

Abstract
The role of cholesterol in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and other coronavirus-host cell interactions is currently being discussed in the context of two main scenarios: i) the presence of the neutral lipid in cholesterol-rich lipid domains involved in different steps of the viral infection and ii) the alteration of metabolic pathways by the virus over the course of infection. Cholesterol-enriched lipid domains have been reported to occur in the lipid envelope membrane of the virus, in the host-cell plasma membrane, as well as in endosomal and other intracellular membrane cellular compartments. These membrane subdomains, whose chemical and physical properties distinguish them from the bulk lipid bilayer, have been purported to participate in diverse phenomena, from virus-host cell fusion to intracellular trafficking and exit of the virions from the infected cell. SARS-CoV-2 recruits many key proteins that participate under physiological conditions in cholesterol and lipid metabolism in general. This review analyses the status of cholesterol and lipidome proteins in SARS-CoV-2 infection and the new horizons they open for therapeutic intervention.
Keywords: Cholesterol, Cholesterol-rich lipid domains, Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), Cholesterol metabolism, Virus infection, Statins, COVID-19 comorbidities, New therapeutic strategies for COVID-19
Abbreviations: ACE2, angiotensin converting enzyme 2; CARC and CRAC, cholesterol-recognition motifs; CDx, methyl-β-cyclodextrin; CMV, human cytomegalovirus; CoV, coronavirus; DMVs, double-membrane vesicles; HCV, hepatitis C virus; HDL, high-density lipoprotein; HIV, human immunodeficiency virus; HMGCR, 3-hydroxy-3-methy-lglutaryl coenzyme A reductase; IAV, influenza A virus; IL, interleukin; LDL, low-density lipoprotein; MERS, Middle-East Respiratory Syndrome; SARS, Severe Acute Respiratory Syndrome; SRE-1, sterol regulatory element 1; SREBP, sterol regulatory element binding protein; TMPRSS2, transmembrane protease serine 2
1. Introduction
Atherosclerosis, hypertension, diabetes mellitus, severe obesity, and other disease conditions directly or indirectly related to lipid metabolism have emerged as major comorbidities of coronavirus disease 2019 (COVID-19) [[1], [2], [3], [4]]. The added risk and severity trends brought about by these comorbidities have a central common substrate: affectation of the endothelial bed [5].
In general, DNA viruses exert pathogenic effects on the infected cells via transcriptional regulation of key metabolic pathways, whereas RNA viruses like the coronaviruses (CoVs) take control of host-cell metabolism via post-transcriptional regulation. Infection by enveloped viruses entails a combination of i) an initial virus-host cell receptor binding, followed by ii) the hemifusion of viral and cell plasma membranes or internalization via endocytosis of the non-fused virus and iii) formation of a fusion pore at the plasma membrane or the endosomal membrane through which the viral genome is transferred into the host cell. These processes are all membrane-associated phenomena, and it is therefore evident that cholesterol, the key neutral lipid and ubiquitous membrane component, is directly or indirectly implicated in such phenomena.
Cholesterol in viral infection can be contemplated from the perspective of the virus and that of the host cell. Having no internal membrane structures, all that matters to an individual virus particle is the ability to obtain the requisite supply of the sterol from the infected cell to maintain the structure of a single macromolecular assembly: its envelope membrane bilayer. For this to occur, the virus must cross the plasma membrane, express its genomic material, assemble new virions, and again cross the plasma membrane to exit the infected cell. Viewed from the host-cell standpoint, the pathogenic viral cycle entails profound changes that reshape its cellular structure and metabolism. These changes involve the sequestration of its biosynthetic machinery, re-addressing it to subserve the production of many viral particles under the control of the viral genome, as well as pathological modifications of its organelles, mainly the membrane-rich endoplasmic reticulum-Golgi complex (the so-called ERGIC) to form the replication-transcription organelle (see below), the site of viral genome replication. The mevalonate-cholesterol biosynthetic pathway, at the very core of the lipid homeostasis in eukaryotic cells (see reviews in refs. [6, 7]), is an obligatory target for enveloped virus infection, as discussed in this review.
The involvement of cholesterol in virus-host cell interactions has been mainly addressed in two contexts: i) its participation in virus entry, on the basis of its occurrence in cholesterol-rich lipid domains, the so-called “lipid rafts” and the purported preferential partition of viruses in such domains [6,[8], [9], [10], [11], [12], [13], [14], [15], [16]], and ii) indirectly, via the alteration of cellular biosynthetic pathways by viral infection. Most of the work published during the last twenty years on the first of these two topics has relied on indirect biochemical approaches such as the use of detergent extraction/centrifugation assays as the main (and often only) criterion to infer the occurrence of such domains in the actual membrane structure. Studies on the second topic have accelerated considerably thanks to the availability of new genetic, biochemical, and biophysical approaches. This review focuses on recent studies underlying the possible participation of cholesterol at different stages of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infectious cycle but is also nurtured by new data stemming from related areas of research. Conversely, developments in current knowledge of cholesterol metabolism and control mechanisms have benefited from new data generated by studies on their dysregulation in viral infection. These advances encompass recent studies on cholesterol uptake and trafficking mechanisms, as well as on the key involvement of the mevalonate-cholesterol biosynthetic pathway and cholesterol-binding proteins at the cell surface and intracellular organelles in viral infection. The new information is shedding light on the interconnectedness of virus-host interactions in the various cholesterol-dependent processes that are critically exploited by SARS-CoV-2 and other viruses. The enhanced metabolic demand of virus-infected cells is associated with dysregulation of the host-cell lipid homeostasis. The overall emerging notion is that cholesterol and cholesterol-recognition proteins play a more important role in virus-receptor interactions, fusion with the host membrane, and the intracellular cycle, than originally anticipated.
2. Sources of cellular cholesterol and its distinct distribution at the subcellular to organism level
Not all cells possess the biosynthetic machinery to produce cholesterol de novo, thus relying on external supplies from dietary sources or from other cells, as is the case with cholesterol in brain neurons. Dietary cholesterol absorption begins by solubilization of the neutral lipid by bile acids, transport across the apical membrane of enterocytes, passage across the basal enterocyte membrane, and incorporation into the blood stream and lymph via chylomicrons [17]. Niemann-Pick C1-like 1 protein (NPC1L1) intervenes in cholesterol absorption at the enterocyte plasmalemma [18]. Even those cells that do possess the enzymes required for cholesterol synthesis employ cholesterol from external sources. Receptor-mediated endocytosis of low-density lipoproteins (LDLs) is the most common mechanism that cells employ to acquire exogenous sterol. LDL-cholesterol is delivered to cells upon binding to the LDL-receptor (LDL-R) and internalized by endocytosis of the holo-particle [18]. Cholesteryl esters carried in by endocytosed LDL are hydrolysed by lysosomal acid lipase, and unesterified (free) cholesterol traffics to the ER, where it is either re-esterified to cholesteryl esters and stored within cytoplasmic lipid droplets or transported from the ER to the plasma membrane, mitochondria or other intracellular membrane compartments [19]. The complex and coordinated homeostatic regulation of cholesterol biosynthesis / external cholesterol uptake is constantly adjusting the cellular cholesterol content and distribution. This results in the peculiar topography of the neutral lipid in the cell: ca. 90% of total cholesterol content is concentrated in the plasma membrane, where it represents ~50% of the total membrane lipids [20,21]. The non-homogeneous distribution in the plane of the membrane -observed in both native and synthetic membranes [22]- and its asymmetry along the axis perpendicular to the membrane normal, i.e., the trans-bilayer cholesterol asymmetry, are two additional manifestations of cholesterol´s remarkable distribution, which carry important functional implications. The former gave rise to the concept of “lipid rafts” [[23], [24], [25]]. A consensus definition was adopted at the 2006 Keystone Symposium [26]: rafts are lipid domains characterized by their relative enrichment in cholesterol intermixed with sphingolipids and glycerophospholipids with saturated acyl chains (relative to the rest of the bilayer lipid), and display the physico-chemical characteristics of liquid-ordered (Lo) gels [27]. It was subsequently found that raft lipid domains are very dynamic structures of nanometric lateral dimensions (10-200 nm) [28,29]. The trans-bilayer asymmetry of cholesterol is less understood; in model membranes made up of cholesterol and phosphatidylcholine with fatty acids of different acyl chain length/degrees of saturation, cholesterol reorients rapidly in the plane perpendicular to the membrane bilayer, adopting a tilted orientation in regions with shorter acyl chains, as assessed by neutron scattering and solid-state 2H NMR, implying that the hydrophobic thickness is the primary driving force of cholesterol topography across the membrane [30,31]. Finally, the ultimate asymmetry of cholesterol is inherent to its molecule proper: it possesses a smooth α face, with which it interacts with other lipids, e.g., with sphingomyelins, and a rough β face, suited to match the cavities, grooves and irregular contours of many transmembrane protein surfaces and of other irregularly shaped lipids like the gangliosides [32].
The unique features of cholesterol distribution are also manifested at the organism level: the brain constitutes roughly 20% of the body´s weight, yet it is the organ with the highest concentration of the neutral lipid (approximately 23% of the body’s cholesterol content), mainly due to the high membrane/volume ratio of neurons and the high proportion of myelin [33]. The source of brain´s cholesterol is not the neuronal cell, but the oligodendrocyte, which synthesizes the sterol to build up the myelin sheath of axons and the biomembranes of neuronal and glial cells alike. A particularly important function of glia-derived cholesterol complexed to apolipoprotein E (ApoE)-containing lipoproteins is brain synaptogenesis [[34], [35], [36], [37]].
3. Cholesterol-rich lipid domains in viral infection
In mixed artificial membrane-mimetic systems, the presence of cholesterol generates the separation and coexistence of liquid-liquid phases, one the liquid-ordered phase (Lo), rich in the sterol and saturated lipids, and the other a liquid-disordered (Ld) phase, enriched in unsaturated acyl chain lipids. These physicochemical characteristics are at the root of the concept of “lipid rafts” [[23], [24], [25]]. According to this concept, lateral membrane heterogeneities like those observed in model systems also occur in biological membranes at sites enriched in cholesterol intermixed with sphingolipids and glycerophospholipids with saturated acyl chains. These specialized lipid domains would exhibit the physico-chemical properties of liquid-ordered (Lo) gels [27]. The functional relevance of these lipid domains is that they serve as signal transduction platforms for a wide palette of proteins involved in various cellular phenomena, and their participation at various stages of the viral infection process at the plasma membrane and at later stages of the virus cycle inside the cell are beginning to be delineated. The lipid raft concept has had an important influence on the field of viral infection, to the point that virus formation has been considered “unlikely to occur from a single raft (quoting the 10-50 nm raft size of ref. [38]) and raft-dependent viruses have to organize their membrane by recruitment or coalescence of pre-existing small rafts into larger microdomains or by de novo assembly of a microdomain”, with suggestions that this may occur at the plasma membrane of the host cell [39].
3.1. Lipid domains in the virus envelope membrane
The lipidomic profile of enveloped virus is acquired at late stages of the virus cycle and not by physical coalescence of membrane patches during the virus-host cell fusion process. The Pestivirus bovine viral diarrhoea virus is among the few viruses whose lipidome has been characterized; its envelope is rich in sphingomyelin, cholesterol and hexosyl-ceramide and although the virus buds from the ER, its lipid composition differs from that of a typical ER membrane, indicating that there is lipid sorting during virion formation [40], further evidence against the coalescence hypothesis.
Another well characterized lipidomic profile is that of the human immunodeficiency virus type 1 (HIV-1), a retrovirus. Brügger and coworkers [41] resorted to mass spectrometry to quantitatively analyze the lipid composition of the viruses budding from the infected cells and of the host cell membranes. They found that inhibition of sphingolipid biosynthesis of the host cell resulted in diminished viral infectivity concomitant with enrichment in an unusual sphingolipid, dihydrosphingomyelin, thus disclosing a critical role of this lipid class in the HIV-1 morphogenesis and infectivity. Furthermore, the lipid composition of the HIV-1 resembles that of detergent-resistant membrane microdomains of the host cell, which was interpreted as strong evidence for the occurrence of lipid rafts in living cells. Chan and coworkers [42] also employed mass spectrometry to study the lipidome of HIV-1 and murine leukaemia virus. They found that the overall lipid content matched that of the plasmalemma, although some lipid classes were enriched in comparison to those of the host membrane: cholesterol, ceramides, the ganglioside GM3, and phosphoinositides. PI(4,5)P2 was enriched in HIV-1, and its depletion reduced virion budding. The conclusion was drawn that HIV-1 and other retroviruses bud from cholesterol-rich regions of the plasmalemma and exploit matrix/ PI(4,5)P2 interactions in this process [42].
