

SUMMARY
Understanding metabolic pathways that regulate Th17 development is important to broaden therapeutic options for Th17-mediated autoimmunity. Here, we report a pivotal role of mitochondrial oxidative phosphorylation (OXPHOS) for lineage specification toward pathogenic Th17 differentiation. Th17 cells rapidly increase mitochondrial respiration during development, and this is necessary for metabolic reprogramming following T cell activation. Surprisingly, specific inhibition of mitochondrial ATP synthase ablates Th17 pathogenicity in a mouse model of autoimmunity by preventing Th17 pathogenic signature gene expression. Notably, cells activated under OXPHOS-inhibited Th17 conditions preferentially express Foxp3, rather than Th17 genes, and become suppressive Treg cells. Mechanistically, OXPHOS promotes the Th17 pioneer transcription factor, BATF, and facilitates T cell receptor (TCR) and mTOR signaling. Correspondingly, overexpression of BATF rescues Th17 development when ATP synthase activity is restricted. Together, our data reveal a regulatory role of mitochondrial OXPHOS in dictating the fate decision between Th17 and Treg cells by supporting early molecular events necessary for Th17 commitment.
Graphical Abstract
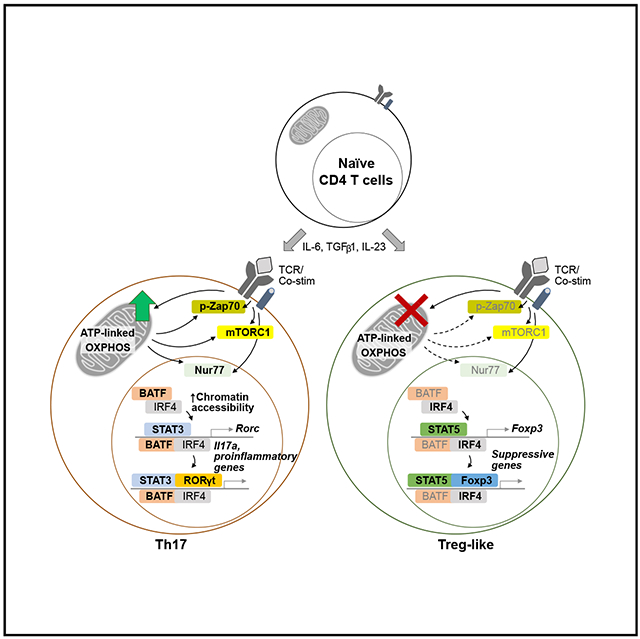
In Brief
Shin et al. report that ATP-linked mitochondrial respiration controls the Th17 and Treg cell fate decision by supporting TCR signaling and Th17-associated molecular events. Inhibition of mitochondrial OXPHOS ablates Th17 pathogenicity in a mouse model of MS and results in generation of functionally suppressive Treg cells under Th17 conditions.
INTRODUCTION
Th17 cells comprise a subset of effector CD4 T cells that provides protection against extracellular pathogens (Patel and Kuchroo, 2015). However, abnormal function of Th17 cells mediates chronic inflammatory disorders, including multiple sclerosis (MS), inflammatory bowel disease (IBD), and rheumatoid arthritis (Gaffen et al., 2014; Ghoreschi et al., 2011). In contrast, Foxp3-expressing regulatory T cells (Treg cells) suppress inflammatory responses and act in a protective role in autoimmune disorders. Consistent with this, an imbalance in the ratio of Th17 and Treg cells is observed in patients with autoimmune disease (Kleinewietfeld and Hafler, 2013). To devise new therapeutic options for autoimmune inflammation, it is critical to understand the mechanism by which Th17 cells obtain pathogenicity and how the Th17-Treg cell equilibrium is regulated.
Th17 development is initiated and specified with both T cell receptor (TCR) and cytokine signaling. A strong TCR signal drives activation of the pioneer transcription factors, basic leucine zipper transcription factor TF-like (BATF) and interferon-regulatory factor 4 (IRF4), which are required to increase chromatin accessibility for the Th17 transcription factors (Ciofani et al., 2012; Schraml et al., 2009). Then, interleukin-6 (IL-6) signaling triggers signal transducer and activator of transcription 3 (STAT3) activation, which together with transforming growth factor β1 (TGF-β1) signaling induces the Th17-lineage-specifying transcription factor, RAR-related orphan receptor γt (RORγt) (Durant et al., 2010; Yang et al., 2007; Yosef et al., 2013). Finally, RORγt binds to the promoter regions of Th17 signature cytokines, including Il17a and Il22 (Gaffen et al., 2014; Ivanov et al., 2006; O’Shea and Paul, 2010). Interestingly, both Th17 and Treg cell development share a similar signaling requirement mediated by TGF-β1, but differential STAT activation influences the bifurcation of Th17 (by STAT3) and Treg cells (by STAT5; O’Shea and Paul, 2010; Yang et al., 2011).
Recent studies demonstrate that distinct metabolic signatures are associated with specific immune cell fate and function (Buck et al., 2017; MacIver et al., 2013; Price et al., 2018). Following TCR activation, T cells rapidly reprogram metabolic processes, including glycolysis and mitochondrial oxidative phosphorylation (OXPHOS) (Chang et al., 2013). Particularly, mitochondria-driven reactive oxygen species (ROS) (Sena et al., 2013) and one carbon metabolism support TCR signaling and nucleic acid synthesis for subsequent transition of quiescent naive T cells into a proliferative state (Ron-Harel et al., 2016; Weinberg et al., 2015). Once activated, T cells show unique metabolic properties that are specific to each T cell subset. In particular, Th17 cells heavily rely on mechanistic target of rapamycin (mTOR) signaling, glycolysis, glutamine metabolism, and fatty acid (FA) synthesis for their differentiation and pro-inflammatory function (Araujo et al., 2017; Berod et al., 2014; Delgoffe et al., 2009, 2011; Gerriets et al., 2015; Shi et al., 2011; Xu et al., 2017). Therefore, deficiency in molecules involved in these pathways, such as Rheb, Rictor (mTOR; Delgoffe et al., 2009, 2011), Hif1a, Pdhk1 (glycolysis; Gerriets et al., 2015; Shi et al., 2011), Got1 (transamination; Xu et al., 2017), or Acc1 (FA synthesis; Berod et al., 2014), profoundly compromises Th17 development. Although immunoregulatory effects of various metabolic processes on Th17 development have been intensively investigated, the precise contribution of mitochondrial OXPHOS to Th17 differentiation is still not defined.
Here, we show that ATP-linked mitochondrial respiration during Th17 differentiation is essential for upregulating glycolysis and TCA cycle as well as Th17-mediated CNS inflammation. Consequently, inhibition of mitochondrial OXPHOS under Th17-skewing conditions impairs differentiation of Th17 cells and results in a functionally suppressive regulatory T cell. Further analysis reveals that ATP synthase supports Th17 lineage commitment in a BATF-dependent manner by regulating TCR signaling. Finally, we show TCR-dependent BATF induction is partially governed by mTORC1 activation, which requires ATP-linked mitochondrial OXPHOS. Together, our data suggest a novel mechanism by which mitochondrial OXPHOS fine-tunes pathogenic Th17 and Treg cell fate decision.
