (6-8 weeks old), associated with a primed profile. This study emphasized that the microenvironment influences microglial profile58. Successive immune stimuli may result in immune memory with microglia reprogramming, which predisposes the cells to either an exaggerated (primed) or absence (tolerant) response to inflammatory stimuli59,60.
Several reports highlight age-related increases in genes associated with innate immune activation in microglia in both mouse and human (Table 3). Analysis of gene expression profiles of immune- and inflammation-related genes conducted across a range of ages in normal and AD human brain found that the major changes in gene expression occurred during the course of cognitively normal aging (64 to 84% of the immune genes, depending on the region) rather than in AD (6% of the genes altered in AD relative to age-matched controls)40. Changes were associated with up-regulation of genes reflecting microglial activation including: (i) the complement components C1QA, C1QB, C1QC, C1S, C3, C3a receptor 1 (C3AR1), C4α, C4β, C5, C5a receptor 1 (C5AR1); (ii) factors modulating complement activation [factor H (CFH), CFH-related 1 (CFHR1) and CLU (clusterin; a risk factor for AD)]; (iii) Toll-like receptors TLR1, TLR2, TLR4, TLR5, TLR7, TLR8 and MYD88 with some regional variation; (iv) inflammasome-related genes such as CASP1 (caspase-1), IL1B, IL18, but not NRLP3 or PYCARD (ASC protein); (v) Fcγ receptors CD64, CD32, CD16 and FCER1A (Fc fragment of IgE receptor for alpha polypeptide); and (vi) up-regulation of the classical MHC Class I and II genes, but also of the non-classical MHC Class I, interpreted as an inhibitory feedback to down-regulate microglial activation40. A study from a different group observed that aged microglia (subjects >50 years old) expressed increases in a number of inflammatory genes with low CX3CR1 and high expression of integrin receptor-binding protein and metabolism genes such as SPP1, APOE and LPL26. Reductions in genes associated with anti-inflammatory M2 microglial profiles (IGF1, PDGFB, PDGFC, TGFB1, CCL13, CCL14, CCL17, CCL22, CCL23, CCL24, CCL26, FN1, IL1RN, RETNLB) were also observed, consistent with a switch towards a more pro-inflammatory profile of microglia during aging48. Of note, these changes were observed mainly in the early adult lifespan (<50 years old). Chemokine ligands and receptors presented disparate results, with CCR1, CXCL5 and CXCL16 genes up-regulated, while CXCL12 and CXCL14 gene expression decreased with aging. CD163 gene, the haptoglobin-hemoglobin receptor, was up-regulated in aging. Interestingly, while CD163 protein is specifically expressed by macrophages normally, microglia presented CD163 in the presence of hemoglobin in the parenchyma as observed after blood-brain barrier breakdown61. Therefore, CD163 expression could reflect blood-brain barrier dysfunction with aging and/or the impact of systemic immunity on the brain. This supports evidence from animal studies that age per se predisposes to inflammation, a concept that has been coined “inflammaging”62, with up-regulation of the innate immune system, including genes coding for inflammasome signaling, Fc-gamma receptors and HLA. Therefore, this concept resonates in the context of microglia, as demonstrated by altered mRNA expression of inflammation-related genes in middle-aged human and mouse brain63, and could be considered a phenomenon associated with “normal” aging.
Genes Associated with Pathogen Recognition, Motility and Phagocytosis
Changes in sensome transcripts with aging in mice included down-regulation for endogenous ligand recognition (most notably changes in P2ry12, P2ry13, Adora3, Trem2, Siglech, Dap12, Ccr5, and Ifngr1) and up-regulation for the ligands involved in microbe recognition and host defense (Tlr2, Cd74, Ltf, Clec7a, Cacl16, and Ifitm family), with an overall shift towards an alternative neuroprotective priming state30. The sensing genes involved in phagocytosis (Cd11b, Cd14, Cd68 and Icam) as well as in sensing microbial ligands were not affected, suggesting that microglia properties in clearing endogenous debris/pathogens are not altered by aging. The gene expression profile of purified microglia from aged human post-mortem parietal cortex identified changes in cell adhesion molecules and cell surface receptors (ICAM3, ROBO2, SEMA3C, SEMA7A), genes involved in actin cytoskeleton dynamics (TLN1, PFN1, EVL, ARPC1A, ARPC1B, CORO1A, CAP1, CTNNA2) and sensome genes (P2RY12, IL6R, TLR10), implying diminished cell motility16, an essential physiological function of homeostatic microglia64. Genes with higher expression during aging encompassed the integrin modulators DOCK1 and DOCK5, the receptors CXCR4, CD163 and IGF2R, the growth factor VEGFA and the transcription factor RUNX3. RNA expression was then confirmed at the protein level from isolated human microglia16. Remarkably, these changes shared limited overlap with the microglial genes regulated during aging in mice. Only 14 increased (e.g. CXCR4, VGFA, TNFAIP2, GP2) and nine decreased (e.g. ETS1, SEMA7A, MRC2, PSTPIP1, EMP2) in both species. Lack of concordance between mice and humans could be explained by intrinsic differences between species, but also by differences in life duration and the presence of infectious events affecting microglia in humans leading to immune memory compared to the specific pathogen free environment of animal houses.
Other Pathways and Risk Factors for Alzheimer’s Disease
Analysis of three independent microarray gene expression data sets from human post-mortem frontal cortex tissue were used to generate gene co-expression modules. Within the microglial module, decrease was reported as an age-related effect for surface receptors associated with neuron crosstalk (e.g. CX3CR1, P2RY12, TREM2, TYROBP) and TLRs48. Although reduced CX3CR1 was also reported by other investigators26, Cribbs et al40 reported that CX3CR1 was not altered with aging, but its ligand fractalkine (CX3CL1) was down-regulated, suggesting a disrupted/malfunctioning communication of microglia with neurons, promoting microglia to respond excessively to environmental changes associated with aging. A recent study investigating the transcriptomic atlas of aging human microglia from the frontal cortex from 10 participants of two prospective studies of aging (> 50 years old, mean at death 95 years old) identified 1054 microglia enriched-genes revealing pathways associated with DNA damage, telomere maintenance, phagocytosis and TGFβ signaling as part of the aged human microglial signature36. The data were confirmed by a proteomic profile consisting of 640 proteins and consistent with the transcriptome. This study indicates that microglial aging manifests as both loss of function and gain of function changes given a unique aged-related microglia phenotype36. Notably, the identified profile of aged microglia was enriched in susceptibility genes for AD but interestingly, independent from the main risk factor APOE4.
Overall, the studies are consistent with microglial profile modified with aging towards an increase in baseline inflammation, in both mice and humans despite some contradictions in human studies. Gene changes consistent with reduced motility, phagocytosis and beneficial neuron cross talk are reported. In general, homeostatic genes appear reduced during aging. Discrepancies are potentially explained by differences in the methods utilized due to the availability of quality human tissue.
“ALZHEIMER’S” MICROGLIA
The DAM/MGnD/ARM Phenotype in Mice
Several investigators reported changes in gene expression in transgenic mouse models displaying Alzheimer-like amyloid pathology (Table 4); these cells were referred to as disease associated microglia (DAMs)65, microglia associated with neurodegenerative disease (MGnD)66 or activated response microglia (ARM)42. These activated cell populations increased in number with age, associated with reduced expression of homeostatic genes such as Cx3cr1, P2ry12, P2ry13, and Tmem119, along with increased expression of genes associated with endocytosis, lysosomal/phagocytic pathways and regulation of immune response such as Apoe, Clec7a, Spp1, and Itgax. Increased expression of Apoe specifically in microglia is a key feature of the transition. Also notable were changes in gene expression for multiple other genes affecting immune function identified by GWAS that are associated with risk of late onset AD, such as Siglech (possibly an ortholog of CD33 in humans), Trem2 and Bin1. This pattern of gene expression changes does not recapitulate microglial gene expression changes in response to lipopolysaccharide (LPS)46. Notably, Trem2, Tyrobp, Ctsd and Hif1a were increased in the DAMs but decreased after LPS. DAMs and MGnDs were localized to amyloid and neuritic plaques, respectively, in human AD specimens. Conversion from the homeostatic to the DAM phenotype was attributed to a two-step process, one of which was dependent upon Trem265. Although details of the proposed mechanisms differ, changes in gene expression in response to amyloid deposition depend strongly on Trem2 expression38,66. Transition from homeostatic microglia to the MGnD phenotype occurred after injection of apoptotic neurons into mouse brain through a Trem2-ApoE-mediated mechanism66 and ARMs cannot form in the absence of Apoe42. A recent review provides more details67.
