

Abstract
Hepatocellular carcinoma (HCC) remains one of the most prevalent and deadliest cancers. The poor outcome associated with HCC is dramatically changing due to the advent of effective systemic therapies. Here we discuss the molecular pathogenesis of HCC, molecular classes and determinants of heterogeneity. In addition, effective single-agent and combination systemic therapies involving immunotherapies as standard of care are analyzed. Finally, we propose a flowchart of sequential therapies, explore mechanisms of resistance and address the need of predictive biomarkers.
Introduction
The incidence of liver cancer is increasing globally, and it is expected to reach 1 million new cases per year by 20211,2. Hepatocellular carcinoma (HCC) accounts for more than 90% of liver cancer cases, and the life expectancy of HCC patients has improved with the implementation of targeted and immune therapies3–6. Although the main features of the molecular pathogenesis, drivers and molecular and immune classes have been identified4,7,8 HCC presents few actionable mutations (only 25% of tumors harbor one druggable target)3,5. As a result, the understanding of oncogenic drivers and molecular classes has not yet been translated into clinical decision-making. Recent findings on immune cell populations6, tumor heterogeneity9,10 and etiology-specific pathogenic traits11 might help to overcome this unmet need favoring the emergence of precision oncology strategies for this cancer.
Advanced HCC is chemo- and radio-resistant2,12, which limited the available therapeutic options for these patients. In 2007, the approval of the tyrosine kinase inhibitor (TKI) sorafenib13, the first systemic treatment for HCC, radically changed its prospects. Several single-agent systemic regimes were subsequently approved in first (lenvatinib)14 and second line (regorafenib15, cabozantinib16 and ramucirumab17). 2020 marks the start of a third era dominated by combination regimens involving immunotherapies6, ignited by the demonstrated superiority of atezolizumab and bevacizumab combination versus sorafenib in all clinical end-points18, including overall survival (OS), progression free survival (PFS) and objective response, and in patient-reported outcomes. This has opened the path to exploring combinations of immunotherapies with TKIs or monoclonal antibodies against VEGFA, combinations of two immunotherapies such as anti-PD1/PD-L1 with anti-CTLA4 inhibitors, and even triplet combinations. In addition, two immune-based regimes (pembrolizumab19 and nivolumab plus ipilimumab20,21) got FDA accelerated approval based on the reporting of positive phase II clinical trials. Current ongoing trials with systemic regimens are expected to further impact the clinical benefits of patients at early-intermediate stages22.
In this Review, we provide an integrated description of the molecular pathogenesis of HCC, critical oncogenic drivers and molecular and immune classes, and the recent developments in systemic therapies. In addition, we discuss how this knowledge could be translated into precision oncology by providing a perspective of the role of systemic therapies in HCC, their current stand in the management of the disease and the optimal transition from loco-regional to systemic regimens. We further dissect the evidence supporting the use of the molecular and immune treatments approved and provide insights on how to navigate through these regimes. Finally, we conduct a critical analysis on emerging clinical trials, biomarkers and trial design for future investigations23.
Molecular pathogenesis
HCC mostly develops in ~80% of cases in the setting of a severely damaged cirrhotic liver that already gathers molecular alterations1. In addition, several etiological (HCV and HBV infection, alcohol use, non-alcoholic steatohepatitis) and environmental factors (aflatoxin, aristolochic acid and tobacco) have been identified with distinct specific paths to cancer development1,6. Specific molecular and immune classes have been defined, which integrate the current molecular knowledge of this cancer6. In this regard, immune11 and epigenetic mechanisms7,24–26 might have major consequences in understanding the onset, evolution and treatment of this malignancy. Overall, the main molecular alterations and pathogenic processes involved in HCC development have been extensively reviewed elsewhere1,4,8
Hepatocarcinogenic process and drivers
Most HCCs develop in patients with cirrhosis1. These neoplasms progress through a sequence of well-defined histopathological phases, starting with emergence of dysplastic nodules which can ultimately transform into HCC (Figure 1)4. Genetic and epigenetic oncogenic alterations likely occur within hepatocytes, a cell type that despite its differentiated features is a facultative stem cell1,27. Although mature hepatocytes are the main cells of origin for HCC, liver stem cells and transit amplifying cell populations have been also implicated in liver oncogenesis8,27–31. In preclinical models, hepatic oncogenesis is favored by cell death with compensatory regeneration of hepatocytes, and blocking apoptosis reduces HCC formation32. Replicative stress within regenerating hepatocytes induces genetic lesions favoring transformation and cancer progression33, especially in the context of inflammation and fibrosis. In human NASH, auto-aggressive CD8+ PD1+ T cells induce hepatocyte cell death, promote NASH pathogenesis and impair immune surveillance, thereby favoring HCC occurrence and progression11.
Figure 1.
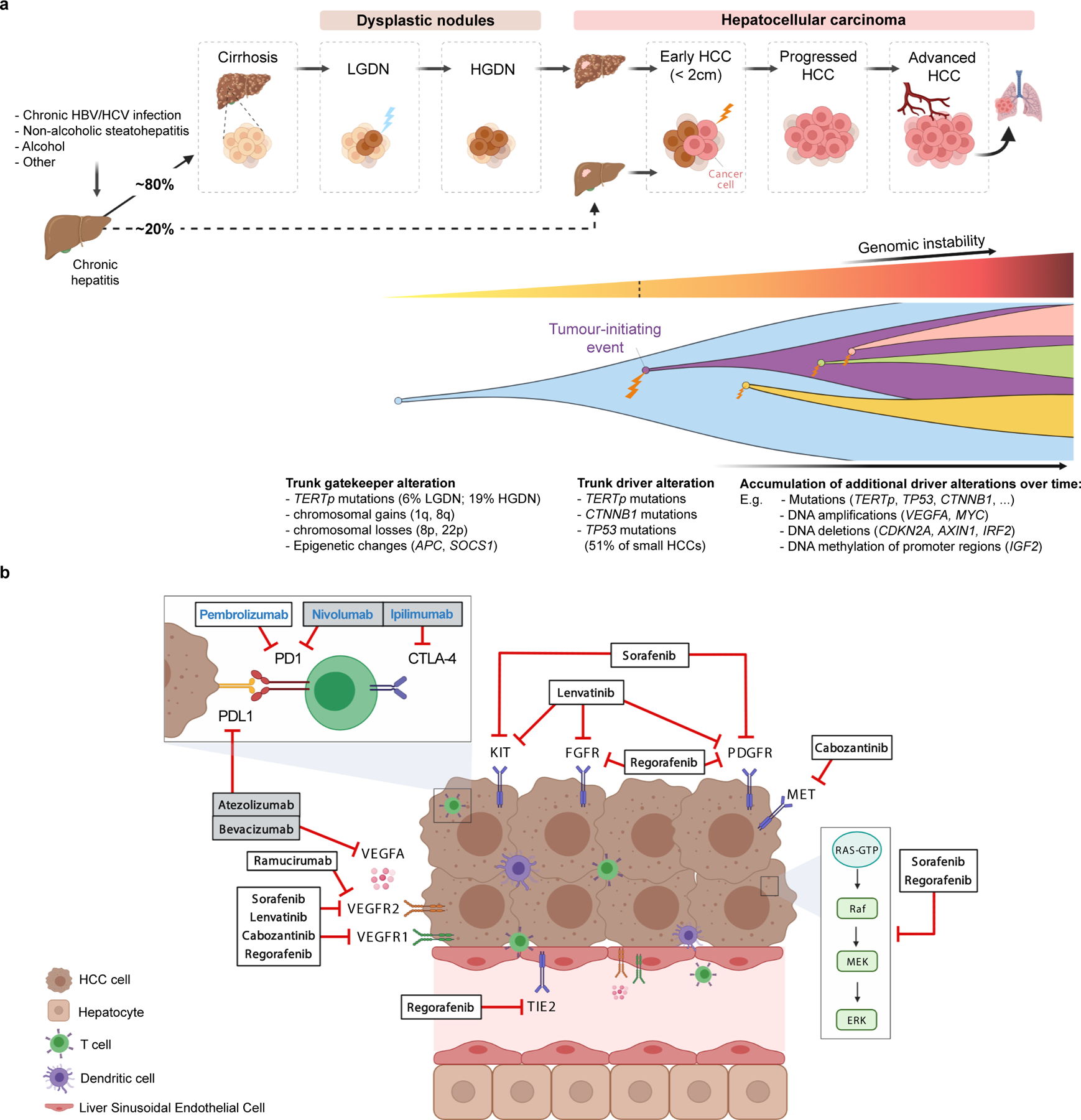
A) Molecular pathogenesis of HCC: step-by-step process, genomic hits and clonal evolution. Both genetic and epigenetic mechanisms (TERT promoter mutations, chromosomal aberrations and methylation events) are thought to function as gatekeepers for malignant transformation of dysplastic nodules. Hepatocarcinogenesis requires a tumour-initiation event such as mutations in TERT, TP53 and CTNNB1, which are already present in 51% of small HCC tumors. Further acquired genetic alterations and changes to the tumour microenvironment enable these tumors to progress to advanced stages under the constant pressure of evolutional selection, leading to vast intratumoral heterogeneity. HBV, hepatitis B virus; HCV, hepatitis C virus; HGDN, high-grade dysplastic nodules; LGDN, low-grade dysplastic nodules; TERTp, TERT promoter.
B) Molecular depiction of systemic therapies in HCC. Tumor cells, liver sinusoidal endothelial cells and lymphocytes are represented in relation to tyrosine kinase inhibitors, immunotherapies and monoclonal antibodies approved in HCC based on phase III data. Therapy names in bold black indicate positive results based on phase III trials, either with a superiority design (atezolizumab plus bevacizumab, sorafenib, regorafenib, cabozantinib and ramucirumab) or with a non-inferiority design (lenvatinib). Therapy names in bold blue designate other FDA-approved drugs based on non-randomized phase II trials (pembrolizumab and nivolumab plus ipilimumab). Grey boxes indicate combination therapies.
On the other hand, the sequential accumulation of somatic genomic and epigenetic alterations has been shown to play a key role in liver carcinogenesis. Single nucleotide polymorphisms (SNP) that predispose to liver disease, including PNPLA3 rs738409, TM6SF2 rs585542926, and HSD17B13 rs72613567 (which encode proteins involved in metabolism) increase the risk for HCC4,34. Aflatoxin B1 and aristolochic acid, are environmental genotoxic compounds inducing somatic mutations in HCC3,35. Aflatoxin B1, an aspergillus metabolite found in maize and nuts, synergistically promotes HCC in patients with HBV infection. Aristolochic acid, found in Chinese herbal teas, causes abundant T to A oncogenic transversions36. On average, HCC tumors have 60–70 somatic mutations. The majority are “passenger mutations” and do not directly participate in the carcinogenetic process, but some mutations occur in the so called “driver genes” and activate signaling pathways that are key for liver carcinogenesis (Table 1). The most prevalent somatic mutation in HCC (60%) affects the promoter region of the telomerase reverse transcriptase gene (TERT), a master regulator of telomere length3. Additionally, integration of hepatitis B virus (HBV) or adeno-associated virus 2 (AAV2) in the TERT promoter has been reported37. TERT-activating mutations occur in 20% of dysplastic nodules, making this molecular feature a putative gatekeeper of HCC38. Subpopulations of hepatocytes expressing high levels of telomerase exist distributed throughout all liver zones39, which may contribute to hepatocarcinogenesis by preventing cellular senescence, thereby providing a mutation-prone source of replicating cells in chronic liver injury. Other epigenetic (e.g. hypermethylation of TSPYL5)24 and genetic alterations (e.g. chr. 8q loss)38 found in dysplastic nodules have been suggested as cancer gatekeepers. The second most frequently altered gene in HCC is CTNNB1 (~30%), a gene that encodes β-catenin and is a critical effector of the Wnt pathway. Wnt/β-catenin signaling is largely limited to zone three of the hepatic lobule40, and hepatocarcinogenesis involving β-catenin mutations likely occurs in this hepatic zone. Other key mutations occur in TP53 (~25%), and AXIN1 (~10%) or in epigenetic regulators, such as BAP1, ARID1A/B and ARID23,41. Mutations in conventional targets for TKIs, such as PDGFR, MET, EGFR, PIK3CA are rare (< 3%).