RESULTS
Th17 Cells Actively Utilize Mitochondrial OXPHOS to Fuel Optimal Cellular Metabolism
Mitochondrial OXPHOS generates ATP by utilizing the electrochemical gradient created by mitochondrial electron transport, and this pathway profoundly affects energy metabolism, calcium homeostasis, and cellular redox balance (Weinberg et al., 2015). Hence, we reasoned that this fundamental process is critical for the biology of Th17 cells. Previous studies report that fully differentiated Th17 cells showed increased mitochondrial respiration in comparison to naive CD4 T cells (Franchi et al., 2017; Gerriets et al., 2015). However, it is unknown whether mitochondrial OXPHOS is induced during the process of Th17 lineage commitment. We examined the kinetics of mitochondrial respiration during Th17 development by analyzing the oxygen consumption rate (OCR) of CD4 T cells at various time points of Th17 differentiation: 0 h; 24 h; 48 h; and 72 h after stimulation. At all time points, CD4 T cells activated under Th17 differentiation conditions showed higher basal and maximal capacity in cellular bioenergetics, including an increased commitment to ATP synthesis in comparison to unstimulated naive CD4 T cells (Figures 1A and 1B). Notably, the basal OCR level was increased at 24 h, reached the maximal level at 48 h, and then decreased at 72 h, suggesting that mitochondrial OXPHOS could be required for critical early events during Th17 differentiation (Figures 1A and 1B). Moreover, Th17 cells exhibited a concordant increase in the extracellular acidification rate (ECAR), consistent with an increase in glycolysis, which also peaked at 48 h post-stimulation (Figure S1A). Together, these data show that Th17 cells vigorously exploit mitochondrial respiration in conjunction with increased glycolytic flux when undergoing lineage specification.
Figure 1. Th17 Cells Increase Mitochondrial OXPHOS during Differentiation, which Promotes Metabolic Reprogramming.

(A and B) Naive CD4 T cells were activated under Th17 conditions in the presence of vehicle (A) oroligomycin (B), and the OCR was measured at the indicated time points (4–7 independent experiments).
(C and D) RNA sequencing was performed on naive CD4 T cells activated under Th17 conditions in the presence of vehicle or oligomycin for 48 h.
(C) Heatmaps illustrate the relative expression of genes in the different groups, oligo, oligomycin; veh, vehicle.
(D) Pathways enriched in vehicle-treated cells using the hallmark gene sets in Molecular Signature Database (MSigDB) are shown with statistical significance.
(E) Proliferation of naive CD4 T cells cultured under Th17 conditions in the presence of vehicle or oligomycin. Representative histogram overlay shows the proliferation of live CD4 T cells on day 5, and frequency of cells in each division is quantitated (3 independent experiments).
Graphs show the average ± SD; (A) two-way ANOVA; (B) unpaired t test *p< 0.05, **p< 0.01 (comparison between vehicle- and oligomycin-treated group at indicated time point), #p < 0.05 (comparison between naive and vehicle-treated group at indicated time point); (E) two-way ANOVA *p < 0.05, **p < 0.01, ***p < 0.001.
Considering that Th17 differentiation is accompanied by a robust increase of anabolic processes, we postulated that upregulation of mitochondrial OXPHOS is linked to the bioenergetic demands of Th17 development. The basal cellular OCR is a combination of ATP-linked (ATP-synthase-dependent) and non-ATP-linked (electron transport chain [ETC]-dependent) respiration. Specifically, at all time points analyzed, 70%–80% of the basal OCR in developing Th17 cells can be attributed to ATP-linked respiration (Figure 1A). Therefore, we hypothesized that ATP synthase activity is required for the anabolic switch during Th17 lineage commitment. To test this, naive CD4 T cells were activated under Th17 conditions in the presence of vehicle or the mitochondrial OXPHOS inhibitor, oligomycin. Oligomycin specifically inhibits the proton channel of ATP synthase in the ETC and results in inhibition of oxidative phosphorylation of ADP to ATP (Symersky et al., 2012). Although prolonged exposure to oligomycin can be toxic to the cells, a low concentration (2.5 ng/mL) of oligomycin allowed for greater than 90% viability during Th17 differentiation (Figure S1F). Importantly, treatment with this low concentration (2.5 ng/mL) of oligomycin was still sufficient to block the increase in the basal OCR and ATP-linked OCR observed at 24 h, 48 h, and 72 h post-activation, reflecting effective suppression of mitochondrial OXPHOS (Figures 1B and S1B-S1D). Intriguingly, oligomycin treatment not only impacted OXPHOS but also resulted in a significant decrease in ECAR at 24 h (Figure S1E), suggesting that the initial increase of glycolysis and mitochondrial flux during Th17 differentiation are closely linked.
To understand the contribution of ATP synthase and mitochondrial OXPHOS to other metabolic and cellular processes in developing Th17 cells, we performed RNA sequencing of CD4 T cells activated under Th17 conditions in the presence of oligomycin or vehicle for 48 h. Consistent with our extracellular metabolic flux data, inhibition of ATP synthase during Th17 differentiation resulted in reduced expression of genes associated with mitochondrial function and glycolysis (Hk1, Hk2, Eno1, and Aldoa, for example). Moreover, oligomycin treatment also caused significant decreases in the expression of hypoxia-induced factor (HIF) pathway (including Ldha and Hif1a) and TCA cycle genes (such as Idh1, Mdh1, Aco1, and Aco2) compared to vehicle-treated, control Th17 cells (Figure 1C).
In addition to the analysis of genes associated with various metabolic processes, we also performed pathway analysis of the RNA sequencing data to determine how else OXPHOS contributes to Th17 development. Among the top ten enriched pathways, cell-cycle-associated events were the most differentially regulated pathways by ATP synthase inhibition (Figure 1D). Consistent with this, treatment with oligomycin resulted in reduced proliferation of CD4 T cells during Th17 differentiation (Figure 1E). These overall alterations in central metabolic and cellular pathways with ATP synthase blockade demonstrate the critical role of mitochondrial OXPHOS in fueling metabolic requirements and proliferative capacity during Th17 differentiation.
Mitochondrial Respiration Is Required for the Induction of Th17 Pathogenicity
Th17 cell responses are associated with the induction of chronic inflammatory disorders, such as MS and IBD (Gaffen et al., 2014; Ghoreschi et al., 2011; Kleinewietfeld and Hafler, 2013; Patel and Kuchroo, 2015). Importantly, Th17 pathogenicity is supported by high rates of cellular metabolism; therefore, genetic or chemical perturbation of glycolysis, FA oxidation, or glutamine metabolism prevents Th17-mediated autoimmune diseases (Araujo et al., 2017; Berod et al., 2014; Gerriets et al., 2015; Shi et al., 2011; Xu et al., 2017). Our previous data showed that Th17 cells rapidly increased their oxygen consumption during differentiation (Figure 1A), and this elevated mitochondrial respiration was required for other metabolic processes (Figure 1C). Intriguingly, these data implicate a possible role of mitochondrial OXPHOS in regulating the pathogenic potential of Th17 cells. To investigate this, we utilized a mouse model of MS, experimental autoimmune encephalomyelitis (EAE), in which Th17 cells mediate disease progression (Gaublomme et al., 2015; Ghoreschi et al., 2010; Jain et al., 2016; Lee et al., 2012). Transgenic 2D2 CD4 T cells, which express a TCR specific for myelin oligodendrocyte glycoprotein (MOG), were differentiated with Th17-polarizing cytokines in the presence of oligomycin or vehicle and subsequently transferred into Rag1-deficient recipient mice that were monitored daily for disease severity. Strikingly, inhibition of ATP synthase during Th17 differentiation significantly delayed the onset of disease and reduced disease severity (Figure 2A). Although ATP synthase was required for proliferation of cells during Th17 differentiation, both vehicle and oligomycin-treated cells showed a robust proliferation upon transfer and exhibited comparable number and frequency of Ki-67+ cells (Figures S1G and S1H). Thus, these data demonstrate that mitochondrial respiration is essential for Th17 cells to cause neuropathology.
Figure 2. Mitochondrial OXPHOS Controls the Fate Decision between Pathogenic Th17 and Treg Cell Development.

(A) 2D2 CD4 T cells were differentiated under Th17-polarizing conditions in the presence of vehicle or oligomycin for 5 days and transferred into Rag1−/− recipients. Disease symptoms were monitored daily (n = 9; 2 independent experiments).
(B) Heatmap illustrates relative expression of RNA sequencing data of genes associated with Th17 cells.
(C and D) Naive CD4 T cells were activated under Th17 conditions in the presence of vehicle or oligomycin for 72 h. Representative plots are gated on live CD4 T cells (C) and graphs (D) show the expression of IL-17A, Foxp3, and RORγt (representative of 12 independent experiments).
