

Abstract
Type 1 diabetes (T1D) is a chronic autoimmune disease characterized by pancreatic islet β cell loss and dysfunction resulting in insulin deficiency and hyperglycemia. During a presymptomatic phase of established β cell autoimmunity, β cell loss may first be evident through assessment of β cell secretory capacity, a measure of functional β cell mass. Reduction in pancreatic islet β cell reserve eventually manifests as impaired first-phase insulin response to glucose and abnormal glucose tolerance, which progresses until the functional capacity for β cell secretion can no longer meet the demand for insulin to control glycemia. A functional β cell mass of ~25% of normal may be required to avoid symptomatic T1D but is already associated with dysregulated glucagon secretion. With symptomatic T1D, stimulated C-peptide levels >0.60 ng/mL (0.200 pmol/mL) indicate the presence of clinically meaningful residual β cell function for contributing to glycemic control, although even higher residual C-peptide appears necessary for evidencing glucose-dependent islet β and α cell function that may contribute to maintaining (near)normal glycemia.β cell replacement by islet transplantation can restore a physiologic reserve capacity for insulin secretion, confirming thresholds for functional β cell mass required for independence from insulin therapy.
Keywords: type 1 diabetes, pancreatic islet, β cell, α cell, insulin secretion
Introduction
Type 1 diabetes (T1D) is a disease of insulin deficiency characterized by autoimmune destruction of pancreatic islet β cells. Now, 100 years since the discovery of insulin in 1921–1922, the clinical course of T1D has been transformed. However, insulin therapy by injection or subcutaneous infusion informed by ongoing glucose monitoring remains an unremitting daily challenge, with a continued risk of complications secondary to chronic high glucose exposure and dangerous acute episodes of hypoglycemia.1–5
T1D initially follows a subclinical course and presents at the point when residual β cell functional capacity can no longer meet the demand required for the maintenance of normal glucose homeostasis.6 The subclinical course of T1D described as early as the 1980s helped to establish the Eisenbarth model of a chronic autoimmune disorder targeting the insulin-producing β cell initiated by an interaction of genetic and environmental factors and progressing at a variable rate until insufficient reserve capacity for insulin secretion results in the clinical presentation of diabetes.7–11 As family history is associated with an increased risk of T1D, with a 1-in-20 lifetime risk in first-degree relatives compared with a 1-in-300 lifetime risk for the general U.S. population,12 family studies have been invaluable in advancing the understanding of diabetes pathogenesis, including its subclinical course to better inform pre-clinical staging.11,13,14 The genetic predisposition is largely mediated through human leukocyte antigen (HLA) genotype DR3/DR4-DQ8 in addition to non-HLA–related genes,15–18 which in combination with one or more still undefined environmental factors can lead to seroconversion through the development of autoantibodies against β cell–specific antigens. These include glutamic acid decarboxylase 65 (GAD-65), the tyrosine phosphatase–related islet antigen 2 (IA-2), insulin (IAA), and zinc transporter 8 (ZnT8), with their autoantibodies representing markers of β cell autoimmunity.
Once autoimmunity is established, subclinical T1D can progress through a presymptomatic phase that can be identified clinically using biomarkers of β cell autoimmunity and dysglycemia before the development of overt, symptomatic diabetes (Table 1 and Ref. 19). During subclinical disease progression, the pancreatic islet reserve capacity for insulin secretion declines as increasing numbers of β cells are lost to autoimmune destruction. This reserve capacity for secretion normally maintains glucose homeostasis during periods of growth and times of stress, and as it becomes limited results in the appearance of glucose intolerance, and once exhausted, the development of overt diabetes. Thus, clinically, T1D may be considered a disease from the time autoimmunity is established before devel oping glucose intolerance (stage 1), progressing to glucose intolerance (stage 2), and leading to typical signs and symptoms of diabetes (stage 3).19
Table 1.
Stages in the pathogenesis of type 1 diabetes19
| Nomeclature | Prestage 1 | Stage 1 | Stage 2 | Stage 3 |
|---|---|---|---|---|
| Natural history | Genetic and environmental susceptibility | Presymptomatic | Presymptomatic | Symptomatic |
| β cell autoimmunity | 0–1 autoantibodiesa | 2 or more autoantibodiesb | 2 or more autoantibodiesb | 2 or more autoantibodiesb |
| Glucose tolerance and glycemia | Normal, normoglycemia | Normal, normoglycemia | Impaired, dysglycemia | Diabetic, dysglycemia |
| First-phase insulin response | Normal | Normal to mild impairment | Impaired | Absent |
| β cell secretory capacity | Normal | Normal to moderate impairment | Variable impairment | Markedly impaired to absent |
T1D-associated autoantibodies include those against the β cell–specific antigens, glutamic acid decarboxylase 65 (GAD-65), the tyrosine phosphatase–related islet antigen 2 (IA-2), insulin (IAA), and zinc transporter 8 (ZnT8).
The risk for disease progression toward diabetes is much less with a single autoantibody; however, some individuals with a single autoantibody do progress, and the clinical diagnosis of symptomatic T1D does not require the presence of multiple autoantibodies.
One goal of staging presymptomatic T1D is to enable early immune intervention strategies aimed at preventing or ameliorating symptomatic diabetes through the preservation of sufficient pancreatic functional β cell mass to maintain (near)normoglycemia.20 Preservation of endogenous β cell function after clinical diagnosis is also important in facilitating optimal metabolic control. The objective of this review is to examine the physiological and clinical correlates of pancreatic islet reserve at each stage of T1D in order to aid understanding of pancreatic islet physiology informing interventions to maintain or restore a functional β cell mass.
Islet reserve and function in health
Pancreatic islets are distributed throughout the pancreatic acinar/exocrine tissue and account for 2–3% of pancreatic mass. Human islets are composed predominantly of insulin-producing β cells and glucagon-producing α cells with the remaining 10% comprising somatostatin-producing δ cells, pancreatic polypeptide producing PP cells, and ghrelin-producing ɛ cells.21 Nervous system innervation, β cell electrical activity, and intraislet paracrine signaling support glucose-dependent insulin and glucagon secretion.22 Insulin secretion is pulsatile in nature, with pulses occurring every ~5–6 min and (in the nondiabetic state) increasing in amplitude in response to glucose.23 In healthy individuals, insulin secretion varies by several orders of magnitude to meet the demand for control of glucose homeostasis. This is accomplished both by biphasic kinetics of insulin secretion and recruitment of additional β cells to an activated state, which provides the reserve capacity for secretion. The first-phase secretion involves rapid exocytosis of insulin granules from a readily releasable pool. Under conditions of a sustained glucose stimulus, this first phase is followed by a subsequent second, more prolonged phase of insulin release from the mobilization of granules within the reserve pool, comprising 95% of β cell insulin granules.24 Heterogeneity of β cell functional states has long been appreciated,25 and more recently, evidence has emerged supporting a subpopulation (<10% of β cells) that functions as a central hub, coordinating integrated β cell response to stimuli.26 Thus, normal insulin secretion appears dependent on coordinated β cell function from intact islets.
Normal insulin secretion is also dependent on the presence of sufficient reserve capacity of functional β cell mass. Functional β cell mass is best estimated from the β cell secretory capacity derived from glucose-potentiation of insulin or C-peptide release in response to injection of a nonglucose secretagogue, such as arginine or glucagon.27 Glucose potentiation creates the condition for second-phase insulin secretion, whereby mobilization of secretory granules from the reserve pool of an increased number of activated β cells results in augmented acute insulin release in response to membrane depolarization induced by the nonglucose secretagogue. Increased islet blood flow and islet oxygenation has been demonstrated in animal models of partial pancreatectomy and gestation, suggesting the recruitment of a functional reserve capacity in response to increasing β cell demand for secretion occurs without an increase in β cell mass.28,29 Nonetheless, the recruitment of senescent β cells and generation of new β cells from neogenesis or self-duplication of existing β cells may also contribute to increasing β cell secretory capacity for increased demand, as shown in rodent models.30 The risk of glucose intolerance secondary to a marked reduction in pancreatic mass is evidenced in healthy hemipancreatectomy donors in whom a residual 40% β cell secretory capacity was associated with reduced first-phase insulin secretion in response to glucose, as well as the development of glucose intolerance, and long-term risk of progression to diabetes, especially in association with weight gain.31,32 Another study involving individuals with exocrine pancreatic disease undergoing pancreatic surgery showed that diabetes was associated with a reduction in relative β cell area (assessed from sections of resected pancreatic tissue) to 30–40% of that in nondiabetic individuals.33 Indeed, both human and animal models suggest an islet mass of ~50% may be the threshold for maintaining sufficient β cell functional reserve for normal glucose control.34–36 Thus, while the range is variable among individuals,37,38 in general, with less than 50% of normal β cell secretory capacity, there may be insufficient islet reserve (including loss of first-phase insulin secretion) to meet demand. Other work reports that even mild hyperglycemia following 90% partial pancreatectomy in animal models is associated with changes in β cell–specific gene expression profiles,39 supporting a role for β cell dysfunction in addition to absolute β cell mass loss in contributing to insulin secretory defects with potential consequences for long-term β cell health.
There exists reciprocal glucose-dependent regulation of β cell insulin secretion and α cell glucagon secretion in the normal pancreatic islet, such that increasing glucose potentiates insulin secretion and inhibits glucagon secretion in order to avoid hyperglycemia, while decreasing glucose inhibits insulin secretion and activates glucagon secretion in order to avoid hypoglycemia.22 Hemipancreatectomy resulting in 40% of normal β cell secretory capacity is associated with 35% of normal acute glucagon response to arginine with and without glucose inhibition,31 in keeping with equivalent reductions in both pancreatic islet β and α cell mass. Importantly, despite reduced pancreatic islet mass, the α cell glucagon secretion in response to hypoglycemia is seemingly normal,40 and it is proposed that the remaining intact islets exhibit normal paracrine activation of glucagon secretion by the intraislet decrement in insulin secretion that occurs with the development and in defense of hypoglycemia,41 leading to physiological secretion despite a substantial decrease in α cell mass.
