

Abstract
Objectives
Idiopathic pulmonary fibrosis (IPF) is a chronic fibrotic lung disease characterized by dry cough, fatigue, and progressive exertional dyspnea. Lung parenchyma and architecture is destroyed, compliance is lost, and gas exchange is compromised in this debilitating condition that leads inexorably to respiratory failure and death within 3–5 years of diagnosis. This review discusses treatment approaches to IPF in current use and those that appear promising for future development.
Data Source
The data were obtained from the Randomized Controlled Trials and scientific studies published in English literature. We used search terms related to IPF, antifibrotic treatment, lung transplant, and management.
Results
Etiopathogenesis of IPF is not fully understood, and treatment options are limited. Pathological features of IPF include extracellular matrix remodeling, fibroblast activation and proliferation, immune dysregulation, cell senescence, and presence of aberrant basaloid cells. The mainstay therapies are the oral antifibrotic drugs pirfenidone and nintedanib, which can improve quality of life, attenuate symptoms, and slow disease progression. Unilateral or bilateral lung transplantation is the only treatment for IPF shown to increase life expectancy.
Conclusion
Clearly, there is an unmet need for accelerated research into IPF mechanisms so that progress can be made in therapeutics toward the goals of increasing life expectancy, alleviating symptoms, and improving well‐being.
Keywords: idiopathic pulmonary fibrosis, lung transplantation, nintedanib, pentraxin, pirfenidone
1. INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is a chronic interstitial lung disease characterized by fibrosis, inflammation, and destruction of lung architecture. 1 , 2 , 3 Damage to the alveolar epithelium and abnormal wound repair are theorized to be key factors in the development of this disease. IPF is thus far incurable with average onset at about age 65 years and a survival rate of 3–5 years after diagnosis. Likelihood of living 5 years from time of diagnosis ranges from 20% to 40%. 4 About 70% of patients are male and history of cigarette smoking is common, with the number of IPF patients estimated at 5.6 per 100 000 per year. 5 , 6 , 7 The causes of IPF remain unknown, although it is thought to result from a combination of genetic and environmental factors. Constant micro‐injuries to aging alveolar epithelium are believed to lead to disrupted epithelial–fibroblast communication, which culminates in recruitment and activation of myofibroblasts that produce collagen‐rich extracellular matrix. Excessive accumulation of this matrix renders alveoli irreversibly collapsed and nonfunctional, and the result is reduced gas exchange and difficulty breathing. 8 , 9 Since there is no cure, treatment has remained focused on slowing progression of fibrosis, maintaining comfort and, in late stages, on palliative care. 5 , 6 , 10 , 11 , 12 , 13 Eventually, IPF patients die from respiratory failure, often during an acute exacerbation or from the effects of another comorbidity such as cardiovascular disease, lung cancer, or thromboembolism. 14 , 15 , 16 The lack of curative treatment demands aggressive pursuit of better choices as described throughout this review.
2. PRESENTATION AND DIAGNOSIS
Early diagnosis of IPF is difficult because initial symptoms, most commonly dyspnea and cough, are mild and nonspecific and overlap many other common conditions. 17 , 18 , 19 Once symptoms become more pronounced, the diagnosis can be made and antifibrotic therapy options may be initiated. 20 Upon physical examination, respiratory auscultation may detect Velcro crackles and clubbing of fingers may be apparent. 21 , 22 , 23 Pulmonary function testing (PFT) shows a restrictive pattern measured as reduced forced vital capacity (FVC), reduced forced expiratory volume in 1 s (FEV1), and reduced efficiency of lung gas transfer estimated with measurement of the diffusion capacity of carbon monoxide (DLCO). The Gender, Age, and Physiology (GAP) Index Score encompasses both FVC and DLCO along with age and gender and is considered a practical index and staging system for predicting IPF mortality. 24
The 6‐min walk test, an easy and straightforward way to determine exercise capacity by having the patient walk as far as possible in 6 min on a flat surface, has prognostic value since faster decline in distance covered is associated with higher mortality. 25 , 26
The histopathological features of IPF are those of usual interstitial pneumonia (UIP) which appears under the microscope as a heterogeneous patchwork of focal areas of fibrosis with hyperplastic alveolar epithelial cells adjacent to these fibroblastic foci, alternating with less affected areas of normal or nearly normal lung tissue. 27 Lung architecture is disrupted leading to impaired gas exchange (Figure 1). 6
FIGURE 1.
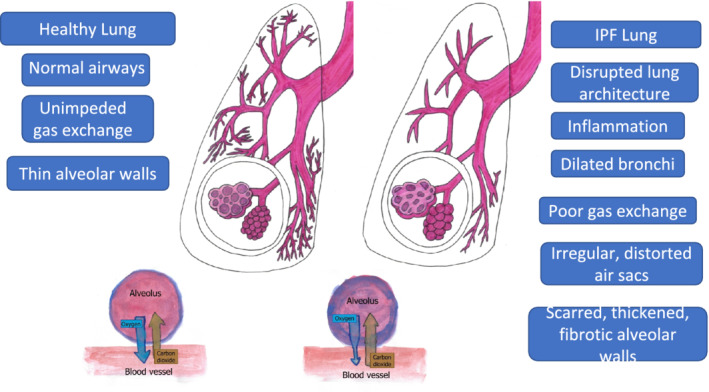
Comparison of healthy lung to idiopathic pulmonary fibrosis (IPF) lung. The healthy lung is characterized by unscarred airways with thin‐walled alveoli and unimpeded gas exchange. Pathological features of the IPF lung include dilated bronchi, airway distortion, and thickened alveolar walls. Inflammation and fibrosis lead to impaired gas exchange within the alveoli
IPF is most often imaged by computer tomography (CT), but a role for magnetic resonance imaging is emerging. 28 , 29 The most typical radiological pattern is UIP. 30 Extent of fibrosis on CT can be assessed visually or quantified more objectively using computer algorithms. 31 , 32 High‐resolution CT often shows honeycomb changes, traction bronchiectasis, and a reticular pattern that is predominantly in the periphery of the lower lobes. 5 , 33 , 34 , 35
In addition to the poor prognosis, IPF patients often have multiple comorbidities including hypertension, chronic obstructive pulmonary disease (COPD) emphysema, diabetes mellitus, and gastroesophageal reflux disease (GERD). 36 , 37 These can decrease quality of life, complicate treatment, increase hospitalization, and contribute to deterioration of the patient's health.
3. PAST TREATMENT STRATEGIES TARGETING INFLAMMATION
Until 2014, the standard practice for treating IPF focused primarily on immunosuppressant therapy using a combination of prednisone, azathioprine, and N‐acetylcysteine. 5 However, after the release of the results from the Evaluating the Effectiveness of Prednisone, Azathioprine and N‐acetylcysteine in Patients with IPF (PANTHER‐IPF) TRIAL in 2012, it became apparent that, compared with placebo, this form of treatment actually increased the likelihood of hospitalization, treatment‐related severe adverse events, and death in IPF patients. 38 In IPF subjects, a post hoc analysis across PANTHER‐IPF and a second large multisite clinical trial (ACE‐IPF AntiCoagulant Effectiveness in IPF [ACE‐IPF]) found an inverse association between leukocyte telomere length and poor outcome in response to immunosuppressants. 39 Immunosuppressants such as cyclophosphamide and mycophenolate showed no benefit in IPF clinical trials and are now considered inadvisable for IPF while steroids are still used to reduce cough. 40 , 41 , 42 , 43
Thalidomide is a drug with an infamous history, originally intended for use as a sedative and an anti‐emetic; it caused serious teratogenic effects and was subsequently removed from the market. 44 However, since then, thalidomide has been found to be an effective treatment for several cancers, such as multiple myeloma and kidney cancer, as well as inflammation‐based diseases. 45 , 46 The anti‐inflammatory properties of thalidomide are what make this glutamic acid derivative a compelling contender for treating IPF, and there has been speculation about its properties being used for such a purpose for many years. 47 However, the information and amount of testing done to explore the potential utility of thalidomide in IPF is still very limited.