(E) Foxp3+ (GFP+) CD4 T cells were isolated after Th17 differentiation in the presence of oligomycin and co-cultured with proliferation dye labeled CD45.1+ WT naive CD4 T cells at a 1:1 ratio. Representative plots show proliferation of CD45.1+ WT CD4 T cells on day 3 (3 independent experiments).
Graphs show the average ± SEM (A) or SD (D and E); (A and E) two-way ANOVA; (D) unpaired t test *p < 0.05, **p < 0.01, ***p < 0.001.
The EAE data suggest that mitochondrial function regulates expression of genes associated with Th17 pathogenicity (Gaublomme et al., 2015; Ghoreschi et al., 2010; Lee et al., 2012). To interrogate this, we examined the RNA sequencing data from oligomycin and vehicle-treated Th17 cells for differential expression of previously described pathogenic and non-pathogenic gene sets. Most of the genes linked to the pathogenicity of Th17 cells, including Tgfb3, Il23r, Stat4, and Gpr65, were downregulated in cells from the oligomycin-treated condition, although the genes inversely correlated with Th17 pathogenicity, such as Socs3 and Il10ra, were elevated (Figures 2B and S2A). Together, these results establish a fundamental role for ATP-synthase-mediated mitochondrial respiration in transcriptional programming of Th17 pathogenicity.
Further examination of the RNA sequencing data showed that Il17a mRNA was significantly decreased when ATP synthase activity was blocked during Th17 generation. Therefore, we assessed protein expression of the Th17 signature molecules IL-17A and RORγt after differentiating naive CD4 T cells toward the Th17 lineage with oligomycin or vehicle. Consistent with the RNA sequencing data, CD4 T cells stimulated in the presence of oligomycin had significant reductions in the frequencies of IL-17A and RORγt-expressing cells (Figures 2C and 2D). This was not due to translational regulation by cellular metabolism, as mRNA expression profile was similar to protein expression patterns (Figure S3A).
Mitochondrial Respiration Determines the Lineage Specification of Pathogenic Th17 over Immunosuppressive Treg Cell Fate
TGF-β signaling is important for the induction of Th17 cells, but this cytokine also directs the differentiation of Treg cells. One key determinant that instructs the Th17/Treg cell fate decision is the IL-2/STAT5 pathway, which skews development in the favor of Treg cells at the expense of Th17 differentiation (Chen et al., 2003; Nakayamada et al., 2012; Veldhoen et al., 2006; Kitagawa and Sakaguchi, 2017; Yang et al., 2011). Interestingly, we detect increased levels of phosphorylated STAT5 in oligomycin-treated Th17 cells (Figure S4A), and RNA sequencing revealed genes associated with the STAT5 pathway are enriched in CD4 T cells activated under these conditions (Figure S4B). These data prompted us to next evaluate the expression of Foxp3 in CD4 T cells activated under Th17 conditions in the presence or absence of oligomycin. Surprisingly, blockade of ATP synthase during Th17 differentiation resulted in the emergence of Foxp3+ CD4 T cells (Figures 2C, 2D, and S3A), and this outcome was also observed when the other OXPHOS inhibitors antimycin A and rotenone were included during Th17 differentiation (Figures S3B and S3C).
The Foxp3+ CD4 T cells that develop as a result of OXPHOS blockade during Th17 differentiation exhibited various properties of induced Treg cells, including a transcriptional signature consistent with Treg cells as opposed to Th17 cells (Figure S2B). One hallmark of Foxp3+ Treg cells is the ability to inhibit T cell proliferation via an in vitro suppression assay. Using this assay, the Foxp3+ CD4 T cells derived from oligomycin-treated Th17 cultures significantly inhibited proliferation of naive CD4 T cells (Figure 2E). Together, these data demonstrate that one function of mitochondrial OXPHOS during Th17 differentiation is to limit the IL-2/STAT5 pathway and the induction of Foxp3+ CD4 T cells.
Mitochondrial OXPHOS Supports STAT3 Signaling during Th17 Differentiation
STAT3 activation via IL-6 signaling is a key fate-determining factor for Th17 differentiation and suppressing Treg cell induction. Activated STAT3 dimer binds to the promoter region of Rorc, which specifies Th17 lineage commitment, in addition to the Il17a, Il21, and Il22 promoter loci. Therefore, defects in STAT3 signaling ablate Th17 development (Durant et al., 2010; Gaffen et al., 2014; Yang et al., 2007). As Th17 cells rely on mitochondrial OXPHOS for development and function, we examined whether mitochondrial respiration promotes STAT3 signaling. Consistent with previous reports (Chen et al., 2006; Durant et al., 2010), vehicle-treated cells robustly induced STAT3 phosphorylation within 24 h after Th17 polarization. In contrast, inhibition of ATP synthase significantly dampened phosphorylation of STAT3 (Figure S4C). This suggests that mitochondrial respiration is essential for optimal STAT3 signaling. As a result, we tested whether forcing STAT3 activation in OXPHOS-suppressed conditions would rescue the phenotype by utilizing transgenic mice that express hyperactive STAT3 mutant proteins in T cells (STAT3Cstopfl/flCD4cre; referred as STAT3C+/+; Fogli et al., 2013). Surprisingly, sustained activation of STAT3 signaling was not sufficient to overcome the requirement for OXPHOS during Th17 specification; oligomycin treatment on STAT3C+/+ CD4 T cells still showed decreased IL-17A production and induction of Foxp3-expressing cells (Figure S4D). Together, these data reveal that, although mitochondrial OXPHOS is critical for STAT3 activation during Th17 differentiation, it regulates additional processes in the cell that dictate Th17 lineage commitment.
OXPHOS Functions Early after TCR Stimulation to Regulate Th17 Differentiation in a BATF-Dependent Manner
We show that mitochondrial respiration was rapidly increased at 24 h and peaked at 48 h during Th17 differentiation (Figure 1A); hence, we postulated that mitochondrial OXPHOS controls early molecular events after the TCR stimulation. To test this, naive CD4 T cells were activated with Th17-differentiating cytokines in the presence of vehicle or oligomycin for only the initial 24 h (Figure 3A). Interestingly, exposure to the OXPHOS inhibitor in the initial 24 h imprinted the fate decision toward Foxp3-expressing cells and resulted in a lower frequency of IL-17A-producing cells, indicating that mitochondrial OXPHOS is crucial early during Th17 development (Figure 3B).
Figure 3. Mitochondrial OXPHOS Regulates Th17 Differentiation by Promoting BATF Expression.

(A and B) Naive CD4 T cells were activated under Th17 conditions in the presence of vehicle or oligomycin for 24 h. Cells were then washed and cultured with Th17-differentiating cytokines for an additional 48 h. (A) Experimental design. (B) Frequency of IL-17A+ and Foxp3+ live CD4 T cells was determined by flow cytometry (4 independent experiments).
(C and D) Naive CD4 T cells were activated under Th17 conditions in the presence of vehicle or oligomycin for 24 h; (C) relative mRNA expression of Batf and Irf4 and (D) BATF and RORγt protein expression are shown (3 to 4 independent experiments).
(E) Vehicle- or oligomycin-treated activated CD4 T cells were retrovirally transduced with hNGFR-BATF overexpressing vector (OE) or hNGFR-empty vector (EV) at 24 h. The expression of IL-17A+ and RORγt+ cells (gated on CD4+ hNGFR+ T cells) were analyzed on days 3 to 4 (4 independent experiments).
Graphs show the average ± SD; (B) unpaired t test; (C) ratio-paired t test; (E) one-way ANOVA *p < 0.05, **p < 0.01, ns, not significant.
