

Abstract
Merkel cell carcinoma (MCC) is a rare neuroendocrine carcinoma. The cellular origin of MCC may include Merkel cell precursors. The incidence of MCC has increased significantly however trends may have been confounded by evolving diagnostic criteria. The two key aetiologies of MCC are ultraviolet radiation and Merkel cell polyoma virus (MCPyV). Both have unique mechanisms of carcinogenesis. MCC presents non‐specifically as a rapidly growing, red‐to‐violet nodule on sun‐exposed areas. Diagnostic accuracy has improved through immunohistochemical markers such as CK‐20. Lymph nodes should be evaluated in MCC through examination and sentinel biopsy. USS, CT, MRI and CT‐PET may be useful in staging. Management depends on tumour location, stage and comorbidities. MCPyV status may guide treatment strategy in the future. Treatment for the primary MCC is commonly wide local excision followed by radiotherapy, guided by anatomical constraints. There is uncertainty about surgical margins. Treatments for nodal disease have not been determined through trials. They include nodal dissection or radiotherapy for clinically or radiologically apparent disease, and adjuvant nodal irradiation for negative nodes, microscopic disease or following nodal dissection for definite disease. Patients with loco‐regional advanced inoperable disease should be considered for combination therapy including chemotherapy, radiotherapy, surgery and immunotherapy. Systemic therapy for advanced disease includes immune checkpoint inhibitors targeting the PD‐1/PD‐L1 pathway. Avelumab can improve survival in metastatic MCC. Immunotherapy may result in longer disease control. Various other immunotherapeutic and molecular agents are undergoing trials. MCC continues to have a high mortality characterized by high recurrence and early metastases.
Merkel cell carcinoma (MCC) is a rare neuroendocrine carcinoma. MCC continues to have a high mortality characterised by high recurrence and early metastases. Diagnostic accuracy has improved through immunohistochemical markers such as CK‐20.
1.
What's already known about this topic?
Merkel cell carcinoma (MCC) is a rare neuroendocrine carcinoma. The two key aetiologies are ultraviolet radiation and Merkel cell polyoma virus (MCPyV). Both have unique mechanisms of carcinogenesis.
MCC presents non‐specifically as a rapidly growing, red‐to‐violet nodule on sun‐exposed areas.
Treatment for the primary MCC is commonly wide local excision followed by radiotherapy. Patients with loco‐regional advanced inoperable disease should be considered for combination therapy including chemotherapy, radiotherapy, surgery and immunotherapy.
What does this study add?
The incidence of MCC has increased significantly however trends may have been confounded by evolving diagnostic criteria. Diagnostic accuracy has improved through immunohistochemical markers such as CK‐20.
Management depends on tumour location, stage and comorbidities. MCPyV status may guide treatment strategy in the future.
Systemic therapy for advanced disease includes immune checkpoint inhibitors targeting the PD‐1/PD‐L1 pathway. Avelumab can improve survival in metastatic MCC.
2. INTRODUCTION
This review article presents a concise update of knowledge in these areas relevant to the clinician. This review will discuss the pathogenesis, epidemiology, diagnosis, staging, treatment and prognosis of Merkel cell carcinoma (MCC).
3. PATHOGENESIS
MCC is a rare, aggressive neuroendocrine carcinoma first described in Baltimore, USA by Cyril Toker in 1972. 1 Merkel cells are postmitotic neuroendocrine cells in the basal epidermis which secrete amine and polypeptide hormones. Despite the name, it is unlikely that MCC are derived from Merkel cells themselves. Merkel cells function mainly as touch receptors and the common sites of MCC do not correlate with the common locations of Merkel cells. However, MCC does share much of its immunophenotype and ultrastructure with Merkel cells such that it could be regarded as differentiating towards the Merkel cell phenotype, presumably acquired during carcinogenesis from a precursor cell or cells. 2 Candidates for the MCC cell of origin include Merkel cell precursors, epithelial progenitors, dermal mesenchymal stem cells, fibroblasts, or even pro‐B or pre‐B cells. 2
The two main identified contributors to carcinogenesis are ultraviolet radiation (UVR) and Merkel cell polyoma virus (MCPyV). MCPyV is a ubiquitous commensal skin microbiota also carried in other organs and peripheral blood. 3 It is carried by up to 80% of adults. 3 A landmark study detected MCPyV in 80% of MCC, although the incidence is lower in other studies perhaps due to an increased proportion of MCC secondary to high UVR exposure. 3 , 4 Clonal integration of MCPyV DNA into the genome may drive carcinogenesis. UVR causes immunosuppression and mutagenesis of regulatory genes such as TP53, MYC‐L and RB1. 5 , 6 UVR may compound viral carcinogenesis through immunosuppression. 5
Recent studies have suggested VP‐MCC and VN‐MCC may arise from different cells of origin. 7 , 8 , 9 VP‐MCC from dermal fibroblasts and VN‐MCC from epidermal keratinocytes. 7 If true, MCC may represent the first malignancy which evolves from cells of origin from two distinct germ layers. Future epigenetic studies may help confirm these distinct lineages. Understanding of the origin, mutational landscape and complex interactions of MCC with the tumour microenvironment has driven the development of targeted immunological and molecular therapies and may give further insight into pathogenesis and therapeutic options.