Table 1:
Key oncogenic drivers and pathways de-regulated in HCC
| Altered pathway | Altered gene | Type of alteration | Prevalence in HCC % (range) |
|---|---|---|---|
| Telomere maintenance | TERT$ | Promoter Activating mutation | 55 (44–59) |
| High-level focal amplification | 6 (1–9) | ||
| Viral insertion | 3 (1 – 5) | ||
| Cell cycle regulation | TP53$ | Loss of function mutation | 27 (18–31) |
| Homozygous deletion | 2 (0–2) | ||
| ATM | Loss of function mutation | 4 (2–5) | |
| RB1 | Loss of function mutation | 4 (3–5) | |
| Homozygous deletion | 5 (4–6) | ||
| CDKN2A | Loss of function mutation | 2 (1–3) | |
| Homozygous deletion | 5 (4–6) | ||
| MYC | High-level focal amplification | 12 (4–18) | |
| CCND1$ | High-level focal amplification | 7 (5–7) | |
| Wnt / β-catenin signaling | CTNNB1$ | Activating mutation | 29 (23–36) |
| AXIN1 | Loss of function mutation | 7 (4–10) | |
| APC | Loss of function mutation | 2 (0–3) | |
| Chromatin remodeling | ARID1A | Loss of function mutation | 8 (4–12) |
| ARID2 | Loss of function mutation | 7 (3–10) | |
| KMT2A | Loss of function mutation | 3 (0–4) | |
| KMT2C | Loss of function mutation | 3 (2–5) | |
| KMT2B | Loss of function mutation | 2 (0–4) | |
| BAP1 | Loss of function mutation | 2 (0–5) | |
| ARID1B | Loss of function mutation | 1 (0–3) | |
| Ras/PI3K/mTOR | RPS6KA3 | Unclassified | 4 (3–6) |
| PIK3CA # | Activating mutation | 2 (1–4) | |
| KRAS # | Activating mutation | 1 (0–1) | |
| NRAS | Activating mutation | 0 (0–1) | |
| PDGFRA # | Mutation | 1 (0–4) | |
| EGFR # | Activating mutation | 1 (0–2) | |
| PTEN | Loss of function mutation | 1 (0–2) | |
| FGF signaling# | FGF19 | High-level focal amplification | 6 (5–6) |
| VEGF pathway# | VEGFA | High-level focal amplification | 5 (1–8) |
| Oxidative stress | NFE2L2& | Activating mutation | 4 (2–6) |
| KEAP1& | Activating mutation | 3 (2–5) | |
| Hepatocyte differentiation | ALB | Mutation | 9 (5–13) |
| APOB | Mutation | 8 (1–10) | |
| JAK–STAT | IL6ST | Mutation | 2 (0–3) |
| JAK1$ | Mutation | 1 (0–3) | |
| TGFβ signaling$ | ACVR2A | Loss of function mutation | 4 (1–10) |
| IGF signaling$ | IGF2R | Mutation | 1 (0–2) |
Mutation frequencies are reported for a total of 1,339 patients included in multiple whole-exome sequencing studies3,41,60,170,171 (modified and updated from5); TERT promoter mutations were assessed using Sanger sequencing (n=1,517 patients)172. Copy number alterations were detected using single-nucleotide polymorphism (SNP) arrays (n=857 patients)3,41,45,60,170. Viral integrations were detected using viral capture and DNA sequencing (n=645 patients). HCC, hepatocellular carcinoma; IGF, insulin growth factor; mTOR, mammalian Target of Rapamycin; STAT, signal transducer and activator of transcription; TGFβ, transforming growth factor β.
: targetable by an FDA-approved drug.
: targetable by a drug in testing phases.
: targetable using mTOR inhibitors in testing phases.
Strikingly, out of the 34 most commonly reported genes in HCC (Table 1), only 6 have been proven targetable by an FDA-approved drug and another 8 are under evaluation in early phase trials. Some examples include the high-level focal amplification of the 11q13 locus containing FGF1942,43, which has led to proof-of-concept studies demonstrating anti-tumoral activity with FGFR4 inhibitors in HCCs with FGF19 overexpression44. Also, the high-level focal amplification in chromosome 6p21 including the VEGFA gene, which has a 5% reported prevalence in HCC45 (Table 1). Drugs targeting VEGFA (such as bevacizumab) or VEGFR2 (such as ramucirumab) have been approved but there is no specific information on whether they are more efficacious in tumors with these amplifications. Other alterations such those in the IGF pathway are also prevalent in HCC46 (Table 1) but drugs blocking them are still in early clinical trials47,48. Finally, although targeting non-enzymatic mutations has proven difficult (e.g., CTNNB1 exon three mutations), newer therapeutic approaches such as proteolysis-targeted chimeras (PROTACS)49, which induces targeted protein degradation by the ubiquitin proteosome pathway, are promising.
Molecular and immune HCC classes
The molecular landscape of each tumor results from the accumulation of genomic and epigenomic alterations and is shaped by the tumor microenvironment. Based on genomic, transcriptomic and epigenomic data, distinct HCC molecular and immune subtypes have been identified45,50–55.
a). Molecular classes.
The most extended HCC molecular classification distinguishes between the proliferation class and the non-proliferation class. HCCs of the proliferation class (~50% of the cases) are associated with high levels of AFP, poor clinical outcome and HBV-related etiology. These tumors present activation of signaling pathways involved in cell proliferation and survival, including MAPK signaling, PI3K/AKT/mTOR signaling and MET signaling45,50,51, are enriched in TP53 mutations and focal chromosomal amplifications in the 11q13 locus including FGF19/CCND145,56, and harbor high chromosomal instability57. Proliferation class tumors can be subdivided into those with TGFβ-non-canonical Wnt activation (S1/Wnt-TGFβ subclass)50, and those with progenitor cell features, overexpression of EPCAM, AFP and IGF2 (S2 subclass)50. A methylation-based signature with prognostic value has been reported to identify a subset of HCCs of the S2 subclass58.
The non-proliferation HCC class is more heterogeneous. It is associated with alcohol- and HCV-related cases and is associated with better clinical outcome. A subgroup of tumors of this class are dominated by canonical Wnt signaling59 (CTNNB1 subclass).
A recent publication analyzing non-alcoholic steatohepatitis (NASH)-related HCC reported an enrichment in bile and fatty acid signaling, oxidative stress and inflammation; a higher proportion of Wnt/TGFβ subclass; and higher immunosuppressive features60. At the genomic level, mutations in ACVR2A gene (10%) were significantly higher in HCCs associated with this etiology60.
b). Immune classes.
Around ~35% of tumors belong to the ‘inflamed class’, and present high immune cell infiltration, high cytolytic activity, increased levels of PD1/PD-L1, activation of interferon signaling and low burden of broad chromosomal alterations, which recapitulate the characteristics of hot tumors, and which includes a small subgroup of tumors dominated by high interferon signaling coexisting with CTNNB1 mutations6,54,55. In principle, this class includes tumors with the highest immune infiltration, a more diverse T cell repertoire and enrichment in signatures predicting response to immune checkpoint inhibitors. Non-inflamed- cold tumors are characterized by T cell exclusion, and either TP53 mutations (intermediate class) or activation of canonical Wnt signaling through CTNNB1 mutations (excluded class)6,55. Whether ‘inflamed’ HCCs or other immune-related biomarkers are associated with response to immune checkpoint blockers is currently being investigated61,62.
Cancer evolution and molecular heterogeneity
The changing tumor microenvironment (TME) imposes a constant selective pressure that leads to intratumor heterogeneity (ITH), a key feature of solid malignancies63,64. Evidence of ITH in HCC via multi-regional DNA sequencing of tumors9,10,38,65,66, reveals the presence of trunk alterations such as TP53, CTNNB1 and TERT during early stages of hepatocarcinogenesis (Figure 1). However, while driver mutations may be positively selected during tumor evolution, putative passenger mutations are also inadvertently introduced67,68. Recent studies have focused on the characterization of non-genetic clonal diversity69–71, but the faithful integrated modeling of tumor evolution still remains a challenge to guide molecular-targeted therapeutics. From the epigenetic perspective, genome-wide DNA methylation studies across normal livers, cirrhotic tissues, dysplastic nodules and HCC point to epigenetic factors as key regulators during the transition between dysplastic nodules and HCC24,72.
Furthermore, single cell studies have provided unique insights into tumor evolution29,73. For instance, scRNAseq has revealed novel T-cell subtypes associated with HCC or with responses to treatment11,74,75, and uncovered significant transcriptomic diversity of cancer stem cell populations within HCC76. Single cell analysis coupled with regional neo-epitope profiling and viral antigen burden evidenced regional clonal immune responses contributing to ITH in HCC10. Another scRNAseq analysis of patients with HCC undergoing immunotherapy revealed that VEGF expression was associated with higher transcriptomic diversity, TME reprogramming and worse overall survival and response to therapy77. Longitudinal multi-region analysis following therapy by scRNAseq provided a molecular portrait of the immune cell landscape of early-relapse HCC78.
Translating molecular knowledge into precision oncology
With the exception of elevated serum AFP level predicting response to ramucirumab17, approved systemic agents lack appropriate biomarkers to identify responders79. Three factors hamper the translation of precision oncology into HCC decision-making. First, the most prevalent molecular alterations -TERT, CTNNB1, and TP53 mutations- are currently undruggable (Table 1)3,41,60, with only 20–25% of tumors hosting a driver actionable mutation3,5. This differs markedly from other cancers, such as melanoma or gastrointestinal stromal tumors (GIST)80,81. For instance, in a study of ~10,000 solid tumors, patients with GIST, thyroid, breast, melanoma or glioma received specific targeted therapies against actionable aberrations in ~60–75% of cases, compared to only 5% of HCC cases82. Second, HCC is clinically diagnosed using non-invasive imaging criteria according to guidelines83–87. Despite clear calls for access to tissue specimens for research purposes in randomized clinical trials (RCT)2,23, systematic collection of tissues to develop biomarkers has been scarce. Finally, the significant intratumoral heterogeneity, present in up to 25% of cases10,66, is the third obstacle to the identification of useful biomarkers88.
Few RCT have been designed enriched for biomarker-based populations, and none based on molecular or immune HCC classes. In the REACH-2 trial, ramucirumab showed better significant outcome in patients with serum AFP levels >400 ng/ml -around 40% of the advanced HCC second–line population- compared to placebo17. Other biomarker-related studies yielded negative or inconclusive results, including the trial of tivantinib, a non-specific MET and tubulin inhibitor89 tested in HCCs with high tumor MET expression (~50% of advanced HCC cases) detected by immunohistochemistry90. A phase II study enriched for RAS mutations the combination of sorafenib and refametinib was inconclusive91. Fisogatinib, a specific inhibitor of the FGFR4 receptor (activated by the oncogenic FGF19 in ~25% of HCCs92), was tested in a proof of concept study leading to 16% of objective response42,44.
Systemic therapies
Systemic therapies have profoundly changed the landscape of management of HCC. It is estimated that 50–60% of patients are treated with systemic therapies, either because they are diagnosed at advanced stages of the disease or because they progress after surgical or loco-regional therapies1. In this section we discuss the timings, selection and prospects of systemic therapies for patients with HCC.
Current systemic therapies in HCC
Since the initial study showing benefits of sorafenib treatment compared to placebo, in the last 15 years we have witnessed the approval by FDA/EMA and most of Asian regulatory agencies of six regimens (atezolizumab plus bevacizumab18, sorafenib13, lenvatinib14, regorafenib15, cabozantinib16 and ramucirumab17) based on phase III data (Figure 1, Table 2). Two additional regimes (pembrolizumab19, nivolumab plus ipilimumab20,21) have been approved by FDA based on the results of phase II trials. Very recently, the combination of tremelimumab and durvalumab has been shown to be superior to sorafenib for OS93, whereas cabozantinib plus atezolizumab showed superiority against sorafenib in terms of PFS94. This unprecedented improvement in treatment armamentarium of the disease has impacted in the expected outcome of patients and in the early transitioning from loco-regional therapies to systemic therapies.