One of the major molecular events that happens during the first 24 h of Th17 differentiation is re-shaping of the chromatin landscape by pioneer transcription factors to promote accessibility to Th17 lineage specification genes (Ciofani et al., 2012; Schraml et al., 2009; Yosef et al., 2013). As mitochondrial OXPHOS is required during the early phase of Th17 development, we examined the expression of Th17 pioneer transcription factors, BATF and IRF4. Although Irf4 mRNA level was slightly increased in oligomycin-treated cells, both mRNA and protein expression of BATF were significantly decreased when ATP-synthase-mediated respiration was inhibited (Figures 3C and 3D). Notably, the failure of BATF induction in oligomycin-treated cells correlated with the profound defect in expression of the signature Th17 transcription factor, RORγt (Figure 3D). Furthermore, comparison of previously published BATF-deficient (Batf−/−) Th17 microarray data (Schraml et al., 2009) with our RNA sequencing datasets indicated that genes downregulated in Batf−/− Th17 cells (denoted as wild-type [WT] up genes) were similarly decreased in oligomycin-treated Th17 cells, such that the vehicle-treated Th17 cells showed a significant enrichment with BATF-sensitive Th17 genes (Figure S2C). These data are consistent with a role for OXPHOS in regulating BATF expression during Th17 differentiation.
Next, we examined whether forced expression of BATF could bypass the requirement for mitochondrial OXPHOS during Th17 commitment. To test this, CD4 T cells were retrovirally transduced to overexpress BATF during Th17 differentiation in the presence of oligomycin or vehicle. Strikingly, BATF overexpression was able to rescue the IL-17A production in Th17 cells cultured in the presence of oligomycin; however, it was not able to prevent the upregulation of Foxp3 in a portion of these cells (Figures 3E and S5A). Together, OXPHOS promotes Th17 differentiation by mediating BATF induction and, in turn, BATF-sensitive transcriptional changes.
ATP-Linked Mitochondrial Respiration Regulates BATF Induction Independent of Th17 Cytokines
BATF expression is controlled and maintained by TCR signaling, as well as cytokine signaling (Iwata et al., 2017; Murphy et al., 2013; Xin et al., 2015). To distinguish between the requirement of OXPHOS for TCR and cytokine signaling to support BATF induction, we activated naive CD4 T cells under Th1 and Th2 conditions with or without concurrent oligomycin treatment. This experimental platform provides the same level of TCR stimulation to the CD4 T cells but alter the cytokine signals it perceives. Strikingly, inhibition of ATP synthase profoundly dampened BATF induction in both Th1 and Th2 lineages similar to Th17 cells, although IRF4 expression was not severely altered (Figure 4A). In particular, expression of the Th2 lineage defining transcription factor GATA3 was also significantly impaired (Figure 4B), which is consistent with a critical role of BATF in Th2 development. These data suggest that mitochondrial OXPHOS regulates BATF expression independent of Th17-polarizing cytokine signaling and functions as a key regulator of BATF induction in other effector CD4 T cells subsets.
Figure 4. ATP-Linked OXPHOS Controls BATF Induction by Regulating TCR Signaling.

(A and B) Naive CD4 T cells were differentiated under Th1 or Th2 polarizing conditions in the presence of vehicle or oligomycin for 24 h, and expression of (A) BATF, IRF4, and (B) GATA3 was determined by flow cytometry (4 to 5 independent experiments).
(C and D) Naive CD4 T cells from Nur77/GFP spleens were activated under Th17 conditions in the presence of vehicle or oligomycin for indicated period of time.
(C) Graph shows the expression of Nur77/GFP. (D) Representative plots are gated on live CD4 T cells (representative of 3 independent experiments).
(E) Naive CD4 T cells were activated under Th17 conditions with vehicle or oligomycin. Representative histogram overlay shows the phosphorylation of ZAP70 at 24 h (representative of 5 independent experiments).
(F) Naive CD4 T cells from Nur77/GFP reporter spleens were stimulated with Th17 cytokines for 6 h. BATF expression in GFP+ and GFP− cells were determined by flow cytometry (3 independent experiments).
Graphs show the average ± SD; (A and C) two-way ANOVA; (B, E and F) unpaired t test *p < 0.05, **p < 0.01, ***p < 0.001.
Mitochondrial Respiration Is Essential for TCR Signaling, which Mediates BATF Induction during Th17 Differentiation
TCR activation is a known factor that induces BATF expression, and the strength of TCR signaling affects the binding ability of BATF-IRF4 complexes to the target loci in Th2 cells (Ciofani et al., 2012; Iwata et al., 2017; Murphy et al., 2013; Schraml et al., 2009; Yosef et al., 2013). In agreement with these reports, BATF induction during Th17 differentiation was also highly dependent on the strength of TCR signaling, suggesting that TCR affinity is a key regulator of BATF expression in Th17 cells (Figures S5B and S5C). These data prompted us to examine whether mitochondrial OXPHOS supports BATF expression by controlling TCR signal strength. Using the Nr4a1-GFP (Nur77-GFP) reporter system, in which GFP expression is indicative of the TCR signal strength (Au-Yeung et al., 2014; Moran et al., 2011), we assessed the kinetics of Nur77 induction during activation of naive CD4 T cells under Th17 conditions in the presence of vehicle or oligomycin. Intriguingly, oligomycin treatment profoundly impaired the induction of Nur77 at all time points, indicating an essential role of OXPHOS in defining the TCR signal strength (Figures 4C and 4D). In addition, phosphorylation of ZAP70 was significantly reduced upon ATP synthase inhibition (Figure 4E). Importantly, Nur77 induction was closely linked to the BATF expression, as Nur77+ cells showed higher BATF expression in comparison to Nur77− cells (Figure 4F). These data together illustrate a pivotal role of mitochondrial respiration in TCR signaling, which in turn is a crucial regulator of BATF induction and effector CD4 T cell fate.
Mitochondrial Respiration Fuels mTOR Signaling, which Is Required for an Optional Expression of BATF
Although it is clear that TCR engagement promotes expression of BATF, molecular mediators bridging TCR signaling and BATF expression are not known. TCR signaling activates the mTOR pathway, and the mTOR pathway is critical for Th17 development; hence, we hypothesized that mTOR complex 1 (mTORC1) is involved in TCR-dependent BATF expression. We find that phosphorylation of S6 subunit (pS6) and 4E-BP1 (p4E-BP1), molecules downstream of mTORC1, positively correlates with increased TCR strength during Th17 differentiation (Figures S5B and S5C), consistent with the previously known role of TCR signaling in mTOR activation. Furthermore, the pS6 levels are closely associated with Nur77 expression (Figure 5A), highlighting a possible role of mTORC1 in TCR-dependent molecular events.
Figure 5. mTORC1 Activation during Th17 Differentiation Requires OXPHOS and Supports BATF Induction.

(A) Naive CD4 T cells from Nur77/GFP mice were activated under Th17 conditions in the presence of vehicle or oligomycin for 1 h. Phosphorylation of S6 is measured by flow cytometry (representative of 3 independent experiments).
(B) Relative expression of indicated genes derived from RNA sequencing is shown (vehicle Th17 versus oligomycin Th17). (C–F) Naive CD4 T cells were activated under Th17 conditions in the presence of vehicle or the indicated inhibitor for 24 h. Phosphorylation of (C) S6 and (D) 4E-BP1 were analyzed.
(E and F) BATF expression was analyzed in (E and F) vehicle- or (F) inhibitor-treated cells (6–9 independent experiments).
Graphs show the average ± SD; (A, C, D, and E) unpaired t test; (F) one-way ANOVA *p < 0.05, **p < 0.01, ***p < 0.001.
These findings led us to interrogate whether OXPHOS is also necessary for TCR-induced mTOR activation. In agreement with a severe defect in TCR signaling, oligomycin-treated cells showed an altered expression of genes involved in mTOR pathway (Figure 1D). More extensive analysis of these data demonstrated that molecules positively associated with mTOR signaling were downregulated in Th17 cells when ATP synthase was inhibited, whereas genes negatively associated with mTOR signaling were increased in these cells (Figure 5B). Moreover, the levels of pS6 and p4E-BP1 were significantly lower in CD4 T cells polarized to Th17 cells with OXPHOS blockade (Figures 5C and 5D), indicating impaired activation of mTORC1.
