

Abstract
Objectives:
Recent transcriptome analyses revealed that 15-fold expanded HLA-DR+CD90+ synovial fibroblasts (SFs) are potential key mediators of inflammation in rheumatoid arthritis (RA). The reasons for the expansion of HLA-DR+CD90+ SFs are unclear, but genetic signatures indicated a central role of IFNɣ in the generation of this fibroblast subset.
In the present study, we investigated the generation of HLA-DR+CD90+ SFs and their function. We hypothesized that infiltrating leukocytes such as NK cells become activated in situ, provide IFNɣ and thus contribute to the generation of arthritic HLA-DR+CD90+ SFs.
Methods:
We combined functional assays using primary human materials and focused bioinformatic analyses of mass cytometry and transcriptomics patient datasets.
Results:
We detected enriched and activated FcɣRIIIA(CD16)+ NK cells in synovia from active RA. CD16 recognized immune complexes in synovial fluid, potentially contributing to NK cell activation in RA. In vitro, NK cell-derived IFNɣ induced HLA-DR and an inflammatory, cytokine secreting, HLA-DR+ phenotype in CD90+ SFs. HLA-DR+CD90+ SFs consecutively activated CD4+ T cells upon receptor crosslinking via superantigens. HLA-DR+CD90+ SFs also activated CD4+ T cells in absence of superantigens, an effect that was boosted by NK cell-derived IFNɣ and that was four times stronger in RA compared to osteoarthritis. Finally, JAK inhibition prevented HLA-DR induction and blocked pro-inflammatory signals to T cells.
Conclusions:
HLA-DR+CD90+ SFs are an activation state that can be induced by IFNɣ, likely provided from infiltrating leukocytes such as activated NK cells. The induction of these pro-inflammatory, IL-6 producing and likely antigen-presenting SFs can be targeted by JAK inhibition.
Keywords: NK cells, Interferon-ɣ, synovial fibroblasts, JAK inhibition, rheumatoid arthritis
INTRODUCTION
Rheumatoid Arthritis (RA) is a systemic inflammatory disease characterized by chronic destructive arthritis (1). Autoantibodies frequently occur in RA and are a risk factor for poor outcome. The genetic risk in RA is based on single-nuclear polymorphisms in genes involved in immune regulation (1, 2), especially in the MHC-II region in seropositive RA, including HLA-DRB1 (3).
Synovial fibroblasts (SFs) are the most abundant cell type in synovia and have been well implicated in the pathogenesis of RA (4–6). SFs are tissue-destructive and recruit and activate immune cells (4). Genetic signatures combined with high-dimensional surface molecule analyses recently revealed a subset of SFs that is more than 15-fold expanded in leukocyte-rich RA (5). This THY1(CD90)+ subset is characterized by a high expression of HLA-DR. The RNA expression profile of HLA-DR+CD90+ SFs identified these cells as pivotal contributors to inflammation in RA by producing cytokines and chemokines (5). As such, HLA-DR+CD90+ SFs are a major source of synovial IL-6 (4, 5). IL-6 is an important pro-inflammatory cytokine in RA and blocking the binding to its receptor is a successful treatment strategy (7).
The functional role of HLA-DR molecules on SFs is not well established (8, 9), but a pathogenic role of HLA-DR-mediated antigen presentation in RA in general is intuitive because of the strong genetic associations and the formation of autoantibodies. It has recently been shown that SFs can internalize, process and present arthritic antigens via HLA-DR (10–12). In line, a gene ontology analysis revealed increased genes involved in ‘antigen processing/presentation of peptide antigen via MHC II’ specifically in HLA-DR+CD90+ SFs (5), suggesting that antigen presentation by HLA-DR CD90+ SFs occurs in vivo in RA.
Importantly, HLA-DR+CD90+ SFs possess a strong IFNɣ signature (5), indicating that IFNɣ may be involved in the generation of its phenotype. However, functional studies implicating IFNɣ in the development of the HLA-DR+CD90+ SF phenotype are pending.
Natural Killer (NK) cells are immediately-acting innate lymphocytes that respond to a variety of stress signals by secreting cytokines, in particular IFNɣ (13). NK cells reside in inflamed synovial fluid (14–17), and an increased IFNɣ-production by synovial fluid NK cells was associated with a more erosive disease course (14). Nevertheless, the role of NK cells in RA is not well defined and data on the presence of NK cells in synovial tissue are scarce. Only few studies with relatively small sample sizes described NK cells in RA synovia (16, 18, 19). Increasing knowledge on NK cells in RA is of interest, as polymorphisms in a prominent activating NK cell receptor, the low affinity Fc-γ-receptor IIIa (FcγRIIIa; CD16), are a risk factor to develop RA (13, 20, 21). CD16 binds to surface-bound antibodies and immune complexes and triggers cytotoxicity and IFNɣ production (22–25). We hypothesized that NK cells may be activated in the inflammatory milieu of RA and provide IFNɣ to SFs.
Together, the generation of HLA-DR+CD90+ SFs may be a key event in RA pathogenesis, given its expansion, its inflammatory profile and the potential to present antigens along with the strong clinical associations with MHC-II alleles. In the present study we investigated the generation of HLA-DR+CD90+ SFs and their function in vitro.
MATERIAL AND METHODS
Synovial fluid analysis by flow cytometry
100μl native synovial fluid (> 1×106 leukocytes) from in total five joints from three patients with highly active chronic arthritis (1 juvenile idiopathic arthritis, 1 seronegative RA, 1 seropositive RA) were processed immediately or kept overnight at 4°C. Cells were washed and incubated with antibodies in PBS + 2% FCS (supplementary methods). After 30min at 4°C, cells were washed, fixed in 2% paraformaldehyde and analyzed on a three-laser flow cytometer (LSR II, BD).
Synovial tissue analysis by mass cytometry
Mass cytometry data were generated in a multi-center resource study (AMP-RA) (5). In brief, synovial tissues from biopsies and arthroplasty surgeries were brought into single cell suspensions, cryo-preserved, incubated with a panel of 35 metal ion-linked monoclonal antibodies and analyzed by mass cytometry. We analyzed normalized FCS files from 15 OA and 26 RA patients (supplementary table 1). FlowJo was used for manual gating. Cytobank was used for unbiased clustering of pre-gated viable cells using the SPADE algorithm. The following 20 clustering channels were chosen: CD19, −64, −16, −8a, −20, −45RO, −38, −279, −14, −185, −4, −3, −11c, −307d, −138, −90, 34, −66b, −106 and −45. The target number of nodes was 300.