Table 2:
Systemic therapies approved for HCC: patients characteristics and outcomes.
| Study name | Treatment | Inhibited molecules | BCLC (0/A/B/C) % |
Previous local therapies % |
MVI % |
EHD % |
ECOG PS (0/1/2) % |
Child-Pugh A % |
Median OS (HR, 95% CI) | Median PFS (HR, 95% CI) | ORR mRECIST; RECIST % |
|---|---|---|---|---|---|---|---|---|---|---|---|
| First-line therapies | |||||||||||
| IMbrave15018,141 | Atezolizumab + bevacizumab | PDL1 (immune checkpoint) VEGF (angiogenesis) | - / 2 / 15 / 82 | 48 | 38 | 63 | 62 / 38 / - | 100 | 19.2mo (0.66, 0.52–0.85) |
6.9mo (0.65, 0.53–0.81) | 35.4; 29.8 |
| SHARP13 (IMbrave150, REFLECT) | Sorafenib | VEGFR, PDGFR (angiogenesis) MAPK (BRAF) |
- / - / 18 / 82 | 67 | 36 | 53 | 54 / 38 / 8 | 95 | 10.7–13.4moa (0.69, 0.55–0.87)b | 3.7–4.3moa (NR)b |
NR; 2 |
| REFLECT14 | Lenvatinib | VEGFR, PDGFR, FGFR (angiogenesis) KIT, RET |
- / - / 22 / 78 | 78 | 23 | 61 | 64 / 36 / - | 99 | 13.6mo (0.92, 0.79–1.06) | 7.4mo (0.66, 0.57–0.77) |
24.1; 18.8 |
| Second-line therapies | |||||||||||
| RESORCE15 | Regorafenib | VEGFR, PDGFR (angiogenesis) MAPK (BRAF) |
- / <1 / 14 / 86 | 85 | 29 | 70 | 65 / 35 / - | 98 | 10.6mo (0.63, 0.5–0.79) | 3.1mo (0.46, 0.37–0.56) | 11; 7 |
| CELESTIAL16 | Cabozantinib | MET (proliferation) VEGFR (angiogenesis) RET |
- / - / 9 / 91 | 44c | 27 | 79 | 52 / 48 / <1 | 98 | 10.2mo (0.76, 0.63–0.92) | 5.2mo (0.44, 0.36–0.52) | NR; 4 |
| REACH-217 | Ramucirumab (AFP>400 ng/dL) | VEGFR2 (angiogenesis) | - / - / 17 / 83 | 62d | 36 | 72 | 57 / 43 / - | 100 | 8.5mo (0.71, 0.53–0.95) | 2.8mo (0.45, 0.34–0.6) | NR; 5 |
| KEYNOTE-240136 | Pembrolizumab | PD1 (immune checkpoint) | - / - / 20 / 80 | NR | 13 | 70 | 58 / 42 / - | 100 | 13.9mo (0.78, 0.61–1) | 3mo (0.78, 0.61–0.99) | NR; 18 |
| KEYNOTE-22419 | Pembrolizumab | PD1 (immune checkpoint) | - / - / 24 / 76 | NR | 17 | 64 | 61 / 39 / - | 94 | 13.2mo | 4.9mo | 15; 18.3 |
| CheckMate 04021 | Nivolumab + ipilimumab (Arm A) |
PD1 and CTLA4 (immune checkpoints) | 2 / 4 / 8 / 86 | ≥72 | 36 | 80 | NR | 100 | 22.8mo | NR | NR; 32 |
This range corresponds to the reported survival data in SHARP (experimental arm), REFLECT and IMbrave150 (control arm).
The Hazard Ratio corresponds to the phase 3 SHARP trial that compared sorafenib with placebo.
Includes only liver-directed non-radiation therapies.
Includes only surgical procedures and radiotherapy. AFP, alpha-fetoprotein; BCLC, Barcelona Clinic Liver Cancer stage; CI, Confidence Interval; CTLA4, cytotoxic T-lymphocyte antigen 4; ECOG PS, Eastern Cooperative Oncology Group performance status; EHD, extrahepatic disease; FGFR, fibroblast growth factor receptor; HCC, hepatocellular carcinoma; MVI, macrovascular invasion; HR, Hazard Ratio; mRECIST, modified RECIST; NR, not reported; RECIST, Response Evaluation Criteria In Solid Tumors; OS, overall survival; ORR, overall response rate; PD1, programmed cell death protein 1; PD-L1, programmed death ligand 1; PDGFR, platelet-derived growth factor receptor; PFS, progression-free survival; VEGFR, vascular endothelial growth-factor receptor.
Placing systemic therapies in the context of HCC management.
Tumor stage, liver dysfunction and performance status underpin clinical practice guidelines of HCC from scientific societies1,2,83–86,95–97. The Barcelona Clinic for Liver Cancer (BCLC) staging algorithm proposed in 199998, endorsed by European and American Hepatology/Oncology-based organizations83–86, classifies patients into five stages (BCLC-0, A, B, C or D) and allocates them into specific treatments1,85 (Figure 2). In principle, patients with HCC at very early stage (BCLC-0, single HCC < 2cm) and early stage (BCLC-A, with a single tumor or 2–3 tumors <3cm in diameter), are considered for curative therapies such as resection, liver transplant (following Milan criteria)99 or ablation83–86,95–97. Downstaging –i.e., reducing the tumor burden with therapies to meet the Milan criteria– is accepted in the US100. Patients with preserved liver function and more advanced multifocal tumors confined to the liver are classed BCLC-B and treated with transarterial chemoembolization (TACE). These patients have median survival times of 26–30 months22. Patients with portal vein invasion or extra-hepatic disease are classed BCLC-C and systemic therapies are recommended. Around 50–70% of the patients receiving systemic therapies are progressing from surgery or locoregional therapies, while 30–50% are treatment-naïve101 (Table 2).
Figure 2.
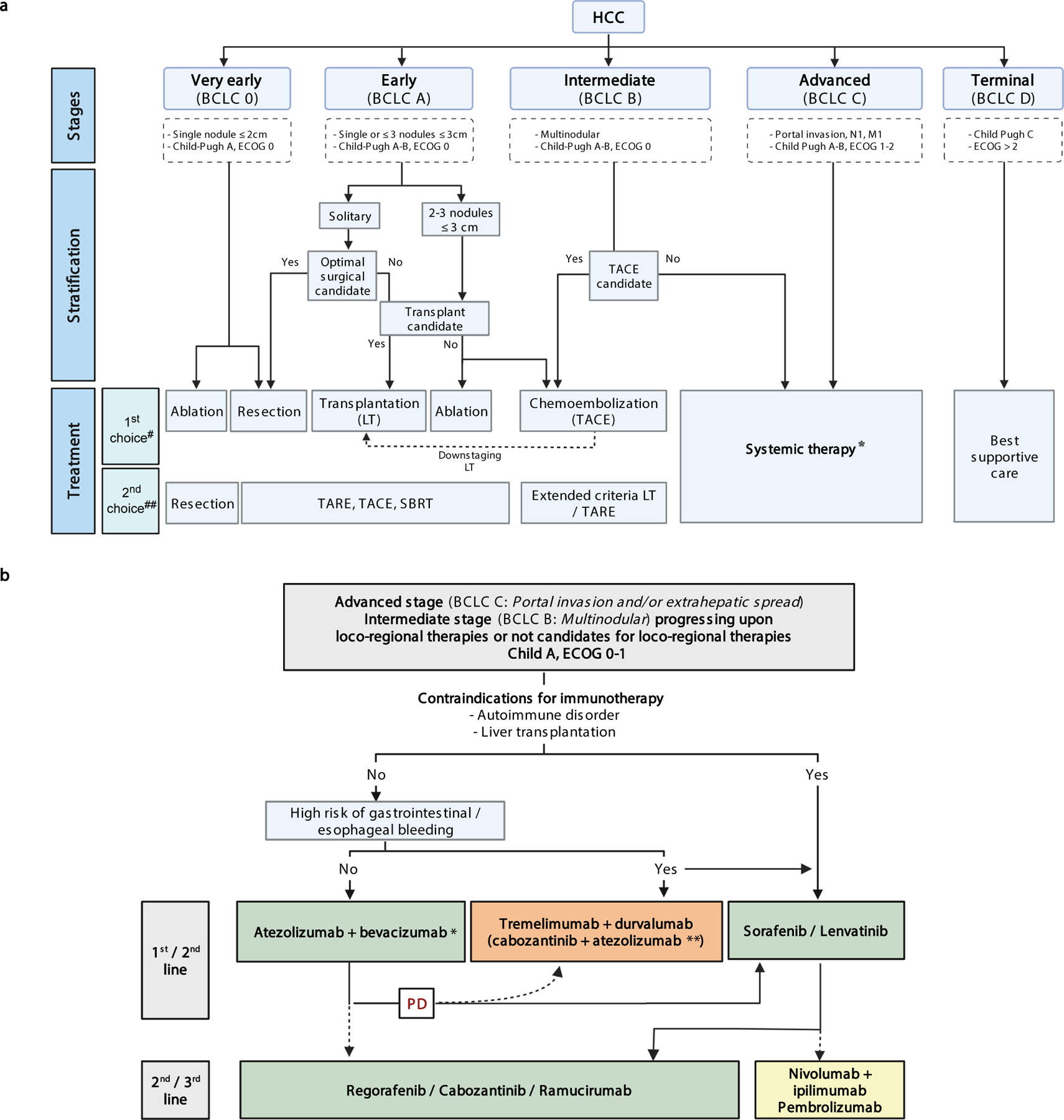
A) BCLC treatment algorithm with new systemic agents. Treatment strategy in the management of HCC is guided by the Barcelona Clinic Liver Cancer (BCLC) staging system, which consists of five stages depending on tumour burden features, liver function and performance status. Asymptomatic patients with low tumour burden and good liver function (BCLC 0/A) should be treated with local curative treatments (resection, ablation or transplantation, depending on the presence of portal hypertension, number of nodules and liver function). Asymptomatic patients with multinodular disease and adequate liver function (BCLC B) should receive chemoembolization and patients with portal thrombosis or extrahepatic spread (BCLC C) should be treated with systemic therapies. HCC, Hepatocellular carcinoma; ECOG PS, Eastern Cooperative Oncology Group performance status; LT, liver transplantation; M1, distant metastasis; N1, lymph node metastasis; SBRT, Stereotactic Body Radiation Therapy; TACE, Transarterial Chemoembolization; TARE; Transarterial Radioembolization. Adapted with permission from [ref. 23], Wiley. #: Based on high level of evidence studies. ##: Based on low/moderate level of evidence studies. *: see Figure 2b. [AU: we have initiated the collection of permissions to adapt and re-use parts of this figure]
B) Treatment strategy for HCC with systemic therapies. Green: Regulatory approved regimes based on phase III studies. Orange: positive combinations vs sorafenib, but drugs not yet approved. Yellow: treatments that got FDA accelerated approval based on phase II studies. (*) Around 70–80% of patients are expected to receive this regime. (**) COSMIC-312 phase III trial reported superior PFS for the combination of cabozantinib plus atezolizumab versus sorafenib, but final analysis on benefit on OS is not yet available94. BCLC, Barcelona Clinic Liver Cancer; ECOG PS, Eastern Cooperative Oncology Group performance status; PD, Progressive Disease.
Timing for systemic therapies in HCC.
Deciding when to transition from loco-regional to systemic treatment in patients at intermediate stage (BCLC-B) is of paramount importance. A late decision to transition might jeopardize gains in OS because only Child Pugh A class patients benefit from systemic therapies22. However, there is no consensus on when to halt local therapies22,102. Score-based selection of patients for treatment and re-treatment with TACE has not been thoroughly validated, and is not widely implemented103–105. Overall, recommendation of transitioning can be made in case of either progressive disease, impairment of liver function or occurrence of technical or other known contraindications for TACE during the ongoing therapy22,105. Lack of objective response after at least two treatment sessions of TACE is a clear predictor of poor survival106. The success of first line combination of atezolizumab plus bevacizumab leading to objective response of 35% and median survival of ~19 months for all patients18, with even better outcomes for patients at intermediate stage, also provides justification of moving to systemic agents when response to TACE is limited. Initial combinations of TACE with single-agent molecular therapies (i.e sorafenib or brivanib) did not yield positive results107–109, a feature that is expected to change with the immunotherapy-based combination regimens (Figure 2).
Selection of patients and expected outcomes
The seminal SHARP trial13 established the benchmark criteria for selection of patients for front-line systemic treatment – including Child-Pugh stage liver dysfunction, tumor burden and ECOG performance status. Table 2 summarizes the overall characteristics of patients included in phase III trials in first and second line. While in almost all studies Child-Pugh A is dominant (97%−100%), differences are observed in 1st vs 2nd line, respectively, regarding advanced tumor staging –BCLC C (~80% vs ~90%), ECOG performance status 0 (~60–70% vs ~50–60%), and extrahepatic spread (50–60% vs 70–80%). Overall, expected survival of patients treated with first line systemic therapies ranges from ~19 months for atezolizumab plus bevacizumab18 to 13–14 months for lenvatinib14 or sorafenib13. This improvement in OS from ~13 months (sorafenib) to ~19 month (atezolizumab plus bevacizumab) reflects not only the higher efficacy of the treatment, but additional nuances. For instance, the increased applicability of effective second line treatments (currently administered in ~30–50% of cases)13,14,18, the better patient selection, the treatment at earlier stages of the natural history of the disease and the better management of adverse events and complications. Other clinical end-points have also improved, such as PFS (ranging from 4–7 months), objective response rates (up to ~35% with combination atezolizumab plus bevacizumab) and patient reported outcomes. In second line therapies after progression on sorafenib, expected survival ranges from 10–11 months for regorafenib15 and cabozantinib16 to 8 months for ramucirumab in patients with aggressive tumors (AFP>400ng/ml)17. Subgroup analysis showed that some treatment-related adverse events are associated with better survival, including skin toxicity for sorafenib110 or hypertension for lenvatinib111, cabozantinib108 and ramucirumab112 (Table 3).
