

Abstract
Pulmonary hypertension (PH) due to chronic lung disease is categorized as Group 3 PH in the most recent classification system. Prevalence of these diseases is increasing over time, creating a growing need for effective therapeutic options. Recent approval of the first pulmonary arterial hypertension (PAH) therapy for the treatment of Group 3 PH related to interstitial lung disease (ILD) represents an encouraging advancement. This review focuses on molecular mechanisms contributing to pulmonary vasculopathy in chronic hypoxia, the pathology and epidemiology of Group 3 PH, the right ventricular dysfunction observed in this population and clinical trial data that inform the use of pulmonary vasodilators in Group 3 PH.
Keywords: Pulmonary Hypertension, Translational Studies
Introduction
Pulmonary hypertension (PH) due to chronic lung disease is categorized as Group 3 PH in the World Symposium on Pulmonary Hypertension (WSPH) classification system1. Group 3 PH deserves special attention as chronic lung diseases are increasing in prevalence and there are a lack of sufficient therapeutic options for PH in lung disease. Currently available therapies for Group 1 pulmonary arterial hypertension (PAH) have been tested in Group 3 PH with mixed results2. This may reflect a lack of understanding of the mechanisms driving Group 3 PH as well as the heterogeneity of patients and diseases involved. There is disproportionate right ventricular (RV) dysfunction in Group 3 PH patients as compared to PAH despite less severe hemodynamic impairment3, highlighting an additional knowledge gap. As RV function is a critical determinant of outcome in pulmonary vascular disease and chronic lung diseases are a major public health problem, Group 3 PH is an important focus for study and novel therapeutic development. The recent approval of a single PAH therapy for Group 3 PH related to interstitial lung disease (ILD) has re-ignited interest in this topic. In this focused review, we will discuss the available experimental models for studying Group 3 PH, currently known molecular mechanisms that contribute to pulmonary vascular remodeling in chronic hypoxia, review the pathology and epidemiology of Group 3 PH and the RV dysfunction observed in this population and highlight experimental and clinical trial data that inform the use of pulmonary vasodilators in Group 3 PH.
Experimental Models and Molecular Mechanisms of Pulmonary Vascular Remodeling in Chronic Hypoxia
Much of the evidence regarding the molecular mechanisms responsible for pulmonary vasculopathy in chronic lung disease comes from pre-clinical animal models with hypoxia exposure4, 5. Common experimental models of Group 3 PH and their strengths and weaknesses are summarized in Table 1. Although chronic hypoxia may mimic high-altitude PH, it is unclear whether it accurately recapitulates chronic lung diseases such as chronic obstructive pulmonary disease (COPD) and ILD6–9. As in Group 1 PAH, endothelial cell dysfunction and aberrant cell signaling with unchecked endothelial, fibroblast and smooth muscle cell growth, proliferation and dysregulated angiogenesis are thought to contribute to the pathobiology of Group 3 PH10. Of particular interest in Group 3 PH is the independent role that cigarette smoke plays in the development of pulmonary vasculopathy (Figure 1A). Emerging themes in chronic hypoxia models of PH include the role of microRNAs (miRs) and progenitor cells in disease pathobiology and altered cellular metabolism and mitochondrial function as novel therapeutic targets (Figure 1B). Experimental models have also elucidated the potential mechanistic benefit of prostacyclins in PH associated with chronic lung disease.
Table 1.
Summary of experimental models of hypoxia in pulmonary hypertension
| Model | Species | Methodology | Histopathology | Strengths | Weaknesses |
|---|---|---|---|---|---|
| Chronic hypoxia219–222 | Rats, mice, Guinea pig, pig, sheep | Chronic exposure to hypoxia | Pulmonary vasoconstriction, muscularization of non-muscular arterioles, increased media thickness and matrix deposition | • Widely used • Simple |
• Variable response to hypoxia |
| SU5416+Hypoxia223–225 | Rats, mice | VEGFR-2 blockade coupled with chronic hypoxia | Development of PH with complex plexiform-like lesions | • Mimics human PAH | • Skill-specific • May not adequately model Group 3 PH • More costly |
| Fawn-hooded rats226, 227 | Rats | Rat strain with deficient serotonin uptake into platelets | Rats have immature development of lungs, reduced number of alveoli, and development of PH | • May be useful to study specific pathways in PH | • Mimics limited features of PH • Cost |
| Broiler chicken228–233 | Chicken | Pulmonary diffusion limitation leading to hypoxemia, possibly inherent mitochondrial dysfunction, | Right ventricular hypertrophy, perivascular leakage, vascular medial hypertrophy, perivascular inflammatory infiltration | • May be useful to study specific pathways in PH | • Mimics limited features of PH • Cost |
| Bleomycin234–236 | Rats, mice, rabbits | Administration of bleomycin, commonly intratracheally | Bleomycin leads to pulmonary fibrosis development with increased lung inflammation and muscularization | • Useful to study PH in ILD • Simple • Relatively inexpensive |
• Mimics only limited features of human PH |
| Term and preterm237–243 | Mice, rats, rabbits, sheep, baboons | Varies depending on model but commonly preterm animals with mechanical ventilation. Hyperoxia and inflammation by bacterial LPS also used. | Alveolar simplification (rarefication) and loss of surfactant | • Useful to study PH in BPD | • Variability in lung and vascular injury dependent on species, age of animal, and mechanisms of injury • Heterogeneity of lung pathologies in human BPD is not accurately recapitulated in animals |
SU5416 = Sugen 5416. VEGFR-2 = vascular endothelial growth factor receptor 2. PH = pulmonary hypertension. PAH = pulmonary arterial hypertension. ILD = interstitial lung disease. LPS = lipopolysaccharide. BPD = bronchopulmonary dysplasia. Adapted from Nogueira-Ferreira, et al. Cardiol and Cardiovasc Med244.
Figure 1.
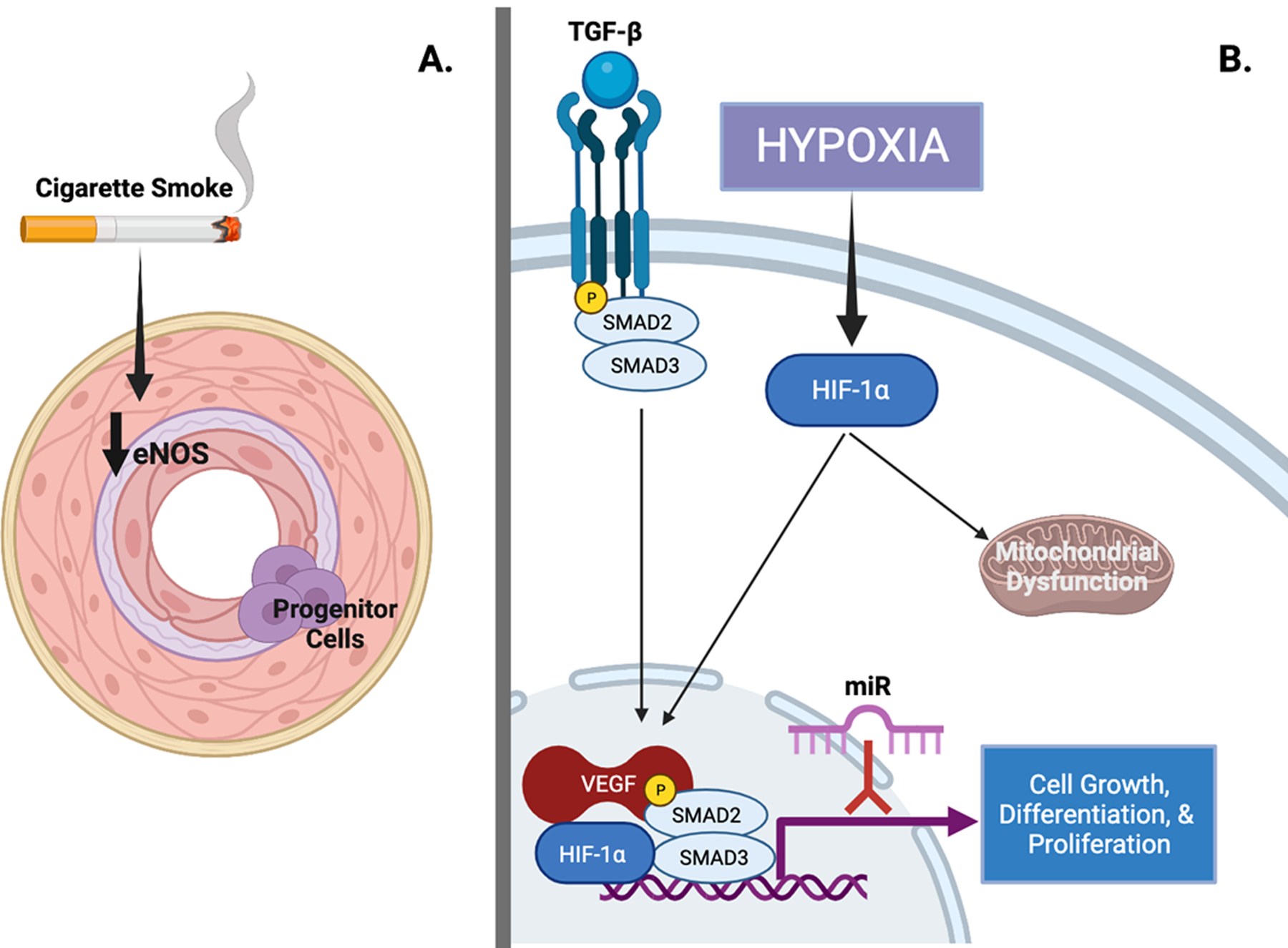
Molecular mechanisms of pulmonary vasculopathy in Group 3 pulmonary hypertension (PH).
A. Cigarette smoke leads to decreased expression of endothelial nitric oxide (eNOS) along with intimal hyperplasia, increased expression of vascular endothelial growth factor (VEGF), inflammatory infiltrate, and metabolic dysfunction. Poorly differentiated progenitor cells have been observed in the intima of pulmonary arteries; their origin and role in the development of pulmonary vasculopathy remains unclear. B. Hypoxia inducible factor-1 alpha (HIF-1α) and transforming growth factor-beta (TGF-β) signaling are recognized to regulate growth, differentiation, and proliferation of almost all cell types in hypoxia-induced PH. VEGF expression is induced by hypoxia via HIF-1α signaling. HIF-1α is also thought to play a central role in the development of mitochondrial dysfunction that may explain a Warburg-like shift to anaerobic glycolysis. microRNAs (miRs) are small noncoding RNAs that regulate gene expression and in mouse models inhibition of miRs prevent development of pulmonary vasculopathy. Figure created with biorender.com. (Illustration Credit: Ben Smith).
Transforming growth factor-beta 1 (TGF-β1) has been recognized to regulate cell growth, differentiation, and proliferation in almost all cell types11, 12 and molecular mechanisms integral to the vascular remodeling observed in PH. TGF-β1 expression is closely linked to hypoxia-inducible factor-1 alpha (HIF-1α) and both signaling pathways are well established in the development of pulmonary vascular disease13–15. In hypoxia exposed rats, TGF-β1 expression was increased as compared to normoxic controls and associated with a decrease in phosphatase and tensin homolog deleted on chromosome 10 (PTEN)16. Vascular endothelial growth factor A (VEGFA) is also induced by HIF-1α; its central role in the development of PAH is reviewed separately17. In rats, chronic hypoxia increases VEGFA expression and its receptor, VEGFR218, 19. Though loss of VEGFB in mice had no effect, its overexpression was beneficial20, suggesting that it plays a protective role alongside VEGFA in hypoxic PH. Platelet-derived growth factor β (PDGFβ) production is stimulated by TGF-β1 and increases expression of VEGFA, augmenting hypoxia-induced endothelial cell proliferation21, 22. Increased expression of angiogenesis mediators and growth factors such as VEGF and receptor-II of TGF-β (TGF-βRII) has been observed in the pulmonary arteries of patients with COPD23. End-stage COPD patients who underwent lung transplantation and patients with mild to moderate COPD have impaired endothelial-dependent relaxation24, 25. As a result, endothelial dysfunction remains a potential therapeutic target in Group 3 PH. Sildenafil abrogated endothelial reactive oxygen species generation along with improving RV/left-ventricle+septum ratio and decreasing pulmonary vascular and RV fibrosis in a bleomycin-induced ILD mouse model26. Fibroblast growth factors (FGFs) contribute to the proliferation of pulmonary artery endothelial cells (PAECs) and smooth muscle cells. Production of FGF2 is increased alongside HIF-1α in a feedforward manner27, 28. While additional mechanistic work is needed to clarify the role of FGF2 in pulmonary vascular remodeling during hypoxia, FGF2 is elevated in mouse models of chronic hypoxia21, 29 and in preterm infants with bronchopulmonary dysplasia (BPD)30.
Cigarette smoke as a toxic insult and cause of pulmonary vascular disease is particularly relevant in Group 3 PH. Independent of airflow obstruction, smoking has been observed to cause intimal hyperplasia31, reduced expression of endothelial nitric oxide synthase (eNOS)32, increased VEGF expression23, infiltration of inflammatory cells33 and an imbalance of mitochondrial fission and fusion with resultant mitochondrial oxidative stress and dysfunction34. It is believed that cigarette smoking plays a central role in the vascular changes that lead to the development of PH in COPD35–37. In vitro work has confirmed this hypothesis. In a dose and time-dependent manner, human PAECs exposed to cigarette smoke demonstrate decreased eNOS activity and protein expression38 and decreased prostacyclin synthase mRNA and protein expression39. Vascular remodeling due to cigarette exposure may precede the development of clinically detectable emphysema or airflow obstruction as observed in animal models40–43.
There is growing interest in the role of miRs in the development of all forms of pulmonary vascular disease. miRs are small noncoding RNAs that regulate gene expression by binding to target mRNAs and promote degradation or inhibit translation44, 45. In mouse models of hypoxia-induced PH, inhibition of miR-21 (A-21) improved vessel muscularization and RV hypertrophy46 and inhibition of miR-20a improved bone morphogenic receptor 2 (BMPR2) expression (a known causal pathway in Group 1 PAH) and reduced luminal occlusion of small pulmonary arteries and RV hypertrophy47.