The S glycoprotein from SARS-CoV contains cysteine-rich amino acids forming clusters at its carboxy-term cytoplasmic domain; cluster 1 is the closest to the short TM domain of the S protein [43]. Mutagenesis of this and adjacent cluster 2 reduces S protein-mediated cell fusion and cell-surface expression. Envelope membrane-proximal cysteines in clusters 1 and 2 are palmitoylated in the wild-type SARS-CoV [43]. This post-translational lipidation can be either a dynamic, transient modification or a stable one. The latter seems to be the case for SARS-CoV, and palmitoylations appears to occur in the pre-medial Golgi compartment in infected HEK293T cells [44]. These authors also found that palmitoylation is necessary for partitioning SARS-CoV S into Triton X-100 detergent-resistant membranes. A more recent study using state-of-the-art fluorescence microscopy and other techniques found that SARS-CoV-2 S glycoprotein does not partition into liquid-ordered domains in giant unilamellar vesicles (GUVs), used as a model counterpart of the plasma membrane [45]. These authors concluded that SARS-CoV-2 infection depends on the presence of cholesterol in the virus membrane envelope and not on the participation of cholesterol-enriched domains in the host cell. In the words of the authors: a raft-independent yet cholesterol-dependent mechanism. It is clear from these studies on various viruses that cholesterol present in the surface membrane bilayer of enveloped virus plays a critical role in its infectivity.
Methyl-β-cyclodextrin (CDx) is a cyclic oligosaccharide formed by glucose subunits linked by glycosidic bonds. The central hydrophobic cavity of the cyclodextrin macrocyclic ring accommodates a great variety of compounds, including sterols, and CDx has been extensively employed to man-tailor cholesterol levels in experimental studies. CDx-mediated cholesterol removal from human immunodeficiency virus 1 (HIV-1) particles reduced viral infectivity [46]. Replenishment of cholesterol, also via CDx preloaded with the sterol, or addition of i) the cholesterol structural analogue cholestenone or ii) exogenous sphingomyelin (another characteristic ordered-lipid domain lipid normally occurring in the HIV-1 envelope) rescued the effects of cholesterol depletion. A subsequent study undertook similar experiments on HIV-1 and simian immunodeficiency virus (SIV), showing that cholesterol depletion by CDx treatment of purified viruses produced patches devoid of membrane bilayer, releasing envelope glycoprotein (gp120) and to a lesser extent the internal capsid protein (p28) and viral RNA content. Cholesterol depletion abrogated infectivity [47]. However, some of the control electron micrographs also showed bilayer-less areas and no other experimental evidence was provided to attest the occurrence of ordered-lipid domains. Cholesterol reduction of the canine CoV virions by CDx treatment is dose-dependent, and at a concentration of 9 mM CDx infectivity was reduced by 73% [16]. In the case of influenza virus, cholesterol interacts with hemagglutinin in the viral envelope to control the rate-limiting step of fusion [48].
3.2. Lipid domains in the plasma membrane of the host cell
The hypothesis that cholesterol-enriched domains at the plasma membrane of cells are a preferential target for enveloped (and to a lesser extent non-enveloped) viruses has been entertained for many years (see reviews by [6,12,13,49]). Probably one of the earliest reports indirectly pointing to the influence of host-cell cholesterol on viral infection is that of Campbell and coworkers, who found that dietary-induced hypercholesterolemia made mice highly susceptible to coxsackievirus B infection [50]. Fusion-resistant L-2 fibroblast mutants regained the ability to fuse with the CoV murine hepatitis virus (MHV) upon supplementation with cholesterol [51]. Fusogenic activity was also related to the fatty acid metabolism of these cells. These authors did not find MHV to be sensitive to hypercholesterolemia as reported by Campbell and coworkers for coxsackievirus B fusion susceptibility. Influenza A virus (IAV) is an enveloped virus belonging to the Orthomixoviridae family. The chemistry and target predilections of the IAV are relatively well understood: the virus hemagglutinin is recognized by a variety of sialylated glycoproteins and glycolipids [52,53] at the surface of e.g., epithelial cells in the mucosae that line the upper respiratory tract. Cholesterol co-clusters with sialic acid-containing glycolipids in the host plasma membrane, augmenting IAV binding affinity; binding exhibits a cooperative dependence on the concentration of receptors. Using single-molecule superresolution microscopy, these authors also show that this entails the formation of nanoclusters of glycosphingolipid receptors [52]. However, as discussed in the section above on lipid domains in the virus envelope, other authors find that it is the cholesterol content in the membrane bilayer of the IAV proper that matters, and not the sterol in the host cell membrane [48].
In the case of the CoVs, the role of cholesterol in plasma membranes has been centred around two related areas: i) the role of cholesterol-rich lipid domains at the plasmalemma as a platform for virus entry and ii) the concentration of the cell-surface metalloprotease, the angiotensin converting enzyme 2 (ACE2), considered to be the canonical receptor for SARS-CoV-2 and SARS-CoV viruses, at these cholesterol-enriched lipid domains. The two issues have been, and remain, matters of controversy. Binding of SARS-CoV-2 to ACE2 is mediated by the receptor-binding domain (RBD) harboured in the S1 subunit of the S spike protein, close to the N-term region [54]. The RBD-ACE2 recognition process is seconded by auxiliary co-receptors like the transmembrane (TM) protease serine 2 precursor (TMPRSS2), furin, or cathepsin B or L, depending on the target host-cell type [55]. Antibodies directed against ACE2 provide only partial suppression of the binding process, whereas some monoclonal antibodies generated in convalescent COVID-19 patients against the N-term domain of the S1 protein neutralize this process but do not bind the RBD [56,57], raising the possibility that the virus could engage other host-cell receptors or co-receptors for cell entry and/or involve alternative mechanistic steps. For instance, acidic glycolipids or glycosphingolipids [52] co-clustered with ACE2 could act as co-receptors, thus explaining the partial suppression of binding by anti-ACE2 antibodies.
Recent data using genome-wide CRISPR (clustered regularly interspaced short palindromic repeats)-based studies have found that several cell lines that are susceptible to SARS-CoV-2 infection have very low ACE2 expression levels, suggesting the possibility of alternative ACE2-independent entry routes [58]. Experimental evidence of an ACE2-independent pathway has recently been reported to operate in H522 human lung cells, which can be infected by the SARS-CoV-2 variant E484 (a spike S protein mutant) even in the absence of ACE2 expression [59]. Infection requires heparan sulphates at the cell surface and involves clathrin-mediated endocytosis. Single-cell transcriptomics has shown that SARS-CoV-2 viral RNA is found in various types of immune cells, including myeloid cells with phagocytic activity (neutrophils and macrophages) and lymphocytes without such phagocytic activity (T, B, and NK cells). Remarkably, SARS-CoV-2 RNA-positive immune cells do not co-express the canonical ACE2 and TMPRSS2 receptor/co-receptor, or other hypothetical entry co-factors [60]. T lymphocytes from COVID-19 patients have been found to contain SARS-CoV-2 RNA and viral antigen, and SARS-CoV-2 induces marked lymphopenia, suggesting that these cells are targets of the coronavirus. Using ACE2 knockdown or receptor blocking experiments, Shen and coworkers recently found that SARS-CoV-2 infection of T lymphocytes is spike-ACE2/TMPRSS2-independent [61]. Another ACE2-independent mechanism of SARS-CoV-2 internalisation has recently been reported by Liu and coworkers, who showed that upon initial contact with ACE2 in cells of the respiratory tract, cell invasion by SARS-CoV-2 proceed via the ubiquitous cell-surface integrin α5β2 [62].
The main argument in support of the participation of cholesterol-rich lipid domains in the initial binding/recognition step is based on the hypothesis that ACE2 or similar cell-surface receptors reside in raft lipid domains that purportedly serve as platforms for viral recognition and entry. Evidence in support of this notion is available for several viruses. One of the human common-cold etiological agents, the coronavirus HCoV-229E, sediments in Triton X-100-resistant membrane fractions isolated from infected human fibroblasts, where the virus is recognized by its receptor, alanine aminopeptidase N, also termed CD13 [9]. Similar experimental approaches were used to arrive to analogous conclusions in the case of SARS-CoV [13]. Another experimental tool frequently used in attempting to demonstrate the lipid domain-dependence of viral infection is acute CDx-mediated cholesterol depletion from the plasma membrane. This procedure reduces porcine rotavirus infection by 99% [63] and infection by canine CoV by up to 68% [16]. Likewise, acute CDx-mediated cholesterol reduction decreases by ca. 50% the binding of the SARS-CoV S protein to ACE2-expressing hamster kidney cells and the entry of the virus into these cells [64]. A reduction of ~90% in the infectivity of Vero E6 cells by pseudo-typed SARS-CoV has also been reported upon CDx acute treatment [13]. However, several authors have specifically investigated whether ACE2 is localized in cholesterol-rich lipid domains and found no support for this claim using CHO cells transiently expressing ACE2 [65] or Vero E6 cells endogenously expressing ACE2 [12]. Interestingly, in the study of Li and coworkers, cholesterol depletion inhibited the release of SARS-CoV-2 particles from the infected cells, and cholesterol supplementation restored virion production, suggesting that cholesterol was implicated in stages of the virus cycle other than initial binding to ACE2 or subsequent viral entry.
Invasion of the brain parenchyma by SARS-CoV-2 virions via the general circulation involves the crossing of the virions through the blood-brain barrier, i.e., traversing the plasma membranes of endothelial cells, pericytes and smooth muscle cells [5]. A recent work employed fluorescently labeled spike protein to follow in vitro the possible pathways employed by the virus to engage with these cell types. Uptake was mediated by ACE2, requiring interaction with ganglioside GM1 in lipid rafts [66], as suggested by the inhibition of S protein uptake by anti-ACE2 or anti-GM1 antibodies.
3.3. Lipid domains in endosomal and other intracellular membranes
Cholesterol is ubiquitously present in cellular membranes, and although most of the neutral lipid occurs at the plasma membrane, the intracellular membrane pool also affects other steps of the viral life cycle, as exemplified by HIV. One of the natural target cells of HIV is the macrophage. Using acute cyclodextrin-mediated depletion, statin-mediated chronic metabolic inhibition (lovastatin), or cholesterol-drug complex formation with the antifungal drug filipin (or the antibiotic nystatin), HIV virion release from macrophages was significantly inhibited [15]. Virion particles co-localized with fluorescent cholera toxin B, a marker of liquid-ordered lipid domains. These results implicate cholesterol-rich domains in the ERGIC membranes involved in virus packing and exit from the cell.
Using a recently developed single-virus fluorescence assay, influenza A virus binding to target membrane vesicles was shown to augment the fraction of influenza virus fusing with the endosomal membrane as the cholesterol content of the endosomes increased [67]. These results are consistent with previous findings from the same authors showing that endosomal cholesterol stabilizes the formation of tight pores through which the influenza A virus can transfer its genome into the target cell as the endosome matures and lowers its pH, thus enabling the viral envelope hemagglutinin to insert its hydrophobic fusion peptide into the endosome [68].
4. Cholesterol-recognition motifs in the SARS-CoV-2 spike S glycoprotein
Cell-surface receptors for neurotransmitters have been instrumental to the discovery of linear amino acid sequences with the capacity to recognize the neutral lipid cholesterol. Studies of the benzodiazepine receptor led to the identification of the sequence (L/V)-X−5-(Y)-X−5-(K/R), currently referred to as CRAC (Cholesterol Recognition/interaction Amino acid Consensus sequence) [69,70]. Working on the nicotinic acetylcholine receptor protein we identified the inverse or “mirror” image of CRAC, which we coined CARC ((K/R)-X1−5-(Y/F/W)-X1−5-(L/V)) [71]. In subsequent work we found that these two cholesterol consensus motifs are present in a great variety of membrane proteins [72], including the superfamily of ligand-gated ion-channel (LGIC) proteins [73] and other channels like the transient receptor potential (TRP) channel [74]. Other groups searched for cholesterol motifs in cell-surface receptors, including the large superfamily of G/protein coupled receptor (GPCR) proteins [[75], [76], [77]]. CRAC/CARC sequences share a central aromatic residue, like tyrosine for CRAC, and tyrosine, phenylalanine, or tryptophan for CARC, flanked on both sides by one to five amino acid residues ending in a basic (arginine or lysine) and an apolar terminus (valine or leucine). Both CARC and CRAC are vectorial motifs, with the CARC motif preferentially, albeit not exclusively, located in the exofacial membrane leaflet, whereas CRAC most often occurs in the cytoplasmic-facing leaflet of the plasma membrane [72], a condition met by single-passage membrane proteins with exofacial N-term and cytoplasmic-facing C-term.
The presence of cholesterol-recognition motifs has recently been reported in the primary sequence of SARS-CoV-2 spike S homotrimeric glycoprotein [78] that interacts with the host cell-surface canonical receptor ACE2 via its receptor binding domain (RBD) [79]. The ACE2 metalloprotease is bound to the host-cell membrane and protrudes into the extracellular space, and the RBD-ACE2 binding reaction occurs in this compartment, outside the bilayer. In contrast, the S2 subunit crosses the viral envelope lipid bilayer, laying its three-footed base inside the host cell. The S2 transmembrane domain consists of three portions: a juxtamembrane aromatic section, a central hydrophobic section, and a cysteine-rich section [80]. The transmembrane domain is highly conserved in the three highly pathogenic members of the heptad human CoVs, SARS-CoV, MERS-CoV, and SARS-CoV-2 [55], all sharing the same cholesterol-recognition motif topography, with CARC deeper in the aromatic amino acid-rich part of the transmembrane domain and CRAC laying immediately adjacent to it, partly embedded in the juxtamembrane portion, in the so-called tail-to-tail disposition [81].
As its name indicates, another portion of the S2 subunit, the so-called fusion peptide, is involved in the fusion of the virus and host-cell membranes during the infection process. The fusion peptide possesses the same cholesterol-recognition motif in SARS-CoV and SARS-CoV-2, but not in MERS-CoV. The pathophysiological implications of this difference are not known.