4. EPIDEMIOLOGY
Knowledge of the epidemiology of MCC is improving but older data are lacking in this rare cancer. The Surveillance of Rare Cancers in Europe (RARECARE) database reported an incidence of 0.13 per 100 000 person‐years between 1995 and 2002. 10 The highest age‐standardized incidence rate globally was reported in Australia between 2012 and 2016 of 2.5 per 100 000. 11 The highest incidence in Europe was reported in a regional UK study between 2004 and 2013 of 1.78 per 100 000 person‐years. 9 This was 12‐fold higher than the previously reported UK mean. 12 By comparison, melanoma was found to be 33‐fold more frequent, but half as fatal as MCC. 13 , 14
Controversy remains about actual incidence rates due to evolving diagnostic criteria, improved cancer registration, increased awareness by clinicians, greater availability of diagnostic markers, disease rarity and considerable variation in incidence between developed countries with majority populations of less pigmented skin types. 12 Since first described in 1972, no epidemiological studies on MCC were reported until 1980. 15 In 1986, MCC was first allocated a histological code. 15 From 1992 onwards, cytokeratin‐staining and immunological profiling was increasingly utilized which helped differentiate MCC. 15 Currently, most cancer registries use the International Classification of Diseases (ICD) 10 coding, which does not have a specific MCC code. MCC is currently coded by ICD‐11, ICD‐oncology‐third edition and some versions of SNOMED. Recent MCC coding expansion may have driven the increased accuracy of epidemiological studies, incorporating data from population‐based registries. 16 Comparison between countries may be unreliable because different studies use different measures (crude rates or age‐standardized rates with different age standards). 17 In addition, studies differ in relation to topographic localizations of MCC, for example MCC with unknown primary, that are excluded from their analyses. 17
Known risk factors include history of UVR, MCPyV, immunosuppression (HIV, transplant, medications and haematological malignancies such as chronic lymphocytic leukaemia), white ethnicity, chronic arsenic exposure, concomitant other malignancies (such as squamous cell carcinoma) and chronic inflammatory disorders. 12 , 18 The median age at diagnosis is approximately 76 years and MCC tends to affect men twice more than women although few small cohort studies reported higher incidence in women. 13 , 19 , 20 MCC is roughly 25 times more prevalent in fair‐skinned individuals. 15 Immunocompromised individuals have earlier onset and higher mortality from MCC. 21
5. DIAGNOSIS
MCC presents as an irregular, red‐to‐violet cutaneous or subcutaneous nodule (Figure 1). 22 The important features of MCC can be condensed in the acronym ‘AEIOU’, which stands for asymptomatic/absence of tenderness; expanding rapidly; immunosuppression; older than age 50; UV (exposed site fair‐skinned individuals). 22 However, alternative presentations have been documented, including cases of the intraepidermal variant of MCC presenting as an erythematous scaly plaque. 9 The non‐specific presentation of MCC may lead to delayed clinical diagnosis. Common differential diagnoses include amelanotic melanoma, squamous cell carcinoma, cutaneous lymphoma, cutaneous metastasis or benign lesions such as cysts.
FIGURE 1.
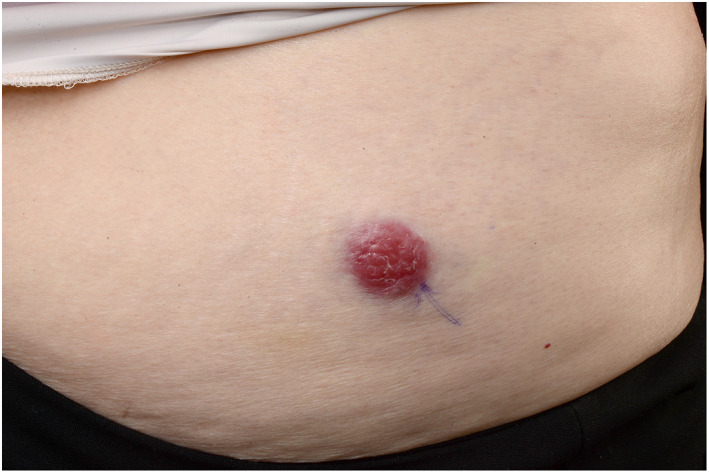
Macroscopic image of a Merkel cell carcinoma showing an erythematous nodule on the right lower abdomen
Due to the wide clinical differential diagnosis, a diagnosis of MCC relies on biopsy and a thorough full body examination of the skin and all lymph nodes. Suggestive features of MCC under dermoscopy include poorly focused vessels, polymorphous vascular pattern with architectural disruption, milky‐red areas on a white sheen, and large calibre arborizing vessels (Figure 2). 23
FIGURE 2.
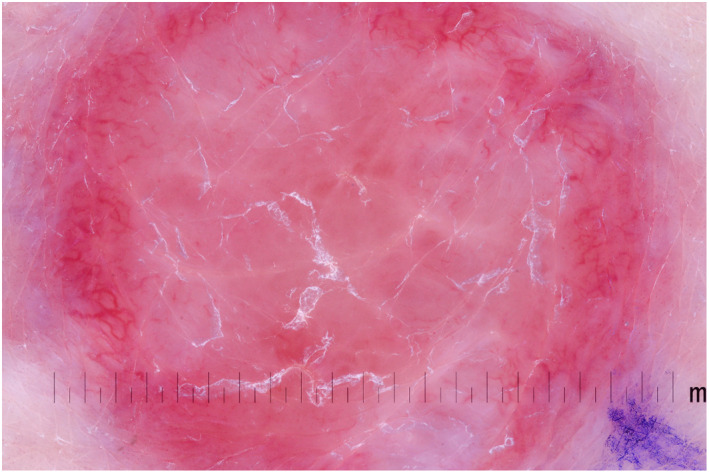
Dermoscopic image of a Merkel cell carcinoma showing a structureless central area with pink and white areas within, peripheral polymorphous and poorly focused vessels are also seen
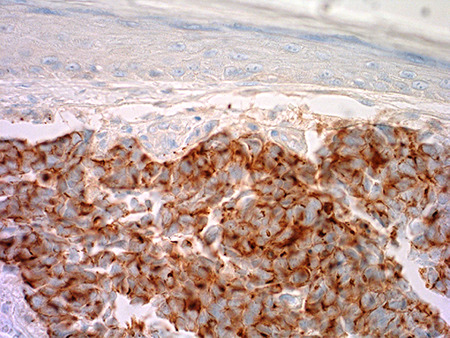
Tissue sampling is undertaken by either punch or full‐thickness incisional biopsy of the skin. Histological diagnosis requires knowledge of the entity and may be more readily made by specialist dermatopathologists. 24 MCC stained with haematoxylin & eosin (Figure 3a) reveals a ‘small‐blue‐round‐cell tumour’ composed of dermal and/or subcutaneous nodules or sheets of small, closely packed, monomorphic, round‐to‐oval basaloid cells with a vesicular nucleus, finely granular chromatin pattern and scanty cytoplasm. 25 , 26 Numerous mitotic figures and apoptotic/necrotic cells are common. Morphology of the MCC component is very similar in combined VN‐MCC tumours, most commonly in association with a squamous cell carcinoma component (Figure 3b). The morphological differential diagnosis of small blue round cell tumour is wide but can be easily sorted by immunohistochemistry. 26 Perinuclear staining with cytokeratin 20 (CK‐20) (Figure 3c) has approximately 95% sensitivity and 60% specificity. 27 , 28 The identification and mainstream production of CK20 as an MCC marker has improved MCC identification since at least 1997. 29 Neurofilament protein (Figure 3d) has lower sensitivity but good specificity and is used to detect CK‐20 negative and VN‐MCC, with 96.7% specificity. 28 Positive immunohistochemistry for MCPyV in VP‐MCC is also a useful diagnostic marker with reliable results using the MCPyV large T‐antigen Antibody (CM2B4) from Santa Cruz Biotechnology (USA). 30 This mouse monoclonal IgG2b (kappa light chain) is raised against large T/57kT exon 2 peptides of MCPyV; however, it is not widely available in many countries. Pathology reporting of MCC is being standardized internationally including in the 2019 first edition of the International Collaboration on Cancer Reporting (ICCR), MCC Histopathology Reporting Guide. 31
FIGURE 3.
