

Abstract
Adrenocortical cancer (ACC) is a rare cancer of the adrenal gland. Several driver mutations have been identified in both primary and metastatic ACCs, but the therapeutic options are still limited. We performed whole-genome and transcriptome sequencing on seven patients with metastatic ACC. Integrative analysis of mutations, RNA expression changes, mutation signature, and homologous recombination deficiency (HRD) analysis was performed. Mutations affecting CTNNB1 and TP53 and frequent loss of heterozygosity (LOH) events were observed in our cohort. Alterations affecting genes involved in cell cycle (RB1, CDKN2A, CDKN2B), DNA repair pathways (MUTYH, BRCA2, ATM, RAD52, MLH1, MSH6), and telomere maintenance (TERF2 and TERT) consisting of somatic and germline mutations, structural variants, and expression outliers were also observed. HRDetect, which aggregates six HRD-associated mutation signatures, identified a subset of cases as HRD. Genomic alterations affecting genes involved in epigenetic regulation were also identified, including structural variants (SWI/SNF genes and histone methyltransferases), and copy gains and concurrent high expression of KDM5A, which may contribute to epigenomic deregulation. Findings from this study highlight HRD and epigenomic pathways as potential therapeutic targets and suggest a subgroup of patients may benefit from a diverse array of molecularly targeted therapies in ACC, a rare disease in urgent need of therapeutic strategies.
Keywords: neoplasm of the adrenal gland
INTRODUCTION
Adrenocortical cancer (ACC) is a rare genitourinary malignancy, with an incidence of 1–2 cases per million patient-years; of these, ∼40% present with metastatic disease (Ng and Libertino 2003). Although most cases are localized, metastatic disease has a poor prognosis, with a 5-yr survival rate of ∼15% (Fassnacht et al. 2009). In addition, in more than one-half of cases, the tumors secrete steroid hormones, which can lead to Cushing's syndrome and medical complications thereof (Ng and Libertino 2003).
The current standard treatment of ACC is limited to cytotoxic chemotherapy (etoposide, doxorubicin, and cisplatin [EDP]) and the adrenolytic agent mitotane. The FIRM-ACT phase 3 clinical trial has established the combination of mitotane and triplet chemotherapy as a standard treatment for advanced ACC, with a median overall survival of 14.8 mo (Fassnacht et al. 2012). There is no established second-line therapy, and small phase 2 trials evaluating different targeted agents in all comers have shown no clear clinical benefit. The vascular endothelial growth factor (VEGF) antibody bevacizumab in combination with capecitabine (Wortmann et al. 2010), VEGF receptor inhibitors axitinib (O'Sullivan et al. 2014) or sunitinib (Kroiss et al. 2012), epidermal growth factor receptor (EGFR) inhibitor erlotinib in combination with gemcitabine (Quinkler et al. 2008), or the insulin-like growth factor type 1 receptor (IGF1R) antibody cixutumumab (Lerario et al. 2014) or inhibitor linsitinib (Fassnacht et al. 2015) have shown limited activity. Immune checkpoint inhibitors have also been investigated. A phase Ib expansion cohort from the JAVELIN solid tumor trial showed a response rate of 6% to the PD-L1 antibody avelumab (Le Tourneau et al. 2018). A phase II study of the PD-1 antibody pembrolizumab had an overall response rate of 23%; two of six patients with mismatch repair–deficient tumors (identified by immunohistochemistry [IHC]) had a response to therapy (Raj et al. 2020). In this context, better treatments are sorely needed.
Although studies of targeted agents have shown poor overall outcomes, molecular characterization of ACC may inform genetic changes involved in cancer progression and reveal potential therapeutic targets. Studies leveraging whole-exome sequencing and single-nucleotide polymorphism (SNP) and expression arrays in ACC have revealed recurrent mutations in CTNNB1, TP53, MEN1, PRKAR1A, RPL22, TERF2, TERT, CCNE1, RB1, and NF1 genes, frequent whole-genome doubling events, large-scale loss of heterozygosity (LOH), and increased ERBB4, IGF2, WNT, retinoic acid receptor, GPCR (Gq, G, Gi), and PDGFR signaling (Giordano et al. 2003; Tissier et al. 2005; Assié et al. 2014; Zheng et al. 2016; Gara et al. 2018; Fojo et al. 2020).
Here we describe the genomic profiles from whole-genome and transcriptome analysis (WGTA) of seven advanced ACC tumors, with detailed clinical and therapeutic annotations. These patients underwent molecular analysis as part of the personalized medicine program at BC Cancer (NCT02155621; Pleasance et al. 2020). In addition to identifying previously described recurrent alterations affecting genes involved in the WNT pathway, DNA repair, and cell cycle regulation, our analysis revealed frequent alterations in genes involved in epigenetic regulation. Specifically, we identified multiple copy gains affecting KDM5A, a lysine demethylase that antagonizes the histone methyltransferase activity associated with the tumor suppressor MEN1 (Feng et al. 2017), a well-described tumor suppressor in ACC. Multiple potentially actionable therapeutic targets were revealed from profiling metastatic ACC tumors, and treatment and response to systemic therapies are reported.
RESULTS
Clinical Presentations and Treatment Outcomes
Baseline characteristics at the time of diagnosis of metastatic ACC are summarized in Table 1. Overall, six of seven patients were female. The median age was 40 (range, 19–54), and five of seven patients had symptoms of cortisol excess, with one patient also having symptoms of aldosterone excess and another also having symptoms of testosterone excess. The clinical course is outlined in Figure 1. Standard chemotherapy and mitotane were given to four of seven patients, with an additional patient receiving this treatment in the adjuvant setting (Case 3). There were no responses to treatment except for Case 5 (response to single-agent mitotane, which had also been used in the adjuvant setting) and Case 6 (response to EDP chemotherapy without mitotane). Case 5 also showed stable disease for 8.5 mo on olaparib. There were no adverse events leading to treatment discontinuation, except for Case 6 in whom an immune checkpoint inhibitor led to autoimmune hepatitis requiring treatment discontinuation and corticosteroids.
Table 1.
Presenting clinical and histopathological characteristics of seven patients with metastatic adrenocortical carcinoma
| Case number | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| Age, gender | 40, M | 19, F | 24, F | 54, F | 46, F | 34, F | 48, F |
| Elevated cortisol | No | Yes | Yes | No | Yes | Yes | Yes |
| Other hormone elevation | No | No | No | No | Aldosterone | Testosterone | No |
| Source of tissue for WGTA | Primary | Primary | Soft tissue metastasis | Lymph node | Lung | Primary | Primary |
| Histopathology score | |||||||
| >5 mitotic figures/50 HPF | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Clear or vacuolated cells <25% | n/a | 1 | 0 | 1 | n/aa | 1 | n/a |
| Atypical mitotic figures | n/a | 1 | 0 | 1 | 0 | 1 | 1 |
| Necrosis | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Capsular invasion | n/a | 1 | 1 | 0 | 0 | 1 | n/a |
| Nuclear grade III or IV | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Diffuse architecture | 1 | 1 | 1 | 1 | 1 | 1 | n/a |
| Venous invasion | n/a | 1 | 0 | 0 | 0 | 1 | n/a |
| Sinusoidal invasion | n/a | 1 | 0 | 1 | 0 | 1 | n/a |
| Specimen type | Biopsy | Resection | Resection | Resection | Resection | Resection | Biopsy |
| Score: Weiss (modified Weiss) | 4/9 (3/7) | 9/9 (7/7) | 5/9 (4/7) | 7/9 (6/7) | * | 9/9 (7/7) | 4/9 (4/7) |
(M) Male, (F) female, (WGTA) whole-genome and transcriptome analysis, (HPF) high-power field, (n/a) not applicable, (*) oncocytic variant.
Figure 1.

Treatment and outcomes. The gray bar represents the time from diagnosis of metastatic disease to the last follow-up or death, with colored sections representing the time on treatment for different systemic agents.
