

Abstract
Metabolic intermediates influence inflammation not only through signaling effects, but also by fueling the production of pro-inflammatory molecules. Microglial production of nitric oxide (NO) requires the consumption of NADPH. NADPH consumed in this process is regenerated from NADP+ primarily through the hexose monophosphate shunt, which can utilize only glucose as a substrate. These factors predict that glucose availability can be rate-limiting for glial NO production. To test this prediction, cultured astrocytes and microglia were incubated with lipopolysaccharide and interferon-γ to promote expression of inducible nitric oxide synthase, and the rate of NO production was assessed at defined glucose concentrations. Increased NO production was detected only in cultures containing microglia. The NO production was markedly slowed at glucose concentrations below 0.5 mM, and comparably reduced by inhibition of the hexose monophosphate shunt with 6-aminonicotinamide. Reduced NO production caused by glucose deprivation was partly reversed by malate, which fuels NADPH production by malate dehydrogenase, and by NADPH itself. These findings highlight the role of the hexose monophosphate shunt in fueling NO synthesis and suggest that microglial NO production in the brain may be limited at sites of low glucose availability, such as abscesses or other compartmentalized infections.
Keywords: abscess, hyperglycemia, iNOS, NADPH, pentose phosphate pathway
1 |. INTRODUCTION
There is growing interest in the interactions between metabolic factors and innate immune responses, and several such interactions have been identified. In most cases, these interactions involve signaling properties of metabolites that, directly or indirectly, influence inflammatory cascades (Bernier et al., 2020; Lauro & Limatola, 2020). However, metabolites may also affect immune responses by fueling the production of inflammatory signaling molecules. Glucose, in addition to being the primary metabolic substrate for the brain, also fuels the hexose monophosphate shunt, which serves to regenerate NADPH from NADP+. NADPH is required for both thioredoxin activity and glutathione recycling, and the importance of the hexose monophosphate shunt for these antioxidant functions is well recognized (Bolanos, 2016; Cherkas et al., 2020). Less well recognized is the role of the hexose monophosphate shunt in producing the oxidant signaling molecule, nitric oxide.
Nitric oxide (NO) is a membrane-permeant pro-inflammatory signaling molecule (Guzik et al., 2003; Nagy et al., 2007) which, in addition, forms the potently antimicrobial and cytotoxic compound peroxynitrite when produced in concert with superoxide (Brown & Neher, 2010; Pacher et al., 2007; Zhu et al., 1992). Nitric oxide production is thus an important aspect of brain responses to microbial infection. The nitric oxide produced by microglia is generated almost exclusively by inducible nitric oxide synthase (iNOS), which is so-named because its expression is induced by pro-inflammatory factors.
iNOS catalyzes the consumption of L-arginine and NADPH to generate citrulline, NADP+, and NO. NADPH consumed in this process is regenerated from NADP+ primarily through the hexose monophosphate shunt (Figure 1). Lesser amounts of NADPH are also produced by cytosolic malic enzyme (Frenkel, 1975). The hexose monophosphate shunt utilizes only glucose as its substrate, and this predicts that glucose availability should limit the rate glial NO production. Here we tested this prediction using cultured mouse glia (astrocytes and microglia) in which iNOS expression was induced by co-incubation with lipopolysaccharide (LPS) and interferon-γ (IFNγ). LPS/IFNγ treatment induced NO production only in cultures containing microglia and the rate of NO production was limited by low glucose concentrations and by inhibition of the hexose monophosphate shunt.
FIGURE 1.
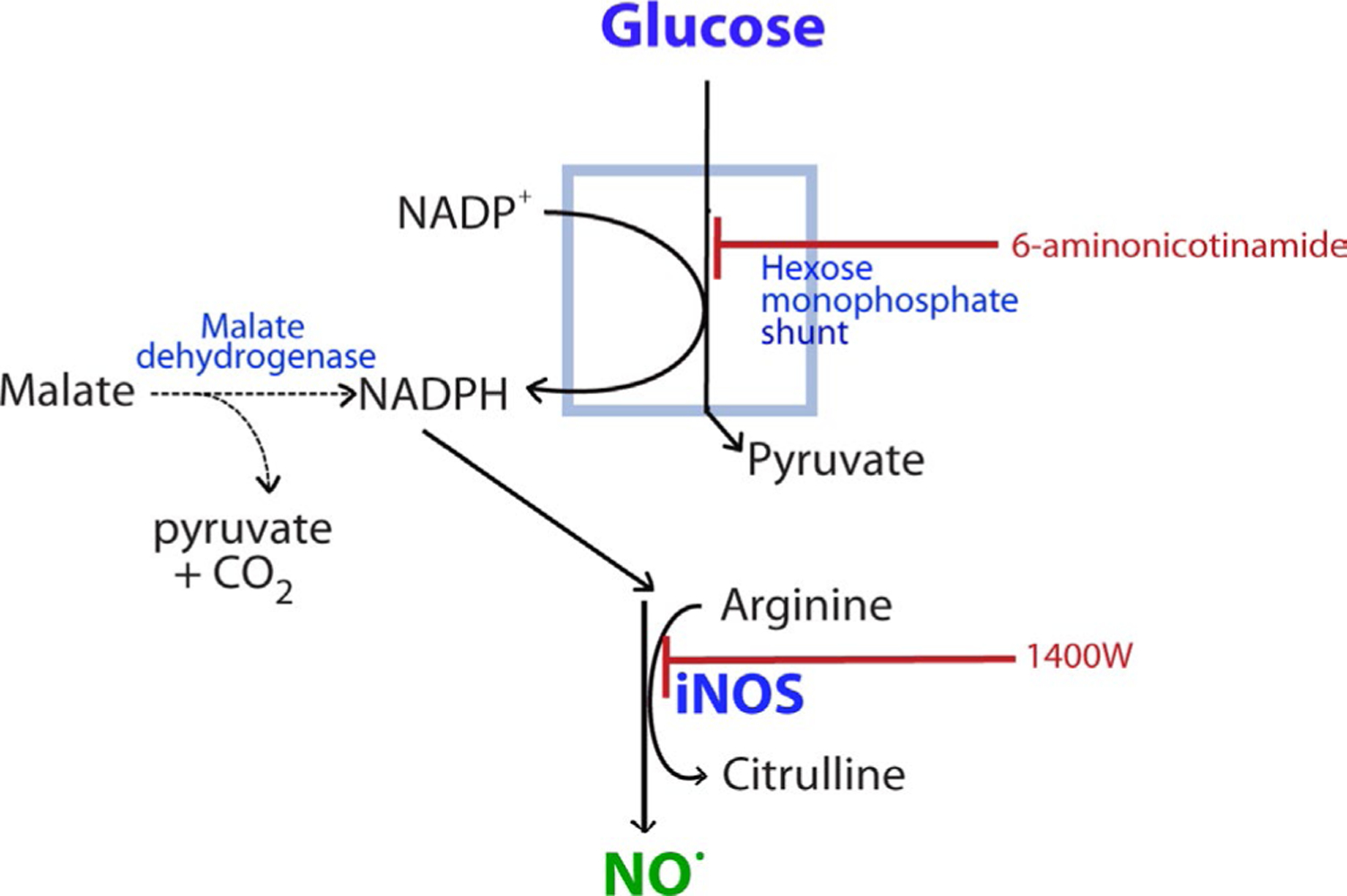
Metabolic relationship between glucose metabolism and NO production. Nitric oxide production by iNOS consumes NADPH. The regeneration of NADPH from NADP+ occurs primarily through the hexose monophosphate shunt, which requires glucose as a substrate. NADPH can also be produced by malic enzyme (malate dehydrogenase). 6-aminonicotinamide blocks flux through the hexose monophosphate shunt. 1400W (N-(3-(aminomethyl)benzyl)acetamidine) is an inhibitor of iNOS
2 |. METHODS
2.1 |. Glial cultures