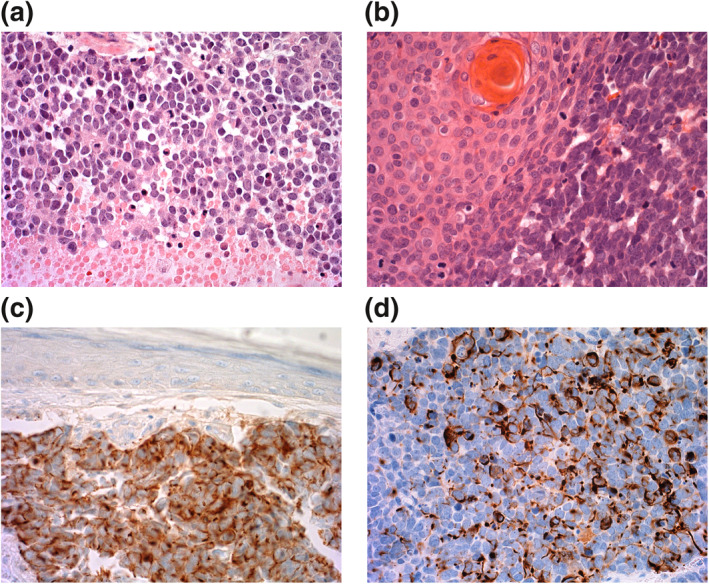
(a) Merkel cell carcinoma (MCC) showing mitotic activity and necrosis of the bottom quarter of the field. H&E ×400. (b) Combined VN‐MCC with MCC component on the left and squamous cell carcinoma component on the right. (c) MCC cytokeratin 20 immunohistochemistry positive (brown cytoplasmic staining) with negative epidermis above. (d) MCC neurofilament protein immunohistochemistry positive (brown cytoplasmic staining)
6. STAGING
Staging may aid diagnosis by ruling out other sources of primary neuroendocrine carcinoma, although MCC is by far the most common type to be biopsied in the skin. Cutaneous metastases of other neuroendocrine carcinomas are rare and likely to be end stage with known primary disease elsewhere. Clinically palpable nodes should be assessed by ultrasound and core biopsy or fine needle aspiration. 32 In those without clinical lymphadenopathy, sentinel lymph node biopsy should be considered because almost one third of patients have occult nodal involvement. 32 It is unclear whether SLNB confers increased disease‐specific survival with conflicting reports in the literature. 33 , 34 CT, MRI and PET‐CT imaging have become increasingly integrated for staging patients who cannot tolerate or decline SLNB, to identify distant metastasis and for surveillance, however imaging cannot substitute SLNB. 35 , 36 The most recent staging system is the American Joint Committee on Cancer consensus (AJCC) staging system updated in 2018 (Table 1). 20 , 37 However, Union for International Cancer Control (UICC) TNM 8 (Table 2), rather than AJCC TNM 8, has been selected by the Royal College of Pathologists, UK, for pathological staging in the UK because this provides TNM staging of the entire skin surface for cutaneous carcinoma compared with only the head and neck in AJCC 8. 38 UICC TNM 8 stage's I and II are no longer characterized by SLNB negativity (A) or not done (B), instead the A/B suffix relate to T stage. 38 Therefore, SLNB has been deprioritised in current staging.
TABLE 1.
American Joint Committee on Cancer consensus (AJCC) staging system of Merkel cell carcinoma 2018
| Stage | Primary tumour | Lymph node | Metastasis |
|---|---|---|---|
| 0 | In situ (within epidermis only) | No regional lymph node metastasis | No distant metastasis |
| I Clinical a | ≤2 cm maximum tumour dimension | Nodes negative by clinical exam (no pathological exam performed) | No distant metastasis |
| I Pathological b | ≤2 cm maximum tumour dimension | Nodes negative by pathological exam | No distant metastasis |
| IIA Clinical | ≥2 cm tumour dimension | Nodes negative by clinical exam (no pathological exam performed) | No distant metastasis |
| IIA Pathological | ≥2 cm tumour dimension | Nodes negative by pathological exam | No distant metastasis |
| IIB Clinical | Primary tumour invades bone, muscle, fascia or cartilage | Nodes negative by clinical exam (no pathological exam performed) | No distant metastasis |
| IIB Pathological | Primary tumour invades bone, muscle, fascia or cartilage | Nodes negative by pathological exam | No distant metastasis |
| III Clinical | Any size/depth tumour | Nodes positive by clinical exam (no pathological exam performed) | No distant metastasis |
| IIIA Pathological | Any size/depth tumour | Nodes positive by pathological exam only (nodal disease not apparent on clinical exam) | No distant metastasis |
| IIIA Pathological | Not detected (unknown primary) | Nodes positive by clinical exam and confirmed by pathological exam | No distant metastasis |
| IIIB Pathological | Any size/depth tumour | Nodes positive by clinical exam and confirmed by pathological exam or in‐transit metastasis c | No distant metastasis |
| IV Clinical | Any | ± Regional nodal involvement | Distant metastasis detected via clinical examination |
| IV Pathological | Any | ± Regional nodal involvement | Distant metastasis confirmed by pathological exam |
Clinical detection of nodal or metastatic disease may be via inspection, palpation and/or imaging.
Pathological detection/confirmation of nodal disease may be via sentinel lymph node biopsy, lymphadenectomy or fine needle biopsy and pathological confirmation of metastatic disease may be via biopsy of the suspected metastasis.