Genomic Analyses
Sequencing coverage, tumor content, and tumor ploidy for all seven cases is shown in Table 2. All patients had advanced disease, with four of the biospecimens for sequencing acquired from the primary site and three from metastatic sites. Somatic alterations including small mutations/indels, copy-number alterations (CNAs), structural variant data, and the gene expression (the number of reads per kilobase of transcript per million mapped reads [RPKM]) data for each case presented in the paper have been deposited at the Michael Smith Genome Sciences Centre at BC Cancer (BCGSC; https://bcgsc.ca/downloads/POG_ACC/) and are highlighted below.
Table 2.
Sequencing coverage, tumor content, and tumor ploidy
| Case ID | Tumor genome sequencing coverage | RNA sequencing coverage (no. of paired reads) | Tumor content (%) | Ploidy | Ts/Tv |
|---|---|---|---|---|---|
| Case1 | 74× | N/A | 40 | Triploid | 0.691 |
| Case2 | 98× | 331M | 86 | Triploid | 0.749 |
| Case3 | 84× | 167M | 54 | Diploid | 0.541 |
| Case4 | 91× | 192M | 80 | Triploid | 0.79 |
| Case5 | 59× | 213M | 84 | Triploid | 1.177 |
| Case6 | 107× | 286M | 80 | Tetraploid | 0.964 |
| Case7 | 96× | 208M | 80 | Diploid | 2.005 |
(Ts/Tv) Transition to transversion ratio, (N/A) not applicable, (M) million.
Somatic Copy-Number Alterations and Loss of Heterozygosity
Large-scale LOH affecting multiple whole chromosomes was observed in six of the seven samples (Fig. 2A). Chromosome 17 was affected in all cases (with lesser extent in Case 2) and Chromosomes 2, 4, and 6 were also largely affected in five cases. Frequent copy-number gains were observed in cases 4, 6, and 7 and these were accompanied by frequent copy-number losses in Cases 4 and 6 (Fig. 2B). Large-scale copy-number losses were also detected in Case 5. Gene amplification events were not seen in Case 7, and Cases 1 and 2 have very few amplification events. In Case 5, arm-level amplification was observed on Chromosome 12 (Ch12p) together with focal amplification events on the other arm (Ch12q).
Figure 2.
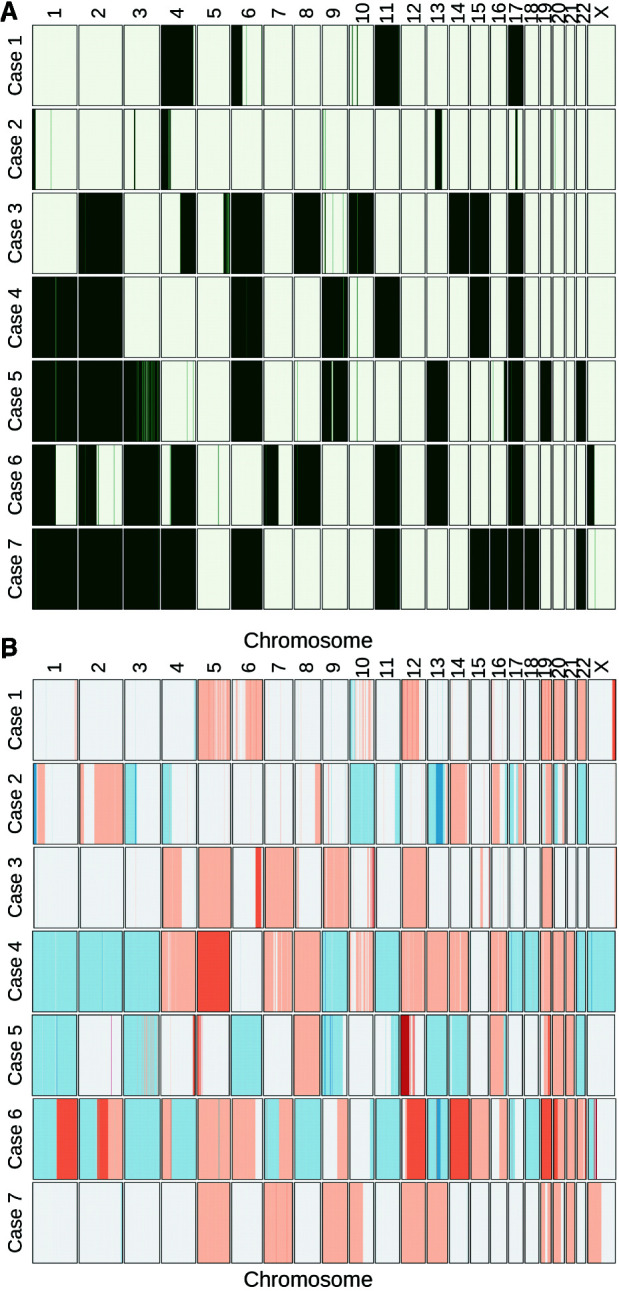
Loss of heterozygosity (LOH) events (A) and copy-number alterations (B) in seven ACC tumors. LOH events are indicated in green, copy gains are indicated in red, and copy losses are indicated in blue.
Evaluation of genes affected by copy gains and losses revealed recurrent gains affecting KDM5A. A gain of two or more copies of the KDM5A gene on Chromosome 12 was observed in four cases and a single copy gain was observed in one case (Case 4) (Fig. 3A). KDM5A encodes a demethylase that antagonizes the histone methyltransferase activity promoted by MEN1, a gene that is recurrently altered in ACC. KDM5A gene expression was higher in cases with multiple copies gains, supporting the hypothesis that KDM5A activity may be elevated and contribute to epigenetic dysregulation in cases with KDM5A copy gains (Fig. 3B). TERT promoter hotspot mutations were frequently identified in primary ACC tumors (Liu et al. 2014). We did not observe TERT hotspot mutations; however, all samples had copy gains affecting TERT on Chromosome 5 and one sample was found to have a somatic variant of unknown significance affecting TERT (R447C) (Fig. 3A). Copy gains on Chromosome 16 affecting TERF2, a member of the telomere nucleoprotein complex involved in telomerase regulation and telomere maintenance, was also observed in four cases.
Figure 3.

(A) OncoPrint describing alterations in previously identified driver genes and in genes involved in signaling processes frequently affected in our cohort. (B) KDM5A expression in cases with (≥2) and without (<2) multiple copy gains. (C) homologous recombination deficiency (HRD) score, single base substitution signatures (SBS3 and SBS8), structural variant signatures (SV signature 3 and 5), fraction of deletions with microhomology, and the aggregate HRDetect score in the cohort. (D) Hierarchical clustering of ACC tumors with transcriptome profiles (biopsy site is indicated for each case).
Copy losses affecting the genes involved in the cell cycle were observed including homozygous loss of CDKN2A and CDKN2B (Case 4) and RB1 (Cases 2 and 6). Copy gains, but not amplification, of CCNE1 were noted in Cases 6 and 7. NF1, another recurrently altered gene in ACC, was subjected to a homozygous deletion in one case (Case 3).
Single-Nucleotide Variants and Indels
The variants highlighted in this section are listed in Table 3. Several alterations predicted to affect diverse DNA repair pathways were observed. Somatic alterations in genes involved in the homologous recombination repair pathways include a homozygous in-frame deletion of ATM (Case 6), a heterozygous BRCA2 nonsense mutation (Case 4); other alterations affecting DNA repair pathways include a pathogenic germline mutation affecting a base excision repair gene, MUTYH (Case 1), and a homozygous frameshift mutation affecting MSH6 and a homozygous somatic mutation in MLH1 (Case 7; Fig. 3A).
Table 3.