Islet reserve and function at early disease stages, before clinical diagnosis
Longitudinal studies continued over several decades of follow-up have confirmed that the presence of β cell autoimmunity, as evidenced by seroconversion of two or more islet autoantibodies, almost inevitably heralds progression to the clinical diagnosis of T1D.42–44 While considered prestage 1 disease, autoimmunity manifested by the presence of only a single autoantibody also confers risk with ~20% developing multiple antibodies within 5 years and ~15% progressing to clinical diabetes despite remaining with single autoantibody positivity.45 The stages of the diabetes framework describe the progression of disease by changes in glycemic status and has proved to be useful for refining inclusion and outcome criteria for clinical trials aiming to slow or delay disease progression. T1D is, however, primarily a disease of β cell dysfunction and loss with the secondary consequence of glucose dysregulation. β cell secretory abnormalities are present long before clinical diagnosis;46 but interestingly, often not well correlated with either the number of autoantibodies or glucose tolerance.
For example, Vandemeulebroucke et al. assessed functional β cell mass using a hyperglycemic clamp with glucagon stimulation to measure β cell secretory capacity in 17 IA-2 antibody–positive first-degree relatives followed over 5 years during which 50% were predicted to develop clinical diabetes.47 Ten of these individuals did not develop clinical diabetes over the 5-year follow-up and had entirely normal oral glucose tolerance and β cell secretory capacity at baseline, not different from a control group with genetic risk for T1D without antibodies.47 The seven individuals who did progress to clinical diabetes from 3 to 63 months after baseline study had oral glucose tolerance that was initially similar to that in the group of nonprogressors and controls, but β cell secretory capacity that was <50% of normal, indicating the presence of a substantial reduction in functional β cell mass.47 This small study suggests that the presence of two or more islet autoantibodies does not, in itself, indicate a reduction in pancreatic islet reserve, and moreover, that identification of a reduced β cell secretory capacity appears to be more sensitive than assessment of oral glucose tolerance (OGTT) for detecting those likely to progress more rapidly to overt diabetes.
β cell function in autoantibody-positive individuals is most often assessed from insulin or C-peptide levels obtained during an oral glucose tolerance test (OGTT) or from the first-phase insulin response (FPIR) during an intravenous glucose tolerance test (IVGTT). Decreased insulin secretion in individuals with early-stage T1D confers an increased risk of eventual clinical disease,48 but strikingly, low FPIR is only moderately associated with glucose intolerance in cross-sectional studies (r2 = 0.114).49 However, a separate study in type 2 diabetes (T2D) found that once fasting glucose exceeds 115 mg/dL, FPIR is absent,50 likely signifying the exhaustion of a reserve capacity for insulin secretion at that time. Studies by Hao et al. have shown significant variation in insulin secretory reserve/functional β cell mass, assessed by the acute insulin response to glucose-potentiated arginine stimulation, in antibody-positive individuals with reduced β cell function, assessed by the FPIR.51 It has previously been shown that the insulin response to glucose is lost earlier than the response to arginine in settings of a reduced β cell mass, including both presymptomatic T1D52 and recipients of islet transplantation,53 groups characterized by a marked reduction in functional β cell mass, where acute insulin responses to arginine are maintained despite marked impairment of or loss of FPIR. Evidence for differences in various surrogate measures of functional β cell mass following oral or intravenous stimulatory challenges further supports underlying heterogeneity in T1D pathogenesis.51
The rate of progression from autoimmunity to clinical diabetes is variable.54 β cell glucose sensitivity, measured by the slope of insulin secretion in response to increasing plasma glucose, may be impaired several years before diagnosis and so might represent an early sign of β cell dysfunction nearing a critical threshold of insulin deficiency beyond which clinical diabetes ensues.55 The Diabetes Prevention Trial-Type 1 risk score (DPTRS) can assess risk for future diabetes in autoantibody-positive individuals using age, body mass index, fasting C-peptide, and the 30-, 60-, 90-, and 120-min C-peptide and glucose values during a 2-h OGTT.56 Using only the 60-min C-peptide and glucose (DPTRS60) was recently shown to predict diabetes development with similar accuracy.57 Importantly, a 1-h OGTT glucose ≥155 mg/dL (8.6 mmol/L) had slightly greater receiver operating characteristics (sensitivity versus 1–specificity) than the standard 2-h OGTT glucose ≥140 mg/dL (7.8 mmol/L) for predicting future diabetes development.57 The potentially greater utility of the 1-h OGTT glucose to reflect the early loss of pancreatic islet secretory reserve in T1D is similar to that observed in individuals with pancreatic insufficient cystic fibrosis, where a 1-h OGTT glucose ≥155 mg/dL (8.6 mmol/L) is associated with an already marked reduction in β cell secretory capacity.58
Follow-up of a cohort of individuals with autoantibody positivity and relatives with T1D by 6-monthly OGTT toward the onset of diabetes in the Diabetes Prevention Trial-Type 1 showed a progressive decline in glucose tolerance over the 2-year period preceding diagnosis, with a more rapid decline in stimulated C-peptide occurring in the last 6 months.59 More recently, Bogun et al. demonstrated that this accelerated rate of fall in insulin secretion persists during and beyond the peridiagnostic period.60 Whether this accelerated decline in β cell function prior and through the peridiagnostic period is related to ongoing immunologic destruction and/or metabolic exhaustion of β cells is unknown.
Some individuals have been reported to have disproportionately increased secretion of proinsulin relative to insulin or C-peptide during disease progression, as assessed by the elevated fasting proinsulin-to-C-peptide ratio before diagnosis.61 Similar to observations in T2D, impaired proinsulin processing before secretion is suggestive of β cell endoplasmic reticulum stress; however, the relationship of the abnormal proinsulin-to-C-peptide ratios to either autoimmunity or glucose tolerance remains unclear. It is likely that both the dysfunction of β cells in intact islets and the loss of islet β cells due to chronic β cell destruction contribute to disease progression toward diabetes. Whether individuals with a greater dysfunctional component underlying reduced insulin secretion but relatively intact β cell secretory capacity indicating preserved β cell mass may be more likely to respond to disease-modifying therapy requires further investigation.51
Islet reserve and function after clinical diagnosis
Stage 3 T1D is defined by the onset of clinical disease through the presence of signs and symptoms of hyperglycemia or when asymptomatic by crossing the diagnostic glucose thresholds established by the World Health Organization/American Diabetes Association. The prospective monitoring of individuals with presymptomatic disease can enable early detection of diabetes through the confirmation of fasting glucose ≥126 mg/dL (≥7.0 mmol/L); 2-h OGTT glucose ≥200 mg/dL (≥11.1 mmol/L); and/or an HbA1c ≥6.5% (48 mmol/mol) before the onset of symptoms. ≥ Individuals with early, but still asymptomatic, stage 3 T1D defined by a 2-h OGTT glucose ≥200 mg/dL (≥ 11.1 mmol/L) who are able to maintain nondiabetic levels of fasting glucose and HbA1c already exhibit markedly impaired insulin secretion in response to oral and intravenous glucose, yet preserved FPIR to the nonglucose β cell secretagogue arginine.62 This may be explained by the potentiation of insulin release to nonglucose secretagogues by increasing fasting glucose concentration. The reduction in overall β cell secretory capacity is, however, observed by reduced insulin release in response to arginine potentiated by submaximal (AIRpot) and maximal (AIRmax) hyperglycemic clamp conditions. Indeed, glucose-potentiated arginine testing demonstrated that the β cell secretory capacity in this cohort of individuals with asymptomatic T1D was only 25% of that in a normal control group.62
In another study of new-onset T1D, the median β cell secretory capacity of 80 individuals was again 25% of that measured in 20 healthy, agematched control subjects using glucose potentiation of glucagon-induced insulin release.63 Here, individuals with β cell secretory capacity at or above the 25th percentile were more likely to experience the preservation of β cell function and a reduction in insulin requirements 18 months after receiving a 6-day course of anti-CD3 antibody therapy.63 Thus, a functional β cell mass of at least 25% normal may be required to avoid symptomatic T1D and may also represent a threshold that allows for the easier demonstration of protection of residual β cell function. However, it is unknown whether individuals with lower pancreatic islet reserve at clinical diagnosis have more severe and potentially treatment-resistant autoimmunity or simply greater and potentially less reversible loss of functional β cell mass. Interestingly, at a β cell secretory capacity <25% of normal adults, insulin release to nonglucose secretagogues is already maximal in the fasted state in the presence of elevated glucose concentrations, such that further glucose potentiation of insulin release to arginine under hyperglycemic clamp conditions is no longer possible (Fig. 1).
Figure 1.