Table 4.
Examples of expression changes in mouse models with Alzheimer-type pathology.
| References | Mouse Model | # Up genes | Examples of ups | Up Pathways | # Down genes | Examples of downs | Methodology and Tools |
|---|---|---|---|---|---|---|---|
| 87 | PS2APP; 7, 13 months | Bulk: 85 (66 imputed to microglia)/Isolated microglia: 215 | CCL3; Clec7a; Trem2; Apoe; Igf1 | Not discussed | Bulk: 0 Isolated microglia: 34 | Bulk: 0 Isolated microglia: Clec4a3; Irf4 | Cortex + HPC; percoll gradient separation; RNAseq |
| 65 | 5xFAD; 1, 3, 6, 8 months | 145 in DAM vs homeostatic microglia | Apoe; Axl; Csf1; Clec7a; Cst7; Gpnmb; Igf1; Itgax; Spp1; Trem2 | lysosomal/phagocytic pathways; endocytosis; regulation of immune response | Cx3cr1; P2ry12; Tmem119 | Percoll gradient separation; Cd45+ cells sorted; single cell RNAseq | |
| 66 | APP-PS1; 9, 24 months | 28 in MGnD vs homeostatic microglia | Apoe; Axl; Ccl2; Csf1; Clec7a; Itgax; Lilrb4; Spp1 | 68 | Cx3Cr1; Csf1r; Egr1; Gpr34; Olfml3; P2ry12; Sall1; Tgfb1; Tmem119 | Percoll gradient separation; Fcrls+ cells sorted; single cell RNAseq or microarray | |
| 76 | CK-p25 inducible neurodegeneration | 202/278 DAM genes | MHC-I; MHC-II; Irf7; Ifitm3 | Proliferation early; immune response later; interferon response | HPC; Cd11b+/Cd45+ cells sorted; single cell RNAseq; | ||
| 75 | PS2APPxCx3cr1-GFP, 14-15 months; hMAPT-P301L, 12 months; hMAPT-P301S, 6 months | Neurodegeneration module with 134 genes | Apoe; Axl; Clec7a; Csf1; Cst7; Igf1; Itgax; Spp1 | Plasma membrane; extracellular space | Homeostatic genes | Cortex or HPC; Cx3cr1+ cells sorted | |
| 71 | rTg4510 (hMAPT-P301L); 2, 4, 6, 8 months | 293-2101 depending on age | Apoe; C1qa, b, c; C3; Clec7a; Itgax; Trem2 | Innate immune activation; lysosome/phagosome | 75-1588 depending on age | Glutamate synapse | Forebrain; percoll gradient separation or Cd11b+ MACS selection; RNAseq |
| 90 | APP-NL-G-F (APP KI); 3, 6, 12, 18 months | 57 amyloid plaque induced genes (PIGs); A majority, but not all, PIGs are microglial genes | 18 in common with DAMs= Apoe; Hexa; Trem2; Tyrobp Others = Axl; C1qa; Clu; Csf1r; Cst3; Cx3cr1; Grn; H2-D1; H2-K1; Hexb; Olfml3 | classical complement cascade activation; endocytosis; lysosomal degradation; antigen processing and presentation; immune response; oxidation/reduction processes | Spatial transcriptomics; in situ sequencing | ||
| 42 | APP-NL-G-F; 3, 6, 12, 21 months | Identified ARM | Cst7; Clec7a; Gpnmb; Itgax; Spp1 | Cortex or HPC; Cd11b+ cells sorted; single cell RNAseq | |||
| 68 | APPswe/PS1-L166P; Tau22; 4, 10-11 months | 287 in APPswe/PS1-L166P; 47 in Tau-22 | Apoe; Clec7a; Cst7; Itgax; Tyrobp | Aβ-induced transcriptional response; immune response; cytokine production; inflammation | P2ry12; Tmem119; Nav2 | 77 in Tau22, primarily neuronal origin | HPC; Cd11b+/Cd45+; RNAseq |
| 38 | 5xFAD; Trem2−/−x5xFAD; 7,10, 15 months | Apoe; Cst7, Csf1; Lpl; Trem2; MHC-1, MHC-II, cathepsins | P2ry12; Selplg; Tmem119; Cx3cr1 | HPC, Cortex; snRNAseq |
Abbreviations: APP, amyloid precursor protein; ARM, activated response microglia; DAM, disease-associated microglia; FACS, fluorescent cell sorting; HPC, hippocampus; KI, knock in; MACS, positive cell selection using magnetic beads; MGnD, microglial neurodegenerative phenotype; PS, presenilin
Studying amyloid (APPswe/PS L166P) and tau (Tau22) transgenic models driven by the same Thy-1 promoter, Sierksma et al (2020)68 argued that the transition from the homeostatic to the DAM/MGnD/ARM signature depends on amyloid more than tau. Amyloid-depositing mice displayed increases in 80% of microglial specific genes, dysregulation in genes associated with GWAS identified risk factors and increased prevalence of DAM/MGnD/ARM. In contrast, tau mice displayed more limited gene expression changes, primarily decreased expression of neuron specific genes. Thus, it appears that microglia respond to amyloid with a consistent program of gene expression changes, at least in mouse models. This conclusion resonates with data that microglia express cell surface receptors allowing internalization of oligomeric and fibrillar Aβ69, inducing production of cytokines70. On the other hand, RNAseq on pooled isolated microglia from a more severe tauopathy model (rTg4510) revealed up-regulation of many microglial genes associated with immune activation and GWAS identified risk factors, including Apoe, Trem2, Clec7a, complement components and scavenger receptors71. Another technical challenge is the extraction of microglia clustered around Aβ deposits which may make them under-represented in the analyses.
However, conversion from the homeostatic to the activated phenotype does not appear to be a stochastic process, but a continual one. Transition or intermediate populations have been described. Subtypes of DAMs were suggested, with “proinflammatory” DAMs identified by increased expression of Cd44, Il1b, Nfkb, Stat1 and Tlr2 emerging earlier, and “anti-inflammatory” DAMs identified by increased expression of Apoe, Atf1, Cxcr4, Igf1 and Lxra/b more prominent at later disease stages72. The localization of DAMs/MGnDs/ARMs to amyloid deposits suggests that this cell population is important for Alzheimer’s etiology. An excellent review of DAMs73 underscores the linkage between gene expression changes in DAMs and multiple AD risk alleles to argue that the DAM phenotype reduces Alzheimer pathology. However, DAMs/MGnDs/ARMs have now been identified in normal aging and many neurodegenerative disease models, suggesting that this phenotype is not uniquely associated with AD. Because sporadic AD affects individuals late in life, there could be no evolutionary natural selection pressure to shape microglial reactivity to cope with Alzheimer-type neuropathology. On the other hand, synaptic pruning of exuberantly produced synapses and neuronal number during development did shape microglial functions. Consequently, it is not surprising that microglia can mount a general response to neurodegeneration, that is not exquisitely tuned to respond directly to amyloid or tau pathology74.
Other subpopulations of activated microglia are also starting to be delineated. A meta-analysis of microglia/myeloid cell profiles from different mouse models of diseases (ischemic, infectious, inflammatory, tumor, demyelination and neurodegeneration) revealed 45 modules of co-regulated genes, which could be clustered into 7 prominent groupings related to [1] microglial specific (homeostatic) genes, [2] proliferation (primarily in response to tumor and virus) [3] core neurodegeneration, [4] interferon response, [5] endotoxin response, [6] macrophage and [7] neutrophil/monocyte75. These authors confirmed the presence of DAMs in 5xFAD brains, demonstrating that DAMS increase expression of core neurodegeneration module genes and decrease expression of homeostatic genes. Additional clusters of microglia were detected, mainly associated with an interferon-related module, a proliferation module and a module consisting of the immediate early genes Fos and Egr175. The proliferation and interferon modules were also described by others42,68,76. Several investigators argue that these microglial subpopulations exist in all individuals, but the relative sizes of the populations change as amyloid and tau pathology increase. Consequently, they have chosen the ARM nomenclature to indicate an activation state that is not necessarily disease specific. Others argue that the DAMs are restricted to pathological conditions65,75. The newer studies have examined more cells/reads so it is possible that earlier studies missed rare populations. However, this important point needs further attention. It is also not known whether conversion to the DAM/MGnD/ARM phenotype is stable or if cells can shift from one cluster to another. Based on trajectory analyses, Sala Frigerio et al (2019)42 argue that homeostatic microglia transition to either the ARM or the interferon-related cluster (IRM), but one activation state does not transition to the other.