To further elucidate the potential link between mTORC1 activation and BATF induction, we analyzed BATF levels in CD4 T cells differentiated under Th17 conditions that expressed pS6. At 24 h after activation, Th17 cell pS6 staining was bimodal and approximately 80% of the pS6+ cells expressed BATF, although the pS6− cells showed significantly less BATF induction (~50%; Figure 5E). Finally, we tested whether activated mTOR signaling is necessary for BATF induction. CD4 T cells were treated with rapa mycin, a pan inhibitor of mTOR signaling, during Th17 differentiation. Strikingly, BATF levels were significantly reduced when mTOR was inhibited, indicating that the mTOR pathway is an important regulator of BATF expression. Of note, blockade ATP synthase activity and OXPHOS resulted in a more severe defect in BATF expression, implying additional regulatory mechanisms for BATF induction via OXPHOS (Figure 5F). Together, these data reveal that mitochondrial OXPHOS fuels Th17 differentiation by promoting a critical Th17 pioneer factor, BATF, and this regulation was associated with the requirement of mitochondrial function for optimal TOR and mTOR signaling.
DISCUSSION
Cellular metabolism regulates T cell development and function through transcriptional, translational, and epigenetic mechanisms (Berod et al., 2014; Chang et al., 2013; Chisolm et al., 2017; Delgoffe et al., 2009; Gerriets et al., 2015; Xu et al., 2017). Here, we show that mitochondrial OXPHOS directs lineage commitment of pathogenic Th17 cells and suppresses Treg cells by orchestrating distinct cellular and molecular events. It supports metabolic reprogramming through glycolysis, the HIF pathway, and TCA cycle, which further impacts cell proliferation. Moreover, mitochondrial OXPHOS is essential for early molecular events associated with Th17 development, including STAT3 activation and BATF induction. In this study, we have used oligomycin as a selective inhibitor of the mitochondrial ATP synthase activity. It is unlikely that the effects we observe are due to bioenergetic impairment of the cells, because processes requiring ATP, such as protein phosphorylation, gene transcription, and protein synthesis, are not globally inhibited.
Strikingly, the OXPHOS-mediated Th17 lineage decision was made early following T cell activation in a BATF-dependent manner. Previous publications demonstrated that BATF expression is rapidly induced after TCR stimulation, and quality of TCR signaling determines binding affinity of BATF and IRF4 heterodimers to the target regulatory loci (Iwata et al., 2017; Murphy et al., 2013; Schraml et al., 2009). Intriguingly, we find that inhibition of ATP synthase regulates key TCR signals, Nur77 induction, and Zap70 phosphorylation. This may represent a mechanism by which mitochondrial OXPHOS directs BATF expression and early transcriptional programming required for Th17 lineage commitment. Furthermore, mitochondrial metabolism may regulate the availability of important intermediates that serve as epigenetic co-factors or regulators, as it is known that cellular metabolism influences histone modification and chromatin remodeling in T cells (Cameron et al., 2016; Chisolm et al., 2017; Xu et al., 2017). Still, we do show a direct connection between mTORC1 activation and BATF expression; hence, it is possible that OXPHOS regulates BATF induction via control of the mTOR pathway. It will be necessary to dissect how mitochondrial respiration selectively fuels Th17-specific events and what mediators contribute to this process.
Recent studies showed that lineage-committed Th17 cells require mitochondrial OXPHOS for synthesis of effector molecules, including IL-17A and IL-21, in vivo and induction of the pathogenic Th17 signature (Franchi et al., 2017; Kaufmann et al., 2019). These findings, together with our data, suggest that mitochondrial respiration plays distinct but consistently supportive roles in Th17 biology during different developmental phases and environmental contexts. Notably, ATP-linked OXPHOS mediates BATF induction in other subsets of T cells, such as Th1 and Th2 cells. It will be of interest to investigate whether OXPHOS is also necessary for their protective and pathogenic functions.
Our data show that the inhibition of mitochondrial ATP synthase during Th17 differentiation biases the fate decision toward functionally suppressive Treg cell development by driving a robust induction of STAT5 signaling. This contrasts with the known phenotype of conventional Treg cells, which heavily rely on mitochondrial energy metabolism for their function and transcriptional programming (Angelin et al., 2017; Gerriets et al., 2015). It is possible that Treg cells generated with OXPHOS inhibition under Th17 conditions may exhibit distinct functional and transcriptional characteristics from classical Treg cells. This could reflect a mechanism of the abnormal generation of Treg cells under a pro-inflammatory environment, such as in the context of the tumor microenvironment. Interestingly, Batf−/− CD4 T cells also preferentially express Foxp3 under Th17 differentiation conditions (Schraml et al., 2009). However, BATF overexpression did not suppress the development of Foxp3+ Treg cells under oligomycin-treated Th17 conditions. These data suggest that mitochondrial OXPHOS supports pathogenic Th17 differentiation through a BATF-dependent mechanism, but the lineage decision favoring Treg cells with ATP synthase blockade may be ascribed to a BATF-independent mechanism.
Certain metabolic sensors are known to regulate the Th17/Treg cell balance, including HIF-1α and mTOR (Delgoffe et al., 2009, 2011; Dang et al., 2011; Shi et al., 2011). In the absence of mTORC1 or HIF-1α, Th17 induction is corrupted and Foxp3+ cells develop, mirroring the phenotype we observe when mitochondrial OXPHOS is inhibited during Th17 differentiation. Interestingly, we find that both mTORC1 activation (pS6 levels) and Hif1a mRNA expression are impaired in oligomycin-treated Th17 cultures. As stated earlier, one potential mechanism by which OXPHOS facilitates Th17 differentiation is by governing mTORC1 activation and subsequent BATF induction. HIF-1α is induced by STAT3 signaling and functions in Th17 cells by directly interacting in a complex with RORγt at the Il17a promoter (Dang et al., 2011), events downstream of OXPHOS-regulated BATF induction. Nevertheless, it remains to be determined whether and how these pathways intersect to promote Th17 development.
In summary, we show that mitochondrial OXPHOS is essential for pathogenic Th17 development; inhibition of OXPHOS during Th17 differentiation was sufficient to ablate Th17-mediated autoimmune pathology in mice. Also, our study demonstrates the profound regulatory roles of mitochondrial metabolism in shaping molecular events during Th17 lineage commitment, such as pioneer factor induction, TCR signaling, and mTOR activation. Our study highlights the possibility that manipulating cellular metabolism, and specifically mitochondrial OXPHOS, may provide a new promising therapeutic intervention for modulating the balance between pathogenic Th17 and Treg cells in chronic autoimmune disorders.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Laurie E. Harrington (lharring@uab.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
C57BL/6J (B6), B6.129S7-Rag1tm1Mom/J (Rag1−/−), B6. Tg(Tcra2D2,Tcrb2D2)1Kuch/J (2D2), B6.129(Cg)-Foxp3tm3(DTR/GFP)Ayr/J (Foxp3-GFP), and B6.SJL-Ptprca Pepcb/BoyJ (CD45.1) mice were purchased from the Jackson Laboratory and bred at the University of Alabama at Birmingham. Both B6 and B6.Ifng/Thy1.1 knock-in mice (Harrington et al., 2008) were used as wild-type (WT) control. STAT3Cstopfl/flCD4cre mice and Nr4a1-GFP mice were described previously (Fogli et al., 2013; Moran et al., 2011). Both male and female mice were used for this study. All animals were bred and maintained under specific pathogen-free conditions at the University of Alabama at Birmingham according to Institutional Animal Care and Use Committee (IACUC) regulations.