5. Cholesterol in the virus-host cell membrane fusion step
In the case of enveloped viruses, release of their genome into the host-cell requires the fusion of their membrane bilayer to either the plasma membrane or the endocytic membrane of the target cell. Despite the high resolution information made available in recent months on the structure of the SARS-CoV-2 spike glycoprotein (see review in [79]), we still lack atomic resolution data on the section of the viral machinery involved in the fusion process, the so-called fusion peptide, a polypeptide region contained in its S2 subunit [82]. Molecular dynamics simulations have attempted to fill this gap [[82], [83], [84], [85], [86], [87], [88], [89]].
One of the clinical features and indication of severity of COVID-19 is the so-called hyperreactive cytokine release syndrome or cytokine storm (see reviews in [5,90,91]). The pathognomonic cytokine and interferon profile elicited by SARS-CoV-2 virosis involves the recruitment of interferon-stimulated genes [92]. One such gene, CH25H, codes for cholesterol 25-hydroxylase, the enzyme that catalyzes the oxidation of cholesterol to 25-hydroxycholesterol [93], a metabolite that represses cholesterol biosynthesis, is an intermediate in the pathway to the GPRC GPR183/EB12 (a chemotactic receptor for lymphoid cells), and also participates in another cholesterol metabolic pathway, acting as a potent suppressor of the sterol regulatory element-binding protein (SREBP) pathway [94] (see Fig. 1 in section below). The oxysterol 25-hydroxycholesterol has been found to exert a broad anti-viral [92] and anti-CoV action [95]. The latter authors disclosed a mechanism involved in SARS-CoV-2 inhibition of fusion at the cell surface via activation of the ER-resident acyl-CoA:cholesterol acyltransferase (ACAT) enzyme, involving the esterification and metabolic depletion of accessible cholesterol from the plasma membrane. The 25-hydroxycholesterol-induced mobilization of cholesterol from the plasma membrane to the ER had been previously found to be dissociable from the ACAT inhibition by 25-hydroxycholesterol [94]. Experimentally, 25-hydroxycholesterol was found to inhibit SARS-CoV-2 pseudovirus infection of lung and colorectal epithelial cell lines and human lung organoids [95]. 7α-hydroxycholesterol, structurally similar to 25-hydroxycholesterol, did not exert anti-SARS-CoV-2 activity via such mechanism [95].
Fig. 1.

Endogenous cholesterol synthesis and monitoring of cellular cholesterol levels. When the amount of accessible cholesterol in the plasma membrane exceeds a threshold level, the excess cholesterol is moved to the endoplasmic reticulum (ER) via a non-vesicular mechanism mediated by the Aster/GRAMD1 family of sterol transporters. The sterol regulatory element-binding protein 2 (SREBP2) is the main regulator of the cholesterol biosynthesis. SREBP2 is proteolytically activated at the Golgi, releasing the N-term transcription factor domain that translocates to the nucleus. Upon migration into the nucleus (long purple arrow), SREBP2 transactivates target genes via the binding of the sterol regulatory elements (SRE) to the gene promoters. In the ER, cholesterol regulates the SREBP2 pathway and undergoes esterification. SCAP and INSIG are sterol-sensing proteins that monitor cholesterol and oxysterol levels in the ER, respectively. When INSIG binds SCAP, the complex retains SREBP2 in the ER. If the SREBP-activating protein (SCAP) is not bound to INSIG, the SCAP-SREBP2 is exported to the Golgi complex (long red arrow). Fluctuations of <5% in cholesterol levels in the ER suffice to trigger SREBBP2 cleavage (short red arrow). Phosphatidylserine is a serine phospholipid present in the inner leaflet of the plasma membrane that is also recognized by the GRAM domain of Aster/GRAMD1 proteins and modulates cholesterol transport from the PM to the ER.
The work of Wang and coworkers also has wider implications on the entry mechanism of SARS-CoV-2 into host cells. To this end Calu-3 and Vero E6 cells were infected with SARS-CoV-2 in the presence of inhibitors of TMPRSS2 and cathepsin B/L and compared with DMSO-treated control cells. Camostat mesylate, a TMPRSS2 inhibitor [96,97], efficiently abrogated viral entry into Calu-3 cells but not into Vero cells, while the cathepsin B/L inhibitor E-64d blocked entry into Vero cells but not Calu-3 infection, confirming the original experiments of Hoffmann and coworkers on TMPRSS2 being required for SARS-CoV-2 entry into Calu-3 cells, and more generally, that the priming of S protein viral fusion is mediated by TMPRSS2 on the plasma membrane and not by the pH-dependent cleavage by the endosomal cysteine protease cathepsin B/L once internalized [54]. The pH-dependent fusion reaction with cholesterol-rich endosomal membranes is a mechanism of viral penetration widely employed by viruses other than CoVs, e.g. the Togaviridae Semliki Forest virus and Sindbis virus, the vesicular stomatitis virus, or murine leukemia virus (reviewed in [6]). In the case of SARS-CoV-2, during the initial months of the COVID-19 pandemic the early variants favored entry activation at the plasma membrane mediated by TMPRSS2, but several most recent studies indicate that the Omicron (B.1.1.529) variant switches to the endosomal route of entry mediated by catepsin B/L [[98], [99], [100]].
ACE2 of both Vero E6 and Caco-2 cells distribute in detergent-resistant membranes [64] and SARS-CoV-2 entry requires cholesterol. Tang et al. [82] proposed that rather than simply organizing ACE2 and SARS-CoV-2 proteins in lipid rafts, cholesterol directly affects membrane fusion dynamics, guaranteeing the fusion intermediate formation. Nardacci et al. [101] reported that accumulation of lipids in SARS-CoV-2 infected cells in vitro and in the lungs of patients suggested that lipids can be involved in SARS-CoV-2 pathogenesis. Cholesterol depletion of cell membranes with methyl-β-cyclodextrin reduced SARS-CoV-2 infection in ACE2-expressing HEK293T cells, highlighting the importance of cholesterol for SARS-CoV-2 infection [102]. More recently, Sanders et al. [45] showed that treatment of SARS-CoV-2 with methyl-β-cyclodextrin prior to exposure of cells to SARS-CoV-2 blocked virus infection, pointing out that the cholesterol content of SARS-CoV-2 viral particles is critical to infectivity. Lai and Freed [84] described that although SARS-CoV-2 fusion peptide and SARS-CoV fusion peptide have ~93% identity, the former induces greater membrane ordering than SARS-CoV fusion peptide, possibly due to its greater hydrophobicity. These authors concluded that this effect could explain, at least in part, why SARS-CoV-2 is better able to infect host cells [84].
6. Cholesterol homeostasis and cholesterol-recognition proteins involved in viral infection
Interconnected metabolic pathways intervene in cholesterol homeostasis and its regulation. Several proteins, including the SREBPs, SREBP cleavage-activating protein (SCAP), HMGCR, and insulin-induced genes (INSIGs) play important roles in this complex protein network that topographically includes the plasma membrane, the ER, the Golgi complex, the ERGIC, and the nucleus, as schematically depicted in Fig. 1. The ER is the central hub of this network, and the SREBP pathway a key cholesterol-level monitoring system. In this section we will dissect the main constitutive elements of the network and analyze their recently suggested participation in SARS-CoV-2 viral infection.
6.1. Sterol regulatory element binding proteins (SREBPs) and SREBP-cleavage activating protein (SCAP)
Key to the intertwined network of cholesterol metabolic pathways is the SREBP pathway. SREBPs are a family of membrane-bound transcription factors involved in the biosynthesis of cholesterol and other lipids. SREBPs stimulate the transcription of more than 30 sterol-regulated genes and genes involved in fatty acid biosynthesis, with the participation of the mitogen-activated protein kinase (MAPK) signalling pathway. SREBPs are in turned controlled by multiple mechanisms involving proteolytic activation, mRNA synthesis, and transcriptional activity (reviewed in [103]). Three SREBP proteins are encoded by two different genes. The Srebf1 gene gives rise, via alternative transcriptional start sites, to SREBP-1a and SREBP-1c, using alternate promoters that yield transcripts in which different first exons are spliced to a common second exon. SREBP-1 was cloned in 1993 by Brown, Goldstein and coworkers, who showed that this leucine-zipper protein binds the sterol regulatory element 1 (SRE-1), a conditional enhancer of the promoters for the LDL-R gene. SREBP-1 activates transcription in sterol-depleted cells and is silenced by sterols [104]. Canonical SREBP1c signalling triggers expression of fatty acid biosynthesis. Sterol regulatory element binding protein-2 (SREBP-2), or sterol regulatory element binding transcription factor 2 (SREBF2), is encoded by a different gene, Srebf2, and the second member of the family that also binds SRE-1 [105]. SREBPs are predominantly involved in the transactivation of genes involved in cholesterol biosynthesis, lipoprotein import, and intracellular lipid trafficking [106]. SREBPs are synthesized as precursor proteins in the ER, where they form a complex with SREBP cleavage activating protein (SCAP), an ER-resident protein with a sterol-sensing domain that detects cholesterol levels (Fig. 1). SCAP is a key regulator of cholesterol and fatty acid biosynthesis (see comprehensive review in [107]).
When sterol concentration is sufficient to sustain cellular homeostasis, it accumulates in the ER and binds to SCAP, inducing a conformational transition that leads to the interaction of this protein with either one of two ER membrane retention proteins, INSIG-1 or INSIG-2 that reduce in turn transport of the SCAP-SREBP to the nucleus [108]. SCAP confines SREBPs to the ER (see Fig. 1). The NH-term of SCAP, containing transmembrane segments 2-6 (the so-called INSIG-binding domain), is totally embedded in the membrane and harbours the sterol-sensing domain. Mutations in this domain abrogate its sensitivity to sterols and inhibit the interaction of SCAP with INSIG [109]. If sterol concentration is reduced below a threshold level, the SCAP-SREBP complex migrates to the Golgi complex, where SREBPs reach their mature form by proteolytically releasing their amino-terminal basic helix-loop-helix domain, i.e. their transcriptionally active forms from the membrane. The biologically active transcription factors translocate to the nucleus, becoming proper nuclear SRBPs, and bind sterol response elements (see Fig. 1 and reviews in [107,110]) that activate the full set of genes involved in cholesterol synthesis and uptake from LDL. In the case of cholesterol biosynthesis, this implies activating transcription of the key reductase HMGCR, the rate-limiting enzyme in the mevalonate-cholesterol biosynthetic pathway.
As mentioned above, cleavage of the SCAP-SRBP-1 complex is precluded by high cholesterol levels. Viral infection by human CMV overrides this inhibition and increased cleavage of the complex, the translocation of SREBPs to the nucleus, and the activation of genes involved in lipogenesis [111] (Fig. 1), a requisite for CMV growth in cells [112].
Pathogens, including viruses, can activate SREBP-2. Epstein-Barr virus induces its expression (but not that of SRBP-1), but, somewhat unexpectedly, instead of synthesizing cholesterol, the mevalonate-cholesterol pathway is geared towards the production of geranylgeranyl pyrophosphate that contributes to the outgrowth of newly infected B-cells and downstream activation of the small Ras superfamily G-protein Rab13 [113]. In the mevalonate-cholesterol pathway, acetyl-CoA can be reduced by HMG-CoA-reductase to produce mevalonate and converted into farnesyl pyrophosphate, which can either be used to synthesize squalene for cholesterol biosynthesis or be diverted toward GGPP synthesis, the route induced by the Epstein-Barr virus.
In the context of COVID-19, SREBPs have been recently identified as targets of AM580 using a lipidomic screening, a retinoid derivative and retinoic acid receptor-α agonist having anti-inflammatory and neuroprotective effect [114] (see Fig. 3 below). In addition to these effects, AM580 displays a potent and broad-spectrum antiviral activity against MERS-CoV, as well as IAV, in vitro and in vivo [115]. SREBP-2 regulates the production of interleukin-1β and tumour necrosis factor α and through these effector molecules is directly related to one of the complications of COVID-19, the hyperreactive cytokine syndrome, involving the disruption of cholesterol biosynthesis [116]. Different CoVs produce different hyperreactive cytokine responses [5]. Transcription of the sestrin-1 gene (SESN1), also known as p-53 regulated protein PA26, which mediates p53 inhibition of cell growth, is regulated by SRBP-2; SESN1 is a causal gene associated with plasma cholesterol levels [117]. Conversely, sestrin-1 inhibits SRBP-2-induced cholesterol biosynthesis [117], thus linking cytokine hyperreactive syndrome to SREBP-2-induced cholesterol biosynthesis. Expression of Sestrin-1 and proprotein convertase subtilisin/kexin type 9 (PCSK9), two regulators of cholesterol synthesis, together with that of SREBP-2, correlate with the severity of COVID-19, but mRNA levels of HMG-CoA-reductase, which acts upstream of cholesterol synthesis, as well as those of the LDL-R did not vary with disease severity. Cytokine production in COVID-19 induces active nuclear translocation of hydroxycholesterol from the ER, and further secretion of IL-6, IL-8, and M-CSF cytokines.
Fig. 3.