In 2007, thalidomide was used on bleomycin‐induced pulmonary fibrosis in mice and in 2015 it was shown to protect against emphysematous changes in mice exposed to cigarette smoke. 48 , 49 In the bleomycin model, C57BL/6 mice received intraperitoneal bleomycin sulfate with and without intervention with intraperitoneal thalidomide. Those given thalidomide showed reduced collagen deposition in the lung and accompanying this histologic benefit, thalidomide mitigated bleomycin‐induced upregulation of interleukin‐6 and transforming growth factor (TGF)‐β.
In 2012 at Johns Hopkins University School of Medicine, a 24‐week double blind randomized trial evaluated the antitussive action of thalidomide in IPF. 50 In this study, 24 IPF patients were randomly assigned to receive 12 weeks of thalidomide or placebo followed by a 2‐week washout and then a crossover for a further 12 weeks. While receiving thalidomide, patients showed a definite increase in comfort and quality of life as measured by the Cough Quality of Life Questionnaire. However, it was a very small study and only 20 patients completed the trial. Subsequently, a panel of experts did not recommend thalidomide treatment for cough in IPF. 51
In 2017, a cell culture study using rat lung epithelial cells (CCL‐149) showed that the presence of thalidomide mitigated TGF‐β1‐stimulated epithelial to mesenchymal transition. 52 Despite documented anti‐inflammatory properties of thalidomide, there have been no significant further attempts to study its effect on IPF in human trials. This lack of momentum and enthusiasm likely reflects long‐held deeply negative associations.
4. CURRENT PHARMACOLOGIC TREATMENTS
Two antifibrotic therapies have been approved for the treatment of IPF: pirfenidone and nintedanib (Table 1). Pirfenidone is a modified pyridine small molecule with antifibrotic, anti‐inflammatory and antioxidant properties. It decreases the production of collagen, slows the fibrotic process by suppressing the cytokine TGF‐β and lowers the rate of decline in FVC. 53 , 54 , 55 This was shown in two phase 3 CAPACITY trials and the ASCEND trial. 56 , 57 , 58 Pirfenidone lowers the likelihood of respiratory‐related hospitalization over the course of 1 year of treatment. 59 A retrospective study of 34 IPF patients in the North of England who were treated with perfenidone found greater benefit to those patients who were experiencing more rapid decline (≥5% decrease in FVC per year) prior to administration of the drug. 60 While pirfenidone is generally well‐tolerated and the side effects are mild (skin rash, weight loss, nausea, and fatigue), there have been cases of abnormal liver function, particularly elevation of serum alanine aminotransferase (ALT)/aspartate aminotransferase (AST) and bilirubin and therefore patients being treated with pirfenidone need regular monitoring of liver function. 61 , 62 , 63
TABLE 1.
IPF treatments
| Treatment | Mechanism of action | Clinical effects |
|---|---|---|
| Current therapies | ||
| Pirfenidone | Antifibrotic and anti‐inflammatory | Slows rate of decline in FVC |
| Nintedanib | Antifibrotic and anti‐inflammatory | Slows rate of decline in FVC |
| Oral corticosteroids, opioids | Antitussive | Decrease cough—improve quality of life |
| Anti‐acids, proton pump inhibitors | Reduces GERD | Benefits unclear |
| Lung transplantation | Surgical replacement of one lung or both lungs | Only available potentially curative therapy |
| Therapies in development | ||
| PRM‐151 | Recombinant human pentraxin‐2; acts as an antifibrotic agent | Slows rate of decline in FVC |
| Pamrevlumab | Fully human recombinant monoclonal antibody against CTGF | Slows rate of decline in FVC |
| TD139 | Small molecule inhibitor of galectin‐3 | Decreases plasma biomarkers of inflammation. Study in progress to asses effect on FVC |
| PLN‐74809 | Blocks activation of the TGFβ pathway | Study in progress with end‐points of safety, tolerability, pharmacokinetics |
| TRK‐250 | Suppresses expression of TGFβ | Study in progress to assess safety and tolerability of single and multiple inhaled doses |
Abbreviations: CTGF, connective tissue growth factor; FVC, forced vital capacity; GERD, gastroesophageal reflux disease; IPF, idiopathic pulmonary fibrosis.
Nintedanib is an intracellular tyrosine kinase inhibitor that binds to adenosine triphosphate binding sites, thus suppressing the signaling pathways linked to vascular endothelial growth factor receptor, fibroblast growth factor receptor 1–3, and platelet‐derived growth factor receptor α and β. These effects on receptor tyrosine kinases lead to decreased fibroblast activity. 5 , 64 , 65 INPULSIS phase 3 trials I and II showed a significant decrease in the rate of FVC deterioration in IPF patients although the death rate remained the same. 66 The side effects, primarily diarrhea and nausea, are manageable with medication, but hepatotoxicity may occur in rare cases and the drug is not recommended for those with severe liver disease. 67 , 68
Either nintedanib or pirfenidone are good choices as the primary treatment for IPF at the present time. 69 , 70 , 71 , 72 , 73 The two drugs are able to slow deterioration in FVC and provide the patient with greater comfort for a noticeably longer time. However, use of these drugs may incur high out‐of‐pocket costs without changing the overall progression of the disease and the high mortality within 3 to 5 years after diagnosis. 74 Some patients may not respond to these drugs, and in light of their expense and side effect profile, studies are underway to determine when these tyrosine kinase inhibitors may not be helpful. 75
5. NONPHARMACOLOGIC THERAPY: LUNG TRANSPLANTATION
Lung transplantation may be a viable treatment option for IPF. 76 , 77 A key advantage compared with other modes of treatment is that it is the only method to improve both symptoms and survival time. 78 , 79 Previously, in the United States, COPD had been the main indication for lung transplant, but interstitial lung disease now supercedes COPD. 80 , 81
Determining the need for transplant is challenging, but in selecting IPF patients for transplant, risk of mortality and likelihood of survival after transplantation are key considerations. Furthermore, other comorbidities that may lead to complications such as cardiac dysfunction, GERD, diabetes, and obesity must be taken into account. 82 , 83 , 84 Generally accepted benchmarks for lung transplant referral as well as contraindications to the procedure are based on guidelines proposed by the International Society of Heart and Lung Transplantation (ISHLT). 77 , 85 Among the key criteria for placement on the transplant list are rapid decline in FVC or DLCO, oxygen desaturation, or decrease in distance covered during 6‐min walk test, pulmonary hypertension, or hospitalization due to respiratory decline, pneumothorax, or acute exacerbation.
Importantly, telomere shortening and mutations in genes that control telomere function such as Telomerase Reverse Transcriptase (TERT) and Telomerase RNA Component (TERC) are associated with familial and sporadic cases of IPF and this may be considered in transplant decision‐making. 86 , 87 , 88 , 89 Although there is no conclusive evidence that transplant outcome is influenced by telomere length, short telomeres may confer vulnerability to bone marrow dysfunction upon administration of immunosuppressive drugs posttransplant. 90 , 91 , 92 At this time, telomere length testing and genetic testing in IPF are not routine, but as more tests are performed, we may be able to better tailor immunosuppressants to the patient. 93
Before transplant, antifibrotic treatment is generally maintained and strict adherence to medication regimen is encouraged, even as clinical trials are being conducted to determine if this affects transplant outcome. 94 , 95
Lung transplant may be either unilateral or bilateral. Benefits of unilateral transplant are the shorter wait times, easier procedure with lower perioperative complication rate, and the potential to improve the health of two patients from one donor. 90 Data from the United Network for Organ Sharing database and the Organ Procurement and Transplantation network show a better long‐term survival rate for bilateral versus unilateral transplant. 96 , 97 A recent meta‐analysis found no survival advantage with bilateral lung transplant but did document better pulmonary function. 98 Bilateral transplants can improve FVC and FEV significantly, and they can reach 100% of predicted values by 6–12 months. These measures also improve in unilateral transplant but reach about 80% of predicted values. Overall 5 year posttransplant survival is about 50%.