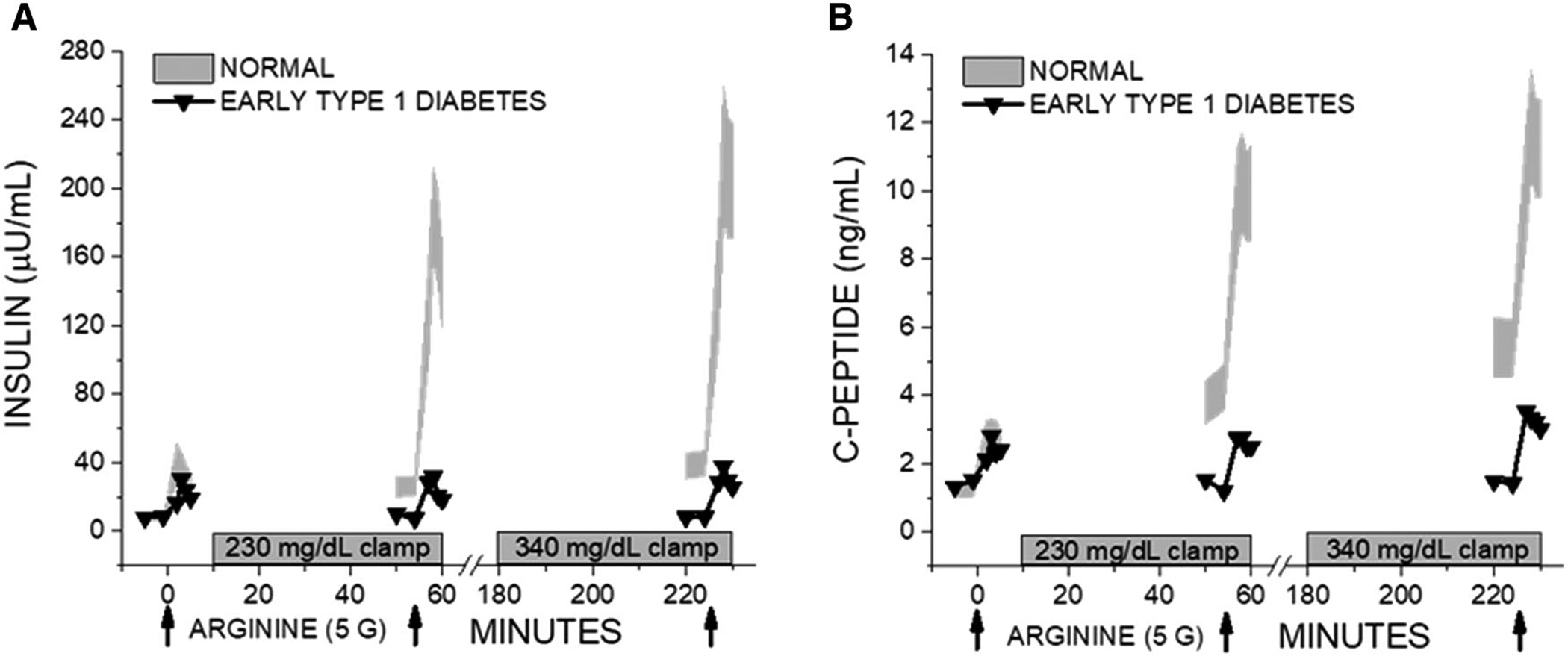
β cell secretory capacity data from an individual with adult-onset T1D, initially diagnosed with T2D and at the time of glucose-potentiated arginine testing before initiating insulin therapy. Under fasting conditions, the acute insulin (A) or C-peptide (B) response to arginine appear “normal” (although not when considering the fasting glucose was elevated at 162.5 mg/dL (not shown)), and further elevation of the glucose cannot further potentiate insulin release as the entire reserve capacity for secretion (~15% of normal) is already engaged under the fasting condition. The normal data are 95% CI based on 28 nondiabetic individuals.
At the point when pancreatic islet reserve for β cell insulin secretion is compromised in T1D, marked abnormalities in α cell regulation of glucagon secretion are also evident. Metabolic testing through OGTT and IVGTT at the earliest identification of stage 3 T1D reveals impaired suppression of glucagon secretion in response to hyperglycemia.62 This impaired regulation of glucagon secretion in T1D may be explained by the loss of paracrine effects of β cells on α cell function in islets, where β cells are destroyed or no longer functional due to β cell autoimmunity. The absence of suppression of glucagon during oral glucose or mixed-meal tolerance testing (MMTT) likely contributes to postprandial hyperglycemia in early T1D.62,64
In addition, glucagon response to hypoglycemia is already markedly impaired at diabetes onset.65,66 Proposed underlying mechanisms include altered islet innervation, abnormal α cell glucose sensing, and reduced intraislet paracrine signaling, whereby β cell deficiency leads to an insufficient decrement of insulin in response to hypoglycemia to act as a paracrine trigger for α cell glucagon release (reviewed by Rickels67). Whatever the mechanism, an impaired glucagon response to hypoglycemia is observed uniformly in recent-onset diabetes regardless of the level of residual C-peptide.66,68
Brissova et al. have provided cellular evidence for an inherent α cell defect in T1D, with the reduction in transcription factors associated with α cell identity and reduced expression of exocytosis and signaling-related proteins demonstrated in islets isolated from individuals with T1D.69 Isolated islets showed the preservation of glucose-stimulated insulin secretion from the low number of β cells present; however, glucagon release to low glucose incubation was deficient despite the observation of an increased number of α cells in islets from the individuals with T1D compared with controls. Although a relative increase in α to β cells is anticipated due to β cell loss, this observation of increased numbers but the impaired function of α cells suggests an additional role for islet cell dedifferentiation in diabetes pathogenesis, a finding that is supported by pathology studies in T2D.70 Similar data available from the Human Pancreas Analysis Program (https://hpap.pmacs.upenn.edu) demonstrate seemingly appropriate dynamics of insulin secretion in response to glucose, albeit at the markedly reduced magnitude, and paradoxically increased glucagon secretion (Fig. 2A). Interestingly, the transplantation of islets from individuals with T1D into a normoglycemic nonautoimmune environment led to partial restoration of gene expression changes toward a normal α cell phenotype.69
Figure 2.
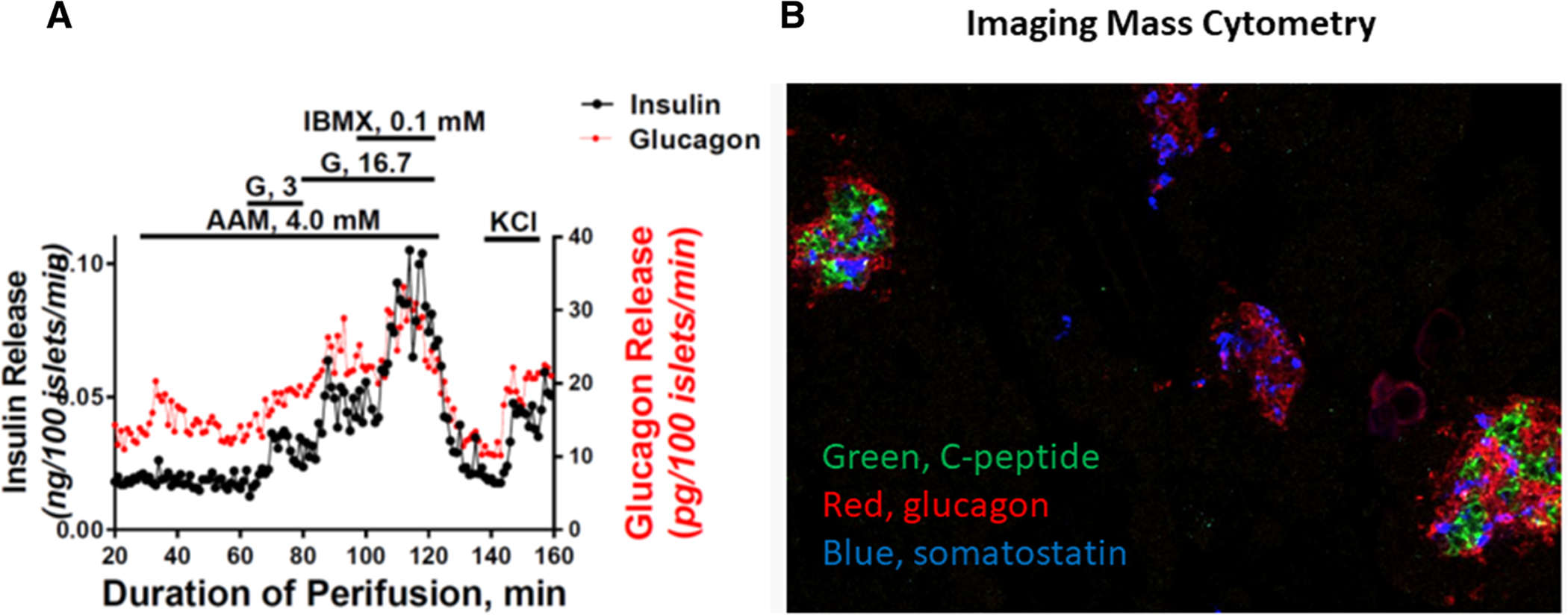
(A) The perifusion of islets isolated from a deceased donor with a 5-year duration of T1D. Insulin release is appropriate in response to change from low glucose (G; 3 mM) to high glucose (G; 16.7 mM), and to phosphodiesterase inhibition with 3-isobutyl-1-methylxanthine (IBMX, 0.1 mM) and membrane depolarization with KCl, albeit at the markedly reduced magnitude. Glucagon release is appropriate in response to amino acid mixture (AAM, 4.0 mM), IBMX, and KCl, but paradoxically increased (failed suppression) in response to change from low to high glucose. (B) Imaging mass cytometry of pancreatic sections from the same deceased donor demonstrates the presence of few islets with normal numbers of β cells (green) and more representative islets containing α (red) and δ (blue) but no β cells. Data are available at https://hpap.pmacs.upenn.edu.
In vitro studies have demonstrated the down-regulation of key β cell transcription factors and changes in β cell phenotype associated with hyperglycemia, which, to some extent, may be reversible with normalization of glycemia.39 However, the extent to which β cell senescence, dedifferentiation, and transdifferentiation are implicated in β cell dysfunction and loss in human T1D remains unclear.39 Intensive insulin use at diagnosis has been associated with a period of preservation of C-peptide production71,72 with the fasting euglycemia attained from exogenous insulin therapy, perhaps contributing to β cell “rest” and possible protection from the phenotypic and functional changes associated with β cell stress. Similar “remission” is seen with aggressive glycemic control in those with T2D. However, the failure of a randomized trial to confirm the preservation of functional β cell mass with intensive insulin therapy in recently diagnosed T1D patients,73 as well as the failure of parenteral insulin to delay disease progression in the early stages of disease,74 suggests that the benefit of insulin therapy on the preservation of secretion is only manifest with marked initial hyperglycemia, supporting the hypothesis that glucotoxicity plays a role in β cell dysfunction with a potentially reversible component.