Increased susceptibility to SARS-CoV-2 viral infection upon expression of scavenger receptor class B type 1 (SR-B1) and rerouting of SREBP pathway to cytokine production. SR-B1 is one of the cell-surface proteins acting as HDL receptor and mainly involved in the uptake of cholesteryl esters (CE). According to recent views, SR-B1 expression facilitates SARS-CoV-2 adhesion to the host-cell membrane via an indirect mechanism whereby SARS-CoV-2 virus (2) binds to the HDL-CE complexes (1) forming HDL-CE-virus supramolecular assemblies. The HDL-CE complexes are recognized by SR-B1 (4) while the virus cargo is recognized by its cognate receptor, ACE2 (step 5) [78]. Under normal conditions, SREBP2 activity increases cholesterol biosynthesis; in SARS-CoV-2 infection the ER-resident protein is translocated to the nucleus (6) and increases the production of pro-inflammatory cytokines and chemokines (7). AM580 (chemical formula shown in top-right) is a stable benzoic derivative of retinoic acid. The drug targets SRBP2.
6.2. Aster/GRAMD1 proteins and phosphatidylserine, a cholesterol partner in viral infection
The Aster/GRAMD1 proteins constitute a family of evolutionarily conserved ER-resident sterol transporter proteins that mediate non-vesicular cholesterol transport from the plasma membrane to the ER. Sandhu and coworkers described the Aster A, B and C cholesterol-binding proteins [118]. The central domain of Aster-A is similar to the sterol-binding fold of mammalian StARD proteins, whereas the Aster N-term GRAM domain, exposed to the cytosol, binds phosphatidylserine and is responsible for Aster recruitment to plasma membrane-ER contact sites when cholesterol accumulates in the plasma membrane. GRAMD1 proteins are required for instance for delivery of HDL-cholesterol to the adrenal cortex, where the biosynthesis of steroid hormones proceeds from their precursor, cholesterol [119]. Under lipid-poor conditions GRAMD1 localizes at the ER membrane; in response to excess cholesterol in the plasma membrane, GRAMD1 is recruited to the ER-plasma membrane contact sites (EPCS) via its GRAM domain (Fig. 1). At the EPCS, the sterol-binding VASt/ASTER domain of GRAM binds to cholesterol at the plasma membrane (Fig. 1) and catalyses its transfer from the cell surface to the ER. GRAMD1 proteins were shown to possess synergistic but distinct sites that can sense free cholesterol and anionic lipids in close vicinity in the plasma membrane, a topographical relationship that is also regulated by sphingomyelins [120]. The crystal structure of AsterA (GramD1a) with bound cholesterol molecules is shown in Fig. 2 .
Fig. 2.
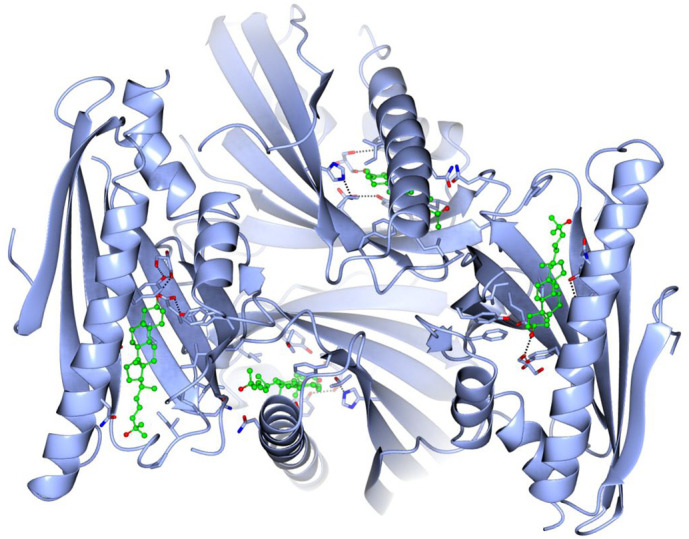
Structure of mouse AsterA (GramD1a) with four bound 25-hydroxy-cholesterol molecules (PDB accession number 6GQF). The crystal structure of the protein (blue ribbon) and sterol molecules (green ball-and-stick rendering, oxygen in red) was obtained by X-ray crystallography at 2.9 Å resolution [118]. Molecular coordinates were downloaded from the PDB data bank and structures produced using the CCP4MG Molecular Graphics Program of the University of York, U.K. [121].
Phosphatidylserine has been shown to modulate the transport of LDL–derived cholesterol from lysosomes to the ER [122] and play a role in the localization of cholesterol in the cell [120,123]. When the plasma membrane cholesterol exceeds a certain threshold, excess cholesterol moves to the ER in a phosphatidylserine-dependent manner [122]. In a homeostatic feedback mechanism, the surplus cholesterol in the ER reduces external cholesterol uptake, preventing further cholesterol accumulation. It becomes apparent that the complex regulation of cholesterol metabolism is intimately related to the fate of other lipid components in the cell, with a close functional and topographical relationship between cholesterol and anionic phosphoglycerides, particularly phosphatidylserine.
6.3. Transmembrane protein 41B (TMEM41B)
The transmembrane protein 41B (TMEM41B) is best known for its participation at early stages of autophagosome biogenesis from ER membranes, in which process TMEM41B mobilizes neutral lipids from lipid droplets. In the context of viral infection, TMEM41B plays a key additional role: it is an absolute requirement to infect the host cell for all members of the Flaviridae family of viruses, which includes the mosquito- and tick-borne human pathogenic yellow fever virus, the Zika virus and the West Nile virus [124]. The mechanism does not involve autophagosome formation; TMEM41B apparently lowers the free-energy required for induction of membrane curvature [124] at the virus-induced membrane complex known as replication-transcription complexes. These complexes are employed by Flaviviruses like the Zika virus [125], and CoVs like MHV [126], SARS-CoV [127], and SARS-CoV-2 [128,129]. Replication complexes can be envisaged as a virus-induced organelle derived from the ER-Golgi complex (ERGIC) characteristically enriched in 200-300 nm double-membrane vesicles (DMVs) [79]. The liquid-ordered domain lipid sphingomyelin is essential for the formation of the DMVs required for the replication of hepatitis C virus and other +-stranded RNA viruses [130]. A recently conducted search using genome-wide CRISPR genetic knockout screens showed that SARS-CoV-2 and three other seasonal Coronaviridae viruses (HCoV-OC43, HCoV-NL63, and HCoV-229E) require TMEM41B for infection and replication [131].
TMEMB1 and VMP1 have recently been found to play a role in the distribution of free cholesterol and phosphatidylserine [132]. In the absence of these two proteins, more free cholesterol becomes available on the cytoplasmic-facing leaflet of the plasma membrane, and an abnormal increase in cell-surface cholesterol content may ensue. Moreover, TMEM41B and VMP1 have phospholipid scramblase enzymatic activity, thus contributing to the maintenance of phospholipid distribution in the cell [132]. Scramblases possess linear cholesterol-recognition motifs [133].
6.4. Scavenger receptor class B type 1 (SR-B1) and SARS-CoV-2 infection
Ubiquitously located at the surface of a great variety of cells, scavenger receptor class B type 1 (SR-B1) is a multifunctional membrane-bound protein mainly expressed in liver and one of the heavy-density lipoprotein (HDL) receptors (see review in [19]). SR-B1 is considered to be a major player in inflammation and atherosclerosis, and as such to be an important modifiable risk factor in hypercholesterolemia, coronary heart disease, hypertension, obesity, and stroke [134]. SR-B1 participates in the selective transport and regulation of cholesteryl esters and other lipids, but it possesses the capacity to bind many other ligands, including proteins, proteoglycans, phospholipids, and carbohydrates, and intervenes in immune surveillance, pathogen infection, and cancer [135].
In terms of sterol transport proper, SR-B1 mediates the selective uptake of HDL-derived cholesteryl esters into the cell and facilitates the efflux of cholesterol from peripheral tissues. Unlike the mechanism of cholesterol uptake mediated by LDL-Rs, which involves the internalization of the entire LDL-cholesterol complex, HDL-cholesterol particles bind to SR-B1 and cholesteryl esters are delivered into the cell without the carrier lipoprotein. The free cholesteryl esters are subsequently hydrolysed by a hormone-sensitive lipase, a mechanism utilized by steroidogenic cells that synthesize their hormones from the cholesterol precursor.
SR-B1 serves as cell-surface receptor for the Flavivirus HCV, together with tetraspanin CD81 and claudin-1 and occludin (two tight junction proteins) [136,137]. Upon recognition by the extracellular domain of SR-B1, the ensuing internalization of the virus proceeds via clathrin-mediated endocytosis and pH-dependent fusion with endosomal membranes [137]. SR-B1 can also interact with virus-associated lipoproteins [19].
The work of Wei and coworkers demonstrates that expression of SR-B1 confers susceptibility to SARS-CoV-2 infection, facilitating the attachment and entry of the virus. Since SR-B1 only enhanced viral uptake in the presence of ACE2, the authors interpreted these last results as an indication that SR-B1 is an entry cofactor of SARS-CoV-2 [78]. Inhibitors of SR-B1 or silencing of its expression abrogates SARS-CoV-2 infection. They further showed that in the presence of HDL viral infection increases significantly. Interestingly, blocking the cholesterol/HDL binding site of SR-B1 strongly inhibited SAR-CoV-2 infectivity. Wei et al. hypothesize that SARS-CoV-2 first binds to HDL, and the complex is then recognized by SR-B1 as an entry cofactor, thus aiding its concentration in host-cell membrane regions where ACE2 is expressed and facilitating encounter with the receptor [116] (Fig. 3 ).
7. Other lipids and host cofactors intervening in the intracellular cycle and exit of SARS-CoV-2
Another genome-wide CRISPR genetic screening of SARS-CoV-2 and HCoV-229E disclosed further associations between viral infection of these two CoVs and virus-specific and/or shared host-cell factors, including the genes coding for the autophagy regulator TMEM41B and the enzyme phosphatidyl-inositol-kinase type 3 (PI3K) [58]. The kinase, which catalyses the biosynthesis of the important inositol signalling molecule phosphatidyl-inositol-3-phosphate, serves as a host-factor for the common-cold CoVs HCoV-229E and HCoV-OC43, and to a lesser extent for SARS-CoV-2. In addition, these authors found that SARS-CoV-2 requires the lysosomal protein TMEM106B to infect human cell lines and primary pulmonary cells in culture. TMEM106B is an endosome/lysosome-resident protein that has gained attention because of its role in frontotemporal dementia, a form of pre-senile neurodegenerative neuropsychiatric disease [138]. TMEM106B was found to be the strongest dependency host factor for SARS-CoV-2 infection, whereas HCoV-229E infection requires lysosomal acidification, but is TMEM106B-independent. TMEM106B normally controls size, number, and trafficking of lysosomes by modulating acidification of this organelle through interaction with ATPase accessory protein 1, a vacuole proton pump. A TMEM106B knock-out murine model showed the downregulation of the proton pump levels and the dysregulation of lysosomal enzymes, with the concomitant normalization of frontotemporal dementia behavioural phenotypes and retinal degeneration [139].
The genes coding for a phosphatidylserine transporter, OSBPL9, the synthase intervening in the biosynthesis of this phospholipid, PTDSS1, and for the cofactor of phosphatidyl flippase, TMEM30A, were also identified in the screening study of Baggen and coworkers as acting as host cofactors for SARS-CoV-2 (Baggen et al., 2021), further reinforcing the view that phospholipid metabolism plays a role in the intracellular membrane-associated cycle of SARS-CoV-2. Furthermore, as reviewed in preceding sections, there is a close link between cholesterol plasma membrane levels and phosphatidylserine. Cells deficient in the gene coding for the phosphatidylserine synthase, PTDSS1, result in a phenotype in which LDL-cholesterol can leave the lysosome but fails to reach the ER, with a concomitant accumulation in the plasma membrane [122].
Budding of viruses from infected host cells has also been associated with cholesterol-rich membrane domains. HIV-1 virions generated by infected T-cell lines incorporate the glycosylphosphatidylinositol (GPI)-linked proteins Thy-1 and CD59, and the ganglioside GM1, all characteristic markers of liquid-ordered (raft) lipid domains, into their envelope membrane [140]. The fluorescent probe di-C(16) (1,1'-didodecyl-3,3,3',3'-tetramethylindocarbocyanine (DiI-C(16))), a marker of liquid-ordered domains, was found to co-localize with HIV-1, Thy-1, CD59 and GM1. Another concurrent report showed that HIV-1 infection alters lipid metabolism, shifting phospholipid synthesis to neutral lipid synthesis, a change that subserves the budding of HIV-1 virions with cholesterol-enriched envelopes stemming from analogously cholesterol-enriched membrane domains in the host cell. This study, however, relied once again entirely on the detergent-extraction criterion [8].