Follow‐up generally involves monitoring of FVC, DLCO, and pulmonary hypertension. 85 Chronic lung allograft dysfunction and noncytomegalovirus infections are early causes of mortality. 99 Other possible complications include immunosuppression, drug toxicity, infection, and neoplasia. 100 Interestingly, a recent meta‐analysis found that several circulating tumor‐associated markers known to be elevated in IPF, including Ca125, Ca15.3, and Ca19.9, decreased significantly posttransplant. 101
6. THE FUTURE: THERAPIES IN DEVELOPMENT
6.1. Stem cells
A nonpharmacological treatment being explored for used in IPF is based on the finding that mesenchymal stem cells (MSC), multipotent, undifferentiated cells, can regulate fibrotic processes and exert control over lung injury and repair. 102 , 103 MSC from IPF patients are dysfunctional and express reduced levels of tissue protective growth factors. 103 , 104 , 105 In the Allogeneic human MSC in patients with IPF via intravenous delivery (AETHER) study, bone marrow‐derived MSC were infused intravenously into patients with IPF. AETHER was a nonrandomized, nonplacebo‐controlled phase 1 trial of a single MSC infusion. In this study, over the course of 6–12 months, the primary endpoint of safety was met. 106 , 107 A high cumulative dose stem cell infusion into patients with rapidly progressive IPF not taking antifibrotic drugs also showed excellent safety and tolerability. 108 Human Autologous Lung Stem Cell Transplant for IPF (HALT‐IPF) (NCT04262167), a randomized trial of intravenous infusion of lung stem cells in persons with IPF, is currently enrolling.
6.2. Pentraxin
The pentraxin protein family consists of three highly phylogenetically conserved acute phase reactant plasma proteins made in the liver which are known to be involved in inflammation and innate immunity. 109 Pentraxin 2 (PTX2, also known as serum amyloid P) is a constitutively synthesized member of the pentraxin protein family that can modulate wound healing and fibrotic remodeling of injured tissue. 110 PTX2 acts to modify neutrophil adhesion and inhibit monocyte differentiation into profibrotic macrophages and fibrocytes. 111 , 112 It also binds to cellular debris, enhancing phagocytosis by leukocytes, and it inhibits the production of TGF‐β, a cytokine long considered a key fibrosis mediator. 113 , 114 , 115 Serum PTX2 levels are significantly lower in IPF patients than in healthy controls. 114 High local levels of PTX2 can delay wound healing via inhibition of fibrocytes and macrophages, which limits scarring and fibrosis around wounds. 116 These antifibrotic properties are being explored for therapeutic benefits in fibrotic disorders such as IPF. 117 , 118 , 119 The rationale for use of PTX2 in IPF is the clinical evidence that circulating fibrocyte concentrations (which correlate with abundance of fibroblastic foci in IPF tissue) above 5% of total blood leukocytes predict lifespan in IPF at an average of 7.5 months, while fibrocyte levels below 5% are associated with lifespan of about 27 months. 120 Furthermore, bleomycin treatment induces prolonged lung inflammation and increased fibrosis in mice with genetic deletion of PTX2 compared with wild type mice. 121
The first human clinical study on the pharmacokinetics and safety of PRM‐151, a recombinant human PTX2 protein, as a treatment for IPF, was a modest randomized, blinded, placebo‐controlled trial in 26 healthy volunteers between the ages of 18 and 53 years and 3 patients, 2 with familial interstitial pneumonia, and 1 with IPF, ages ranging between 29 and 72 years. 122 The enrollees were given a single intravenous infusion varying from 0.1 to 20 mg/kg, and the drug was well‐tolerated with mild adverse effects in both placebo and treatment groups. No safety concerns were raised.
In a randomized, double blind, placebo‐controlled, multiple ascending dose trial of PRM‐151 conducted in 3 centers, 21 patients between the ages of 40 and 80 years diagnosed with IPF were given PRM‐151 in 5 intravenous infusions over 15 days at doses of 1, 5, or 10 mg/kg or placebo. All doses were well‐tolerated and raised circulating levels of PTX2. DLCO was not affected by treatment, but 6‐min walking distance and FVC showed a trend toward an increase. 118
In a second randomized, double‐blind placebo‐controlled phase 2 study (PRM‐151‐202) conducted at 18 sites in 7 countries, 117 patients with IPF between the ages of 40 and 80 years received placebo or 10 mg/kg PRM‐151 every 4 weeks for 24 weeks after a three‐dose loading regimen, along with usual IPF treatment in 78%. Compared with placebo, the mean percentage of predicted FVC and 6‐min walking distance declined significantly less in the PRM‐151 group. 123
An open label extension study to week 76 in which 74 patients continued treatment with PRM‐151 and 37 placebo‐treated patients crossed over to receive the drug showed persistent positive effect of treatment in those who continued and slowed rate of decline of FVC in those who crossed over. As expected in an IPF population, adverse events accumulated over time and most were not deemed to be related to treatment. 124 A phase 3 efficacy and safety study of PRM‐151 (NCT04552899) is in progress with estimated completion date of March 2023.
6.3. Pamrevlumab
Pamrevlumab is a fully human recombinant monoclonal antibody that targets connective tissue growth factor (CTGF) and thus can possibly limit fibrotic progression. 125 It is being evaluated for use in locally advanced unresectable pancreatic cancer and in IPF. 126 In PRAISE a randomized, double‐blind, placebo‐controlled phase 2 clinical trial, 103 IPF patients age 40 to 80 years were split into 2 groups, 50 received the drug, while 53 were given the placebo every 3 weeks for 48 weeks in total. 127 , 128 Of the original 103 patients, 78 (38 on pamrevlumab and 40 on placebo) completed the study. Pamrevlumab safely and effectively slowed lung function decline in IPF patients with mean change from baseline to week 48 in percentage of predicted FVC of −2.9% in the pamrevlumab group compared with a change of −7.2% in the placebo group. This represents a relative reduction in percentage of predicted FVC decline of 60.3% in the arm treated with pamrevlumab. Disease progression, measured as decline in percentage of predicted FVC ≥ 10%, or death, was lower in the pamrevlumab group (10.0%) than in the placebo group (31.4%), with a significant between‐group difference at week 48 (p = 0·013). Quantitative lung fibrosis scores obtained with high‐resolution computed tomography at 48 weeks showed significantly less fibrotic progression in the pamrevlumab group. The PRAISE findings are considered promising, but they are still limited by the small sample size. The ZEPHYRUS study (clinicaltrials.gov identifier NCT03955146), a phase 3 randomized, double‐blind, placebo‐controlled, multicenter clinical trial with plans to enroll 340 subjects, is ongoing and will evaluate efficacy and safety of pamrevlumab over 52 weeks. 129 , 130
6.4. Autotaxin inhibitors
Autotaxin (ATX) is an enzyme that hydrolyzes lysophosphatidyl choline into the signaling molecule lysophosphatidic acid (LPA). 131 , 132 ATX is expressed by bronchial epithelial cells and alveolar macrophages and is upregulated in IPF. 133 It has been postulated that increased ATX generates more LPA in the lung, which then has profibrotic effects on epithelial cells, endothelial cells, and fibroblasts, thus contributing to IPF. 134 , 135 GLPG1690 is an orally available inhibitor of ATX, also known as lysophospholipase D. Based on this sequence of events, by inhibiting ATX, GLPG1690 may be of benefit in IPF. Initial studies in humans indicate that dosages of 200 and 600 mg per day of GLPG1690 will yield at least an 80% reduction in LPA in clinical trials. 136 Two phase 3, randomized, placebo‐controlled trials of GLPG1690 have completed recruitment with a primary endpoint of rate of decline of FVC over 52 weeks. 137 Unfortunately, in February 2021, the trials were discontinued due to the conclusion by the Independent Data Monitoring Committee that the benefit–risk profile did not support continuation. 138 It is now uncertain whether other ATX inhibitors, such as BBT‐877, will continue to be evaluated as candidate treatments for IPF. 139
6.5. Galectin‐3
Galectin‐3 is a beta‐galactoside‐binding protein with pro‐fibrotic properties that is present at elevated levels in bronchoalveolar lavage fluid from patients with IPF. 140 TD139, a small molecule galectin‐3 inhibitor, was shown to be safe and well‐tolerated in IPF subjects and healthy controls in a phase 1/2 clinical trial (ClinicalTrials.gov Identifier: NCT02257177). 141 In addition, IPF patients showed improvements in several plasma biomarkers of inflammation, including platelet‐derived growth factor and the chemokine CCL18. A phase 2b clinical trial (NCT03832946) with FVC as the primary outcome measure is now enrolling IPF patients.