Islet reserve and function in established clinical disease
Functional β cell mass continues to decline following a diagnosis such that the majority of individuals have a stimulated C-peptide <0.60 ng/mL (0.200 pmol/mL) within 5 years of symptomatic diagnosis.75 This decline is dependent on the age at symptomatic disease onset, with individuals presenting as adults >18 years old tending to maintain residual β cell function longer than those presenting as children.76 In the Diabetes Control and Complications Trial (DCCT), those individuals with a mixed-meal stimulated C-peptide >0.60 ng/mL (0.200 pmol/mL) within 5 years of symptomatic diagnosis had a lower incidence of retinopathy and nephropathy and a decreased prevalence of severe hypoglycemia than those with lower amounts of residual C-peptide detection regardless of treatment intensity.77,78 These results fairly conclusively support that preservation of residual β cell function characterized by stimulated C-peptide >0.60 ng/mL (0.200 pmol/mL) is clinically meaningful and supports immunotherapy approaches toward the preservation of functional β cell mass (reviewed by Lord and Greenbaum79). A more recent analysis of the DCCT suggests that any level of stimulated C-peptide above the detection threshold of 0.09 ng/mL (0.03 pmol/mL) used at trial entry may be associated with better clinical outcomes.80
The development of highly sensitive C-peptide assays led to the detection of very low levels of C-peptide (that would have been “undetectable” in the DCCT) in the majority of individuals over the first 10 years of symptomatic T1D and in a declining minority of individuals more than a decade after clinical diagnosis.81–83 These observations are consistent with Yu et al., who identified few scattered insulin-positive cells in all individuals with T1D at autopsy.84 Follow-up of individuals at least 5 years after diagnosis showed that circulating proinsulin may persist even in the absence of detectable C-peptide. Whether this is caused by ongoing (immunologically or metabolically mediated) β cell stress or represents an irreversible shift in β cell phenotype precluding normal proinsulin processing remains unclear.85,86 Assessment of type 1 donor islets from pancreatic sections obtained from the Network for Pancreatic Organ Donors with Diabetes demonstrated the reduced expression of the proinsulin processing enzymes, prohormone convertase 1/3, and carboxypeptidase, compared with nondiabetic controls, which may account for the elevated proinsulin:C-peptide ratios in individuals with the lowest detectable β cell peptides.87
Sustained C-peptide, >0.60 ng/mL (0.200 pmol/mL) in a random sample, has been observed in members of the Joslin Medalist group despite more than 50 years of insulin-dependent diabetes. In this cohort, sustained C-peptide was associated with a later age of diagnosis and the presence of DR3 risk alleles.88 Postmortem examination of pancreata from nine Medalists showed significant numbers of persisting insulin-positive cells scattered as single extra-islet cells or as single cells within some islets or many cells in some islets in several lobes among many insulin-negative islets despite the long duration of diabetes.88 Insulin-positive islets were more likely to be present in later-onset diabetes. However, even Medalists without detectable C-peptide on MMTT could have evidence of residual insulin-positive β cells present as single cells within islets.84,88 Nevertheless, the number of residual insulin-positive cells correlated with the preceding MMTT C-peptide response such that those who experienced a physiologically significant doubling of C-peptide on stimulation had considerably more insulin-positive cells found within islets of certain lobules of the pancreas.88 This finding suggests that physiological inducible insulin secretion may depend on the presence of residual β cells in intact islets rather than occur from scattered insulin-positive cells (Fig. 2B).
A subsequent study of 68 individuals in the Joslin Medalist cohort by Yu et al. identified three patterns of residual insulin staining, including scattered insulin-positive cells present in all samples; few insulin-positive cells within some islets in ~87%; and many insulin-positive cells in some islets within certain lobes of tissue present in ~20% of samples.84 Prospective follow-up of C-peptide and autoantibody status of Medalists over 4 years showed low levels of detectable C-peptide (random sample >0.05 ng/mL (0.017 pmol/mL)) were present in ~32% of individuals. Detectable C-peptide was associated with IA2 autoantibody positivity, with GAD positivity more commonly observed in those with undetectable C-peptide. However, antibody titers and indeed C-peptide levels fluctuated over follow-up, suggestive of possible episodic autoimmune aggravation alternating with at least minimal recovery of some β cell function despite long-standing disease duration.84 Preserved lobes containing insulin-positive islets were associated with greater C-peptide, lower HbA1c, and older age at diagnosis compared with individuals with only residual scattered insulin-positive cells (within and outside islets). These findings further suggest the importance of β cell contact/islet microenvironment in the maintenance of physiological insulin secretion.84
Rickels et al. assessed the physiological significance of varying levels of residual C-peptide in individuals with established T1D stratified by MMTT peak C-peptide as negative (<0.02 ng/mL (0.007 pmol/mL)), low (0.05–0.60 ng/mL (0.017–0.200 pmol/mL)), intermediate (0.60–1.20 ng/mL(0.200–0.400 pmol/mL)), and high (>1.20 ng/mL (>0.400 pmol/mL)).89 Functional β cell mass, assessed by the acute C-peptide response to glucose-potentiated arginine, was shown to be highly associated with MMTT peak C-peptide,89 a relationship also seen with the MMTT C-peptide area-under-the-curve (Fig. 3). Despite this continuous association between MMTT peak C-peptide and β cell secretory capacity as a measure of functional β cell mass, only the high C-peptide group showed β cell responsivity to glucose during a hyperglycemic clamp before the injection of arginine (Fig. 4A) and α cell responsivity to insulin-induced hypoglycemia during a hyperinsulinemic hypoglycemic clamp (Fig. 4B). The increased glucagon response to hypoglycemia observed in the high C-peptide group may depend on functional β cell suppression within intact islets activating neighboring α cells. However, even with high residual C-peptide, β cell secretion of insulin in response to meal or arginine stimulation does not suppress α cell glucagon secretion.89 This dysregulation of α cell function may be explained by the failure of glucagon suppression from the majority of diseased islets that do not contain β cells.
Figure 3.
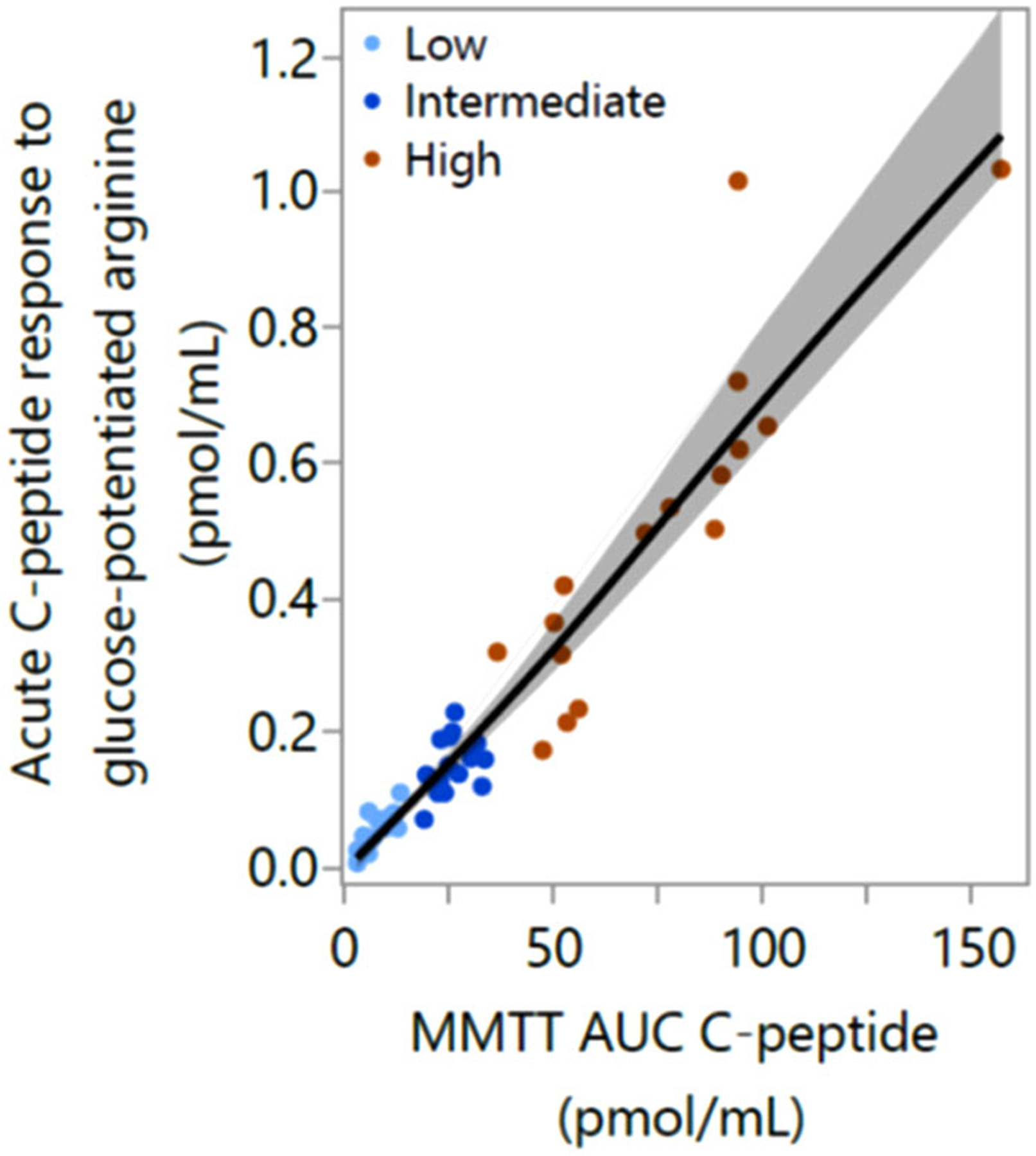
Relationship between β cell secretory capacity measured from the acute C-peptide response to glucose-potentiated arginine and the mixed-meal tolerance test (MMTT) area-under-the-curve (AUC) C-peptide response over 120 min in individuals with MMTT peak C-peptide defined as: low, 0.05–0.60 ng/mL (0.17–0.20 pmol/mL); intermediate, >0.60–1.20 ng/mL (0.20–0.40 pmol/mL); or high, >1.20 ng/mL(>0.40 pmol/mL). Data are from Ref. 89.