8. Clinical studies disclose prognostic value of SREBP2 in COVID-19
A recent retrospective analysis of 861 patients with mild, moderate, severe, or critical (the latter with respiratory failure, shock or organ failure) clinical presentations of COVID-19 evaluated the possible association of plasma cholesterol and HDL levels on morbidity and prognosis of the disease [141]. The levels of total cholesterol and HDL were found to be inversely correlated with severity, and severely ill patients with high HDL had a better prognosis. Survivors also had higher cholesterol and HDL levels. Furthermore, pre-treatment with ITX 5061, a potent antagonist of SR-B1 that increases HDL levels, competitively inhibited in vitro the SARS-CoV-2 infection of HEK-293T cells [141]. ITX-5061 and TX 7650 are two small organic molecules that inhibit HCV entry into cells. They do so by competing for HDL-mediated lipid transfer acting as a pharmacological antagonist of SR-B1 [142]. Recent studies confirm the lower-than-normal levels of plasma cholesterol, HDL and LDL in COVID-19 patients [143]. Reduced levels of total cholesterol, HDL-cholesterol, and LDL-cholesterol are observed in COVID-19 patients despite the increased expression levels of the SREBP-2 transcription factor for cholesterol and lipid biosynthesis in the plasma of these patients [116]. The inverse correlation and the fact that SREBP-2 also enhances the production of inflammatory cytokines like interleukin-1β and tumour necrosis factor α, led these authors to suggest the use of SREBP-2 levels in plasma as an indicator of COVID-19 severity and dysregulated vasculature. The alteration of the endothelial cells is at the centre of the multisystemic syndrome of COVID-19, and these cytokines and interleukins are characteristic noxa of the injured endothelium [5].
9. Therapeutic targeting of the lipidome in COVID-19
One can envisage two approaches to anti-viral therapy: interfering with the virus proper or addressing the host-cell, in the latter case either inhibiting the pathways employed by the virus or reinforcing the endogenous defense mechanisms. The use of statins in COVID-19 has been pondered in three contexts: i) in an attempt to inhibit SARS-CoV-2 entry and endocytic trafficking; ii) to mitigate the vascular compromise observed in the disease [5] and iii) to increase the therapeutic levels in COVID-19 patients with hyperlipidemia, history of congestive heart disease or myocardial infarction, frequent comorbidities and risk factors of worse outcomes of COVID-19.
9.1. Statins as inhibitors of SARS-CoV-2 entry and trafficking
In the case of both SARS-CoV-2 and SARS-CoV, S1 glycoprotein-ACE2 recognition/ binding is the first therapeutically addressable mechanistic step. Based on the purported location of ACE2 in cholesterol-enriched lipid domains, statin-mediated reduction of cholesterol levels in the plasma membrane has been attempted in several viral diseases, and, indeed, found to lower viral titres and, in some cases, arrest the internalization of viruses [7]. By the same token, cholesterol analogues have been proposed as small drugs to interfere with SARS-CoV-2 entry [144]. Lovastatin is reported to decrease viral load and increase CD4+ cell counts in models of acute infection and in chronically HIV-1 patients [145].The polyomavirus BK virus infection is abrogated by pravastatin [146]. Simvastatin (or CDx) inhibits poliovirus [147] or Epstein-Barr virus [113] infection; however, simvastatin-mediated abrogation of poliovirus infection appears not to be mediated by inhibition of cholesterol biosynthesis but rather is due to the production of vesicles that cannot sustain viral RNA transcription at a later stage of the virus cycle [147]. This and other studies reinforce the view that besides inhibiting HMG-CoA-reductase, statins have pleiotropic effects, upregulating LDL receptors, increasing the clearance of LDL-cholesterol, and acting as anti-inflammatory, anti-thrombotic and immunomodulatory drugs (see recent review in [148]). Even when they operate on the mevalonate pathway statins may exert effects other than inhibition of cholesterol synthesis, as exemplified by the effect of Lovastatin on HIV-1 infection: the mechanism impeding HIV-1 entry and budding involves prenylation of small G-proteins and downregulation of Rho GTPase activity [145]. Attempting to establish cause-effect links based exclusively on the main effect of statins on the rate-limiting enzymatic step of the mevalonate-cholesterol biosynthetic pathway is an oversimplification.
The notion that cholesterol is involved in virus trafficking and exit from the host cell in association with cholesterol-rich platforms has prompted several authors to suggest the use of drugs that interfere with these pathways as possible therapeutic agents against SARS-CoV-2 [7,149,150], sometimes in combination with a variety of drugs of still unproven efficacy such as chloroquine or hydroxychloroquine, with the argument that such drugs destabilize the order of liquid-ordered lipid domains [149]. Targeting lipid “rafts” has also been the argument behind suggesting the use of cholesterol-sequestering cyclodextrins as the “best candidate to improve complex therapies” in COVID-19 [149]. Cyclodextrins have a dose-dependent toxicity on the human organism, particularly on the renal parenchyma, and require chemical modifications to be used as anti-viral agents. Mercaptoundecane sulfonic acids have been recently employed to produce biocompatible broad-spectrum antivirals, tested on herpes simplex virus, dengue virus, respiratory syncytial virus, and Zika virus [151]. Modified cyclodextrins are also finding applications as carriers, adjuvants, cryo-preservation agents, and stabilizers of anti-viral drugs [152].
Niemann-Pick disease type C is a neurodegenerative lysosomal storage metabolic disease caused by loss-of-function mutations in the NPC1 gene that codes for NPC1, a endosomal-lysosomal cholesterol transporter, causing the accumulation of lipoprotein-derived cholesterol in late endosomes and lysosomes [153,154]. Genome-wide genetic screening of human cells was used to detect the host factors required for the Filovirus Ebola virus entry into host cells [155]. Nearly all the genes identified were related to the endosomal-lysosomal trafficking, including cathepsin B, some of which involve the NPC1 cholesterol transporter protein. Cells defective for this factor, as well as cells from Niemann-Pick C1 disease patients [155] and, curiously, reptiles [156] are resistant to Ebola virus infection. An adamantane derivative was identified as a pharmacologically active drug that inhibits Ebola infection and affects cholesterol uptake by a mechanism that involves cathepsin B, exposing the N-term domain of the Ebola glycoprotein, which binds to NPC1, and regulates membrane fusion to the endosomal membrane [157]. The compound U18666A, an inhibitor of cholesterol trafficking from late endosomes to lysosomes known to mimic the loss of NPC1 [158], diminished Ebola virus infection [155].
9.2. Statins as anti-thrombotic agents
Thrombotic and thromboembolic complications have increasingly been documented as the COVID-19 pandemic evolved [159,2]. Sepsis with affectation of the microcirculatory endothelium has also been reported [160,161]. Post-mortem studies of COVID-19 patients showed (micro)thrombotic/thromboembolic lesions in the CNS and olfactory mucosae [162]. Pulmonary arterial thrombosis of mid-size and small vessels has been described as a cause of death [163]. In another study 35% of hospitalized critically ill patients were found to present thrombotic complications [164]. As analyzed in more detail in a recent review [5], endothelial dysregulation appears to be at the root of the thrombogenic endothelial dysregulation in COVID-19 [165]. It is in this context that statins have been considered as pharmacological agents for the prevention of thrombosis and the hyper-inflammatory complications and severe forms of the disease, and to potentially minimize tissue injury through production of angiotensin [[1], [2], [3], [4], [5], [6], [7],166,159,167]. Concerns were raised, however, about the main statin-mediated cholesterol lowering effects that according to these authors upregulate ACE2 expression at the plasmalemma and may thus enhance SARS-CoV-2 entry. The use of statins in COVID-19 is thus still an open subject: propensity-scores matching retrospective studies, which match COVID-19 patients who were on statin therapy with those who were not, suggest that statin medication is mainly associated with a reduction of mortality, other evidence being largely anecdotal (reviewed in [168]). This outcome is observed, however, with high doses of statins and not with low dosage; no link has been found between antecedent statin therapy and admission to intensive care units [169]. We must therefore wait for randomized controlled trials of statin use in COVID-19 [170] to assess its therapeutic value, appropriate dosage, and mechanism of action.
10. Expanding the therapeutic arsenal in COVID-19
The identification of SR-B1 as a SARS-CoV-2 entry cofactor (see Fig. 3) has opened new therapeutic horizons for COVID-19 treatment. As recently proposed ( [78,141], drugs that target SR-B, such as AM580 (Fig. 3) could be employed as SARS-CoV-2 antivirals. As a proof of concept, these authors showed that ITX 5601, a clinically approved inhibitor of HCV infection, inhibits SARS-CoV-2 infection in vitro. Other metabolic steps offer prophylactic or therapeutic possibilities for inhibiting SARS-CoV-2, such as 25-hydroxycholesterol [95].
11. Concluding remarks
As analyzed in the different sections of this review, cholesterol plays key roles in the SARS-CoV-2 life cycle, from initial entry into the host cell to the virion exit into the extracellular compartment. Cholesterol-rich lipid domains are also intimately related to the fate of the SARS-CoV-2 in the infected organism. They serve as platforms for the recruitment of host receptors like ACE2 and TMPRSS2 to interact with the spike proteins, participate in the intracellular trafficking and egress of virions from the infected cells. Biosynthesis and regulation of cholesterol are mediated by proteins in the plasma membrane, ER, endosome and lysosome, which constitute potential therapeutic targets. Concepts raised in this review shed light on novel inhibitors and treatments which need to be further investigated.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Declaration of Competing Interest
The author declares no conflicts of interest.
Acknowledgements
Thanks are due to Phyllis Johnson for critical reading of the manuscript and to Sofía Vallés for help with the illustrations.
References
- 1.Argenziano M.G., Bruce S.L., Slater C.L., Tiao J.R., Baldwin M.R., Barr R.G., et al. Characterization and clinical course of 1000 patients with coronavirus disease 2019 in New York: retrospective case series. BMJ (Clinical research ed) 2020;369 doi: 10.1136/bmj.m1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jain V., Yuan J.-M. Systematic review and meta-analysis of predictive symptoms and comorbidities for severe COVID-19 infection. Int J Public Health. 2020;25:1–14. doi: 10.1007/s00038-020-01390-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hussain A., Bhowmik B., do Vale Moreira N.C. COVID-19 and diabetes: Knowledge in progress. Diabetes Res Clin Pract. 2020;162 doi: 10.1016/j.diabres.2020.108142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fridman S., Bullrich M.B., Jimenez-Ruiz A., Costantini P., Shah P., Just C., et al. Stroke Risk, phenotypes, and death in COVID-19: Systematic review and newly reported cases. Neurology. 2020;95(24):e3373–e3385. doi: 10.1212/WNL.0000000000010851. [DOI] [PubMed] [Google Scholar]
- 5.Barrantes F.J. The unfolding palette of COVID-19 multisystemic syndrome and its neurological manifestations. Brain, Behavior, & Immunity - Health. 2021;14 doi: 10.1016/j.bbih.2021.100251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chazal N., Gerlier D. Virus entry, assembly, budding, and membrane rafts. Microbiol and Mol Biol Rev : MMBR. 2003;67(2):226–237. doi: 10.1128/MMBR.67.2.226-237.2003. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Proto M.C., Fiore D., Piscopo C., Pagano C., Galgani M., Bruzzaniti S., et al. Lipid homeostasis and mevalonate pathway in COVID-19: Basic concepts and potential therapeutic targets. Prog Lipid Res. 2021;82 doi: 10.1016/j.plipres.2021.101099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raulin J. Human immunodeficiency virus and host cell lipids. Interesting pathways in research for a new HIV therapy. Prog Lipid Res. 2002;41(1):27–65. doi: 10.1016/s0163-7827(01)00019-4. [DOI] [PubMed] [Google Scholar]