6.6. Human antigen R
Human antigen R (HuR), a member of the Hu/embryonic lethal, abnormal vision family of RNA binding proteins, is known to promote excessive inflammation and fibrogenesis in murine models of diabetic cardiovascular disease and liver disease. 142 , 143 This has led to an interest in HuR as a contributing factor to IPF. Al‐Habeeb et al. recently found that levels of HuR are increased in the lungs of IPF patients and that TGFβ drives translocation of HuR from nucleus to cytoplasm in human lung fibroblasts. Knockdown of HuR in human lung fibroblasts mitigated induction of α‐smooth muscle actin protein as well as a number of extracellular matrix proteins by TGFβ. Further, HuR knockdown reduced TGFβ‐driven morphological changes indicative of myofibroblast differentiation. 144 This link between HuR and myofibroblast transformation may indicate a potential new target for IPF treatment.
6.7. Cell‐based therapies
The IPF Cell Atlas is a major initiative designed to make available single‐cell sequencing data on multiple cell types present in the lung with the ability to compare control and IPF cell transcriptomes. 145 , 146 The creators of this website intend it to be used to achieve insight into pathogenesis that can lead to novel treatment approaches. The Atlas has already yielded information on a cell type enriched in the IPF lung known as aberrant basaloid cells. 145 These cells express epithelial and basal cell markers as well as a cellular senescence signature and mesenchymal markers. They are found in distal lung parenchyma of IPF subjects in areas of fibroblastic foci and were also found in persons with a diagnosis of systemic sclerosis‐associated interstitial lung disease. 147 It is hypothesized that these aberrant basaloid cells represent a common disease mechanism and that therapies may be directed toward this cell type in IPF and other fibrotic states in which they appear.
7. BIOMARKERS AND ASSESSING TREATMENT RESPONSE
Predicting the clinical course of IPF based on respiratory function and radiologic imaging is unreliable and, at this time, we cannot predict disease trajectory accurately. 148 , 149 Monitoring of IPF progression is most commonly accomplished by measuring change in FVC over 12 months, and a decline in FVC ≥ 10% is generally considered the threshold for affirming progression. 150 There are no well‐established blood‐based biomarkers for IPF. Developing this type of biomarker for clinical use is a high priority that would be useful in early diagnosis and assessment of treatment efficacy. A number of possible blood biomarkers are being considered and evaluated. These include the receptor for advanced glycation end products (RAGE), which is highly expressed in alveolar epithelial type I cells and is significantly decreased in the lungs of patients with IPF. 151 Decreased circulating RAGE levels correlate with declining lung function in IPF. 152 Another possible biomarker assesses type I and III collagen turnover, which likely reflects the increase in interstitial collagen found in the lung in IPF. Measures of the serum levels of matrix metalloproteases that break down collagens type I and III using enzyme‐linked immunosorbent assays may lead to a clinically relevant IPF biomarker. 153 As we try new IPF treatment approaches, the importance of accurate, reliable, reasonably priced assays to monitor the efficacy for clinical trial and real‐world application will be increasingly important.
8. CONCLUSION
There is an urgent need for more attention and in‐depth research into development of targeted treatments that prolong life and improve quality of life for persons with IPF. 154 Pirfenidone and nintedanib are the two antifibrotic agents currently available for the treatment IPF. At this time, only lung transplant can alter its relentless course. Novel treatments under evaluation include pentraxin, pamrevulmab (monoclonal antibody against CTGF), and ATX inhibitors (Table 1). Also under study are agents that control TGFβ. One of these is PLN‐74809, a small molecule, dual selective inhibitor of the integrins αVβ1/αVβ6. These integrins are known to activate TGFβ. A phase 2a randomized, double‐blind, dose‐ranging, placebo‐controlled study of PLN‐74809 in IPF is ongoing (clinicaltrials.gov identifier NCT04396756). Another approach to targeting TGFβ is via silencing RNA as exemplified by TRK‐250, a single‐stranded oligonucleotide that produces siRNA targeting human TGFβ mRNA. A phase 1 study of this inhaled nucleic acid medication is in progress with an estimated completion date of April 2022 (clinicaltrials.gov identifier NCT03727802).
Gene and protein expression profiling is beginning to generate information on the mechanisms that result in lung damage in IPF. 155 , 156 Lipidomics are also being analyzed to expand our knowledge of how IPF progresses. 157 The clinical and research communities must come together to find better ways to regulate fibrotic and inflammatory responses in the IPF lung.
CONFLICT OF INTEREST
All authors have no conflict of interest to declare.
ETHICS STATEMENT
No ethics approval was needed, no human subjects were involved in this review paper, and no consent to participate and publish was needed.
AUTHOR CONTRIBUTIONS
Conception: Reiss, Glass. Literature review: Renna, Grossfeld, Agarwala. Figures and manuscript writing: Reiss, Glass, Spiegler, De Leon. All authors provided critical review of the manuscript and approved this draft.
ACKNOWLEDGMENTS
The authors would like to thank The Mother Mary Breathe Easy Foundation and The Herb and Evelyn Abrams Family Amyloid Research Fund. We thank Mr. Robert Buescher for his support. Original artwork watercolor on paper was by Samantha M. Steiner.
No funding was provided for this review.
Glass DS, Grossfeld D, Renna HA, et al. Idiopathic pulmonary fibrosis: Current and future treatment. Clin Respir J. 2022;16(2):84-96. doi: 10.1111/crj.13466
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
REFERENCES
- 1. King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949‐1961. [DOI] [PubMed] [Google Scholar]
- 2. Olson AL, Swigris JJ, Lezotte DC, Norris JM, Wilson CG, Brown KK. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med. 2007;176:277‐284. [DOI] [PubMed] [Google Scholar]
- 3. Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: evidence‐based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788‐824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: Incidence, prevalence, and survival, 2001‐11. Lancet Respir Med. 2014;2:566‐572. [DOI] [PubMed] [Google Scholar]
- 5. Pleasants R, Tighe RM. Management of idiopathic pulmonary fibrosis. Ann Pharmacother. 2019;53:1238‐1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389:1941‐1952. [DOI] [PubMed] [Google Scholar]
- 7. Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: A risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1997;155:242‐248. [DOI] [PubMed] [Google Scholar]
- 8. Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 2011;208:1339‐1350, 133950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Todd NW, Atamas SP, Luzina IG, et al. Permanent alveolar collapse is the predominant mechanism in idiopathic pulmonary fibrosis. Expert Rev Respir Med. 2015;9:411‐418. [DOI] [PubMed] [Google Scholar]
- 10. Martinez FJ, Safrin S, Weycker D, et al. IPF study group. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med. 2005;142:S963‐S967. [DOI] [PubMed] [Google Scholar]
- 11. Smith ML. Update on pulmonary fibrosis: Not all fibrosis is created equally. Arch Pathol Lab Med. 2016;140:221‐229. [DOI] [PubMed] [Google Scholar]