Replication of these specific activation profiles in humans remains to be ascertained. Friedman et al (2018)75 argue that the core neurodegeneration, LPS and neutrophil/monocyte modules are increased in AD, but their data are based on bulk RNA measurements with imputed microglial expression. The rapid proliferation of single nucleus RNAseq from human specimens will be needed to answer this question definitively.
Human Studies
Transcriptomic analysis of nuclear RNA indicated that all major cell types are affected at the transcriptional level by AD pathology38,76. Comparison of early (pathology with no cognition problem) vs. no pathology subgroups revealed that large-scale transcriptional changes occur before individuals develop severe pathological features and in a similar pattern to those observed between the no-AD vs. AD pathology groups. A cell population (Mic1) was found with increased expression of AD risk genes (APOE, TREM2, MEF2C, PICALM, HLA-DRBI and HLA-DRB5), many of them expressed in microglia, and associated with AD pathology76. The Mic1 microglial subpopulation was also distinct from a population identified in aged microglia and thus appeared to be AD-specific76.
Concordance between genes differentially expressed in human AD and the mouse DAM/MGnD/ARM signature is poor. Only 28 of the 229 genes of the DAM profile were identified in humans, including APOE, SPP1 and TYROBP, while 49 AD-associated genes were specific to humans including complement components, HLA components and MS4A6A (membrane spanning 4-domains A6A)76. The dichotomy between the mouse and human data was consistent with another study investigating autosomal-dominant and sporadic AD77. Similarly, although some DAM/MGnD/ARM profile genes were up-regulated in AD dorsolateral prefrontal cortex (APOE, CD68, MHCII, TREM2), others were not changed (TYROBP), not detected (CST7) or even decreased (SPP1)38. Using CD11B to select myeloid cells from post-mortem human brains, instead of exploring AD-associated transcriptome in all brain cells, a similar lack of overlap with the DAM profile was reported, with APOE being the only common gene significantly increased in humans, maybe reflecting the difference in the innate immune response between humans and mice78. Human specific gene changes included up-regulation of PLXNC1 (plexin C1), TGFB1, ADAM8 (disintegrin and metalloproteinase domain-containing protein 8) and APOE, and down-regulation of SERPINF1 (serpin family F member 1, also known as pigment epithelium-derived factor [PEDF])78 or up-regulation of A2M, CHI3L1, SORL1, and genes associated with iron homeostasis38. In addition, none of the homeostatic microglial genes (e.g. P2RY12, CX3CR1) were down-regulated78 or were even increased in AD (CX3CR1, IRF8, P2RY12, TMEM119)38. Instead, a human Alzheimer’s microglia (HAM) population was defined79, and included a mixture of age-associated gene expression changes reported as “enhancing aging” (CECR2, IGSF10, HIST2H2BA, MOV10L1, PDCD6IPP2, TLN2, SELENBP1, MEIS1, TNFRRSF21, ZNF662, ASTN1, SERPINF1, ZNF532, ANKRD26P3), consistent with previous studies15,16, and an age-independent AD specific disease-related phenotype (ADAMTS13, ULK3, ZNF843, GYPC, APOE, KCNJ5, SMAD7, LSR, SLC38A7, STEAP3, ZNF703, TM9SF1, CLDN15, ARSA, PTPRG, ZNF696, TTYH3, ATOH8)79.
An important question in the field of AD, based on animal data, is whether the changes in microglial profile detected in disease are specific to the disease pathogenesis or simply reflect the ongoing neurodegeneration, and thus potentially common to neurodegenerative diseases. A recent study evaluated protein co-expression modules in AD, frontotemporal dementia-TDP43, progressive supranuclear palsy, corticobasal degeneration, Parkinson’s disease, Parkinson’s disease dementia, amyotrophic lateral sclerosis, and multiple system atrophy80. Three modules were detected specific to AD (synaptic processes, immune response [astrocytes] and cell-cell interaction [microglia/endothelial cells]) and three other modules associated with diagnosis independently of the disease (electron transport chain [GABAergic neuron], MAPK signaling, protein localization and transport). Focusing on the early (pathology with no cognitive problem) vs. late AD cases, up-regulation of two glial modules was reported early in the disease: the immune response (astrocytes) and cell-cell interaction (microglia/endothelial cells), consistent with the transcriptomic analyses. These two modules, independently of the severity of the cases, were positively associated with pathology, negatively correlated with cognitive status, and up-regulated in all neurodegenerative conditions with dementia, but not in Parkinson’s disease, amyotrophic lateral sclerosis and multiple system atrophy where dementia is not a key feature80.
A recent meta-analysis of coexpression network analysis of 9 human published datasets highlighted the high variability within the human studies in terms of number of genes identified, with no common genes detected81! Using gene-coexpression based analysis, the authors identified a core human microglial signature of 249 genes centered around CX3CR1, AIF1, and CSFR1, and containing APBB1IP (amyloid beta precursor protein binding family B member 1 interacting protein), ABI3, FCER1G (high affinity IgE receptor), ARHGDIB (rho GDP dissociation inhibitor beta), TLR signaling (TLR1, TLR2), complement pathway (C3AR1, C1QA, C2), TYROBP signaling (TREM2, TYROBP), cytoskeletal organization (CAPG, WAS), and the homeostatic genes GPR34, P2RY12, P2RY13, and TMEM119. In the context of AD, another set of 165 microglial-associated genes was identified co-expressed with the core signature, related to cell activation, wound healing, angiogenesis, apoptosis, and immune defense response. Taking in consideration publications on microglial cell numbers and state activation (based on the vulnerability of the regions to aging and AD), 52 genes were reported differentially expressed in AD vs age-matched controls, when the younger cases were excluded (<60 years). These genes were related to cell activation [PYCARD (ASC), PIK3CG], wound healing (A2M, SERPING1), innate immune response [TLR5, ITGAM, PYCARD (ASC)], and pathways associated with phagocytosis, TLR cascade, cell activation linked with neuronal survival and TYROBP signaling pathway (SAMSN1, SIRPB2, CD37, IL10RA, PIK3CG, and BIN2). Only 11 of the 52 genes were microglia specific including LYZ (lysozyme), RPS6KA1, and SLA (Src like adaptor). The homeostatic genes were down-regulated, consistent with the animal models, whereas other genes were up-regulated81.
Overall, the transcriptomic studies highlight that the underlying pathophysiological pathway leading to AD appears to be different from the one associated with aging. A consensus has not emerged delineating a microglial activation response specific to Alzheimer’s disease.
CONCLUSIONS
Since the GWAS studies identifying microglia as a key component of disease onset and progression in AD82–86, technological advances have allowed recent single cell -omic analyses of microglia under homeostatic conditions and with aging and AD-associated pathology (Figure 1). Transcriptomic studies have identified a core signature of genes specifically associated with non-activated microglia in the healthy brain. Despite surprising heterogeneity, there are 7 genes that repeatedly appear in mouse microglia, including Cx3cr1, Gpr34, Gpr84, Olfml3, Sall1, Siglech and Tmem119. Proteomic analyses have confirmed only a small fraction of these markers (P2ry12, Tmem119, Fcrls and Slc2a5)25. Although some homeostatic genes are conserved across species (CX3CR1, TMEM119), overall concordance is not high and there appear to be human-specific homeostatic genes (APOC1, MP2L1, SORL1, CD58, ERAP2, GNLY and S100A12)51. Aging is associated with increased gene expression for genes associated with innate immune activation and reduced gene profiles believed to underlie homeostatic functions, motility, phagocytosis and neuronal “calming” signals. A gene expression pattern resulting from neurodegeneration has emerged in mouse models with Alzheimer-like pathology, but a consensus has not yet developed whether a specific gene set can identify Alzheimer’s disease associated microglia. Gene expression changes responsible for morphological phenotypes remain elusive. It is not known whether microglial phenotypes identified by transcriptomics can be accommodated within the M1 vs M2 concept. Despite the amount of data generated from mouse and human brains, these cells retain their mysteries.