RNAseq data analysis
Transcripts of the IFNɣ signaling pathway were individually analyzed taking advantage of the online accessible database from the AMP-RA Phase I dataset (immunogenomics.org, (5)). Furthermore, we downloaded raw data and obtained log2 normalized transcript per million (TPM) RNA expression profiles from sorted populations of synovial fibroblasts, T cells, B cells and monocytes (5). The median expression of HLA-DRB1 for each respective cell type within the OA control group was subtracted from the RA conditions to obtain fold changes in inflammation. The Kruskal-Wallis test followed by unpaired t-tests for all pairwise comparisons was used to assess statistical significance. Log2 TPM values for JAK1, JAK2 and JAK3 were plotted as heatmap.
Generation of primary human cells
Fibroblasts were isolated after written informed consent from synovial tissue (from OA and RA patients that underwent either arthroscopic synovectomy or joint replacement surgery) and from skin (from mamma reduction surgery from healthy volunteers and punch biopsies from affected skin from patients with morphea). Synovial tissue was digested with collagenase and dispase (Cellsystems, Troisdorf, Germany) at 37°C for 2h. The cell suspension was cultured in medium (DMEM/F12, Merck, Darmstadt, Germany) supplied with 10% heat-inactivated fetal calf serum (Thermo Fisher Scientific, Waltham, Massachusetts, United States).
Peripheral blood mononuclear cells (PBMCs) were isolated from heparinized blood from healthy volunteers by density gradient centrifugation using Biocoll separating solution (Biochrom Ltd, Cambourne, UK). NK cells and CD4+ T cells were isolated from BPMCs using MojoSort™ Human NK cells/CD4+ T cells isolation Kits (BioLegend, 480054 and 480010, respectively). The purity of isolated cells was > 90%. Cells were counted manually and checked for viability (trypan blue).
Cell culture
All cells were cultured in 5% CO2 at 37°C. Fibroblasts were cultured in DMEM/Ham F-12 supplemented with 10% FCS, 1% L-Glutamine and 1% Penicillin/Streptomycin and used from passage 4 to 10. PBMCs, NK cells, T cells, the NK cell-sensitive myeloid leukemia cell line K562 (ATCC®CCL-243™), K562 “feeder” cells and co-cultures were maintained in RPMI-1640 supplemented with 10% FCS, 1% L-glutamine, 15mM HEPES buffer and 1% penicillin/streptomycin (“R10”). K562 “feeder” cells were engineered to express GFP, 41BB-ligand and membrane-bound IL-15 (K562–41BBL-mbIL-15; kind gift from Dario Campana; St. Jude Children’s Research Hospital, Memphis, TN) (26).
Co-culture of synovial fibroblasts and NK cells
100,000 SFs were seeded per 24-well. After attachment, 200IU/mL IL-2 (Peprotech, 200–02) with or without 500,000 freshly isolated NK cells were added. After 3 or 7 days, cells were incubated with fluorochrome-labelled antibodies (supplementary methods) and directly analyzed by flow cytometry. The procedure of intracellular IL-6 staining is described in the supplementary methods.
Generation of supernatant from freshly isolated NK cells
For the generation of supernatant from NK cell/SFs co-cultures, NK cells were cultured on 15,000 SFs per 96-well for 3 days. For stimulation by cytokines, 1×106 NK cells/ml were cultured with 200IU/ml IL-2 (Peprotech, 200–02) for 3 days. For stimulation via Fcɣ-receptor-IIIa (CD16), 2×106 NK cells/ml were cultured overnight on plates coated with anti-CD16 (1μg/ml; 3G8; BioLegend, 302014). For stimulation by target cells, 1×106 or 2×106 NK cells/ml were cultured with log-phase K562 or K562 “feeder” cells in a ratio of 1:1 overnight. For the generation of pooled supernatant, NK cells were cultured for 3 days with irradiated (30Gy X-ray) K562 “feeder” (K562 “feeder”: NK cells = 1: 2) in presence of 200U/ml IL-2 and 100ng/ml IL-21 (27). Supernatants were harvested, centrifuged and stored at −20°C.
Fibroblasts culture with rIFNɣ and supernatants from activated NK cells
15,000 SFs per 96-well were treated with 100ng/mL recombinant (r)IFNɣ (Peprotech, 300–02), 20% NK cell supernatant or controls [100ng/mL IL-21 (Biosource, PHC0214) and 200 IU/mL IL-2 (Peprotech, 200–02)]. In order to neutralize IFNɣ, 50μg/mL anti-IFNɣ antibody (BD Pharmingen™ 554698) was added to medium containing rIFNɣ or NK cell supernatants 30min before addition to fibroblasts. Upadacitinib or 10μg/mL adalimumab were added to SFs the day before and maintained during the treatment with rIFNɣ or NK-SN. After 3 days, live SFs were analyzed by flow cytometry (supplementary methods).
Co-culture of synovial fibroblasts and CD4+ T cells
Treated SFs were washed, incubated with or without 5µg staphylococcus aureus enterotoxin B (SEB, Sigma-Aldrich, S4881) per ml R10 at 37°C for 30min and washed twice with PBS. 150,000 CD4+ T cells were added overnight and analyzed by flow cytometry (supplementary methods).
Enzyme-linked immunosorbent assay (ELISA)
For the detection of IFNɣ and IL-6 in cell culture supernatants, the following kits were used: Human IFNγ DuoSet® ELISA Development kit (R&D Systems, DY285) and Human IL-6 DuoSet® ELISA Development kit (R&D Systems, DY206).
Western Blot
To measure JAK protein content and phosphorylation, synovial fibroblasts were cultured on 6-well plates for the indicated periods of time and directly lysed using a standard lysing buffer (RIPA) supplemented with a phosphatase inhibitor cocktail (Sigma). JAK protein content and phosphorylation was determined using the Phospho-JAK family antibody sampler kit from Cell Signaling Technology (#97999). Quantification of western blot bands was performed by densitometric analysis with ImageJ2.
FcγR activation reporter assay (25)
BW5147-FcγRIIIA reporter cell line (CD16+ BW5147) and wildtype (parental) BW5147 mouse thymoma cells (BW, obtained from ATCC: TIB-47) were maintained at 3×105 to 9×105 cells/ml in RPMI (RPMI GlutaMAX, Gibco) supplemented with 10% FCS, sodium pyruvate (1x, Gibco) and β-mercaptoethanol (0.1 mM, Gibco). FcγRIIIA activation was measured using a previously described cell-based assay (23). The assay was adapted to measure FcγRIIIA activation in solution. Briefly, 2×105 BW5147-FcγRIIIa reporter cells were incubated with synovial fluid from RA patients in a total volume of 100 μl for 16h at 37°C/5% CO2. Incubation was performed in a 96-well ELISA plate (Nunc Maxisorp) pretreated with PBS/10% FCS for 1h at 4°C. Reporter cell mIL-2 secretion was quantified via ELISA. Recombinant human TNFα (Stem cell technologies) and Infliximab® were mixed in a 1:1 ratio to generate synthetic immune complexes as positive control (Starting dilution = 1:100).