In transit metastasis: a tumour distinct from the primary lesion and located either (1) between the primary lesion and the draining regional lymph node or (2) distal to the primary lesion.
TABLE 2.
Union for International Cancer Control (UICC) TNM 8
| Stage | Primary tumour | Regional lymph nodes | Distant metastasis |
|---|---|---|---|
| Stage 0 | Tis | N0 | M0 |
| Stage I | T1 | N0 | M0 |
| Stage IIA | T2, T3 | N0 | M0 |
| Stage IIB | T4 | N0 | M0 |
| Stage IIIA | T0 | N1b | M0 |
| T1, T2, T3, T4 | N1a(sn), N1a | M0 | |
| Stage IIIB | T1, T2, T3, T4 | N1b, N2, N3 | M0 |
| Stage IV | T0, T1, T2, T3, T4 | Any N | M1 |
TX – Primary tumour cannot be assessed.
T0 – No evidence of primary tumour.
Tis – In situ primary tumour.
T1 – mm maximal clinical dimension of tumour.
T2 – >20 mm to mm maximal clinical dimension of tumour.
T3 – >50 mm maximal clinical dimension of tumour.
T4 – Primary tumour invades fascia, muscle, bone or cartilage.
NX – Regional nodes cannot be assessed.
N0 – Regional nodes negative by pathological exam.
N1 – Regional nodes positive by pathological exam.
N1a(sn) – Clinically occult but regional node positive by SLNB.
N1a – Clinically occult but regional nodes positive by lymphadenectomy.
N1b – Clinically detected regional nodes.
N2 – In‐transit metastasis without lymph node metastasis.
N3 – In‐transit metastasis with lymph node metastasis.
M0 – No distant metastases.
M1 – Metastasis beyond regional lymph nodes.
M1a – Metastasis to distant skin, subcutaneous tissues or distant lymph nodes confirmed microscopically.
M1b – Metastases to lung confirmed microscopically.
M1c – Metastasis to other visceral sites conformed.
7. GENERAL RULES OF TREATMENT
The choice of treatment depends on the stage at presentation, location of the disease, comorbidities and performance status of the patient. Variation in MCPyV incidence in MCC in different countries may lead to different management strategies. 39 There is no vaccine available against MCPyV and it is unlikely that this will be soon developed given the low incidence of MCC. The interaction between the tumour, the tumour microenvironment and the immune system is likely to be of high importance in response to therapy in this highly immunogenic tumour, stimulating much current research. 40 MCC management strategy should be established through a multi‐disciplinary approach. 41 , 42
8. SURGERY
Locoregional disease is commonly treated with wide surgical excision with or without SLNB and adjuvant radiotherapy. 43 Recurrence following excision ranged from 25% to 40%. 44 Studies have highlighted a poorer prognosis in larger (>2 cm) primary lesions and in selected small lesions radiotherapy may not be warranted. 45 , 46 Clinical excision margins are not proven in trials, with recommended wide local excision margins ranging between 1 and 3 cm down to either muscle fascia or pericranium. 47 Controversy remains as to the macrographic or micrographic margins required alongside adjuvant radiotherapy. Mohs micrographic surgery may offer reduced local persistence and regional metastasis however no prospective trials have been performed. 48
9. RADIOTHERAPY
Radiotherapy is a treatment option in inoperable patients with primary MCC as monotherapy, with wide‐margin radiotherapy offering in‐field disease control in 75%–100%, however recurrence, disease‐specific and overall mortality are higher compared to operable lesions. 49 , 50 Adjuvant radiation to the primary site has become standard of care to reduce local recurrence risk and including the nodal basin if SLNB positive, though trials in this area are lacking. 51 A retrospective analysis of 4843 patients with localized disease demonstrated improved overall survival with adjuvant radiotherapy compared to surgery alone. 52 Retrospective studies suggest non‐inferiority of nodal irradiation to complete lymph node dissection (CLND) in SLNB‐positive MCC. 42 , 53 , 54 Randomized trials are required to identify the superior modality. Radiotherapy to the nodal drainage basin may reduce recurrence following CLND. 52 Radiotherapy may also help reduce pain from bone and other metastasis. 55 Radiotherapy works by direct dose‐related cytotoxicity. 56 However, it may also prime T cells augmenting presentation of viral and tumour antigens. 56 This may explain the poorer outcomes of radiotherapy in immunosuppressed individuals. 57 Further evidence is required to evaluate the appropriate radiation dose based on patient comorbidities and high competing risk of distant metastases. 58 Radiotherapy techniques for MCC are discussed further in a recent review. 38
10. CHEMOTHERAPY
MCC is a chemosensitive malignancy with reported overall response rates (ORR) to a range of agents (commonly cisplatin or carboplatin plus etoposide, or anthracycline combinations) up to 75%. 59 Responses are durable in a minority of patients and survival benefit in the metastatic setting has not been tested in randomized trials compared to no treatment or newer monoclonal antibodies. In particular, it has a role in multi‐modality treatment for challenging MCC loco‐regional disease in which rapidity of response is a priority, enabling downstream surgery and radiotherapy. 60 A routine role as adjuvant or neo‐adjuvant therapy for operable disease would have to be defined in a randomized trial.
11. IMMUNOTHERAPY
There is evidence from uncontrolled trials supporting the use of immune checkpoint inhibitors to treat metastatic or inoperable MCC. The therapeutic target is the programmed death receptor‐1/programmed death ligand‐1 (PD1‐PDL1) immune‐checkpoint pathway that otherwise inhibits effector activity by differentiated T lymphocytes. 61 , 62 Both Avelumab (anti‐PDL1 IgG1 monoclonal antibody) and Pembrolizumab (anti‐PD1 IgG4 monoclonal antibody) were licensed by European Medicines Agency (EMA) and Food and Drug Administration (FDA) for metastatic MCC in 2017 and 2018 respectively. Avelumab received UK funding approval in February 2018, as an option for metastatic MCC unresponsive to chemotherapy. In small trials of patients with metastatic MCC naïve to chemotherapy, Pembrolizumab had an ORR of 56% (95% CI 35%–76%) and 6‐month progression free survival (PFS) of 67% (95% CI 49%–86%). 63 In a similar population, Avelumab, gave an ORR of 62% with >80% ongoing at 6‐months. 64 In chemotherapy refractory patients, the ORR was lower, 32.8% (95% CI 22%–43%) but with 82% of responses sustained over an average of 10 months. 65 Though PD‐1/PD‐L1 targeting agents are generally well tolerated, they can trigger a wide range of auto‐immune adverse events, some steroid responsive such as colitis and pneumonitis, and others with life changing consequences, such as optic neuritis and type 1 diabetes. 66
Intra‐tumoural infiltration by active T lymphocytes and by MCV‐specific T cells is associated with better MCC‐specific survival. 67 Virus‐negative MCC tumours have a molecular signature characterized by ultraviolet‐induced DNA damage and the presence of tumour‐associated neoantigens. 68 MCC viral status and mutation burden may influence response to therapy however evidence is required for this to guide treatment choices. Downregulation of MHC class I (MHC I) expression in MCC can be reversed by radiotherapy, IFNβ and chemotherapies, potentially increasing the sensitivity of tumours to subsequent immunotherapy. 69 , 70 Radiotherapy may induce the release of tumour‐associated antigens and increase inflammation, thus further synergizing with immunotherapy. 71 None‐the‐less, routine combination of cytotoxic modalities with immune checkpoint blockade requires evidence from trials.