Variants of interest
| Gene | Chromosome | HGVS DNA reference | HGVS protein reference | Variant type | Predicted effect | dbSNP/dbVar ID | Genotype | ClinVar accession number |
|---|---|---|---|---|---|---|---|---|
| CTNNB1 | 3 | c.133T > G | p.Ser45Ala | SNV | Missense | rs121913407 | Het | |
| CTNNB1 | 3 | c.133T > G | p.Ser45Ala | SNV | Missense | rs121913407 | Hom | |
| TERT | 5 | c.1339C > T | p.Arg447Cys | SNV | Missense | rs1304418053 | Het | |
| ATM | 11 | c.1837_1839delGTG | p.Val613del | DEL | In-frame deletion | Hom | SCV002062047 | |
| BRCA2 | 13 | c.2368G > T | p.Glu790* | SNV | Nonsense | rs398122746 | Het | |
| MUTYH | 1 | c.1145G > A | p.G382D | SNV | Missense | rs36053993 | Het | |
| MSH6 | 2 | c.3644T > A | p.Leu1215* | SNV | Nonsense | Hom | SCV002062052 | |
| MLH1 | 3 | c.884G > T | p.Ser295Ile | SNV | Splice region variant | Hom | SCV002062048 | |
| TP53 | 17 | c.499C > T | p.Gln167* | SNV | Nonsense | rs1555526097 | Het | |
| TP53 | 17 | c.742C > T | p.Arg248Trp | SNV | Missense | rs121912651 | Hom | |
| ARID1B | 6 | c.4084C > T | p.Gln1362* | SNV | Nonsense | Hom | SCV002062056 | |
| SMARCA4 | 19 | c.276G > C | p.Gln92His | SNV | Missense | Het | SCV002062051 | |
| SMARCC2 | 12 | c.3221delC | p.Pro1074fs | SNV | Frameshift variant | rs752788954 | Het | |
| KMT2C | 7 | c.8390delA | p.Lys2797fs | SNV | Frameshift variant | rs747256476 | Het | |
| KMT2A | 11 | c.2318dupC | p.Ser774fs | SNV | Frameshift variant | Hom | SCV002062055 | |
| ATRX | X | c.6235C > T | p.Arg2079* | SNV | Nonsense | Het | SCV002062053 | |
| ATRX | X | c.4377_4379delGGA | p.Glu1460del | DEL | Disruptive in-frame deletion | rs587780286 | Het | |
| ATRX | X | c.3904delA | p.Arg1302fs | SNV | Frameshift variant | Het | SCV002062054 | |
| PIK3CA | 3 | c.2086G > A | p.Gly696Arg | SNV | Missense | Het | SCV002062049 | |
| JAK2 | 9 | c.2385G > T | p.Arg795Ser | SNV | Missense | Hom | SCV002062050 |
(HGVS) Human Genome Variation Society, (SNV) single-nucleotide variant, (Het) heterozygous, (Hom) homozygous.
Somatic mutations affecting CTNNB1 and TP53 are the most recurrent driver events in ACC (Zheng et al. 2016) and were observed in Cases 1, 5, 6, and 7, and co-occurred in Case 1 (Fig. 3A). The TP53 variants comprised a nonsense mutation, a missense mutation associated with loss of function and a homozygous deletion. The recurrent gain-of-function S45P mutation affecting CTNNB1 was detected in two samples, with a third sample affected by an in-frame deletion of exons 2–3 of CTNBB1 that deletes the first 80 amino acids of the protein, leading to the loss of inhibitory amino-terminal phosphorylation sites (Maharjan et al. 2018).
Mutations affecting MEN1, a gene involved in epigenetic regulation through its association with the histone methyltransferase MLL1/KMT2A, are recurrent in ACC but were not observed in this cohort. On the other hand, we observed several somatic mutations affecting SWI/SNF chromatin remodeling genes (ARID1B, SMARCA4, SMARCC2) and histone methyltransferase genes (KMT2C, KMT2A, and ATRX) (Fig. 3A).
Alterations affecting genes involved in oncogenic signaling pathways, including a somatic missense mutation of unknown significance in PIK3CA (Case 2), were detected (Fig. 3A). A novel homozygous missense mutation affecting the kinase domain of JAK2 was also observed in Case 5 and may affect JAK2-mediated cytokine signaling (Fig. 3A).
Structural Variant Analysis
Few structural variants affecting cancer genes were observed in this case series. Case 1 was found to harbor a translocation between CDKN2C and FAF1 (1:50978798|1:51436870), which may disrupt the function of CDKN2C involved in cell cycle inhibition. Case 5 harbored a structural variant in RAD52 (12:1029459|12:1095898), which is predicted to cause a duplication of one-half of the gene and the creation of an additional truncated RAD52 protein that lacks the carboxy-terminal 181–412 residues (Fig. 3A). The same case was also found to harbor an interchromosomal translocation affecting STK11 and a noncoding region that was predicted to disrupt the first 300 residues of STK11 and is an inferred loss-of-function mutation. A complete list of high-quality structural variants (see Methods) have been made publicly accessible for download (https://bcgsc.ca/downloads/POG_ACC/).
Transcriptome Analysis
EZH2, histone methyltransferase, and catalytic subunit of the PRC2 complex were highly expressed in two patients compared to the Cancer Genome Atlas (TCGA) ACC data set (96th percentile in Cases 2 and 91st percentile in Case 6). Both cases also had homozygous loss of RB1, in agreement with previous findings that loss of this tumor suppressor promotes EZH2 expression in ACC (median EZH2 expression is 7.4 RPKM in RB1 mutant vs. 2.6 RPKM in RB1 wild-type cases in our cohort) (Drelon et al. 2016).
IGF1R signaling has been shown to be frequently activated in ACCs (Weber et al. 1997), and high expression of IGF1R (Cases 3 and 7; 100th and 95th percentile, respectively), IGF2R (Cases 3, 6, and 7; 100th, 85th, and 89th percentile, respectively), IGF2 (Case 5; 82th percentile) and IGF1 (Cases 2, 3, and 5; 86th, 91st, and 85th percentile, respectively) compared to the TCGA ACC data set was observed, indicating potential increased activation of the IGF1R signaling axis.
Clustering based on genomic, methylation, and transcriptome profiles has revealed molecular subgroups of ACC as well as signatures associated with adrenocortical differentiation (Zheng et al. 2016). Transcriptome clustering using previously defined gene sets identified three different expression subtypes, with the rarest steroid-phenotype-low + proliferation expression subtype lacking in our cohort (Fig. 3A). Three patients (Cases 4, 5, and 7) clustered with the steroid-phenotype-high group, two patients (Cases 2 and 6) with the steroid-phenotype-high + proliferation group, and one patient with the steroid-phenotype-low group. Evaluation of the adrenocortical differentiation gene expression signature score (ADS) revealed four of six patients with positive ADS values supporting the differentiated state of these cases, one of which harbored a CTNNB1 somatic mutation and exhibited a very high ADS value.
Previously described expression signatures were not closely aligned with driver mutations in our cohort; thus, we sought to determine if hierarchical clustering in our cohort may reveal expression profiles associated with driver status. Hierarchical clustering of transcriptome profiles from six patients with available RNA sequencing (RNA-seq) distinguished three pairs (Fig. 3D). The three pairs were characterized by different driver gene mutations, TP53 mutation or loss was detected in pair 1 (Cases 7 and 3), CTNNB1 mutation or deletion was identified in pair 2 (Cases 5 and 6), whereas tumors in pair 3 (Cases 2 and 4) had no mutations in either TP53 or CTNNB1 but had other gene alterations. In addition, pair 1 and 2 were clustered together and were distinguished from tumors with no TP53 and CTNNB1 alterations. These data suggest genomic alterations might lead to gene expression profiles that are characteristic of tumors with specific driver gene mutations.