- 9.Nomura R., Kiyota A., Suzaki E., Kataoka K., Ohe Y., Miyamoto K., et al. Human coronavirus 229E binds to CD13 in rafts and enters the cell through caveolae. J Virol. 2004;78(16):8701–8708. doi: 10.1128/JVI.78.16.8701-8708.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bollinger C.R., Teichgraber V., Gulbins E. Ceramide-enriched membrane domains. Biochim.Biophys.Acta. 2005;1746(3):284–294. doi: 10.1016/j.bbamcr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 11.Hambleton S., Steinberg S.P., Gershon M.D., Gershon A.A. Cholesterol dependence of varicella-zoster virion entry into target cells. J Virol. 14, 2007;81:7548–7558. doi: 10.1128/JVI.00486-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li G.M., Li Y.G., Yamate M., Li S.M., Ikuta K. Lipid rafts play an important role in the early stage of severe acute respiratory syndrome-coronavirus life cycle. Microbes Infect. 2007;9(1):96–102. doi: 10.1016/j.micinf.2006.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu Y., Liu D.X., Tam J.P. Lipid rafts are involved in SARS-CoV entry into Vero E6 cells. Biochem Biophys Res Commun. 2008;369(2):344–349. doi: 10.1016/j.bbrc.2008.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glende J., Schwegmann-Wessels C., Al-Falah M., Pfefferle S., Qu X., Deng H., et al. Importance of cholesterol-rich membrane microdomains in the interaction of the S protein of SARS-coronavirus with the cellular receptor angiotensin-converting enzyme 2. Virology. 2008;381(2):215–221. doi: 10.1016/j.virol.2008.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carter G.C., Bernstone L., Sangani D., Bee J.W., Harder T., James W. HIV entry in macrophages is dependent on intact lipid rafts. Virology. 2009;386(1):192–202. doi: 10.1016/j.virol.2008.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pratelli A., Colao V. Role of the lipid rafts in the life cycle of canine coronavirus. J Gen Virol. 2015;96(Pt 2):331–337. doi: 10.1099/vir.0.070870-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Betters J.L., Yu L. NPC1L1 and cholesterol transport. FEBS Lett. 2010;584(13):2740–2747. doi: 10.1016/j.febslet.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jia L., Betters J.L., Yu L. Niemann-pick C1-like 1 (NPC1L1) protein in intestinal and hepatic cholesterol transport. Annu Rev Physiol. 2011;73:239–259. doi: 10.1146/annurev-physiol-012110-142233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen W.J., Azhar S., Kraemer F.B. SR-B1: A Unique Multifunctional Receptor for Cholesterol Influx and Efflux. Annu Rev Physiol. 2018;80:95–116. doi: 10.1146/annurev-physiol-021317-121550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lange Y., Swaisgood M.H., Ramos B.V., Steck T.L. Plasma membranes contain half the phospholipid and 90% of the cholesterol and sphingomyelin in cultured human fibroblasts. J.Biol.Chem. 1989;264:3786–3793. [PubMed] [Google Scholar]
- 21.van Meer G., Voelker D.R., Feigenson G.W. Membrane lipids: where they are and how they behave. NatRevMolCell Biol. 2008;9(2):112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fiorini R., Gratton E., Curatola G. Effect of cholesterol on membrane microheterogeneity: A study using 1,6-diphenyl-1,3,5-hexatriene fluorescence lifetime distributions. Biochim.Biophys.Acta. 1989;1006:198–202. doi: 10.1016/0005-2760(89)90196-3. [DOI] [PubMed] [Google Scholar]
- 23.Simons K., Ikonen E. Functional rafts in cell membranes. Nature. 1997;387(6633):569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 24.Ahmed S.N., Brown D.A., London E. On the origin of sphingolipid/cholesterol-rich detergent-insoluble cell membranes: physiological concentrations of cholesterol and sphingolipid induce formation of a detergent-insoluble, liquid-orderer lipid phase in model membranes. Biochem. 36, 1997;36:10944–10953. doi: 10.1021/bi971167g. [DOI] [PubMed] [Google Scholar]
- 25.Jacobson K., Mouritsen O.G., Anderson R.G. Lipid rafts: at a crossroad between cell biology and physics. Nat Cell Biol. 2007;9(1):7–14. doi: 10.1038/ncb0107-7. [DOI] [PubMed] [Google Scholar]
- 26.Pike L.J. Rafts defined: a report on the Keystone Symposium on Lipid Rafts and Cell Function. J Lipid Res. 2006;47(7):1597–1598. doi: 10.1194/jlr.E600002-JLR200. [DOI] [PubMed] [Google Scholar]
- 27.Samsonov A.V., Mihalyov I., Cohen F.S. Characterization of cholesterol-sphingomyelin domains and their dynamics in bilayer membranes. Biophys.J. 2001;81(3):1486–1500. doi: 10.1016/S0006-3495(01)75803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mayor S., Rao M. Rafts: scale-dependent, active lipid organization at the cell surface. Traffic. 2004;5(4):231–240. doi: 10.1111/j.1600-0854.2004.00172.x. [DOI] [PubMed] [Google Scholar]
- 29.Sezgin E., Levental I., Mayor S., Eggeling C. Nature reviews; Molecular cell biology: 2017. The mystery of membrane organization: composition, regulation and roles of lipid rafts. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marquardt D., Heberle F.A., Greathouse D.V., Koeppe R.E., Standaert R.F., Van Oosten B.J., et al. Lipid bilayer thickness determines cholesterol's location in model membranes. Soft Matter. 2016;12(47):9417–9428. doi: 10.1039/c6sm01777k. [DOI] [PubMed] [Google Scholar]
- 31.Marquardt D., Kucerka N., Wassall S.R., Harroun T.A., Katsaras J. Cholesterol's location in lipid bilayers. Chem Phys Lipids. 2016;199:17–25. doi: 10.1016/j.chemphyslip.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 32.Fantini J., Barrantes F.J. Sphingolipid/cholesterol regulation of neurotransmitter receptor conformation and function. Biochim Biophys Acta. 2009;1788(11):2345–2361. doi: 10.1016/j.bbamem.2009.08.016. [DOI] [PubMed] [Google Scholar]
- 33.Dietschy J.M., Turley S.D. Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res. 2004;45(8):1375–1397. doi: 10.1194/jlr.R400004-JLR200. [DOI] [PubMed] [Google Scholar]
- 34.Mauch D.H., Nägler K., Schumacher S., Göritz C., Müller E.C., Otto A., et al. CNS synaptogenesis promoted by glia-derived cholesterol. Science (New York, NY) 2001;294(5545):1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- 35.Pfrieger F.W. Cholesterol homeostasis and function in neurons of the central nervous system. Cell Mol Life Sci : CMLS. 2003;60(6):1158–1171. doi: 10.1007/s00018-003-3018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pfrieger F.W. Role of cholesterol in synapse formation and function. Biochim Biophys Acta. 2003;1610(2):271–280. doi: 10.1016/s0005-2736(03)00024-5. [DOI] [PubMed] [Google Scholar]
- 37.Slezak M., Pfrieger F.W. New roles for astrocytes: regulation of CNS synaptogenesis. Trends Neurosci. 2003;26(10):531–535. doi: 10.1016/j.tins.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 38.Simons K., Sampaio J.L. Membrane organization and lipid rafts. Cold Spring Harb Perspect Biol. 2011;3(10):a004697. doi: 10.1101/cshperspect.a004697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lorizate M., Kräusslich H.G. Role of lipids in virus replication. Cold Spring Harb Perspect Biol. 10, 2011;3:a004820. doi: 10.1101/cshperspect.a004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Callens N., Brügger B., Bonnafous P., Drobecq H., Gerl M.J., Krey T., et al. Morphology and Molecular Composition of Purified Bovine Viral Diarrhea Virus Envelope. PLoS Pathog. 2016;12(3) doi: 10.1371/journal.ppat.1005476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brügger B., Glass B., Haberkant P., Leibrecht I., Wieland F.T., Kräusslich H.-G. The HIV lipidome: a raft with an unusual composition. Proc Natl Acad Sci U S A. 2006;103(8):2641–2646. doi: 10.1073/pnas.0511136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chan R., Uchil P.D., Jin J., Shui G., Ott D.E., Mothes W., et al. Retroviruses human immunodeficiency virus and murine leukemia virus are enriched in phosphoinositides. J Virol. 2008;82(22):11228–11238. doi: 10.1128/JVI.00981-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petit C.M., Chouljenko V.N., Iyer A., Colgrove R., Farzan M., Knipe D.M., et al. Palmitoylation of the cysteine-rich endodomain of the SARS-coronavirus spike glycoprotein is important for spike-mediated cell fusion. Virology. 2007;360(2):264–274. doi: 10.1016/j.virol.2006.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McBride C.E., Machamer C.E. Palmitoylation of SARS-CoV S protein is necessary for partitioning into detergent-resistant membranes and cell–cell fusion but not interaction with M protein. Virology. 2010;405(1):139–148. doi: 10.1016/j.virol.2010.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanders D.W., Jumper C.C., Ackerman P.J., Bracha D., Donlic A., Kim H., et al. SARS-CoV-2 requires cholesterol for viral entry and pathological syncytia formation. Elife. 2021;10 doi: 10.7554/eLife.65962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Campbell S.M., Crowe S.M., Mak J. Virion-associated cholesterol is critical for the maintenance of HIV-1 structure and infectivity. AIDS (London, England) 2002;16(17):2253–2261. doi: 10.1097/00002030-200211220-00004. [DOI] [PubMed] [Google Scholar]
- 47.Graham D.R., Chertova E., Hilburn J.M., Arthur L.O., Hildreth J.E. Cholesterol depletion of human immunodeficiency virus type 1 and simian immunodeficiency virus with beta-cyclodextrin inactivates and permeabilizes the virions: evidence for virion-associated lipid rafts. J Virol. 2003;77(15):8237–8248. doi: 10.1128/JVI.77.15.8237-8248.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zawada K.E., Wrona D., Rawle R.J., Kasson P.M. Influenza viral membrane fusion is sensitive to sterol concentration but surprisingly robust to sterol chemical identity. Sci Rep. 2016;6:29842. doi: 10.1038/srep29842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rawat S.S., Viard M., Gallo S.A., Rein A., Blumenthal R., Puri A. Modulation of entry of enveloped viruses by cholesterol and sphingolipids (Review) Mol Membr Biol. 2003;20(3):243–254. doi: 10.1080/0968768031000104944. [DOI] [PubMed] [Google Scholar]
- 50.Campbell A.E., Loria R.M., Madge G.E., Kaplan A.M. Dietary hepatic cholesterol elevation: effects on coxsackievirus B infection and inflammation. Infect Immun. 1982;37(1):307–317. doi: 10.1128/iai.37.1.307-317.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cervin M., Anderson R. Modulation of coronavirus-mediated cell fusion by homeostatic control of cholesterol and fatty acid metabolism. J Med Virol. 1991;35(2):142–149. doi: 10.1002/jmv.1890350213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goronzy I.N., Rawle R.J., Boxer S.G., Kasson P.M. Cholesterol enhances influenza binding avidity by controlling nanoscale receptor clustering. Chem Sci. 2018;9(8):2340–2347. doi: 10.1039/c7sc03236f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang J., Pekosz A., Lamb R.A. Influenza virus assembly and lipid raft microdomains: a role for the cytoplasmic tails of the spike glycoproteins. J Virol. 2000;74(10):4634–4644. doi: 10.1128/jvi.74.10.4634-4644.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181(2):271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barrantes F.J. While We Wait for a Vaccine Against SARS-CoV-2, Why Not Think About Available Drugs? Front Physiol. 2020;11:820. doi: 10.3389/fphys.2020.00820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chi X., Yan R., Zhang J., Zhang G., Zhang Y., Hao M., et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science (New York, NY) 2020;369(6504):650–655. doi: 10.1126/science.abc6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suryadevara N., Shrihari S., Gilchuk P., VanBlargan L.A., Binshtein E., Zost S.J., et al. Neutralizing and protective human monoclonal antibodies recognizing the N-terminal domain of the SARS-CoV-2 spike protein. Cell. 2021;184(9):2316–2331. doi: 10.1016/j.cell.2021.03.029. e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baggen J., Persoons L., Vanstreels E., Jansen S., Van Looveren D., Boeckx B., et al. Genome-wide CRISPR screening identifies TMEM106B as a proviral host factor for SARS-CoV-2. Nat Genet. 2021;53(4):435–444. doi: 10.1038/s41588-021-00805-2. [DOI] [PubMed] [Google Scholar]
- 59.Puray-Chavez M., LaPak K.M., Schrank T.P., Elliott J.L., Bhatt D.P., Agajanian M.J., et al. Systematic analysis of SARS-CoV-2 infection of an ACE2-negative human airway cell. Cell Rep. 2021;36(2):109364. doi: 10.1016/j.celrep.2021.109364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ren X., Wen W., Fan X., Hou W., Su B., Cai P., et al. COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas. Cell. 2021;184(7):1895–1913. doi: 10.1016/j.cell.2021.01.053. e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shen X.-R., Geng R., Li Q., Chen Y., Li S.-F., Wang Q., et al. ACE2-independent infection of T lymphocytes by SARS-CoV-2. Signal Transduct Target Ther. 2022;7(1):83. doi: 10.1038/s41392-022-00919-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu J., Lu F., Chen Y., Plow E., Qin J. Integrin mediates cell entry of the SARS-CoV-2 virus independent of cellular receptor ACE2. J Biol Chem. 2022;298(3) doi: 10.1016/j.jbc.2022.101710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dou X., Li Y., Han J., Zarlenga D.S., Zhu W., Ren X., et al. Cholesterol of lipid rafts is a key determinant for entry and post-entry control of porcine rotavirus infection. BMC Vet Res. 2018;14(1):45. doi: 10.1186/s12917-018-1366-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Glende J., Schwegmann-Wessels C., Al-Falah M., Pfefferle S., Qu X., Deng H., et al. Importance of cholesterol-rich membrane microdomains in the interaction of the S protein of SARS-coronavirus with the cellular receptor angiotensin-converting enzyme 2. Virology. 2008;381(2):215–221. doi: 10.1016/j.virol.2008.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Warner F.J., Lew R.A., Smith A.I., Lambert D.W., Hooper N.M., Turner A.J. Angiotensin-converting enzyme 2 (ACE2), but not ACE, is preferentially localized to the apical surface of polarized kidney cells. J Biol Chem. 2005;280(47):39353–39362. doi: 10.1074/jbc.M508914200. [DOI] [PubMed] [Google Scholar]