Approximately 50% of patients with metastatic MCC do not respond or experience disease progression after their initial response to treatment, delineating the need for novel strategies to broaden antitumour immune responses. 72 Immunosuppressed and elderly individuals are excluded from most clinical trials so the applicability of findings from small trials to real world populations is uncertain. There is evidence suggesting some benefit of re‐exposure to checkpoint inhibitors in a reported series of 13 cases. 73 Further treatments under investigation include non‐classic immunotherapies such as intratumoural interferon, IL‐12 DNA electroporation, TLR‐4 agonists, adoptive T cell transfer and HLA upregulation.
12. MOLECULAR THERAPIES
Treatments targeting mutated or oncogenic driver pathways might have a role in MCC treatment, perhaps immunosuppressed populations. Tyrosine kinase inhibitors and somatostatin analogues have shown promise in small case series but results are yet to be replicated in larger clinical trials. 74 , 75
13. PROGNOSIS
MCC has a high risk of local‐regional recurrence and early distant metastasis. Five‐year disease‐specific survival ranges from 60% to 87% for those presenting with local disease, 39% to 62% for nodal disease and 11% to 20% for metastatic disease. 17 , 20 , 34 , 76 Future studies may show that survival has improved since the widespread availability of immunotherapy. MCC usually spreads first to lymph nodes with distant metastases to lungs, adrenal glands, liver and bones. 20 Three monthly clinical follow up for at least 2 years is recommended. Rarely, spontaneous regression may occur: the mechanism is not understood but this could guide new therapies. 77 The only reliable prognostic indicator is MCC stage at diagnosis. A large primary tumour and the presence of locoregional or distant metastases are all associated with a poorer prognosis. 20 Prognostic biomarkers are an area of investigation. Studies of the prognostic role of MCPyV status have, thus far, had mixed results, predominantly finding either a worsened prognosis in patients with VN‐MCC tumours or no difference relative to VP‐MCC tumours. 78 In VP‐MCC, higher anti‐VP1 and anti‐ST antibodies have been associated with a better prognosis. 79 Other negative prognostic markers include immunosuppression. 80
14. CONCLUSION
With increasing incidence and high mortality, MCC is likely to be an increasing burden on healthcare resources. However, a lack of international epidemiological data exists for this rare disease. International collaboration and homogeneity in reporting is required to reliably compare epidemiological data. The cellular origin of MCC remains unclear. Identification of the cellular origin may guide management similarly to the discovery of MCPyV and its carcinogenic mechanisms. Clinical diagnosis remains challenging although improvements in cancer registration and immunohistochemistry with increased specialization in cellular pathology and the adoption of a multidisciplinary approach are improving diagnosis, staging and data collection.
Following an International Workshop on MCC research in 2018, the following areas were identified as the highest‐priority research questions 2 : identification of the MCC cell of origin; distinctions between MCPyV positive (VP‐MCC) and MCPyV negative (VN‐MCC) MCC; and the role of these subtypes in guiding management, including combination immunotherapy. Progress in our understanding of MCC tumour immunobiology has led to a rapidly evolving therapeutic landscape and novel immunotherapies have improved prognosis. Five‐year mortality and morbidity are high. Immunotherapy as part of combination therapy may improve outcomes. Monitoring of MCPyV T antigen may prove a useful screening tool for recurrence. The low incidence makes it challenging to power prospective clinical trials. As a result, most management recommendations are based on case series, retrospective reviews, and expert opinion. An international collaborative network is required to establish prospective clinical trials and better establish optimal management.
CONFLICT OF INTERESTS
No conflict of interests have been declared.
AUTHOR CONTRIBUTIONS
K. Mistry: Conceptualization; Data curation; Formal analysis; Methodology; Visualization; Writing – original draft; Writing – review & editing. N. J. Levell: Conceptualization; Supervision; Writing – review & editing. P. Craig: Visualization; Writing – original draft; Writing – review & editing. N. M. Steven: Validation; Writing – review & editing. Z. C. Venables: Conceptualization; Methodology; Supervision; Writing – original draft; Writing – review & editing.
ACKNOWLEDGEMENTS
Thank you to the Medical Illustration Department, Norfolk & Norwich University Hospital NHS Foundation Trust for producing Figures 1 and 2.