Integrative Analysis
Our observations, and previous reports in ACC (Zheng et al. 2016; Fojo et al. 2020), have indicated a significant subset of ACC may be associated with homologous recombination (HR) repair deficiency, although the impact of mutations affecting several HR genes is unclear. To investigate the impact of HR mutations in our cohort, we performed mutation signature analysis and investigated homologous recombination repair using HRDetect that aggregates four single-nucleotide variant (SNV) and structural variant (SV) mutation signatures, homologous recombination deficiency (HRD) index, and the proportion of small deletions associated with microhomology (Fig. 3C; Davies et al. 2017; Zhao et al. 2017). Case 5 had a high proportion of mutations attributed to Catalog Of Somatic Mutations in Cancer (COSMIC) single base substitution 3 (SBS3) signature, which previously has been associated with HRD (Figs. 3A and 4A), and HRDetect results were consistent with this tumor being deficient in homologous recombination repair (Fig. 3C), despite the absence of a BRCA1 or BRCA2 pathogenic mutation (Davies et al. 2017). In addition to the structural variant in RAD52, copy losses of several DNA repair genes including BRCA2, CHEK2, and ATM were detected in this case (Fig. 3A). The patient received the PARP inhibitor olaparib and had disease stabilization for 8.5 mo. Case 2 was also predicted to be HRD by HRDetect and similarly lacked a BRCA1/2 alteration. Interestingly, Case 4 with the heterozygous BRCA2 mutation and strong SBS3 signature was predicted to be HR-proficient by HRDetect, in line with the presence of a functional wild-type allele. Apart from HRD-associated signatures, Case 3 and Case 6 had a high proportion of mutations attributed to SBS31, SBS35, and DBS5, all previously associated with prior platinum drug treatment (Fig. 4A,B). Case 3 received platinum treatment prior to metastatic disease (86 d), whereas Case 6, which exhibited very high SBS31 and SBS35 exposures (ranked second and first, respectively, compared against 570 cancer genomes of mixed origins), was treated with platinum for 163 d prior to biopsy. The presence of genome signatures consistent with HRD provides support for involvement of the HR pathway in ACC pathogenesis and may represent a compelling target in ACC as in other tumors of the endocrine system.
Figure 4.

The contribution of previously reported single base substitution (SBS) (A), doublet base substitution (DBS) (B), and indel (ID) (C) mutational signatures (Catalog of Somatic Mutations in Cancer [COSMIC v3.1]) in ACC tumors are compared against 570 cancer genomes of mixed origins (Pleasance et al. 2020).
Case 1 with the pathogenic germline MUTYH mutation was characterized by a high proportion of mutations attributed to SBS18 (ranked 20th compared against 570 cancer genomes), previously associated with MUTYH deficiency (Fig. 4A; Thibodeau et al. 2019). Case 7 with deleterious mutations in MSH6 and MLH1 demonstrated a high mutation burden (whole-genome tumor mutation burden of 28.6 mutations/Mb), and microsatellite instability (39% of microsatellites were unstable; Niu et al. 2014). Mutation signature analysis also revealed a high proportion of mutations attributed to SBS26, SBS44, DBS7, ID2, and ID7, all previously associated with mismatch repair deficiency (MMRd) (Fig. 4; Drost et al. 2017). SBS44 (ranked eighth), DBS7 (ranked sixth), ID2 (ranked 11th), and ID7 (ranked fourth) were particularly high compared to a large, previously sequenced cancer cohort. Interestingly, Case 7 also harbored mutations consistent with signatures SBS7c and SBS38 (Fig. 4A), both of which are primarily observed in melanomas (Saini et al. 2016). We and others have reported SBS38 in non-UV-associated melanoma subtypes, raising the possibility of a common underlying mutational process in ACC and a subset of non-UV-exposed melanomas (Newell et al. 2020; Pleasance et al. 2020). After progressing on EDP chemotherapy and mitotane, the patient was initiated on avelumab, but unfortunately died before completing one cycle because of rapidly progressing disease.
DISCUSSION
There are relatively few studies of comprehensive genomic characterizations of ACC (Zheng et al. 2016; Fojo et al. 2020). Current exome sequencing studies have revealed a few recurrently altered genes in ACC, with examples of variants of uncertain significance impacting multiple genes aligned to several cancer associated pathways. Here we describe a cohort of patients with metastatic ACC profiled using whole-genome and transcriptome analysis (WGTA) and provide detailed clinical annotation.
Our study is limited by the small sample size and heterogeneity of the cohort. Four out of seven samples were obtained from primary adrenal tumors, whereas the remaining three were obtained through biopsies of metastatic lesions. This introduces significant heterogeneity. The fact that four patients received systemic therapy prior to biopsy, including two patients who received cytotoxic chemotherapy, also creates significant bias. Our analysis highlights the genomic diversity of ACC, which makes inferring specific conclusions about the management of this rare disease challenging. The clinical–genomic correlates are presented as hypothesis-generating findings.
Whole-genome sequencing allows for detection of potentially disruptive structural variants, including the STK11 and RAD52 variants detected here, and evaluation of genome-wide mutation signature phenotypes including HRDetect and microsatellite instability. Transcriptome sequencing informs on the impact of observed genomic alterations, differentiation, and revealed distinct expression profiles that were correlated with driver gene mutation status.
Consistent with previous studies, somatic mutations in CTNNB1 and/or TP53 were observed in our cohort, including an exon level deletion affecting CTNNB1 similar to that described in 7% of ACC cases (Maharjan et al. 2018). Although no somatic mutations in other previously reported driver genes were detected in our cohort, we observed the activation of pathways implicated in ACCs. We observed copy-number gains of CCNE1 in six cases and detected homozygous loss of RB1 in two cases and homozygous loss of CDKN2A and CDKN2B in one case, suggesting the deregulation of the cell cycle and suggesting that targeting the cell cycle pathway may be beneficial in a subset of patients. We did not detect MEN1 mutations in our cohort; however, we did observe multiple copy gains and concurrent high expression of KDM5A, a demethylase that antagonizes MEN1-mediated methylation. A single TCGA ACC sample harbored a KDM5A amplification, and this case was also negative for MEN1 mutation (Zheng et al. 2016). Increased KDM5A expression may represent an alternate mechanism of epigenetic dysregulation compared to MEN1 loss of function and is the subject of active investigation as an oncology target (Yang et al. 2021).
In addition to alterations in ACC driver genes, alterations consistent with deregulation of epigenetic signaling and DNA repair processes were observed. We identified somatic mutations in SMARCA4, SMARCC2, and ARID1B, suggesting deregulation of the chromatin remodeling complex, SWI/SNF. Inactivating mutations in genes encoding SWI/SNF subunits are frequently observed in various cancer types and have been associated with deficiency in DNA repair as well as response to multiple targeted therapies (Mittal and Roberts 2020; Ribeiro-Silva et al. 2019), but the role of the SWI/SNF complex in ACC tumorigenesis and therapeutic response has not yet been investigated. In addition, somatic variants were detected in KMT2A and KMT2C histone methyltransferases and in ATRX and DAXX as previously reported in primary ACCs (Zheng et al. 2016). We also observed increased EZH2 expression, particularly in cases with RB1 loss. EZH2 inhibitors are approved for use in EZH2 mutant follicular lymphoma and in SMARCB1/INI1 deficient epitheliod sarcoma (Groisberg 2020) and may be therapeutically beneficial in a subset of SWI/SNF deficient, or EZH2 overexpressing ACCs.
Four out of the seven cases were associated with mutations in DNA repair genes, including somatic alterations in BRCA2, MLH1, MSH6, ATM, and a germline pathogenic mutation affecting MUTYH. Mutations in the DNA-mismatch repair pathway have been observed in primary ACCs (Zheng et al. 2016) and ACC has been shown to be a Lynch syndrome–associated cancer (Raymond et al. 2013). Germline MUTYH mutations are also known to be associated with ACC predisposition; however, germline alterations affecting HR-related genes have only been rarely attributed to ACC (El Ghorayeb et al. 2016; Xie et al. 2018), suggesting a potential subset of ACCs is characterized by HRD derived from somatic alterations as opposed to germline mutation. Two cases in our cohort demonstrating a strong HRD signature lacked mutations in BRCA1/2, suggesting either epigenomic dysregulation or alterations affecting other HR genes may contribute to the HRD in ACC. Profiling mutational signatures associated with HRD may reveal ACC patients that can benefit from platinum-based therapy or PARP inhibitors, as was observed in other tumor types (Zhao et al. 2017; Chopra et al. 2020; Golan et al. 2021).