Figure 1.
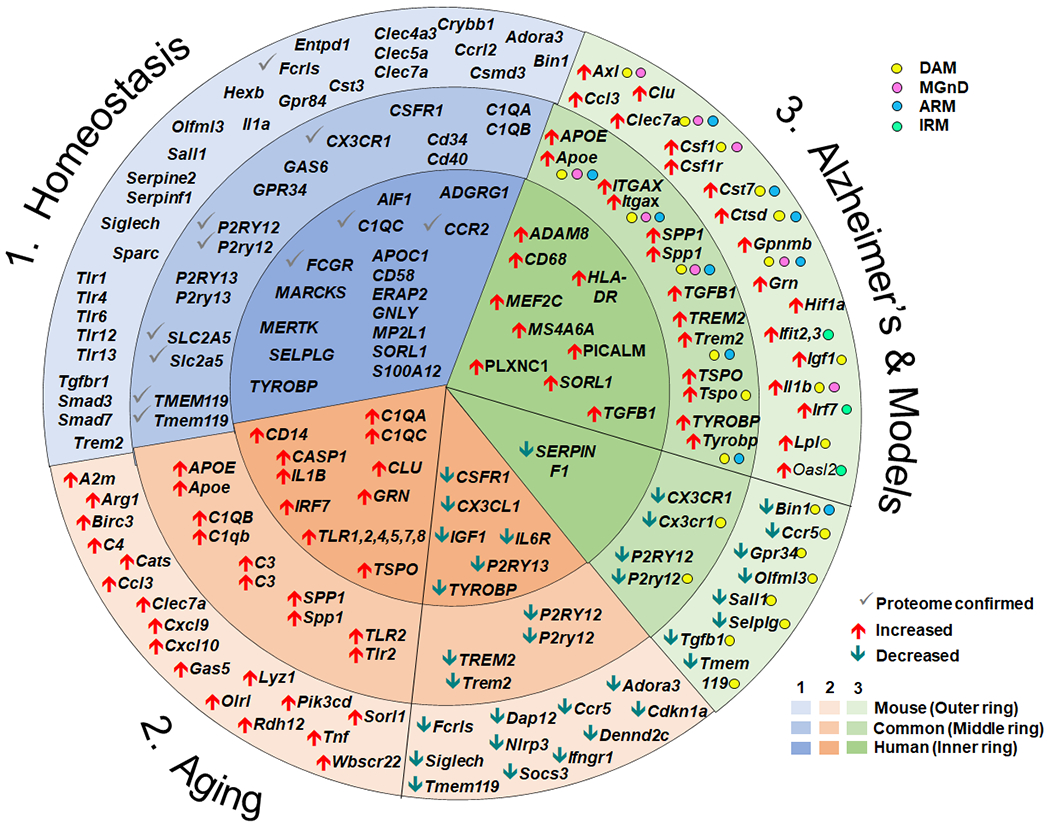
Summary of select gene expression changes in health, aging and Alzheimer’s disease. The circle represents gene expression. Data from mice are presented in the outer ring (light shading), while data from human are presented in the interior circle (darkest shading). Gene changes that are expressed by both human and mouse are presented in the middle ring with medium shading. Genes associated with homeostatic microglia are presented within the blue wedge (1) with the gene expression changes confirmed at the proteomic level indicated with a grey check mark. Changes in gene expression accompanying aging are presented in the beige wedge (2). Genes that are increased with aging relative to young adult are indicated with red up arrows, while genes that decrease are indicated with green down arrows. Changes in gene expression accompanying Alzheimer’s disease or Alzheimer-like pathology in mouse models are presented in the green wedge (3). In humans, increases and decreases compared with participants with no cognitive impairment are displayed in the green inner ring with red up or green down arrows, respectively. Within the wedge and ring for mouse models with Alzheimer-like pathology, the genes associated with the activated microglial subtypes DAM65, MGnD66, ARM42 or IRM42 are identified with color dots and the increase or decrease expression relative to the homeostatic profile indicated with red or green arrows. Not all gene changes can be depicted in the diagram.
Characterising the microglial phenotype in humans is challenging due to logistical (difficulty in accessing healthy brain tissue) and technical (requirement for fresh tissue) methodology. Transcriptomic and epigenetics have been performed on isolated human microglia sourced from either resected brain tissue for treatment of epilepsy, brain tumors, multiple sclerosis, or acute ischemia and from frozen samples15,16,36. Therefore, the physiological status of the cells isolated from a diseased tissue and after isolation could be questioned. Indeed, one of the limitations of the human studies exploring isolated microglia from alive patients (resected tissue) or post-mortem tissue has been the small number of subjects available to be assessed. Consequently, tissue with different pathologies must be combined, making difficult the identification of a clear disease-specific cluster. In addition, humans are exposed to more environmental pathogens than are laboratory mice. These lifelong exposures activating immune function could generate significant differences in microglial responses between mice and humans. While many measures of cortical architecture are similar across species, differences in cell composition, laminar distribution, and morphology contribute to species differences in single nucleus RNAseq87. Similarly, the neuropathological characteristics of humans are not fully replicated in mouse models. The majority of amyloid-depositing mice fail to show the significant neuronal death and brain atrophy observed in late stage AD, although they do display synaptic dysfunction and loss. Finally, mice are usually perfused to remove circulating myeloid cells prior to analyses, so it seems likely that human specimens will include more circulating blood cells among brain myeloid populations. Different cellular components would affect bulk RNA isolated from tissue more than single cell measurements.
Consequently, differences in the microglial profile between mouse and human, in the tools and resources used, and the modest concordance between gene expression and proteomic changes do not facilitate our understanding of the role of microglia. Many key questions remain to be answered. Are microglia losing their protective function with aging? How do microglia respond to early vs. late stage of the disease? Is microglial response to protein accumulation and/or neurodegeneration a common pathway between the neurodegenerative diseases or specific to the disease? Nevertheless, the identified genes in mice and humans support a broad role for microglia in homeostasis (synaptogenesis, chemotaxis, neurogenesis), host defense and response to injury, emphasizing that microglia have specialised functions not performed by other CNS cells and myeloid populations. In order to decipher the role of microglia in AD, additional transcriptomic research is needed.
RECOMMENDATIONS FOR FUTURE RESEARCH.
Consensus must be achieved regarding the number of microglial subtypes. Harmonization of the gene sets characterizing these subtypes is necessary. Only then may definitive studies of sources of biological variability such as sex or brain region be integrated.
Longitudinal human studies are needed to identify the dynamic complexity of microglial phenotypes over the Alzheimer’s disease trajectory.
Gene expression changes observed using bulk tissues must be confirmed at the cellular level.
Transcriptomic changes may not change microglial function if they do not drive changes in proteins. It will be essential to verify that detected transcriptional changes reach the protein level.
Future studies should endeavour to use unbiased or alternative cell selection methods.
Experiments to associate gene expression changes with Alzheimer-type pathology and microglial morphology using digital spatial profiling, mass cytometry (CyTOF), multiplexed single-molecule fluorescent in situ hybridization or laser capture microdissection might allow identification of disease-specific microglial phenotypes.
RESEARCH IN CONTEXT.
Systematic Review.
This Perspective reviews microglial gene expression profiles in healthy brain, aging, Alzheimer’s disease and its mouse models. Published reports of microglial transcriptomics were identified through traditional searches of archived data sets (BioRxiv) and life sciences journal literature (PubMed and Web of Science). Special attention is given to fundamental differences between mouse and human biology.
Interpretation.
Microglia in the healthy brain express a homeostatic signature of canonical genes. However, this signature is modulated by many biological variables, making it difficult to develop a consensus set of defining genes. The microglial profile shifts with aging towards an increase in baseline inflammation and a reduction in homeostatic genes. It is not clear if changes in Alzheimer microglia result specifically from Alzheimer pathology or from more general stimuli such as neurodegeneration.
Future Directions.