METHOD DETAILS
In vitro Th17 differentiation
Naive CD4 T cells from WT, STAT3C+/+, Foxp3-GFP, Nur77-GFP, and CD45.1 mice were isolated by FACS sorting as described previously (Yeh et al., 2011) or by magnetic bead enrichment using the mouse naive CD4 isolation kit (Stem Cell Technology) according to the manufacturer’s protocol. Naive CD4 T cells were stimulated in R10 media (RPMI 1640 with 10% FCS, 2 mM L-glutamine, 100 IU/ml penicillin, 100 μg/ml streptomycin, 1 × nonessential amino acids, 1 μM sodium pyruvate, and 50 μM β-mercaptoethanol) with plate-bound 10 μg/ml anti-CD3ε (BioXCell, clone 145C-11) and soluble 1 μg/ml anti-CD28 (eBioscience, clone 37.51), 20 ng/ml rmIL-6 (Biolegend), 2.5 ng/ml rhTGFβ1 (Peprotech), 10 ng/ml rmIL-23 (Biolegend), 10 μg/ml anti-IL-4 (BioXCell, 11B11), and 10 μg/ml anti-IFNγ (BioXCell, XMG1.2) for indicated amount of time. The following inhibitors were used in this study:
2.5 ng/ml oligomycin (Sigma-Aldrich),
250 nM antimycin A and 250 nM rotenone (Agilent technologies),
50 nM rapamycin (LC Laboratories).
EAE induction
Naive CD4 T cells from 2D2 mice were differentiated under Th17 conditions in the presence of vehicle or 2.5 ng/ml oligomycin for 5 days, and 1x106 CD4 T cells were transferred into age- and sex- matched Rag1−/− mice. Recipient mice were injected with 200 ng of pertussis toxin (List Biological Laboratories) intraperitoneally on day 0 and day 2. Disease was monitored daily by the following criteria: 0, no disease; 1, tail paralysis; 2, hind limb paresis; 2.5, one hind limb paralysis; 3, both hind limbs paralysis; 4, forelimbs paralysis; 5, moribund.
Retroviral transduction
The open reading frame of mouse Batf was amplified and sub-cloned into the retroviral vector MSCV-IRES-hNGFR plasmid. Retrovirus containing sequence encoding Batf was produced in Plat-E cells by cotransfection with retroviral vectors and helper plasmids. Naive CD4 T cells were activated in Th17 conditioned media with vehicle or oligomycin for 24h, and transduced with virus containing medium with polybrene (6 μg/ml) following centrifugation for 2h at 2000rpm. After centrifugation, cells were incubated at 37°C for 1h, and then the medium was replaced with Th17 conditioned media with vehicle or oligomycin. Cells were cultured for 2-3 additional days.
Flow cytometry analysis
Cell surface and intracellular staining was performed following PMA and ionomycin stimulation as described previously (Yeh et al., 2011). A viability dye (Life Technologies, Aqua) was applied to exclude dead cells. Intracellular staining for IL-17A, RORγt, Foxp3, and BATF was performed using the Foxp3 Permeabilization/Fixation kit (eBioscience). To determine the phosphorylation of STAT3, STAT5, and S6, cells were fixed with 4% paraformaldehyde for 15 min at room temperature and permeablized with 100% methanol at −20°C for 30 min prior to the intracellular staining. Samples were acquired using LSRII flow cytometer (BD Biosciences) and data was analyzed with FlowJo version 10 (Tree Star).
RNA purification, RNA sequencing, and analysis
For RNA sequencing, total RNA was isolated from in vitro cultured CD4 T cells at 48h using the RNeasy Plus Mini Kit (QIAGEN) according to the manufacturer’s protocols and submitted to GENEWIZ (South Plainfield, NJ). Total RNA was sequenced on Illumina HiSeq 2500 (1x50 base pair, single-read) and aligned to the mouse mm10 reference genome using TopHat. The gene expression values (FPKM) were calculated with Cufflinks and significant changes in transcript expression were determined with Cuffdiff. RNA sequencing data have been submitted to the Gene Expression Omnibus (GEO) Repository (GSE115282).
For gene set enrichment analysis (GSEA), RNA sequencing data was pre-ranked according to an adjusted p value and the sign of differential expression. The gene sets from the hallmark gene sets (H) of the Molecular Signatures Database (MSigDB) were used for computing enrichment. The normalized enrichment score (NES), nominal p value, and false detection rate (FDR) q-value were assessed using GSEA software from Broad Institute by running in pre-ranked list mode with 1,000 permutations. The significant genes obtained by q value < 0.05 were visualized as heatmaps using Gitools software version 2.3.1.
cDNA synthesis and real-time PCR
cDNA synthesis and real-time PCR was described previously (Yeh et al., 2011). Relative gene expression was calculated according to the ΔΔ threshold cycle (Ct) method by utilizing β2-microglobulin as a housekeeping gene.
In vitro suppression assay
Naive CD4 T cells from Foxp3-GFP mice were cultured in Th17 polarizing conditions in the presence of oligomycin. After 72h of culture, GFP+ (Foxp3+) CD4+ T cells were FACS sorted. FACS sorted cells were co-cultured with proliferation dye (eFluor 450 proliferation dye, eBioscience) labeled CD45.1 naive CD4 at a 1:1 ratio with plate-bound anti-CD3ε and anti-CD28 in R10. Dilution of proliferation dye was assessed at 72h by flow cytometry.
Seahorse extracellular flux analysis
To determine oxygen consumption rate (OCR) and extracellular acidification rate (ECAR), CD4 T cells cultured under Th17 conditions for 0h (naive CD4 T cells), 24h, 48h, and 72h were harvested, resuspended in XF-DMEM medium (pH 7.4, 37°C), and plated at 2x105 cells/well on Cell-Tak (Corning) pre-coated extracellular flux (XF) 96-well assay plate. The OCR and ECAR were assessed using the Seahorse XFe96 analyzer (Agilent) by simultaneous assessment upon 1 μg/ml oligomycin, 1.5 μM FCCP, 10 μg/ml antimycin A, and 120 mM 2DG. Basal OCR was calculated by the difference between OCRinitial and OCRantimycinA. ATP-liked OCR was determined by the difference between OCRinitial and OCRoligomycin. Basal ECAR was measured by the difference between ECARinitial and ECAR2DG (Hill et al., 2012).
QUANTIFICATION AND STATISTICAL ANALYSIS
Unpaired t test was used to calculate statistical significance for individual groups. The ratio paired t test was performed to analyze statistical significance of real-time PCR data. To compare the average of three or more groups, One-way ANOVA with Tukey’s multiple comparisons test was used. Two-way ANOVA with Tukey’s multiple comparison test was performed to compare the mean differences between oligomycin and vehicle treated group at multiple time points. *p < 0.05; **p < 0.01; ***p < 0.001. All statistical analyses (excluding RNaseq, described above) were performed using Prism 6.0c software (GraphPad).