Despite numerous shared alterations, this cohort also illustrates the high degree of heterogeneity that exists within ACC and represents a challenge to successfully propose targeted therapy for ACCs. One patient (Case 6) had a partial response to combination chemotherapy (etoposide, doxorubicin, and cisplatin), and another patient (Case 5) has had sustained benefit from mitotane and subsequently from olaparib, whereas other treatments only led to minimal clinical benefit. Therapy informed by WGTA analysis was given for Case 5, Case 6, and Case 7. Case 5 had evidence of HRD despite the absence of a BRCA1 or BRCA2 mutation and received the PARP inhibitor olaparib resulting in disease stabilization for 8.5 mo. This patient had a potential disruptive structural variant in RAD52, which participates in HR. In mammals RAD52 deletion alone does not compromise HR repair; however, depletion of human RAD52 is synthetically lethal with mutations in other members of the HR pathways including BRCA1, BRCA2, PALB2, and RAD51 paralogs (Feng et al. 2011; Chun et al. 2013; Lok et al. 2013). In this case the structural variant in RAD52 together with the copy losses of other HR genes might have resulted in the HRD phenotype and partial response to PARPi. The patient showed mixed response to the subsequent carboplatin and paclitaxel combination with disease progression 5 mo after initiation of treatment. Case 6 was enrolled in a phase 1 clinical trial of a TTK inhibitor based on the presence of a CTNNB1 activating mutation and preclinical evidence (Zaman et al. 2017), but did not demonstrate clinical benefit. This patient subsequently received the PARP inhibitor olaparib based on the presence of an in-frame deletion affecting ATM with loss of heterozygosity and low ATM gene expression based on transcriptome and immunohistochemistry, although this sample was not classified as HRD by HRDetect. A phase 2 study in gastric cancer (Bang et al. 2015) suggested improved overall survival for patients treated with a PARP inhibitor and a taxane, when compared to a taxane alone. This benefit was preferentially seen in a cohort of patients with low ATM expression by immunohistochemistry, although a confirmatory phase 3 study was negative (Bang et al. 2017). In this case, the PARP inhibitor olaparib was combined with temozolomide based on the recommended dose and schedule derived from a phase 1 clinical trial in small-cell lung cancer (Farago et al. 2018), but unfortunately the patient did not benefit from this combination. Case 7 had evidence of microsatellite instability and mismatch repair deficiency; and based on these findings the patient was initiated on an immune checkpoint inhibitor (Le et al. 2017), but the patient's clinical status had rapidly deteriorated and she passed away within a few days of initiating treatment without a response assessment.
Overall, the findings of this cohort suggest that molecular profiling of ACC could be utilized to target specific treatments to certain alterations affecting several potentially actionable pathways including DNA repair, cell cycle, oncogenic signaling pathways, and genes involved in epigenetic regulation found in specific subpopulations. Comprehensive genomic and transcriptome analysis of ACC has the potential to reveal therapeutic strategies tailored to patients’ individual tumors. Although the lack of response seen in previous studies of targeted agents may be related to the poor activity of these drugs, it is also possible that the heterogeneity of the underlying disease contributed to the disappointing results observed. To better guide future studies, more information is required on the molecular makeup of metastatic ACC.
METHODS
Sample Collection and Preparation
Whole-genome sequencing of tumors and matched normal tissue was performed in seven cases; four of the biopsies were obtained from primary adrenal tumors, whereas the remaining three were biopsies of metastatic lesions, as outlined in Table 1. Whole-transcriptome sequencing was performed on six of the seven tumors. Patients with locally advanced or metastatic ACC with a life expectancy of greater than 6 mo were enrolled on the Personalized OncoGenomics (POG) project (ClinicalTrials.gov identified: NCT02155621) at BC Cancer, in Vancouver, Canada (Laskin et al. 2015; Jones et al. 2017).
Tumor sections were reviewed by a pathologist to determine tumor content and cellularity, and remaining sections were used for DNA and RNA preparation. Peripheral blood was collected for use as a normal tissue reference and pathogenic and likely pathogenic germline variants in 98 known cancer predisposition genes were analyzed as outlined in Dixon et al. (2020). DNA and RNA were coextracted for the construction of polymerase chain reaction (PCR)-free DNA libraries and poly(A) selected RNA libraries as described previously (Sheffield et al. 2015).
Sequencing and Bioinformatics
Tumor genomes sequencing was performed to 80× coverage and normal genomes to 40× coverage on Illumina HiSeq platform with 125- or 150-bp paired-end reads. Illumina HiSeq2500 or NextSeq500 were used for RNA sequencing with 150–200 million 75-bp paired-end reads. Sequence reads were aligned to the human reference genome (hg19) by the BWA tool (Li and Durbin 2010) and somatic SNVs and small indels were identified using SAMtools (v0.1.17) (Li et al. 2009) and Strelka (v1.0.6) (Saunders et al. 2012). Regions of copy-number alteration (CNA) and LOHs were determined using CNAseq (v0.0.6) (Jones et al. 2010) and APOLLOH (v0.1.1) (Ha et al. 2012), respectively. Amplification was defined as the ploidy-corrected copy-number gain being greater than the estimated tumor ploidy. Copy gains are defined as an increase in gene copies equivalent to or less than the estimated average ploidy. SVs in RNA-seq data were detected by ABySS v1.3.4 and TransABySS (v1.4.10) (Birol et al. 2009; Simpson et al. 2009) and Chimerascan (v0.4.5) (Iyer et al. 2011) and DeFUSE (v0.6.2) (McPherson et al. 2011); SVs in the DNA were identified using ABySS and TransABySS followed by Manta v1.0.0 (Chen et al. 2016) and Delly v0.7.3 (Rausch et al. 2012). SV calls from multiple algorithms were merged into a consensus caller MAVIS (v2.1.1) that performs a subsequent validation by local assembly (Reisle et al. 2019). Structural variants were filtered for events that were called by multiple tools and were supported by an assembled contig and were deposited for download (https://bcgsc.ca/downloads/POG_ACC/). The contribution of previously reported mutational signatures in COSMIC v3.1 (https://cancer.sanger.ac.uk/signatures; Alexandrov et al. 2020) was calculated using nonnegative least squares optimization. HRDetect scores were computed using a logistic regression model described previously in Davies et al. (2017). Six HRD-associated mutation signatures were used in the algorithm: (i) Signature 3, (ii) Signature 8, (iii) SV signature 3, (iv) SV signature 5, (v) the HRD index, and (vi) the fraction of deletions with microhomology. Somatic SNVs called by Strelka (v1.0.6) were used for single base substitution signature calculation. The contribution of previously reported mutational signatures in COSMIC v3.1 (https://cancer.sanger.ac.uk/cosmic/signatures) was calculated using Monte Carlo Markov chain (MCMC) sampling (https://github.com/eyzhao/SignIT). Somatic, DNA-derived SVs identified by the consensus caller MAVIS were used for SV signature calculation (Reisle et al. 2019). MAVIS calls that were detected by more than one tool and for which the contig could be assembled were included in the analysis, and the contribution of the previously reported SV mutational signatures was calculated using MCMC sampling (https://github.com/eyzhao/SignIT) (Nik-Zainal et al. 2016). HRD index was computed as the arithmetic sum of LOH, telomeric-allelic imbalance (TAI), and large scale transitions (LST) scores (Zhao et al. 2017). All signatures were normalized and log transformed, and a threshold of 0.7 was used as previously reported (Davies et al. 2017; Zhao et al. 2017).
Microsatellite instability (MSI) was calculated by MSIsensor (v0.2) (Niu et al. 2014).
Gene Expression Analysis
A transcriptome of an average depth of 200 million reads was generated from the tumor sample. The RNA sequencing reads were analyzed with JAGuaR (Butterfield et al. 2014) to include alignments to a database of exon junction sequences and subsequent repositioning onto the genomic reference. The RNA sequencing data were processed using the Genome Sciences Centre's wtss (whole-transcriptome shotgun sequencing) pipeline coverage analysis (version 1.1) with the “stranded” option to determine gene and exon read counts and normalized expression level. The level of expression of each gene was determined as the number of RPKM.