Figure 4.
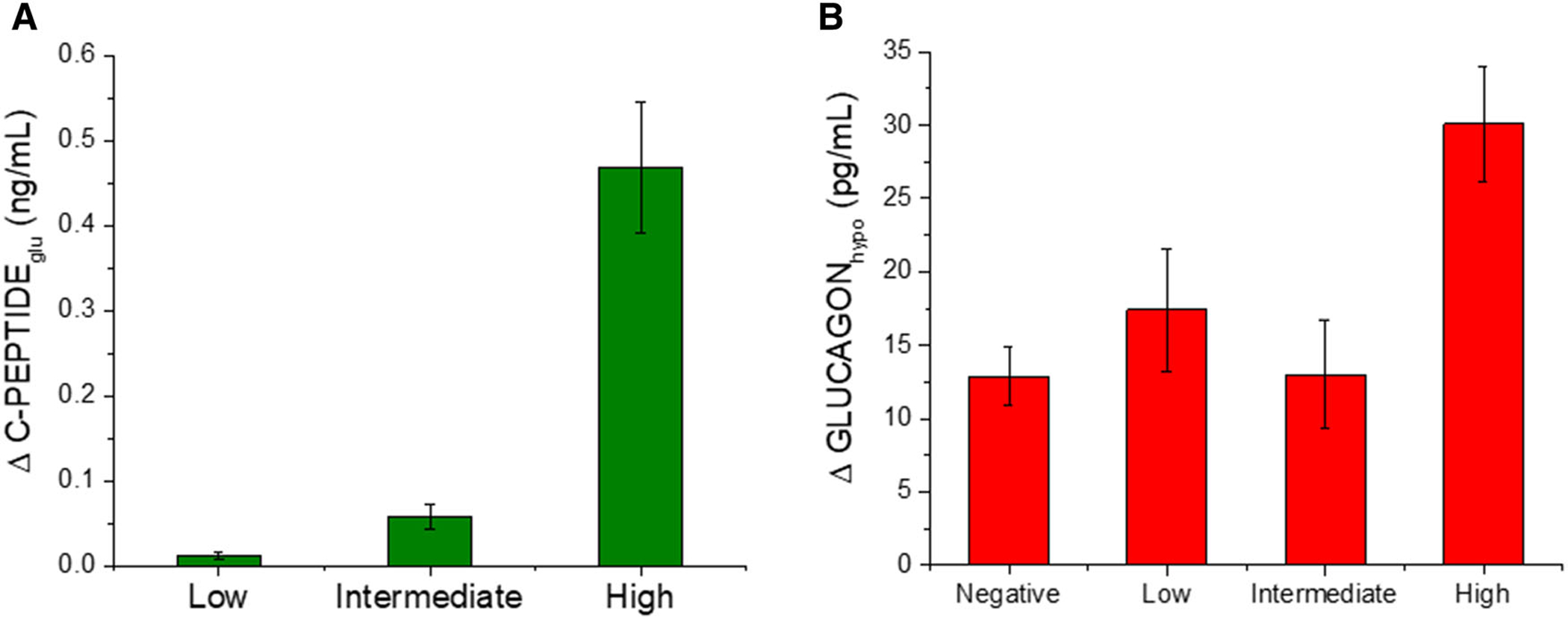
(A) β cell response to an increase in glucose during a hyperglycemic clamp, and (B) α cell response to insulin-induced hypoglycemia during a hypoglycemic clamp in individuals with T1D by peak C-peptide during a mixed-meal tolerance test (negative, <0.02 ng/mL; low, 0.05–0.60 ng/mL; intermediate, >0.60–1.20 ng/mL; and high, >1.20 ng/mL). An increase in C-peptide in response to hyperglycemia and an increase in glucagon in response to hypoglycemia were observed only in the group with the highest residual C-peptide production that may represent a minimal functional islet reserve. Data are from Ref. 89.
Importantly, individuals with high MMTT peak C-peptide who have evidence of preserved islet β and α cell responsivity to glucose also exhibit lower mean glucose and greater time in range on continuous glucose monitoring (Fig. 5A).89 Consistent with these findings, a study investigating the glycemic effects of 45 min of aerobic exercise in T1D found that individuals with higher MMTT peak C-peptide (>0.60 ng/mL (0.200 pmol/mL)) spent more time in range on continuous glucose monitoring during the subsequent 24 h than those with low or undetectable C-peptide, and while not statistically significant given the small sample size, spent twofold less time with hypoglycemia during the postexercise period.90 Thus, improved glycemic control in individuals with high residual C-peptide may be explained by a valuable functional reserve of preserved β and α cell responsivity to glucose, which can refine the effects of exogenous insulin, but which is less apparent with lower residual C-peptide production. Nevertheless, the Scottish Diabetes Research Network Type 1 Bioresource following over 6000 individuals with T1D has shown that after adjusting for diabetes duration, even low levels of residual C-peptide (0.09–0.60 ng/mL (0.030–0.200 pmol/mL)) in a random sample provide protection from severe hypoglycemia independent of HbA1c or insulin dose.91
Figure 5.
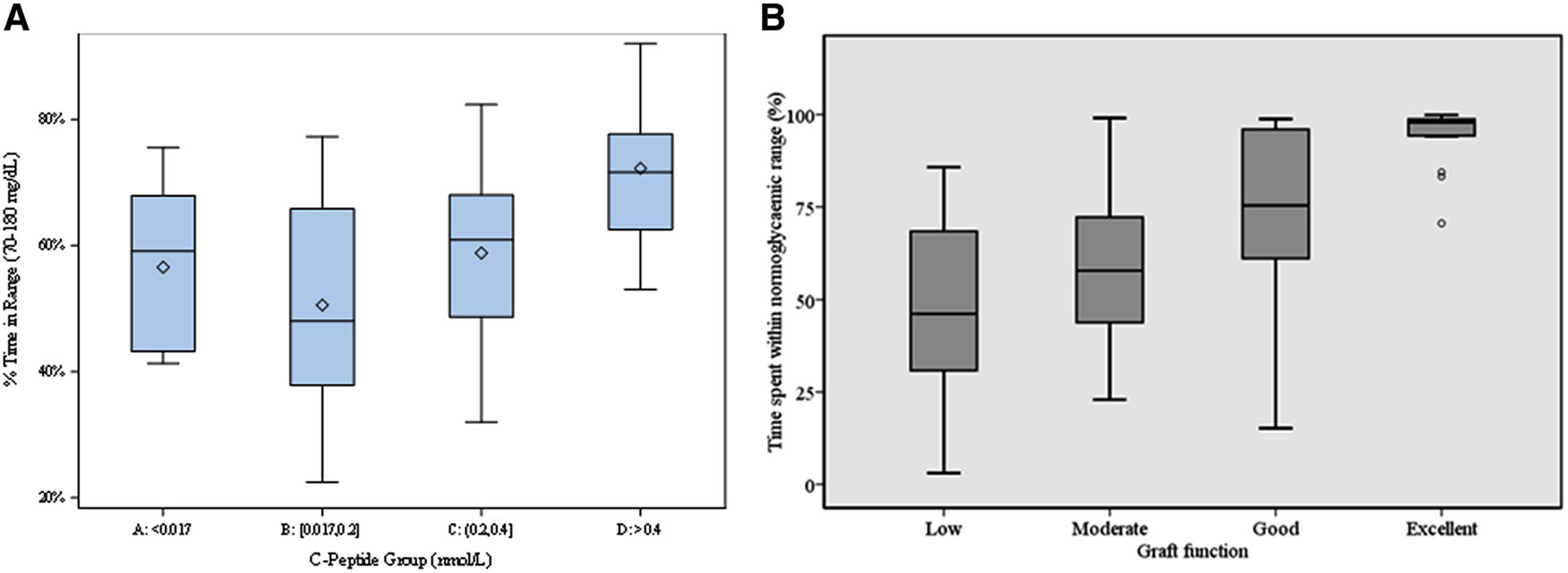
(A) Relationship of residual MMTT peak C-peptide (negative, <0.02 ng/mL; low, 0.05–0.60 ng/mL; intermediate, >0.60–1.20 ng/mL; and high, >1.20 ng/mL) and time spent in glucose range 70–180 mg/dL by continuous glucose monitoring. (B) Relationship of islet transplant MMTT peak C-peptide (low, <0.60 ng/mL; moderate, 0.60–<1.50 ng/mL; good, 1.5-to–<3.0 ng/mL; and excellent, ≥3.0 ng/mL) and time spent in the glucose range of 54–180 mg/dL by continuous glucose monitoring. Data are from Refs. 89 (A) and 108 (B).