Progenitor cells may be important in the development of PH in chronic lung diseases. Poorly differentiated cells have been observed in the intima of pulmonary arteries of patients with Group 3 PH. While their origin and role have not been defined, dedifferentiation of smooth muscle cells, transdifferentiation during endothelial-to-mesenchymal-transition, resident precursor cells, or recruitment from bone marrow-derived progenitor cells are all potential sources.35, 48–50. Bone marrow-derived progenitor cells have been identified in the intima of pulmonary arteries from COPD patients51 and are associated with endothelial dysfunction and the degree of vascular remodeling51, 52, lending support to this hypothesis.
Metabolic reprogramming and mitochondrial dysfunction are likely critical to the pathogenesis of all subtypes of PH. Increased expression of HIF-1α during hypoxia is theorized to alter cellular metabolism and partially explain the Warburg-like shift to anaerobic glycolysis in PH53, 54. When mitochondrial fission is inhibited in a rat model of hypoxia, pulmonary artery smooth muscle cell proliferation is abrogated and exercise capacity, RV function and hemodynamics improve55.
As discussed below, inhaled trepostinil was recently approved for ILD-PH and was associated with improved measures of both pulmonary vascular and lung function in the landmark INCREASE trial56. Experimental models provide timely mechanistic insight into these results. Prostacyclin acts largely via the IP receptors to increase cAMP which, in the vasculature, promotes relaxation and limits proliferation and, in platelets, regulates calcium levels to reduce thrombosis57–59. Prostacyclin appears to also have antifibrotic properties via the IP receptor60 and activation of peroxisome proliferator-activated receptors (PPAR)61, abrogating both parenchymal lung and vascular fibrosis in murine models of bleomycin-induced pulmonary fibrosis and Fra-2 transgenic mice62–65.
Histopathology of Group 3 PH
Most of our knowledge of human histopathologic changes in Group 3 PH comes from explanted lungs from lung transplantation and autopsies. Idiopathic pulmonary fibrosis (IPF)/usual interstitial pneumonia (UIP) and COPD are the most frequently transplanted lung conditions that may be complicated by PH, usually at an advanced stage. Other than chronic hypoxic vasoconstriction, which is an inherent factor in the early evolution of obstructive and restrictive lung disease, other mechanisms that display a morphologic correlate in histology may be the loss of microvessels and capillaries in the progressive destruction of alveolar septa in COPD and IPF, compression of peripheral or central lung vessels due to solid collagen-rich fibrosis in advanced IPF, or direct remodeling of lung vessels with hyperplasia of smooth muscle cells and fibroblasts, leading to constrictive thickening of the media and the intima. IPF is an interstitial process that affects the lung parenchyma very heterogeneously, and interestingly the remodeling of muscular pulmonary arteries and arterioles appears to be more or less restricted to the fibrotic lung areas (Figure 2A). In contrast, it has been shown that the distribution of microvascular changes in IPF lungs is as heterogenous as the fibrotic process, only in an inverse and somehow compensatory fashion, with the highest density of capillaries within non-fibrotic areas, and low microvascular density in fibrotic areas with excessive collagen deposition66. Microvascular heterogeneity has been specifically associated with IPF disease evolution by others67, 68.
Fig. 2.
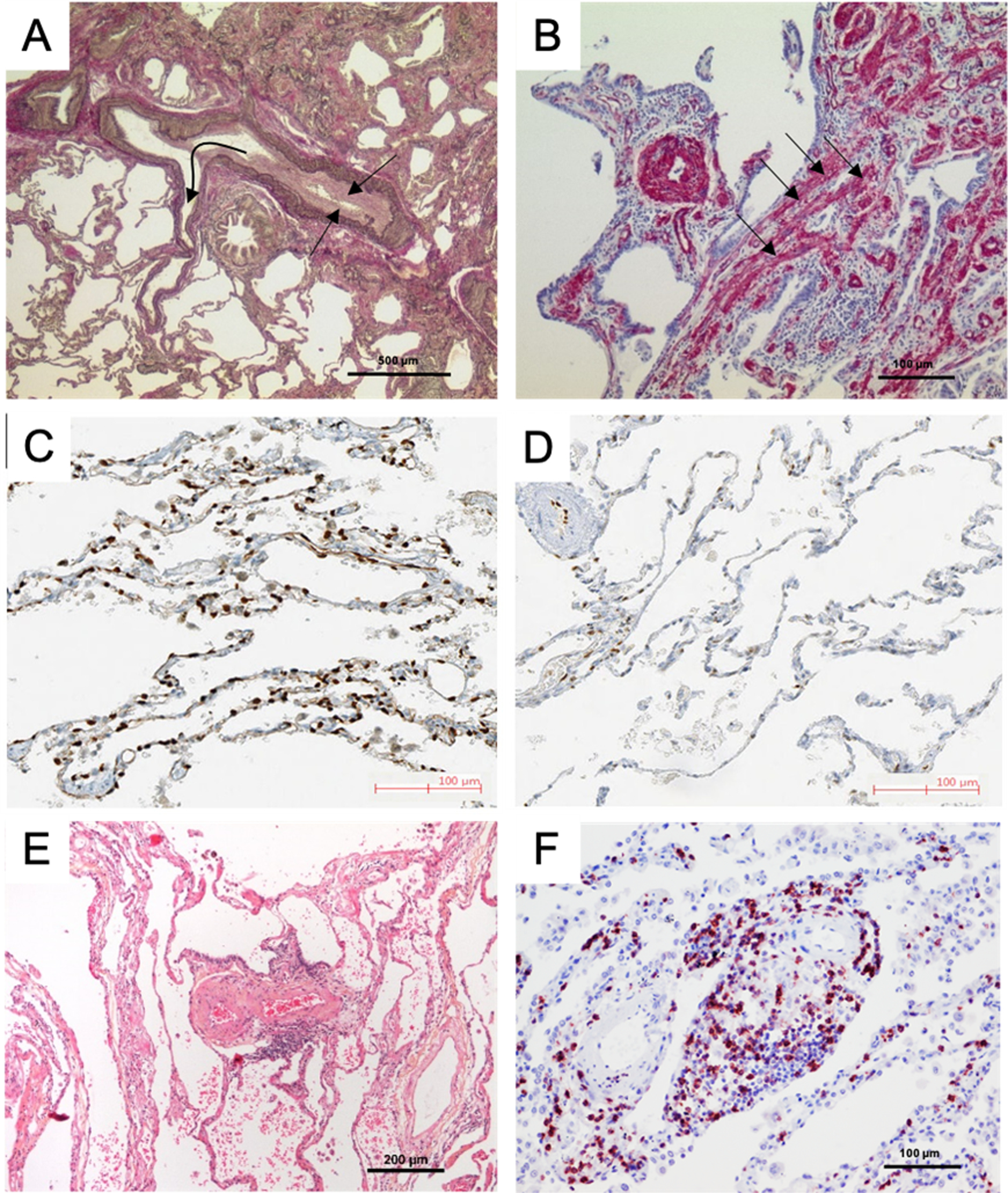
Pulmonary vascular changes in patients with lung disease and Group 3 pulmonary hypertension (PH). A. Pulmonary artery (PA) and its adjacent airway in a patient with idiopathic pulmonary fibrosis (IPF) and PH; note the constrictive intimal fibrosis of the vessel (straight arrows) within the fibrosing area (upper part) and the slender walls of the PA branching out of this area into preserved lung areas (flexuous arrow) with near-normal architecture (lower part). B. Smooth muscle cell hyperplasia within the interstitium (arrows) and within the wall of a small PA, in a patient with IPF-PH; smooth muscle actin-staining. C and D: Capillary density of a patient with chronic obstructive pulmonary disease (COPD) displaying moderate PH (C) and a patient with COPD and severe PH (D): note the loss of ERG-stained endothelial cells within the alveolar septa, corresponding to a decrease of vascular density; photos reprinted with regard to the STM permission guidelines 2022 from Bunel et al. CHEST. 201972. E. PA in a patient with COPD-PH, with severe intimal fibrosis; note the perivascular lymphocytic infiltrate (small blue dots, center). F. Remodeled arterioles in a COPD-PH patient with dense CD5-positive peri-vascular lymphocytic infiltrate (red dots), a feature that can also be observed in pulmonary arterial hypertension.
The partial loss of pulmonary capillaries in Group 3 PH compared to other subtypes of PH has served as an explanation for largely negative clinical trials of pulmonary vasodilators in Group 3 PH. These observations were initially reinforced by animal models demonstrating a reduction in alveolar capillary density with increasing smoke exposure69. However these findings have been contested in other experimental settings70, 71. It has been recently shown that lungs from patients with COPD lacking PH and COPD with moderate PH can be discriminated from COPD with severe PH by the degree of microvascular muscularization and the loss of capillaries in the latter group (Figure 2C and 2D)72.
Heterogeneous distribution of capillary density with foci of proliferation is known from PAH with overt involvement of pulmonary veins and capillaries, known as pulmonary veno-occlusive disease/pulmonary capillary hemangiomatosis. It has been reported that in lungs from patients with IPF, pulmonary veins show constrictive remodeling in preserved areas as well as in fibrotic areas, in the absence of pulmonary artery remodeling73. Taken together, it appears that all compartments of the lung vasculature may be involved in the natural history of Group 3 PH, even if it is still unclear to what extent and with what hemodynamic consequences.
Genetic Predisposition in Group 3 PH
Recent studies have identified genetic variants that predispose to or are associated with the development of Group 3 PH in chronic lung disease. Variants of kinase insert domain receptor (KDR), which encodes for VEGFR2 has been shown by several groups to correlate with impaired gas transfer, parenchymal lung disease and older age74–76. In neonates, single nucleotide polymorphisms (SNPs) of dual-specificity phosphatases (DUSPs) can predict PH in patients with BPD77 and variants of endothelin-1 (EDN1)78, carbamoyl phosphate synthetase I (CPS1), neurogenic locus notch homolog protein 3 (NOTCH3), and SMAD family member 9 (SMAD9)79 increase risk for development of persistent pulmonary hypertension of the newborn (PPHN). Taken together, these data suggest that genetic background may explain some of the heterogeneity observed in chronic lung diseases and their variable and patient-specific propensity for pulmonary vascular complications.
Genetic variability may also provide insight into mechanisms of disease development. Patients with IPF-PH have a unique genetic signature including expression of mediators of pulmonary artery smooth muscle cell (PASMC) and endothelial cell proliferation, Wnt signaling, complement system activation and extracellular matrix (ECM) remodeling and apoptosis80, 81. This genetic signature was observed to be similar to IPF patients with and without PH, suggesting that genetic reprogramming may occur prior to the development of PH or that additional mechanisms or “hits” predispose to the development of PH in IPF82. Similarly, microarray analysis of laser capture microdissected pulmonary arteries from patients with COPD shows that expression of genes related to retinol metabolism and ECM remodeling is uniquely characteristic of COPD-PH81.
Epidemiology of Group 3 PH Due to Lung Disease and/or Hypoxia
Following left-sided heart disease, Group 3 PH is the second leading cause of PH83, 84 and both the prevalence and incidence are increasing84. Patients afflicted with PH as a complication of parenchymal lung disease have the worst long-term survival as compared to all other PH groups3, 84, 85. Group 3 PH patients have higher medical costs86 and increased morbidity and mortality87, 88 as compared to patients with chronic lung disease without PH.
Obstructive and restrictive lung diseases and chronic hypoxia can all lead to PH (Table 2). COPD is the most common type of obstructive lung disease and is highly prevalent in the general population. Restrictive lung diseases include primarily diffuse parenchymal lung diseases (DPLDs), a heterogeneous group of disorders that includes idiopathic interstitial pneumonias (IIPs), connective tissue disease associated (CTD) ILDs, and unusual ILDs like lymphangioleiomyomatosis (LAM) (Figure 3)89, 90. Not all parenchymal lung diseases with PH are classified as Group 3 PH. For example, pulmonary Langerhans cell histiocytosis and sarcoidosis are in Group 5 PH due to multifactorial mechanisms1. In the absence of parenchymal lung disease, chronic hypoxia (e.g., altitude) is another known cause of PH. Additional etiologies for Group 3 PH include sleep disordered breathing (SDB) and developmental lung disease, namely BPD.
Table 2.
Clinical classification of pulmonary hypertension (PH) due to lung diseases and/or hypoxia
| Group 3 PH due to lung diseases and/or hypoxia |
| 3.1 Obstructive lung disease |
| 3.2 Restrictive lung disease |
| 3.3 Other lung disease with mixed restrictive/obstructive pattern |
| 3.4 Hypoxia without lung disease |
| 3.5 Developmental lung disorders |
|
|
| Group 5 PH with unclear and/or multifactorial mechanisms |
| 5.2 Systemic and metabolic disorders |
| Pulmonary Langerhans cell histiocytosis |
| Sarcoidosis |
Adapted from Simonneau, et al. ERJ. 2019.1
Figure 3.