DATA AND CODE AVAILABILITY
The accession number for the RNA sequencing datasets reported in this paper is GSE115282.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-mouse CD4 PerCP-Cy5.5 (clone RM4-5) | eBioscience | Cat# 45-0042-82 |
| anti-mouse CD4 PE-Cy7 (clone RM4-5) | eBioscience | Cat# 25-0042-82 |
| anti-mouse CD4 APC (clone RM4-5) | eBioscience | Cat# 17-0042-82 |
| anti-mouse CD4 APC780 (clone RM4-5) | eBioscience | Cat# 47-0042-82 |
| anti-mouse CD3ε PE-Cy7 (clone 145-2C11) | eBioscience | Cat# 25-0031-82 |
| anti-mouse CD45.1 APC (clone A20) | eBioscience | Cat# 17-0453-82 |
| anti-mouse CD45.2 PE (clone 104) | eBioscience | Cat# 14-0454-82 |
| anti-human NGFR APC (clone ME20.4) | Biolegend | Cat# 345107 |
| anti-mouse IL-17A PE (clone TC11-18H10) | BD Biosciences | Cat# 559502 |
| anti-mouse IL-17A eFluor 450 (clone eBio17B7) | eBioscience | Cat# 48-7177-82 |
| anti-mouse RORγt PerCP e710 (clone B2D) | eBioscience | Cat# 46-6981-82 |
| anti-mouse RORγt APC (clone B2D) | eBioscience | Cat# 17-6981-82 |
| anti-mouse Foxp3 FITC (clone FJK-16 s) | eBioscience | Cat# 11-5773-82 |
| anti-mouse Foxp3 eFluor 450 (clone FJK-16 s) | eBioscience | Cat# 48-5773-82 |
| anti-mouse BATF PE (clone D7C5) | Cell Signaling Technology | Cat# 27120S |
| anti-mouse IRF4 FITC (clone 3E4) | eBioscience | Cat# 11-9858-82 |
| anti-GATA3 PE (clone TWAJ) | eBioscience | Cat# 12-9966-42 |
| anti-pSTAT3 (pY705) PE (clone 4/p-STAT3) | BD Biosciences | Cat# 612569 |
| anti-pSTAT5 (pY694) Alexa Fluor 647 (clone 47/Stat5) | BD Biosciences | Cat# 612599 |
| anti-pS6 ribosomal protein (pS235/236) Pacific Blue (clone D57.2.2E) | Cell Signaling Technology | Cat# 8520S |
| anti-p4E-BP (pT37/46) Alexa Fluor 647 (clone 236B4) | Cell Signaling Technology | Cat# 5123S |
| anti-pZAP70 (pY319)/Syk(pY352) PE (clone 65E4) | Cell Signaling Technology | Cat# 14791S |
| anti-CD3ε (clone 145C-11) | BioXCell | Cat# BE0001-1 |
| anti-CD28 (clone 37.51) | eBioscience | Cat# 14-0281-82 |
| anti-IL-4 (clone 11B11) | BioXCell | Cat# BE0045 |
| anti-IFNγ (clone XMG1.2) | BioXCell | Cat# BE0055 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant mouse IL-6 | Biolegend | Cat# 575706 |
| Recombinant mouse IL-23 | Biolegend | Cat# 589006 |
| Recombinant human TGFβ1 | Peprotech | Cat# 100-21 |
| Pertussis toxin | List Biological Laboratories | Cat# 181 |
| eBioscience Cell Proliferation Dye eFluor 450 | eBioscience | Cat# 65-0842-85 |
| Oligomycin from Streptomyces diastatochromogenes | Sigma-Aldrich | Cat# O4876 |
| Antimycin A | Agilent | Cat# 103015-100 |
| Rotenone | Agilent | Cat# 103015-100 |
| Rapamycin | LC Laboratories | Cat# R-5000 |
| Critical Commercial Assays | ||
| Seahorse XF Cell Mito Stress Test Kit | Agilent | Cat# 103015-100 |
| Foxp3/Transcription Factor Staining Buffer Set | eBioscience | Cat# 00-5523-00 |
| EasySep Mouse Naive CD4+ T Cell Isolation Kit | Stem Cell Technologies | Cat# 19765 |
| RNeasy Plus Mini Kit | QIAGEN | Cat# 74134 |
| LIVE/DEAD Fixable Aqua Dead Cell Stain Kit | Life Technologies | Cat# L34966 |
| Deposited Data | ||
| RNA sequencing | This paper | GSE115282 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Cat# 000664 |
| Mouse: B6.129S7-Rag1tm1Mom/J (Rag1−/−) | The Jackson Laboratory | Cat# 002216 |
| Mouse: B6. Tg(Tcra2D2,Tcrb2D2)1Kuch/J (2D2) | The Jackson Laboratory | Cat# 006912 |
| Mouse: B6.129(Cg)-Foxp3tm3(DTR/GFP)Ayr/J (Foxp3-GFP) | The Jackson Laboratory | Cat# 016958 |
| Mouse: B6.SJL-Ptprca Pepcb/BoyJ (CD45.1) | The Jackson Laboratory | Cat# 002014 |
| Mouse: STAT3Cstopfl/flCD4cre | (Fogli et al., 2013) | N/A |
| Mouse: Nr4a1-GFP | (Moran et al., 2011) | N/A |
| Oligonucleotides | ||
| Il17a forward CTCCAGAAGGCCCTCAGACTAC | (Yeh et al., 2011) | N/A |
| Il17a reverse GGGTCTTCATTGCGGTGG | (Yeh et al., 2011) | N/A |
| Rorc forward CCGCTGAGAGGGCTTCAC | (Yeh et al., 2011) | N/A |
| Rorc reverse TGCAGGAGTAGGCCACATTACA | (Yeh et al., 2011) | N/A |
| Foxp3 forward TTCATGCATCAGCTCTCCAC | (Yeh et al., 2011) | N/A |
| Foxp3 reverse CTGGACACCCATTCCAGACT | (Yeh et al., 2011) | N/A |
| Batf forward CTCCTCCCCCTGGCAAAC | This paper | N/A |
| Batf reverse GGGCAGCGATGCGATTCT | This paper | N/A |
| Irf4 forward GAAGCCTTGGCGCTCTCA | This paper | N/A |
| Irf4 reverse CACGAGGATGTCCCGGTAAT | This paper | N/A |
| Software and Algorithms | ||
| FlowJo 10 | Tree Star | https://www.flowjo.com |
| Seahorse Wave | Agilent | https://www.agilent.com |
| GraphPad Prism 8 | GraphPad Software | https://www.graphpad.com |
| TopHat | PennState Galaxy; (Kim et al., 2013) | Ver 2.1.0 |
| Cufflinks | PennState Galaxy; (Trapnell et al., 2010) | Ver 2.2.1.0 |
| Cuffdiff | PennState Galaxy; (Trapnell et al., 2010) | Ver 2.2.1.3 |
| GSEA | MIT Broad Institute; (Liberzon et al., 2015; Subramanian et al., 2005) | http://software.broadinstitute.org/gsea |
| Gitools | (Perez-Llamas and Lopez-Bigas, 2011) | Ver 2.3.1 |
| Other | ||
| BD FACSAria Sorter | BD Biosciences | N/A |
| BD LSR II | BD Biosciences | N/A |
| Seahorse XFe96 Analyzers | Agilent | N/A |
Highlights.