Islet reserve and function with replacement in long-standing clinical disease
Long-standing (>10–15 years) symptomatic T1D is inextricably linked not only to decreasing prevalence of physiologically significant pancreatic islet cell reserve but also the development of progressive defects in glucose counterregulation to defend against the development of hypoglycemia. Initially, once insulin-deficient diabetes is established, the maintenance of an endogenous glucose production (EGP) response to defend against the development of low blood glucose appears most dependent on an intact epinephrine response rather than a relative increase in glucagon secretion.89 However, it is well established that exposure to iatrogenic hypoglycemia associated with intensive insulin therapy reduces the glycemic threshold and magnitude of subsequent sympathoadrenal responses to hypoglycemia92,93 such that with repeated hypoglycemia exposure, these may fail to reverse low blood glucose and contribute to a cycle whereby hypoglycemia begets further hypoglycemia with the development of impaired awareness and severe events.94
Impairment of counterregulatory epinephrine and autonomic symptom responses to hypoglycemia contributes to impaired awareness of hypoglycemia, which combined with defective glucose counterregulation (impaired EGP response), leads to the development of hypoglycemia-associated autonomic failure (HAAF), also known as hypoglycemia unawareness,95 which increases the risk of experiencing life-threatening severe hypoglycemia 20-fold.96 Replacement of islet mass through allogeneic β cell transplantation in individuals with long-standing symptomatic T1D complicated by hypoglycemia unawareness restores physiologic islet cell responses to hypoglycemia that are associated with recovery of glucose counterregulation97 and reversal of HAAF.98 Thus, islet allotransplantation can provide additional insight into the relationship between functional islet reserve and glycemic control in T1D.
Islet allotransplantation is indicated for individuals with T1D and hypoglycemia unawareness experiencing severe hypoglycemia and/or excessive glycemic variability refractory to medical management with intensive insulin therapy. Islet allotransplantation involves the isolation of human islets from one or more deceased donor pancreata, which are transplanted by intraportal infusion during a percutaneous or minilaparotomy procedure for islet engraftment within the liver parenchyma.99,100 Insulin independence is achieved when functional β cell mass is restored to >25–40% of normal based on measures of β cell secretory capacity derived from glucose potentiation of glucagon- or arginine-induced insulin secretion.101,102 Lower estimates of functional β cell mass/secretory capacity following islet transplantation, while associated with ongoing insulin requirements, are related to proportionate improvement in glycemic control and variability relative to pretransplantation. Islet grafts with a functional β cell mass <5% of nondiabetic controls do not demonstrate any benefit on glycemic variability.103 Continued islet graft function with reduction in severe hypoglycemia and glycemic variability is observed in 90% of recipients at 5 years; however, a decline in islet graft reserve capacity is often observed over time, with the majority returning to exogeneous insulin therapy by 5 years.104 By contrast, although associated with significantly greater periprocedure morbidity, allogeneic whole pancreas transplantation more often results in insulin independence, and when transplanted simultaneously with the kidney from the same deceased donor is associated with a 5-year insulin independence rate >70%.105 Assessment of β cell secretory capacity may require consideration for the systemic hyperinsulinemia that results from systemic venous drainage of the pancreas graft (reviewed by Rickels106), although functional β cell mass/secretory capacity has been shown to be normal in recipients with portal venous drainage of the pancreas graft.107 Restoration of a normal reserve capacity for insulin secretion likely explains the more durable insulin independence experienced by recipients of the whole pancreas when compared with isolated islet transplantation.
Incremental improvements in MMTT-stimulated C-peptide following islet allotransplantation are associated with a progressive reduction in glycemic variability and increasing time spent in the target glucose range by continuous glucose monitoring.108 A continuous relationship was seen between stimulated C-peptide and all continuous glucose monitoring parameters, with a 0.100-pmol/mL increment in C-peptide associated with a 5% reduction in glucose standard deviation and a 2.5% reduction in interstitial glucose. Improved glycemic control (HbA1c) with a simultaneous reduction in exogenous insulin requirement was also observed across C-peptide groupings. Low levels (<0.6 ng/mL (<0.200 pmol/mL)) of MMTT-stimulated C-peptide following islet allotransplantation were associated with poor glycemic control and excessive glucose variability, with time in the range of 54–180 mg/dL (3–10 mmol/L) improving with moderate (0.6–1.5 ng/mL (0.200–0.500 pmol/mL)) and more significantly with good (1.5–3.0 ng/mL (0.500–1.000 pmol/mL)) levels (Fig. 5B). MMTT-stimulated C-peptide (>3.0 ng/mL (>1.000 pmol/mL)) was associated with 95% of the time within the 54–180 mg/dL glucose range and attainment of insulin independence.108 Importantly, the restoration of only moderate levels of MMTT-stimulated C-peptide above the DCCT threshold of 0.6 ng/mL (0.200 pmol/mL) was associated in this high-risk cohort with absolute prevention of biochemical hypoglycemia (glucose <54 mg/dL).108 Thus, the goal for interventions targeting preservation or restoration of β cell function in T1D should consider aiming for a stimulated C-peptide level of at least 0.6 ng/mL (0.200 pmol/mL) as suggested by recent consensus guidelines for defining outcomes of β cell replacement therapies.109
Conclusions
Abnormalities in pancreatic islet β cell function and secretory capacity can be detected before the clinical presentation of T1D and progress over the years following diagnosis. It is now clear that residual islet β cells may persist for many decades with evidence in some cases of physiologic islet β cell function and that individuals with high residual C-peptide production may benefit from reduced glucose variability and improved overall glycemic control. Effective approaches to both preserve and restore pancreatic islet reserve before or at diabetes onset and sustain or replace the functional β cell mass in established diabetes are likely to lead to reduced diabetes-related complications with direct implications for quality of life.110 Transplant studies have shown that even partial restoration of functional islet β cell mass can enable insulin independence, setting a target mass to attain through nontrans-plant approaches designed to maintain or regenerate endogenous pancreatic islet reserve.
Acknowledgments
The authors thank Henry T. Bahnson and Alyssa Ylescupidez of the Benaroya Research Institute for help with the creation of Figure 3. M.R.R. is supported in part by Public Health Services Research Grants R01 DK091331 and UC4 DK112217 (Human Pancreas Analysis Program). This manuscript used data for Figure 2 acquired from the Human Pancreas Analysis Program (HPAP-RRID:SCR_016202) Database (https://hpap.pmacs.upenn.edu), a Human Islet Research Network (RRID:SCR_014393) consortium (UC4-DK-112217, U01-DK-123594, UC4-DK-112232, and U01-DK-123716).111
Competing interests
M.R.R. has received consulting honoraria from Semma Therapeutics and Sernova Corp., and has received research support from Xeris Pharmaceuticals.
References
- 1.Gubitosi-Klug RA; DCCT/EDIC Research Group. 2014. The diabetes control and complications trial/epidemiology of diabetes interventions and complications study at 30 years: summary and future directions. Diabetes Care 37: 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hendrieckx C, Halliday JA, Bowden JP, et al. 2014. Severe hypoglycaemia and its association with psychological well-being in Australian adults with type 1 diabetes attending specialist tertiary clinics. Diabetes Res. Clin. Pract 103: 430–436. [DOI] [PubMed] [Google Scholar]
- 3.Lind M, Svensson A, Kosiborod M, et al. 2014. Glycemic control and excess mortality in type 1 diabetes. N. Engl. J. Med 371: 1972–1982. [DOI] [PubMed] [Google Scholar]
- 4.Rawshani A, Sattar N, Franzén S, et al. 2018. Excess mortality and cardiovascular disease in young adults with type 1 diabetes in relation to age at onset: a nationwide, register-based cohort study. Lancet 392: 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foster NC, Beck RW, Miller KM, et al. 2019. State of type 1 diabetes management and outcomes from the T1D exchange in 2016–2018. Diabetes Technol. Ther 21: 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weir GC & Bonner-Weir S. 2004. Five of stages of evolving β-cell dysfunction during progression to diabetes. Diabetes 53: S16–S21. [DOI] [PubMed] [Google Scholar]
- 7.Eisenbarth GS, Nayak RC & Rabinowe SL. 1988. Type I diabetes as a chronic autoimmune disease. J. Diabet. Complications 2: 54–58. [DOI] [PubMed] [Google Scholar]
- 8.Palmer JP 1987. Insulin autoantibodies: their role in the pathogenesis of IDDM. Diabetes Metab. Rev 3: 1005–1015. [DOI] [PubMed] [Google Scholar]
- 9.Bottazzo GF, Florin-Christensen A & Doniach D. 1974. Islet-cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiencies. Lancet 304: 1279–1283. [DOI] [PubMed] [Google Scholar]
- 10.Srikanta S, Ganda OP, Jackson RA, et al. 1983. Type 1 diabetes mellitus in monozygotic twins: chronic progressive beta cell dysfunction. Ann. Intern. Med 99: 320–326. [DOI] [PubMed] [Google Scholar]
- 11.Ziegler AG, Ziegler R, Vardi P, et al. 1989. Life-table analysis of progression to diabetes of anti-insulin autoantibody-positive relatives of individuals with type 1 diabetes. Diabetes 38: 1320–1325. [DOI] [PubMed] [Google Scholar]
- 12.Maahs DM, West NA, Lawrence JM, et al. 2010. Epidemiology of type 1 diabetes. Endocrinol. Metab. Clin. North Am 39: 481–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riley WJ, Maclaren NK, Krischer J, et al. 1990. A prospective study of the development of diabetes in relatives of patients with insulin-dependent diabetes. N. Engl.J. Med 323: 1167–1172. [DOI] [PubMed] [Google Scholar]