Graphics
Graphics were created with BioRender.com.
Statistics
Exploratory statistical analysis was performed using Graph Pad Prism (versions 5 and 8). p values < 0.05 were considered significant and have to be interpreted descriptively. If not stated otherwise, normal distribution was not assumed and tests were two-sided. The specific tests used are indicated in the figure legends. *, ** and *** in graphs represent p values of <0.05, <0.01 and <0.001, respectively.
Study approval
Written informed consent was received from participants prior to inclusion in the study. The local institutional review boards approved this study (ethics committee of the University of Heidelberg). Patient data and materials were treated in a pseudonymized way.
RESULTS
HLA-DR+CD90+ SFs are manifold expanded in RA and possess a strong IFNɣ signature (5), suggesting that IFNɣ is relevant for their generation. We hypothesized that activated NK cells may be a source of synovial IFNɣ and participate in the development of the inflammatory HLA-DR+CD90+ SF phenotype.
IFNɣ derived from activated NK cells induces HLA-DR on fibroblasts from multiple origins and conditions
We first tested in vitro whether recombinant IFNɣ (rIFNɣ) and soluble factors derived from activated NK cells can induce MHC molecules on human fibroblasts from different origins (healthy and sclerotic skin and joints from osteoarthritis (OA) and RA) in our experimental setup. Both rIFNɣ and supernatant (NK-SN) from IFNɣ-producing NK cells (Suppl. Fig. S1) increased the surface expression of HLA-ABC and induced HLA-DR (Suppl. Fig. S2; Fig. 1). The levels of HLA-DR induced by NK-SN were not different between OA and RA (Suppl. Fig. S2). Once induced, HLA-DR persisted on SFs over at least one week (Fig. 1B). A monoclonal anti-IFNɣ antibody inhibited HLA-DR-induction by rIFNɣ and NK-SN (Fig. 1C). These data confirmed that NK cell-derived IFNɣ induces HLA-DR on fibroblasts from different origins and diseases, including SFs from RA.
Fig. 1. (A-C) Induction of HLA-DR on fibroblasts by recombinant and NK cell-derived IFNɣ.

SFs were cultured over 3 days in medium (w/o) +/− recombinant (r)IFNɣ or pooled NK-SN and analyzed by flow cytometry. FI, fluorescence intensity. (A) Example histograms and percentages of HLA-DR+ RA-SFs, p=0.028. (B) HLA-DR kinetic (n=7 experiments; 3 OA-SFs; 4 RA-SFs); p<0.0001. (C) Effect of anti(α)-IFNɣ antibody on HLA-DR induction. Left: titration, n=1 experiment; middle: IFNɣ concentration in NK-SNs; right: 50μg/ml α-IFNɣ; n=7 experiments with three different RA-SFs, p=0.003. Statistics: Friedman test; significant post-tests are indicated. (D-F) Synovial fluid from seropositive RA activates CD16 (FcγRIIIA). Mouse-CD16+BW5147 reporter cells were cultured with synovial fluid (SFl) from two joints from an active seropositive RA patient or from each one joint from two patients with seronegative joint swelling (seronegative chronic polyarthritis and OA). (D) CD16 expression on BW5147 cells. Empty histograms: autofluorescence. (E) CD16 activation by SFl at a dilution of 1:1200, assessed by mouse-IL-2-ELISA (A450nm) (1 experiment in duplicates). TNFα + infliximab (TNF/Ifx) served as positive control. (F) CD16-activation by titrated SFl (n=3 technical replicates, each performed in triplicates). Three-way ANOVA confirmed significant effects of dilution and seropositivity (p<0.0001 and p=0.002). Bars: means +/− standard deviations.
Synovial immune complexes bind to and activate CD16
NK cells secrete IFNɣ in response to cytokines, target cells or triggering FcɣRIIIA(CD16) (Suppl. Fig. S1). We hypothesized that synovial immune complexes may bind to CD16 and thus contribute to NK cell activation. Leveraging a reporter model for CD16 stimulation (25), we found that synovial fluid from seropositive RA contains immune complexes that can be recognized by CD16 (Fig. 1D-F). In contrast, synovial fluid from seronegative patients did not trigger CD16. Therefore, immune complexes may contribute to CD16+ NK cell activation in seropositive RA.
Activated NK cells reside in synovial tissues from RA patients
We detected by flow cytometry that about 80% of synovial fluid NK cells expressed the activation marker CD69 in a bright fashion (Fig. 2A). To investigate the presence of NK cells in synovial tissue, we analyzed mass cytometry data derived from 15 OA and 26 RA patients (Suppl. Table 1) in the AMP-RA trial (5) (Fig. 2B-D). Using the unbiased SPADE algorithm, a cell cluster that was CD16 positive and negative for all other lineage markers emerged, being most likely CD16+ NK cells (Fig. 2B,D, Suppl. Fig. S3). We confirmed the finding of CD16+ NK cells in synovia with a manual gating strategy (Fig. 2C). The number of NK cells in relation to fibroblasts and the expression of CD69 on NK cells were increased in leukocyte-rich RA (Fig. 2C,D). These data showed the presence of CD16+ NK cells in synovial tissues, and indicated activation of NK cells in RA.
Fig. 2. Activated NK cells in synovial tissues.

(A) Flow cytometry analysis of synovial NK cells (CD45+CD3−CD56+CD19−CD14− lymphocytes). Left: gating strategy, right: percentage of CD69bright NK cells. (B-D) Mass cytometry data from synovial tissues from n=26 RA and n=15 OA patients. (B) Unbiased clustering with SPADE. Colors represent CD16 expression. Clustering channels are shown in Suppl. Fig. S3. Each one representative sample from leukocyte-rich or -poor RA and OA are shown. (C) Leveraging manual gating strategies, we defined leukocyte-poor RA (percentages of CD45+ live cells were similar to OA; n=17) and leukocyte-rich RA (percentages higher than the upper range of OA + 1 standard deviation; n=9) (left), identified HLA-DR+CD90+ cells among CD45-podoplanin(PDPN)+ fibroblasts (middle) and determined the ratio of NK cell / fibroblast counts (right). (D) CD69-positive CD16+ NK cells. Left: manual gating strategy; donors with less than twenty CD16+NK cells were excluded (n=9, 8 and 9 for OA, leukocyte-poor and -rich RA). CD69 on CD16+NK cells was controlled and visualized by SPADE; right: shows sections from clustering trees shown in (B) containing CD16+ NK cells. Box and whiskers: median and range. Significance was determined using Kruskal-Wallis test (p<0.003, in C and D, respectively); significant post tests are indicated.