Mistry K, Levell NJ, Craig P, Steven NM, Venables ZC. Merkel cell carcinoma. Skin Health Dis. 2021;1(4):e55. 10.1002/ski2.55
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
REFERENCES
- 1. Toker C. Trabecular carcinoma of the skin. Arch Dermatol. 1972;105(1):107–10. [PubMed] [Google Scholar]
- 2. Harms PW, Harms KL, Moore PS, Moore PS, DeCaprio JA, Nghiem P, et al. The biology and treatment of Merkel cell carcinoma: current understanding and research priorities. Nat Rev Clin Oncol. 2018;15(12):763–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319(5866):1096–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Garneski KM, Warcola AH, Feng Q, Kiviat NB, Leonard JH, Nghiem P. Merkel cell polyomavirus is more frequently present in North American than Australian Merkel cell carcinoma tumors. J Invest Dermatol. 2009;129(1):246–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Popp S, Waltering S, Herbst C, Moll I, Boukamp P. UV‐B‐type mutations and chromosomal imbalances indicate common pathways for the development of Merkel and skin squamous cell carcinomas. Int J Cancer. 2002;99(3):352–60. [DOI] [PubMed] [Google Scholar]
- 6. Paulson KG, Lemos BD, Feng B, Jaimes N, Peñas PF, Bi X, et al. Array‐CGH reveals recurrent genomic changes in Merkel cell carcinoma including amplification of L‐Myc. J Invest Dermatol. 2009;129(6):1547–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sunshine JC, Jahchan NS, Sage J, Choi J. Are there multiple cells of origin of Merkel cell carcinoma? Oncogene. 2018;37(11):1409–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang A, Cordoba C, Cheung K, Konya J. Merkel cell carcinoma in situ: new insights into the cells of origin. Australas J Dermatol. 2019;60(4):e311–e313. [DOI] [PubMed] [Google Scholar]
- 9. Navarrete‐Dechent C, Cordova M, Aleissa S, Battle LR, Ganly I, Pulitzer M, Rossi AM. Dermoscopy and reflectance confocal microscopy of intraepidermal Merkel cell carcinoma. Australas J Dermatol. 2021;62 (2):238–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van der Zwan JM, Trama A, Otter R, Larrañaga N, Tavilla A, Marcos‐Gragera R, et al. Rare neuroendocrine tumours: results of the surveillance of rare cancers in Europe project. Eur J Cancer. 2013;49(11):2565–78. [DOI] [PubMed] [Google Scholar]
- 11. Garbutcheon‐Singh KB, Curchin DJ, McCormack CJ, Smith SD. Trends in the incidence of Merkel cell carcinoma in Victoria, Australia, between 1986 and 2016. Australas J Dermatol. 2020;61(1):e34–e38. [DOI] [PubMed] [Google Scholar]
- 12. Goon PK, Greenberg DC, Igali L, Levell NJ. Merkel cell carcinoma: rising incidence in the East of England. J Eur Acad Dermatol Venereol. 2016;30(12):2052–5. [DOI] [PubMed] [Google Scholar]
- 13. Howlader N, Noone AM, Krapcho M, Miller D, Bishop K, Altekruse SF, et al. Surveillance, Epidemiology and End Results (SEER) Cancer Statistics Review. Bethesda, MD: National Cancer Institute, National Cancer Institute; 1975‐2013. [Google Scholar]
- 14. Miller RW, Rabkin CS. Merkel cell carcinoma and melanoma: etiological similarities and differences. Cancer Epidemiol Biomarkers Prev. 1999;8(2):153–8. Erratum in: Cancer Epidemiol Biomarkers Prev 1999;8(5):485. [PubMed] [Google Scholar]
- 15. Agelli M, Clegg LX, Becker JC, Rollison DE. The etiology and epidemiology of merkel cell carcinoma. Curr Probl Cancer. 2010;34(1):14–37. [DOI] [PubMed] [Google Scholar]
- 16. Moshiri AS, Nghiem P. Milestones in the staging, classification, and biology of Merkel cell carcinoma. J Natl Compr Canc Netw. 2014;12(9):1255–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Becker JC, Stang A, DeCaprio JA, Cerroni L, Lebbé C, Veness M, et al. Merkel cell carcinoma. Nat Rev Dis Primers. 2017;26(3):17077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sahi H, Sihto H, Artama M, Koljonen V, Böhling T, Pukkala E. History of chronic inflammatory disorders increases the risk of Merkel cell carcinoma, but does not correlate with Merkel cell polyomavirus infection. Br J Cancer. 2017;17(2):260–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Song PI, Liang H, Wei WQ, Jiang YQ, Smith JS, Qiao YL. The clinical profile of Merkel cell carcinoma in mainland China. Int J Dermatol. 2012;51(9):1054–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harms KL, Healy MA, Nghiem P, Sober AJ, Johnson TM, Bichakjian CK, Wong SL. Analysis of prognostic factors from 9387 Merkel cell carcinoma cases forms the basis for the new 8th edition AJCC staging system. Ann Surg Oncol. 2016;23(11):3564–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paulson KG, Iyer JG, Blom A, Warton EM, Sokil M, Yelistratova L, et al. Systemic immune suppression predicts diminished Merkel cell carcinoma‐specific survival independent of stage. J Invest Dermatol. 2013;133(3):642–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heath M, Jaimes N, Lemos B, Mostaghimi A, Wang LC, Peñas PF, Nghiem P. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58(3):375–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dalle S, Parmentier L, Moscarella E, Phan A, Argenziano G, Thomas L. Dermoscopy of Merkel cell carcinoma. Dermatology. 2012;224(2):140–4. [DOI] [PubMed] [Google Scholar]
- 24. Slater D. Dataset for histopathological reporting of primary cutaneous Merkel cell carcinoma and regional lymph nodes. Royal College of Pathologists; 2019. [Google Scholar]
- 25. Fried I, Cerroni L. Merkel cell carcinoma. Pathologe. 2014;35(5):467–75. [DOI] [PubMed] [Google Scholar]
- 26. Harms PW. Update on Merkel cell carcinoma. Clin Lab Med. 2017;37(3):485–501. [DOI] [PubMed] [Google Scholar]
- 27. Bobos M, Hytiroglou P, Kostopoulos I, Karkavelas G, Papadimitriou CS. Immunohistochemical distinction between Merkel cell carcinoma and small cell carcinoma of the lung. Am J Dermatopathol. 2006;28(2):99–104. [DOI] [PubMed] [Google Scholar]
- 28. Stanoszek LM, Chan MP, Palanisamy N, Carskadon S, Siddiqui J, Patel RM, et al. Neurofilament is superior to cytokeratin 20 in supporting cutaneous origin for neuroendocrine carcinoma. Histopathology. 2019;74(3):504–13. [DOI] [PubMed] [Google Scholar]