CD4 T cells rapidly increase mitochondrial respiration during Th17 differentiation
OXPHOS is essential for Th17 cell pathogenicity in a mouse model of MS
Mitochondrial respiration shapes the Th17 and Treg cell fate decision
OXPHOS facilitates TCR and mTOR signaling, which in turn support BATF induction
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health (NIH) grants R01 AI113007 (L.E.H.) and O’Brien Center P30 DK079337 (V.M.D.-U.), American Heart Association grant 16PRE29650004 (B.S.), AMC21 Award from the UAB School of Medicine (L.E.H.), and the Blue Sky Award from the UAB School of Medicine (V.M.D.-U.). We thank members of the Harrington, Zajac, and Elson laboratories for comments and technical assistance. We thank Haiguang Wang and Kristin A. Hogquist at the University of Minnesota for the Nr4a1-GFP mice. We acknowledge Marion Spell and Enid Keyser of the UAB comprehensive flow cytometry core for cell sorting (NIH P30 AI27667 and NIH P30 AR048311).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.01.022.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, Wang Z, Quinn WJ 3rd, Kopinski PK, Wang L, et al. (2017). Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. 25, 1282–1293.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo L, Khim P, Mkhikian H, Mortales CL, and Demetriou M (2017). Glycolysis and glutaminolysis cooperatively control T cell function by limiting metabolite supply to N-glycosylation. eLife 6, e21330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au-Yeung BB, Zikherman J, Mueller JL, Ashouri JF, Matloubian M, Cheng DA, Chen Y, Shokat KM, and Weiss A (2014). A sharp T-cell antigen receptor signaling threshold for T-cell proliferation. Proc. Natl. Acad. Sci. USA 111, E3679–E3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, Sandouk A, Hesse C, Castro CN, Bähre H, et al. (2014). De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med 20, 1327–1333. [DOI] [PubMed] [Google Scholar]
- Buck MD, Sowell RT, Kaech SM, and Pearce EL (2017). Metabolic instruction of immunity. Cell 169, 570–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron AM, Lawless SJ, and Pearce EJ (2016). Metabolism and acetylation in innate immune cell function and fate. Semin. Immunol 28, 408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, et al. (2013). Post-transcriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, and Wahl SM (2003). Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med 198, 1875–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu BM, Tato C, Yoshimura A, Hennighausen L, and O’Shea JJ (2006). Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc. Natl. Acad. Sci. USA 103, 8137–8142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisolm DA, Savic D, Moore AJ, Ballesteros-Tato A, León B, Crossman DK, Murre C, Myers RM, and Weinmann AS (2017). CCCTC-binding factor translates interleukin 2- and α-ketoglutarate-sensitive metabolic changes in T cells into context-dependent gene programs. Immunity 47, 251–267.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkhurst CN, Muratet M, et al. (2012). A validated regulatory network for Th17 cell specification. Cell 151, 289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR, et al. (2011). Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 146, 772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, and Powell JD (2009). The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 30, 832–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, Xiao B, Worley PF, and Powell JD (2011). The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol 12, 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L, Takahashi H, Sun HW, Kanno Y, Powrie F, and O’Shea JJ (2010). Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity 32, 605–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogli LK, Sundrud MS, Goel S, Bajwa S, Jensen K, Derudder E, Sun A, Coffre M, Uyttenhove C, Van Snick J, et al. (2013). T cell-derived IL-17 mediates epithelial changes in the airway and drives pulmonary neutrophilia. J. Immunol. 191, 3100–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi L, Monteleone I, Hao LY, Spahr MA, Zhao W, Liu X, Demock K, Kulkarni A, Lesch CA, Sanchez B, et al. (2017). Inhibiting oxidative phosphorylation in vivo restrains Th17 effector responses and ameliorates murine colitis. J. Immunol 198, 2735–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffen SL, Jain R, Garg AV, and Cua DJ (2014). The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat. Rev. Immunol 14, 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaublomme JT, Yosef N, Lee Y, Gertner RS, Yang LV, Wu C, Pandolfi PP, Mak T, Satija R, Shalek AK, et al. (2015). Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell 163, 1400–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, et al. (2015). Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Invest 125, 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, et al. (2010). Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature 467, 967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoreschi K, Laurence A, Yang XP, Hirahara K, and O’Shea JJ (2011). T helper 17 cell heterogeneity and pathogenicity in autoimmune disease. Trends Immunol. 32, 395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington LE, Janowski KM, Oliver JR, Zajac AJ, and Weaver CT (2008). Memory CD4 T cells emerge from effector T-cell progenitors. Nature 452, 356–360. [DOI] [PubMed] [Google Scholar]
- Hill BG, Benavides GA, Lancaster JR Jr., Ballinger S, Dell’Italia L, Jianhua Z, and Darley-Usmar VM (2012). Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol. Chem 393, 1485–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, and Littman DR (2006). The orphan nuclear receptor ROR-gammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133. [DOI] [PubMed] [Google Scholar]
- Iwata A, Durai V, Tussiwand R, Briseño CG, Wu X, Grajales-Reyes GE, Egawa T, Murphy TL, and Murphy KM (2017). Quality of TCR signaling determined by differential affinities of enhancers for the composite BATF-IRF4 transcription factor complex. Nat. Immunol 18, 563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R, Chen Y, Kanno Y, Joyce-Shaikh B, Vahedi G, Hirahara K, Blumenschein WM, Sukumar S, Haines CJ, Sadekova S, et al. (2016). Interleukin-23-induced transcription factor Blimp-1 promotes pathogenicity of T helper 17 cells. Immunity 44, 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann U, Kahlfuss S, Yang J, Ivanova E, Koralov SB, and Feske S (2019). Calcium signaling controls pathogenic Th17 cell-mediated inflammation by regulating mitochondrial function. Cell Metab. 29, 1104–1118.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa Y, and Sakaguchi S (2017). Molecular control of regulatory T cell development and function. Curr. Opin. Immunol 49, 64–70. [DOI] [PubMed] [Google Scholar]
- Kleinewietfeld M, and Hafler DA (2013). The plasticity of human Treg and Th17 cells and its role in autoimmunity. Semin. Immunol 25, 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, et al. (2012). Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol 13, 991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIver NJ, Michalek RD, and Rathmell JC (2013). Metabolic regulation of T lymphocytes. Annu. Rev. Immunol 31, 259–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, and Hogquist KA (2011). T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J. Exp. Med 208, 1279–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TL, Tussiwand R, and Murphy KM (2013). Specificity through cooperation: BATF-IRF interactions control immune-regulatory networks. Nat. Rev. Immunol 13, 499–509. [DOI] [PubMed] [Google Scholar]
- Nakayamada S, Takahashi H, Kanno Y, and O’Shea JJ (2012). Helper T cell diversity and plasticity. Curr. Opin. Immunol 24, 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea JJ, and Paul WE (2010). Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science 327, 1098–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel DD, and Kuchroo VK (2015). Th17 cell pathway in human immunity: lessons from genetics and therapeutic interventions. Immunity 43, 1040–1051. [DOI] [PubMed] [Google Scholar]
- Perez-Llamas C, and Lopez-Bigas N (2011). Gitools: analysis and visualisation of genomic data using interactive heat-maps. PLoS ONE 6, e19541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MJ, Patterson DG, Scharer CD, and Boss JM (2018). Progressive upregulation of oxidative metabolism facilitates plasmablast differentiation to a T-independent antigen. Cell Rep. 23, 3152–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron-Harel N, Santos D, Ghergurovich JM, Sage PT, Reddy A, Lovitch SB, Dephoure N, Satterstrom FK, Sheffer M, Spinelli JB, et al. (2016). Mitochondrial biogenesis and proteome remodeling promote one-carbon metabolism for T cell activation. Cell Metab. 24, 104–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schraml BU, Hildner K, Ise W, Lee WL, Smith WA, Solomon B, Sahota G, Sim J, Mukasa R, Cemerski S, et al. (2009). The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature 460, 405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, Wang CR, Schumacker PT, Licht JD, Perlman H, et al. (2013). Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38, 225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, and Chi H (2011). HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med 208, 1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symersky J, Osowski D, Walters DE, and Mueller DM (2012). Oligomycin frames a common drug-binding site in the ATP synthase. Proc. Natl. Acad. Sci. USA 109, 13961–13965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, and Stockinger B (2006). TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24, 179–189. [DOI] [PubMed] [Google Scholar]
- Weinberg SE, Sena LA, and Chandel NS (2015). Mitochondria in the regulation of innate and adaptive immunity. Immunity 42, 406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin G, Schauder DM, Lainez B, Weinstein JS, Dai Z, Chen Y, Esplugues E, Wen R, Wang D, Parish IA, et al. (2015). A critical role of IL-21-induced BATF in sustaining CD8-T-cell-mediated chronic viral control. Cell Rep. 13, 1118–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Stewart KM, Wang X, Liu K, Xie M, Ryu JK, Li K, Ma T, Wang H, Ni L, et al. (2017). Metabolic control of TH17 and induced Treg cell balance by an epigenetic mechanism. Nature 548, 228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, and Dong C (2007). STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem 282, 9358–9363. [DOI] [PubMed] [Google Scholar]
- Yang XP, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, Hirahara K, Sun HW, Wei L, Vahedi G, et al. (2011). Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat. Immunol 12, 247–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh WI, McWilliams IL, and Harrington LE (2011).Autoreactive Tbet-positive CD4 T cells develop independent of classic Th1 cytokine signaling during experimental autoimmune encephalomyelitis. J. Immunol 187, 4998–5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A, Wu C, Karwacz K, Xiao S, Jorgolli M, et al. (2013). Dynamic regulatory network controlling TH17 cell differentiation. Nature 496, 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA sequencing datasets reported in this paper is GSE115282.