Increased HLA-DRB1 expression is restricted to synovial fibroblasts in leukocyte-rich RA
Given the presence of activated NK cells in synovia and their potential to produce IFNɣ and to induce HLA-DR, we were interested in general changes of HLA-DR expression in RA. While several cell types intrinsically express HLA-DRB1, the gene with the highest risk association in RA (3), we investigated whether cell-type specific expression patterns of HLA-DRB1 are skewed in RA synovia. We re-analyzed RNAseq data from sorted synovial tissue T cells, B cells, monocytes and SFs (5). HLA-DRB1 RNA was increased in SFs from leukocyte-rich RA, compared to leukocyte-poor RA and OA (Fig. 3A). In contrast, HLA-DRB1 was unaltered in T cells, B cells and monocytes.
Fig. 3. HLA-DRB1 is upregulated on fibroblasts in leukocyte-rich RA.

(A) HLA-DRB1 RNA expression profiles from sorted populations of T cells, B cells, monocytes and synovial fibroblasts (methods). Log2 fold change in expression of HLA-DRB1 RNA in leukocyte-poor (poor) and leukocyte-rich (rich) RA compared to osteoarthritis (OA). N for each cell type in the order of OA, RA poor and RA rich: T cells = 14; 13; 15. B cells = 7; 6; 12. Monocytes = 13; 14; 16. Fibroblasts = 12; 15; 14. Statistical analysis using Kruskal-Wallis test revealed significance only in the fibroblasts group (p=2.7×10−5). Post tests: ***, p=2.5×10−6; #, p=5.3×10-5. (B, C) Protein expression of HLA-DR in the mass cytometry dataset introduced in fig. 3 (manual gating). Left graphs: Median fluorescence intensities (FI) of HLA-DR on CD45+CD14+ monocytes and CD45-PDPN+ synovial fibroblasts between patient groups (Fig. 3). Kruskal-Wallis test revealed significance in the fibroblasts group. Significant post tests are indicated by stars. ns, not significant. Right XY graphs: Median HLA-DR FI on monocytes and fibroblasts as well as percentage of HLA-DR+ fibroblasts in relation to the percentage of CD69+CD16+ NK cells. r, Spearman’s correlation coefficient. Percentages of HLA-DR+ monocytes are not shown (all cells were positive).
In line, HLA-DR protein was significantly increased on SFs, but not on monocytes, in the mass cytometry dataset. Furthermore, HLA-DR protein on SFs correlated with the percentage of activated (CD69+)CD16+ synovial NK cells (Fig. 3B, C).
These observations linked two important pathogenicity factors in RA: MHC-II/HLA-DRB1 (genetic risk) and inflammatory fibroblasts. They further emphasized the question about the mechanism of HLA-DR induction on SFs, and indicated interactions of SFs with IFNɣ-producing cells in leukocyte-rich RA. This prompted us to investigate NK cell/RA-SF interactions in more detail.
Activated NK cells induce inflammatory HLA-DR+CD90+ synovial fibroblasts
To investigate the direct effect of activated NK cells on SFs, we co-cultured NK cells and SFs in vitro. It is known that SFs lose their surface MHC-II (8) and positional identity (28) after only a few passages in vitro. Instead, cultured RA-SFs converge to a homogenous mixed phenotype (28). We therefore determined surface expressions of HLA-DR and CD90, a surrogate of positional identity (28), by flow cytometry. Our cultured SFs consistently expressed CD90 but lacked HLA-DR (Fig. 4A, B), indicating that CD90 and positional identity may be differently regulated than HLA-DR. In monocultures with the NK cell-activating cytokine IL-2, surface markers on CD90+ SFs were unchanged, including HLA-DR, HLA-ABC, CD54 (ICAM1) and CD90 (Fig. 4A-D and Suppl. Fig. S4). In contrast, addition of NK cells was associated with increased levels of CD54 and HLA-ABC and with a de-novo induction of HLA-DR (Fig. 4A-D). Furthermore, we detected IFNɣ in the supernatant of IL-2-treated co-cultures, which was most likely produced by NK cells (Suppl. Fig. S1).
Fig. 4. Induction of HLA-DR and IL-6 in CD90+ synovial fibroblasts by activated NK cells in co-cultures.

(A-D) Synovial fibroblasts (SFs) from 3 OA patients were cultured in the absence or presence of IL-2-activated NK cells. The surface expressions of CD90, HLA-DR, HLA-ABC and CD54 (ICAM1) were analyzed by flow cytometry. Example histograms and means of median fluorescence intensities (FI) or means of percentages of positive cells and standard deviations are shown (n=3 identical experiments). Day 0: Untreated SFs. Statistical analysis: two-way ANOVA. *P<0.05, **P<0.01, ***P<0.001. Autofl.: autofluorescence. (E) Intracellular staining of IL-6 in OA-SF after direct co-culture with NK cells over three days. One of two similar experiments is shown. (F) For quantification of secreted IL-6, OA-SF and RA-SF (n=3 each) were cultured for 3 days +/− 20% pooled NK-SN. Supernatants were analyzed by ELISA. Significance was confirmed by Mann Whitney test (p=0.0022). In the same ELISA run, relevant amounts of IL-6 in NK-SN were excluded (n=6).
Moreover, SFs co-cultured with IL-2-activated NK cells produced IL-6, as determined by intracellular flow cytometry (Fig. 4E). IL-6 secretion was confirmed by treating SFs with NK-SN (Fig. 4F).
In order to further evaluate the inflammatory character of HLA-DR+CD90+ SFs, we established a co-culture system in which SFs were first treated with NK-SN and then co-cultured overnight with freshly isolated CD4+ T cells from healthy donors. Significantly more T cells expressed the activation marker CD69 when co-cultured on SFs that were pre-treated with NK-SN (Suppl. Fig. S5).