- 29. Chan JK, Suster S, Wenig BM, Tsang WY, Chan JB, Lau AL. Cytokeratin 20 immunoreactivity distinguishes Merkel cell (primary cutaneous neuroendocrine) carcinomas and salivary gland small cell carcinomas from small cell carcinomas of various sites. Am J Surg Pathol. 1997;21(2):226–34. [DOI] [PubMed] [Google Scholar]
- 30. Pasternak S, Carter MD, Ly TY, Doucette S, Walsh NM. Immunohistochemical profiles of different subsets of Merkel cell carcinoma. Hum Pathol. 2018;82:232–8. [DOI] [PubMed] [Google Scholar]
- 31. Busam K, Bichakjian C, Coit D, Kutzner H, Requena L, Scolyer R, et al. Merkel cell carcinoma, histopathology reporting guide. 1st edition. International Collaboration on Cancer Reporting; 2019. [DOI] [PubMed] [Google Scholar]
- 32. Lemos B, Nghiem P. Merkel cell carcinoma: more deaths but still no pathway to blame. J Invest Dermatol. 2007;127(9):2100–3. [DOI] [PubMed] [Google Scholar]
- 33. Fields RC, Busam KJ, Chou JF, Panageas KS, Pulitzer MP, Allen PJ, et al. Recurrence after complete resection and selective use of adjuvant therapy for stage I through III Merkel cell carcinoma. Cancer. 2012;118(13):3311–20. [DOI] [PubMed] [Google Scholar]
- 34. Medina‐Franco H, Urist MM, Fiveash J, Heslin MJ, Bland KI, Beenken SW. Multimodality treatment of Merkel cell carcinoma: case series and literature review of 1024 cases. Ann Surg Oncol. 2001;8(3):204–8. [DOI] [PubMed] [Google Scholar]
- 35. Llombart B, Kindem S, Chust M. Merkel cell carcinoma: an update of key imaging techniques, prognostic factors, treatment, and follow‐up. Actas Dermosifiliogr. 2017;108(2):98–107. [DOI] [PubMed] [Google Scholar]
- 36. Concannon R, Larcos GS, Veness M. The impact of (18)F‐FDG PET‐CT scanning for staging and management of Merkel cell carcinoma: results from Westmead Hospital, Sydney, Australia. J Am Acad Dermatol. 2010;62(1):76–84. [DOI] [PubMed] [Google Scholar]
- 37. Amin MB, Edge S, Greene F, Byrd DR, Brookland RK, Washington MK, et al. AJCC cancer staging manual. 8th edition. Springer International Publishing: American Joint Commission on Cancer; 2017. [Google Scholar]
- 38. Brierley JD, Gospodarowicz MK, Wittekind CH. TNM classification of malignant tumours. 8th edition. Oxford: Wiley‐Blackwell; 2017. [Google Scholar]
- 39. Kok DL, Wang A, Wen X, Chua MSR, Guminski A, Veness M, et al. The changing paradigm of managing Merkel cell carcinoma in Australia: An expert commentary. Asia Pac J Clin Oncol. 2020;16(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Blank CU, Haanen JB, Ribas A, Schumacher TN. The cancer immunogram. Science. 2016;352(6286):658–60. [DOI] [PubMed] [Google Scholar]
- 41. Nghiem P, Park SY. Less toxic, more effective treatment‐a win‐win for patients with Merkel cell carcinoma. JAMA Dermatol. 2019;155:1223. 10.1001/jamadermatol.2019.2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Steven N, Lawton P, Poulsen M. Merkel cell carcinoma ‐ current controversies and future directions. Clin Oncol (R Coll Radiol). 2019;31(11):789–96. [DOI] [PubMed] [Google Scholar]
- 43. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) Merkel cell carcinoma version 2 2019. https://merkelcell.org/wp‐content/uploads/2017/10/MCC_v.2.2019‐2.pdf [Google Scholar]
- 44. Villani A, Fabbrocini G, Costa C, Carmela Annunziata M, Scalvenzi M. Merkel cell carcinoma: therapeutic update and emerging therapies. Dermatol Ther (Heidelb). 2019;9(2):209–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Allen PJ, Zhang ZF, Coit DG. Surgical management of Merkel cell carcinoma. Ann Surg. 1999;229(1):97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Albores‐Saavedra J, Batich K, Chable‐Montero F, Sagy N, Schwartz AM, Henson DE. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol. 2010;37(1):20–7. [DOI] [PubMed] [Google Scholar]
- 47. Lebbe C, Becker JC, Grob JJ, Malvehy J, del Marmol V, Pehamberger H, et al. Diagnosis and treatment of Merkel cell carcinoma. European consensus‐based interdisciplinary guideline. Eur J Cancer. 2015;51(16):2396–403. [DOI] [PubMed] [Google Scholar]
- 48. O’Connor WJ, Roenigk RK, Brodland DG. Merkel cell carcinoma. Comparison of Mohs micrographic surgery and wide excision in eighty‐six patients. Dermatol Surg. 1997;23:929–33. [PubMed] [Google Scholar]
- 49. Pape E, Rezvoy N, Penel N, Salleron J, Martinot V, Guerreschi P, et al. Radiotherapy alone for Merkel cell carcinoma: a comparative and retrospective study of 25 patients. J Am Acad Dermatol. 2011;65(5):983–90. [DOI] [PubMed] [Google Scholar]
- 50. Harrington C, Kwan W. Outcomes of Merkel cell carcinoma treated with radiotherapy without radical surgical excision. Ann Surg Oncol. 2014;21(11):3401–5. [DOI] [PubMed] [Google Scholar]
- 51. Tello TL, Coggshall K, Yom SS, Yu SS. Merkel cell carcinoma: An update and review: current and future therapy. J Am Acad Dermatol. 2018;78(3):445–54. [DOI] [PubMed] [Google Scholar]
- 52. Bhatia S, Storer BE, Iyer JG, Moshiri A, Parvathaneni U, Byrd D , et al. Adjuvant radiation therapy and chemotherapy in Merkel cell carcinoma: survival analyses of 6908 cases from the National Cancer Data Base. J Natl Cancer Inst. 2016;108(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fang LC, Lemos B, Douglas J, Iyer J, Nghiem P. Radiation monotherapy as regional treatment for lymph node‐positive Merkel cell carcinoma. Cancer. 2010;116(7):1783–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee JS, Durham AB, Bichakjian CK, Harms PW, Hayman JA, McLean SA, et al. Completion lymph node dissection or radiation therapy for sentinel node metastasis in Merkel cell carcinoma. Ann Surg Oncol. 2019;26(2):386–94. [DOI] [PubMed] [Google Scholar]