Procedures were approved by the San Francisco Veterans Affairs Medical Center Institutional Animal Care and Use Committee (protocol Cat. # 20–009). This study was not pre-registered with any study database. Mixed glial cultures containing astrocytes and microglia were prepared from postnatal day 0 or day 1 C57BL/6 wild-type mice of both sexes as described (Lian et al., 2016). The mouse colony was originally obtained from Jackson Labs (Cat. # 000664) and was maintained in an AAALAC—accredited facility. The mice were housed 3–4 per cage with access to food and water ad libitum. The mouse pups were killed for brain harvest by decapitation under isoflurane anesthesia. The brain cortices and hippocampi were removed, dissociated with papain, triturated, and suspended in Dulbecco’s minimal essential medium (DMEM; ThermoFisher, Cat. # A1443001) supplemented with 10% fetal bovine serum (FBS; ThermoFisher, Cat. # 10438026), 100 U/ml penicillin, 100 µg/ml streptomycin, 2 mM l-glutamine, 5 mM glucose, and 1 mM sodium pyruvate. Cells from multiple pups were pooled for each dissection. The dissociated cells were plated in this culture medium into flasks coated with poly-d-lysine (PDL; ThermoFisher, Cat. # A3800401) and maintained in a 5% CO2 37°C incubator. The culture medium was exchanged on the following day to remove cell debris, and the cultures were maintained until confluency (8–12 days). At confluency, the cells were lifted by trypsinization and re-plated into PDL-coated 24-well culture plates at a density of 2 × 105 cells/ml.
Astrocyte monocultures (devoid of microglia) were prepared as described (Hamby et al., 2006). At confluency, the cultures were treated with 5 µM cytosine arabinoside (MilliporeSigma, Cat. # C6645) with medium exchanges each day for 5 days. On the following day, cultures were treated with 50 mM l-leucyl methyl ester MilliporeSigma Cat. # L1002) for 90 min. Twenty-four hours later the cells were lifted by trypsinization and re-plated into PDL-coated 24-well culture plates at a density of 2 × 105 cells/ml. Experiments were performed 3 days after re-plating. The total number of mice used for these studies was approximately 65, excluding breeders.
2.2 |. Experimental procedures
Experiments were initiated by replacing the culture medium with FBS-free culture medium, with or without 500 ng/ml lipopolysaccharide (LPS; MiliporeSigma, Cat. # L2880) and 50 ng/ml interferon gamma (IFNγ; BioLegend, Cat. # 575306) to induce expression of iNOS (Chao et al., 1992; Saha & Pahan, 2006), and incubated for 18 h. The cultures were then placed in glucose-free DMEM (ThermoFisher, Cat. # A14430) (supplemented with 1 mM pyruvate and 2 mM glutamine) for 15 min to deplete intracellular glucose and glycogen stores, and subsequently incubated with the DMEM containing defined glucose concentrations ranging from 0 to 15 mM for an additional 90 min before assessment of NO production or cellular adenosine triphosphate (ATP) levels, or for longer periods to assess cell viability. (Note DMEM contains arginine, which along with NADPH is required for iNOS production of NO). Some cultures were additionally treated with the 6-phosphogluconate dehydrogenase inhibitor 6-aminonicotinamide (MilliporeSigma, Cat. # A68203), added at the beginning of LPS/INFγ treatment; or with 1400W dihydrochloride (R&D Systems, Cat. # 1415), L-malic acid (MilliporeSigma, Cat. # 02288, NADPH (MilliporeSigma, Cat. # N1630), NADP+ (MilliporeSigma, Cat. # 481972), or sodium azide (MilliporeSigma, Cat. # 71289). Culture wells were assigned to treatment conditions in an alternating manner, such that the pattern on the plate varied from experiment to experiment.
2.3 |. Immunofluorescence
Cells were fixed in a 4% paraformaldehyde solution, permeabilized with 0.3% Triton-X for 10 min, and incubated for 1 h with blocking buffer. Cells were incubated overnight at 4°C with primary antibodies diluted in blocking buffer: mouse anti-GFAP (MilliporeSigma, Cat. # MAB360; 1:1000) and goat anti-Iba1 (Abcam, Cat. # ab107159; 1:500). After washing, the cells were incubated 1 h with Alexa Fluor-conjugated secondary antibodies. 4′,6-diamidino-2-phenylindole (DAPI) was included in the coverslip mounting medium (ThermoFisher, Cat. # P36962) for nuclear counterstaining. Images were acquired using a fluorescence microscope. Three images were obtained from each culture well by an observer who was blinded to the experimental conditions. Where assessed, the relative number of microglia and astrocytes in the cultures were calculated from 3 photomicrographs taken from each of three culture wells of each treatment condition from each of three independent experiments.
2.4 |. Griess assay
NO production was assessed by measuring the accumulated nitrate and nitrite in the culture medium (Bryan & Grisham, 2007) with a commercial Griess assay kit that (Cayman, Cat. # 780001). Nitrate was enzymatically reduced to nitrite, and the NO concentration in each sample was determined using a standard curve of nitrite standards. Absorbance at 540 nm was measured with a plate reader. NO values in the medium of each well were normalized to protein content of the culture wells as determined by the bicinchoninic acid method.