Harmonization of microglial subtypes must be achieved. Longitudinal human studies are needed to elucidate the dynamic complexity of microglial phenotypes over the Alzheimer’s disease trajectory. Finally, it will be essential to verify that detected transcriptional changes reach the protein level to affect microglial function.
ACKNOWLEDGMENTS:
This manuscript was facilitated by the Alzheimer’s Association International Society to Advance Alzheimer’s Research and Treatment (ISTAART), through the Immunity and Neurodegeneration Professional Interest Area (PIA). The views and opinions expressed by authors in this publication represent those of the authors and do not necessarily reflect those of the PIA membership, ISTAART or the Alzheimer’s Association. The authors thank Michael Heneka, MD, PhD, University of Bonn Medical Center and the German Center for Neurodegenerative Disease (DZNE) for critical reading of the manuscript. MG is supported by AG062217 and the Spectrum Health-MSU Alliance Corporation. DB is supported by the Alzheimer’s Research UK (ARUK-PPG2018B-019, ARUK-PG2018A-012), and the Alzheimer’s Society (AS-PG-16b-012).
Footnotes
Declarations of interest: none
REFERENCES
- 1.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010; 330: 841–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, Wieghofer P, Heinrich A, Riemke P, Holscher C, Muller DN, Luckow B, Brocker T, Debowski K, Fritz G, Opdenakker G, Diefenbach A, Biber K, Heikenwalder M, Geissmann F, Rosenbauer F, Prinz M. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci 2013; 16: 273–80. [DOI] [PubMed] [Google Scholar]
- 3.McCarter JC. A silver carbonate staining method for oligodendrocytes and microglia for routine use. Am J Pathol. 1940. Mar;16(2):233–235. [PMC free article] [PubMed] [Google Scholar]
- 4.Mori S, Leblond CP. Identification of microglia in light and electron microscopy. J Comp Neurol 1969; 135: 57–80. [DOI] [PubMed] [Google Scholar]
- 5.Grassivaro F, Menon R, Acquaviva M, Ottoboni L, Ruffini F, Bergamaschi A, Muzio L, Farina C, Martino G. Convergence between microglia and peripheral macrophages phenotype during development and neuroinflammation. J Neurosci 2020; 40: 784–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu CH, Chien HF, Chang CY, Ling EA. Heterogeneity of antigen expression and lectin labeling on microglial cells in the olfactory bulb of adult rats. Neurosci Res 1997; 28: 67–75. [DOI] [PubMed] [Google Scholar]
- 7.Ginhoux F, Prinz M. Origin of microglia: Current concepts and past controversies. Cold Spring Harb Perspect Biol 2015; 7: a020537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boche D, Perry VH, Nicoll JAR. Review: activation patterns of microglia and their identification in the human brain. Neuropath Appl Neurobiol 2013; 39: 3–18. [DOI] [PubMed] [Google Scholar]
- 9.Ransohoff RM. A polarizing question: Do M1 and M2 microglia exist? Nature Neurosci 2016; 19: 987–91. [DOI] [PubMed] [Google Scholar]
- 10.Jurga AM, Paleczna M, Kuter KZ: Overview of general and discriminating markers of differential microglia phenotypes. Front Cell Neurosci 2020; 14: 198–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX3CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 2000; 20: 4106–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saederup N, Cardona AE, Croft K, Mizutani M, Cotleur AC, Tsou CL, Ransohoff RM, Charo IF. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS One 2010; 5: e13693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Korin B, Ben-Shaanan TL, Schiller M, Dubovik T, Azulay-Debby H, Boshnak NT, Koren T, Rolls A. High-dimensional, single-cell characterization of the brain’s immune compartment. Nat Neurosci 2017; 20: 1300–9. [DOI] [PubMed] [Google Scholar]
- 14.Gerrits E, Heng Y, Boddeke EWGM, Eggen BJL. Transcriptional profiling of microglia; current state of the art and future perspectives. Glia 2020; 68: 740–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, Jaeger BN, O’Connor C, Fitzpatrick C, Pasillas MP, Pena M, Adair A, Gonda DD, Levy ML, Ransohoff RM, Gage FH, Glass CK. An environment-dependent transcriptional network specifies human microglia identity. Science 2017; 356: 1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galatro TF, Holtman IR, Lerario AM, Vainchtein ID, Brouwer N, Sola PR, Veras MM, Pereira TF, Leite REP, Moller T, Wes PD, Sogayar MC, Laman JD, den Dunnen W, Pasqualucci CA, Oba-Shinjo SM, Boddeke E, Marie SKN, Eggen BJL. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat Neurosci 2017; 20: 1162–71. [DOI] [PubMed] [Google Scholar]
- 17.van der Poel M, Ulas T, Mizee MR, Hsiao CC, Miedema SSM, Adelia, Schuurman KG, Helder B, Tas SW, Schultze JL, Hamann J, Huitinga I. Transcriptional profiling of human microglia reveals grey-white matter heterogeneity and multiple sclerosis-associated changes. Nat Commun. 2019; 10: 1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bakken TE, Hodge RD, Miller JA, Yao Z, Nguyen TN, Aevermann B, Barkan E, Bertagnolli D, Casper T, Dee N, Garren E, Goldy J, Graybuck LT, Kroll M, Lasken RS, Lathia K, Parry S, Rimorin C, Scheuermann RH, Schork NJ, Shehata SI, Tieu M, Phillips JW, Bernard A, Smith KA, Zeng H, Lein ES, Tasic B. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS One 2018; 13: e0209648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lake BB, Codeluppi S, Yung YC, Gao D, Chun J, Kharchenko PV, Linnarsson S, Zhang K. A comparative strategy for single-nucleus and single-cell transcriptomes confirms accuracy in predicted cell-type expression from nuclear RNA. Sci Rep 2017; 7: 6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis, Barres BA, Wu JQ. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 2014; 34: 11929–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharma K, Schmitt S, Bergner CG, Tyanova S, Kannaiyan N, Manrique-Hoyos N, Kongi K, Cantuti L, Hanisch U, Philips M, Rossner MJ, Mann M, Simons M. Cell type- and brain region-resolved mouse brain proteome. Nat Neurosci 2015; 18: 1819–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP, Weiner HL. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 2014; 17: 131–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, Mazloom AR, Ma’ayan A, Chua WJ, Hansen TH, Turley SJ, Merad M, Randolph GJ, Best AJ, Knell J, Goldrath A, Brown B, Jojic V, Koller D, Cohen N, Brennan P, Brenner M, Regev A, Fletcher A, Elpek K, Bellemare-Pelletier A, Malhotra D, Turley S, Jianu R, Laidlaw D, Collins J, Narayan K, Sylvia K, Kang J, Gazit R, Garrison BS, Rossi DJ, Kim F, Rao TN, Wagers A, Shinton SA, Hardy RR, Monach P, Bezman NA, Sun JC, Kim CC, Lanier LL, Heng T, Kreslavsky T, Painter M, Ericson J, Davis S, Mathis D, Benoist C. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol 2012; 13: 1118–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee M, Lee Y, Song J, Lee J, Chang SY. Tissue-specific role of CX3CR1 expressing immune cells and their relationships with human disease. Immune Netw 2018; 18: e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haage V, Semtner M, Vidal RO, Hernandez DP, Pong WW, Chen Z, Hambardzumyan D, Magrini V, Ly A, Walker J, Mardis E, Mertins P, Sauer S, Kettenmann H, Gutmann DH. Comprehensive gene expression meta-analysis identifies signature genes that distinguish microglia from peripheral monocytes/macrophages in health and glioma. Acta Neuropathol Commun 2019; 7: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sankowski R, Bottcher C, Masuda T, Geirsdottir L, Sagar, Sindram E, Seredenina T, Muhs A, Scheiwe C, Shah MJ, Heiland DH, Schnell O, Grun D, Priller J, Prinz M. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat Neurosci 2019; 22: 2098–110. [DOI] [PubMed] [Google Scholar]