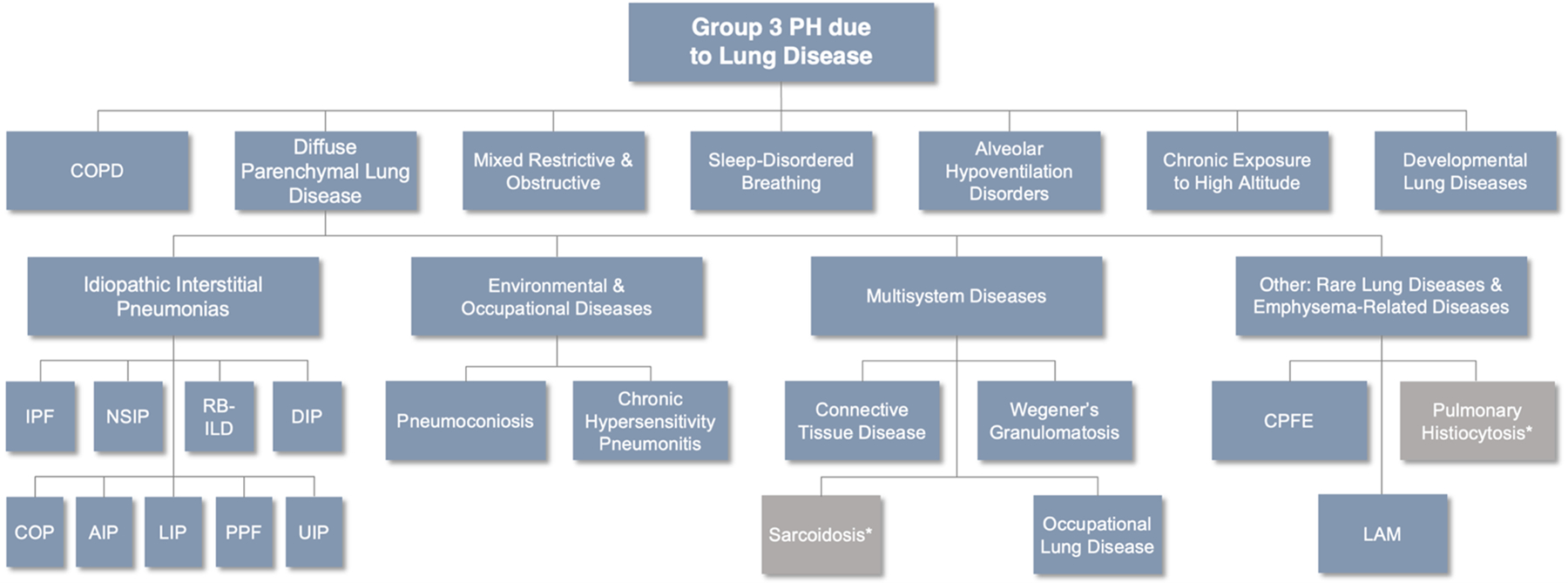
Clinical classification of diseases causing Group 3 pulmonary hypertension (PH). Aside from chronic obstructive pulmonary disease (COPD), many diseases responsible for Group 3 PH belong to the diffuse parenchymal lung diseases (DPLDs) and are further classified based on their etiology. Idiopathic interstitial pneumonias (IIPs) are idiopathic diseases of varying prevalence but represent an important group of diseases that contribute to the development of PH. *Belongs to Group 5 PH. IPF = idiopathic pulmonary fibrosis. NSIP = nonspecific interstitial pneumonitis. RB-ILD = respiratory bronchiolitis-interstitial lung disease. DIP = desquamative interstitial pneumonia. COP = cryptogenic organizing pneumonia. AIP = acute interstitial pneumonia. LIP = lymphocytic interstitial pneumonia. PPF = pleuroparenchymal fibroelastosis. UIP = usual interstitial pneumonia. CPFE = combined pulmonary fibrosis and emphysema. LAM = lymphangioleiomyomatosis. Adapted from Nathan, SD. “Inhaled Treprostinil in Interstitial Lung Disease Associated Pulmonary Hypertension: The INCREASE Study.” Breaking News: Clinical Trial Results in Pulmonary Medicine. ATS Meeting. May 28, 2020.
COPD is the most prevalent chronic respiratory disease worldwide and the third leading cause of death in the United States91. Most patients with COPD will develop mild PH, as characterized by a mild elevation in pulmonary pressures. Approximately 1 – 5% of COPD patients develop severe PH, with a mean pulmonary artery pressure (mPAP) > 35 – 40 mmHg at rest92. Though the rate of progression of PH in COPD is recognized to be slow (< 1 mmHg increase in mPAP per year)93, the concomitant presence of PH with COPD is a strong predictor of mortality. There is an inverse relationship between mPAP and pulmonary vascular resistance (PVR) and survival in COPD94–96. Recent data from the COMPERA (Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension) and GRAPHIC (GRAz Pulmonary Hypertension in COPD) registries demonstrate that both mPAP and PVR are strong predictors of survival in COPD associated PH; a threshold PVR > 5 Woods units (WU) discriminates mortality in these registries97, 98. The presence of PH in COPD patients is more strongly associated with mortality than forced expiratory volume in the first second (FEV1) or gas exchange99. Similar to idiopathic PAH, the morphology of vascular lesions in COPD correlates with the severity of PH100.
DPLDs include the IIPs, combined pulmonary fibrosis and emphysema (CPFE) and CTD-associated ILDs. Most data about IIP-associated PH comes from studies of IPF, the most common form of IIP. Other common DPLDs include chronic hypersensitivity pneumonitis, nonspecific interstitial pneumonitis (NSIP), unclassifiable ILD and LAM. The IIPs have differing radiographic and pathologic features; however, all of them, especially the ones characterized by elements of fibrosis, may be complicated by PH. Approximately 8 – 15% of IPF patients have a mPAP ≥ 25 mmHg during initial workup101. Limited data are available regarding the prevalence of PH in other fibrotic ILDs. PH was reported in 31% of a cohort of 35 patients with idiopathic NSIP and 44% of a cohort of 50 patients with chronic hypersensitivity pneumonitis102, 103. More advanced parenchymal lung disease is associated with worse hemodynamics104–106 and even echocardiographic PH107 or minimal elevations in mPAP (> 17 mmHg)108 are associated with reduced survival in IPF patients. Interestingly, the degree of parenchymal lung involvement, based on PFTs109 and high-resolution CT fibrosis score107, 110–113, does not correlate with PH severity. In COMPERA, a PVR > 5 WU outperformed the now accepted definition of chronic lung disease with severe PH (mPAP ≥ 35 mmHg or mPAP ≥ 25 mmHg with CI < 2.0 L/min/m2) in discriminating survival114.
With the advent of wide-scale computed tomography imaging, CPFE has been recognized as a unique phenotype of ILD that is associated with poor outcomes and severe concurrent PH. As many as 30 – 50% of patients with CPFE are estimated to develop PH115, 116 and survival is poor115–117. Incident PH in this population is severe in approximately one-half of patients (68% with mPAP > 35 mmHg, 48% with mPAP > 40 mmHg) at the time of right heart catheterization115. These patients are often characterized by profound diffusion limitation, which may be a marker of pulmonary vascular involvement34, 118–120.
CTD-associated ILD presents a particular challenge, as CTD-related vasculopathy is an important cause of Group 1 PAH, making differentiation between Group 1 and Group 3 disease more difficult. Moreover, the presence of both PH and ILD is associated with significantly higher mortality than either of the processes alone121.
In the absence of lung disease, chronic hypoxia predisposes individuals to the development of PH, particularly at high altitude (defined as an elevation > 2500 m above sea level109). Though reliable prevalence data are lacking, high-altitude PH is a significant problem in certain regions of the world. Chronic hypoxia leads to pulmonary vasoconstriction and over time can cause pulmonary vascular remodeling and elevations in PVR122, 123. This process along with erythrocytosis are hallmark features of high-altitude PH124. Though there are some data on the use of pulmonary vasodilators in this population, high-altitude PH is entirely reversible on re-exposure to normal inspired oxygen tension109.
Sleep disordered breathing (SDB) includes obstructive sleep apnea (OSA) and obesity hypoventilation syndrome (OHS). Both OSA and OHS are characterized by nocturnal hypoventilation, however PH in isolated OSA is uncommon and typically mild109. In contrast, patients with OHS or overlap syndrome (OSA and COPD) are at high risk for pulmonary vascular complications125, 126. Though prevalence estimates of PH in OHS vary, OHS itself is common in the general population (about 0.4% of the US population)127 and PH as a complication of OHS is frequently severe and associated with RV failure and poor outcomes128. As obesity becomes more prevalent in the general population, pulmonary vascular disease as a complication of OHS or overlap syndrome may become much more common.
There are several developmental lung disorders associated with PH, including alveolar capillary dysplasia (ACD) with or without misalignments of the veins (MPV), congenital diaphragmatic hernia (CDH), lung hypoplasia, pulmonary alveolar proteinosis (PAP) and abnormalities in surfactant protein129, 130. BPD is a clinical syndrome of injury to preterm lungs characterized by disruption of alveolarization and microvascular development and caused by multiple antenatal and postnatal exposures, including early gestational age, low birth weight, maternal infections and the use of mechanical ventilation131. The disorder causes impaired proximal airway and bronchoalveolar development and often abnormal vascular remodeling and vascular growth arrest (or rarefication of the pulmonary vasculature)132. BPD is the most frequent complication of extreme preterm birth (defined as infants born < 28 weeks gestational age)131, 133 and the strongest risk factors for BPD remain prematurity and low birth weight134–140. The prevalence of BPD is increasing, likely due to the increased survival of extremely low gestational age newborns141, 142. Pulmonary vascular disease can result from extrapulmonary right-to-left shunting across a patent foramen ovale and/or ductus arteriosus causing hypoxia and a sustained elevation in PVR (PPHN)143 to later and more persistent PH after hospital discharge and in young adults. Although BPD is a syndrome of lung injury, its relationship to PH is not sequential – antenatal stresses like oligohydramnios and prolonged premature rupture of membranes cause primary vascular abnormalities and lead to elevations in PVR and subsequent development of BPD143–146.
Right Ventricular Dysfunction in Group 3 PH
Though the term “cor pulmonale” has no consensus definition, it is commonly used to describe RV dysfunction as a result of chronic lung disease associated PH147, 148. RV dysfunction is a major determinant of outcome in all forms of pulmonary vascular disease149–151. A recent study utilizing echocardiographic RV fractional area change (FAC) assessment in a cohort of 147 patients with Group 3 PH showed that RVFAC < 28% discriminated between long term outcomes152. Because of the typically slow increases over time in PVR in Group 3 PH153, the RV has time to compensate, often resulting in RV hypertrophy without systolic dysfunction154, 155. The prevalence of RV hypertrophy in chronic lung disease varies from 50% on echocardiogram of patients with restrictive lung disease156 to 76% at autopsy of COPD patients157. Diastolic dysfunction is the predominant form of RV dysfunction in patients with chronic lung disease as has been demonstrated both in COPD158 and in healthy individuals exposed to acute hypoxia159. This increase in RV thickness is accompanied by cardiomyocyte hypertrophy, cardiac ECM remodeling, alterations in cellular metabolism, and in some models, an increase in RV capillary density160–163.
Whether by impaired ventilation/perfusion matching from alveolar destruction in COPD, loss of pulmonary capillary beds from fibrosis in ILD, hypoventilation in OHS, or hypoxic vasoconstriction due to low inspired oxygen tension at high-altitude, a common pathway linking chronic lung diseases and hypoxia to RV dysfunction in Group 3 PH is the elevation in RV afterload. In addition, pathologic changes in the lungs directly affect RV mechanics. Lung hyperinflation in COPD leads to decreased venous return and RV preload164 along with mechanical compression of the ventricles165. Similarly, the increased intrapleural pressure required for inspiration in COPD and during apneic episodes of sleep disordered breathing likely increase left atrial pressure and RV afterload166.
A persistently elevated RV afterload will inevitably cause systolic dysfunction. Systolic dysfunction may be limited to the most severe cases of PH and underscores the tremendous ability of the cardiovascular system to compensate to stress167. As in Group 1 PAH, there is sexual dimorphism in RV systolic function in Group 3 PH. Males had worse RV systolic function assessed by transthoracic echocardiography as compared to females in one study152, despite females having a significantly higher PVR and a trend toward lower pulmonary artery compliance. As PVR increased in males, RV contractility worsened, whereas it was preserved in females, suggesting a sex-based differential RV response to pulmonary afterload in Group 3 PH152.
Diagnosis of Group 3 PH
Echocardiography is the initial test of choice in the diagnosis of PH109 as it is noninvasive and can identify other etiologies of PH including left-heart disease, which is highly prevalent in patients with chronic lung disease168, 169. However, pulmonary artery pressure estimates from echocardiography alone are often inaccurate and may overdiagnose PH in patients with chronic lung disease170. Plasma levels of brain natriuretic peptide (BNP) or NT-proBNP may also be useful and were strong predictors of mortality in a mixed ILD cohort171. However, BNP levels lack sensitivity and can be confounded by the presence of left-heart disease172.
Pulmonary function tests establish obstructive, restrictive and mixed ventilatory defects in patients with lung disease. The diffusing capacity of carbon monoxide (DLCO) may be particularly revealing in patients with Group 3 PH and is associated with lower survival173. In COPD and ILD, PH is associated with a lower DLCO than expected based on ventilatory impairments107, 110, 112, 174, 175. The six-minute walk test (6MWT) has several features which may help detect PH complicating primary lung disease. A reduction in six-minute walk distance (6MWD) and pronounced desaturation during the walk are associated with the presence of PH and reduced survival in chronic lung disease176. Reduced heart rate recovery (maximal heart rate achieved minus heart rate at one minute post exercise < 13 beats/min) has also been shown to predict PH in patients with IPF177. Computed tomography (CT) is widely used in patients with lung disease to assess the burden of parenchymal lung abnormalities and/or to screen for lung cancer. A ratio > 1 (range 0.9 – 1.1) of the main pulmonary artery to ascending aorta diameter on CT imaging may predict PH in both COPD and IPF178–180 and combined with other non-invasive measures improves the accuracy of predicting hemodynamic PH in this population174, 179. Finally, an RV to LV ratio > 1 measured on CT is associated with increased PVR and predicts mortality in ILD patients181. Table 3 lists findings that when present increase the likelihood of PH in patients with chronic lung diseases.
Table 3.
Findings suggestive of pulmonary hypertension in chronic lung disease
| Pulmonary function testing | • DLCO < 30% or worsening DLCO in the setting of preserved lung volumes |
| 6-Minute Walk Test | • Marked or worsening exertional desaturation • Severely reduced or worsening 6MWT distance • Impaired (decreased) heart rate recovery following exercise (< 13 beats per minute) |
| CT scan | • Increased pulmonary artery to aorta ratio (> 0.9) • RV to LV ratio > 1 |
| Echocardiogram | • RVSP elevation > 45 • RV dilation • TAPSE < 1.8cm • RVOT diameter > 3.4cm • Reduced RV fractional area change • Reduced RV ejection fraction on 3D ECHO |
| Laboratory studies | • Elevated BNP |
ILD = interstitial lung disease. DLCO = diffusion capacity of carbon monoxide. 6MWT = 6-minute walk test. RV = right ventricle. LV = left ventricle. RVSP = right ventricular systolic pressure. TAPSE = tricuspid annular plane systolic exertion. RVOT = right ventricular outflow tract. ECHO = echocardiogram. BNP = brain natriuretic peptide. Adapted from King, et al. CHEST. 2020.245
Right heart catheterization (RHC) remains the gold standard for the diagnosis of pulmonary vascular disease including Group 3 PH. RHC is required in the evaluation of Group 3 PH to assess for left ventricular fluid and/or pressure overload – i.e., to distinguish pre- vs. postcapillary PH – and to define PH severity. Hemodynamics are needed and often repeated to evaluate patients with clinical worsening or progressive gas exchange abnormalities disproportionate to obstructive or ventilatory impairments, to evaluate for lung transplant and for inclusion in clinical trials or registries182. In cases where off-label therapy is being considered to treat non-ILD forms of Group 3 PH, or to initiate inhaled treprostinil for ILD-associated PH, RHC is required.