2.5 |. ATP assay
ATP levels were measured using a luminescence detection kit (Abcam, Cat. # ab113849). The ATP content in each sample was determined using a standard curve of ATP standards. Luminescence was measured with a plate reader. Values from each well were normalized to those of control wells from the same experiment.
2.6 |. Cell viability
Propidium iodide (2 µg/ml) (ThermoFisher, Cat. # P1304MP) and Hoechst 33342 (5 µM) (ThermoFisher, Cat. # H3570) were added at the designated time intervals after glucose deprivation and incubated for an additional 10 min before photomicrographs were taken. Cells which were unable to exclude propidium iodide were considered non-viable. Hoechst staining was used to identify cell nuclei. Quantified data were obtained using three photomicrographs from each well taken in a triangular pattern around the well center by an observer blinded to the treatment conditions. Percent non-viable cells were calculated as PI-(positive nuclei/total nuclei) × 100.
2.7 |. Statistics
The “n” for each study was defined as the number of experiments, each performed with independent culture preparations. Data were analyzed with GraphPad Prism 8 and expressed as mean ± standard error of the mean (SEM). Statistical comparisons were performed using analysis of variance (ANOVA) and the Brown–Forsyth test of equal variances, followed by either Sidak’s multiple comparison test or Dunnett’s test for multiple comparisons against a common group. Data were considered statistically significant when p < 0.05. Sample sizes were based on a prior study using similar culture preparations and endpoints (Ghosh et al., 2018). No inclusion/exclusion criteria were set, and no data points were excluded as “outliers”.
3 |. RESULTS
The experimental design for these studies of NO production is illustrated in Figure 2a. Cultures were treated overnight with 500 ng/ml LPS plus 50 ng/ml IFNγ to induce up-regulation and activation of iNOS (Chao et al., 1992). The culture medium was then replaced with glucose-free DMEM for 15 min to deplete intracellular glucose and glycogen, and then replaced again with a medium containing 0–15 mM glucose for 90 min. The amount of nitrite and nitrate that accumulated in the medium during the 90 min interval was measured as assay of nitric oxide production (Bryan & Grisham, 2007). Immunostaining for the astrocyte marker GFAP and the microglial marker Iba1 showed that both cell types were present in the mixed glial cultures and that both types remained present under the treatment conditions (Figure 2b). Microglia in these cultures exhibited the typical hypertrophy associated with LPS exposure. There was no significant decrement in ATP content at the 90-min time point (Figure S1a), as would be expected given that glucose-free DMEM contained other substrates for energy metabolism including 1 mM pyruvate, 2 mM glutamine, and lower concentrations of other amino acids. Cell viability, as assessed by PI staining showed less than 0.5% cell death over the 90-min interval used for measures of NO production (n = 4; Figure S1b). However, a significant (>10%) loss of cell viability was observed in the LPS/IFNγ treated cultures when the glucose deprivation interval was extended to 4 h (Figure S1b).
FIGURE 2.
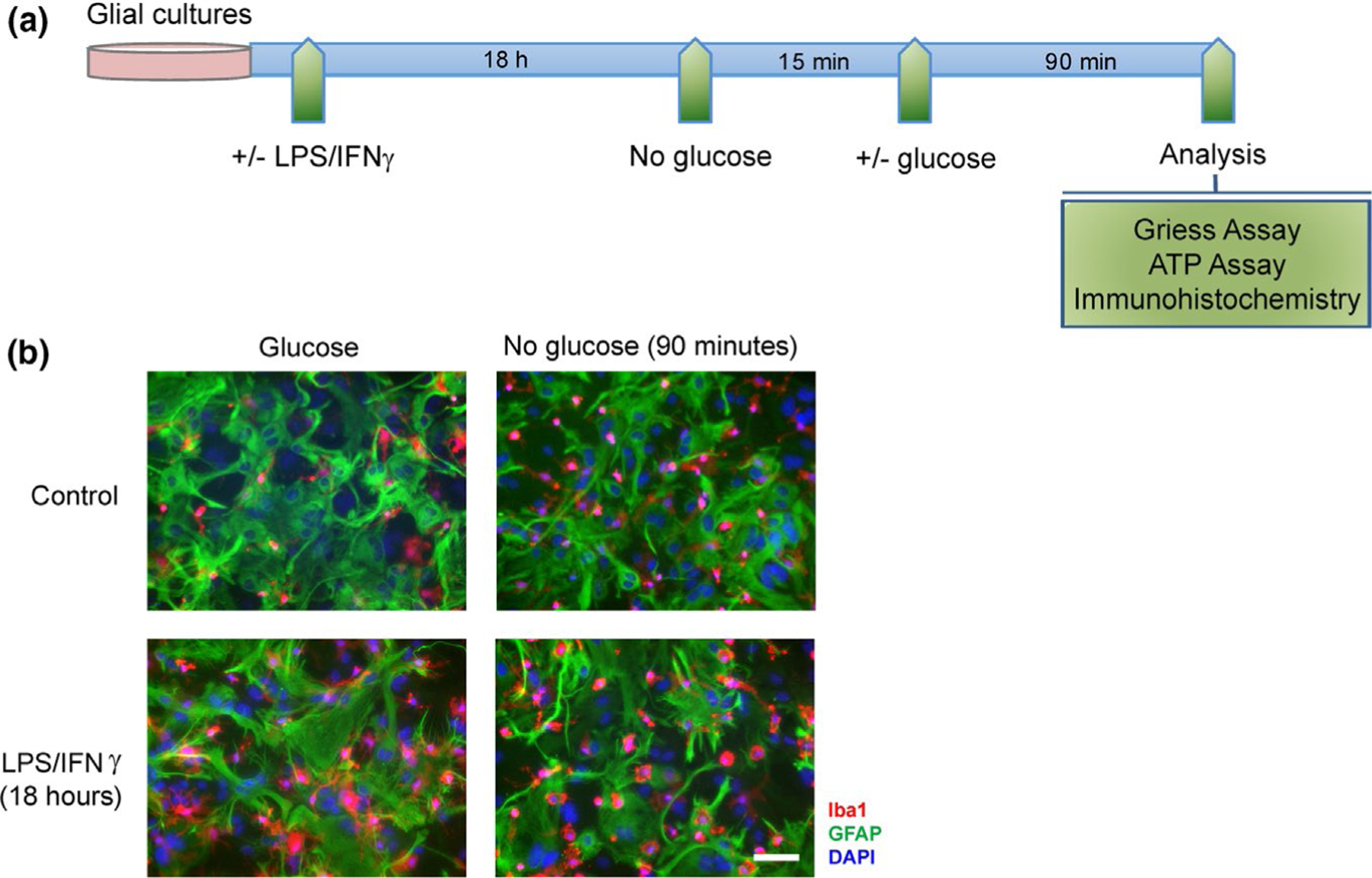
LPS/INFγ treatments and glucose deprivation. (a) Timeline of experimental procedures. (b) Cultures immunostained for the microglial marker Iba1 (red) and the astrocyte marker GFAP (green). Nuclei are labeled with DAPI (blue). Scale bar = 50 µm
Cultures that had been treated with LPS/IFNγ showed a large increase in nitric oxide production over the 90-min observation period (Figure 3a). This increase was completely prevented by the iNOS inhibitor 1400W (Garvey et al., 1997), thus confirming the specificity of the Griess assay for NO under these conditions and also confirming iNOS as the source of NO production (Figure 3a). Most notably, cultures in medium containing no glucose during the assay period also showed reduced NO production. This reduction was also observed in cultures provided with 5 mM glucose but treated with the hexose monophosphate shunt inhibitor 6-aminonicotinamide (Bender & Van Epps, 1985), consistent with a requirement for glucose flux specifically through the hexose monophosphate shunt for NO production.