- 12. van Manen MJ, Geelhoed JJ, Tak NC, Wijsenbeek MS. Optimizing quality of life in patients with idiopathic pulmonary fibrosis [published correction appears in Ther Adv Respir Dis. 2017;11:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Senanayake S, Harrison K, Lewis M, McNarry M, Hudson J. Patients' experiences of coping with Idiopathic Pulmonary Fibrosis and their recommendations for its clinical management. PLoS One. 2018;13:e0197660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kulkarni T, Duncan SR. Acute exacerbation of idiopathic pulmonary fibrosis: Who to treat, how to treat. Curr Pulmonol Rep. 2019;8:123‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sprunger DB, Olson AL, Huie TJ, et al. Pulmonary fibrosis is associated with an elevated risk of thromboembolic disease. Eur Respir J. 2012;39(1):125‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Suzuki Y, Aono Y, Kono M, et al. Cause of mortality and sarcopenia in patients with idiopathic pulmonary fibrosis receiving antifibrotic therapy. Respirology. 2021;26:171‐179. [DOI] [PubMed] [Google Scholar]
- 17. Mori Y, Kondoh Y. What parameters can be used to identify early idiopathic pulmonary fibrosis? Respir Investig. 2021;59:53‐65. [DOI] [PubMed] [Google Scholar]
- 18. Davidsen JR, Lund LC, Laursen CB, Hallas J, Henriksen DP. Dynamics in diagnoses and pharmacotherapy before and after diagnosing idiopathic pulmonary fibrosis. ERJ Open Res. 2020;6:00479‐02020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thickett D, Voorham J, Ryan R, et al. Historical database cohort study addressing the clinical patterns prior to idiopathic pulmonary fibrosis (IPF) diagnosis in UK primary care. BMJ Open. 2020;10:e034428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Martinez FJ, Chisholm A, Collard HR, et al. The diagnosis of idiopathic pulmonary fibrosis: Current and future approaches. Lancet Respir Med. 2017;5:61‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sellarés J, Hernández‐González F, Lucena CM, et al. Auscultation of velcro crackles is associated with usual interstitial pneumonia. Medicine (Baltimore). 2016;95:e2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jafri S, Ahmed N, Saifullah N, Musheer M. Epidemiology and clinico‐radiological features of interstitial lung diseases. Pak J Med Sci. 2020;36:365‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van Manen MJG, Vermeer LC, Moor CC, et al. Clubbing in patients with fibrotic interstitial lung diseases. Respir Med. 2017;132:226‐231. [DOI] [PubMed] [Google Scholar]
- 24. Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156:684‐691. [DOI] [PubMed] [Google Scholar]
- 25. Lancaster L, Fieuw A, Meulemans J, Ford P, Nathan SD. Standardization of the 6‐min walk test in clinical trials of idiopathic pulmonary fibrosis. Contemp Clin Trials. 2020;100:106227. [DOI] [PubMed] [Google Scholar]
- 26. Pastre J, Barnett S, Ksovreli I, et al. Idiopathic pulmonary fibrosis patients with severe physiologic impairment: Characteristics and outcomes. Respir Res. 2021;22:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Selman M, Pardo A. Role of epithelial cells in idiopathic pulmonary fibrosis: From innocent targets to serial killers. Proc am Thorac Soc. 2006;3:364‐372. [DOI] [PubMed] [Google Scholar]
- 28. Romei C, Turturici L, Tavanti L, et al. The use of chest magnetic resonance imaging in interstitial lung disease: A systematic review. Eur Respir Rev. 2018;27:180062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hobbs S, Chung JH, Leb J, Kaproth‐Joslin K, Lynch DA. Practical imaging interpretation in patients suspected of having idiopathic pulmonary fibrosis: Official recommendations from the radiology working group of the pulmonary fibrosis foundation. Radiol Cardiothorac Imaging. 2021;3:e200279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Park J, Jung J, Yoon SH, et al. CT quantification of the heterogeneity of fibrosis boundaries in idiopathic pulmonary fibrosis. Eur Radiol. 2021;13:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Watadani T, Sakai F, Johkoh T, et al. Interobserver variability in the CT assessment of honeycombing in the lungs. Radiology. 2013;266:936‐944. [DOI] [PubMed] [Google Scholar]
- 32. Bartholmai BJ, Raghunath S, Karwoski RA, et al. Quantitative CT imaging of interstitial lung diseases. J Thorac Imaging. 2013;28:298‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: A Fleischner society white paper. Lancet Respir Med. 2018;6:138‐153. [DOI] [PubMed] [Google Scholar]
- 34. Cosgrove GP, Bianchi P, Danese S, Lederer DJ. Barriers to timely diagnosis of interstitial lung disease in the real world: The INTENSITY survey. BMC Pulm Med. 2018;18:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim GHJ, Goldin JG, Hayes W, Oh A, Soule B, Du S. The value of imaging and clinical outcomes in a phase II clinical trial of a lysophosphatidic acid receptor antagonist in idiopathic pulmonary fibrosis. Ther Adv Respir Dis. 2021;15. 17534666211004238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alhamad EH, Cal JG, Alrajhi NN, Aharbi WM, AlRikabi AC, AlBoukai AA. Clinical characteristics, comorbidities, and outcomes in patients with idiopathic pulmonary fibrosis. Ann Thorac Med. 2020;15:208‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fisher JH, Kolb M, Algamdi M, et al. Baseline characteristics and comorbidities in the CAnadian REgistry for Pulmonary Fibrosis. BMC Pulm Med. 2019;19:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Idiopathic pulmonary fibrosis clinical research network. Prednisone, azathioprine, and N‐acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366:1968‐1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Newton CA, Zhang D, Oldham JM, et al. Telomere length and use of immunosuppressive medications in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2019;200:336‐347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moon SW, Kim SY, Chung MP, et al. Longitudinal changes in clinical features, management, and outcomes of idiopathic pulmonary fibrosis: A nationwide cohort study. Ann am Thorac Soc. 2021;18:780‐787. [DOI] [PubMed] [Google Scholar]
- 41. Vigeland CL, Hughes AH, Horton MR. Etiology and treatment of cough in idiopathic pulmonary fibrosis. Respir Med. 2017;123:98‐104. [DOI] [PubMed] [Google Scholar]
- 42. Kondoh Y, Taniguchi H, Yokoi T, et al. Cyclophosphamide and low‐dose prednisolone in idiopathic pulmonary fibrosis and fibrosing nonspecific interstitial pneumonia. Eur Respir J. 2005;25:528‐533. [DOI] [PubMed] [Google Scholar]
- 43. Tzouvelekis A, Bouros E, Oikonomou A, et al. Effect and safety of mycophenolate mofetil in idiopathic pulmonary fibrosis. Pulm Med. 2011:849035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Franks ME, MacPherson GR, Figg WD. Thalidomide. Lancet. 2004;363:1802‐1811. [DOI] [PubMed] [Google Scholar]
- 45. Bartlett JB, Dredge K, Dalgleish AG. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat Rev Cancer. 2004;4:314‐322. [DOI] [PubMed] [Google Scholar]
- 46. Nemer G, Khalil A. A cautious note on thalidomide usage in cancer treatment: Genetic profiling of the TBX2 sub‐family gene expression is required. Drug Res (Stuttg). 2019;69:512‐518. [DOI] [PubMed] [Google Scholar]
- 47. Horton MR, Hallowell RW. Revisiting thalidomide: Fighting with caution against idiopathic pulmonary fibrosis. Drugs Today (Barc). 2012;48:661‐671. [DOI] [PubMed] [Google Scholar]
- 48. Tabata C, Tabata R, Kadokawa Y, et al. Thalidomide prevents bleomycin‐induced pulmonary fibrosis in mice. J Immunol. 2007;179:708‐714. [DOI] [PubMed] [Google Scholar]
- 49. Tabata C, Tabata R, Takahashi Y, Nakamura K, Nakano T. Thalidomide prevents cigarette smoke extract‐induced lung damage in mice. Int Immunopharmacol. 2015;25:511‐517. [DOI] [PubMed] [Google Scholar]
- 50. Horton MR, Santopietro V, Mathew L, et al. Thalidomide for the treatment of cough in idiopathic pulmonary fibrosis: A randomized trial. Ann Intern Med. 2012;157:398‐406. [DOI] [PubMed] [Google Scholar]
- 51. Birring SS, Kavanagh JE, Irwin RS, et al. Treatment of interstitial lung disease associated cough: CHEST guideline and expert panel report. Chest. 2018;154:904‐917. [DOI] [PubMed] [Google Scholar]