Table 3:
Systemic therapies approved for HCC: adverse events and regulatory status.
| Treatment (Study name) | Treatment dose (baseline) | Treatment-related adverse events (AE) (Grade 3–5) | Strategy dose reduction | Regulatory approval | |||||
|---|---|---|---|---|---|---|---|---|---|
| Overall prevalence | Prevalence of most common | % of patients undergoing dose reduction/ interruption | % of patients undergoing treatment withdrawal | % of patients with adverse events leading to death | Year (FDA/EMA) |
Country | |||
| First-line therapies | |||||||||
| Atezolizumab + bevacizumab (IMbrave15018) | 1200mg + 15mg/kg every 3w | 36% | Hypertension 10%; increased AST 4%; proteinuria 3% | Reduction: not allowed Interruption: 50% |
Withdrawal of atezolizumab or bevacizumab: 16%; Withdrawal of atezolizumab +bevacizumab: 7% |
2% | Not recommended | 2020 | EU US AWJPa JP |
| Sorafenib [SHARP13 (IMbrave150, REFLECT)] | 400mg every 12h | 45% | Diarrhea 8%; HFS 8%; fatigue 4% | Reduction: 26% Interruption: 44% |
11% | NR | Reduce 1 level (400mg q24h) if persistent G2 or G3. Discontinue if G4. | 2007 | EU US AWJP JP |
| Lenvatinib (REFLECT14) | 12mg≥ 60kg every 24h 8mg<60kg every 24h |
57% | Hypertension 23%; weight loss 8%; increased BR 7% | Reduction: 37% Interruption: 40% |
9% | 2% | Reduce 1 level (8 or 4mg q24h in ≥ 60kg and < 60kg, respectively) if persistent G2 or G3. Discontinue if G4 | 2018 | EU US AWJP JP |
| Second-line therapies | |||||||||
| Regorafenib (RESORCE15) | 160mg every 24h |
50% | Hypertension 13%; HFS 13%; fatigue 9% | Reduction/interruption: 68% | 10% | 2% | Reduce 1 level (120mg q24h) for persistent G2 and G3 AEs. Discontinue if G4. | 2017 | EU US AWJP JP |
| Cabozantinib (CELESTIAL16) | 60mg every 24h |
68%b | HFS 17%; hypertension 16%; increased AST 12% | Reduction: 62% | 16% | 1% | Reduce 1 level (40mg q24h) for persistent G2 and G3 AEs. Discontinue if G4. | 2018c | EU US AWJP |
| Ramucirumab (REACH-217) | 8mg/kg every 2w |
57% | Hypertension 8%; liver injury or failure 4%; proteinuria 2% | Reduction: 5% Interruption: 35% |
11% | 2% | Reduce 1 level (6mg/kg q2w) for G3 AEs. Discontinue if G4. | 2019 | EU US AWJPa JP |
| Pembrolizumab (KEYNOTE-240136) | 200mg every 3w |
53%b | Increased AST 13%; increased BR 8%; Increased ALT 6% | Reduction: not allowed Interruption: 30% |
17% | 3% | Not recommended | 2018 | US AWJPa |
| Pembrolizumab (KEYNOTE-22419) | 200mg every 3w |
26% | Increased AST 7%; increased ALT 4%; fatigue 4% | Reduction: not allowed Interruption: 25% |
5% | 1% | Not recommended | 2018 | US AWJPa |
| Nivolumab + ipilimumab (CheckMate 040 (Arm A) 21) | 1mg/kg + 3mg/kg every 3w (4 doses), followed by nivolumab 240mg every 2w | 53% | Increased AST 16%; increased lipase 12%; increased ALT 8%; fatigue 4% | Reduction: not allowed Interruption: NR |
22% | 2% | Not recommended | 2020 | US AWJPa |
Not in all AWJP countries.
Adverse events owing to all causes are shown.
FDA approval on 2019. AEs, adverse events; ALT, alanine aminotransferase; AST, aspartate aminotransferase; AWJP, Asia without Japan; BR, bilirubin; EMA, European Medicines Agency; EU, European Union; FDA, Food and Drug Administration; h, hours; HCC, hepatocellular carcinoma; HFS, hand–foot syndrome; NR, not reported; US, United States; w, weeks.
Although OS is recommended as the best endpoint for phase III trials testing systemic therapies, the fact that patients are exposed to effective second line treatments in ~30–50% of cases14,18,113 has posed a concern on whether OS should be the sole end-point in front-line research23. Previous concerns using PFS as end-point in HCC due to competing risk with cirrhosis-related death have been diminished by the universal selection of Child-Pugh A patients in phase III investigations, thus reducing the 1-year risk of death due to decompensation to <5%23. Nowadays, PFS has been proposed as primary end point using restrictive rules supported by results of RCT showing that a hazard ratio (HR) for PFS ≤0.6 is a good surrogate of OS benefit23. Conversely, HR for PFS of >0.6 are considered to have uncertain association with survival benefit, particularly in cases of HR >0.7 where almost all RCT have not shown any survival benefit of the tested drug23,79,114.
Evidence-based knowledge for systemic therapies in HCC
The treatment of advanced HCC has been limited to sorafenib for almost a decade13.The panorama of HCC therapy is constantly evolving and now a plethora of first and second line therapies are available, either as monotherapy or in combination with different agents, including immunotherapies. Here we discuss the available options and how are they continuously evolving.
Single-agent, targeted, first-line therapies
The seminal SHARP trial was a placebo-controlled, double-blinded study that randomized 602 patients to sorafenib or placebo13. This was the first systemic therapy approved as a result of improvements in overall survival (10.7 vs. 7.9 months) 13. The magnitude of effect was confirmed in an Asia-Pacific trial115. Sorafenib has been widely used globally, and subsequent studies suggested that it is more effective in liver-only disease, in HCV etiology and in patients with low neutrophil/lymphocyte ratio116. Initial recommended dose is of 800 mg per day, but it may be reduced (30% of cases) or withdrawn (in 10–15% of cases) due to treatment-related adverse events (Table 3), particularly hand-foot skin reaction, a feature that has been associated with better outcomes117. Treatment related death is less than 2%. In Child-Pugh B patients it is not well tolerated and leads to median overall survival of 5–6 months118.
A second era of first-line studies started after the approval of sorafenib14,18,119 (Table 2). Lenvatinib, a multikinase inhibitor blocking FGFR1–4 (Figure 1) was compared to sorafenib in the open-label REFLECT trial with a non-inferiority design, and demonstrated comparable efficacy with a HR of 0.92 and median overall survival of 13.6 versus 12.3 months14. Treatment dose is 8 or 12mg daily depending on the body weight (above or below 60 Kg). The most common adverse events are hypertension, weight loss and fatigue leading to treatment reduction in ~40% of cases and withdrawal in ~10% of cases. Subgroup analysis yielded better outcomes for lenvatinib in patients with high tumoral burden, aggressive disease, and HBV infection. Lenvatinib is currently tested in phase III in combination with pembrolizumab in patients at intermediate stages and in first line at advanced stages (Figure 2).
Other regimes tested in first line resulted in negative results, such as brivanib (a selective VEGFR and FGF receptor (FGFR) TKI)120, sunitinib (a multi-target TKI with activity against VEGFRs, PDGFRs, and KIT)121 and linifanib (a VEGFR and PDGFR TKI)122, as well as the combinations of sorafenib with erlotinib (an EGFR inhibitor)123, doxorubicin124, pravastatin125 or TACE126. The reasons behind these negative results are reviewed elsewhere127. Systemic doxorubicin, showed lack of survival benefits and was discarded from the treatment armamentarium of advanced HCC124. The STAH trial126 tested the combination of TACE and sorafenib compared to sorafenib alone in advanced HCC in Asian patients and did not meet its primary endpoint, survival (9.3 vs 9.4 months, HR 0.91). Only the SoraHAIC open-label trial reported superior efficacy of the combination of sorafenib and hepatic intraarterial chemotherapy (HAIC) of oxaliplatin, fluorouracil, and leucovorin (i.e. FOLFOX) vs sorafenib alone in advanced HCC with portal vein invasion119. This treatment regime that led to a significant increase in overall survival from 7.1 months to 13.3 months [HR of 0.35] has not been adopted by Western guidelines due to methodological concerns84,85,95,97. Finally, the phase III trials SARAH128 and SIRveNIB129 testing transarterial radioembolization (TARE) with Yttrium-90 (Y90) in first-line advanced HCC reported negative results compared to sorafenib. Based on these results, TARE is not recommended as an alternative to systemic therapy in the advanced setting.
Second-line targeted therapies
In second-line advanced HCC, regorafenib improved overall survival compared to placebo in the randomized, phase III RESORCE trial15. Regorafenib is a multikinase inhibitor, but with a broader range of angiogenic -including TIE2- and oncogenic targets than sorafenib (Figure 1)130. A key eligibility criterion was the requirement for prior treatment with first-line sorafenib at a dosage of at least 400 mg per day for at least 20, a fact that selected for patients with increased likelihood of tolerating regorafenib. In these patients regorafenib improved OS beyond placebo with a HR of 0.63 (95% CI: 0.50, 0.79) and median OS of 10.6 versus 7.8 months (p<0.0001). The sequential treatment strategy of sorafenib follow by regorafenib yield a OS of 26.0 months compared to 19.0 for sorafenib followed by placebo131. The most common grade 3 or 4 adverse events for regorafenib are hypertension, hand-foot skin reaction, fatigue, and diarrhea. In a retrospective multicenter analysis, regorafenib was associated with higher rates of grade 3 or 4 adverse events and shorter OS and PFS in patients with Child-Pugh B hepatic dysfunction than in Child-Pugh A patients132 (Table 3).
Cabozantinib is another multikinase inhibitor targeting anti-angiogenic pathways, MET, AXL, TYRO3, and MER, members of a family of proteins that contribute to a suppressed tumor immune microenvironment (Figure 1)133. In the randomized, phase III CELESTIAL trial, cabozantinib improved overall survival over placebo in patients who had received one or two prior systemic therapies for HCC, with a hazard ratio of 0.76 and a median overall survival of 10.2 months by comparison to 8.0 months for placebo (p=0.005)16 (Table 2). Cabozantinib also prolonged PFS, compared to placebo with medians of 5.2 months and 1.9 months, respectively (HR 0.44). The most frequent grade 3 or 4 adverse events were hand-foot skin reaction, hypertension, elevated transaminase levels, fatigue, and diarrhea. Finally, though the VEGFR2-targeted antibody ramucirumab did not improve survival compared to placebo in an unselected patient population, ramucirumab improved OS over placebo in patients with elevated AFP (>400 ng/ml) after progression on sorafenib (Figure 1)17,134. The median OS was 8.5 versus 7.3 months for ramucirumab and sorafenib, respectively (HR 0.71. The most common grade 3 or higher adverse events were hyponatremia and hypertension.
These positive clinical trials of regorafenib, cabozantinib, and ramucirumab were preceded by a multitude of negative studies in first- and second-line treatment settings90,120–124,135. The success of the more recent trials is credited to these agents’ distinct inhibitor profiles but may also reflect contributions from more favorable therapeutic indices and evolving supportive care for underlying hepatic dysfunction.
Immune checkpoint inhibitor monotherapies
Single agent Immune checkpoint inhibitors (ICIs) targeting PD1 were evaluated in advanced HCC and showed a safety profile similar to other solid tumors. In the phase I/II trials CheckMate 040 and KEYNOTE-224, nivolumab and pembrolizumab resulted in an objective response rate (ORR) ranging between 14 and 20%19,113. The confirmatory phase III trials for both agents failed to show a statistically significant improvement in OS. KEYNOTE-240 compared pembrolizumab to best supportive care in second line post sorafenib with median overall survival of 13.9 vs. 10.6 months but the pre-specified p value to reach significance was not reached136. Nonetheless, the Asian phase III trial comparing pembrolizumab versus placebo (KEYNOTE-394137) has rendered a positive OS outcomes with similar magnitude of benefit. In CheckMate 459, a randomized study of nivolumab versus sorafenib in first line HCC, the median OS was 16.4 months versus 14.7 months respectively with a HR of 0.85138. While both phase III studies suffered from some statistical design limitations and from cross-over to the ICI-treatment in the control arm, an important conclusion was that single agent ICIs may not have sufficient activity to show significant improvements in median OS in an unselected population.