FIGURE 3.
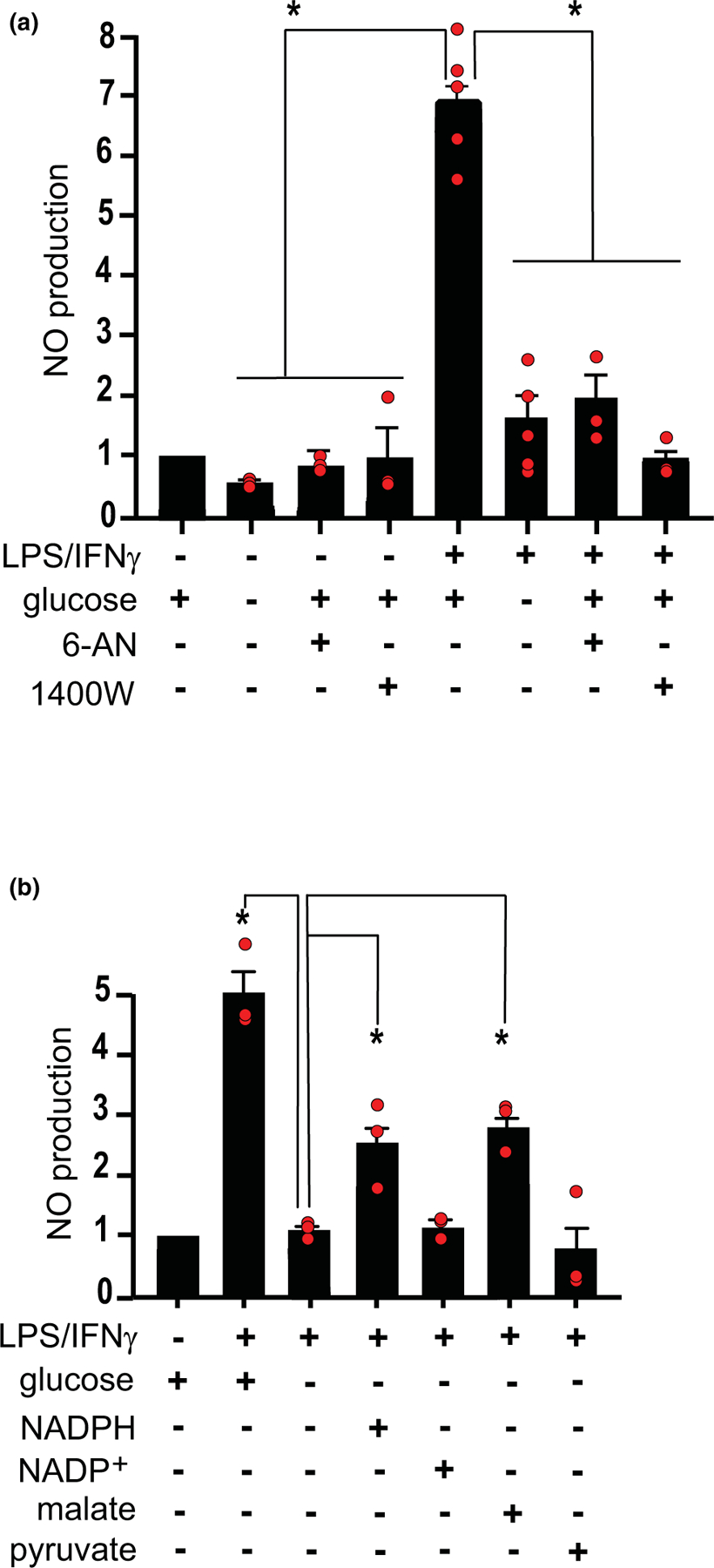
Effects of glucose deprivation on NO production. (a) NO production was increased in cultures treated with LPS/INFγ, and this increase was suppressed by the iNOS inhibitor 1400W (25 µM); by glucose deprivation; and by the hexose monophosphate shunt inhibitor 6-aminonicotinamide (6-AN, 10 µM). Data from each experiment are normalized to the control condition (5 mM glucose, not stimulated by LPS/INFγ) and expressed as means ± SEM. n = 3–5 experiments, each performed with independent culture preparations. *p < 0.05 by ANOVA and Dunnett’s test [F(7, 22) = 46.01, p < 0.0001, r2 = 0.936; Browne–Forsythe F = 1.862, p = 0.125]. (b) The effect of glucose-deprivation was partially reversed by addition of 500 µM malate or 100 µM NADPH to the medium, but not by equal concentrations of pyruvate or NADP+ evaluated as controls. Data from each experiment are normalized to the control condition (5 mM glucose, not stimulated by LPS/INFγ) and expressed as means ± SEM. n = 3 experiments, each performed with independent culture preparations. *p < 0.05 by ANOVA and Dunnett’s test. [F(5, 12) = 27.41, p < 0.0001, r2 = 0.920; Browne–Forsythe F = 1.353, p = 0.87]
To further test whether glucose-fueled NADPH production is required for NO production, we evaluated the effects of adding malate and NADPH to the medium of the glucose-deprived cultures. Malate is a substrate for cytosolic malic enzyme, which generates NADPH and can thereby provide this metabolite in the absence of hexose monophosphate shunt activity (Figure 1). Malate (500 µM) was found to partially reversed the effect of glucose deprivation (Figure 3b). The addition of 100 µM NADPH to the culture medium also partially reversed the effect of glucose deprivation, whereas NADP+ did not (Figure 3b).