We next wanted to know whether surface MHC-II on HLA-DR+CD90+ SFs is stable and potentially functionally relevant. We found that crosslinking NK-SN-induced MHC-II with T cell receptors through superantigen staphylococcus aureus enterotoxin B (SEB) up-regulated the activation marker CD69 on CD4+ T cells (Suppl. Fig. S5, Fig. 5). We have chosen this approach to circumvent issues secondary to the allogenic experimental design. This surrogate of CD4+ T cell activation was not observed when SFs were treated with NK-SN in presence of monoclonal anti-IFNɣ antibody. In contrast, blocking TNFα in NK-SN with adalimumab had no effect on HLA-DR expression and the surrogate of CD4+ T cell activation (Suppl. Fig. S5). These data indicated that NK cell-derived IFNɣ induces MHC-II on SFs in sufficient quantity and stability to activate CD4+ T cells by means of superantigens. In line, when summarizing all SEB experiments, the MHC-II expression on SFs correlated with the percentage of CD69+ CD4+ T cells in presence of SEB (Fig. 6).
Fig. 5. Effect of upadacitinib on the induction of pro-inflammatory HLA-DR+CD90+ RA synovial fibroblasts by NK cell-derived IFNɣ.

(A) Relative mRNA expression of JAK1/2/3 in SFs in vivo. Heat map displaying log2 transformed transcripts per million of JAK1/2/3 in sorted SFs from OA, leukocyte-poor and leukocyte-rich RA (n=12;15;14) (methods). (B-F) SFs were cultured with pooled NK cell supernatant (NK-SN) +/− anti-IFNɣ or JAK1-inhibitor upadacitinib, and subsequently incubated with staphylococcus aureus enterotoxin B (SEB). After washing, freshly isolated CD4+ T cells were added overnight. CD69 as a correlate of CD4+ T cell activation was analyzed by flow cytometry. (B) Schematic experimental design. (C) HLA-DR fluorescence intensities (FI) on SFs depending on upadacitinib concentrations (n=2 RA and n=3 OA for 4µM; n=3 RA and n=4 OA in remaining concentrations). Recombinant (r)IFNɣ and anti(α)-IFNɣ (50μg/ml) served as controls. Statistics: Kruskal-Wallis test, significant post tests are indicated. (D-F) In n=6 parallel experiments, SFs from 6 OA and 4 RA patients were treated with NK-SN +/−4μM upadacitinib (Upa.). (D) Representative dot plots showing CD4+ T cells. SSC, sideward scatter. (E, F) HLA-DR on SFs (left); CD69 on CD4+ T cells (right). Groups of biological interest were compared using Wilcoxon test. *, p<0.05; ns, not significant.
Fig. 6. HLA-DR+CD90+ rheumatoid arthritis fibroblasts induced by NK-SN possess an intrinsically enhanced capacity to stimulate CD4+ T cells.

(A-D) For a summarizing statistical analysis and direct comparison of the effect of OA and RA SFs on CD4+ T cells, we pooled data derived from all series of SEB experiments with identical conditions (n=12). (A, B) percentage of CD69-positive CD4+ T cells after co-culture with OA-SF and RA-SF under variable conditions. (C, D) Direct comparison of the effect of OA-SF and RA-SF on CD4+ T cell activation in absence of SEB. (D) Percentage of CD69+ CD4+ T cells in relation to HLA-DR expression on OA-SF and RA-SF that were pre-treated with pooled NK-SN (OA: n=11, the outlier in C was excluded without relevant alteration of statistical results, r=0.160, p=0.6. RA: n=12, r=0.165, p=0.6). In A, B and C, Wilcoxon test was used to compare selected groups of biological interest. ***, p<0.001. (E, F) Pooled data across all conditions derived from all experiments in which the median fluorescence intensities (FI) of HLA-DR on SFs and the percentage of CD69-positive CD4+ T cells after co-culture overnight were determined in parallel. Correlation analyses were performed using Spearman’s test (r).
Together, these data showed on the protein level that NK cells induce an inflammatory, cytokine secreting, T cell activating, HLA-DR+ phenotype in CD90+ SFs.
IFNɣ-induced HLA-DR on synovial fibroblasts relies on JAK signaling
The IFNɣ receptor chains 1 and 2 intracellularly bind to JAK1 and JAK2 which activate STAT1 to dimerize and act as transcription factor (29). We queried expression levels of these IFNɣ pathway transcripts in the AMP-RA database (methods). The receptor chains were unaltered in RA (not shown). Almost all RA-SFs expressed JAK1 (about 90% in most subsets). JAK2 was significantly increased in leukocyte-rich RA as compared to leukocyte-poor RA and OA (p=2e-05), with the numerically highest percentage of positive cells in the HLA-DR+CD90+ subset (62.9%). The remaining JAK3 and TYK2 were expressed in <20% and 31.2 to 50.1% of SFs, respectively. STAT1 was increased in leukocyte-rich RA (p=4e-05), and 91% of HLA-DR+CD90+ SFs expressed STAT1. We confirmed the dominating expression of JAK1 mRNA over JAK2 and JAK3 mRNA by re-analyzing AMP-RA bulk RNAseq raw data (Fig. 5A). These data indicated unaltered or increased signaling capacities of the IFNɣ pathway in HLA-DR+CD90+ SFs in vivo, at least on the mRNA level.
JAK-inhibition is a new and effective treatment strategy in RA. The approved JAK inhibitors tofacitinib, baricitinib and upadacitinib possess a relative specificity for JAK1&3, JAK1&2 and JAK1, respectively. Upadacitinib had a significant and dose-dependent inhibitory effect on HLA-DR on SFs induced by both rIFNɣ and NK-SN (Fig. 5C-F). Similar to the blocking anti-IFNɣ antibody, treatment of SFs with upadacitinib prevented CD4+ T cell activation by SEB (after stimulation with NK-SN) (Fig. 5E, F). Tofacitinib and baricitinib also tended to inhibit NK-SN-induced HLA-DR-induction on SFs (Fig. S6).
Because of the increased mRNA levels of JAK2 in vivo in leukocyte-rich RA (see above), we stimulated RA synovial fibroblasts with NK-SN and measured JAK2 protein and its phosphorylation by western blot. We found that JAK2 protein was increased after stimulation over 3 days. JAK2 was phosphorylated after 30min of stimulation, but not after continued stimulation over 3 days (Fig. S6).
Together, NK-SN stimulated the JAK1/2 pathway. The induction of HLA-DR and the inflammatory character of SFs by NK cell-derived IFNɣ could be blocked by the JAK inhibitor upadacitinib.