- 14.Tarn AC, Dean BM, Schwarz G, et al. 1988. Predicting insulin-dependent diabetes. Lancet 331: 845–850. [DOI] [PubMed] [Google Scholar]
- 15.Deschamps I, Boitard C, Hors J, et al. 1992. Life table analysis of the risk of type 1 (insulin-dependent) diabetes mellitus in siblings according to islet cell antibodies and HLA markers. An 8-year prospective study. Diabetologia 35: 951–957. [DOI] [PubMed] [Google Scholar]
- 16.Eringsmark Regnéll S & Lernmark Å. 2013. The environment and the origins of islet autoimmunity and type 1 diabetes. Diabet. Med 30: 155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vehik K, Lynch KF, Wong MC, et al. 2019. Prospective virome analysis in young children at increased genetic risk for type 1 diabetes. Nat. Med 25: 1865–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Craig ME, Kim KW, Isaacs SR, et al. 2019. Early-life factors contributing to type 1 diabetes. Diabetologia 62: 1823–1834. [DOI] [PubMed] [Google Scholar]
- 19.Insel RA, Dunne JL, Atkinson MA, et al. 2015. Staging presymptomatic type 1 diabetes: a scientific statement of JDRF, the Endocrine Society and the American Diabetes Association. Diabetes Care 38: 1964–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dayan C, Korah M, Tatovic D, et al. 2019. Changing the landscape for type 1 diabetes: the first step to prevention. Lancet 394: 1286–1296. [DOI] [PubMed] [Google Scholar]
- 21.Da Silva Xavier G 2018. The cells of the islets of Langer-hans. J. Clin. Med 7. 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moede T, Leibiger IB & Berggren PO. 2020. Alpha cell regulation of beta cell function. Diabetologia 63: 2064–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song SH, Kjems L, Ritzel R, et al. 2002. Pulsatile insulin secretion by human pancreatic islets. J. Clin. Endocrinol. Metab 87: 213–221. [DOI] [PubMed] [Google Scholar]
- 24.Rorsman P, Eliasson L, Renstrom E, et al. 2000. The cell physiology of biphasic insulin secretion. News Physiol. Sci 15: 72–77. [DOI] [PubMed] [Google Scholar]
- 25.Pipeleers D, Mesmaeker ID, Robert T, et al. 2017. Heterogeneity in the beta-cell population: a guided search into its significance in pancreas and in implants. Curr. Diab. Rep 17: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson NR, Mitchell RK, Haythorne E, et al. 2016. Beta cell hubs dictate pancreatic islet responses to glucose. Cell Metab. 24: 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robertson RP, Raymond RH, Lee DS, et al. 2014. Arginine is preferred to glucagon for stimulation testing of β-cell function. Am. J. Physiol. Endocrinol. Metab 307: E720–E727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olsson R & Carlsson PO. 2011. A low-oxygenated subpopulation of pancreatic islets constitutes a functional reserve of endocrine cells. Diabetes 60: 2068–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jansson L, Barbu A, Bodin B, et al. 2016. Pancreatic islet blood flow and its measurement. Ups J. Med. Sci 121: 81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weir GC & Bonner-Weir S. 2011. Sleeping islets and the relationship between β-cell mass and function. Diabetes 60: 2018–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seaquist ER & Paul Robertson R. 1992. Effects of hemipancreatectomy on pancreatic alpha and beta cell function in healthy human donors. J. Clin. Invest 89: 1761–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robertson RP, Lanz KJ, Sutherland DE, et al. 2002. Relationship between diabetes and obesity 9 to 18 years after hemipancreatectomy and transplantation in donors and recipients. Transplantation 73: 736–741. [DOI] [PubMed] [Google Scholar]
- 33.Meier JJ, Menge BA, Breuer TGK, et al. 2009. Functional assessment of pancreatic beta-cell area in humans. Diabetes 58: 1595–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ritzel RA, Butler AE, Rizza RA, et al. 2006. Relationship between beta-cell mass and fasting blood glucose concentration in humans. Diabetes Care 29: 717–718. [DOI] [PubMed] [Google Scholar]
- 35.Saisho Y, Butler AE, Manesso E, et al. 2010. Relationship between fractional pancreatic beta cell area and fasting plasma glucose concentration in monkeys. Diabetologia 53: 111–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kjems LL, Kirby BM, Welsh EM, et al. 2001. Decrease in beta-cell mass leads to impaired pulsatile insulin secretion, reduced postprandial hepatic insulin clearance, and relative hyperglucagonemia in the minipig. Diabetes 50: 2001–2012. [DOI] [PubMed] [Google Scholar]
- 37.Seino S, Shibasaki T & Minami K. 2011. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J. Clin. Invest 121: 2118–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weir GC, Gaglia J & Bonner-Weir S. 2020. Inadequate β-cell mass is essential for the pathogenesis of type 2 diabetes. Lancet Diabetes Endocrinol. 8: 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ebrahimi AG, Hollister-Lock J, Sullivan BA, et al. 2020. Beta cell identity changes with mild hyperglycemia: implications for function, growth and vulnerability. Mol. Metab 35. 100959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robertson RP, Sutherland DER, Seaquist ER, et al. 2003. Glucagon, catecholamine, and symptom responses to hypoglycemia in living donors of pancreas segments. Diabetes 52: 1689–1694. [DOI] [PubMed] [Google Scholar]
- 41.Cooperberg BA & Cryer PE. 2010. Insulin reciprocally regulates glucagon secretion in humans. Diabetes 59: 2936–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ziegler AG, Rewers M, Simell O, et al. 2013. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 309: 2473–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Redondo MJ, Geyer S, Steck AK, et al. 2018. A type 1 diabetes genetic risk score predicts progression of islet autoimmunity and development of type 1 diabetes in individuals at risk. Diabetes Care 41: 1887–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beyerlein A, Bonifacio E, Vehik K, et al. 2019. Progression from islet autoimmunity to clinical type 1 diabetes is influenced by genetic factors: results from the prospective TEDDY study. J. Med. Genet 56: 602–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Redondo MJ, Sosenko J, Libman I, et al. 2020. Single islet autoantibody at diagnosis of clinical type 1 diabetes is associated with older age and insulin resistance. J. Clin. Endocrinol. Metab 105: 1629–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Evans-Molina C, Sims EK, DiMeglio LA, et al. 2018. β Cell dysfunction exists more than 5 years before type 1 diabetes diagnosis. JCI Insight 3. e120877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vandemeulebroucke E, Keymeulen B, Decochez K, et al. 2010. Hyperglycaemic clamp test for diabetes risk assessment in IA-2-antibody-positive relatives of type 1 diabetic patients. Diabetologia 53: 36–44. [DOI] [PubMed] [Google Scholar]
- 48.Chase HP, Cuthbertson DD, Dolan LM, et al. 2001. First-phase insulin release during the intravenous glucose tolerance test as a risk factor for type 1 diabetes. J. Pediatr 138: 244–249. [DOI] [PubMed] [Google Scholar]
- 49.Greenbaum CJ, Cuthbertson D, Krischer JP, et al. 2001. Type 1 diabetes manifested solely by 2-h oral glucose tolerance test criteria. Diabetes 50: 470–476. [DOI] [PubMed] [Google Scholar]
- 50.Brunzell JD, Robertson RP, Lerner RL, et al. 1976. Relationships between fasting plasma glucose levels and insulin secretion during intravenous glucose tolerance tests. J. Clin. Invest 42: 222–229. [DOI] [PubMed] [Google Scholar]
- 51.Hao W, Woodwyk A, Beam C, et al. 2017. Assessment of β cell mass and function by AIRmax and intravenous glucose in high-risk subjects for type 1 diabetes. J. Clin. Endocrinol. Metab 102: 4428–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ganda OP, Srikanta S, Brink SJ, et al. 1984. Differential sensitivity to beta-cell secretagogues in ‘early,’ type 1 diabetes mellitus. Diabetes 33: 516–521. [DOI] [PubMed] [Google Scholar]
- 53.Rickels MR, Naji A & Teff KL. 2007. Acute insulin responses to glucose and arginine as predictors of β-cell secretory capacity in human islet transplantation. Transplantation 84: 1357–1360. [DOI] [PubMed] [Google Scholar]
- 54.Atkinson MA, Eisenbarth GS & Michels AW. 2014. Type 1 diabetes. Lancet 383: 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ferrannini E, Mari A, Nofrate V, et al. 2010. Progression to diabetes in relatives of type 1 diabetic patients: mechanisms and mode of onset. Diabetes 59: 679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sosenko JM, Skyler JS & Palmer JP. 2015. The development, validation, and utility of the Diabetes Prevention Trial-Type 1 Risk Score (DPTRS). Curr. Diab. Rep 15: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Simmons KM, Sosenko JM, Warnock M, et al. 2020. One-hour oral glucose tolerance tests for the prediction and diagnostic surveillance of type 1 diabetes. J. Clin. Endocrinol. Metab 105: e4094–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nyirjesy SC, Sheikh S, Hadjiliadis D, et al. 2018. β-Cell secretory defects are present in pancreatic insufficient cystic fibrosis with 1-hour oral glucose tolerance test glucose ≥155 mg/dL. Pediatr. Diabetes 19: 1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sosenko JM, Palmer JP, Greenbaum CJ, et al. 2006. Patterns of metabolic progression to type 1 diabetes in the diabetes prevention trial-type 1. Diabetes Care 29: 643–649. [DOI] [PubMed] [Google Scholar]
- 60.Bogun MM, Bundy BN, Goland RS, et al. 2020. C-peptide levels in subject followed longitudinally before and after type 1 diabetes diagnosis in TrialNet. Diabetes Care 43: 1836–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sims EK, Chaudhry Z, Watkins R, et al. 2016. Elevations in the fasting serum proinsulin-to-C-peptide ratio precede the onset of type 1 diabetes. Diabetes Care 39: 1519–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Greenbaum CJ, Prigeon RL & D’Alessio DA. 2002. Impaired β-cell function, incretin effect, and glucagon suppression in patients with type 1 diabetes who have normal fasting glucose. Diabetes 51: 951–957. [DOI] [PubMed] [Google Scholar]
- 63.Keymeulen B, Vandemeulebroucke E, Ziegler AG, et al. 2005. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N. Engl. J. Med 352: 2598–2608. [DOI] [PubMed] [Google Scholar]