- 52. Zhou XL, Xu P, Chen HH, et al. Thalidomide inhibits TGF‐β1‐induced epithelial to mesenchymal transition in alveolar epithelial cells via Smad‐dependent and Smad‐independent signaling pathways. Sci Rep. 2017;7:14727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Glassberg MK. Overview of idiopathic pulmonary fibrosis, evidence‐based guidelines, and recent developments in the treatment landscape. Am J Manag Care. 2019;25:S195‐S203. [PubMed] [Google Scholar]
- 54. Saito S, Alkhatib A, Kolls JK, Kondoh Y, Lasky JA. Pharmacotherapy and adjunctive treatment for idiopathic pulmonary fibrosis (IPF). J Thorac Dis. 2019;11:S1740‐S1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Collins BF, Raghu G. Antifibrotic therapy for fibrotic lung disease beyond idiopathic pulmonary fibrosis. Eur Respir Rev. 2019;28:190022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Noble PW, Albera C, Bradford WZ, et al. Pirfenidone for idiopathic pulmonary fibrosis: Analysis of pooled data from three multinational phase 3 trials. Eur Respir J. 2016;47:243‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet. 2011;377:1760‐1769. [DOI] [PubMed] [Google Scholar]
- 58. King TE, Bradford WZ, Castro‐Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083‐2092. [DOI] [PubMed] [Google Scholar]
- 59. Ley B, Swigris J, Day BM, et al. Pirfenidone reduces respiratory‐related hospitalizations in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2017;196:756‐761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Eaden JA, Barber CM, Renshaw SA, Chaudhuri N, Bianchi SM. Real world experience of response to pirfenidone in patients with idiopathic pulmonary fibrosis: A two centre retrospective study. Sarcoidosis Vasc Diffuse Lung Dis. 2020;37:218‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cameli P, Refini RM, Bergantini L, et al. Long‐term follow‐up of patients with idiopathic pulmonary fibrosis treated with pirfenidone or nintedanib: A real‐life comparison study. Front Mol Biosci. 2020;7:581828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Feng H, Zhao Y, Li Z, Kang J. Real‐life experiences in a single center: Efficacy of pirfenidone in idiopathic pulmonary fibrosis and fibrotic idiopathic non‐specific interstitial pneumonia patients. Ther Adv Respir Dis. 2020;14:1753466620963015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Verma N, Kumar P, Mitra S, et al. Drug idiosyncrasy due to pirfenidone presenting as acute liver failure: Case report and mini‐review of the literature. Hepatol Commun. 2018;2:142‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lamb YN. Nintedanib: A review in fibrotic interstitial lung diseases. Drugs. 2021;81:575‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wollin L, Wex E, Pautsch A, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J. 2015;45:1434‐1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071‐2082. [DOI] [PubMed] [Google Scholar]
- 67. Librero Jiménez M, Heredia Carrasco C, Fernández Cano MC. Severe Hepatotoxicity Secondary to Nintedanib. Rev Esp Enferm Dig. 2020. Dec 29. Epub ahead of print [DOI] [PubMed] [Google Scholar]
- 68. Rahaghi F, Belperio JA, Fitzgerald J, et al. Delphi consensus recommendations on management of dosing, adverse events, and comorbidities in the treatment of idiopathic pulmonary fibrosis with nintedanib. Clin Med Insights Circ Respir Pulm Med. 2021;15:11795484211006050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bargagli E, Piccioli C, Rosi E, et al. Pirfenidone and Nintedanib in idiopathic pulmonary fibrosis: Real‐life experience in an Italian referral centre. Pulmonology. 2019;25:149‐153. [DOI] [PubMed] [Google Scholar]
- 70. Rochwerg B, Neupane B, Zhang Y, et al. Treatment of idiopathic pulmonary fibrosis: A network meta‐analysis. BMC Med. 2016;14:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wright WA, Crowley LE, Parekh D, et al. Real‐world retrospective observational study exploring the effectiveness and safety of antifibrotics in idiopathic pulmonary fibrosis. BMJ Open Respir Res. 2021;8:e000782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Justet A, Klay D, Porcher R, et al. Safety and efficacy of pirfenidone and nintedanib in patients with idiopathic pulmonary fibrosis and carrying a telomere‐related gene mutation. Eur Respir J. 2021;57:2003198. [DOI] [PubMed] [Google Scholar]
- 73. Schmid U, Weber B, Magnusson MO, Freiwald M. Exposure‐efficacy analyses of nintedanib in patients with chronic fibrosing interstitial lung disease. Respir Med. 2021;180:106369. [DOI] [PubMed] [Google Scholar]
- 74. Dempsey TM, Payne S, Sangaralingham L, et al. Adoption of the anti‐fibrotic medications pirfenidone and nintedanib for patients with idiopathic pulmonary fibrosis. Ann am Thorac Soc. 2021;18:1121‐1128. [DOI] [PubMed] [Google Scholar]
- 75. Nemoto M, Zaizen Y, Kataoka K, et al. Histologic factors associated with nintedanib efficacy in patients with idiopathic pulmonary fibrosis. PLoS One. 2021;16:e0245147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Somogyi V, Chaudhuri N, Torrisi SE, Kahn N, Müller V, Kreuter M. The therapy of idiopathic pulmonary fibrosis: What is next? Eur Respir Rev. 2019;28:190021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Weill D, Benden C, Corris PA, et al. A consensus document for the selection of lung transplant candidates: 2014—An update from the pulmonary transplantation council of the international society for heart and lung transplantation. J Heart Lung Transplant. 2015;34:1‐15. [DOI] [PubMed] [Google Scholar]
- 78. Amor MS, Rosengarten D, Shitenberg D, Pertzov B, Shostak Y, Kramer MR. Lung transplantation in idiopathic pulmonary fibrosis: Risk factors and outcome. Isr Med Assoc J. 2020;22:741‐746. [PubMed] [Google Scholar]
- 79. Riddell P, Kleinerova J, Eaton D, et al. Meaningful survival benefit for single lung transplantation in idiopathic pulmonary fibrosis patients over 65 years of age. Eur Respir J. 2020;56:1902413. [DOI] [PubMed] [Google Scholar]
- 80. Yusen RD, Edwards LB, Kucheryavaya AY, et al. The registry of the international society for heart and lung transplantation: Thirty‐first adult lung and heart‐lung transplant report—2014; focus theme: Retransplantation. J Heart Lung Transplant. 2014;33:1009‐1024. [DOI] [PubMed] [Google Scholar]
- 81. Chambers DC, Cherikh WS, Goldfarb SB, et al. The international thoracic organ transplant registry of the international society for heart and lung transplantation: Thirty‐fifth adult lung and heart‐lung transplant report‐2018; focus theme: Multiorgan transplantation. J Heart Lung Transplant. 2018;37:1169‐1183. [DOI] [PubMed] [Google Scholar]
- 82. Hackman KL, Bailey MJ, Snell GI, Bach LA. Diabetes is a major risk factor for mortality after lung transplantation. Am J Transplant. 2014;14:438‐445. [DOI] [PubMed] [Google Scholar]
- 83. Patti MG, Vela MF, Odell DD, Richter JE, Fisichella PM, Vaezi MF. The intersection of GERD, aspiration, and lung transplantation. J Laparoendosc Adv Surg Tech A. 2016;26:501‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. George PM, Patterson CM, Reed AK, Thillai M. Lung transplantation for idiopathic pulmonary fibrosis. Lancet Respir Med. 2019;7:271‐282. [DOI] [PubMed] [Google Scholar]
- 85. Balestro E, Cocconcelli E, Tinè M, et al. Idiopathic pulmonary fibrosis and lung transplantation: When it is feasible. Medicina (Kaunas). 2019;55:702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bilgili H, Białas AJ, Górski P, Piotrowski WJ. Telomere abnormalities in the pathobiology of idiopathic pulmonary fibrosis. J Clin Med. 2019;8:1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kumar A, Kapnadak SG, Girgis RE, Raghu G. Lung transplantation in idiopathic pulmonary fibrosis. Expert Rev Respir Med. 2018;12:375‐385. [DOI] [PubMed] [Google Scholar]