Patients with chronic lung disease are at risk for both precapillary and postcapillary PH due to concurrent left heart disease. As patients with chronic lung disease tend to have significant respiratory variation, several breath cycles should be captured to measure hemodynamics including the pulmonary capillary wedge pressure (PCWP) which should be measured at end-expiration109. In the setting of underlying parenchymal lung disease, Group 3 PH is diagnosed when the mPAP is > 20 mmHg, the PCWP is ≤ 15 mmHg, and the PVR is ≥ 3 WU1. Patients with risk factors for post-capillary PH, such as obesity, systemic hypertension, atrial fibrillation, diabetes mellitus and OSA, and echocardiographic evidence of diastolic dysfunction, should be considered for provocative maneuvers such as fluid loading and exercise, to determine a dominant driver of pulmonary pressures. The 2011 German Cologne PH Conference and the most recent WSPH statement introduced a concept of chronic lung disease with severe PH, defined as mPAP ≥ 35 mmHg or mPAP ≥ 25 mmHg with a low cardiac index (< 2.0 L/min/m2)109, 183. This distinction is supported by evidence that patients with COPD and PH with a mPAP ≥ 40 mmHg demonstrate functional impairment due to circulatory-limitation as compared to COPD patients with moderate PH (mean mPAP of 31 mmHg)184. These observations have been recapitulated in IPF patients with mPAP ≥ 35 mmHg87, 105, 185. As discussed above, recent registry data suggests that PVR thresholds may provide improved prognostication over mPAP values97, 114 which may reflect the degree that combined assessment of pulmonary pressures and the RV response are more prognostic in this population. There are no data to support routine vasodilator testing in Group 3 PH99, although in patients with chronic hypoxia it may be important to assess for a hemodynamic response to oxygen at the time of catheterization.
Differentiating Group 1 PAH from Group 3 PH can be challenging in many patients, particularly in those with underlying CTD. The degree of pulmonary vascular disease and parenchymal lung involvement exists on a continuum for many patients, however there are distinguishing features that may help to define the predominant pathophenotype (Table 4)109.
Table 4.
Distinguishing features of Group 1 pulmonary arterial hypertension and Group 3 pulmonary hypertension
| Criteria Favoring Group 1 PAH | Parameter | Criteria Favoring Group 3 PH |
|---|---|---|
| Extent of Lung Disease | ||
| • Normal or mildly impaired spirometry (FEV1 >60% predicted in COPD; FVC >70% predicted in IPF) • Low DLCO in relation to obstructive/restrictive changes (i.e. DLCO “out of proportion” to spirometry/lung volumes) |
Pulmonary function testing | • Moderate to severely impaired spirometry (FEV1 <60% predicted in COPD; FVC <70% predicted in IPF) • DLCO corresponds to obstructive/restrictive changes (i.e. DLCO “in proportion” to spirometry/lung volumes) |
| Absence of or only moderate parenchymal abnormalities | High resolution CT scan | Characteristic parenchymal abnormalities |
| Hemodynamic Profile | ||
| Moderate-to-severe PH | Right Heart Catheterization or Echocardiogram | Mild-to-moderate PH |
| Ancillary Testing | ||
| Present | Additional PAH risk factors (e.g. family history or identified mutations associated with PAH, connective tissue disease, HIV, history of drug/toxin exposure, etc) | Absent |
| Features of circulatory limitation to exercise: • Preserved breathing reserve • Reduced O2 pulse • Low CO/VO2 slope • Mixed venous oxygen saturation at lower limit • No change or decrease in PaCO2 during exercise |
Cardiopulmonary exercise test | Features of ventilatory limitation to exercise: • Reduced breathing reserve • Normal O2 pulse • Normal CO/VO2 slope • Mixed venous oxygen saturation above lower limit • Increase in PaCO2 during exercise |
PAH = pulmonary arterial hypertension. PH = pulmonary hypertension. FEV1 = forced expiratory volume in the first second. COPD = chronic obstructive pulmonary disease. FVC = forced vital capacity. IPF = idiopathic pulmonary fibrosis. DLCO = diffusion capacity of carbon monoxide. CT = computed tomography. HIV = human immunodeficiency virus. Adapted from Nathan, et al. ERJ. 2019.109
Therapeutic Strategies
Treatment of the underlying lung disease remains the initial therapeutic strategy in Group 3 PH including bronchodilators in COPD, immunosuppression and antifibrotics in ILDs, and nocturnal positive pressure therapy in sleep disordered breathing125, 126, 128. Appropriate and timely lung transplant referral is essential in patients with advanced lung disease, given the lack of effective therapies and that the development of PH is associated with worse outcomes. As such, the presence of moderate to severe PH in COPD and evidence of PH by echocardiography or RHC in ILD is a specific indication for lung transplantation listing186. Long-term oxygen therapy is frequently prescribed for PH patients who are hypoxic, however this has only been prospectively evaluated in COPD where it was noted to prevent an increase in mPAP and slightly decrease mPAP when used for >18 hours per day187, 188. There are no data examining the benefit of long-term oxygen on clinical outcomes or survival in PH and this remains a significant knowledge gap. RV dysfunction and pulmonary artery pressures (as measured by transthoracic echocardiography) appear to transiently increase during COPD exacerbations, physiology that should be taken into account when initiating therapy in this patient population189.
Pulmonary Vasodilators
Pulmonary vasodilators have long been theorized to worsen gas exchange in patients with chronic lung disease due to worsening of hypoxic vasoconstriction and ventilation/perfusion mismatch, although this has not been substantiated in clinical studies99. When delivered via an inhaled route, pulmonary vasodilators may access better ventilated areas of the lung, a feasible paradigm for the treatment of Group 3 PH190–192. On balance, PAH therapies have modest and inconsistent benefits in Group 3 PH patients193. Because of the complexities of adequately phenotyping patients, who may have overlapping features from multiple WSPH groups, and the potential for harm with pulmonary vasodilators, consideration of PH-specific therapy should be done at expert centers.
Endothelin receptor antagonists (ERAs) have no evidence of benefit and may be harmful in ILD-PH patients. Bosentan was evaluated in a randomized clinical trial (RCT) of 60 participants with fibrotic ILD and PH confirmed by invasive hemodynamics. Neither the primary endpoint (reduction in PVR index by 20% at 16 weeks) nor secondary endpoints including functional capacity, symptoms, adverse events or death were significantly different between the bosentan group and placebo194. The ARTEMIS-IPF trial enrolled 492 participants with IPF to ambrisentan or placebo. The trial was terminated early due to an increase in IPF disease progression and hospitalizations and a trend toward increased mortality and decreased lung function in the ambrisentan group195.
There have been numerous RCTs of phosphodiesterase-type 5 (PDE5) inhibitors in patients with chronic lung disease. A study of tadalafil in 120 COPD participants failed to improve 6MWD or quality of life196. A pilot double-blind, placebo-controlled RCT of sildenafil in COPD-PH (SPHERIC-1) demonstrated a decrease in PVR and improved quality of life metrics with no effect on 6MWD197. STEP-IPF examined the role of sildenafil in 180 IPF participants regardless of the presence of PH. Although there was no improvement in the primary endpoint of 6MWD, a post-hoc subgroup analysis demonstrated that IPF patients with RV dysfunction randomized to sildenafil had a lower decline in 6MWD and improvements in two quality of life measurements198, 199. A more recent controlled trial examined the addition of sildenafil to pirfenidone in severe IPF patients (DLCO ≤ 40%) at risk of developing PH, but when compared to placebo, found no difference in prevention of disease progression200.
Riociguat, a soluble guanylate cyclase stimulator, was studied in RISE-IIP, a randomized, double-blind placebo-controlled trial of 147 ILD-associated PH participants with a primary endpoint of 6MWD. This trial was stopped early due to an increase in adverse events and mortality in the riociguat group with no improvement in the 6MWD201.
Parenteral prostanoid use has only been reported in case reports or small series in Group 3 PH. In an open-label study of parenteral treprostinil in severe PH associated with ILD (mPAP > 35 mmHg), there was a modest improvement in 6MWD, RV function, and hemodynamics202.
Results of the recently published INCREASE trial56 of inhaled treprostinil support the hypothesis that inhaled pulmonary vasodilators could cause less ventilation-perfusion mismatch in Group 3 PH compared to systemic therapies. This trial was a randomized, double-blind, placebo-controlled multicenter study that evaluated inhaled treprostinil administered by an iNEB (adaptive aerosol delivery system) device in 326 participants with ILD-associated PH with change in 6MWD at week 16 as the primary endpoint. The least-squares mean difference in the change from baseline 6MWD with treprostinil compared to placebo was 31 m (95% CI, 17 – 45; p < 0.0001) and treprostinil reduced clinical worsening (hazard ratio 0.61, 95% CI 0.40 – 0.92; p < 0.04). The reduction in clinical worsening was driven by the improvement in 6MWD and treprostinil did not improve quality of life. There tended to be more women and CTD patients in the treprostinil group, which may have biased the results as these groups tend to respond more favorably to PAH therapy. A post-hoc analysis demonstrated a placebo-corrected 169 mL improvement in forced vital capacity (FVC) in a subgroup of participants with IPF and a 108 mL improvement in FVC in a subgroup of participants with IIPs at week 16. Participants with IIPs on inhaled treprostinil had numerically fewer exacerbations203. These results, along with preclinical data suggesting that treprostinil has direct antifibrotic effects on the lung204, 205, have led to the ongoing TETON trial of IIP without PH (NCT04708782). In another post hoc analysis of INCREASE, treatment with inhaled treprostinil resulted in decreased likelihood of disease progression defined as either ≥ 5% decline in 6MWD, ≥ 10% decline in FVC, acute exacerbation, cardiopulmonary hospitalization, lung transplantation, or death (22 vs 36% multiple progression events in treatment vs placebo groups, respectively; p = 005)206. The United States Food and Drug Administration approved inhaled treprostinil for Group 3 PH due to ILD on April 1, 2021, marking the first and only approved PAH therapy for Group 3 PH due to ILD in the United States.
Registry data mirrors what has been learned from clinical trials to-date in Group 3 PH. Hoeper and colleagues compared the safety and efficacy of off-label PAH therapy in 151 ILD-associated PH patients with severe PH and RV dysfunction (mPAP 37 ± 9 mmHg and cardiac index 2.1 ± 0.61 L/min/m2) in COMPERA207. The majority of this group was treated with PDE5 inhibitors and showed a modest improvement in 6MWD and functional class. A subsequent analysis of COPD-associated PH in COMPERA demonstrated a 30 m increase in 6MWD208. Despite these improvements in intermediate end points, both analyses confirmed that these groups had a significantly worse survival as compared to idiopathic PAH patients. Group 3 PH patients in the Scottish Pulmonary Vascular Unit treated with pulmonary vasodilators (primarily PDE5 inhibitors) demonstrated no improvement in 6MWD, but a significant decrease in serum NT-proBNP levels209. Patients with CPFE had a significant reduction in serum NT-proBNP levels and an improvement in 6MWD of 41 m (above the minimally important difference of 33 m210), whereas this observation was not seen in patients with ILD or emphysema. Thenappan and colleagues performed a meta-analysis of five RCTS in COPD-PH, two RCTs in ILD-PH, and four single-arm clinical trials in ILD-PH2. Although there was a signal toward improved hemodynamics, PAH therapy (the majority with PDE5 inhibitors in COPD-PH and PDE5 inhibitor, ERA, riociguat, and parenteral treprostinil in ILD-PH) showed no symptomatic benefit in COPD-PH or ILD-PH though treatment did not worsen hypoxemia. The single-arm ILD-PH studies demonstrated improvement in 6MWD, however these findings were not recapitulated in either the COPD-PH or ILD-PH RCTs.
In summary, data supporting the use of PAH therapy in Group 3 PH are limited and further study is needed. Vasoactive medications may have a benefit in parenchymal lung disease patients with severe hemodynamic impairment with depressed cardiac output, which may identify a target population for future studies. Riociguat and ambrisentan are now contraindicated in PH associated with ILD, other ERAs should be avoided and evidence is conflicting or limited for the use of PDE5 inhibitors. Inhaled treprostinil is now approved for use in patients with PH complicating ILDs in the United States.
Emerging Therapies
Inhaled treprostinil is currently being studied in the PERFECT trial in patients with COPD-associated PH (NCT03496623). Inhaled treprostinil requires at least four nebulized inhalations each day and its major side effect is cough. A treprostinil-prodrug based nanoparticle with slower time release is in development and may have less cough associated with its use211–214. PULSE, a clinical trial of inhaled nitric oxide in PH-ILD, has demonstrated favorable patient reported outcomes215 and increased physical activity in a Phase II trial216 and is currently being studied in a Phase III trial (NCT03267108). Bardoxylone methyl, a novel agent with broad ranging anti-inflammatory effects, is also being studied in PH-ILD (NCT02036970).
Since Group 3 PH patients have worse RV dysfunction than other subgroups for a given RV afterload, therapies that target RV remodeling and dysfunction may be of particular benefit to Group 3 PH patients. Early work examined digoxin as one such agent, without evidence of benefit217. As in left ventricular dysfunction, mechanical assist devices for the RV may one day provide symptomatic relief in end-stage patients and serve as destination therapy or a bridge to transplant. Initial data is promising218 and a feasibility trial is currently underway to examine the safety of a device targeting pulmonary arterial compliance in Group 3 PH patients (NCT05001711).
Conclusion
Group 3 PH encompasses a group of patients with highly prevalent lung diseases and poor outcomes. Discrete pathobiological mechanisms in this population remain an active area of study as does the development of novel therapeutic targets. Recent advances in therapeutic options include more robust approaches to clinical phenotyping and the benefit of inhaled treprostinil in ILD-associated PH, which will hopefully stimulate further research for effective therapies in all patients suffering from Group 3 PH.
Table 5.