HLA-DR+CD90+ RA-SFs possess an intrinsically enhanced capacity to stimulate CD4+ T cells
Interestingly, we observed that SFs treated with NK-SN can induce CD69 on CD4+ T cells also in absence of SEB (Fig. 6). This effect was about four times stronger in RA compared to OA. The allogenic setting may contribute to this T cell activation, but alloreaction does not explain differences between RA and OA. Induced HLA-DR levels were comparable in RA and OA (e.g., Fig. 5). The SEB-independent CD69 up-regulation on CD4+ T cells was initiated by NK-cell-derived IFNɣ and could be blocked by anti-IFNɣ and upadacitinib (Figs. 6B and 5F).
Together, NK cell-derived IFNɣ boosted the crosstalk between RA-SFs and CD4+ T cells. This inflammatory boost could be prevented by JAK blockade.
DISCUSSION
SFs are an attractive therapeutic target in RA. A recent resource study identified SF subsets in RA (5). HLA-DR+CD90+ SFs are expanded and characterized by a pro-inflammatory phenotype and a strong IFNɣ signature. These cells were suggested to be major producers of IL-6 and expressed genes that were involved in antigen processing and presentation. However, some of these findings have not yet been verified on the protein level. The signals relevant for the generation of HLA-DR+CD90+ SFs have not been validated in functional assays.
To address these issues, we combined focused analyses of mass cytometry and transcriptomics patient datasets with a series of functional assays on human primary cells in vitro. Our study confirms on the protein level that IFNɣ induces an inflammatory HLA-DR+CD90+ phenotype in SFs. This includes the expression of HLA-DR and IFNɣ-induced genes like ICAM1, and the secretion of IL-6. Importantly, we show that HLA-DR+CD90+ SFs possess a functional pro-inflammatory phenotype by physically interacting with cells from the adaptive immune system.
We hypothesized that NK cells may provide IFNɣ to SFs. The mass cytometry analyses showed that CD16+ NK cells are physically present in synovial tissue and activated in active RA. Even though CD16+ NK cells include only one of two major NK cell subsets, these cells may be of importance in seropositive RA, given that CD16 can be triggered by local immune complexes. CD16 triggers IFNɣ secretion by NK cells, which may be enhanced by an inflammatory cytokine milieu (24). Evidence of synovial (CD16+) NK cells was also supplied by studies with smaller sample sizes applying bulk RNA sequencing with CIBERSORTX (19) or using clustering strategies of single cell-RNA sequencing data to detect NK cells (18).
Indeed, NK cell-derived IFNɣ induces HLA-DR on CD90+ SFs in vitro. Our findings indicate that activated NK cells induce a global pro-inflammatory fibroblast phenotype characterized by HLA-DR+CD90+IL6+ICAM1+HLA-ABCbright,JAK2bright cells possessing an inflammatory character and (per its activation stimulus) an IFNγ signature that resembles the phenotype of HLA-DR+CD90+ SFs in vivo (5). Thus, the in vivo RNA expression profile of HLA-DR+CD90+ SFs is likely related to an inducible, functional phenotype with pro-inflammatory character, consistent with findings in mouse models (30).
It is important to consider NK cells as providers of IFNɣ despite potential redundancy with CD8+ T cells (5). Both cell types secrete IFNɣ, but the stimuli for IFNɣ secretion are different. Therefore, both cell types may be important depending on environmental conditions. The exact triggers of NK cell activation in vivo remain poorly characterized, but we show that immune complexes within synovial fluid from seropositive RA can stimulate CD16 signaling in our reporter model. CD16 is a potential risk gene for RA (20, 31). These data sustain a role for immune complexes in RA (32–34), and point towards immune complex-mediated activation of NK cells. Cytokines and direct cell-cell interaction involving innate NK cell receptor ligands may also contribute to activation of synovial NK cells, and most likely a combination of these factors is found in vivo. We suggest that immune-complex-activated NK cells may contribute to the clinically more severe disease course in seropositive compared to seronegative RA. Despite these functional considerations, our study does not allow statements on the quantitative contribution of NK cells to synovial IFNɣ in RA.
Apart from being a suitable surface marker to distinguish HLA-DR+CD90+ SFs from other SFs subsets (5), we show that surface MHC-II on SFs is stable, enabling superantigen presentation (see also (11)). Even though our study did not investigate autoantigen presentation or antigen-specific T cell reactions, the finding of NK-SN-induced stable MHC-II on SFs is important, as it has been shown by other groups that SFs can present (auto)antigens via MHC-II (10–12). It is difficult to quantify antigen presentation by SFs in comparison to other APCs like monocytes. However, SFs are the most numerous synovial cell type in OA and leukocyte-poor RA, and remain an important cell fraction upon leukocyte infiltration, with the subpopulation of HLA-DR+CD90+ SFs being 15-fold increased in active RA (5). We therefore suggest that antigen presentation by SFs may be relevant, the more so as the key MHC-II allele HLA-DRB1 was significantly increased only on SFs, and restricted to active RA.
Further studies sustain a role of local antigen presentation to CD4+ T cells in RA (18). Synovial CD4+ T “peripheral helper” cells (TPH) are expanded in seropositive RA and express activation markers like CD69 (35). TPH strongly express IL-6-receptor and promote B cell differentiation and antibody production (18, 35). Beyond this, not much is known about the impact of SFs on the activation of T cells, even though the release of chemokines and cytokines as well as several membrane-bound molecules like CD54 were involved in the recruitment and activation of CD4+ T cells in RA, pointing towards complex reciprocal interactions (36–39). Together, several lines of evidence indicate a role of HLA-DR+CD90+ SFs in synovial antigen presentation and T cell support in RA. Our study adds that this can be boosted by IFNɣ.
HLA-DR+CD90+ SFs from RA patients possess an intrinsically increased capability to activate CD4+ T cells in response to IFNɣ. While it is well-described that SFs lose certain characteristics during in vitro culture (28), it is also broadly accepted that SFs possess an imprinted aggressive phenotype (40). Epigenetic factors may account for a SFs phenotype “primed for inflammation”, probably paving the way for memory or recall inflammatory responses that may be kicked off by IFNɣ. Together with the study from Wei et al. showing that the positional identity of SFs can be induced by NOTCH signals (28), our data indicate that HLA-DR+CD90+ SFs are rather an activation state than a classic, per se persistent cellular subset. Given the potential central role in RA pathogenesis, blocking the induction of the HLA-DR+CD90+ phenotype may be a promising new, fibroblast-centered therapeutic strategy. So far, it has not been investigated whether HLA-DR+CD90+ SFs can be specifically targeted.