- 88. Alvarez D, Cardenes N, Sellares J, et al. IPF lung fibroblasts have a senescent phenotype. Am J Physiol Lung Cell Mol Physiol. 2017;313:L1164‐L1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Merck SJ, Armanios M. Shall we call them “telomere‐mediated”? Renaming the idiopathic after the cause is found. Eur Respir J. 2016;48:1556‐1558. [DOI] [PubMed] [Google Scholar]
- 90. Le Pavec J, Dauriat G, Gazengel P, et al. Lung transplantation for idiopathic pulmonary fibrosis. Presse Med. 2020;49:104026. [DOI] [PubMed] [Google Scholar]
- 91. Silhan LL, Shah PD, Chambers DC, et al. Lung transplantation in telomerase mutation carriers with pulmonary fibrosis. Eur Respir J. 2014;44:178‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Tokman S, Singer JP, Devine MS, et al. Clinical outcomes of lung transplant recipients with telomerase mutations. J Heart Lung Transplant. 2015;34:1318‐1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kannengiesser C, Borie R, Renzoni EA. Pulmonary fibrosis: Genetic analysis of telomere‐related genes, telomere length measurement‐or both? Respirology. 2019;24:97‐98. [DOI] [PubMed] [Google Scholar]
- 94. Veit T, Leuschner G, Sisic A, et al. Pirfenidone exerts beneficial effects in patients with IPF undergoing single lung transplantation. Am J Transplant. 2019;19:2358‐2365. [DOI] [PubMed] [Google Scholar]
- 95. Tuyls S, Verleden SE, Wuyts WA, et al. Determinants of survival in lung transplantation patients with idiopathic pulmonary fibrosis: A retrospective cohort study. Transplant Int. 2019;32:399‐409. [DOI] [PubMed] [Google Scholar]
- 96. Force SD, Kilgo P, Neujahr DC, et al. Bilateral lung transplantation offers better long‐term survival, compared with single‐lung transplantation, for younger patients with idiopathic pulmonary fibrosis. Ann Thorac Surg. 2011;91:244‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Villavicencio MA, Axtell AL, Osho A, et al. Single‐ versus double‐lung transplantation in pulmonary fibrosis: Impact of age and pulmonary hypertension. Ann Thorac Surg. 2018;106:856‐863. [DOI] [PubMed] [Google Scholar]
- 98. Li D, Liu Y, Wang B. Single versus bilateral lung transplantation in idiopathic pulmonary fibrosis: A systematic review and meta‐analysis. PLoS One. 2020;15(5):e0233732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Snell GI, Yusen RD, Weill D, et al. Report of the ISHLT working group on primary lung graft dysfunction, part I: Definition and grading‐A 2016 consensus group statement of the international society for heart and lung transplantation. J Heart Lung Transplant. 2017;36:1097‐1103. [DOI] [PubMed] [Google Scholar]
- 100. Laporta Hernandez R, Aguilar Perez M, Lázaro Carrasco MT, Ussetti GP. Lung transplantation in idiopathic pulmonary fibrosis. Med Sci (Basel). 2018;6:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. d'Alessandro M, Bergantini L, Torricelli E, et al. Systematic review and metanalysis of oncomarkers in ipf patients and serial changes of oncomarkers in a prospective Italian real‐life case series. Cancers (Basel). 2021;13:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lama VN, Smith L, Badri L, et al. Evidence for tissue‐resident mesenchymal stem cells in human adult lung from studies of transplanted allografts. J Clin Invest. 2007;117:989‐996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Chanda D, Kurundkar A, Rangarajan S, et al. Developmental reprogramming in mesenchymal stromal cells of human subjects with idiopathic pulmonary fibrosis. Sci Rep. 2016;6:37445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Toonkel RL, Hare JM, Matthay MA, Glassberg MK. Mesenchymal stem cells and idiopathic pulmonary fibrosis. Potential for clinical testing. Am J Respir Crit Care Med. 2013;188:133‐140. [DOI] [PubMed] [Google Scholar]
- 105. Tzouvelekis A, Toonkel R, Karampitsakos T, et al. Mesenchymal stem cells for the treatment of idiopathic pulmonary fibrosis. Front Med (Lausanne). 2018;5:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Glassberg MK, Minkiewicz J, Toonke RL, et al. Allogeneic human mesenchymal stem cells in patients with idiopathic pulmonary fibrosis via intravenous delivery (AETHER) A phase I safety clinical trial. Chest. 2017;151:971‐981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Fishman JE, Kim GJ, Kyeong NY, Goldin JG, Glassberg MK. Intravenous stem cell dose and changes in quantitative lung fibrosis and DLCO in the AETHER trial: A pilot study. Eur Rev Med Pharmacol Sci. 2019;23:7568‐7572. [DOI] [PubMed] [Google Scholar]
- 108. Averyanov A, Koroleva I, Konoplyannikov M, et al. First‐in‐human high‐cumulative‐dose stem cell therapy in idiopathic pulmonary fibrosis with rapid lung function decline. Stem Cells Transl Med. 2020;9:6‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Deban L, Bottazzi B, Garlanda C, de la Torre YM, Mantovani A. Pentraxins: Multifunctional proteins at the interface of innate immunity and inflammation. Biofactors. 2009;35:138‐145. [DOI] [PubMed] [Google Scholar]
- 110. Cox N, Pilling D, Gomer RH. Serum amyloid P: A systemic regulator of the innate immune response. J Leukoc Biol. 2014;96:739‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Pilling D, Galvis‐Carvajal E, Karhadkar TR, Cox N, Gomer RH. Monocyte differentiation and macrophage priming are regulated differentially by pentraxins and their ligands. BMC Immunol. 2017;18:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Cox N, Pilling D, Gomer RH. Distinct Fcγ receptors mediate the effect of serum amyloid p on neutrophil adhesion and fibrocyte differentiation. J Immunol. 2014;193:1701‐1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Bharadwaj D, Mold C, Markham E, Du Clos TW. Serum amyloid P component binds to Fc gamma receptors and opsonizes particles for phagocytosis. J Immunol. 2001;166:6735‐6741. [DOI] [PubMed] [Google Scholar]
- 114. Murray LA, Chen Q, Kramer MS, et al. TGF‐beta driven lung fibrosis is macrophage dependent and blocked by Serum amyloid P. Int J Biochem Cell Biol. 2011;43:154‐162. [DOI] [PubMed] [Google Scholar]
- 115. Castaão AP, Lin SL, Surowy T, et al. Serum amyloid P inhibits fibrosis through Fc gamma R‐dependent monocyte‐macrophage regulation in vivo. Sci Transl Med. 2009;1:5ra13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Naik‐Mathuria B, Pilling D, Crawford JR, et al. Serum amyloid P inhibits dermal wound healing. Wound Repair Regen. 2008;16:266‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Tzouvelekis A, Tzilas V, Antoniou KM, Bouros D. Human pentraxin 2 protein treatment for IPF. Lancet Respir Med. 2019;7:640‐641. [DOI] [PubMed] [Google Scholar]
- 118. van den Blink B, Dillingh MR, Ginns LC, et al. Recombinant human pentraxin‐2 therapy in patients with idiopathic pulmonary fibrosis: Safety, pharmacokinetics and exploratory efficacy. Eur Respir J. 2016;47:889‐897. [DOI] [PubMed] [Google Scholar]
- 119. Duffield JS, Lupher ML Jr. PRM‐151 (recombinant human serum amyloid P/pentraxin 2) for the treatment of fibrosis. Drug News Perspect. 2010;23:305‐315. [DOI] [PubMed] [Google Scholar]
- 120. Moeller A, Gilpin SE, Ask K, et al. Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179:588‐594. [DOI] [PubMed] [Google Scholar]
- 121. Pilling D, Gomer RH. Persistent lung inflammation and fibrosis in serum amyloid P component (APCs−/−) knockout mice. PLoS One. 2014;9:e93730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Dillingh MR, van den Blink B, Moerland M, et al. Recombinant human serum amyloid P in healthy volunteers and patients with pulmonary fibrosis. Pulm Pharmacol Ther. 2013;26:672‐676. [DOI] [PubMed] [Google Scholar]