- 27.Chiu IM, Morimoto ET, Goodarzi H, Liao JT, O’Keeffe S, Phatnani HP, Muratet M, Carroll MC, Levy S, Tavazoie S, Myers RM, Maniatis T. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep 2013; 4: 385–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A, Tucker A, Weissman IL, Chang EF, Li G, Grant GA, Hayden Gephart MG, Barres BA. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A 2016; 113: E1738–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beutner C, Linnartz-Gerlach B, Schmidt SV, Beyer M, Mallmann MR, Staratschek-Jox A, Schultze JL, Neumann H. Unique transcriptome signature of mouse microglia. Glia 2013; 61: 1429–42. [DOI] [PubMed] [Google Scholar]
- 30.Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 2013; 16: 1896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crotti A, Ransohoff RM. Microglial physiology and pathophysiology: Insights from genome-wide transcriptional profiling. Immunity 2016; 44: 505–15. [DOI] [PubMed] [Google Scholar]
- 32.Davies CL, Miron VE. Distinct origins, gene expression and function of microglia and monocyte-derived macrophages in CNS myelin injury and regeneration. Clin Immunol 2018; 189: 57–62. [DOI] [PubMed] [Google Scholar]
- 33.Satoh J, Kino Y, Asahina N, Takitani M, Miyoshi J, Ishida T, Saito Y. TMEM119 marks a subset of microglia in the human brain. Neuropathol 2016; 36: 39–49. [DOI] [PubMed] [Google Scholar]
- 34.Minett T, Classey J, Matthews FE, Fahrenhold M, Taga M, Brayne C, Ince PG, Nicoll JA, Boche D. Microglial immunophenotype in dementia with Alzheimer’s pathology. J Neuroinflam 2016; 13: 135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rakic S, Hung YMA, Smith M, So D, Tayler HM, Varney W, Wild J, Harris S, Holmes C, Love S, Stewart W, Nicoll JAR, Boche D. Systemic infection modifies the neuroinflammatory response in late stage Alzheimer’s disease. Acta Neuropath Commun 2018; 6: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olah M, Patrick E, Villani AC, Xu J, White CC, Ryan KJ, Piehowski P, Kapasi A, Nejad P, Cimpean M, Connor S, Yung CJ, Frangieh M, McHenry A, Elyaman W, Petyuk V, Schneider JA, Bennett DA, De Jager PL, Bradshaw EM. A transcriptomic atlas of aged human microglia. Nature Comm 2018; 9: 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Franco-Bocanegra DK, George B, Lau LC, Holmes C, Nicoll JAR, Boche D. Microglial motility in Alzheimer’s disease and after Abeta42 immunotherapy: a human post-mortem study. Acta Neuropath Comm 2019; 7: 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, Poliani PL, Cominelli M, Grover S, Gilfillan S, Cella M, Ulland TK, Zaitsev K, Miyashita A, Ikeuchi T, Sainouchi M, Kakita A, Bennett DA, Schneider JA, Nichols MR, Beausoleil SA, Ulrich JD, Holtzman DM, Artyomov MN, Colonna M. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med 2020; 26: 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, Freeman TC, Summers KM, McColl BW. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci 2016; 19: 504–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cribbs DH, Berchtold NC, Perreau V, Coleman PD, Rogers J, Tenner AJ, Cotman CW. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. J Neuroinflamm 2012; 9: 179–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Villa A, Gelosa P, Castiglioni L, Cimino M, Rizzi N, Pepe G, Lolli F, Marcello E, Sironi L, Vegeto E, Maggi A. Sex-specific features of microglia from adult mice. Cell Rep 2018; 23: 3501–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sala Frigerio C, Wolfs L, Fattorelli N, Thrupp N, Voytyuk I, Schmidt I, Mancuso R, Chen W-T, Woodbury ME, Srivastava G, Möller T, Hudry E, Das S, Saido T, Karran E, Hyman B, Perry VH, Fiers M, De Strooper B. The major risk factors for Alzheimer’s Disease: Age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep 2019; 27: 1293–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kodama L, Guzman E, Etchegaray JI, Li Y, Sayed FA, Zhou L, Zhou Y, Zhan L, Le D, Udeochu JC, Clelland CD, Cheng Z, Yu G, Li Q, Kosik KS, Gan L. Microglial microRNAs mediate sex-specific responses to tau pathology. Nat Neurosci 2020; 23:167–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Galea I, Palin K, Newman TA, Van Rooijen N, Perry VH, Boche D. Mannose receptor expression specifically reveals perivascular macrophages in normal, injured, and diseased mouse brain. Glia 2005; 49: 375–84. [DOI] [PubMed] [Google Scholar]
- 45.Bottcher C, Schlickeiser S, Sneeboer MAM, Kunkel D, Knop A, Paza E, Fidzinski P, Kraus L, Snijders GJL, Kahn RS, Schulz AR, Mei HE, Hol EM, Siegmund B, Glauben R, Spruth EJ, de Witte LD, Priller J. Human microglia regional heterogeneity and phenotypes determined by multiplexed single-cell mass cytometry. Nat Neurosci 2019; 22: 78–90. [DOI] [PubMed] [Google Scholar]
- 46.Masuda T, Sankowski R, Staszewski O, Bottcher C, Amann L, Sagar, Scheiwe C, Nessler S, Kunz P, van Loo G, Coenen VA, Reinacher PC, Michel A, Sure U, Gold R, Grun D, Priller J, Stadelmann C, Prinz M. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019; 566: 388–92. [DOI] [PubMed] [Google Scholar]
- 47.Olah M, Menon V, Habib N, Taga M, Yung C, Cimpean M, Khairalla A, Dionne D, Hopp S, Frosch MP, Hyman BT, Beach TG, Sarkis R, Cosgrove GR, Helgager J, Golden JA, Pennell PB, Schneider JA, Bennett DA, Regev A, Elyaman W, Bradshaw EM, De Jager PL. A single cell-based atlas of human microglial states reveals associations with neurological disorders and histopathological features of the aging brain. bioRxiv 2018: 343780. [Google Scholar]
- 48.Wehrspaun CC, Haerty W, Ponting CP. Microglia recapitulate a hematopoietic master regulator network in the aging human frontal cortex. Neurobiol Aging 2015; 36: 2443 e9–e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fahrenhold M, Rakic S, Classey J, Brayne C, Ince PG, Nicoll JAR, Boche D. TREM2 expression in the human brain: a marker of monocyte recruitment? Brain Pathol 2018; 28: 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kang SS, Baker KE, Wang X, Kocher J-P, Fryer JD. Translational profiling of microglia reveals artifacts of cell sorting. bioRxiv 2017: 135566. [Google Scholar]
- 51.Dubbelaar ML, Kracht L, Eggen BJL, Boddeke EWGM. The kaleidoscope of microglial phenotypes. Front Immunol 2018; 9: 1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu H, Leak RK, Hu X. Neurotransmitter receptors on microglia. Stroke Vasc Neurol 2016; 1: 52–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Verbitsky M, Yonan AL, Malleret G, Kandel ER, Gilliam TC, Pavlidis P. Altered hippocampal transcript profile accompanies an age-related spatial memory deficit in mice. Learning & Memory (Cold Spring Harbor, NY) 2004; 11: 253–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zahn JM, Poosala S, Owen AB, Ingram DK, Lustig A, Carter A, Weeraratna AT, Taub DD, Gorospe M, Mazan-Mamczarz K, Lakatta EG, Boheler KR, Xu X, Mattson MP, Falco G, Ko MS, Schlessinger D, Firman J, Kummerfeld SK, Wood WH 3rd, Zonderman AB, Kim SK, Becker KG. AGEMAP: a gene expression database for aging in mice. PLoS genetics 2007; 3: e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee DC, Rizer J, Hunt JB, Selenica ML, Gordon MN, Morgan D. Review: experimental manipulations of microglia in mouse models of Alzheimer’s pathology: activation reduces amyloid but hastens tau pathology. Neuropathol Appl Neurobiol 2013; 39: 69–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol 2014; 10: 217–24. [DOI] [PubMed] [Google Scholar]