- 66.McQuaid C., Solorzano A., Dickerson I., Deane R. SARS-CoV-2 spike proteins uptake mediated by lipid raft ganglioside GM1 in human cerebrovascular cells. bioRxiv. 2022 doi: 10.3389/fnins.2023.1117845. 2022.03.20.485050, preprint (biorXiv) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu K.N., Boxer S.G. 2021. Single-virus content mixing assay reveals cholesterol-enhanced influenza membrane fusion efficiency. 2021.04.26.441491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu K.N., Boxer S.G. Target Membrane Cholesterol Modulates Single Influenza Virus Membrane Fusion Efficiency but Not Rate. Biophys J. 2020;118(10):2426–2433. doi: 10.1016/j.bpj.2020.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li H., Papadopoulos V. Peripheral-type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology. 1998;139(12):4991–4997. doi: 10.1210/endo.139.12.6390. [DOI] [PubMed] [Google Scholar]
- 70.Jamin N., Neumann J.M., Ostuni M.A., Vu T.K., Yao Z.X., Murail S., et al. Characterization of the cholesterol recognition amino acid consensus sequence of the peripheral-type benzodiazepine receptor. Mol.Endocrinol. 2005;19(3):588–594. doi: 10.1210/me.2004-0308. [DOI] [PubMed] [Google Scholar]
- 71.Baier C.J., Fantini J., Barrantes F.J. Disclosure of cholesterol recognition motifs in transmembrane domains of the human nicotinic acetylcholine receptor. Sci Rep. 2011;1:69. doi: 10.1038/srep00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fantini J., Barrantes F.J. How cholesterol interacts with membrane proteins: an exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front Physiol. 2013;4:31. doi: 10.3389/fphys.2013.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Di Scala C., Baier C.J., Evans L.S., Williamson P.T.F., Fantini J., Barrantes F.J. Relevance of CARC and CRAC Cholesterol-Recognition Motifs in the Nicotinic Acetylcholine Receptor and Other Membrane-Bound Receptors. Curr Top Membr. 2017;80:3–23. doi: 10.1016/bs.ctm.2017.05.001. [DOI] [PubMed] [Google Scholar]
- 74.Lee A.G. Interfacial Binding Sites for Cholesterol on TRP Ion Channels. Biophys J. 2019;117(10):2020–2033. doi: 10.1016/j.bpj.2019.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jafurulla M., Tiwari S., Chattopadhyay A. Identification of cholesterol recognition amino acid consensus (CRAC) motif in G-protein coupled receptors. Biochem Biophys Res Commun. 2011;404(1):569–573. doi: 10.1016/j.bbrc.2010.12.031. [DOI] [PubMed] [Google Scholar]
- 76.Jafurulla M., Chattopadhyay A. In: Cholesterol Homeostasis: Methods and Protocols. Gelissen I.C., Brown A.J., editors. Springer; New York, New York, NY: 2017. Structural Stringency of Cholesterol for Membrane Protein Function Utilizing Stereoisomers as Novel Tools: A Review; pp. 21–39. [DOI] [PubMed] [Google Scholar]
- 77.Prasanna X., Sengupta D., Chattopadhyay A. Cholesterol-dependent Conformational Plasticity in GPCR Dimers. Sci Rep. 2016;6:31858. doi: 10.1038/srep31858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wei C., Wan L., Yan Q., Wang X., Zhang J., Yang X., et al. HDL-scavenger receptor B type 1 facilitates SARS-CoV-2 entry. Nat Metab. 2020;2:1391–1400. doi: 10.1038/s42255-020-00324-0. [DOI] [PubMed] [Google Scholar]
- 79.Barrantes F.J. The Contribution of Biophysics and Structural Biology to Current Advances in COVID-19. Annu Rev Biophys. 2021;50:493–523. doi: 10.1146/annurev-biophys-102620-080956. [DOI] [PubMed] [Google Scholar]
- 80.Xia X. Domains and Functions of Spike Protein in Sars-Cov-2 in the Context of Vaccine Design. Viruses. 2021;13(1) doi: 10.3390/v13010109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fantini J., Di Scala C., Evans L.S., Williamson P.T.F., Barrantes F.J. A mirror code for protein-cholesterol interactions in the two leaflets of biological membranes. Sci Rep. 2016;6:21907. doi: 10.1038/srep21907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tang T., Bidon M., Jaimes J.A., Whittaker G.R., Daniel S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antiviral Res. 2020;178 doi: 10.1016/j.antiviral.2020.104792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Khelashvili G., Plante A., Doktorova M., Weinstein H. Ca(2+)-dependent mechanism of membrane insertion and destabilization by the SARS-CoV-2 fusion peptide. Biophys J. 2021;120(6):1105–1119. doi: 10.1016/j.bpj.2021.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lai A.L., Freed J.H. SARS-CoV-2 Fusion Peptide has a Greater Membrane Perturbating Effect than SARS-CoV with Highly Specific Dependence on Ca(2) J Mol Biol. 2021;433(10) doi: 10.1016/j.jmb.2021.166946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pattnaik G.P., Bhattacharjya S., Chakraborty H. 2021. Enhanced Cholesterol-dependent Hemifusion by Internal Fusion Peptide 1 of SARS Coronavirus-2 Compared to its N-terminal Counterpart. 2021.01.14.426613. [DOI] [PubMed] [Google Scholar]
- 86.Schaefer S.L., Jung H., Hummer G. 2021. Binding of SARS-CoV-2 fusion peptide to host membranes. 2021.05.10.443474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shekhar N., Sarma P., Prajapat M., Avti P., Kaur H., Raja A., et al. In Silico Structure-Based Repositioning of Approved Drugs for Spike Glycoprotein S2 Domain Fusion Peptide of SARS-CoV-2: Rationale from Molecular Dynamics and Binding Free Energy Calculations. mSystems. 2020;5(5) doi: 10.1128/mSystems.00382-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Su R., Zeng J., O'Shaughnessy B. 2021. Large fluctuations of the fusion intermediate help SARS-CoV-2 capture host cell membranes. 2021.04.09.439051. [Google Scholar]
- 89.Gorgun D., Lihan M., Kapoor K., Tajkhorshid E. 2021. Binding Mode of SARS-CoV2 Fusion Peptide to Human Cellular Membrane. 2020.10.27.357350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Behrens E.M., Koretzky G.A. Review: Cytokine Storm Syndrome: Looking Toward the Precision Medicine Era. Arthritis & rheumatology (Hoboken, NJ) 2017;69(6):1135–1143. doi: 10.1002/art.40071. [DOI] [PubMed] [Google Scholar]
- 91.Channappanavar R., Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. 2017;39(5):529–539. doi: 10.1007/s00281-017-0629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liu S.Y., Aliyari R., Chikere K., Li G., Marsden M.D., Smith J.K., et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity. 2013;38(1):92–105. doi: 10.1016/j.immuni.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Holmes R.S., Vandeberg J.L., Cox L.A. Genomics and proteomics of vertebrate cholesterol ester lipase (LIPA) and cholesterol 25-hydroxylase (CH25H), 3. Biotech. 2011;1(2):99–109. doi: 10.1007/s13205-011-0013-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Du X., Pham Y.H., Brown A.J. Effects of 25-hydroxycholesterol on cholesterol esterification and sterol regulatory element-binding protein processing are dissociable: implications for cholesterol movement to the regulatory pool in the endoplasmic reticulum. J Biol Chem. 2004;279(45):47010–47016. doi: 10.1074/jbc.M408690200. [DOI] [PubMed] [Google Scholar]
- 95.Wang S., Li W., Hui H., Tiwari S.K., Zhang Q., Croker B.A., et al. Cholesterol 25-Hydroxylase inhibits SARS-CoV-2 and other coronaviruses by depleting membrane cholesterol. EMBO J. 2020;39(21) doi: 10.15252/embj.2020106057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kawase M., Shirato K., van der Hoek L., Taguchi F., Matsuyama S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J Virol. 2012;86(12):6537–6545. doi: 10.1128/JVI.00094-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hoffmann M., Hofmann-Winkler H., Smith J.C., Krüger N., Arora P., Sørensen L.K., et al. Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and its metabolite GBPA exerts antiviral activity. EBioMedicine. 2021;65 doi: 10.1016/j.ebiom.2021.103255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Meng B., Abdullahi A., Ferreira I.A.T.M., Goonawardane N., Saito A., Kimura I., et al. The Genotype to Phenotype Japan, C.C. Ecuador, Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature. 2022;603(7902):706–714. doi: 10.1038/s41586-022-04474-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Peacock T.P., Brown J.C., Zhou J., Thakur N., Newman J., Kugathasan R., et al. The SARS-CoV-2 variant, Omicron, shows rapid replication in human primary nasal epithelial cultures and efficiently uses the endosomal route of entry. bioRxiv. 2022 2021.12.31.474653. [Google Scholar]
- 100.Willett B.J., Grove J., MacLean O.A., Wilkie C., Logan N., Lorenzo G.D., et al. The hyper-transmissible SARS-CoV-2 Omicron variant exhibits significant antigenic change, vaccine escape and a switch in cell entry mechanism. medRxiv : the preprint server for health sciences. 2022 2022.01.03.21268111. [Google Scholar]
- 101.Nardacci R., Colavita F., Castilletti C., Lapa D., Matusali G., Meschi S., et al. Evidences for lipid involvement in SARS-CoV-2 cytopathogenesis. Cell Death Dis. 2021;12(3):263. doi: 10.1038/s41419-021-03527-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li X., Zhu W., Fan M., Zhang J., Peng Y., Huang F., et al. Dependence of SARS-CoV-2 infection on cholesterol-rich lipid raft and endosomal acidification, Comput Struct. Biotechnol J. 2021;19:1933–1943. doi: 10.1016/j.csbj.2021.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.DeBose-Boyd R.A., Ye J. SREBPs in Lipid Metabolism, Insulin Signaling, and Beyond. Trends Biochem Sci. 2018;43(5):358–368. doi: 10.1016/j.tibs.2018.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yokoyama C., Wang X., Briggs M.R., Admon A., Wu J., Hua X., et al. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell. 1993;75(1):187–197. [PubMed] [Google Scholar]
- 105.Hua X., Yokoyama C., Wu J., Briggs M.R., Brown M.S., Goldstein J.L., et al. SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc Natl Acad Sci U S A. 1993;90(24):11603–11607. doi: 10.1073/pnas.90.24.11603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Horton J.D., Goldstein J.L., Brown M.S. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109(9):1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Brown M.S., Radhakrishnan A., Goldstein J.L. Retrospective on Cholesterol Homeostasis: The Central Role of Scap. Annu Rev Biochem. 2018;87:783–807. doi: 10.1146/annurev-biochem-062917-011852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang T., Espenshade P.J., Wright M.E., Yabe D., Gong Y., Aebersold R., et al. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110(4):489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 109.Yabe D., Xia Z.P., Adams C.M., Rawson R.B. Proceedings of the National Academy of Sciences of the United States of America. 99(26) 2002. Three mutations in sterol-sensing domain of SCAP block interaction with insig and render SREBP cleavage insensitive to sterols; pp. 16672–16677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Weber L.W., Boll M., Stampfl A. Maintaining cholesterol homeostasis: sterol regulatory element-binding proteins. World J Gastroenterol. 2004;10(21):3081–3087. doi: 10.3748/wjg.v10.i21.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yu Y., Maguire T.G., Alwine J.C. Human cytomegalovirus infection induces adipocyte-like lipogenesis through activation of sterol regulatory element binding protein 1. J Virol. 2012;86(6):2942–2949. doi: 10.1128/JVI.06467-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kotzamanis K., Angulo A., Ghazal P. Infection homeostasis: implications for therapeutic and immune programming of metabolism in controlling infection. Med Microbiol Immunol. 2015;204(3):395–407. doi: 10.1007/s00430-015-0402-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang L.W., Wang Z., Ersing I., Nobre L., Guo R., Jiang S., et al. Epstein-Barr virus subverts mevalonate and fatty acid pathways to promote infected B-cell proliferation and survival. PLoS Pathog. 2019;15(9) doi: 10.1371/journal.ppat.1008030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang Q., Thome R., Rostami A., Ma C.-G., Zhang G.-X. The selective retinoic acid receptor-α agonist AM580 fails to control autoimmune neuroinflammation. Cell Mol Immunol. 2019;16(8):727–729. doi: 10.1038/s41423-019-0238-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yuan S., Chu H., Chan J.F., Ye Z.W., Wen L., Yan B., et al. SREBP-dependent lipidomic reprogramming as a broad-spectrum antiviral target. Nat Commun. 2019;10(1):120. doi: 10.1038/s41467-018-08015-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lee W., Ahn J.H., Park H.H., Kim H.N., Kim H., Yoo Y., et al. COVID-19-activated SREBP2 disturbs cholesterol biosynthesis and leads to cytokine storm. Signal Transduct Target Ther. 2020;5(1):186. doi: 10.1038/s41392-020-00292-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li Z., Votava J.A., Zajac G.J.M., Nguyen J.N., Leyva Jaimes F.B., Ly S.M., et al. Integrating Mouse and Human Genetic Data to Move beyond GWAS and Identify Causal Genes in Cholesterol Metabolism. Cell Metab. 2020;31(4):741–754.e5. doi: 10.1016/j.cmet.2020.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sandhu J., Li S., Fairall L., Pfisterer S.G., Gurnett J.E., Xiao X., et al. Aster Proteins Facilitate Nonvesicular Plasma Membrane to ER Cholesterol Transport in Mammalian Cells. Cell. 2018;175(2):514–529.e20. doi: 10.1016/j.cell.2018.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Naito T., Ercan B., Krshnan L., Triebl A., Koh D.H.Z., Wei F.Y., et al. Movement of accessible plasma membrane cholesterol by the GRAMD1 lipid transfer protein complex. Elife. 2019;8 doi: 10.7554/eLife.51401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ercan B., Naito T., Koh D.H.Z., Dharmawan D., Saheki Y. Molecular basis of accessible plasma membrane cholesterol recognition by the GRAM domain of GRAMD1b. EMBO J. 2021;40(6) doi: 10.15252/embj.2020106524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.McNicholas S., Potterton E., Wilson K.S., Noble M.E. Presenting your structures: the CCP4mg molecular-graphics software. Acta crystallographica Section D, Biol Crystallograph. 2011;67(Pt 4):386–394. doi: 10.1107/S0907444911007281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Trinh M.N., Brown M.S., Goldstein J.L., Han J., Vale G., McDonald J.G., et al. Proceedings of the National Academy of Sciences of the United States of America. 117(31) 2020. Last step in the path of LDL cholesterol from lysosome to plasma membrane to ER is governed by phosphatidylserine; pp. 18521–18529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Maekawa M., Fairn G.D. Complementary probes reveal that phosphatidylserine is required for the proper transbilayer distribution of cholesterol. J Cell Sci. 2015;128(7):1422–1433. doi: 10.1242/jcs.164715. [DOI] [PubMed] [Google Scholar]