- 55. Iyer JG, Parvathaneni U, Gooley T, Miller NJ, Markowitz E, Blom A, et al. Single‐fraction radiation therapy in patients with metastatic Merkel cell carcinoma. Cancer Med. 2015;4(8):1161–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood. 2009;114(3):589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cook M, Baker K, Redman M, Lachance K, Nguyen MH, Parvathaneni U, et al. Differential outcomes among immunosuppressed patients with Merkel cell carcinoma: impact of immunosuppression type on cancer‐specific and overall survival. Am J Clin Oncol. 2019;42(1):82–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Goff P, Cook M, Schaub S, Park S, Hippe D, Liao J, et al. Efficacy and toxicity of hypofractionated adjuvant radiotherapy in Merkel cell carcinoma. Int J Radiation Onc. 2020;108(2). [Google Scholar]
- 59. Tai PT, Yu E, Winquist E, Hammond A, Stitt L, Tonita J, Gilchrist J. Chemotherapy in neuroendocrine/Merkel cell carcinoma of the skin: case series and review of 204 cases. J Clin Oncol. 2000;18(12):2493–9. [DOI] [PubMed] [Google Scholar]
- 60. Poulsen M, Rischin D, Walpole E, Harvey J, Mackintosh J, Ainslie J, et al. High‐risk Merkel cell carcinoma of the skin treated with synchronous carboplatin/etoposide and radiation: a Trans‐Tasman Radiation Oncology Group Study‐‐TROG 96:07. J Clin Oncol. 2003;21(23):4371–6. [DOI] [PubMed] [Google Scholar]
- 61. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Canc Cell. 2015;27(4):450–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Twyman‐Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non‐redundant immune mechanisms in cancer. Nature. 2015;16(7547):373–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nghiem PT, Bhatia S, Lipson EJ, Kudchadkar RR, Miller NJ, Annamalai L, et al. PD‐1 blockade with pembrolizumab in advanced Merkel‐cell carcinoma. N Engl J Med. 2016;374(26):2542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. D'Angelo SP, Russell J, Lebbé C, Chmielowski B, Gambichler T, Grob JJ, et al. Efficacy and safety of first‐line avelumab treatment in patients with stage IV metastatic Merkel cell carcinoma: a preplanned interim analysis of a clinical trial. JAMA Oncol. 2018;4(9):e180077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P, D'Angelo SP, et al. Avelumab in patients with chemotherapy‐refractory metastatic Merkel cell carcinoma: a multicentre, single‐group, open‐label, phase 2 trial. Lancet Oncol. 2016;17(10):1374–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Brahmer JR, Lacchetti C, Schneider BJ, Atkins MB, Brassil KJ, Caterino JM, et al. Management of immune‐related adverse events in patients treated with immune checkpoint inhibitor therapy: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol. 2018;36(17):1714–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Miller NJ, Church CD, Dong L, Crispin D, Fitzgibbon MP, Lachance K, et al. Tumor‐infiltrating Merkel cell polyomavirus‐specific T cells are diverse and associated with improved patient survival. Cancer Immunol Res. 2017;5(2):137–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wong SQ, Waldeck K, Vergara IA, Schröder J, Madore J, Wilmott JS, et al. UV‐associated mutations underlie the etiology of MCV‐negative Merkel cell carcinomas. Cancer Res. 2015;75 (24):5228–34. [DOI] [PubMed] [Google Scholar]
- 69. Paulson KG, Tegeder A, Willmes C, Iyer JG, Afanasiev OK, Schrama D, et al. Downregulation of MHC‐I expression is prevalent but reversible in Merkel cell carcinoma. Cancer Immunol Res. 2014;2(11):1071–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chapuis AG, Afanasiev OK, Iyer JG, Paulson KG, Parvathaneni U, Hwang JH, et al. Regression of metastatic Merkel cell carcinoma following transfer of polyomavirus‐specific T cells and therapies capable of re‐inducing HLA class‐I. Cancer Immunol Res. 2014;2(1):27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cassler NM, Merrill D, Bichakjian CK, Brownell I. Merkel cell carcinoma therapeutic update. Curr Treat Options Oncol. 2016;17(7):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Colunga A, Pulliam T, Nghiem P. Merkel cell carcinoma in the age of immunotherapy: facts and hopes. Clin Cancer Res. 2018;24(9):2035–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. LoPiccolo J, Schollenberger MD, Dakhil S, Rosner S, Ali O, Sharfman WH, et al. Rescue therapy for patients with anti‐PD‐1‐refractory Merkel cell carcinoma: a multicenter, retrospective case series. J Immunother Cancer. 2019;7(1):170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Akaike T, Qazi J, Anderson A, Behnia FS, Shinohara MM, Akaike G, et al. High somatostatin receptor expression and efficacy of somatostatin analogues in patients with metastatic Merkel cell carcinoma. Br J Dermatol. 2020;2:319–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tarabadkar ES, Thomas H, Blom A, Parvathaneni U., Olencki T., Nghiem P., Bhatia S. Clinical benefit from tyrosine kinase inhibitors in metastatic Merkel cell carcinoma: a case series of 5 patients. The American Journal of Case Reports. 2018;19:505–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lemos BD, Storer BE, Iyer JG, Phillips JL, Bichakjian CK, Fang LC, et al. Pathologic nodal evaluation improves prognostic accuracy in Merkel cell carcinoma: analysis of 5823 cases as the basis of the first consensus staging system. J Am Acad Dermatol. 2010;63(5):751–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Walsh NM. Complete spontaneous regression of Merkel cell carcinoma (1986‐2016): a 30 year perspective. J Cutan Pathol. 2016;43(12):1150–4. [DOI] [PubMed] [Google Scholar]
- 78. Moshiri AS, Doumani R, Yelistratova L, Blom A, Lachance K, Shinohara MM, et al. Polyomavirus‐negative Merkel cell carcinoma: a more aggressive subtype based on analysis of 282 cases using multimodal tumor virus detection. J Invest Dermatol. 2017;137(4):819–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Samimi M, Molet L, Fleury M, Laude H, Carlotti A, Gardair C, et al. Prognostic value of antibodies to Merkel cell polyomavirus T antigens and VP1 protein in patients with Merkel cell carcinoma. Br J Dermatol. 2016;174(4):813–22. [DOI] [PubMed] [Google Scholar]
- 80. Ma JE, Brewer JD. Merkel cell carcinoma in immunosuppressed patients. Cancers (Basel). 2014;6(3):1328–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.