Importantly, our data and another study show that JAK inhibition suppresses the impact of IFNɣ on SFs in vitro (41), including HLA-DR induction and the boosting effect on fibroblast/CD4+ T cell interactions. The link between JAK inhibition and the pathogenic role of IFNɣ in RA has so far been rather hypothetical (42). However, our data suggest that JAK1 inhibition with upadacitinib may block the development of HLA-DR+CD90+ SFs. Vice versa, preventing SFs from acquiring this activation state may be an important, yet underappreciated, mechanism of action of upadacitinib in RA.
Conclusions
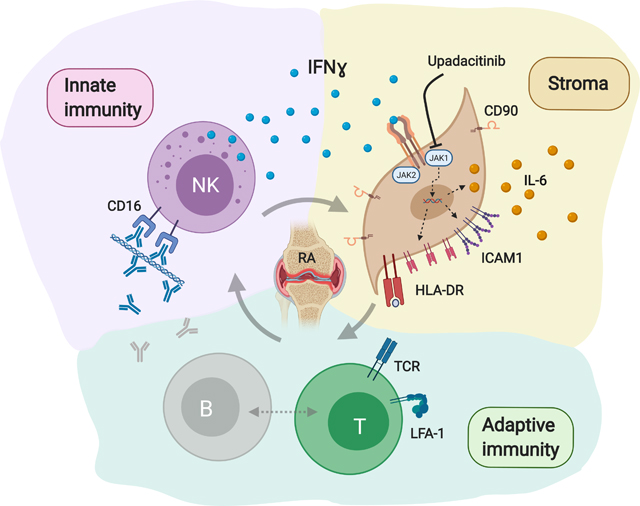
We conclude that HLA-DR+CD90+ SFs are an activation state that can be induced by IFNɣ, potentially provided from tissue resident NK cells. The generation of these pro-inflammatory, IL-6 producing and likely antigen-presenting SFs can be targeted by the JAK1 inhibitor upadacitinib. We propose a pro-inflammatory vicious circle within the complex network of innate, stromal and adaptive immunity in seropositive RA (graphical abstract).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Accelerating Medicines Partnership (AMP) RA/SLE for the generation of publicly accessible data on synovial tissues.
We thank Peter Nigrovic for connecting people, Xin Guan for helping with IL-6 ELISAs, Stefan Krienke and Sabine Wingert for technical support, and Doris Urlaub and Maren Claus for valuable advice.
The study was supported by: Medical Faculty of University of Heidelberg (to W.M. and to R.G.-B.), Eva Luise and Horst Köhler Foundation (to W.M.), German Society for Rheumatology (DGRh, to R.G.-B.), Roche Pharma AG (to W.M.), Third Affiliated Hospital of Nantong University (to S.Z.), NIH K08 AR072791 from NIAMS (to D.A.R.), EQUIP - Funding for Medical Scientists, Faculty of Medicine, University of Freiburg (to P.K.), Deutsche Forschungsgemeinschaft (DFG FOR2830 HE 2526/9–1, to P.K.), NaFoUniMedCovid19 (FKZ: 01KX2021) – COVIM (to P.K.) and Joint Biology Consortium (JBC, funded by National Institute of Arthritis and Musculosceletal and Skin Diseases at NIH, P30 AR070253).
(Disclosures: W.M. has received consulting fees, speaking fees, and/or honoraria from Novartis and Roche (less than $10,000 each), and unallocated funds for research from Roche (15.000€). R.G.-B. received research funding from Gilead for work outside this study. D.A.R. reports personal fees from Pfizer, Merck, Scipher Medicine, and Bristol-Myers Squibb (all <$10,000) and research funding Celgene for work outside the submitted work. H.-M.L. received consultancy fees and/or honoraria for lectures and/or travel reimbursements and/or educational seminars and/or clinical studies from Abbvie, Astra-Zeneca, Actelion, Alexion, Amgen, Bayer Vital, Baxter, Biogen, Boehringer Ingelheim, BMS, Celgene, Fresenius, Genzyme, GSK, Gilead, Hexal, Janssen-Cilag, Lilly, Medac, MSD, Mundipharm, Mylan, Novartis, octapharm, Pfizer, Roche/Chugai, Sandoz, Sanofi, Shire, SOBI, Thermo Fisher and UCB (less than $10,000 each), and support for scientific projects from Pfizer and Novartis (less than $50,000 each/year). L.-O.T. received support for scientific projects from Pfizer (less than $50,000 each/year).)
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors declare no competing financial interests.
Publisher's Disclaimer: This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. Please cite this article as doi: 10.1002/ART.41958
REFERENCES
- 1.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205–19. [DOI] [PubMed] [Google Scholar]
- 2.Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506(7488):376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee HS, Jia X, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet. 2012;44(3):291–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev. 2010;233(1):233–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang F, Wei K, Slowikowski K, Fonseka CY, Rao DA, Kelly S, et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat Immunol. 2019;20(7):928–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chu CQ. Fibroblasts in Rheumatoid Arthritis. N Engl J Med. 2020;383(17):1679–81. [DOI] [PubMed] [Google Scholar]
- 7.Schett G Physiological effects of modulating the interleukin-6 axis. Rheumatology (Oxford). 2018;57(suppl_2):ii43-ii50. [DOI] [PubMed] [Google Scholar]
- 8.Duraes FV, Thelemann C, Sarter K, Acha-Orbea H, Hugues S, Reith W. Role of major histocompatibility complex class II expression by non-hematopoietic cells in autoimmune and inflammatory disorders: facts and fiction. Tissue Antigens. 2013;82(1):1–15. [DOI] [PubMed] [Google Scholar]
- 9.Kambayashi T, Laufer TM. Atypical MHC class II-expressing antigen-presenting cells: can anything replace a dendritic cell? Nat Rev Immunol. 2014;14(11):719–30. [DOI] [PubMed] [Google Scholar]
- 10.Tran CN, Davis MJ, Tesmer LA, Endres JL, Motyl CD, Smuda C, et al. Presentation of arthritogenic peptide to antigen-specific T cells by fibroblast-like synoviocytes. Arthritis Rheum. 2007;56(5):1497–506. [DOI] [PubMed] [Google Scholar]
- 11.Carmona-Rivera C, Carlucci PM, Moore E, Lingampalli N, Uchtenhagen H, James E, et al. Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis. Sci Immunol. 2017;2(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sugawara E, Kato M, Kudo Y, Lee W, Hisada R, Fujieda Y, et al. Autophagy promotes citrullination of VIM (vimentin) and its interaction with major histocompatibility complex class II in synovial fibroblasts. Autophagy. 2020;16(5):946–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9(5):503–10. [DOI] [PubMed] [Google Scholar]
- 14.Yamin R, Berhani O, Peleg H, Aamar S, Stein N, Gamliel M, et al. High percentages and activity of synovial fluid NK cells present in patients with advanced stage active Rheumatoid Arthritis. Sci Rep. 2019;9(1):1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pridgeon C, Lennon GP, Pazmany L, Thompson RN, Christmas SE, Moots RJ. Natural killer cells in the synovial fluid of rheumatoid arthritis patients exhibit a CD56bright,CD94bright,CD158negative phenotype. Rheumatology (Oxford). 2003;42(7):870–8. [DOI] [PubMed] [Google Scholar]