In Checkmate 040, there was an association between PD-L1 expression ≥ 1% and OS in the overall trial population. In a subset of 37 patients, associations with ORR or overall survival were noted for 7 out of 10 evaluated inflammatory gene signatures61. A recent study also identified a gene signature able to predict response to either nivolumab or pembrolizumab62. If validated, such emerging biomarkers may be utilized for patient selection in the future.
ICI combination with anti-VEGF antibody
There is strong scientific rationale to combine ICIs with targeted therapies or other immune-oncology agents5,6. Anti-angiogenic therapies targeting VEGF ligands can mitigate the local immunosuppressive effects of VEGF signaling and promote T cell infiltration6,139. Effectively, combining bevacizumab, a monoclonal antibody targeting VEGF-A, with the anti-PD-L1 inhibitor atezolizumab (Figure 1) demonstrated safety and ORRs of 36% of patients in a large phase 1b study140 leading to the positive randomized, open-label, sorafenib-controlled trial phase III IMbrave150 trial of this combination with co-primary endpoints of OS and PFS18. This regimen represents a paradigm shift in the management of HCC due to the absolute gains in survival and has become a new standard of care for first-line treatment of advanced HCC. This study was halted at the interim analysis due to positive results favoring the combination arm, with a HR of 0.5818. Follow-up mature survival analysis confirmed the survival benefit with a median of 19.2 months for the combination arm compared to 13.2 month for sorafenib141. The trial required to perform an upper gastrointestinal endoscopy within the 6 months prior to randomization to exclude the presence of high risk esophageal/gastric varices, given the increased risk of bleeding associated with bevacizumab. The co-primary endpoint of PFS was also positive, with a HR of 0.59. In addition, patient-reported outcomes were significantly better for the combination compared to sorafenib alone (time to deterioration 11.2 months vs 3.6 months, respectively). Finally, objective response rate was significantly better in the combo arm (27–33% vs 12–13%). Thirty percent of patients treated with atezolizumab/ bevacizumab experienced durable objective responses, including 8% with confirmed complete responses. The most common grade 3 and 4 adverse event was hypertension with rare events of bleeding in the study population.
Combinations of ICI with TKIs
Multikinase inhibitors with anti-angiogenic activity as well as a diverse array of other kinase targets also hold the potential to modulate the tumor immune microenvironment in varying ways that could augment response to ICI142,143. In a large phase 1b study, the combination of lenvatinib with pembrolizumab achieved objective responses in 36%, with median PFS of 8.6 months and OS of ~22 months142. These findings prompted the ongoing randomized, phase III trial of this combination compared to lenvatinib monotherapy (LEAP-002). Another ongoing approach is the combination of cabozantinib with atezolizumab. The interim analysis of the randomized, phase III trial, COSMIC-312, comparing the efficacy of cabozantinib plus atezolizumab versus sorafenib revealed significant improvement of PFS (HR=0.63) but not of overall survival94. Other combinations of targeted therapies plus ICI are being investigated in earlier phase trials in advanced stages of HCC (Table 4, Figure 2).
Table 4:
Selected ongoing phase I-III trials for advanced HCC
| Agent(s) (Targets) | Primary endpoint | Line of treatment | Phase | Sample size | NCT |
|---|---|---|---|---|---|
| ICI combinations with targeted therapies | |||||
| Pembrolizumab (PD1), lenvatinib (VEGFR1–3, PDGFR, FGFR1–4, RET) | OS, PFS | 1st | III | 750 | NCT03713593 |
| Atezolizumab (PD-L1), cabozantinib (VEGFR1–3, MET, RET) | OS, PFS | 1st | III | 740 | NCT03755791 |
| AK105 (PD1), anlotinib (VEGFR1–3, FGFR1–4, PDGFR, KIT receptor) | OS | 1st | III | 648 | NCT04344158 |
| Camrelizumab (PD1), apatinib (VEGFR2) | OS, PFS | 1st | III | 510 | NCT03764293 |
| Tislelizumab (PD1), lenvatinib (VEGFR1–3, PDGFR, FGFR1–4, RET) | ORR | 1st | II | 66 | NCT04401800 |
| Nivolumab (PD1), sorafenib (VEGFR1–3, PDGFR, RAF kinase, KIT receptor) | ORR, MTD | 1st | II | 12 | NCT03439891 |
| Pembrolizumab (PD1), sorafenib (VEGFR1–3, PDGFR, RAF kinase, KIT receptor) | ORR | 1st | I/II | 27 | NCT03211416 |
| HX008 (PD1), bevacizumab (VEGFA), lenvatinib (VEGFR1–3, PDGFR, FGFR1–4, RET) | ORR | 1st | II | 72 | NCT04741165 |
| CS1001 (PD-L1), fisogatinib (FGFR4) | ORR, DLT | 1st or 2nd | Ib/II | 52 | NCT04194801 |
| Pembrolizumab (PD1), regorafenib (VEGFR1–3, PDGFR, RAF kinase, FGFR1–2) | ORR | 2nd | II | 119 | NCT04696055 |
| Atezolizumab (PD-L1), sorafenib (VEGFR1–3, PDGFR, RAF kinase, KIT receptor), lenvatinib (VEGFR1–3, PDGFR, FGFR1–4, RET) | OS | 2nd | III | 554 | NCT04770896 |
| PDR001 PD1, INC280/campatinib (MET) | ORR, DLT | 2nd | Ib/II | 90 | NCT02795429 |
| Tislelizumab (PD1), sitravatinib (TYRO3, AXL, MERTK, VEGFR2, KIT receptor, MET) | ORR, incidence of AEs/SAEs | Refractory to standard therapies | I/II | 104 | NCT03941873 |
| ICI combinations with other ICI | |||||
| Durvalumab (PD-L1) plus tremelimumab (CTLA4) | OS | 1st | III | 1504 | NCT03298451 |
| Nivolumab (PD1) plus ipilimumab (CTLA4) | OS | 1st | III | 650 | NCT04039607 |
| Nivolumab (PD1), relatlimab (LAG-3) | ORR | 2nd | II | 250 | NCT04567615 |
| Triplet combinations involving ICI plus targeted therapies | |||||
| Atezolizumab (PD-L1), bevacizumab (VEGFA), tiragolumab (TIGIT), tocilizumab (IL6R), SAR439459 (TGFβ), TPST-1120 (PPARα), RO7247669 (PD1 + LAG-3) | ORR | 1st | Ib/II | 280 | NCT04524871 |
| Pembrolizumab (PD1), quavonlimab (CTLA4), lenvatinib (VEGFR1–3, PDGFR, FGFR1–4, RET) | ORR, DLT, incidence of AEs/SAEs, hepatic AEs, discontinuation due to AEs. | 1st | II | 110 | NCT04740307 |
| Nivolumab (PD1), ipilimumab (CTLA4), cabozantinib (VEGFR1–3, MET, RET) | ORR, incidence of AEs/SAEs | 1st or 2nd | I/II | 1097 | NCT01658878 |
| Novel immunologic targets | |||||
| Voyager V1 (VSV oncolytic virus), cemiplimab (PD1) | ORR | 2nd | II | 152 | NCT04291105 |
| Talimogene laherparepvec (T-VEC, HSV oncolytic virus), pembrolizumab (PD1) | DLT, ORR | 2nd | I/II | 206 | NCT02509507 |
| GNOS-PVO2 (personalized neoantigen), INO-9012 (IL-12), pembrolizumab (PD1) | Incidence of AEs, immunogenicity | 2nd | I/II | 24 | NCT04251117 |
| ET140203 T cells (AFP) | Incidence of AEs, DLTs, RP2D | 3rd+ | I/II | 50 | NCT04502082 |
| ECT204 T cells (GPC3) | Incidence of AEs, DLTs, RP2D | 3rd+ | I/II | 12 | NCT04864054 |
| Other targeted therapies | |||||
| Icaritin (Stem cells) | OS | 1st | III | 200 | NCT03236649 |
| CVM-1118 (Vascular mimicry), sorafenib (VEGFR1–3, PDGFR, RAF kinase, KIT receptor) | ORR | TKI-naïve | II | 40 | NCT03582618 |
| Sorafenib (VEGFR1–3, PDGFR, RAF kinase, KIT receptor), YIV-906 (Unknown) | PFS | 1st | II | 125 | NCT04000737 |
| MTL-CEPBA (C/EBP-alpha transcription factor), sorafenib (VEGFR1–3, PDGFR, RAF kinase, KIT receptor) | ORR, incidence of AEs | TKI-naïve | II | 70 | NCT04710641 |
| ATG-008/CC-223 (mTORC1/2) | ORR, Cmax, AUC, Incidence TEAEs/SAEs | 2nd | II | 75 | NCT03591965 |
| DKN-01 (DKK1), sorafenib (VEGFR1–3, PDGFR, RAF kinase, KIT receptor) | TTP, AEs | 1st | I/II | 70 | NCT03645980 |
| MLN0128 (mTORC1/2) | MTD, TTP | 2nd | I/II | 11 | NCT02575339 |
Note: If multiple studies exist of the same regimen, the latest-phase study is presented. AEs, Adverse events; AFP, alpha-fetoprotein; AUC, Area under the curve; DLT: Dose-limiting toxicity; GPC3, glypican 3; ICI, immune checkpoint inhibitor; MTD, Maximum tolerated dose; mTORC, mammalian target of rapamycin; ORR, objective response rate; PD1, programmed cell death protein 1; PD-L1, programmed death ligand 1; RP2D, Recommended phase 2 dose; SAEs, Severe adverse events; TEAEs, Treatment-emergent adverse events; TKI, tyrosine kinase inhibitor; TTP, Time to progression; VEGFR2, vascular endothelial growth factor receptor 2; VSV, vesicular stomatitis virus.
Immuno-oncology combinations
Co-targeting CTLA4 synergizes with anti-PD1 activity through regulation of T-cell activation in lymph nodes and tissues144. Preclinical studies have shown that anti-CTLA4 inhibition results in expansion of an ICOS+ Th1-like CD4 effector population in addition to engaging specific subsets of exhausted-like CD8 T cells145. In HCC, the combination of nivolumab and ipilimumab has shown promising efficacy with an overall response rate (ORR) of 32% and median overall survival of 22.8 months in second line, which resulted in accelerated approval by the US FDA21. There were no new safety signals but the higher dose of ipilimumab was associated with increased frequency of immune mediated events21. A phase III trial (checkmate 9DW) of the combination of nivolumab and ipilimumab vs. sorafenib or lenvatinib is ongoing. A similar promising signal of activity was seen in Study 22 of durvalumab (anti-PD-L1) with a single loading dose of tremelimumab (anti-CTLA4); the combination resulted in an ORR of 24% and a median overall survival of 18.7 months, along with a manageable safety profile146. More recently, the phase III Himalaya trial has shown that durvalumab with a single, high priming dose of tremelimumab is able to significantly improve OS versus sorafenib as a first line treatment (HR: 0.78; 16.4mo vs 13.8 mo)93.
Selection of first line therapies and treatment sequencing
In principle, if a given treatment is not available or contraindicated for a specific BCLC stage (for instance TACE for intermediate HCC), systemic treatment is recommended (Figure 2). This concept is known as treatment stage migration. A bigger challenge is how to sequentially apply different systemic therapies. Among systemic regimens approved (Figures 1 and 2, Table 2), only a few were compared face-to-face, and none of the approved single agents has been explored after progression of atezolizumab plus bevacizumab.
There is a general agreement that the standard of care in first-line advanced HCC is atezolizumab plus bevacizumab (Figures 1 and 2). There are some restrictions for the use of this combination according to the inclusion criteria reported in the phase III trial: Child-Pugh class A and ECOG PST 0–1, in the absence of other organ/hematology dysfunction, autoimmune disease, active co-infection with HCV or HBV, or untreated varices. Specifically, an upper gastrointestinal endoscopy (within 6 months prior) is required to discard high risk varices. If present, endoscopic band ligation is recommended147. If this decision is taken, it is advised to start the systemic treatment after ~2–6 weeks according to institutional guidelines. In case of untreated varices, durvalumab plus tremelimumab can be considered93. Other major contraindications are prior liver transplantation treated with immunosuppressive drugs due to the risk of graft rejection. In all these circumstances that are estimated to affect ~20% of patients, the treatment of choice in first line should be either sorafenib or lenvatinib1,23,83,97,148,149. A recent meta-analysis concluded that immune therapies are more effective in viral than non-viral etiologies11,101. Collectively, these data suggest that differences in the tumor microenvironment, likely etiology-related, can impact response to systemic therapies and underscore the importance of clinical annotation and stratification for etiology of liver disease in clinical trials for HCC.