We next evaluated the concentration-response relationship between glucose and NO production. This showed a reduction in NO production at all glucose concentrations of 0.1 mM or less, and no effect over the range of 0.5–15 mM (Figure 4a). We also evaluated the time course of NO production in the 5 and 0 mM glucose conditions (Figure 4b). There was a time-dependent increase in NO production over the entire 90-min interval in the presence of glucose, but a plateau in NO production after 30 min in the no glucose condition (Figure 4b). This result suggests that NO production observed in the absence of glucose may have been fueled by residual carbohydrate reserves, despite 15-min glucose-free pre-incubation period, and that the true capacity of glia to generate NO in the true absence of glucose may be closer to zero.
FIGURE 4.
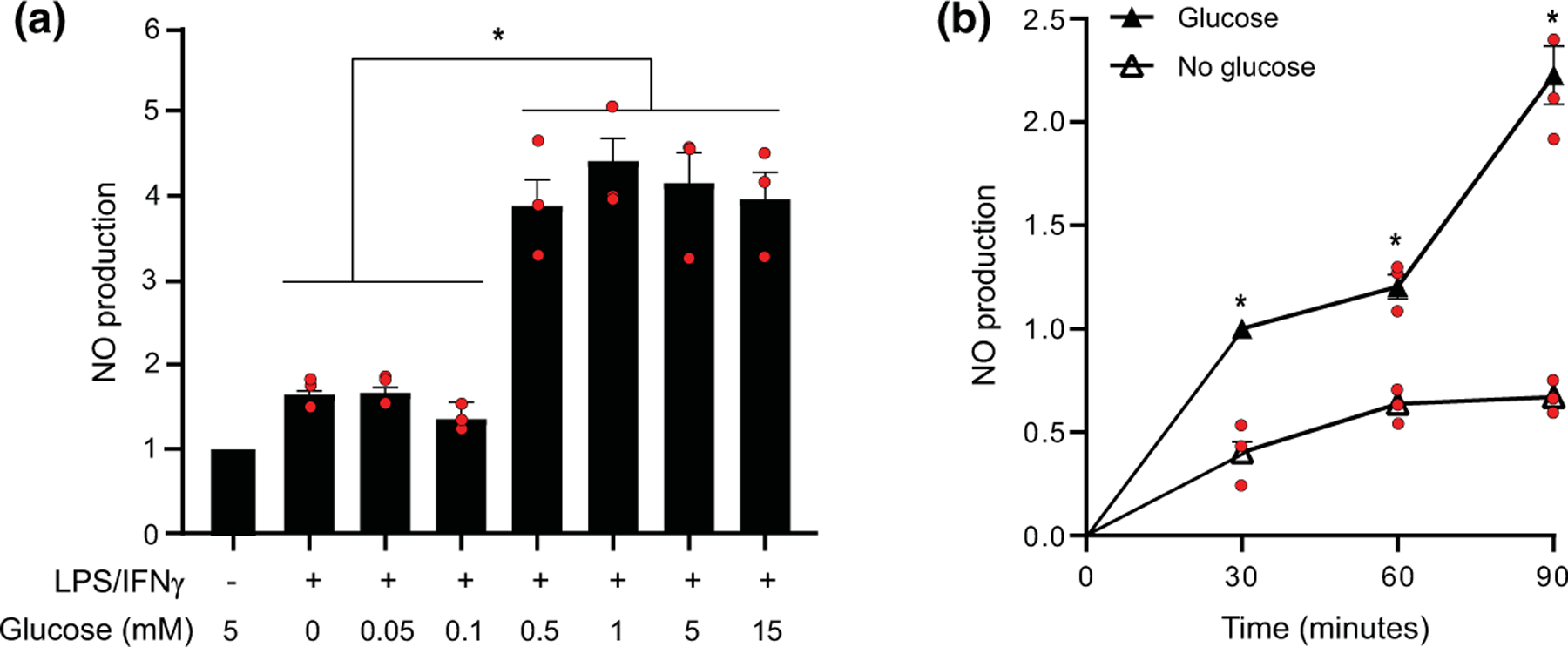
Effects of glucose concentration and incubation times on NO production. (A) Reductions in NO production were observed at medium glucose concentrations of 0.1 mM and lower. Data from each experiment are normalized to the control condition (5 mM glucose, not stimulated by LPS/INFγ) and expressed as means ± SEM. n = 3 experiments, each performed with independent culture preparations. *p < 0.05 by ANOVA and Sidak’s test [F(6, 14) = 21.16, p < 0.0001’ r2 = 0.900; Browne–Forsythe F = 0.671, p = 0.675]. (b) Cumulative NO production measured at 30 min intervals in cultures incubated with and without 5 mM glucose after LPS/INFγ stimulation. Values are expressed relative to the control condition (not stimulated by LPS/INFγ or deprived of glucose). Data from each experiment are normalized to the control condition (5 mM glucose, not stimulated by LPS/INFγ) and expressed as means ± SEM. n = 3 experiments, each performed with independent culture preparations. *p < 0.05 by ANOVA and Sidak’s test. [F(5, 12) = 64.8’ p < 0.0001, r2 = 0.964; Browne–Forsythe F = 1.259, p = 0.342]
A limitation of the mixed glial culture system is that the contributions of astrocytes versus microglia to NO production cannot be assessed. To circumvent this limitation, we initially attempted studies using monocultures of microglial cells; however, in the absence of astrocytes the glucose-deprived microglia quickly rounded up and detached from the plating surface. As an alternative approach, we compared results obtained in mixed astrocyte-microglial cultures and pure astrocyte cultures in which microglia had been eliminated with leucine methyl ester (Figure 5a). Cultures treated with leucine methyl ester contained 0.11 ± 0.05 percent microglia, as compared to 51.9 ± 2.0% microglia in the untreated cultures (n = 3). No significant differences were observed in these ratios after 90 min of treatment with LPS/IFNγ, with or without concomitant glucose deprivation. A comparison of mixed and microglia-depleted cultures showed that LPS/IFNγ induced NO production only in the microglia-containing cultures, and glucose deprivation suppressed this NO production (Figure 5b). The observation that NO was produced only in the cultures containing microglia permitted an estimate of the absolute microglial NO production rate. Assuming that microglia account for roughly 50% of the protein in the mixed cultures, and that astrocyte NO production is unaffected by the presence of microglia, the rate of NO production from the LPS/IFNγ—stimulated microglia can be estimated as 3.2 ± 0.2 nmol/mg protein/min in the presence of glucose, and 0.3 ± 0.2 nmol/mg protein/min in the absence of glucose.