- 57.Lee CD, Ruiz CR, Lebson L, Selenica MLB, Rizer J, Rojiani R, Reid P, Kammath S, Nash K, Dickey CA, Gordon MN, Morgan D. Aging enhances classical activation but mitigates alternative activation in the CNS. Neurobiol Aging 2013; 34: 1610–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.O’Neil SM, Witcher KG, McKim DB, Godbout JP. Forced turnover of aged microglia induces an intermediate phenotype but does not rebalance CNS environmental cues driving priming to immune challenge. Acta Neuropathol Commun 2018; 6: 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Netea MG, Latz E, Mills KH, O’Neill LA. Innate immune memory: a paradigm shift in understanding host defense. Nat Immunol 2015; 16: 675–9. [DOI] [PubMed] [Google Scholar]
- 60.Neher JJ, Cunningham C. Priming microglia for innate immune memory in the brain. Trends Immunol 2019; 40: 358–74. [DOI] [PubMed] [Google Scholar]
- 61.Galea J, Cruickshank G, Teeling JL, Boche D, Garland P, Perry VH, Galea I. The intrathecal CD163-haptoglobin-hemoglobin scavenging system in subarachnoid hemorrhage. J Neurochem 2012; 121: 785–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013; 153: 1194–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lopez-Gonzalez I, Schluter A, Aso E, Garcia-Esparcia P, Ansoleaga B, LLorens F, Carmona M, Moreno J, Fuso A, Portero-Otin M, Pamplona R, Pujol A, Ferrer I. Neuroinflammatory signals in Alzheimer disease and APP/PS1 transgenic mice: Correlations with plaques, tangles, and oligomeric species. J Neuropathol Exp Neurol 2015; 74: 319–44. [DOI] [PubMed] [Google Scholar]
- 64.Franco-Bocanegra DK, McAuley C, Nicoll JAR, Boche D. Molecular mechanisms of microglial motility: Changes in ageing and Alzheimer’s disease. Cells 2019; 8: 639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, Itzkovitz S, Colonna M, Schwartz M, Amit I. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 2017; 169: 1276–90. [DOI] [PubMed] [Google Scholar]
- 66.Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, Greco DJ, Smith ST, Tweet G, Humulock Z, Zrzavy T, Conde-Sanroman P, Gacias M, Weng Z, Chen H, Tjon E, Mazaheri F, Hartmann K, Madi A, Ulrich JD, Glatzel M, Worthmann A, Heeren J, Budnik B, Lemere C, Ikezu T, Heppner FL, Litvak V, Holtzman DM, Lassmann H, Weiner HL, Ochando J, Haass C, Butovsky O. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 2017; 47: 566–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qin Q, Teng Z, Liu C, Li Q, Yin Y, Tang Y. TREM2, microglia, and Alzheimer’s disease. Mech Ageing Dev 2021; 195: 111438. [DOI] [PubMed] [Google Scholar]
- 68.Sierskma A, Lu A, Mancuso R, Fattorelli N, Thrupp N, Salta E, Zoco J, Blum D, Buée L, De Strooper B, Fiers M. Novel Alzheimer risk genes determine the microglia response to amyloid-β but not to TAU pathology. EMBO Mol Med 2020; 12: e10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar β-amyloid mediates microglial activation. J Neurosci 2003; 23: 2665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Petzold GC, Town T, Morgan D, Shinohara ML, Perry VH, Holmes C, Bazan NG, Brooks DJ, Hunot S, Joseph B, Deigendesch N, Garaschuk O, Boddeke E, Dinarello CA, Breitner JC, Cole GM, Golenbock DT, Kummer MP. Neuroinflammation in Alzheimer’s disease. Lancet Neurol 2015; 14: 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang H, Li Y, Ryder JW, Hole JT, Ebert PJ, Airey DC, Qian HR, Logsdon B, Fisher A, Ahmed Z, Murray TK, Cavallini A, Bose S, Eastwood BJ, Collier DA, Dage JL, Miller BB, Merchant KM, O’Neill MJ, Demattos RB. Genome-wide RNAseq study of the molecular mechanisms underlying microglia activation in response to pathological tau perturbation in the rTg4510 tau transgenic animal model. Mol Neurodegen 2018; 13: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rangaraju S, Dammer EB, Raza SA, Rathakrishnan P, Xiao H, Gao T, Duong DM, Pennington MW, Lah JJ, Seyfried NT, Levey AI. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol Neurodegen 2018; 13: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I. Disease-associated microglia: A universal immune sensor of neurodegeneration. Cell 2018; 173: 1073–81. [DOI] [PubMed] [Google Scholar]
- 74.Morgan D, Mielke M. Knowledge gaps in Alzheimer’s disease immune biomarker research. Alzheimer’s Dementia; 2021; in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, Cao Y, Lee SH, Haddick PCG, Ngu H, Modrusan Z, Larson JL, Kaminker JS, van der Brug MP, Hansen DV. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep 2018; 22: 832–47. [DOI] [PubMed] [Google Scholar]
- 76.Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, Martorell AJ, Ransohoff RM, Hafler BP, Bennett DA, Kellis M, Tsai LH. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019; 570: 332–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Del-Aguila JL, Li Z, Dube U, Mihindukulasuriya KA, Budde JP, Fernandez MV, Ibanez L, Bradley J, Wang F, Bergmann K, Davenport R, Morris JC, Holtzman DM, Perrin RJ, Benitez BA, Dougherty J, Cruchaga C, Harari O. A single-nuclei RNA sequencing study of Mendelian and sporadic AD in the human brain. Alzheimers Res Ther 2019; 11: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Franco-Bocanegra DK, Nicoll JAR, Boche D. Innate immunity in Alzheimer’s disease: the relevance of animal models? J Neural Transmission (Vienna, Austria : 1996) 2018; 125: 827–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Srinivasan K, Friedman BA, Etxeberria A, Huntley MA, van der Brug MP, Foreman O, Paw JS, Modrusan A, Beach TG, Serrano GE, Hansen DV. Alzheimer’s patient microglia exhibit enhanced aging and unique transcriptional activation. Cell Rep 2020; 31: 107843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Swarup V, Chang TS, Duong DM, Dammer EB, Lah JJ, Johnson ECB, Seyfried NT, Levey AI, Geschwind DH. Identification of conserved proteomic networks in neurodegenerative dementia Cell Rep 2020; 31:107807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Patir A, Shih B, McColl BW, Freeman TC. A core transcriptional signature of human microglia: Derivation and utility in describing region-dependent alterations associated with Alzheimer’s disease. Glia 2019; 67: 1240–53. [DOI] [PubMed] [Google Scholar]
- 82.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schurmann B, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Hull M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans PA, O’Donovan M, Owen MJ, Williams J. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 2009; 41: 1088–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fievet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A, Helisalmi S, Porcellini E, Hanon O, de Pancorbo MM, Lendon C, Dufouil C, Jaillard C, Leveillard T, Alvarez V, Bosco P, Mancuso M, Panza F, Nacmias B, Bossu P, Piccardi P, Annoni G, Seripa D, Galimberti D, Hannequin D, Licastro F, Soininen H, Ritchie K, Blanche H, Dartigues JF, Tzourio C, Gut I, Van Broeckhoven C, Alperovitch A, Lathrop M, Amouyel P. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet 2009; 41: 1094–9. [DOI] [PubMed] [Google Scholar]