The main controversy is how to sequence therapies after progression to atezolizumab plus bevacizumab, due to the lack of phase III investigations assessing the efficacy of second line therapies in this scenario. Most updated guidelines support the view that sorafenib or lenvatinib should be offered first, thus maintaining the previously established evidence-based hierarchy prior to atezolizumab/bevacizumab becoming the first line preferred treatment1,23,97,149. The most valuable clinical variables for decision-making are the magnitude of clinical benefit in OS, then PFS or ORR, patient comorbidities, patient quality of life and drug adverse event profile, and finally local availability and/or reimbursement. A summary of these factors is detailed in Table 2 and Table 3 to facilitate decision-making. Re-imbursement plays a significant role in certain regions and in the absence of evidence that any is superior, sorafenib is the most commonly offered second line agent after atezolizumab/bevacizumab150. Upon progression to lenvatinib or sorafenib, conventional second line therapies can be administered. Specifically, regorafenib is indicated in patients that tolerate sorafenib, whereas cabozantinib and ramucirumab were assessed upon progression to sorafenib, the latter indicated only in patients with AFP > 400 ng/ml. There are no head to head comparisons between regorafenib, cabozantinib or ramucirumab and their reported response rates after TKI are similar15–17. Dose modifications and grade 3 adverse events were reported less frequently for ramucirumab, compared to the other agents, indicative that ramucirumab may be better tolerated in elderly patients with cirrhosis or ECOG PST>017. Pembrolizumab is FDA approved and can be considered in second-line scenarios in the US, particularly if adverse events and comorbidities might be detrimental with other agents. The role of durvalumab plus tremelimumab in second line needs to be established. There is not enough data to recommend a specific therapy for patients with liver dysfunction (Child-Pugh B class).
Novel therapeutic strategies
Most of therapeutic strategies in phase II-III trials are involving ICIs in combination with TKIs, other ICI or triplet combinations including all the above (Table 4). Nonetheless, novel therapeutic approaches are being explored in the setting of phase I-II investigations. The advent of single-cell genomic technologies151 has been instrumental to improve cell taxonomy152, assess cellular functional states153 and decipher cell-cell interactions154. This has revealed that, for instance, CAFs are critical in tumor progression or are involved in chemoresistance by sustaining stemness in cancer155,156,157. Clinical trials of novel agents targeting cancer stem cells, such as icaritin and DKN-01, are ongoing in HCC (Table 4).
Tumor infiltrating lymphocytes (TILs) are highly heterogeneous , and as many as eleven subsets of unique subpopulation of CD8+FOXP3+ cells have been identified using single cell RNA sequencing and single cell T-cell receptor sequencing in HCC74. The degree of tumor lymphocyte infiltration is geographically different within the same tumor nodule, with some areas heavily infiltrated while others have minimal TILs. As expected, these differences in TIL burden correlate with predicted tumor neoantigen distribution10, which suggest interaction between cancer and cytotoxic immune158. Given that most biomarker studies use single tissue biopsies as source material, these intratumoral differences in TIL burden could interfere with biomarker discovery and validation. This was addressed in a comprehensive multidimensional study of a small cohort of twelve HCC patients159. Integrated transcriptomic and immunohistochemistry data demonstrated that most patients (60–70%) had consistent signals in terms of immune activation throughout different areas of the same tumor nodule. A variety of approaches including oncolytic viruses coupled with immune checkpoint inhibition as well as personalized neoantigen vaccines are being studied to induce lymphocyte infiltration in the tumor microenvironment (Table 4). Single cell technologies have been applied to study mechanism of resistance in HCC, including patients with paired biopsies before and after treatment with combined durvalumab and tremelimumab160.
Finally, a leading candidate target for both peptide vaccines and engineered T cell receptor (TCR) or chimeric antigen receptor (CAR)-T cell therapies is glypican-3, a cell-surface glycoprotein over-expressed in over 70% of HCC but marginally expressed in cirrhotic liver161–163. AFP is another candidate target for both vaccine and T cell therapies based upon its expression prevalence of around 50% in advanced HCC, without significant expression in non-tumor liver164,165. Current ongoing clinical trials testing CAR-T immunotherapy, TCR engineered T cells, CAR-NK cells or HCC vaccines, among others, have been extensively reviewed elsewhere166 (Table 4).
Future directions
There is a high expectation on the impact of ongoing phase III studies in the clinical decision-making for the next years at all stages of the disease (Figure 2). Neoadjuvant and adjuvant therapies in HCC are still an unmet need, and future studies will explore their utility in depth. These advancements will have implications in the composition of multidisciplinary teams, since the presence of experts in managing systemic therapies will be routinely requested for management of early stages of HCC. In addition, there is a need to identify biomarkers predicting response to single ICI or combinations. Post-hoc analysis of PD-L1 expression did not predict response to single agent ICI s19,113, while gene signatures are in need of further validation61,62. Liquid biopsy has emerged as a non-invasive technology for biomarker discovery in HCC167. Although there are reports correlating mutation and copy number alteration analysis of ctDNA with HCC tissue, further research is needed to validate these biomarkers for surveillance or treatment allocation168,169. Finally, from the regulatory and reimbursement perspective, studies addressing the cost-effectiveness of sequential expensive therapies would need to be considered. Overall, there is an expected shift in the landscape of management that should be accompanied by the identification of biomarkers to guide precision oncology, and to adapt trial design and endpoints to the new clinical scenarios.
Acknowledgements
We thank Marta Piqué, PhD student, and Florian Castet, MD, members of Prof. Llovet’s Lab for their support in the production of this manuscript. J.M.L is supported by grants from Cancer Research UK, Fondazione AIRC and Fundación Científica de la Asociación Española Contra el Cáncer (HUNTER, Ref. C9380/A26813), the NIH (RO1DK56621 and RO1DK128289), the Samuel Waxman Cancer Research Foundation, EIT Health (CRISH2, Ref. 18053), Generalitat de Catalunya (AGAUR, SGR-1358), the Spanish National Health Institute (MICINN, PID2019–105378RB-I00) and the Acadèmia de Ciències Mèdiques i de la Salut de Catalunya i de Balears (Ref. BECA_ACADEMIA21_001). X.W.W. is supported by grants (Z01 BC 010877, Z01 BC 010876, Z01 BC 010313 and ZIA BC 011870) from the intramural research program of the Center for Cancer Research, National Cancer Institute of the United States.
Footnotes
Competing interests
J.M.L received research support from Bayer HealthCare Pharmaceuticals, Eisai Inc, Boehringer-Ingelheim and Ipsen, and consulting fees from Merck, Eli Lilly, Eisai Inc, Bayer HealthCare Pharmaceuticals, Bristol-Myers Squibb, Ipsen, Genentech, Roche, Glycotest, Nucleix, Omega Therapeutics, Iylon, Mina Alpha Ltd, Boston Scientific and AstraZeneca. H.R. has acted in an advisory capacity for Boston Scientific and Sirtex, as well as receiving speaker fees from Eisai and Bayer. A.V. has received consulting fees from Boehringer Ingelheim, FirstWorld, Natera, Cambridge Healthcare Research and Genentech; advisory board fees from Bristol Myers-Squibb, Gilead and NGM Pharmaceuticals; and research support from Eisai. R.K.K. received research support from Agios, Astra Zeneca, Bayer, Bristol Myers-Squibb, Eli Lilly, EMD Serono, Exelixis, Genentech/Roche, Merck, Partner Therapeutics, Novartis, QED, Relay Therapeutics, Surface Oncology and Taiho; and consulting/advisory fees from Exact Sciences, Genentech/Roche and Gilead. A.E-K. has received research support from Astex, Astrazeneca, and Fulgent; advisory or consulting fees from Bayer, Bristol Myers-Squibb, EISAI, Merck, Exelixis, Astrazeneca, Roche/Genentech, Agenus, ABL Bio, QED, Gilead, Cytomx, Pieris, and EMD Serono. G.G., X.W.W. and R.P. have no competing interests.
References
- 1.Llovet JM et al. Hepatocellular carcinoma. Nat. Rev. Dis. Prim 7, 7 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Villanueva A Hepatocellular Carcinoma. N. Engl. J. Med 380, 1450–1462 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Schulze K et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet 47, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zucman-Rossi J, Villanueva A, Nault JC & Llovet JM Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 149, 1226–1239 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Llovet JM, Montal R, Sia D & Finn RS Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol 15, 599–616 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Llovet JM et al. Immunotherapies for hepatocellular carcinoma. Nat. Rev. Clin. Oncol (2021) doi: 10.1038/s41571-021-00573-2. [DOI] [PubMed] [Google Scholar]
- 7.Ding X et al. Genomic and Epigenomic Features of Primary and Recurrent Hepatocellular Carcinomas. Gastroenterology 157, 1630–1645.e6 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Sia D, Villanueva A, Friedman SL & Llovet JM Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 152, 745–761 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xue R et al. Variable Intra-Tumor Genomic Heterogeneity of Multiple Lesions in Patients with Hepatocellular Carcinoma. Gastroenterology 150, 998–1008. (2016). [DOI] [PubMed] [Google Scholar]
- 10.Losic B et al. Intratumoral heterogeneity and clonal evolution in liver cancer. Nat. Commun 11, 1–15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pfister D et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 592, 450–456 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Llovet JM, Burroughs A & Bruix J Hepatocellular carcinoma. Lancet 362, 1907–1917 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Llovet JM et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med 359, 378–390 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Kudo M et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet 391, 1163–1173 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Bruix J et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 389, 56–66 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Abou-Alfa GK et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med 379, 54–63 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu AX et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 20, 282–296 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Finn RS et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med 382, 1894–1905 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Zhu AX et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol 19, (2018). [DOI] [PubMed] [Google Scholar]
- 20.[Press Release] FDA grants accelerated approval to nivolumab and ipilimumab combination for hepatocellular carcinoma (2020). [Google Scholar]
- 21.Yau T et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol 6, e204564 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Llovet JM et al. Locoregional therapies in the era of molecular and immune treatments for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol 18, 293–313 (2021). [DOI] [PubMed] [Google Scholar]
- 23.Llovet JM et al. Trial Design and Endpoints in Hepatocellular Carcinoma: AASLD Consensus Conference. Hepatology 73 Suppl 1, 158–191 (2021). [DOI] [PubMed] [Google Scholar]
- 24.Hernandez‐Meza G et al. DNA-Methylation Profiling of Human Hepatocarcinogenesis. Hepatology 74, 183–199 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meunier L et al. DNA Methylation Signatures Reveal the Diversity of Processes Remodeling Hepatocellular Carcinoma Methylomes. Hepatology 74, 816–834 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hlady RA et al. Interferon drives hepatitis C virus scarring of the epigenome and creates targetable vulnerabilities following viral clearance. Hepatology hep.32111 (2021) doi: 10.1002/hep.32111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michalopoulos GK & Bhushan B Liver regeneration: biological and pathological mechanisms and implications. Nat Rev Gastroenterol Hepatol 18, 40–55 (2021). [DOI] [PubMed] [Google Scholar]
- 28.Bruno S et al. Human Liver Stem Cells: A Liver-Derived Mesenchymal Stromal Cell-Like Population With Pro-regenerative Properties. Front. Cell Dev. Biol 9, 1–14 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramachandran P, Matchett KP, Dobie R, Wilson-Kanamori JR & Henderson NC Single-cell technologies in hepatology: new insights into liver biology and disease pathogenesis. Nat. Rev. Gastroenterol. Hepatol 17, 457–472 (2020). [DOI] [PubMed] [Google Scholar]
- 30.Lee TK-W, Guan X-Y & Ma S Cancer stem cells in hepatocellular carcinoma — from origin to clinical implications. Nat. Rev. Gastroenterol. Hepatol (2021) doi: 10.1038/s41575-021-00508-3. [DOI] [PubMed] [Google Scholar]