- 123. Raghu G, van den Blink B, Hamblin MJ, et al. Effect of recombinant human pentraxin 2 vs placebo on change in forced vital capacity in patients with idiopathic pulmonary fibrosis: A randomized clinical trial. Jama. 2018;319:2299‐2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Raghu G, van den Blink B, Hamblin MJ, et al. Long‐term treatment with recombinant human pentraxin 2 protein in patients with idiopathic pulmonary fibrosis: An open‐label extension study. Lancet Respir Med. 2019;7:657‐664. [DOI] [PubMed] [Google Scholar]
- 125. Lipson KE, Wong C, Teng Y, Spong S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair. 2012;5(Suppl 1):S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Picozzi V, Alseidi A, Winter J, et al. Gemcitabine/nab‐paclitaxel with pamrevlumab: A novel drug combination and trial design for the treatment of locally advanced pancreatic cancer. ESMO Open. 2020;5:e000668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Richeldi L, Fernández Pérez ER, Costabel U, et al. Pamrevlumab, an anti‐connective tissue growth factor therapy, for idiopathic pulmonary fibrosis (PRAISE): A phase 2, randomised, double‐blind, placebo‐controlled trial. Lancet Respir Med. 2020;8:25‐33. [DOI] [PubMed] [Google Scholar]
- 128. Leask A. Breathe, breathe in the air: The anti‐CCN2 antibody pamrevlumab (FG‐3019) completes a successful phase II clinical trial for idiopathic pulmonary fibrosis. J Cell Commun Signal. 2019;13:441‐442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Sgalla G, Franciosa C, Simonetti J, Richeldi L. Pamrevlumab for the treatment of idiopathic pulmonary fibrosis. Expert Opin Investig Drugs. 2020;29:771‐777. [DOI] [PubMed] [Google Scholar]
- 130. Di Martino E, Provenzani A, Vitulo P, Polidori P. Systematic review and meta‐analysis of pirfenidone, nintedanib, and pamrevlumab for the treatment of idiopathic pulmonary fibrosis. Ann Pharmacother. 2021;55:723‐731. [DOI] [PubMed] [Google Scholar]
- 131. Ninou I, Magkrioti C, Aidinis V. Autotaxin in pathophysiology and pulmonary fibrosis. Front Med (Lausanne). 2018;5:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Desroy N, Housseman C, Bock X, et al. Discovery of 2‐[[2‐Ethyl‐6‐[4‐[2‐(3‐hydroxyazetidin‐1‐yl)‐2‐oxoethyl]piperazin‐1‐yl]‐8‐methylimidazo[1,2‐a]pyridin‐3‐yl]methylamino]‐4‐(4‐fluorophenyl)thiazole‐5‐carbonitrile (GLPG1690), a first‐in‐class autotaxin inhibitor undergoing clinical evaluation for the treatment of idiopathic pulmonary fibrosis. J Med Chem. 2017;60:3580‐3590. [DOI] [PubMed] [Google Scholar]
- 133. Oikonomou N, Mouratis MA, Tzouvelekis A, et al. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am J Respir Cell Mol Biol. 2012;47:566‐574. [DOI] [PubMed] [Google Scholar]
- 134. Tager AM. Autotaxin emerges as a therapeutic target for idiopathic pulmonary fibrosis: Limiting fibrosis by limiting lysophosphatidic acid synthesis. Am J Respir Cell Mol Biol. 2012;47:563‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Tager AM, LaCamera P, Shea BS, et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat Med. 2008;14:45‐54. [DOI] [PubMed] [Google Scholar]
- 136. Taneja A, Desrivot J, Diderichsen PM, et al. Population pharmacokinetic and pharmacodynamic analysis of GLPG1690, an autotaxin inhibitor, in healthy volunteers and patients with idiopathic pulmonary fibrosis. Clin Pharmacokinet. 2019;58:1175‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Maher TM, Kreuter M, Lederer DJ, et al. Rationale, design and objectives of two phase III, randomised, placebo‐controlled studies of GLPG1690, a novel autotaxin inhibitor, in idiopathic pulmonary fibrosis (ISABELA 1 and 2). BMJ Open Respir Res. 2019;6:e000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. https://www.clinicaltrialsarena.com/comment/gilead-galapagos-discontinue-ipf-front-runner/ accessed April 21, 2021
- 139. Tan Z, Lei H, Guo M, Chen Y, Zhai X. An updated patent review of autotaxin inhibitors (2017‐present). Expert Opin Ther Pat. 2021;31:421‐434. [DOI] [PubMed] [Google Scholar]
- 140. Jia W, Wang Z, Gao C, Wu J, Wu Q. Trajectory modeling of endothelial‐to‐mesenchymal transition reveals galectin‐3 as a mediator in pulmonary fibrosis. Cell Death Dis. 2021;12:327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Hirani N, MacKinnon AC, Nicol L, et al. Target‐inhibition of galectin‐3 by inhaled TD139 in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2020;57:2002559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Govindappa PK, Patil M, Garikipati VNS, et al. Targeting exosome‐associated human antigen R attenuates fibrosis and inflammation in diabetic heart. FASEB j. 2020;34:2238‐2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Woodhoo A, Iruarrizaga‐Lejarreta M, Beraza N, et al. Human antigen R contributes to hepatic stellate cell activation and liver fibrosis. Hepatology. 2012;56:1870‐1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Al‐Habeeb F, Aloufi N, Traboulsi H, et al. Human antigen R promotes lung fibroblast differentiation to myofibroblasts and increases extracellular matrix production. J Cell Physiol. 2021;236:6836‐6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Adams TS, Schupp JC, Poli S, et al. Single‐cell RNA‐seq reveals ectopic and aberrant lung‐resident cell populations in idiopathic pulmonary fibrosis. Sci Adv. 2020;6:eaba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Neumark N, Cosme C Jr, Rose KA, Kaminski N. The idiopathic pulmonary fibrosis cell atlas. Am J Physiol Lung Cell Mol Physiol. 2020;319:L887‐L893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Valenzi E, Tabib T, Papazoglou A, et al. Disparate interferon signaling and shared aberrant basaloid cells in single‐cell profiling of idiopathic pulmonary fibrosis and systemic sclerosis‐associated interstitial lung disease. Front Immunol. 2021;12:595811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Drakopanagiotakis F, Wujak L, Wygrecka M, Markart P. Biomarkers in idiopathic pulmonary fibrosis. Matrix Biol. 2018;68‐69:404‐421. [DOI] [PubMed] [Google Scholar]
- 149. Todd JL, Neely ML, Overton R, et al. Peripheral blood proteomic profiling of idiopathic pulmonary fibrosis biomarkers in the multicentre IPF‐PRO Registry. Respir Res. 2019;20:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Wuyts WA, Wijsenbeek M, Bondue B, et al. Idiopathic pulmonary fibrosis: Best practice in monitoring and managing a relentless fibrotic disease. Respiration. 2020;99:73‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Machahua C, Montes‐Worboys A, Planas‐Cerezales L, et al. Serum AGE/RAGEs as potential biomarker in idiopathic pulmonary fibrosis. Respir Res. 2018;19:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Yamaguchi K, Iwamoto H, Mazur W, et al. Reduced endogenous secretory RAGE in blood and bronchoalveolar lavage fluid is associated with poor prognosis in idiopathic pulmonary fibrosis. Respir Res. 2020;2:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Jessen H, Hoyer N, Prior TS, et al. Turnover of type I and III collagen predicts progression of idiopathic pulmonary fibrosis. Respir Res. 2021;22:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Ghumman M, Dhamecha D, Gonsalves A, et al. Emerging drug delivery strategies for idiopathic pulmonary fibrosis treatment. Eur J Pharm Biopharm. 2021;164:1‐12. Apr 18:S0939‐6411(21)00099‐0. doi: 10.1016/j.ejpb.2021.03.017. Epub ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Xu F, Tanabe N, Vasilescu DM, et al. The transition from normal lung anatomy to minimal and established fibrosis in idiopathic pulmonary fibrosis (IPF). EBioMedicine. 2021;66:103325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Konigsberg IR, Borie R, Walts AD, et al. Molecular signatures of idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2021;65:430‐441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Nambiar S, Clynick B, How BS, et al. There is detectable variation in the lipidomic profile between stable and progressive patients with idiopathic pulmonary fibrosis (IPF). Respir Res. 2021;22:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.