- 124.Hoffmann H.H., Schneider W.M., Rozen-Gagnon K., Miles L.A., Schuster F., Razooky B., et al. TMEM41B Is a Pan-flavivirus Host Factor. Cell. 2021;184(1):133–148.e20. doi: 10.1016/j.cell.2020.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cortese M., Goellner S., Acosta E.G., Neufeldt C.J., Oleksiuk O., Lampe M., et al. Ultrastructural Characterization of Zika Virus Replication Factories. Cell Rep. 2017;18(9):2113–2123. doi: 10.1016/j.celrep.2017.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Prentice E., Jerome W.G., Yoshimori T., Mizushima N., Denison M.R. Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem. 2004;279(11):10136–10141. doi: 10.1074/jbc.M306124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Knoops K., Kikkert M., Worm S.H., Zevenhoven-Dobbe J.C., van der Meer Y., Koster A.J., et al. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008;6(9) doi: 10.1371/journal.pbio.0060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Snijder E.J., Limpens R., de Wilde A.H., de Jong A.W.M., Zevenhoven-Dobbe J.C., Maier H.J., et al. A unifying structural and functional model of the coronavirus replication organelle: Tracking down RNA synthesis. PLoS Biol. 2020;18(6) doi: 10.1371/journal.pbio.3000715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yan L., Ge J., Zheng L., Zhang Y., Gao Y., Wang T., et al. Cryo-EM Structure of an Extended SARS-CoV-2 Replication and Transcription Complex Reveals an Intermediate State in Cap Synthesis. Cell. 2021;184(1):184–193.e10. doi: 10.1016/j.cell.2020.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gewaid H., Aoyagi H., Arita M., Watashi K., Suzuki R., Sakai S., et al. Sphingomyelin Is Essential for the Structure and Function of the Double-Membrane Vesicles in Hepatitis C Virus RNA Replication Factories. J Virol. 2020;94(23) doi: 10.1128/JVI.01080-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schneider W.M., Luna J.M., Hoffmann H.H., Sánchez-Rivera F.J., Leal A.A., Ashbrook A.W., et al. Genome-Scale Identification of SARS-CoV-2 and Pan-coronavirus Host Factor Networks. Cell. 2021;184(1):120–132.e14. doi: 10.1016/j.cell.2020.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Li Y.E., Wang Y., Du X., Zhang T., Mak H.Y., Hancock S.E., et al. TMEM41B and VMP1 are scramblases and regulate the distribution of cholesterol and phosphatidylserine. J Cell Biol. 2021;220(6) doi: 10.1083/jcb.202103105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Posada I.M., Fantini J., Contreras F.X., Barrantes F., Alonso A., Goni F.M. A cholesterol recognition motif in human phospholipid scramblase 1. Biophys J. 2014;107(6):1383–1392. doi: 10.1016/j.bpj.2014.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lenahan C., Huang L., Travis Z.D., Zhang J.H. Scavenger Receptor Class B type 1 (SR-B1) and the modifiable risk factors of stroke. Chin Neurosurg J. 2019;5(1):30. doi: 10.1186/s41016-019-0178-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zani I.A., Stephen S.L., Mughal N.A., Russell D., Homer-Vanniasinkam S., Wheatcroft S.B., et al. Scavenger receptor structure and function in health and disease. Cells. 2015;4(2):178–201. doi: 10.3390/cells4020178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Burlone M.E., Budkowska A. Hepatitis C virus cell entry: role of lipoproteins and cellular receptors. J Gen Virol. 2009;90(Pt 5):1055–1070. doi: 10.1099/vir.0.008300-0. [DOI] [PubMed] [Google Scholar]
- 137.Eyre N.S., Drummer H.E., Beard M.R. The SR-BI partner PDZK1 facilitates hepatitis C virus entry. PLoS Pathog. 2010;6(10) doi: 10.1371/journal.ppat.1001130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nicholson A.M., Rademakers R. What we know about TMEM106B in neurodegeneration. Acta Neuropathol. 2016;132(5):639–651. doi: 10.1007/s00401-016-1610-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Klein Z.A., Takahashi H., Ma M., Stagi M., Zhou M., Lam T.T., et al. Loss of TMEM106B Ameliorates Lysosomal and Frontotemporal Dementia-Related Phenotypes in Progranulin-Deficient Mice. Neuron. 2017;95(2):281–296.e6. doi: 10.1016/j.neuron.2017.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nguyen D.H., Hildreth J.E. Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J Virol. 2000;74(7):3264–3272. doi: 10.1128/jvi.74.7.3264-3272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wei C., Wan L., Zhang Y., Fan C., Yan Q., Yang X., et al. Cholesterol Metabolism--Impact for SARS-CoV-2 Infection Prognosis. Entry and Antiviral Therapies. MedrXiv. 2020 2020.04.16.20068528. [Google Scholar]
- 142.Syder A.J., Lee H., Zeisel M.B., Grove J., Soulier E., Macdonald J., et al. Small molecule scavenger receptor BI antagonists are potent HCV entry inhibitors. J Hepatol. 2011;54(1):48–55. doi: 10.1016/j.jhep.2010.06.024. [DOI] [PubMed] [Google Scholar]
- 143.Kočar E., Režen T., Rozman D. Cholesterol, lipoproteins, and COVID-19: Basic concepts and clinical applications, Biochimica et biophysica acta. Molecular and cell biology of lipids. 2021;1866(2):158849. doi: 10.1016/j.bbalip.2020.158849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Baglivo M., Baronio M., Natalini G., Beccari T., Chiurazzi P., Fulcheri E., et al. Natural small molecules as inhibitors of coronavirus lipid-dependent attachment to host cells: a possible strategy for reducing SARS-COV-2 infectivity? Acta bio-medica : Atenei Parmensis. 2020;91(1):161–164. doi: 10.23750/abm.v91i1.9402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.del Real G., Jiménez-Baranda S., Mira E., Lacalle R.A., Lucas P., Gómez-Moutón C.N., et al. Mañes Statins Inhibit HIV-1 Infection by Down-regulating Rho Activity. J Exp Med. 2004;200(4):541–547. doi: 10.1084/jem.20040061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Moriyama T., Sorokin A. Repression of BK virus infection of human renal proximal tubular epithelial cells by pravastatin. Transplantation. 2008;85(9):1311–1317. doi: 10.1097/TP.0b013e31816c4ec5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Liu S., Rodriguez A.V., Tosteson M.T. Role of simvastatin and methyl-beta-cyclodextrin [corrected] on inhibition of poliovirus infection. Biochem Biophys Res Commun. 2006;347(1):51–59. doi: 10.1016/j.bbrc.2006.06.107. [DOI] [PubMed] [Google Scholar]
- 148.Sodero A.O., Barrantes F.J. Pleiotropic effects of statins on brain cells. Biochimica et Biophysica Acta (BBA) -Biomembranes. 2020;1862(9) doi: 10.1016/j.bbamem.2020.183340. [DOI] [PubMed] [Google Scholar]
- 149.Sorice M., Misasi R., Riitano G., Manganelli V., Martellucci S., Longo A., et al. Targeting Lipid Rafts as a Strategy Against Coronavirus. Front Cell Dev Biol. 2020;8 doi: 10.3389/fcell.2020.618296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Abu-Farha M., Thanaraj T.A., Qaddoumi M.G., Hashem A., Abubaker J., Al-Mulla F. The Role of Lipid Metabolism in COVID-19 Virus Infection and as a Drug Target. Int J Mol Sci. 2020;21(10) doi: 10.3390/ijms21103544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Jones S.T., Cagno V., Janeček M., Ortiz D., Gasilova N., Piret J., et al. Modified cyclodextrins as broad-spectrum antivirals. Sci Adv. 2020;6(5):eaax9318. doi: 10.1126/sciadv.aax9318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Braga S.S., Barbosa J.S., Santos N.E., El-Saleh F., Paz F.A.A. Cyclodextrins in Antiviral Therapeutics and Vaccines. Pharmaceutics. 2021;13(3) doi: 10.3390/pharmaceutics13030409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Peake K.B., Vance J.E. Defective cholesterol trafficking in Niemann-Pick C-deficient cells. FEBS Lett. 2010;584(13):2731–2739. doi: 10.1016/j.febslet.2010.04.047. [DOI] [PubMed] [Google Scholar]
- 154.Wüstner D., Solanko K. How cholesterol interacts with proteins and lipids during its intracellular transport. Biochim Biophys Acta. 2015;1848(9):1908–1926. doi: 10.1016/j.bbamem.2015.05.010. [DOI] [PubMed] [Google Scholar]
- 155.Carette J.E., Raaben M., Wong A.C., Herbert A.S., Obernosterer G., Mulherkar N., et al. Ebola virus entry requires the cholesterol transporter Niemann–Pick C1. Nature. 2011;477(7364):340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ndungo E., Herbert A.S., Raaben M., Obernosterer G., Biswas R., Miller E.H., et al. A Single Residue in Ebola Virus Receptor NPC1 Influences Cellular Host Range in Reptiles. mSphere. 2016;1(2) doi: 10.1128/mSphere.00007-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Côté M., Misasi J., Ren T., Bruchez A., Lee K., Filone C.M., et al. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature. 2011;477(7364):344–348. doi: 10.1038/nature10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Cenedella R.J. Cholesterol synthesis inhibitor U18666A and the role of sterol metabolism and trafficking in numerous pathophysiological processes. Lipids. 2009;44(6):477–487. doi: 10.1007/s11745-009-3305-7. [DOI] [PubMed] [Google Scholar]
- 159.Bikdeli B., Madhavan M.V., Jimenez D., Chuich T., Dreyfus I., Driggin E., et al. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-up. J Am Coll Cardiol. 2020;75(23):2950–2973. doi: 10.1016/j.jacc.2020.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Colantuoni A., Martini R., Caprari P., Ballestri M., Capecchi P.L., Gnasso A., et al. COVID-19 sepsis and microcirculation dysfunction. Front Physiol. 2020;11:747. doi: 10.3389/fphys.2020.00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Magro C., Mulvey J.J., Berlin D., Nuovo G., Salvatore S., Harp J., et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. 2020;220:1–13. doi: 10.1016/j.trsl.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Radke J., Dittmayer C., Franz J., Thomas C., Mothes R., et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat Neurosci. 2021;24:168–175. doi: 10.1038/s41593-020-00758-5. [DOI] [PubMed] [Google Scholar]
- 163.Lax S.F., Skok K., Sechner P., Kessler H.H., Kaufmann N., Koelblinger C., et al. Pulmonary Arterial Thrombosis in COVID-19 With Fatal Outcome: Results From a Prospective. Single-Center, Clinicopathologic Case Series, Ann Internal med. 2020;173(5):350–361. doi: 10.7326/M20-2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Piazza G., Morrow D.A. Diagnosis, Management, and Pathophysiology of Arterial and Venous Thrombosis in COVID-19. JAMA. 24, 2020;324:2548–2549. doi: 10.1001/jama.2020.23422. [DOI] [PubMed] [Google Scholar]
- 165.Marini J.J., Gattinoni L. Management of COVID-19 Respiratory Distress. JAMA. 2020:2329–2330. doi: 10.1001/jama.2020.6825. [DOI] [PubMed] [Google Scholar]
- 166.Minz M.M., Bansal M., Kasliwal R.R. Statins and SARS-CoV-2 disease: Current concepts and possible benefits. Diabetes & metabolic syndrome. 2020;14(6):2063–2067. doi: 10.1016/j.dsx.2020.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bikdeli B., Madhavan M.V., Gupta A., Jimenez D., Burton J.R., Der Nigoghossian C., et al. The Global, Pharmacological Agents Targeting Thromboinflammation in COVID-19: Review and Implications for Future Research. Thromb Haemost. 2020;120(07):1004–1024. doi: 10.1055/s-0040-1713152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Rubin R. Could Statins Do More Than Lower Cholesterol in Patients With COVID-19? JAMA. 24, 2021;325:2424–2425. doi: 10.1001/jama.2021.8201. [DOI] [PubMed] [Google Scholar]
- 169.Lohia P., Kapur S., Benjaram S., Mir T. Association between antecedent statin use and severe disease outcomes in COVID-19: A retrospective study with propensity score matching. J Clin Lipidol. 2021;15(3):451–459. doi: 10.1016/j.jacl.2021.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kashour T., Halwani R., Arabi Y.M., Sohail M.R., O'Horo J.C., Badley A.D., et al. Statins as an adjunctive therapy for COVID-19: the biological and clinical plausibility. Immunopharmacol Immunotoxicol. 2021;43(1):37–50. doi: 10.1080/08923973.2020.1863984. [DOI] [PubMed] [Google Scholar]