- 64.Sherr J, Tsalikian E, Fox L, et al. 2014. Evolution of abnormal plasma glucagon responses to mixed-meal feedings in youth with type 1 diabetes during the first 2 years after diagnosis. Diabetes Care 37: 1741–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Siafarikas A, Johnston RJJ, Bulsara MK, et al. 2012. Early loss of the glucagon response to hypoglycemia in adolescents with type 1 diabetes. Diabetes Care 35: 1757–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Arbelaez AM, Xing D, Cryer PE, et al. 2014. Blunted glucagon but not epinephrine responses to hypoglycemia occurs in youth with less than 1 year duration of type 1 diabetes mellitus. Pediatr. Diabetes 15: 127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rickels MR 2019. Hypoglycemia-associated autonomic failure counterregulatory responses, and therapeutic options in type 1 diabetes. Ann. N.Y. Acad. Sci 1454: 68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sherr J, Xing D, Ruedy KJ, et al. 2013. Lack of association between residual insulin production and glucagon response to hypoglycemia in youth with short duration of type 1 diabetes. Diabetes Care 36: 1470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brissova M, Haliyur R, Saunders D, et al. 2018. α Cell function and gene expression are compromised in type 1 diabetes. Cell Rep. 22: 2667–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Armour SL, Anderson SJ, Richardson SJ, et al. 2020. Reduced expression of the co-regulator TLE1 in type 2 diabetes is associated with increased islet α-cell number. Endocrinology 161. bqaa011. [DOI] [PubMed] [Google Scholar]
- 71.Ludvigsson J, Heding LG, Larsson Y, et al. 1977. C-peptide in juvenile diabetics beyond the postinitial remission period. Relation to clinical manifestations at onset of diabetes, remission and diabetic control. Acta Paediatr. Scand 66: 177–184. [DOI] [PubMed] [Google Scholar]
- 72.Gubitosi-Klug RA, Lachin JM, Backlund JYC, et al. 2016. Intensive diabetes treatment and cardiovascular outcomes in type1 diabetes: the DCCT/EDIC study 30-year follow-up. Diabetes Care 39: 686–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Buckingham B, Beck RW, Ruedy KJ, et al. 2015. Effectiveness of early intensive therapy on b-cell preservation in type 1 diabetes. Diabetes Technol. Ther 17: S104–S105. [Google Scholar]
- 74.Diabetes Prevention Trial-Type 1 Diabetes Study Group. 2002. Effects of insulin in relatives of patients with type 1 diabetes mellitus. N. Engl. J. Med 346: 1685–1691. [DOI] [PubMed] [Google Scholar]
- 75.Tsai EB, Sherry NA, Palmer JP, et al. 2006. The rise and fall of insulin secretion in type 1 diabetes mellitus. Diabetologia 49: 261–270. [DOI] [PubMed] [Google Scholar]
- 76.Hao W, Gitelman S, DiMeglio LA, et al. 2016. Fall in C-peptide during first 4 years from diagnosis of type 1 diabetes: variable relation to age, HbA1c, and insulin dose. Diabetes Care 39: 1664–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Steffes MW, Sibley S, Jackson M, et al. 2003. Beta-cell function and the development of diabetes-related complications in the diabetes control and complications trial. Diabetes Care 26: 832–836. [DOI] [PubMed] [Google Scholar]
- 78.The Diabetes Control and Complications Trial Research Group. 1997. Hypoglycemia in the diabetes control and complications trial. Diabetes 46: 271–286. [PubMed] [Google Scholar]
- 79.Lord S & Greenbaum CJ. 2020. Insulin is necessary but not sufficient: changing the therapeutic paradigm in type 1 diabetes. F1000Res. 9. F1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lachin JM, McGee P, Palmer JP, et al. 2014. Impact of C-peptide preservation on metabolic and clinical outcomes in the Diabetes Control and Complications Trial. Diabetes 63: 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang L, Lovejoy NF & Faustman DL. 2012. Persistence of prolonged C-peptide production in type 1 diabetes as measured with an ultrasensitive C-peptide assay. Diabetes Care 35: 465–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Oram RA, Jones AG, Besser REJ, et al. 2014. The majority of patients with long-duration of type 1 diabetes are insulin microsecretors and have functioning beta cells. Diabetologia 57: 187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Davis AK, DuBose SN, Haller MJ, et al. 2015. Prevalence of detectable C-peptide according to age at diagnosis and duration of type 1 diabetes. Diabetes Care 38: 476–481. [DOI] [PubMed] [Google Scholar]
- 84.Yu MG, Keenan HA, Shah HS, et al. 2019. Residual β cell function and monogenic variants in long-duration type 1 diabetes patients. J. Clin. Invest 129: 3252–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Leete P, Oram RA, McDonald JT, et al. 2020. Studies of insulin and proinsulin in pancreas and serum support the existence of aetiopathological endotypes of type 1 diabetes associated with age at diagnosis. Diabetologia 63: 1258–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sims EK, Bahnson HT, Nyalwidhe J, et al. 2019. Proinsulin secretion is a persistent feature of type 1 diabetes. Diabetes Care 42: 258–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sims EK, Syed F, Nyalwidhe J, et al. 2019. Abnormalities in proinsulin processing in islets from individuals with longstanding T1D. Transl. Res 213: 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Keenan HA, Sun JK, Levine J, et al. 2010. Residual insulin production and pancreatic β-cell turnover after 50 years of diabetes: Joslin Medalist study. Diabetes 59: 2846–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rickels MR, Evans-Molina C, Bahnson HT, et al. 2020. High residual C-peptide likely contributes to glycemic control in type 1 diabetes. J. Clin. Invest 130: 1850–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Taylor GS, Smith K, Capper TE, et al. 2020. Postexercise glycemic control in type 1 diabetes is associated with residual β-cell function. Diabetes Care 43: 2362–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jeyam A, Colhoun H, McGurnaghan S, et al. 2021. Clinical impact of residual C-peptide secretion in type 1 diabetes on glycemia and microvascular complications. Diabetes Care 44: 390–398. [DOI] [PubMed] [Google Scholar]
- 92.Heller SR & Cryer PE. 1991. Reduced neuroendocrine and symptomatic responses to subsequent hypoglycemia after 1 episode of hypoglycemia in nondiabetic humans. Diabetes 40: 223–226. [DOI] [PubMed] [Google Scholar]
- 93.Cryer PE 1997. Hypoglycemia-associated autonomic failure in insulin-dependent diabetes mellitus. Adv. Pharmacol 42: 620–622. [DOI] [PubMed] [Google Scholar]
- 94.Cryer PE 1993. Hypoglycemia begets hypoglycemia in IDDM. Diabetes 42: 1691–1693. [DOI] [PubMed] [Google Scholar]
- 95.Cryer PE 2013. Hypoglycemia-associated autonomic failure in diabetes. Handb. Clin. Neurol 117: 295–307. [DOI] [PubMed] [Google Scholar]
- 96.Pedersen-Bjergaard U, Pramming S, Heller SR, et al. 2004. Severe hypoglycaemia in 1076 adult patients with type 1 diabetes: influence of risk markers and selection. Diabetes Metab. Res. Rev 20: 479–486. [DOI] [PubMed] [Google Scholar]
- 97.Rickels MR, Fuller C, Dalton-Bakes C, et al. 2015. Restoration of glucose counterregulation by islet transplantation in long-standing type 1 diabetes. Diabetes 64: 1713–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rickels MR, Peleckis AJ, Markmann E, et al. 2016. Long-term improvement in glucose control and counterregulation by islet transplantation for type 1 diabetes. J. Clin. Endocrinol. Metab 101: 4421–4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Flatt AJS, Bennett D, Counter C, et al. 2019. β-Cell and renal transplantation options for diabetes. Diabet. Med 37: 580–592. [DOI] [PubMed] [Google Scholar]
- 100.Rickels MR & Robertson PR. 2019. Pancreatic islet transplantation in humans: recent progress and future directions. Endocr. Rev 40: 631–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Keymeulen B, Gillard P, Mathieu C, et al. 2006. Correlation between beta cell mass and glycemic control in type 1 diabetic recipients of islet cell graft. Proc. Natl. Acad. Sci. USA 103: 17444–17449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rickels MR, Liu C, Shlansky-Goldberg RD, et al. 2013. Improvement in β-cell secretory capacity after human islet transplantation according to the CIT07 protocol. Diabetes 62: 2890–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gillard P, Hilbrands R, Van de Velde U, et al. 2013. Minimal functional β-cell mass in intraportal implants that reduces glycemic variability in type 1 diabetic recipients. Diabetes Care 36: 3483–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vantyghem MC, de Koning EJP, Pattou F, et al. 2019. Advances in β-cell replacement therapy for the treatment of type 1 diabetes. Lancet 394: 1274–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gruessner AC & Gruessner RW. 2016. Long-term outcome after pancreas transplantation: a registry analysis. Curr. Opin. Organ Transpl 21: 377–385. [DOI] [PubMed] [Google Scholar]
- 106.Rickels MR 2012. Recovery of endocrine function after islet and pancreas transplantation. Curr. Diab. Rep 12: 587–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rickels MR, Mueller R, Teff KL, et al. 2010. Beta-cell secretory capacity and demand in recipients of islet, pancreas, and kidney transplants. J. Clin. Endocrinol. Metab 95: 1238–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Brooks AM, Oram R, Home P, et al. 2015. Demonstration of an intrinsic relationship between endogenous C-peptide concentration and determinants of glycemic control in type 1 diabetes following islet transplantation. Diabetes Care 38: 105–112. [DOI] [PubMed] [Google Scholar]
- 109.Rickels MR, Stock PG, de Koning EJP, et al. 2018. Defining outcomes for β-cell replacement therapy in the treatment of diabetes: a consensus report on the Igls criteria from the IPITA/EPITA opinion leaders workshop. Transplantation 102: 1479–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Foster ED, Bridges ND, Feurer ID, et al. 2018. Improved health-related quality of life in a phase 3 islet transplantation trial in type 1 diabetes complicated by severe hypoglycemia. Diabetes Care 41: 1001–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kaestner KH, Powers AC, Naji A, et al. 2019. NIH initiative to improve understanding of the pancreas, islet, and autoimmunity in type 1 diabetes: the Human Pancreas Analysis Program (HPAP). Diabetes 68: 1394–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]