Gene expression level was evaluated by converting RPKM of select genes into percentile ranks against transcriptomes of ACC tumors from The Cancer Genome Atlas project (https://portal.gdc.cancer.gov/). Expression subtypes classification shown in Figure 3A (Exp subtype track) was inferred by calculating spearman correlation factor between individual TCGA samples and POG samples. Hierarchical clustering shown in Figure 3D was performed after selecting the top 2500 genes according to the magnitude (from largest to smallest in absolute values) of their coefficients contributing to the first principal component analysis (PCA) component and after conversion of the RPKM values to z-scores using the expression data of six patients with RNA-seq results. Agglomerative clustering method was used to obtain clusters, computing the distance matrix with the Euclidean distance. The adrenocortical differentiation score (ADS) shown in Figure 3A (ADS score track) was calculated by using 25 gene expression as described by Zheng et al. (2016). To obtain the expression subtypes described in Zheng et al. (2016), kmeans clustering was performed on the publicly available mRNA data of 76 TCGA ACC samples with expression subtypes classification (downloaded from https://pancanatlas.xenahubs.net) and the POG mRNA data that was calculated using the TOIL RNA-seq pipeline (Vivian et al. 2017) to minimize computational batch effects between POG samples and recomputed TCGA samples. The TOIL pipeline uses CutAdapt for adapter trimming and STAR (Dobin et al. 2013) to generate alignments and performs quantification using RSEM (Li and Dewey 2011) and Kallisto (Bray et al. 2016).
ADDITIONAL INFORMATION
Data Deposition and Access
The data sets generated and analyzed during the current study have been deposited at the Michael Smith Genome Sciences Centre at BC Cancer (BCGSC; https://bcgsc.ca/downloads/POG_ACC/). The genomic and transcriptome sequencing data sets have been deposited at the European Genome-phenome Archive (EGA; https://ega-archive.org/) as part of the study EGAS00001001159 (Patient 1 EGAD00001002037, Patient 2 EGAD00001002049, Patient 3 EGAD00001003053, Patient 4 EGAD00001004905, Patient 5 EGAD00001003676, Patient 6 EGAD00001005844, Patient 7 EGAD00001005897).
Ethics Statement
The study was conducted in accordance with the principles of the Declaration of Helsinki and with approval of the Research Ethics Board at BC Cancer, and all participants provided written consent.
Acknowledgments
This work would not be possible without the participation of our patients and families, the POG team, Canada's Michael Smith Genome Sciences Centre technical platforms, the generous support of the BC Cancer Foundation and their donors and Genome British Columbia. The results published here are in part based upon analyses of data generated by the following projects and obtained from dbGaP (http://www.ncbi.nlm.nih.gov/gap): TCGA managed by the National Cancer Institute and National Human Genome Research Institute (http:// cancergenome.nih.gov). Data from the Pan-Cancer Analysis of Whole Genomes (PCAWG) managed by the International Cancer Genome Consortium was retrieved from https://dcc.icgc.org/pcawg.
Author Contributions
J.-M.L. and V.C. conceptualized the study. Analyses were performed by J.-M.L., V.C., L.M.W., L.C., and G.W. Provision of patient samples and curation of data was conducted by J.-M.L., C.K.K., D.J.R., and J.L. The original draft was written by J.-M.L., V.C., L.M.W., and M.A.M., J.L., C.K.K., D.J.R., and S.J.M.J. reviewed and edited the manuscript. Funding was acquired by M.M., J.L., and S.J.M.J.
Funding
This work was supported by the BC Cancer Foundation and Genome British Columbia (project B20POG).
Competing Interest Statement
The authors have declared no competing interest.
REFERENCES
- Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, et al. 2020. The repertoire of mutational signatures in human cancer. Nature 578: 94–101. 10.1038/s41586-020-1943-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assié G, Letouzé E, Fassnacht M, Jouinot A, Luscap W, Barreau O, Omeiri H, Rodriguez S, Perlemoine K, René-Corail F, et al. 2014. Integrated genomic characterization of adrenocortical carcinoma. Nat Genet 46: 607–612. 10.1038/ng.2953 [DOI] [PubMed] [Google Scholar]
- Bang YJ, Im SA, Lee KW, Cho JY, Song EK, Lee KH, Kim YH, Park JO, Chun HG, Zang DY, et al. 2015. Randomized, double-blind phase II trial with prospective classification by ATM protein level to evaluate the efficacy and tolerability of olaparib plus paclitaxel in patients with recurrent or metastatic gastric cancer. J Clin Oncol 33: 3858–3865. 10.1200/JCO.2014.60.0320 [DOI] [PubMed] [Google Scholar]
- Bang YJ, Xu RH, Chin K, Lee KW, Park SH, Rha SY, Shen L, Qin S, Xu N, Im SA, et al. 2017. Olaparib in combination with paclitaxel in patients with advanced gastric cancer who have progressed following first-line therapy (GOLD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 18: 1637–1651. 10.1016/S1470-2045(17)30682-4 [DOI] [PubMed] [Google Scholar]
- Birol I, Jackman SD, Nielsen CB, Qian JQ, Varhol R, Stazyk G, Morin RD, Zhao Y, Hirst M, Schein JE, et al. 2009. De novo transcriptome assembly with ABySS. Bioinformatics 25: 2872–2877. 10.1093/bioinformatics/btp367 [DOI] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L. 2016. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34: 525–527. 10.1038/nbt.3519 [DOI] [PubMed] [Google Scholar]
- Butterfield YS, Kreitzman M, Thiessen N, Corbett RD, Li Y, Pang J, Ma YP, Jones SJ, Birol I. 2014. JAGuaR: junction alignments to genome for RNA-seq reads. PLoS ONE 9: e102398. 10.1371/journal.pone.0102398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S, Saunders CT. 2016. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32: 1220–1222. 10.1093/bioinformatics/btv710 [DOI] [PubMed] [Google Scholar]
- Chopra N, Tovey H, Pearson A, Cutts R, Toms C, Proszek P, Hubank M, Dowsett M, Dodson A, Daley F, et al. 2020. Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple negative breast cancer. Nat Commun 11: 2662. 10.1038/s41467-020-16142-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J, Buechelmaier ES, Powell SN. 2013. Rad51 paralog complexes BCDX2 and CX3 act at different stages in the BRCA1-BRCA2-dependent homologous recombination pathway. Mol Cell Biol 33: 387–395. 10.1128/MCB.00465-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, Ramakrishna M, Martin S, Boyault S, Sieuwerts AM, et al. 2017. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med 23: 517–525. 10.1038/nm.4292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon K, Young S, Shen Y, Thibodeau ML, Fok A, Pleasance E, Zhao E, Jones M, Aubert G, Armstrong L, et al. 2020. Establishing a framework for the clinical translation of germline findings in precision oncology. JNCI Cancer Spectr 4: pkaa045. 10.1093/jncics/pkaa045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drelon C, Berthon A, Mathieu M, Ragazzon B, Kuick R, Tabbal H, Septier A, Rodriguez S, Batisse-Lignier M, Sahut-Barnola I, et al. 2016. EZH2 is overexpressed in adrenocortical carcinoma and is associated with disease progression. Hum Mol Genet 25: 2789–2800. 10.1093/hmg/ddw136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost J, van Boxtel R, Blokzijl F, Mizutani T, Sasaki N, Sasselli V, de Ligt J, Behjati S, Grolleman JE, van Wezel T, et al. 2017. Use of CRISPR-modified human stem cell organoids to study the origin of mutational signatures in cancer. Science 358: 234–238. 10.1126/science.aao3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Ghorayeb N, Grunenwald S, Nolet S, Primeau V, Côté S, Maugard CM, Lacroix A, Gaboury L, Bourdeau I. 2016. First case report of an adrenocortical carcinoma caused by a BRCA2 mutation. Medicine (Baltimore) 95: e4756. 10.1097/MD.0000000000004756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farago AF, Drapkin BJ, Charles A, Yeap BY, Heist RS, Azzoli CG, Jackman DM, Marcoux JP, Barbie DA, Myers DT, et al. 2018. Safety and efficacy of combination olaparib (O) and temozolomide (T) in small cell lung cancer (SCLC). JCO 36: 8571. 10.1200/JCO.2018.36.15_suppl.8571 [DOI] [Google Scholar]