FIGURE 5.
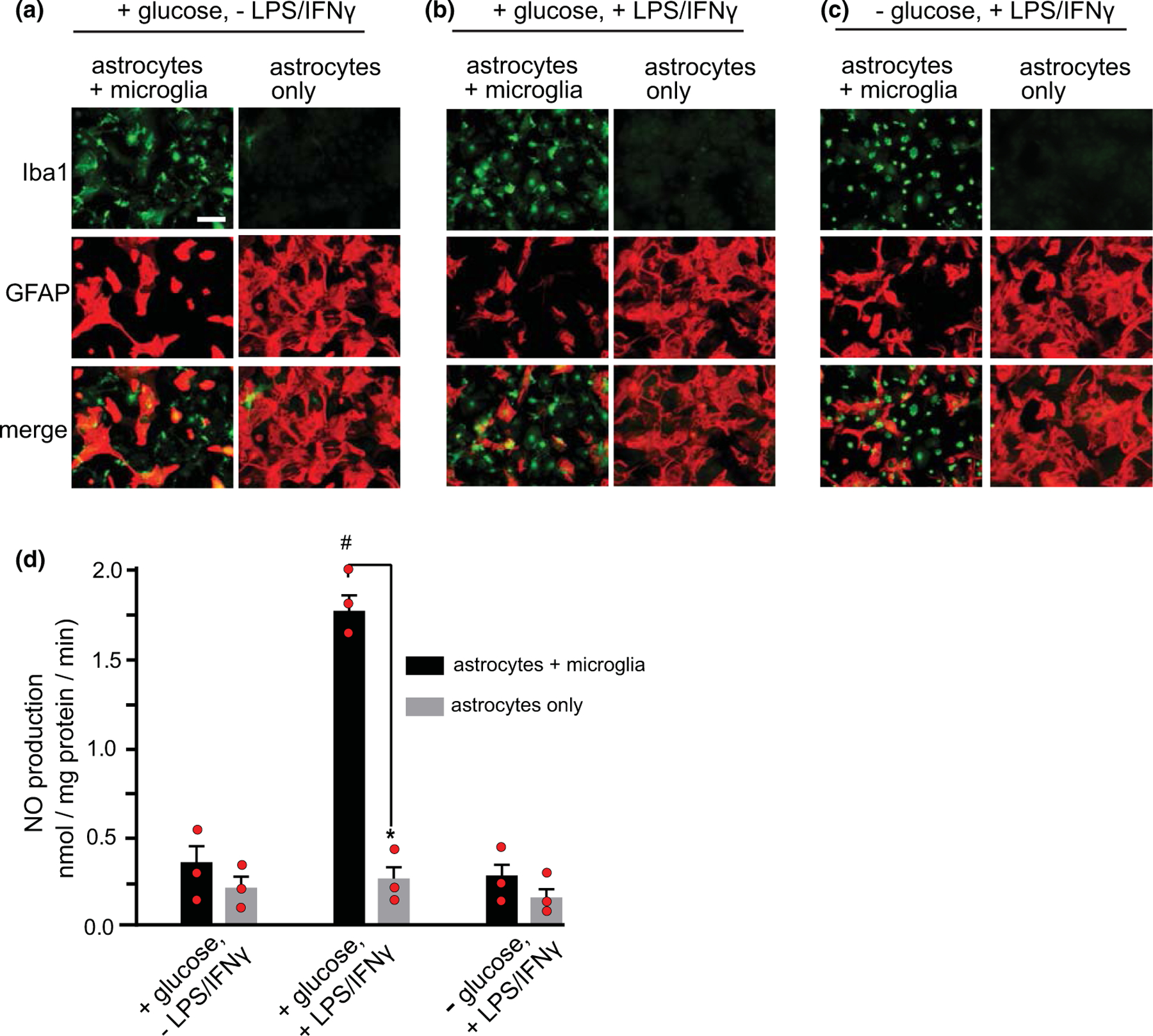
Comparisons between mixed glial cultures and astrocyte monocultures. (a–c) Cultures are immunostained for the microglial marker Iba1 (green) and the astrocyte marker GFAP (red). Scale bar = 50 µm. Cultures were fixed after 18 h treatment with or without LPS/IFNγ and 90 min incubation in 0 glucose or 5 mM glucose medium. (d) NO production under the conditions (a–c). Data are means ± SEM; n = 3 experiments, each performed with independent culture preparations of both types. *p < 0.01 versus astrocytes plus microglia; #p < 0.01 versus astrocytes +microglia under the other treatment conditions, by ANOVA and Sidak’s test. [F(5, 12) = 56.6, p < 0.0001, r2 = 0.959; Browne–Forsythe F = 0.039, p = 0.999]
4 |. DISCUSSION
The results provide evidence that glucose flux through the hexose monophosphate shunt is required for microglial production of NO. The NO production induced by LPS/IFNγ was markedly reduced in medium lacking glucose (but containing other energy substrates), and likewise reduced by the hexose monophosphate shunt inhibitor 6-aminonicotinamide. In addition, the reduction in NO production caused by glucose deprivation was partially reversed in cultures provided with malate or exogenous NADPH. Microglia were identified as the primary source of NO production by the lack of NO produced in cultures in which microglia had been eliminated.
Given the central role of glucose metabolism in many cell functions, glucose deprivation could, in principle, influence NO production by indirect mechanisms unrelated to its role as a substrate for the hexose monophosphate shunt. However, the observation that 6-phosphogluconate dehydrogenase inhibitor 6-aminonicotinamide reduced the NO production suggests that it is glucose flux specifically through the hexose monophosphate shunt that is required for NO production. Likewise, the observations that malate and NADPH both partially reversed the effect of glucose deprivation also argue against an indirect effect. While malate dehydrogenase is normally a relatively minor source of NADPH production, this can be increased by incubation with supra-physiological concentrations of malate as performed here. Malate is taken up into mammalian cells by a ubiquitously expressed family of sodium-dependent dicarboxylic acid transporters (Pajor, 1999). The route of NADPH uptake is less defined (Watkins & Moore, 1977), though NADH is known to be taken by astrocytes via a P2X7 receptor channel mechanism (Lu et al., 2007).