- 84.Guerreiro R, Wojtas A, Bras J, Carrasquillo MM, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin SG, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate AM, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton AB, Hardy J, Group ftAGA. TREM2 Variants in Alzheimer’s Disease. N Engl J Med 2012; 368: 117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JL, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, Van Duijn CM, Thorsteinsdottir U, Kong A, Stefansson H. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 2012; 368: 107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, Kunkle BW, Boland A, Raybould R, Bis JC, Martin ER, Grenier-Boley B, Heilmann-Heimbach S, Chouraki V, Kuzma AB, Sleegers K, Vronskaya M, Ruiz A, Graham RR, Olaso R, Hoffmann P, Grove ML, Vardarajan BN, Hiltunen M, Nothen MM, White CC, Hamilton-Nelson KL, Epelbaum J, Maier W, Choi SH, Beecham GW, Dulary C, Herms S, Smith AV, Funk CC, Derbois C, Forstner AJ, Ahmad S, Li H, Bacq D, Harold D, Satizabal CL, Valladares O, Squassina A, Thomas R, Brody JA, Qu L, Sanchez-Juan P, Morgan T, Wolters FJ, Zhao Y, Garcia FS, Denning N, Fornage M, Malamon J, Naranjo MCD, Majounie E, Mosley TH, Dombroski B, Wallon D, Lupton MK, Dupuis J, Whitehead P, Fratiglioni L, Medway C, Jian X, Mukherjee S, Keller L, Brown K, Lin H, Cantwell LB, Panza F, McGuinness B, Moreno-Grau S, Burgess JD, Solfrizzi V, Proitsi P, Adams HH, Allen M, Seripa D, Pastor P, Cupples LA, Price ND, Hannequin D, Frank-Garcia A, Levy D, Chakrabarty P, Caffarra P, Giegling I, Beiser AS, Giedraitis V, Hampel H, Garcia ME, Wang X, Lannfelt L, Mecocci P, Eiriksdottir G, Crane PK, Pasquier F, Boccardi V, Henandez I, Barber RC, Scherer M, Tarraga L, Adams PM, Leber M, Chen Y, Albert MS, Riedel-Heller S, Emilsson V, Beekly D, Braae A, Schmidt R, Blacker D, Masullo C, Schmidt H, Doody RS, Spalletta G, Jr WTL, Fairchild TJ, Bossu P, Lopez OL, Frosch MP, Sacchinelli E, Ghetti B, Yang Q, Huebinger RM, Jessen F, Li S, Kamboh MI, Morris J, Sotolongo-Grau O, Katz MJ, Corcoran C, Dunstan M, Braddel A, Thomas C, Meggy A, Marshall R, Gerrish A, Chapman J, Aguilar M, Taylor S, Hill M, Fairen MD, Hodges A, Vellas B, Soininen H, Kloszewska I, Daniilidou M, Uphill J, Patel Y, Hughes JT, Lord J, Turton J, Hartmann AM, Cecchetti R, Fenoglio C, Serpente M, Arcaro M, Caltagirone C, Orfei MD, Ciaramella A, Pichler S, Mayhaus M, Gu W, Lleo A, Fortea J, Blesa R, Barber IS, Brookes K, Cupidi C, Maletta RG, Carrell D, Sorbi S, Moebus S, Urbano M, Pilotto A, Kornhuber J, Bosco P, Todd S, Craig D, Johnston J, Gill M, Lawlor B, Lynch A, Fox NC, Hardy J, Albin RL, Apostolova LG, Arnold SE, Asthana S, Atwood CS, Baldwin CT, Barnes LL, Barral S, Beach TG, Becker JT, Bigio EH, Bird TD, Boeve BF, Bowen JD, Boxer A, Burke JR, Burns JM, Buxbaum JD, Cairns NJ, Cao C, Carlson CS, Carlsson CM, Carney RM, Carrasquillo MM, Carroll SL, Diaz CC, Chui HC, Clark DG, Cribbs DH, Crocco EA, DeCarli C, Dick M, Duara R, Evans DA, Faber KM, Fallon KB, Fardo DW, Farlow MR, Ferris S, Foroud TM, Galasko DR, Gearing M, Geschwind DH, Gilbert JR, Graff-Radford NR, Green RC, Growdon JH, Hamilton RL, Harrell LE, Honig LS, Huentelman MJ, Hulette CM, Hyman BT, Jarvik GP, Abner E, Jin LW, Jun G, Karydas A, Kaye JA, Kim R, Kowall NW, Kramer JH, LaFerla FM, Lah JJ, Leverenz JB, Levey AI, Li G, Lieberman AP, Lunetta KL, Lyketsos CG, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Morris JC, Murrell JR, Myers AJ, O’Bryant S, Olichney JM, Pankratz VS, Parisi JE, Paulson HL, Perry W, Peskind E, Pierce A, Poon WW, Potter H, Quinn JF, Raj A, Raskind M, Reisberg B, Reitz C, Ringman JM, Roberson ED, Rogaeva E, Rosen HJ, Rosenberg RN, Sager MA, Saykin AJ, Schneider JA, Schneider LS, Seeley WW, Smith AG, Sonnen JA, Spina S, Stern RA, Swerdlow RH, Tanzi RE, Thornton-Wells TA, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Van Eldik LJ, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Wilhelmsen KC, Williamson J, Wingo TS, Woltjer RL, Wright CB, Yu CE, Yu L, Garzia F, Golamaully F, Septier G, Engelborghs S, Vandenberghe R, De Deyn PP, Fernadez CM, Benito YA, Thonberg H, Forsell C, Lilius L, Kinhult-Stahlbom A, Kilander L, Brundin R, Concari L, Helisalmi S, Koivisto AM, Haapasalo A, Dermecourt V, Fievet N, Hanon O, Dufouil C, Brice A, Ritchie K, Dubois B, Himali JJ, Keene CD, Tschanz J, Fitzpatrick AL, Kukull WA, Norton M, Aspelund T, Larson EB, Munger R, Rotter JI, Lipton RB, Bullido MJ, Hofman A, Montine TJ, Coto E, Boerwinkle E, Petersen RC, Alvarez V, Rivadeneira F, Reiman EM, Gallo M, O’Donnell CJ, Reisch JS, Bruni AC, Royall DR, Dichgans M, Sano M, Galimberti D, St George-Hyslop P, Scarpini E, Tsuang DW, Mancuso M, Bonuccelli U, Winslow AR, Daniele A, Wu CK, Peters O, Nacmias B, Riemenschneider M, Heun R, Brayne C, Rubinsztein DC, Bras J, Guerreiro R, Al-Chalabi A, Shaw CE, Collinge J, Mann D, Tsolaki M, Clarimon J, Sussams R, Lovestone S, O’Donovan MC, Owen MJ, Behrens TW, Mead S, Goate AM, Uitterlinden AG, Holmes C, Cruchaga C, Ingelsson M, Bennett DA, Powell J, Golde TE, Graff C, De Jager PL, Morgan K, Ertekin-Taner N, Combarros O, Psaty BM, Passmore P, Younkin SG, Berr C, Gudnason V, Rujescu D, Dickson DW, Dartigues JF, DeStefano AL, Ortega-Cubero S, Hakonarson H, Campion D, Boada M, Kauwe JK, Farrer LA, Van Broeckhoven C, Ikram MA, Jones L, Haines JL, Tzourio C, Launer LJ, Escott-Price V, Mayeux R, Deleuze JF, Amin N, Holmans PA, Pericak-Vance MA, Amouyel P, van Duijn CM, Ramirez A, Wang LS, Lambert JC, Seshadri S, Williams J, Schellenberg GD. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet 2017; 49: 1373–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, Close JL, Long B, Johansen N, Penn O, Yao Z, Eggermont J, Höllt T, Levi BP, Shehata SI, Aevermann B, Beller A, Bertagnolli D, Brouner K, Casper T, Cobbs C, Dalley R, Dee N, Ding SL, Ellenbogen RG, Fong O, Garren E, Goldy J, Gwinn RP, Hirschstein D, Keene CD, Keshk M, Ko AL, Lathia K, Mahfouz A, Maltzer Z, McGraw M, Nguyen TN, Nyhus J, Ojemann JG, Oldre A, Parry S, Reynolds S, Rimorin C, Shapovalova NV, Somasundaram S, Szafer A, Thomsen ER, Tieu M, Quon G, Scheuermann RH, Yuste R, Sunkin SM, Lelieveldt B, Feng D, Ng L, Bernard A, Hawrylycz M, Phillips JW, Tasic B, Zeng H, Jones AR, Koch C, Lein ES. Conserved cell types with divergent features in human versus mouse cortex. Nature 2019; 573: 61–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Orre M, Kamphuis W, Osborn LM, Jansen AHP, Kooijman L, Bossers K, Hol EM. Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol Aging 2014; 35: 2746–60. [DOI] [PubMed] [Google Scholar]
- 89.Srinivasan K, Friedman BA, Etxeberria A, Huntley MA, van der Brug MP, Foreman O, Paw JS, Modrusan Z, Beach TG, Serrano GE, Hansen DV. Alzheimer’s patient brain myeloid cells exhibit enhanced aging and unique transcriptional activation. bioRxiv 2019: 610345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen WT, Lu A, Craessaerts K, Pavie B, Sala Frigerio C, Corthout N, Qian X, Laláková J, Kühnemund M, Voytyuk I, Wolfs L, Mancuso R, Salta E, Balusu S, Snellinx A, Munck S, Jurek A, Fernandez Navarro J, Saido TC, Huitinga I, Lundeberg J, Fiers M, De Strooper B. Spatial transcriptomics and in situ sequencing to study Alzheimer’s disease. Cell 2020;182: 976–991. [DOI] [PubMed] [Google Scholar]