Clinical trials evaluating pulmonary vasodilators in Group 3 pulmonary hypertension
| Drug Class | Author (year) | Number of Participants | PH Definition | Lung Disease | Treatment | Duration | Primary Outcome | Result |
|---|---|---|---|---|---|---|---|---|
| ERA | Stolz et al246 (2008) | 30 | Echo PASP ≥ 30mmHg | COPD | Bosentan 125mg BID | 12 weeks | Change in 6MWD | No difference |
| Valerio et al247 (2009) | 32 | RHC mPAP ≥ 25mmHg | COPD | Bosentan 125mg BID | 18 months | PFTs, hemodynamics, 6MWD, dyspnea ratings/quality of life | Improvement in mPAP, PVR, 6MWD, dyspnea ratings | |
| Raghu et al195 (2013) | 492 | RHC mPAP ≥ 25mmHg | ILD | Ambrisentan 10mg daily | 16 weeks | Time to ILD progression | No difference; stopped early due to increased adverse events | |
| Corte et al194 (2014) | 60 | RHC mPAP ≥ 25mmHg | ILD | Bosentan 125mg BID | 16 weeks | Decrease in PVRi from baseline ≥20% | No difference | |
| PDE5 inhibitor | Collard et al248 (2007) | 11 | Echo PASP > 35mmHg or RHC mPAP ≥ 25mmHg | ILD | Sildenafil 20 – 50mg TID | 12 weeks | Change in 6MWD | Significant improvement |
| Han et al198, 199 (2010) | 180 | N/A | ILD | Sildenafil 20mg TID | 12 weeks | 20% improvement in 6MWD | No difference except in subgroup with RV systolic dysfunction | |
| Rao et al249 (2011) | 33 | Echo PASP > 40mmHg | COPD | Sildenafil 20mg TID | 12 weeks | Change in 6MWD | Significant improvement | |
| Goudie et al196 (2014) | 120 | Echo PASP > 30mmHg or PA-AT < 120ms | COPD | Tadalafil 10mg daily | 12 weeks | Change in 6MWD | No difference | |
| Vitulo et al197 (2017) | 28 | RHC mPAP ≥ 35mmHg and post-bronchodilator FEV1 < 30% or mPAP ≥ 30mmHg and post-bronchodilator FEV1 > 30% | COPD | Sildenafil 20mg TID | 16 weeks | Reduction in PVR | Significant improvement | |
| Behr et al200 (2021) | 177 | RHC mPAP ≥ 20mmHg | ILD | Sildenafil 20mg TID + Pirfenidone 801mg TID | 52 weeks | Disease progression | No difference | |
| Prostacyclin analogue | Saggar et al202 (2014) | 15 | RHC mPAP ≥ 35mmHg | ILD | Treprostinil 34 ± 21ng/kg/min | 12 weeks | Hemodynamics, 6MWD, PFTs, ECHO, dyspnea ratings/quality of life | Improvement in hemodynamics, 6MWD, dyspnea ratings |
| Waxman et al56, 203 (2021) | 326 | RHC mPAP ≥ 25mmHg | ILD | Treprostinil 72 μg QID | 16 weeks | Change in 6MWD | Significant improvement; Improvement in FVC in IPF subgroup | |
| sGC stimulator | Nathan et al201 (2019) | 147 | RHC mPAP ≥ 25mmHg | ILD | Riociguat 0.5 – 2.5mg TID | 26 weeks | Change in 6MWD | No difference; stopped early due to increased adverse events |
PH = pulmonary hypertension. ERA = endothelin receptor antagonist. RHC = right heart catheterization. mPAP = mean pulmonary artery pressure. ILD = interstitial lung disease. BID = two times per day. PVRi = pulmonary vascular resistance index. PVR = pulmonary vascular resistance. PFT = pulmonary function test. 6MWD = 6 minute walk distance. ECHO = echocardiogram. PDE5 = phosphodiesterase type-5. PASP = pulmonary artery systolic pressure. TID = three times per day. PA-AT = pulmonary artery acceleration time. FEV1 = forced expiratory volume in the first second. QID = four times per day. sGC = soluble guanyl cyclase.
References
- 1.Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG and Souza R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. European Respiratory Journal. 2019;53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prins KW, Duval S, Markowitz J, Pritzker M and Thenappan T. Chronic use of PAH-specific therapy in World Health Organization Group III Pulmonary Hypertension: a systematic review and meta-analysis. Pulmonary circulation. 2017;7:145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prins KW, Rose L, Archer SL, Pritzker M, Weir EK, Kazmirczak F, Misialek JR and Thenappan T. Disproportionate Right Ventricular Dysfunction and Poor Survival in Group 3 Pulmonary Hypertension. Am J Respir Crit Care Med. 2018;197:1496–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen L, Einbinder E, Zhang Q, Hasday J, Balke CW and Scharf SM. Oxidative stress and left ventricular function with chronic intermittent hypoxia in rats. American journal of respiratory and critical care medicine. 2005;172:915–920. [DOI] [PubMed] [Google Scholar]
- 5.Silverman NA, Kohler J, Levitsky S, Pavel DG, Fang RB and Feinberg H. Chronic hypoxemia depresses global ventricular function and predisposes to the depletion of high-energy phosphates during cardioplegic arrest: implications for surgical repair of cyanotic congenital heart defects. The Annals of thoracic surgery. 1984;37:304–308. [DOI] [PubMed] [Google Scholar]
- 6.Colvin KL and Yeager ME. Animal models of pulmonary hypertension: matching disease mechanisms to etiology of the human disease. Journal of pulmonary & respiratory medicine. 2014;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Das M, Fessel J, Tang H and West J. A process-based review of mouse models of pulmonary hypertension. Pulmonary circulation. 2012;2:415–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gomez-Arroyo J, Saleem SJ, Mizuno S, Syed AA, Bogaard HJ, Abbate A, Taraseviciene-Stewart L, Sung Y, Kraskauskas D and Farkas D. A brief overview of mouse models of pulmonary arterial hypertension: problems and prospects. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2012;302:L977–L991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ryan J, Bloch K and Archer S. Rodent models of pulmonary hypertension: harmonisation with the world health organisation’s categorisation of human PH. International journal of clinical practice. 2011;65:15–34. [DOI] [PubMed] [Google Scholar]
- 10.Woo KV, Ornitz DM and Singh GK. Diagnosis and Pathophysiological Mechanisms of Group 3 Hypoxia-Induced Pulmonary Hypertension. Curr Treat Options Cardiovasc Med. 2019;21:16. [DOI] [PubMed] [Google Scholar]
- 11.Jiang Y, Dai A, Li Q and Hu R. Hypoxia induces transforming growth factor-β1 gene expression in the pulmonary artery of rats via hypoxia-inducible factor-1α. Acta biochimica et biophysica Sinica. 2007;39:73–80. [DOI] [PubMed] [Google Scholar]
- 12.Kajdaniuk D, Marek B, Borgiel-Marek H and Kos-Kudła B. Transforming growth factor beta1 (TGFbeta1) in physiology and pathology. Endokrynologia Polska. 2013;64:384–396. [DOI] [PubMed] [Google Scholar]
- 13.Lu A, Zuo C, He Y, Chen G, Piao L, Zhang J, Xiao B, Shen Y, Tang J and Kong D. EP3 receptor deficiency attenuates pulmonary hypertension through suppression of Rho/TGF-β1 signaling. The Journal of clinical investigation. 2015;125:1228–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gilbane AJ, Derrett-Smith E, Trinder SL, Good RB, Pearce A, Denton CP and Holmes AM. Impaired bone morphogenetic protein receptor II signaling in a transforming growth factor-β–dependent mouse model of pulmonary hypertension and in systemic sclerosis. American journal of respiratory and critical care medicine. 2015;191:665–677. [DOI] [PubMed] [Google Scholar]
- 15.Tuder RM, Davis LA and Graham BB. Targeting energetic metabolism: a new frontier in the pathogenesis and treatment of pulmonary hypertension. American journal of respiratory and critical care medicine. 2012;185:260–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y, Cao Y, Sun S, Zhu J, Gao S, Pang J, Zhu D and Sun Z. Transforming growth factor-beta1 upregulation triggers pulmonary artery smooth muscle cell proliferation and apoptosis imbalance in rats with hypoxic pulmonary hypertension via the PTEN/AKT pathways. The international journal of biochemistry & cell biology. 2016;77:141–154. [DOI] [PubMed] [Google Scholar]
- 17.Voelkel NF and Gomez-Arroyo J. The role of vascular endothelial growth factor in pulmonary arterial hypertension. The angiogenesis paradox. American journal of respiratory cell and molecular biology. 2014;51:474–484. [DOI] [PubMed] [Google Scholar]
- 18.Christou H, Yoshida A, Arthur V, Morita T and Kourembanas S. Increased vascular endothelial growth factor production in the lungs of rats with hypoxia-induced pulmonary hypertension. American journal of respiratory cell and molecular biology. 1998;18:768–776. [DOI] [PubMed] [Google Scholar]
- 19.Tuder RM, Flook BE and Voelkel NF. Increased gene expression for VEGF and the VEGF receptors KDR/Flk and Flt in lungs exposed to acute or to chronic hypoxia. Modulation of gene expression by nitric oxide. The Journal of clinical investigation. 1995;95:1798–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Louzier V, Raffestin B, Leroux A, Branellec D, Caillaud JM, Levame M, Eddahibi S and Adnot S. Role of VEGF-B in the lung during development of chronic hypoxic pulmonary hypertension. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2003;284:L926–L937. [DOI] [PubMed] [Google Scholar]
- 21.Gore B, Izikki M, Mercier O, Dewachter L, Fadel E, Humbert M, Dartevelle P, Simonneau G, Naeije R and Lebrin F. Key role of the endothelial TGF-β/ALK1/endoglin signaling pathway in humans and rodents pulmonary hypertension. PloS one. 2014;9:e100310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang S, Yu H, Chen X, Shen T, Cui Z, Zhang J, Cheng Y, Jia S, Song S and Zhang X. PDGF-BB/KLF4/VEGF signaling axis in pulmonary artery endothelial cell angiogenesis. Cellular Physiology and Biochemistry. 2017;41:2333–2349. [DOI] [PubMed] [Google Scholar]
- 23.Santos S, Peinado VI, Ramirez J, Morales-Blanhir J, Bastos R, Roca J, Rodriguez-Roisin R and Barbera JA. Enhanced expression of vascular endothelial growth factor in pulmonary arteries of smokers and patients with moderate chronic obstructive pulmonary disease. American journal of respiratory and critical care medicine. 2003;167:1250–1256. [DOI] [PubMed] [Google Scholar]
- 24.Peinado VI, Barberà JA, Ramírez J, Gómez FP, Roca J, Jover L, Gimferrer JM and Rodriguez-Roisin R. Endothelial dysfunction in pulmonary arteries of patients with mild COPD. American Journal of Physiology-Lung Cellular and Molecular Physiology. 1998;274:L908–L913. [DOI] [PubMed] [Google Scholar]
- 25.Dinh-Xuan AT, Higenbottam TW, Clelland CA, Pepke-Zaba J, Cremona G, Butt AY, Large SR, Wells FC and Wallwork J. Impairment of endothelium-dependent pulmonary-artery relaxation in chronic obstructive lung disease. New England Journal of Medicine. 1991;324:1539–1547. [DOI] [PubMed] [Google Scholar]
- 26.Hemnes AR, Zaiman A and Champion HC. PDE5A inhibition attenuates bleomycin-induced pulmonary fibrosis and pulmonary hypertension through inhibition of ROS generation and RhoA/Rho kinase activation. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2008;294:L24–L33. [DOI] [PubMed] [Google Scholar]
- 27.Conte C, Riant E, Toutain C, Pujol F, Arnal J-F, Lenfant F and Prats A-C. FGF2 translationally induced by hypoxia is involved in negative and positive feedback loops with HIF-1α. PloS one. 2008;3:e3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schultz K, Fanburg BL and Beasley D. Hypoxia and hypoxia-inducible factor-1α promote growth factor-induced proliferation of human vascular smooth muscle cells. American Journal of Physiology-Heart and Circulatory Physiology. 2006. [DOI] [PubMed] [Google Scholar]
- 29.Chang Y-T, Tseng C-N, Tannenberg P, Eriksson L, Yuan K, de Jesus Perez VA, Lundberg J, Lengquist M, Botusan IR and Catrina S-B. Perlecan heparan sulfate deficiency impairs pulmonary vascular development and attenuates hypoxic pulmonary hypertension. Cardiovascular research. 2015;107:20–31. [DOI] [PubMed] [Google Scholar]
- 30.Ambalavanan N and Novak ZE. Peptide growth factors in tracheal aspirates of mechanically ventilated preterm neonates. Pediatric research. 2003;53:240–244. [DOI] [PubMed] [Google Scholar]
- 31.Santos S, Peinado VI, Ramirez J, Melgosa T, Roca J, Rodriguez-Roisin R and Barbera J. Characterization of pulmonary vascular remodelling in smokers and patients with mild COPD. European Respiratory Journal. 2002;19:632–638. [DOI] [PubMed] [Google Scholar]
- 32.Barberá JA, Peinado VI, Santos S, Ramirez J, Roca J and Rodriguez-Roisin R. Reduced expression of endothelial nitric oxide synthase in pulmonary arteries of smokers. American journal of respiratory and critical care medicine. 2001;164:709–713. [DOI] [PubMed] [Google Scholar]
- 33.Peinado VI, Barberà JA, Abate P, Ramírez J, Roca J, Santos S and Rodriguez-roisin R. Inflammatory reaction in pulmonary muscular arteries of patients with mild chronic obstructive pulmonary disease. American journal of respiratory and critical care medicine. 1999;159:1605–1611. [DOI] [PubMed] [Google Scholar]
- 34.Wang Z, White A, Wang X, Ko J, Choudhary G, Lange T, Rounds S and Lu Q. Mitochondrial Fission Mediated Cigarette Smoke–induced Pulmonary Endothelial Injury. American journal of respiratory cell and molecular biology. 2020;63:637–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barberà JA. Mechanisms of development of chronic obstructive pulmonary disease-associated pulmonary hypertension. Pulmonary circulation. 2013;3:160–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peinado VI, Pizarro S and Barbera JA. Pulmonary vascular involvement in COPD. Chest. 2008;134:808–814. [DOI] [PubMed] [Google Scholar]