- 16.Dalbeth N, Gundle R, Davies RJ, Lee YC, McMichael AJ, Callan MF. CD56bright NK cells are enriched at inflammatory sites and can engage with monocytes in a reciprocal program of activation. J Immunol. 2004;173(10):6418–26. [DOI] [PubMed] [Google Scholar]
- 17.Dalbeth N, Callan MF. A subset of natural killer cells is greatly expanded within inflamed joints. Arthritis Rheum. 2002;46(7):1763–72. [DOI] [PubMed] [Google Scholar]
- 18.Stephenson W, Donlin LT, Butler A, Rozo C, Bracken B, Rashidfarrokhi A, et al. Single-cell RNA-seq of rheumatoid arthritis synovial tissue using low-cost microfluidic instrumentation. Nat Commun. 2018;9(1):791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orange DE, Agius P, DiCarlo EF, Robine N, Geiger H, Szymonifka J, et al. Identification of Three Rheumatoid Arthritis Disease Subtypes by Machine Learning Integration of Synovial Histologic Features and RNA Sequencing Data. Arthritis Rheumatol. 2018;70(5):690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee YH, Bae SC, Song GG. FCGR2A, FCGR3A, FCGR3B polymorphisms and susceptibility to rheumatoid arthritis: a meta-analysis. Clin Exp Rheumatol. 2015;33(5):647–54. [PubMed] [Google Scholar]
- 21.Bryceson YT, March ME, Ljunggren HG, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood. 2006;107(1):159–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Urlaub D, Zhao S, Blank N, Bergner R, Claus M, Tretter T, et al. Activation of natural killer cells by rituximab in granulomatosis with polyangiitis. Arthritis Res Ther. 2019;21(1):277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corrales-Aguilar E, Trilling M, Reinhard H, Merce-Maldonado E, Widera M, Schaal H, et al. A novel assay for detecting virus-specific antibodies triggering activation of Fcgamma receptors. J Immunol Methods. 2013;387(1–2):21–35. [DOI] [PubMed] [Google Scholar]
- 24.Campbell AR, Regan K, Bhave N, Pattanayak A, Parihar R, Stiff AR, et al. Gene expression profiling of the human natural killer cell response to Fc receptor activation: unique enhancement in the presence of interleukin-12. BMC Med Genomics. 2015;8:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen H, Maul-Pavicic A, Holzer M, Salzer U, Chevalier N, Voll RE, et al. Immune complex solubility and size govern Fc-gamma receptor responses. 2021:2020.11.11.378232.
- 26.Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106(1):376–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prager I, Liesche C, van Ooijen H, Urlaub D, Verron Q, Sandstrom N, et al. NK cells switch from granzyme B to death receptor-mediated cytotoxicity during serial killing. J Exp Med. 2019;216(9):2113–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei KV, Korsunsky I, Marshall JL, Gao AQ, Watts GFM, Major T, et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature. 2020. [DOI] [PMC free article] [PubMed]
- 29.Alspach E, Lussier DM, Schreiber RD. Interferon gamma and Its Important Roles in Promoting and Inhibiting Spontaneous and Therapeutic Cancer Immunity. Cold Spring Harb Perspect Biol. 2019;11(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Croft AP, Campos J, Jansen K, Turner JD, Marshall J, Attar M, et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature. 2019;570(7760):246–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morgan AW, Barrett JH, Griffiths B, Subramanian D, Robinson JI, Keyte VH, et al. Analysis of Fcgamma receptor haplotypes in rheumatoid arthritis: FCGR3A remains a major susceptibility gene at this locus, with an additional contribution from FCGR3B. Arthritis Res Ther. 2006;8(1):R5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nielen MM, van Schaardenburg D, Reesink HW, van de Stadt RJ, van der Horst-Bruinsma IE, de Koning MH, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. 2004;50(2):380–6. [DOI] [PubMed] [Google Scholar]
- 33.Arend WP, Firestein GS. Pre-rheumatoid arthritis: predisposition and transition to clinical synovitis. Nat Rev Rheumatol. 2012;8(10):573–86. [DOI] [PubMed] [Google Scholar]
- 34.Pfeifle R, Rothe T, Ipseiz N, Scherer HU, Culemann S, Harre U, et al. Regulation of autoantibody activity by the IL-23-TH17 axis determines the onset of autoimmune disease. Nat Immunol. 2017;18(1):104–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature. 2017;542(7639):110–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoshitomi H Regulation of Immune Responses and Chronic Inflammation by Fibroblast-Like Synoviocytes. Front Immunol. 2019;10:1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bombara MP, Webb DL, Conrad P, Marlor CW, Sarr T, Ranges GE, et al. Cell contact between T cells and synovial fibroblasts causes induction of adhesion molecules and cytokines. J Leukoc Biol. 1993;54(5):399- [DOI] [PubMed] [Google Scholar]
- 38.Haynes BF, Grover BJ, Whichard LP, Hale LP, Nunley JA, McCollum DE, et al. Synovial microenvironment-T cell interactions. Human T cells bind to fibroblast-like synovial cells in vitro. Arthritis Rheum. 1988;31(8):947–55. [DOI] [PubMed] [Google Scholar]
- 39.Tykocinski LO, Lauffer AM, Bohnen A, Kaul NC, Krienke S, Tretter T, et al. Synovial Fibroblasts Selectively Suppress Th1 Cell Responses through IDO1-Mediated Tryptophan Catabolism. J Immunol. 2017;198(8):3109–17. [DOI] [PubMed] [Google Scholar]
- 40.Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. 2013;9(1):24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karonitsch T, Beckmann D, Dalwigk K, Niederreiter B, Studenic P, Byrne RA, et al. Targeted inhibition of Janus kinases abates interfon gamma-induced invasive behaviour of fibroblast-like synoviocytes. Rheumatology (Oxford). 2018;57(3):572–7. [DOI] [PubMed] [Google Scholar]
- 42.Kato M New insights into IFN-gamma in rheumatoid arthritis: role in the era of JAK inhibitors. Immunol Med. 2020;43(2):72–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.