- 31.Li W, Li L & Hui L Cell Plasticity in Liver Regeneration. Trends Cell Biol 30, 329–338 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Hirsova P et al. Hepatocyte apoptosis is tumor promoting in murine nonalcoholic steatohepatitis. Cell Death Dis 11, 80 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bayard Q et al. Cyclin A2/E1 activation defines a hepatocellular carcinoma subclass with a rearrangement signature of replication stress. Nat. Commun 9, 5235 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gellert-Kristensen H et al. Combined Effect of PNPLA3, TM6SF2, and HSD17B13 Variants on Risk of Cirrhosis and Hepatocellular Carcinoma in the General Population. Hepatology 72, 845–856 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Yang JD et al. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol 16, 589–604 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nault JC & Letouze E Mutational Processes in Hepatocellular Carcinoma: The Story of Aristolochic Acid. Semin Liver Dis 39, 334–340 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Ningarhari M et al. Telomere length is key to hepatocellular carcinoma diversity and telomerase addiction is an actionable therapeutic target. J Hepatol 74, 1155–1166 (2021). [DOI] [PubMed] [Google Scholar]
- 38.Torrecilla S et al. Trunk mutational events present minimal intra- and inter-tumoral heterogeneity in hepatocellular carcinoma. J. Hepatol 67, 1222–1231 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Lin S et al. Distributed hepatocytes expressing telomerase repopulate the liver in homeostasis and injury. Nature 556, 244–248 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun T et al. ZNRF3 and RNF43 cooperate to safeguard metabolic liver zonation and hepatocyte proliferation. Cell Stem Cell 28, 1822–1837.e10 (2021). [DOI] [PubMed] [Google Scholar]
- 41.Wheeler DA & Roberts LR Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 169, 1327–1341.e23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sawey ET et al. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by Oncogenomic screening. Cancer Cell 19, 347–358 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beroukhim R et al. The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim RD et al. First-in-human phase i study of fisogatinib (BLU-554) validates aberrant FGF19 signaling as a driver event in hepatocellular carcinoma. Cancer Discov 9, 1696–1707 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Chiang DY et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res 68, 6779–88 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martinez-Quetglas I et al. IGF2 Is Up-regulated by Epigenetic Mechanisms in Hepatocellular Carcinomas and Is an Actionable Oncogene Product in Experimental Models. Gastroenterology 151, 1192–1205 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Noonan A & Pawlik TM Hepatocellular carcinoma: an update on investigational drugs in phase I and II clinical trials. Expert Opin. Investig. Drugs 28, 941–949 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Luo X-Y, Wu K-M & He X-X Advances in drug development for hepatocellular carcinoma: clinical trials and potential therapeutic targets. J. Exp. Clin. Cancer Res 40, 172 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schapira M, Calabrese MF, Bullock AN & Crews CM Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug Discov 18, 949–963 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Hoshida Y et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res 69, 7385–7392 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boyault S et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 45, 42–52 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Lee JS et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med 12, 410–416 (2006). [DOI] [PubMed] [Google Scholar]
- 53.Toffanin S et al. MicroRNA-based classification of hepatocellular carcinoma and oncogenic role of miR-517a. Gastroenterology 140, 1618–28.e16 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sia D et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 153, 812–826 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Montironi C et al. Inflamed and non-inflamed classes of HCC: a revised immunogenomic classification. Gut (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang K et al. Genomic landscape of copy number aberrations enables the identification of oncogenic drivers in hepatocellular carcinoma. Hepatology 58, 706–17 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Bassaganyas L et al. Copy-Number Alteration Burden Differentially Impacts Immune Profiles and Molecular Features of Hepatocellular Carcinoma. Clin. Cancer Res 26, 6350–6361 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Villanueva A et al. DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Hepatology 61, 1945–1956 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lachenmayer A et al. Wnt-pathway activation in two molecular classes of hepatocellular carcinoma and experimental modulation by sorafenib. Clin. Cancer Res 18, 4997–5007 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pinyol R et al. Molecular characterisation of hepatocellular carcinoma in patients with non-alcoholic steatohepatitis. J. Hepatol 75, 865–878 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sangro B et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab-treated patients with advanced hepatocellular carcinoma. J. Hepatol 73, 1460–1469 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haber PK et al. Molecular markers of response to anti-PD1 therapy in advanced hepatocellular carcinoma. Oral Abstracts. Hepatology 74, 1–156 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Greaves M & Maley CC Clonal evolution in cancer. Nature 481, 306–13. (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Khatib S, Pomyen Y, Dang H & Wang XW Understanding the Cause and Consequence of Tumor Heterogeneity. Trends in cancer 6, 267–271 (2020). [DOI] [PubMed] [Google Scholar]
- 65.Tao Y et al. Rapid growth of a hepatocellular carcinoma and the driving mutations revealed by cell-population genetic analysis of whole-genome data. Proc. Natl. Acad. Sci. U. S. A 108, 12042–7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang A et al. Circumventing intratumoral heterogeneity to identify potential therapeutic targets in hepatocellular carcinoma. J. Hepatol 67, 293–301. (2017). [DOI] [PubMed] [Google Scholar]
- 67.Maley CC et al. Classifying the evolutionary and ecological features of neoplasms. Nat. Rev. Cancer 17, 605–619 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McGranahan N & Swanton C Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 168, 613–628 (2017). [DOI] [PubMed] [Google Scholar]
- 69.Nam AS, Chaligne R & Landau DA Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat. Rev. Genet 22, 3–18 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Black JRM & McGranahan N Genetic and non-genetic clonal diversity in cancer evolution. Nat. Rev. Cancer 21, 379–392 (2021). [DOI] [PubMed] [Google Scholar]
- 71.Marjanovic ND et al. Emergence of a High-Plasticity Cell State during Lung Cancer Evolution. Cancer Cell 38, 229–246.e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Um TH et al. Aberrant CpG island hypermethylation in dysplastic nodules and early HCC of hepatitis B virus-related human multistep hepatocarcinogenesis. J. Hepatol 54, 939–947 (2011). [DOI] [PubMed] [Google Scholar]
- 73.Heinrich S et al. Understanding tumour cell heterogeneity and its implication for immunotherapy in liver cancer using single-cell analysis. J. Hepatol 74, 700–715 (2021). [DOI] [PubMed] [Google Scholar]
- 74.Zheng C et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 169, 1342–1356.e16 (2017). [DOI] [PubMed] [Google Scholar]
- 75.Zhang Q et al. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 179, 829–845.e20 (2019). [DOI] [PubMed] [Google Scholar]
- 76.Zheng H et al. Single-cell analysis reveals cancer stem cell heterogeneity in hepatocellular carcinoma. Hepatology 68, 127–140 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ma L et al. Tumor Cell Biodiversity Drives Microenvironmental Reprogramming in Liver Cancer. Cancer Cell 36, 418–430.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sun Y et al. Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma. Cell 184, 404–421.e16 (2021). [DOI] [PubMed] [Google Scholar]
- 79.Llovet JM, Montal R & Villanueva A Randomized trials and endpoints in advanced HCC: Role of PFS as a surrogate of survival. J. Hepatol 70, 1262–1277 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hyman DM, Taylor BS & Baselga J Implementing Genome-Driven Oncology. Cell 168, 584–599 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.de Gramont A et al. Pragmatic issues in biomarker evaluation for targeted therapies in cancer. Nat. Rev. Clin. Oncol 12, 197–212 (2015). [DOI] [PubMed] [Google Scholar]
- 82.Zehir A et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med 23, 703–713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vogel A et al. Hepatocellular carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol 29, iv238–iv255 (2018). [DOI] [PubMed] [Google Scholar]
- 84.Marrero JA et al. Diagnosis, Staging, and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 68, 723–750 (2018). [DOI] [PubMed] [Google Scholar]
- 85.Galle PR et al. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J Hepatol 69, 182–236 (2018). [DOI] [PubMed] [Google Scholar]
- 86.Omata M et al. Asia-Pacific clinical practice guidelines on the management of hepatocellular carcinoma: a 2017 update. Hepatol. Int 11, 317–370 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Llovet JM & Ducreux M EASL-EORTC Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol 56, 908–943 (2012). [DOI] [PubMed] [Google Scholar]
- 88.Singal AG et al. International Liver Cancer Association (ILCA) White Paper on Biomarker Development for Hepatocellular Carcinoma. Gastroenterology 160, 2572–2584 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rebouissou S et al. Proliferation markers are associated with MET expression in hepatocellular carcinoma and predict tivantinib sensitivity in vitro. Clin. Cancer Res 23, 4364–4375 (2017). [DOI] [PubMed] [Google Scholar]
- 90.Rimassa L et al. Tivantinib for second-line treatment of MET-high, advanced hepatocellular carcinoma (METIV-HCC): a final analysis of a phase 3, randomised, placebo-controlled study. Lancet Oncol 19, 682–693 (2018). [DOI] [PubMed] [Google Scholar]
- 91.Lim HY et al. Phase II Studies with Refametinib or Refametinib plus Sorafenib in Patients with RAS-Mutated Hepatocellular Carcinoma. Clin. Cancer Res 24, 4650–4661 (2018). [DOI] [PubMed] [Google Scholar]
- 92.Hatlen MA et al. Acquired on-target clinical resistance validates fgfr4 as a driver of hepatocellular carcinoma. Cancer Discov 9, 1686–1695 (2019). [DOI] [PubMed] [Google Scholar]
- 93.Abou-Alfa GK et al. Phase 3 randomized, open-label, multicenter study of tremelimumab (T) and durvalumab (D) as first-line therapy in patients (pts) with unresectable hepatocellular carcinoma (uHCC): HIMALAYA. J. Clin. Oncol 40, 379 (2022). [Google Scholar]
- 94. [Press Release-28/06/2021] Exelixis and Ipsen Announce Cabozantinib in Combination with an Immune Checkpoint Inhibitor Significantly Improved Progression-Free Survival in Phase 3 COSMIC-312 Pivotal Trial in Patients with Previously Untreated Advanced Live. [Google Scholar]
- 95.Vogel A et al. Updated treatment recommendations for hepatocellular carcinoma (HCC) from the ESMO Clinical Practice Guidelines. Ann. Oncol 32, 801–805 (2021). [DOI] [PubMed] [Google Scholar]
- 96.Chen LT et al. Pan-Asian adapted ESMO Clinical Practice Guidelines for the management of patients with intermediate and advanced/relapsed hepatocellular carcinoma: a TOS-ESMO initiative endorsed by CSCO, ISMPO, JSMO, KSMO, MOS and SSO. Ann Oncol 31, 334–351 (2020). [DOI] [PubMed] [Google Scholar]
- 97.Gordan JD et al. Systemic Therapy for Advanced Hepatocellular Carcinoma: ASCO Guideline. J. Clin. Oncol 38, 4317–4345 (2020). [DOI] [PubMed] [Google Scholar]
- 98.Llovet JM, Brú C & Bruix J Prognosis of hepatocellular carcinoma: the BCLC staging classification. Semin. Liver Dis 19, 329–338 (1999). [DOI] [PubMed] [Google Scholar]
- 99.Mazzaferro V et al. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N Engl J Med 334, 693–699 (1996). [DOI] [PubMed] [Google Scholar]
- 100.Yao FY et al. Downstaging of hepatocellular cancer before liver transplant: Long-term outcome compared to tumors within Milan criteria. Hepatology 61, 1968–1977 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Haber PK et al. Evidence-Based Management of Hepatocellular Carcinoma: Systematic Review and Meta-analysis of Randomized Controlled Trials (2002–2020). Gastroenterology 161, 879–898 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Raoul J-L et al. Updated use of TACE for hepatocellular carcinoma treatment: How and when to use it based on clinical evidence. Cancer Treat. Rev 72, 28–36 (2019). [DOI] [PubMed] [Google Scholar]