- 37.Barbera J, Peinado V and Santos S. Pulmonary hypertension in chronic obstructive pulmonary disease. European Respiratory Journal. 2003;21:892–905. [DOI] [PubMed] [Google Scholar]
- 38.Su Y, Han W, Giraldo C, De Li Y and Block ER. Effect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cells. American journal of respiratory cell and molecular biology. 1998;19:819–825. [DOI] [PubMed] [Google Scholar]
- 39.Nana-Sinkam SP, Lee JD, Sotto-Santiago S, Stearman RS, Keith RL, Choudhury Q, Cool C, Parr J, Moore MD and Bull TM. Prostacyclin prevents pulmonary endothelial cell apoptosis induced by cigarette smoke. American Journal of Respiratory and Critical Care Medicine. 2007;175:676–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferrer E, Peinado VI, Díez M, Carrasco JL, Musri MM, Martínez A, Rodríguez-Roisin R and Barberà JA. Effects of cigarette smoke on endothelial function of pulmonary arteries in the guinea pig. Respiratory research. 2009;10:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seimetz M, Parajuli N, Pichl A, Veit F, Kwapiszewska G, Weisel FC, Milger K, Egemnazarov B, Turowska A and Fuchs B. Inducible NOS inhibition reverses tobacco-smoke-induced emphysema and pulmonary hypertension in mice. Cell. 2011;147:293–305. [DOI] [PubMed] [Google Scholar]
- 42.Weissmann N, Lobo B, Pichl A, Parajuli N, Seimetz M, Puig-Pey R, Ferrer E, Peinado VI, Domínguez-Fandos D and Fysikopoulos A. Stimulation of soluble guanylate cyclase prevents cigarette smoke–induced pulmonary hypertension and emphysema. American journal of respiratory and critical care medicine. 2014;189:1359–1373. [DOI] [PubMed] [Google Scholar]
- 43.Wright JL and Churg A. Effect of long-term cigarette smoke exposure on pulmonary vascular structure and function in the guinea pig. Experimental lung research. 1991;17:997–1009. [DOI] [PubMed] [Google Scholar]
- 44.Bushati N and Cohen SM. microRNA functions. Annu Rev Cell Dev Biol. 2007;23:175–205. [DOI] [PubMed] [Google Scholar]
- 45.Du T and Zamore PD. Beginning to understand microRNA function. Cell research. 2007;17:661–663. [DOI] [PubMed] [Google Scholar]
- 46.Pullamsetti SS, Doebele C, Fischer A, Savai R, Kojonazarov B, Dahal BK, Ghofrani HA, Weissmann N, Grimminger F and Bonauer A. Inhibition of microRNA-17 improves lung and heart function in experimental pulmonary hypertension. American journal of respiratory and critical care medicine. 2012;185:409–419. [DOI] [PubMed] [Google Scholar]
- 47.Brock M, Samillan VJ, Trenkmann M, Schwarzwald C, Ulrich S, Gay RE, Gassmann M, Ostergaard L, Gay S and Speich R. AntagomiR directed against miR-20a restores functional BMPR2 signalling and prevents vascular remodelling in hypoxia-induced pulmonary hypertension. European heart journal. 2014;35:3203–3211. [DOI] [PubMed] [Google Scholar]
- 48.Pu X, Du L, Hu Y, Fan Y and Xu Q. Stem/Progenitor Cells and Pulmonary Arterial Hypertension. Arteriosclerosis, thrombosis, and vascular biology. 2021;41:167–178. [DOI] [PubMed] [Google Scholar]
- 49.Lanzola E, Farha S, Erzurum SC and Asosingh K. Bone marrow–derived vascular modulatory cells in pulmonary arterial hypertension. Pulmonary circulation. 2013;3:781–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Péchoux C, Bogaard HJ, Dorfmüller P, Remy S, Lecerf F and Planté S. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation. 2015;131:1006–1018. [DOI] [PubMed] [Google Scholar]
- 51.Peinado VI, Ramirez J, Roca J, Rodriguez-Roisin R and Barbera JA. Identification of vascular progenitor cells in pulmonary arteries of patients with chronic obstructive pulmonary disease. American journal of respiratory cell and molecular biology. 2006;34:257–263. [DOI] [PubMed] [Google Scholar]
- 52.Peinado V, Santos S, Ramirez J, Roca J, Rodriguez-Roisin R and Barberà J. Response to hypoxia of pulmonary arteries in chronic obstructive pulmonary disease: an in vitro study. European Respiratory Journal. 2002;20:332–338. [DOI] [PubMed] [Google Scholar]
- 53.Chen Z, Liu M, Li L and Chen L. Involvement of the Warburg effect in non-tumor diseases processes. Journal of cellular physiology. 2018;233:2839–2849. [DOI] [PubMed] [Google Scholar]
- 54.Mermis J, Gu H, Xue B, Li F, Tawfik O, Buch S, Bartolome S, O'Brien-Ladner A and Dhillon NK. Hypoxia-inducible factor-1 α/platelet derived growth factor axis in HIV-associated pulmonary vascular remodeling. Respiratory research. 2011;12:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang Y-H, Thenappan T, Piao L, Zhang HJ and Pogoriler J. Dynamin-related protein 1–mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circulation research. 2012;110:1484–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Waxman A, Restrepo-Jaramillo R, Thenappan T, Ravichandran A, Engel P, Bajwa A, Allen R, Feldman J, Argula R and Smith P. Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. New England Journal of Medicine. 2021;384:325–334. [DOI] [PubMed] [Google Scholar]
- 57.Mitchell JA, Ahmetaj-Shala B, Kirkby NS, Wright WR, Mackenzie LS, Reed DM and Mohamed N. Role of prostacyclin in pulmonary hypertension. Global Cardiology Science and Practice. 2015;2014:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McGiff JC. Prostaglandins, prostacyclin, and thromboxanes. Annual review of pharmacology and toxicology. 1981;21:479–509. [DOI] [PubMed] [Google Scholar]
- 59.Frölich J Prostacyclin in hypertension. Journal of hypertension Supplement: official journal of the International Society of Hypertension. 1990;8:S73–8. [PubMed] [Google Scholar]
- 60.Samokhin AO, Stephens T, Wertheim BM, Wang R-S, Vargas SO, Yung L-M, Cao M, Brown M, Arons E and Dieffenbach PB. NEDD9 targets COL3A1 to promote endothelial fibrosis and pulmonary arterial hypertension. Science translational medicine. 2018;10:eaap7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lakatos HF, Thatcher TH, Kottmann RM, Garcia TM, Phipps RP and Sime PJ. The role of PPARs in lung fibrosis. PPAR research. 2007;2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pietra GG, Capron F, Stewart S, Leone O, Humbert M, Robbins IM, Reid LM and Tuder R. Pathologic assessment of vasculopathies in pulmonary hypertension. Journal of the American College of Cardiology. 2004;43:S25–S32. [DOI] [PubMed] [Google Scholar]
- 63.Avouac J, Konstantinova I, Guignabert C, Pezet S, Sadoine J, Guilbert T, Cauvet A, Tu L, Luccarini J-M and Junien J-L. Pan-PPAR agonist IVA337 is effective in experimental lung fibrosis and pulmonary hypertension. Annals of the rheumatic diseases. 2017;76:1931–1940. [DOI] [PubMed] [Google Scholar]
- 64.Murakami S, Nagaya N, Itoh T, Kataoka M, Iwase T, Horio T, Miyahara Y, Sakai Y, Kangawa K and Kimura H. Prostacyclin agonist with thromboxane synthase inhibitory activity (ONO-1301) attenuates bleomycin-induced pulmonary fibrosis in mice. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2006;290:L59–L65. [DOI] [PubMed] [Google Scholar]
- 65.Aytemur ZA, Hacievliyagil SS, Iraz M, Samdanci E, Ozerol E, Kuku I, Nurkabulov Z and Yildiz K. Effects of iloprost on bleomycin-induced pulmonary fibrosis in rats compared with methyl-prednisolone. Revista Portuguesa de Pneumologia (English Edition). 2012;18:272–277. [DOI] [PubMed] [Google Scholar]
- 66.Ebina M, Shimizukawa M, Shibata N, Kimura Y, Suzuki T, Endo M, Sasano H, Kondo T and Nukiwa T. Heterogeneous increase in CD34-positive alveolar capillaries in idiopathic pulmonary fibrosis. American journal of respiratory and critical care medicine. 2004;169:1203–1208. [DOI] [PubMed] [Google Scholar]
- 67.Renzoni EA, Walsh DA, Salmon M, Wells AU, Sestini P, Nicholson AG, Veeraraghavan S, Bishop AE, Romanska HM and Pantelidis P. Interstitial vascularity in fibrosing alveolitis. American journal of respiratory and critical care medicine. 2003;167:438–443. [DOI] [PubMed] [Google Scholar]
- 68.Cosgrove GP, Brown KK, Schiemann WP, Serls AE, Parr JE, Geraci MW, Schwarz MI, Cool CD and Worthen GS. Pigment epithelium–derived factor in idiopathic pulmonary fibrosis: a role in aberrant angiogenesis. American journal of respiratory and critical care medicine. 2004;170:242–251. [DOI] [PubMed] [Google Scholar]
- 69.Yamato H, Sun J-P, Churg A and Wright JL. Cigarette smoke-induced emphysema in guinea pigs is associated with diffusely decreased capillary density and capillary narrowing. Laboratory investigation; a journal of technical methods and pathology. 1996;75:211–219. [PubMed] [Google Scholar]
- 70.Jamal K, Fleetham J and Thurlbeck W. Cor pulmonale: correlation with central airway lesions, peripheral airway lesions, emphysema, and control of breathing. Am Rev Respir Dis. 1990;141:1172–1177. [DOI] [PubMed] [Google Scholar]
- 71.Yamato H, Sun J, Churg A and Wright J. Guinea pig pulmonary hypertension caused by cigarette smoke cannot be explained by capillary bed destruction. Journal of Applied Physiology. 1997;82:1644–1653. [DOI] [PubMed] [Google Scholar]
- 72.Bunel V, Guyard A, Dauriat G, Danel C, Montani D, Gauvain C, Thabut G, Humbert M, Castier Y and Dorfmüller P. Pulmonary arterial histologic lesions in patients with COPD with severe pulmonary hypertension. Chest. 2019;156:33–44. [DOI] [PubMed] [Google Scholar]
- 73.Colombat M, Mal H, Groussard O, Capron F, Thabut G, Jebrak G, Brugière O, Dauriat G, Castier Y and Lesèche G. Pulmonary vascular lesions in end-stage idiopathic pulmonary fibrosis: histopathologic study on lung explant specimens and correlations with pulmonary hemodynamics. Human pathology. 2007;38:60–65. [DOI] [PubMed] [Google Scholar]
- 74.Swietlik EM, Greene D, Zhu N, Megy K, Cogliano M, Rajaram S, Pandya D, Tilly T, Lutz KA and Welch CC. Bayesian inference associates rare KDR variants with specific phenotypes in pulmonary arterial hypertension. Circulation: Genomic and Precision Medicine. 2021;14:e003155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Swietlik EM, Prapa M, Martin JM, Pandya D, Auckland K, Morrell NW and Gräf S. ‘There and back again’—Forward genetics and reverse phenotyping in pulmonary arterial hypertension. Genes. 2020;11:1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Eyries M, Montani D, Girerd B, Favrolt N, Riou M, Faivre L, Manaud G, Perros F, Gräf S and Morrell NW. Familial pulmonary arterial hypertension by KDR heterozygous loss of function. European Respiratory Journal. 2020;55. [DOI] [PubMed] [Google Scholar]
- 77.Chen LL, Zmuda EJ, Talavera MM, Frick J, Brock GN, Liu Y, Klebanoff MA and Trittmann JK. Dual-specificity phosphatase (DUSP) genetic variants predict pulmonary hypertension in patients with bronchopulmonary dysplasia. Pediatric research. 2020;87:81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mei M, Cheng G, Sun B, Yang L, Wang H, Sun J and Zhou W. EDN1 gene variant is associated with neonatal persistent pulmonary hypertension. Scientific reports. 2016;6:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu X, Mei M, Chen X, Lu Y, Dong X, Hu L, Hu X, Cheng G, Cao Y and Yang L. Identification of genetic factors underlying persistent pulmonary hypertension of newborns in a cohort of Chinese neonates. Respiratory research. 2019;20:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mura M, Anraku M, Yun Z, McRae K, Liu M, Waddell TK, Singer LG, Granton JT, Keshavjee S and de Perrot M. Gene expression profiling in the lungs of patients with pulmonary hypertension associated with pulmonary fibrosis. Chest. 2012;141:661–673. [DOI] [PubMed] [Google Scholar]
- 81.Hoffmann J, Wilhelm J, Olschewski A and Kwapiszewska G. Microarray analysis in pulmonary hypertension. European Respiratory Journal. 2016;48:229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Patel NM, Kawut SM, Jelic S, Arcasoy SM, Lederer DJ and Borczuk AC. Pulmonary arteriole gene expression signature in idiopathic pulmonary fibrosis. European Respiratory Journal. 2013;41:1324–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Strange G, Playford D, Stewart S, Deague JA, Nelson H, Kent A and Gabbay E. Pulmonary hypertension: prevalence and mortality in the Armadale echocardiography cohort. Heart. 2012;98:1805–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wijeratne DT, Lajkosz K, Brogly SB, Lougheed MD, Jiang L, Housin A, Barber D, Johnson A, Doliszny KM and Archer SL. Increasing incidence and prevalence of World Health Organization groups 1 to 4 pulmonary hypertension: a population-based cohort study in Ontario, Canada. Circulation: Cardiovascular Quality and Outcomes. 2018;11:e003973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gall H, Felix JF, Schneck FK, Milger K, Sommer N, Voswinckel R, Franco OH, Hofman A, Schermuly RT, Weissmann N, Grimminger F, Seeger W and Ghofrani HA. The Giessen Pulmonary Hypertension Registry: Survival in pulmonary hypertension subgroups. J Heart Lung Transplant. 2017;36:957–967. [DOI] [PubMed] [Google Scholar]
- 86.Barbera JA and Blanco I. Management of Pulmonary Hypertension in Patients with Chronic Lung Disease. Curr Hypertens Rep. 2015;17:62. [DOI] [PubMed] [Google Scholar]