- Fassnacht M, Johanssen S, Quinkler M, Bucsky P, Willenberg HS, Beuschlein F, Terzolo M, Mueller HH, Hahner S, Allolio B, et al. 2009. Limited prognostic value of the 2004 International Union Against Cancer staging classification for adrenocortical carcinoma: proposal for a revised TNM classification. Cancer 115: 243–250. 10.1002/cncr.24030 [DOI] [PubMed] [Google Scholar]
- Fassnacht M, Terzolo M, Allolio B, Baudin E, Haak H, Berruti A, Welin S, Schade-Brittinger C, Lacroix A, Jarzab B, et al. 2012. Combination chemotherapy in advanced adrenocortical carcinoma. N Engl J Med 366: 2189–2197. 10.1056/NEJMoa1200966 [DOI] [PubMed] [Google Scholar]
- Fassnacht M, Berruti A, Baudin E, Demeure MJ, Gilbert J, Haak H, Kroiss M, Quinn DI, Hesseltine E, Ronchi CL, et al. 2015. Linsitinib (OSI-906) versus placebo for patients with locally advanced or metastatic adrenocortical carcinoma: a double-blind, randomised, phase 3 study. Lancet Oncol 16: 426–435. 10.1016/S1470-2045(15)70081-1 [DOI] [PubMed] [Google Scholar]
- Feng Z, Scott SP, Bussen W, Sharma GG, Guo G, Pandita TK, Powell SN. 2011. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc Natl Acad Sci 108: 686–691. 10.1073/pnas.1010959107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Ma J, Hua X. 2017. Epigenetic regulation by the menin pathway. Endocr Relat Cancer 24: T147–T159. 10.1530/ERC-17-0298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fojo T, Huff L, Litman T, Im K, Edgerly M, Del Rivero J, Pittaluga S, Merino M, Bates SE, Dean M. 2020. Metastatic and recurrent adrenocortical cancer is not defined by its genomic landscape. BMC Med Genomics 13: 165–167. 10.1186/s12920-020-00809-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gara SK, Lack J, Zhang L, Harris E, Cam M, Kebebew E. 2018. Metastatic adrenocortical carcinoma displays higher mutation rate and tumor heterogeneity than primary tumors. Nat Commun 9: 4172. 10.1038/s41467-018-06366-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano TJ, Thomas DG, Kuick R, Lizyness M, Misek DE, Smith AL, Sanders D, Aljundi RT, Gauger PG, Thompson NW, et al. 2003. Distinct transcriptional profiles of adrenocortical tumors uncovered by DNA microarray analysis. Am J Pathol 162: 521–531. 10.1016/S0002-9440(10)63846-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golan T, O'Kane GM, Denroche RE, Raitses-Gurevich M, Grant RC, Holter S, Wang Y, Zhang A, Jang GH, Stossel C, et al. 2021. Genomic features and classification of homologous recombination deficient pancreatic ductal adenocarcinoma. Gastroenterology 160: 2119–2132.e9. 10.1053/j.gastro.2021.01.220 [DOI] [PubMed] [Google Scholar]
- Groisberg R. 2020. EZH2 inhibition for epithelioid sarcoma and follicular lymphoma. Lancet Oncol 21: 1388–1390. 10.1016/S1470-2045(20)30530-1 [DOI] [PubMed] [Google Scholar]
- Ha G, Roth A, Lai D, Bashashati A, Ding J, Goya R, Giuliany R, Rosner J, Oloumi A, Shumansky K, et al. 2012. Integrative analysis of genome-wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple-negative breast cancer. Genome Res 22: 1995–2007. 10.1101/gr.137570.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer MK, Chinnaiyan AM, Maher CA. 2011. ChimeraScan: a tool for identifying chimeric transcription in sequencing data. Bioinformatics 27: 2903–2904. 10.1093/bioinformatics/btr467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SJ, Laskin J, Li YY, Griffith OL, An J, Bilenky M, Butterfield YS, Cezard T, Chuah E, Corbett R, et al. 2010. Evolution of an adenocarcinoma in response to selection by targeted kinase inhibitors. Genome Biol 11: R82. 10.1186/gb-2010-11-8-r82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MR, Lim H, Shen Y, Pleasance E, Ch'ng C, Reisle C, Leelakumari S, Zhao C, Yip S, Ho J, et al. 2017. Successful targeting of the NRG1 pathway indicates novel treatment strategy for metastatic cancer. Ann Oncol 28: 3092–3097. 10.1093/annonc/mdx523 [DOI] [PubMed] [Google Scholar]
- Kroiss M, Quinkler M, Johanssen S, van Erp NP, Lankheet N, Pöllinger A, Laubner K, Strasburger CJ, Hahner S, Müller HH, et al. 2012. Sunitinib in refractory adrenocortical carcinoma: a phase II, single-arm, open-label trial. J Clin Endocrinol Metab 97: 3495–3503. 10.1210/jc.2012-1419 [DOI] [PubMed] [Google Scholar]
- Laskin J, Jones S, Aparicio S, Chia S, Ch'ng C, Deyell R, Eirew P, Fok A, Gelmon K, Ho C, et al. 2015. Lessons learned from the application of whole-genome analysis to the treatment of patients with advanced cancers. Cold Spring Harb Mol Case Stud 1: a000570. 10.1101/mcs.a000570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et al. 2017. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357: 409–413. 10.1126/science.aan6733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerario AM, Worden FP, Ramm CA, Hesseltine EA, Stadler WM, Else T, Shah MH, Agamah E, Rao K, Hammer GD. 2014. The combination of insulin-like growth factor receptor 1 (IGF1R) antibody cixutumumab and mitotane as a first-line therapy for patients with recurrent/metastatic adrenocortical carcinoma: a multi-institutional NCI-sponsored trial. Horm Cancer 5: 232–239. 10.1007/s12672-014-0182-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Tourneau C, Hoimes C, Zarwan C, Wong DJ, Bauer S, Claus R, Wermke M, Hariharan S, von Heydebreck A, Kasturi V, et al. 2018. Avelumab in patients with previously treated metastatic adrenocortical carcinoma: phase 1b results from the JAVELIN solid tumor trial. J Immunother Cancer 6: 111–119. 10.1186/s40425-018-0424-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12: 323. 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26: 589–595. 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Brown TC, Juhlin CC, Andreasson A, Wang N, Bäckdahl M, Healy JM, Prasad ML, Korah R, Carling T, et al. 2014. The activating TERT promoter mutation C228T is recurrent in subsets of adrenal tumors. Endocr Relat Cancer 21: 427–434. 10.1530/ERC-14-0016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok BH, Carley AC, Tchang B, Powell SN. 2013. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene 32: 3552–3558. 10.1038/onc.2012.391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharjan R, Backman S, Akerstrom T, Hellman P, Bjorklund P. 2018. Comprehensive analysis of CTNNB1 in adrenocortical carcinomas: identification of novel mutations and correlation to survival. Sci Rep 8: 8610–8612. 10.1038/s41598-018-26799-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson A, Hormozdiari F, Zayed A, Giuliany R, Ha G, Sun MG, Griffith M, Heravi Moussavi A, Senz J, Melnyk N, et al. 2011. deFuse: an algorithm for gene fusion discovery in tumor RNA-seq data. PLoS Comput Biol 7: e1001138. 10.1371/journal.pcbi.1001138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal P, Roberts CWM. 2020. The SWI/SNF complex in cancer—biology, biomarkers and therapy. Nat Rev Clin Oncol 17: 435–448. 10.1038/s41571-020-0357-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell F, Wilmott JS, Johansson PA, Nones K, Addala V, Mukhopadhyay P, Broit N, Amato CM, Van Gulick R, Kazakoff SH, et al. 2020. Whole-genome sequencing of acral melanoma reveals genomic complexity and diversity. Nat Commun 11: 5259–5253. 10.1038/s41467-020-18988-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng L, Libertino JM. 2003. Adrenocortical carcinoma: diagnosis, evaluation and treatment. J Urol 169: 5–11. 10.1097/01.ju.0000030148.59051.35 [DOI] [PubMed] [Google Scholar]
- Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, et al. 2016. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534: 47–54. 10.1038/nature17676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, Wendl MC, Ding L. 2014. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics 30: 1015–1016. 10.1093/bioinformatics/btt755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan C, Edgerly M, Velarde M, Wilkerson J, Venkatesan AM, Pittaluga S, Yang SX, Nguyen D, Balasubramaniam S, Fojo T. 2014. The VEGF inhibitor axitinib has limited effectiveness as a therapy for adrenocortical cancer. J Clin Endocrinol Metab 99: 1291–1297. 10.1210/jc.2013-2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasance E, Titmuss E, Williamson L, Kwan H, Culibrk L, Zhao EY, Dixon K, Fan K, Bowlby R, Jones MR, et al. 2020. Pan-cancer analysis of advanced patient tumors reveals interactions between therapy and genomic landscapes. Nat Cancer 1: 452–468. 10.1038/s43018-020-0050-6 [DOI] [PubMed] [Google Scholar]
- Quinkler M, Hahner S, Wortmann S, Johanssen S, Adam P, Ritter C, Strasburger C, Allolio B, Fassnacht M. 2008. Treatment of advanced adrenocortical carcinoma with erlotinib plus gemcitabine. J Clin Endocrinol Metab 93: 2057–2062. 10.1210/jc.2007-2564 [DOI] [PubMed] [Google Scholar]
- Raj N, Zheng Y, Kelly V, Katz SS, Chou J, Do RKG, Capanu M, Zamarin D, Saltz LB, Ariyan CE, et al. 2020. PD-1 blockade in advanced adrenocortical carcinoma. J Clin Oncol 38: 71–80. 10.1200/JCO.19.01586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch T, Zichner T, Schlattl A, Stutz AM, Benes V, Korbel JO. 2012. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28: i333–i339. 10.1093/bioinformatics/bts378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond VM, Everett JN, Furtado LV, Gustafson SL, Jungbluth CR, Gruber SB, Hammer GD, Stoffel EM, Greenson JK, Giordano TJ, et al. 2013. Adrenocortical carcinoma is a Lynch syndrome–associated cancer. J Clin Oncol 31: 3012–3018. 10.1200/JCO.2012.48.0988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisle C, Mungall KL, Choo C, Paulino D, Bleile DW, Muhammadzadeh A, Mungall AJ, Moore RA, Shlafman I, Coope R, et al. 2019. MAVIS: merging, annotation, validation, and illustration of structural variants. Bioinformatics 35: 515–517. 10.1093/bioinformatics/bty621 [DOI] [PubMed] [Google Scholar]
- Ribeiro-Silva C, Vermeulen W, Lans H. 2019. SWI/SNF: complex complexes in genome stability and cancer. DNA Repair (Amst) 77: 87–95. 10.1016/j.dnarep.2019.03.007 [DOI] [PubMed] [Google Scholar]
- Saini N, Roberts SA, Klimczak LJ, Chan K, Grimm SA, Dai S, Fargo DC, Boyer JC, Kaufmann WK, Taylor JA, et al. 2016. The impact of environmental and endogenous damage on somatic mutation load in human skin fibroblasts. PLoS Genet 12: e1006385. 10.1371/journal.pgen.1006385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. 2012. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28: 1811–1817. 10.1093/bioinformatics/bts271 [DOI] [PubMed] [Google Scholar]
- Sheffield BS, Tinker AV, Shen Y, Hwang H, Li-Chang HH, Pleasance E, Ch'ng C, Lum A, Lorette J, McConnell YJ, et al. 2015. Personalized oncogenomics: clinical experience with malignant peritoneal mesothelioma using whole genome sequencing. PLoS ONE 10: e0119689. 10.1371/journal.pone.0119689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. 2009. ABySS: a parallel assembler for short read sequence data. Genome Res 19: 1117–1123. 10.1101/gr.089532.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibodeau ML, Zhao EY, Reisle C, Ch'ng C, Wong HL, Shen Y, Jones MR, Lim HJ, Young S, Cremin C, et al. 2019. Base excision repair deficiency signatures implicate germline and somatic MUTYH aberrations in pancreatic ductal adenocarcinoma and breast cancer oncogenesis. Cold Spring Harb Mol Case Stud 5: a003681. 10.1101/mcs.a003681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissier F, Cavard C, Groussin L, Perlemoine K, Fumey G, Hagneré AM, René-Corail F, Jullian E, Gicquel C, Bertagna X, et al. 2005. Mutations of beta-catenin in adrenocortical tumors: activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res 65: 7622–7627. 10.1158/0008-5472.CAN-05-0593 [DOI] [PubMed] [Google Scholar]
- Vivian J, Rao AA, Nothaft FA, Ketchum C, Armstrong J, Novak A, Pfeil J, Narkizian J, Deran AD, Musselman-Brown A, et al. 2017. Toil enables reproducible, open source, big biomedical data analyses. Nat Biotechnol 35: 314–316. 10.1038/nbt.3772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber MM, Auernhammer CJ, Kiess W, Engelhardt D. 1997. Insulin-like growth factor receptors in normal and tumorous adult human adrenocortical glands. Eur J Endocrinol 136: 296–303. 10.1530/eje.0.1360296 [DOI] [PubMed] [Google Scholar]
- Wortmann S, Quinkler M, Ritter C, Kroiss M, Johanssen S, Hahner S, Allolio B, Fassnacht M. 2010. Bevacizumab plus capecitabine as a salvage therapy in advanced adrenocortical carcinoma. Eur J Endocrinol 162: 349–356. 10.1530/EJE-09-0804 [DOI] [PubMed] [Google Scholar]
- Xie C, Tanakchi S, Raygada M, Davis JL, Del Rivero J. 2018. Case report of an adrenocortical carcinoma associated with germline CHEK2 mutation. J Endocr Soc 3: 284–290. 10.1210/js.2018-00343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang GJ, Zhu MH, Lu XJ, Liu YJ, Lu JF, Leung CH, Ma DL, Chen J. 2021. The emerging role of KDM5A in human cancer. J Hematol Oncol 14: 30–31. 10.1186/s13045-021-01041-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman GJR, de Roos JADM, Libouban MAA, Prinsen MBW, de Man J, Buijsman RC, Uitdehaag JCM. 2017. TTK inhibitors as a targeted therapy for CTNNB1 (β-catenin) mutant cancers. Mol Cancer Ther 16: 2609–2617. 10.1158/1535-7163.MCT-17-0342 [DOI] [PubMed] [Google Scholar]
- Zhao EY, Shen Y, Pleasance E, Kasaian K, Leelakumari S, Jones M, Bose P, Ch'ng C, Reisle C, Eirew P, et al. 2017. Homologous recombination deficiency and platinum-based therapy outcomes in advanced breast cancer. Clin Cancer Res 23: 7521–7530. 10.1158/1078-0432.CCR-17-1941 [DOI] [PubMed] [Google Scholar]
- Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA, Lerario AM, Else T, Knijnenburg TA, Ciriello G, et al. 2016. Comprehensive pan-genomic characterization of adrenocortical carcinoma. Cancer Cell 29: 723–736. 10.1016/j.ccell.2016.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data sets generated and analyzed during the current study have been deposited at the Michael Smith Genome Sciences Centre at BC Cancer (BCGSC; https://bcgsc.ca/downloads/POG_ACC/). The genomic and transcriptome sequencing data sets have been deposited at the European Genome-phenome Archive (EGA; https://ega-archive.org/) as part of the study EGAS00001001159 (Patient 1 EGAD00001002037, Patient 2 EGAD00001002049, Patient 3 EGAD00001003053, Patient 4 EGAD00001004905, Patient 5 EGAD00001003676, Patient 6 EGAD00001005844, Patient 7 EGAD00001005897).