- 103.Kadalayil L et al. A simple prognostic scoring system for patients receiving transarterial embolisation for hepatocellular cancer. Ann. Oncol 24, 2565–2570 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Attallah AM et al. HCC-ART score, a simple, highly sensitive and specific test for early diagnosis of hepatocellular carcinoma: a large-scale, multicentre study. Br. J. Cancer 109, 1657–1665 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Palmer DH, Malagari K & Kulik LM Role of locoregional therapies in the wake of systemic therapy. J. Hepatol 72, 277–287 (2020). [DOI] [PubMed] [Google Scholar]
- 106.Vincenzi B et al. Prognostic Relevance of Objective Response According to EASL Criteria and mRECIST Criteria in Hepatocellular Carcinoma Patients Treated with Loco-Regional Therapies: A Literature-Based Meta-Analysis. PLoS One 10, e0133488 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lencioni R et al. Sorafenib or placebo plus TACE with doxorubicin-eluting beads for intermediate stage HCC: The SPACE trial. J. Hepatol 64, 1090–1098 (2016). [DOI] [PubMed] [Google Scholar]
- 108.Meyer T et al. Sorafenib in combination with transarterial chemoembolisation in patients with unresectable hepatocellular carcinoma (TACE 2): a randomised placebo-controlled, double-blind, phase 3 trial. Lancet Gastroenterol. Hepatol 2, 565–575 (2017). [DOI] [PubMed] [Google Scholar]
- 109.Kudo M et al. Brivanib as adjuvant therapy to transarterial chemoembolization in patients with hepatocellular carcinoma: A randomized phase III trial. Hepatology 60, 1697–1707 (2014). [DOI] [PubMed] [Google Scholar]
- 110.Reig M et al. Early dermatologic adverse events predict better outcome in HCC patients treated with sorafenib. J. Hepatol 61, 318–24 (2014). [DOI] [PubMed] [Google Scholar]
- 111.Sung MW et al. Association between overall survival and adverse events with lenvatinib treatment in patients with hepatocellular carcinoma (REFLECT). J. Clin. Oncol 37, 317–317 (2019). [Google Scholar]
- 112.Llovet JM et al. Prognostic and predictive factors in patients with advanced hepatocellular carcinoma and elevated alpha-fetoprotein treated with ramucirumab in two randomized Phase III trials. Gastroenterology (Submitted) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.El-Khoueiry AB et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 389, 2492–2502 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Llovet JM & Lencioni R mRECIST for HCC: Performance and novel refinements. J. Hepatol 72, 288–306 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cheng A-LL et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol 10, 25–34 (2009). [DOI] [PubMed] [Google Scholar]
- 116.Bruix J et al. Efficacy and safety of sorafenib in patients with advanced hepatocellular carcinoma: Subanalyses of a phase III trial. J. Hepatol 57, 821–829 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Díaz-González Á et al. Systematic review with meta-analysis: the critical role of dermatological events in patients with hepatocellular carcinoma treated with sorafenib. Aliment. Pharmacol. Ther 49, 482–491 (2019). [DOI] [PubMed] [Google Scholar]
- 118.Marrero JA et al. Observational registry of sorafenib use in clinical practice across Child-Pugh subgroups: The GIDEON study. J. Hepatol 65, 1140–1147 (2016). [DOI] [PubMed] [Google Scholar]
- 119.He MK et al. Sorafenib Plus Hepatic Arterial Infusion of Oxaliplatin, Fluorouracil, and Leucovorin vs Sorafenib Alone for Hepatocellular Carcinoma With Portal Vein Invasion: A Randomized Clinical Trial. JAMA Oncol 5, 953–960 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Johnson PJ et al. Brivanib Versus Sorafenib As First-Line Therapy in Patients With Unresectable, Advanced Hepatocellular Carcinoma: Results From the Randomized Phase III BRISK-FL Study. J Clin Oncol 31, 3517–3524 (2013). [DOI] [PubMed] [Google Scholar]
- 121.Cheng A-L et al. Sunitinib versus sorafenib in advanced hepatocellular cancer: results of a randomized phase III trial. J. Clin. Oncol 31, 4067–75 (2013). [DOI] [PubMed] [Google Scholar]
- 122.Cainap C et al. Linifanib Versus Sorafenib in Patients With Advanced Hepatocellular Carcinoma: Results of a Randomized Phase III Trial. J. Clin. Oncol 33, 172–179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhu AX et al. SEARCH: a phase III, randomized, double-blind, placebo-controlled trial of sorafenib plus erlotinib in patients with advanced hepatocellular carcinoma. J. Clin. Oncol 33, 559–66 (2015). [DOI] [PubMed] [Google Scholar]
- 124.Abou-Alfa GK et al. Assessment of Treatment With Sorafenib Plus Doxorubicin vs Sorafenib Alone in Patients With Advanced Hepatocellular Carcinoma: Phase 3 CALGB 80802 Randomized Clinical Trial. JAMA Oncol 5, 1582–1588 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jouve JL et al. Pravastatin combination with sorafenib does not improve survival in advanced hepatocellular carcinoma. J. Hepatol 71, 516–522 (2019). [DOI] [PubMed] [Google Scholar]
- 126.Park JW et al. Sorafenib with or without concurrent transarterial chemoembolization in patients with advanced hepatocellular carcinoma: The phase III STAH trial. J. Hepatol 70, 684–691 (2019). [DOI] [PubMed] [Google Scholar]
- 127.Llovet JM & Hernandez-Gea V Hepatocellular Carcinoma: Reasons for Phase III Failure and Novel Perspectives on Trial Design. Clin. Cancer Res 20, 2072–9 (2014). [DOI] [PubMed] [Google Scholar]
- 128.Vilgrain V et al. Efficacy and safety of selective internal radiotherapy with yttrium-90 resin microspheres compared with sorafenib in locally advanced and inoperable hepatocellular carcinoma (SARAH): an open-label randomised controlled phase 3 trial. Lancet Oncol 18, 1624–1636 (2017). [DOI] [PubMed] [Google Scholar]
- 129.Chow PKH et al. SIRveNIB: Selective Internal Radiation Therapy Versus Sorafenib in Asia-Pacific Patients With Hepatocellular Carcinoma. J. Clin. Oncol 36, 1913–1921 (2018). [DOI] [PubMed] [Google Scholar]
- 130.Wilhelm SM et al. Regorafenib (BAY 73–4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. cancer 129, 245–55 (2011). [DOI] [PubMed] [Google Scholar]
- 131.Finn RS et al. Outcomes of sequential treatment with sorafenib followed by regorafenib for HCC: Additional analyses from the phase III RESORCE trial. J. Hepatol 69, 353–358 (2018). [DOI] [PubMed] [Google Scholar]
- 132.Kim H-D et al. Regorafenib in patients with advanced Child-Pugh B hepatocellular carcinoma: A multicentre retrospective study. Liver Int 40, 2544–2552 (2020). [DOI] [PubMed] [Google Scholar]
- 133.Yakes FM et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther 10, 2298–308 (2011). [DOI] [PubMed] [Google Scholar]
- 134.Zhu AX et al. Ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (REACH): a randomised, double-blind, multicentre, phase 3 trial. Lancet. Oncol 16, 859–70 (2015). [DOI] [PubMed] [Google Scholar]
- 135.Llovet JM et al. Brivanib in Patients With Advanced Hepatocellular Carcinoma Who Were Intolerant to Sorafenib or for Whom Sorafenib Failed: Results From the Randomized Phase III BRISK-PS Study. J Clin Oncol 31, 3509–3516 (2013). [DOI] [PubMed] [Google Scholar]
- 136.Finn RS, Ryoo B, Merle P, Kudo M & Bouattour M Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240 : A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol 38, 1–11 (2019). [DOI] [PubMed] [Google Scholar]
- 137. [Press Release] Merck Announces KEYTRUDA® (pembrolizumab) Met Primary Endpoint of Overall Survival (OS) in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib - Merck.com. [Google Scholar]
- 138.Yau T et al. CheckMate 459: A randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Ann. Oncol 30, v874–v875 (2019). [Google Scholar]
- 139.Rahma OE & Hodi FS The Intersection between Tumor Angiogenesis and Immune Suppression. Clin. Cancer Res 25, 5449–5457 (2019). [DOI] [PubMed] [Google Scholar]
- 140.Lee MS et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): an open-label, multicentre, phase 1b study. Lancet. Oncol 21, 808–820 (2020). [DOI] [PubMed] [Google Scholar]
- 141.Cheng A-L et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J. Hepatol (2021) doi: 10.1016/j.jhep.2021.11.030. [DOI] [PubMed] [Google Scholar]
- 142.Finn RS et al. Phase Ib Study of Lenvatinib Plus Pembrolizumab in Patients With Unresectable Hepatocellular Carcinoma. J. Clin. Oncol 38, 2960–2970 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kelley RK et al. Cabozantinib in combination with atezolizumab versus sorafenib in treatment-naive advanced hepatocellular carcinoma: COSMIC-312 Phase III study design. Future Oncol 16, 1525–1536 (2020). [DOI] [PubMed] [Google Scholar]
- 144.Rotte A Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res 38, 255 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wei SC et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 170, 1120–1133.e17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kelley RK et al. Safety, Efficacy, and Pharmacodynamics of Tremelimumab Plus Durvalumab for Patients With Unresectable Hepatocellular Carcinoma: Randomized Expansion of a Phase I/II Study. J. Clin. Oncol 39, 2991–3001 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.de Franchis R Expanding consensus in portal hypertension. J. Hepatol 63, 743–752 (2015). [DOI] [PubMed] [Google Scholar]
- 148.Benson AB et al. Hepatobiliary Cancers, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw 19, 541–565 (2021). [DOI] [PubMed] [Google Scholar]
- 149.Bruix J, Chan SL, Galle PR, Rimassa L & Sangro B Systemic treatment of hepatocellular carcinoma: An EASL position paper. J. Hepatol 75, 960–974 (2021). [DOI] [PubMed] [Google Scholar]
- 150.Yau T et al. Outcomes of tyrosine kinase inhibitors after immunotherapy in advanced hepatocellular carcinoma: A multi-center study. J. Clin. Oncol 39, e16181–e16181 (2021). [Google Scholar]
- 151.Stuart T & Satija R Integrative single-cell analysis. Nat. Rev. Genet 20, 257–272 (2019). [DOI] [PubMed] [Google Scholar]
- 152.Rozenblatt-Rosen O, Stubbington MJT, Regev A & Teichmann SA The Human Cell Atlas: from vision to reality. Nature 550, 451–453 (2017). [DOI] [PubMed] [Google Scholar]
- 153.Rizvi AH et al. Single-cell topological RNA-seq analysis reveals insights into cellular differentiation and development. Nat. Biotechnol 35, 551–560 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Armingol E, Officer A, Harismendy O & Lewis NE Deciphering cell–cell interactions and communication from gene expression. Nat. Rev. Genet 22, 71–88 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Costa A et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 33, 463–479.e10 (2018). [DOI] [PubMed] [Google Scholar]
- 156.Su S et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 172, 841–856.e16 (2018). [DOI] [PubMed] [Google Scholar]
- 157.Affo S et al. Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell 39, 866–882.e11 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Schreiber RD, Old LJ & Smyth MJ Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331, 1565–70 (2011). [DOI] [PubMed] [Google Scholar]
- 159.Shen Y-C et al. Reliability of a single-region sample to evaluate tumor immune microenvironment in hepatocellular carcinoma. J. Hepatol 72, 489–497 (2020). [DOI] [PubMed] [Google Scholar]
- 160.Ma L et al. Single-cell atlas of tumor cell evolution in response to therapy in hepatocellular carcinoma and intrahepatic cholangiocarcinoma. J. Hepatol (2021) doi: 10.1016/j.jhep.2021.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ikeda M et al. Phase I studies of peptide vaccine cocktails derived from GPC3, WDRPUH and NEIL3 for advanced hepatocellular carcinoma. Immunotherapy 13, 371–385 (2021). [DOI] [PubMed] [Google Scholar]
- 162.Guo M, Zhang H, Zheng J & Liu Y Glypican-3: A New Target for Diagnosis and Treatment of Hepatocellular Carcinoma. J. Cancer 11, 2008–2021 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Shih T-C, Wang L, Wang H-C & Wan Y-JY Glypican-3: A molecular marker for the detection and treatment of hepatocellular carcinoma. Liver Res 4, 168–172 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Rochigneux P et al. Adoptive Cell Therapy in Hepatocellular Carcinoma: Biological Rationale and First Results in Early Phase Clinical Trials. Cancers (Basel) 13, 271 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Docta RY et al. Tuning T-Cell Receptor Affinity to Optimize Clinical Risk-Benefit When Targeting Alpha-Fetoprotein-Positive Liver Cancer. Hepatology 69, 2061–2075 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Sangro B, Sarobe P, Hervás-Stubbs S & Melero I Advances in immunotherapy for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol 18, 525–543 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.von Felden J, Garcia-Lezana T, Schulze K, Losic B & Villanueva A Liquid biopsy in the clinical management of hepatocellular carcinoma. Gut 69, 2025–2034 (2020). [DOI] [PubMed] [Google Scholar]
- 168.Kaseb AO et al. Molecular Profiling of Hepatocellular Carcinoma Using Circulating Cell-Free DNA. Clin. Cancer Res 25, 6107–6118 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Labgaa I et al. A pilot study of ultra-deep targeted sequencing of plasma DNA identifies driver mutations in hepatocellular carcinoma. Oncogene 37, 3740–3752 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ahn et al. A genomic portrait of resectable hepatocellular carcinomas: Implications of RB1 and FGF19 aberrations for patient stratification. Hepatology 2–807 (2014) doi: 10.1002/hep.27198. [DOI] [PubMed] [Google Scholar]
- 171.Totoki Y et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet 46, 1–10 (2014). [DOI] [PubMed] [Google Scholar]
- 172.Nault JC et al. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat. Commun 4, 2218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]