- 87.Andersen KH, Iversen M, Kjaergaard J, Mortensen J, Nielsen-Kudsk JE, Bendstrup E, Videbaek R and Carlsen J. Prevalence, predictors, and survival in pulmonary hypertension related to end-stage chronic obstructive pulmonary disease. J Heart Lung Transplant. 2012;31:373–80. [DOI] [PubMed] [Google Scholar]
- 88.Kimura M, Taniguchi H, Kondoh Y, Kimura T, Kataoka K, Nishiyama O, Aso H, Sakamoto K and Hasegawa Y. Pulmonary hypertension as a prognostic indicator at the initial evaluation in idiopathic pulmonary fibrosis. Respiration. 2013;85:456–63. [DOI] [PubMed] [Google Scholar]
- 89.Travis WD, Costabel U, Hansell DM, King TE Jr, Lynch DA, Nicholson AG, Ryerson CJ, Ryu JH, Selman M and Wells AU. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. American journal of respiratory and critical care medicine. 2013;188:733–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Society AT. ATS/ERS international multidisciplinary consensus classification of idiopathic interstitial pneumonia. Am J Respir crit care med. 2002;165:277–304. [DOI] [PubMed] [Google Scholar]
- 91.Soriano JB, Kendrick PJ, Paulson KR, Gupta V, Abrams EM, Adedoyin RA, Adhikari TB, Advani SM, Agrawal A and Ahmadian E. Prevalence and attributable health burden of chronic respiratory diseases, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. The Lancet Respiratory Medicine. 2020;8:585–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chaouat A, Bugnet A-S, Kadaoui N, Schott R, Enache I, Ducoloné A, Ehrhart M, Kessler R and Weitzenblum E. Severe pulmonary hypertension and chronic obstructive pulmonary disease. American journal of respiratory and critical care medicine. 2005;172:189–194. [DOI] [PubMed] [Google Scholar]
- 93.Weitzenblum E, Sautegeau A, Ehrhart M, Mammosser M, Hirth C and Roegel E. Long-term course of pulmonary arterial pressure in chronic obstructive pulmonary disease. American Review of Respiratory Disease. 1984;130:993–998. [DOI] [PubMed] [Google Scholar]
- 94.Andersen KH, Iversen M, Kjaergaard J, Mortensen J, Nielsen-Kudsk JE, Bendstrup E, Videbaek R and Carlsen J. Prevalence, predictors, and survival in pulmonary hypertension related to end-stage chronic obstructive pulmonary disease. The Journal of heart and lung transplantation. 2012;31:373–380. [DOI] [PubMed] [Google Scholar]
- 95.Burrows B, Kettel LJ, Niden AH, Rabinowitz M and Diener CF. Patterns of cardiovascular dysfunction in chronic obstructive lung disease. New England Journal of Medicine. 1972;286:912–918. [DOI] [PubMed] [Google Scholar]
- 96.Oswald-Mammosser M, Weitzenblum E, Quoix E, Moser G, Chaouat A, Charpentier C and Kessler R. Prognostic factors in COPD patients receiving long-term oxygen therapy: importance of pulmonary artery pressure. Chest. 1995;107:1193–1198. [DOI] [PubMed] [Google Scholar]
- 97.Zeder K, Avian A, Bachmaier G, Douschan P, Foris V, Sassmann T, Troester N, Brcic L, Fuchsjaeger M and Marsh LM. Elevated pulmonary vascular resistance predicts mortality in COPD patients. European Respiratory Journal. 2021. [DOI] [PubMed] [Google Scholar]
- 98.Vizza CD, Hoeper MM, Huscher D, Pittrow D, Benjamin N, Olsson KM, Ghofrani HA, Held M, Klose H and Lange T. Pulmonary Hypertension in Patients With COPD: Results From COMPERA. Chest. 2021. [DOI] [PubMed] [Google Scholar]
- 99.Seeger W, Adir Y, Barberà JA, Champion H, Coghlan JG, Cottin V, De Marco T, Galiè N, Ghio S and Gibbs S. Pulmonary hypertension in chronic lung diseases. Journal of the American College of Cardiology. 2013;62:D109–D116. [DOI] [PubMed] [Google Scholar]
- 100.Carlsen J, Andersen KH, Boesgaard S, Iversen M, Steinbrüchel D and Andersen CB. Pulmonary arterial lesions in explanted lungs after transplantation correlate with severity of pulmonary hypertension in chronic obstructive pulmonary disease. The Journal of Heart and Lung Transplantation. 2013;32:347–354. [DOI] [PubMed] [Google Scholar]
- 101.Kimura M, Taniguchi H, Kondoh Y, Kimura T, Kataoka K, Nishiyama O, Aso H, Sakamoto K and Hasegawa Y. Pulmonary hypertension as a prognostic indicator at the initial evaluation in idiopathic pulmonary fibrosis. Respiration. 2013;85:456–463. [DOI] [PubMed] [Google Scholar]
- 102.King CS, Brown AW, Shlobin OA, Weir N, Libre M, Franco-Palacios D, Ahmad S and Nathan SD. Prevalence and impact of WHO group 3 pulmonary hypertension in advanced idiopathic nonspecific interstitial pneumonia. European Respiratory Journal. 2018;52. [DOI] [PubMed] [Google Scholar]
- 103.Oliveira RK, Pereira CA, Ramos RP, Ferreira EV, Messina CM, Kuranishi LT, Gimenez A, Campos O, Silva CM and Ota-Arakaki JS. A haemodynamic study of pulmonary hypertension in chronic hypersensitivity pneumonitis. European Respiratory Journal. 2014;44:415–424. [DOI] [PubMed] [Google Scholar]
- 104.Behr J and Ryu J. Pulmonary hypertension in interstitial lung disease. European Respiratory Journal. 2008;31:1357–1367. [DOI] [PubMed] [Google Scholar]
- 105.Minai OA, Santacruz JF, Alster JM, Budev MM and McCarthy K. Impact of pulmonary hemodynamics on 6-min walk test in idiopathic pulmonary fibrosis. Respiratory medicine. 2012;106:1613–1621. [DOI] [PubMed] [Google Scholar]
- 106.Nathan SD, Shlobin OA, Ahmad S, Koch J, Barnett SD, Ad N, Burton N and Leslie K. Serial development of pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Respiration. 2008;76:288–294. [DOI] [PubMed] [Google Scholar]
- 107.Lettieri CJ, Nathan SD, Barnett SD, Ahmad S and Shorr AF. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest. 2006;129:746–752. [DOI] [PubMed] [Google Scholar]
- 108.Hamada K, Nagai S, Tanaka S, Handa T, Shigematsu M, Nagao T, Mishima M, Kitaichi M and Izumi T. Significance of pulmonary arterial pressure and diffusion capacity of the lung as prognosticator in patients with idiopathic pulmonary fibrosis. Chest. 2007;131:650–656. [DOI] [PubMed] [Google Scholar]
- 109.Nathan SD, Barbera JA, Gaine SP, Harari S, Martinez FJ, Olschewski H, Olsson KM, Peacock AJ, Pepke-Zaba J and Provencher S. Pulmonary hypertension in chronic lung disease and hypoxia. European Respiratory Journal. 2019;53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Raghu G, Nathan SD, Behr J, Brown KK, Egan JJ, Kawut SM, Flaherty KR, Martinez FJ, Wells AU and Shao L. Pulmonary hypertension in idiopathic pulmonary fibrosis with mild-to-moderate restriction. European Respiratory Journal. 2015;46:1370–1377. [DOI] [PubMed] [Google Scholar]
- 111.Shorr AF, Wainright JL, Cors CS, Lettieri CJ and Nathan SD. Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. European Respiratory Journal. 2007;30:715–721. [DOI] [PubMed] [Google Scholar]
- 112.Nadrous HF, Pellikka PA, Krowka MJ, Swanson KL, Chaowalit N, Decker PA and Ryu JH. Pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Chest. 2005;128:2393–2399. [DOI] [PubMed] [Google Scholar]
- 113.Alhamad EH, Al-Boukai AA, Al-Kassimi FA, Alfaleh HF, Alshamiri MQ, Alzeer AH, Al-Otair HA, Ibrahim GF and Shaik SA. Prediction of pulmonary hypertension in patients with or without interstitial lung disease: reliability of CT findings. Radiology. 2011;260:875–883. [DOI] [PubMed] [Google Scholar]
- 114.Olsson KM, Hoeper MM, Pausch C, Grünig E, Huscher D, Pittrow D, Rosenkranz S and Gall H. Pulmonary vascular resistance predicts mortality in patients with pulmonary hypertension associated with interstitial lung disease: results from the COMPERA registry. European Respiratory Journal. 2021;58. [DOI] [PubMed] [Google Scholar]
- 115.Cottin V, Le Pavec J, Prévot G, Mal H, Humbert M, Simonneau G and Cordier J-F. Pulmonary hypertension in patients with combined pulmonary fibrosis and emphysema syndrome. European respiratory journal. 2010;35:105–111. [DOI] [PubMed] [Google Scholar]
- 116.Cottin V, Nunes H, Brillet P, Delaval P, Devouassoux G, Tillie-Leblond I, Israel-Biet D, Valeyre D and Cordier J-F. Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. European Respiratory Journal. 2005;26:586–593. [DOI] [PubMed] [Google Scholar]
- 117.Mejía M, Carrillo G, Rojas-Serrano J, Estrada A, Suárez T, Alonso D, Barrientos E, Gaxiola M, Navarro C and Selman M. Idiopathic pulmonary fibrosis and emphysema: decreased survival associated with severe pulmonary arterial hypertension. Chest. 2009;136:10–15. [DOI] [PubMed] [Google Scholar]
- 118.Kitaguchi Y, Fujimoto K, Hanaoka M, Honda T, Hotta J and Hirayama J. Pulmonary function impairment in patients with combined pulmonary fibrosis and emphysema with and without airflow obstruction. International journal of chronic obstructive pulmonary disease. 2014;9:805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kiakouama L, Cottin V, Glerant J-C, Bayle J-Y, Mornex J-F and Cordier J-F. Conditions associated with severe carbon monoxide diffusion coefficient reduction. Respiratory medicine. 2011;105:1248–1256. [DOI] [PubMed] [Google Scholar]
- 120.Cottin V and Cordier J-F. The syndrome of combined pulmonary fibrosis and emphysema. Chest. 2009;136:1–2. [DOI] [PubMed] [Google Scholar]
- 121.Mathai SC, Hummers LK, Champion HC, Wigley FM, Zaiman A, Hassoun PM and Girgis RE. Survival in pulmonary hypertension associated with the scleroderma spectrum of diseases: impact of interstitial lung disease. Arthritis & Rheumatism: Official Journal of the American College of Rheumatology. 2009;60:569–577. [DOI] [PubMed] [Google Scholar]
- 122.Mirrakhimov AE and Strohl KP. High-altitude pulmonary hypertension: an update on disease pathogenesis and management. The open cardiovascular medicine journal. 2016;10:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.León-Velarde F, Maggiorini M, Reeves JT, Aldashev A, Asmus I, Bernardi L, Ge R-L, Hackett P, Kobayashi T and Moore LG. Consensus statement on chronic and subacute high altitude diseases. High altitude medicine & biology. 2005;6:147–157. [DOI] [PubMed] [Google Scholar]
- 124.Wilkins MR, Ghofrani H-A, Weissmann N, Aldashev A and Zhao L. Pathophysiology and treatment of high-altitude pulmonary vascular disease. Circulation. 2015;131:582–590. [DOI] [PubMed] [Google Scholar]
- 125.Held M, Walthelm J, Baron S, Roth C and Jany B. Functional impact of pulmonary hypertension due to hypoventilation and changes under noninvasive ventilation. European Respiratory Journal. 2014;43:156–165. [DOI] [PubMed] [Google Scholar]
- 126.Masa JF, Corral J, Caballero C, Barrot E, Terán-Santos J, Alonso-Álvarez ML, Gomez-Garcia T, González M, López-Martín S and De Lucas P. Non-invasive ventilation in obesity hypoventilation syndrome without severe obstructive sleep apnoea. Thorax. 2016;71:899–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nowbar S, Burkart KM, Gonzales R, Fedorowicz A, Gozansky WS, Gaudio JC, Taylor MR and Zwillich CW. Obesity-associated hypoventilation in hospitalized patients: prevalence, effects, and outcome. The American journal of medicine. 2004;116:1–7. [DOI] [PubMed] [Google Scholar]
- 128.CASTRO-AÑÓN O, Golpe R, PÉREZ-DE-LLANO LA, Lopez Gonzalez MJ, Escalona Velasquez EJ, Perez Fernandez R, Testa Fernandez A and Gonzalez Quintela A. Haemodynamic effects of non-invasive ventilation in patients with obesity-hypoventilation syndrome. Respirology. 2012;17:1269–1274. [DOI] [PubMed] [Google Scholar]
- 129.Hansmann G, Sallmon H, Roehr CC, Kourembanas S, Austin ED and Koestenberger M. Pulmonary hypertension in bronchopulmonary dysplasia. Pediatric Research. 2021;89:446–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rosenzweig EB, Abman SH, Adatia I, Beghetti M, Bonnet D, Haworth S, Ivy DD and Berger RM. Paediatric pulmonary arterial hypertension: updates on definition, classification, diagnostics and management. European Respiratory Journal. 2019;53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Thébaud B, Goss KN, Laughon M, Whitsett JA, Abman SH, Steinhorn RH, Aschner JL, Davis PG, McGrath-Morrow SA and Soll RF. Bronchopulmonary dysplasia. Nature reviews Disease primers. 2019;5:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lignelli E, Palumbo F, Myti D and Morty RE. Recent advances in our understanding of the mechanisms of lung alveolarization and bronchopulmonary dysplasia. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2019;317:L832–L887. [DOI] [PubMed] [Google Scholar]
- 133.Goldenberg RL, Culhane JF, Iams JD and Romero R. Epidemiology and causes of preterm birth. The lancet. 2008;371:75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ambalavanan N, Van Meurs KP, Perritt R, Carlo WA, Ehrenkranz RA, Stevenson DK, Lemons JA, Poole WK and Higgins RD. Predictors of death or bronchopulmonary dysplasia in preterm infants with respiratory failure. Journal of perinatology. 2008;28:420–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lemons JA, Bauer CR, Oh W, Korones SB, Papile L-A, Stoll BJ, Verter J, Temprosa M, Wright LL and Ehrenkranz RA. Very low birth weight outcomes of the National Institute of Child health and human development neonatal research network, January 1995 through December 1996. Pediatrics. 2001;107:e1–e1. [DOI] [PubMed] [Google Scholar]
- 136.Marshall DD, Kotelchuck M, Young TE, Bose CL, Kruyer L, O'Shea TM and Association NCN. Risk factors for chronic lung disease in the surfactant era: a North Carolina population-based study of very low birth weight infants. Pediatrics. 1999;104:1345–1350. [DOI] [PubMed] [Google Scholar]