We observed no decrement in NO production at medium glucose concentrations at 0.5 mM or higher, but significant decrements at 0.1 mM glucose and below. These observations are in agreement with established kinetics of cellular glucose utilization. The rate-limiting step for glucose utilization is, under most conditions, the hexokinase reaction, whereby glucose is phosphorylated to glucose-6-phosphate. In mammalian cells, this enzyme has a glucose Km of approximately 0.3 mM (Wilson, 2003). Glucose concentrations well in excess of this value are saturating and would not be expected to increase glucose metabolism. Conversely, glucose concentrations near or below the Km become rate limiting for glucose metabolism. In principle, it is also possible that glucose flux through the hexose monophosphate shunt could be limited by regulation at the steps of glucose uptake or glucose phosphorylation.
A limitation of the mixed glial culture preparation is that it is difficult to define the relative contribution of astrocytes and microglia to NO production. Microglia cultured in the absence of astrocytes may respond in exaggerated or non-physiological ways to signaling and glucose deprivation. Our finding that mixed cultures depleted of microglia did not increase NO production in response to LPS/IFNγ, suggests that the NO measurements obtained with the mixed cultures reflect exclusively microglial metabolism. Of note, there is disagreement in the literature as to whether astrocytes are a significant source of NO production. Some authors have reported a robust response of astrocytes to LPS/IFNγ (e.g. Hamby et al., 2010; Hua et al., 2002), whereas others report no response (Possel et al., 2000; Sheng et al., 2011; Zhang et al., 2020), as observed here. The reasons for these differences are not clear but could stem from contaminating microglia in putatively pure astrocyte cultures or to differing culture conditions.
The present observations linking glucose availability to nitric oxide production are parallel to previous reports pertaining to superoxide production by NADPH oxidase. NADPH oxidase uses NADPH as an electron donor for the production of the superoxide radical in the same manner that iNOS uses NADPH to generate the nitric oxide radical, and superoxide production was similarly shown to be limited by glucose availability and by inhibition of the hexose monophosphate shunt (Brennan et al., 2009; Decoursey & Ligeti, 2005; Ghosh et al., 2018). The dependence of both nitric oxide and superoxide production on the hexose monophosphate shunt stands in contrast to the more widely recognized role of this pathway in supporting cell antioxidant functions (Bolanos, 2016; Cherkas et al., 2020). Given that endothelial nitric oxide synthase and neuronal nitric oxide synthase are also require NADPH as a substrate, it is likely that the current findings pertain to production of nitric oxide by those isoforms of NOS as well.
With the exception of insulin-induced hypoglycemia, blood glucose concentrations do not fall to range found here to be rate limiting for NO production. However, infections where blood delivery of substrates is impaired, such as joint capsules, pleural fluid, and ascites fluid often lead to large reductions in glucose levels (Chavalittamrong et al., 1979; Heidari et al., 2013; Kinugasa et al., 2020; Sahn et al., 1979). The limited data available from central nervous system infections suggests that glucose levels may approach zero in brain abscesses (Dahlberg et al., 2016). In these settings, glucose availability may become a critical factor in supporting bactericidal nitric oxide (and superoxide) production. An additional consideration in extrapolating these findings to brain in vivo is that low glucose availability in conjunction with pro-inflammatory signaling promotes pyroclastic microglial and macrophage demise, in which neighboring cells are also killed (Bergsbaken et al., 2009; Sanman et al., 2016). In these respects, it is possible that the hyperglycemic response that typically accompanies severe infection serves to maintain capacity to produce these responses in areas of restricted blood.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the U.S. Department of Veterans Affairs (1IO1 BX004884) and National Institutes of Health (R01NS105774). The authors thank Rebecca Fong for technical assistance with these studies.
All experiments were conducted in compliance with the ARRIVE guidelines.
Funding information
U.S. Department of Veterans Affairs, Grant/Award Number: 1IO1 BX004884; National Institutes of Health, Grant/Award Number: R01NS105774
Abbreviations:
- ANOVA
analysis of variance
- ATP
adenosine triphosphate
- DAPI
4′,6-diamidino-2-phenylindole
- DMEM
Dulbecco’s Modified Eagle Medium
- FBS
fetal bovine serum
- GFAP
glial fibrillary acidic protein
- Iba1
ionized calcium-binding adapter molecule 1
- IFNγ
interferon gamma
- iNOS
inducible nitric oxide synthase
- LPS
lipopolysaccharide
- NADP+
nicotinamide adenine dinucleotide phosphate
- NADPH
reduced form of nicotinamide adenine dinucleotide phosphate
- NO
nitric oxide
Footnotes
CONFLICT OF INTEREST
The authors state no conflicts of interest.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of the article at the publisher’s website.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Bender JG, & Van Epps DE (1985). Inhibition of human neutrophil function by 6-aminonicotinamide: The role of the hexose monophosphate shunt in cell activation. Immunopharmacology, 10, 191–199. 10.1016/0162-3109(85)90025-6 [DOI] [PubMed] [Google Scholar]
- Bergsbaken T, Fink SL, & Cookson BT (2009). Pyroptosis: Host cell death and inflammation. Nature Reviews Microbiology, 7, 99–109. 10.1038/nrmicro2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier LP, York EM, & MacVicar BA (2020). Immunometabolism in the brain: How metabolism shapes microglial function. Trends in Neurosciences,43,854–869. 10.1016/j.tins.2020.08.008. [DOI] [PubMed] [Google Scholar]
- Bolanos JP (2016). Bioenergetics and redox adaptations of astrocytes to neuronal activity. Journal of Neurochemistry, 139(Suppl 2), 115–125. 10.1111/jnc.13486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, & Swanson RA (2009). NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nature Neuroscience, 12, 857–863. 10.1038/nn.2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GC, & Neher JJ (2010). Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Molecular Neurobiology, 41, 242–247. 10.1007/s12035-010-8105-9 [DOI] [PubMed] [Google Scholar]
- Bryan NS, & Grisham MB (2007). Methods to detect nitric oxide and its metabolites in biological samples. Free Radical Biology and Medicine, 43, 645–657. 10.1016/j.freeradbiomed.2007.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao CC, Hu S, Molitor TW, Shaskan EG, & Peterson PK (1992). Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. The Journal of Immunology, 149, 2736–2741. [PubMed] [Google Scholar]
- Chavalittamrong B, Angsusingha K, Tuchinda M, Habanananda S, Pidatcha P, & Tuchinda C (1979). Diagnostic significance of pH, lactic acid dehydrogenase, lactate and glucose in pleural fluid. Respiration, 38, 112–120. 10.1159/000194067 [DOI] [PubMed] [Google Scholar]
- Cherkas A, Holota S, Mdzinarashvili T, Gabbianelli R, & Zarkovic N (2020). Glucose as a major antioxidant: when, what for and why it fails? Antioxidants, 9, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlberg D, Ivanovic J, & Hassel B (2016). Toxic levels of ammonia in human brain abscess. Journal of Neurosurgery, 124, 854–860. 10.3171/2015.1.JNS142582 [DOI] [PubMed] [Google Scholar]
- Decoursey TE, & Ligeti E (2005). Regulation and termination of NADPH oxidase activity. Cellular and Molecular Life Sciences, 62, 2173–2193. 10.1007/s00018-005-5177-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel R (1975). Regulation and physiological functions of malic enzymes. Current Topics in Cellular Regulation, 9, 157–181. [DOI] [PubMed] [Google Scholar]
- Garvey EP, Oplinger JA, Furfine ES, Kiff RJ, Laszlo F, Whittle BJ, & Knowles RG (1997). 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. Journal of Biological Chemistry, 272, 4959–4963. 10.1074/jbc.272.8.4959 [DOI] [PubMed] [Google Scholar]
- Ghosh S, Castillo E, Frias ES, & Swanson RA (2018). Bioenergetic regulation of microglia. Glia, 66, 1200–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzik TJ, Korbut R, & Adamek-Guzik T (2003). Nitric oxide and superoxide in inflammation and immune regulation. Journal of Physiology and Pharmacology, 54, 469–487. [PubMed] [Google Scholar]
- Hamby ME, Hewett JA, & Hewett SJ (2010). Smad3-dependent signaling underlies the TGF-beta1-mediated enhancement in astrocytic iNOS expression. Glia, 58, 1282–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamby ME, Uliasz TF, Hewett SJ, & Hewett JA (2006). Characterization of an improved procedure for the removal of microglia from confluent monolayers of primary astrocytes. Journal of Neuroscience Methods, 150, 128–137. 10.1016/j.jneumeth.2005.06.016 [DOI] [PubMed] [Google Scholar]
- Heidari K, Amiri M, Kariman H, Bassiri M, Alimohammadi H, & Hatamabadi HR (2013). Differentiation of exudate from transudate ascites based on the dipstick values of protein, glucose, and pH. American Journal of Emergency Medicine, 31, 779–782. 10.1016/j.ajem.2013.01.010 [DOI] [PubMed] [Google Scholar]
- Hua LL, Kim MO, Brosnan CF, & Lee SC (2002). Modulation of astrocyte inducible nitric oxide synthase and cytokine expression by interferon beta is associated with induction and inhibition of interferon gamma-activated sequence binding activity. Journal of Neurochemistry, 83, 1120–1128. [DOI] [PubMed] [Google Scholar]
- Kinugasa M, Kobayashi D, Satsuma S, Sakata R, Shinada Y, & Kuroda R (2020). The predictive value of synovial glucose level in septic arthritis. Journal of Pediatric Orthopedics. Part B, 29, 292–296. 10.1097/BPB.0000000000000628 [DOI] [PubMed] [Google Scholar]
- Lauro C, & Limatola C (2020). Metabolic reprograming of microglia in the regulation of the innate inflammatory response. Frontiers in Immunology, 11, 493. 10.3389/fimmu.2020.00493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian H, Roy E, & Zheng H (2016). Protocol for primary microglial culture preparation. Bio-Protocol, 6, 3–4. 10.21769/BioProtoc.1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Burns D, Garnier P, Wei G, Zhu K, & Ying W (2007). P2X7 receptors mediate NADH transport across the plasma membranes of astrocytes. Biochemical and Biophysical Research Communications, 362, 946–950. 10.1016/j.bbrc.2007.08.095 [DOI] [PubMed] [Google Scholar]
- Nagy G, Clark JM, Buzas EI, Gorman CL, & Cope AP (2007). Nitric oxide, chronic inflammation and autoimmunity. Immunology Letters, 111, 1–5. 10.1016/j.imlet.2007.04.013 [DOI] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, & Liaudet L (2007). Nitric oxide and peroxynitrite in health and disease. Physiological Reviews, 87, 315–424. 10.1152/physrev.00029.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajor AM (1999). Sodium-coupled transporters for Krebs cycle intermediates. Annual Review of Physiology, 61, 663–682. 10.1146/annurev.physiol.61.1.663 [DOI] [PubMed] [Google Scholar]
- Possel H, Noack H, Putzke J, Wolf G, & Sies H (2000). Selective upregulation of inducible nitric oxide synthase (iNOS) by lipopolysaccharide (LPS) and cytokines in microglia: in vitro and in vivo studies.Glia,32,51–59. [DOI] [PubMed] [Google Scholar]
- Saha RN, & Pahan K (2006). Regulation of inducible nitric oxide synthase gene in glial cells. Antioxidants and Redox Signaling, 8, 929–947. 10.1089/ars.2006.8.929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahn SA, Taryle DA, & Good JT Jr (1979). Experimental empyema. Time course and pathogenesis of pleural fluid acidosis and low pleural fluid glucose. American Review of Respiratory Disease, 120, 355–361. [DOI] [PubMed] [Google Scholar]
- Sanman LE, Qian Y, Eisele NA, Ng TM, van der Linden WA, Monack DM, Weerapana E, & Bogyo M (2016). Disruption of glycolytic flux is a signal for inflammasome signaling and pyroptotic cell death. Elife, 5, e13663. 10.7554/eLife.13663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng W, Zong Y, Mohammad A, Ajit D, Cui J, Han D, Hamilton JL, Simonyi A, Sun AY, Gu Z, Hong J-S, Weisman GA, & Sun GY (2011). Pro-inflammatory cytokines and lipopolysaccharide induce changes in cell morphology, and upregulation of ERK1/2, iNOS and sPLA(2)-IIA expression in astrocytes and microglia. Journal of Neuroinflammation, 8, 121. 10.1186/1742-2094-8-121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins DT, & Moore M (1977). Uptake of NADPH by islet secretion granule membranes. Endocrinology, 100, 1461–1467. [DOI] [PubMed] [Google Scholar]
- Wilson JE (2003). Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. Journal of Experimental Biology, 206, 2049–2057. 10.1242/jeb.00241 [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhu LB, He JH, Zhang HQ, Ji SY, Zhang CN, Hou NN, Huang CP, & Zhu JH (2020). Paroxetine suppresses reactive microglia-mediated but not lipopolysaccharide-induced inflammatory responses in primary astrocytes. Journal of Neuroinflammation, 17, 50. 10.1186/s12974-020-1712-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Gunn C, & Beckman JS (1992). Bactericidal activity of peroxynitrite. Archives of Biochemistry and Biophysics, 298, 452–457. 10.1016/0003-9861(92)90434-X [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.