- 137.Oh W, Poindexter BB, Perritt R, Lemons JA, Bauer CR, Ehrenkranz RA, Stoll BJ, Poole K and Wright LL. Association between fluid intake and weight loss during the first ten days of life and risk of bronchopulmonary dysplasia in extremely low birth weight infants. The Journal of pediatrics. 2005;147:786–790. [DOI] [PubMed] [Google Scholar]
- 138.Rojas MA, Gonzalez A, Bancalari E, Claure N, Poole C and Silva-Neto G. Changing trends in the epidemiology and pathogenesis of neonatal chronic lung disease. The Journal of pediatrics. 1995;126:605–610. [DOI] [PubMed] [Google Scholar]
- 139.Stoll BJ, Hansen NI, Bell EF, Walsh MC, Carlo WA, Shankaran S, Laptook AR, Sánchez PJ, Van Meurs KP and Wyckoff M. Trends in care practices, morbidity, and mortality of extremely preterm neonates, 1993–2012. Jama. 2015;314:1039–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Young TE, Kruyer LS, Marshall DD, Bose CL and Association NCN. Population-based study of chronic lung disease in very low birth weight infants in North Carolina in 1994 with comparisons with 1984. Pediatrics. 1999;104:e17–e17. [DOI] [PubMed] [Google Scholar]
- 141.Stoll BJ, Hansen NI, Bell EF, Shankaran S, Laptook AR, Walsh MC, Hale EC, Newman NS, Schibler K and Carlo WA. Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics. 2010;126:443–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wadhawan R, Vohr BR, Fanaroff AA, Perritt RL, Duara S, Stoll BJ, Goldberg R, Laptook A, Poole K and Wright LL. Does labor influence neonatal and neurodevelopmental outcomes of extremely-low-birth-weight infants who are born by cesarean delivery? American journal of obstetrics and gynecology. 2003;189:501–506. [DOI] [PubMed] [Google Scholar]
- 143.Mirza H, Garcia JA, Crawford E, Pepe J, Zussman M, Wadhawan R and Oh W. Natural history of postnatal cardiopulmonary adaptation in infants born extremely preterm and risk for death or bronchopulmonary dysplasia. The Journal of pediatrics. 2018;198:187–193. e1. [DOI] [PubMed] [Google Scholar]
- 144.Aikio O, Metsola J, Vuolteenaho R, Perhomaa M and Hallman M. Transient defect in nitric oxide generation after rupture of fetal membranes and responsiveness to inhaled nitric oxide in very preterm infants with hypoxic respiratory failure. The Journal of pediatrics. 2012;161:397–403. e1. [DOI] [PubMed] [Google Scholar]
- 145.Chandrasekharan P, Kozielski R, Kumar VH, Rawat M, Manja V, Ma C and Lakshminrusimha S. Early use of inhaled nitric oxide in preterm infants: is there a rationale for selective approach? American journal of perinatology. 2017;34:428–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Giesinger RE, More K, Odame J, Jain A, Jankov RP and McNamara PJ. Controversies in the identification and management of acute pulmonary hypertension in preterm neonates. Pediatric research. 2017;82:901–914. [DOI] [PubMed] [Google Scholar]
- 147.Weitzenblum E and Chaouat A. Cor pulmonale. Chronic respiratory disease. 2009;6:177–185. [DOI] [PubMed] [Google Scholar]
- 148.Weitzenblum E Chronic cor pulmonale. Heart. 2003;89:225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Thenappan T, Shah S, Rich S, Tian L, Archer S and Gomberg-Maitland M. Survival in pulmonary arterial hypertension: a reappraisal of the NIH risk stratification equation. European Respiratory Journal. 2010;35:1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Humbert M, Sitbon O, Yaïci A, Montani D, O'callaghan D, Jaïs X, Parent F, Savale L, Natali D and Günther S. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. European Respiratory Journal. 2010;36:549–555. [DOI] [PubMed] [Google Scholar]
- 151.Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, Frost A, Barst RJ, Badesch DB and Elliott CG. Predicting survival in pulmonary arterial hypertension: insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management (REVEAL). Circulation. 2010;122:164–172. [DOI] [PubMed] [Google Scholar]
- 152.Prins KW, Rose L, Archer SL, Pritzker M, Weir EK, Olson MD and Thenappan T. Clinical Determinants and Prognostic Implications of Right Ventricular Dysfunction in Pulmonary Hypertension Caused by Chronic Lung Disease. J Am Heart Assoc. 2019;8:e011464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Party MW. Long-term domiciliary oxygen therapy in chronic hypoxic cor pulmonale complicating chronic bronchitis. Lancet. 1981;1:681–685. [PubMed] [Google Scholar]
- 154.Kolb TM and Hassoun PM. Right ventricular dysfunction in chronic lung disease. Cardiology clinics. 2012;30:243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.MacNee W Right ventricular function in cor pulmonale. Cardiology. 1988;75:30–40. [DOI] [PubMed] [Google Scholar]
- 156.Shivkumar K, Ravi K, Henry JW, Eichenhorn M and Stein PD. Right ventricular dilatation, right ventricular wall thickening, and Doppler evidence of pulmonary hypertension in patients with a pure restrictive ventilatory impairment. Chest. 1994;106:1649–1653. [DOI] [PubMed] [Google Scholar]
- 157.Vonk-Noordegraaf A, Marcus JT, Holverda S, Roseboom B and Postmus PE. Early changes of cardiac structure and function in COPD patients with mild hypoxemia. Chest. 2005;127:1898–1903. [DOI] [PubMed] [Google Scholar]
- 158.Caso P, Galderisi M, Cicala S, Cioppa C, D'andrea A, Lagioia G, Liccardo B, Martiniello AR and Mininni N. Association between myocardial right ventricular relaxation time and pulmonary arterial pressure in chronic obstructive lung disease: analysis by pulsed Doppler tissue imaging. Journal of the American Society of Echocardiography. 2001;14:970–977. [DOI] [PubMed] [Google Scholar]
- 159.Huez S, Retailleau K, Unger P, Pavelescu A, Vachiéry J-L, Derumeaux G and Naeije R. Right and left ventricular adaptation to hypoxia: a tissue Doppler imaging study. American Journal of Physiology-Heart and Circulatory Physiology. 2005;289:H1391–H1398. [DOI] [PubMed] [Google Scholar]
- 160.Bogaard HJ, Abe K, Noordegraaf AV and Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest. 2009;135:794–804. [DOI] [PubMed] [Google Scholar]
- 161.Kayar SR and Banchero N. Myocardial capillarity in acclimation to hypoxia. Pflügers Archiv. 1985;404:319–325. [DOI] [PubMed] [Google Scholar]
- 162.Partovian C, Adnot S, Eddahibi S, Teiger E, Levame M, Dreyfus P, Raffestin B and Frelin C. Heart and lung VEGF mRNA expression in rats with monocrotaline-or hypoxia-induced pulmonary hypertension. American Journal of Physiology-Heart and Circulatory Physiology. 1998;275:H1948–H1956. [DOI] [PubMed] [Google Scholar]
- 163.Zhang P, Vang A, da Silva Goncalves Bos D, Feord J, Mancini T, Park A and Choudhary G. Impaired Mitochondrial Bioenergetics is Associated With Progression of Right Ventricular Dysfunction in Pulmonary Arterial Hypertension. Circulation. 2021;144:A12388–A12388. [Google Scholar]
- 164.Fessler H Heart-lung interactions: applications in the critically ill. European Respiratory Journal. 1997;10:226–237. [DOI] [PubMed] [Google Scholar]
- 165.Visca D, Aiello M and Chetta A. Cardiovascular function in pulmonary emphysema. BioMed research international. 2013;2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Buda AJ, Pinsky MR, Ingels NB Jr, Daughters GT, Stinson EB and Alderman EL. Effect of intrathoracic pressure on left ventricular performance. New England Journal of Medicine. 1979;301:453–459. [DOI] [PubMed] [Google Scholar]
- 167.Sato T, Tsujino I, Oyama-Manabe N, Ohira H, Ito YM, Yamada A, Ikeda D, Watanabe T and Nishimura M. Right atrial volume and phasic function in pulmonary hypertension. International journal of cardiology. 2013;168:420–426. [DOI] [PubMed] [Google Scholar]
- 168.Le Jemtel TH, Padeletti M and Jelic S. Diagnostic and therapeutic challenges in patients with coexistent chronic obstructive pulmonary disease and chronic heart failure. Journal of the American College of Cardiology. 2007;49:171–180. [DOI] [PubMed] [Google Scholar]
- 169.Kizer JR, Zisman DA, Blumenthal NP, Kotloff RM, Kimmel SE, Strieter RM, Arcasoy SM, Ferrari VA and Hansen-Flaschen J. Association between pulmonary fibrosis and coronary artery disease. Archives of internal medicine. 2004;164:551–556. [DOI] [PubMed] [Google Scholar]
- 170.Arcasoy SM, Christie JD, Ferrari VA, Sutton MSJ, Zisman DA, Blumenthal NP, Pochettino A and Kotloff RM. Echocardiographic assessment of pulmonary hypertension in patients with advanced lung disease. American journal of respiratory and critical care medicine. 2003;167:735–740. [DOI] [PubMed] [Google Scholar]
- 171.Corte T, Wort S, Gatzoulis M, Engel R, Giannakoulas G, Macdonald P and Wells A. Elevated brain natriuretic peptide predicts mortality in interstitial lung disease. European Respiratory Journal. 2010;36:819–825. [DOI] [PubMed] [Google Scholar]
- 172.Leuchte HH, Baumgartner RA, Nounou ME, Vogeser M, Neurohr C, Trautnitz M and Behr J. Brain natriuretic peptide is a prognostic parameter in chronic lung disease. American journal of respiratory and critical care medicine. 2006;173:744–750. [DOI] [PubMed] [Google Scholar]
- 173.Rose L, Prins KW, Archer SL, Pritzker M, Weir EK, Misialek JR and Thenappan T. Survival in pulmonary hypertension due to chronic lung disease: influence of low diffusion capacity of the lungs for carbon monoxide. The Journal of Heart and Lung Transplantation. 2019;38:145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Furukawa T, Kondoh Y, Taniguchi H, Yagi M, Matsuda T, Kimura T, Kataoka K, Johkoh T, Ando M and Hashimoto N. A scoring system to predict the elevation of mean pulmonary arterial pressure in idiopathic pulmonary fibrosis. European Respiratory Journal. 2018;51. [DOI] [PubMed] [Google Scholar]
- 175.Gläser S, Obst A, Koch B, Henkel B, Grieger A, Felix SB, Halank M, Bruch L, Bollmann T and Warnke C. Pulmonary hypertension in patients with idiopathic pulmonary fibrosis–the predictive value of exercise capacity and gas exchange efficiency. PloS one. 2013;8:e65643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lettieri CJ, Nathan SD, Browning RF, Barnett SD, Ahmad S and Shorr AF. The distance-saturation product predicts mortality in idiopathic pulmonary fibrosis. Respiratory medicine. 2006;100:1734–1741. [DOI] [PubMed] [Google Scholar]
- 177.Swigris JJ, Olson AL, Shlobin OA, Ahmad S, Brown KK and Nathan SD. Heart rate recovery after six-minute walk test predicts pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Respirology. 2011;16:439–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wells JM, Washko GR, Han MK, Abbas N, Nath H, Mamary AJ, Regan E, Bailey WC, Martinez FJ and Westfall E. Pulmonary arterial enlargement and acute exacerbations of COPD. New England journal of medicine. 2012;367:913–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Alkukhun L, Wang X-F, Ahmed MK, Baumgartner M, Budev MM, Dweik RA and Tonelli AR. Non-invasive screening for pulmonary hypertension in idiopathic pulmonary fibrosis. Respiratory medicine. 2016;117:65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Yagi M, Taniguchi H, Kondoh Y, Ando M, Kimura T, Kataoka K, Furukawa T, Suzuki A, Johkoh T and Hasegawa Y. CT-determined pulmonary artery to aorta ratio as a predictor of elevated pulmonary artery pressure and survival in idiopathic pulmonary fibrosis. Respirology. 2017;22:1393–1399. [DOI] [PubMed] [Google Scholar]
- 181.Bax S, Jacob J, Ahmed R, Bredy C, Dimopoulos K, Kempny A, Kokosi M, Kier G, Renzoni E and Molyneaux PL. Right ventricular to left ventricular ratio at CT pulmonary angiogram predicts mortality in interstitial lung disease. Chest. 2020;157:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Nathan SD and Cottin V. Pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Eur Respir Monogr. 2012;57:148–60. [Google Scholar]
- 183.Hoeper MM, Andreas S, Bastian A, Claussen M, Ghofrani HA, Gorenflo M, Grohé C, Günther A, Halank M and Hammerl P. Pulmonary hypertension due to chronic lung disease: updated Recommendations of the Cologne Consensus Conference 2011. International journal of cardiology. 2011;154:S45–S53. [DOI] [PubMed] [Google Scholar]
- 184.Roger N, Barbera JA, Roca J, Rovira I, Gomez FP and Rodriguez-Roisin R. Nitric oxide inhalation during exercise in chronic obstructive pulmonary disease. American journal of respiratory and critical care medicine. 1997;156:800–806. [DOI] [PubMed] [Google Scholar]
- 185.Boutou AK, Pitsiou GG, Trigonis I, Papakosta D, Kontou PK, Chavouzis N, Nakou C, Argyropoulou P, Wasserman K and Stanopoulos I. Exercise capacity in idiopathic pulmonary fibrosis: the effect of pulmonary hypertension. Respirology. 2011;16:451–458. [DOI] [PubMed] [Google Scholar]
- 186.Leard LE, Holm AM, Valapour M, Glanville AR, Attawar S, Aversa M, Campos SV, Christon LM, Cypel M and Dellgren G. Consensus document for the selection of lung transplant candidates: An update from the International Society for Heart and Lung Transplantation. The Journal of Heart and Lung Transplantation. 2021;40:1349–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.TIMMS RM, KHAJA FU and WILLIAMS GW. Hemodynamic response to oxygen therapy in chronic obstructive pulmonary disease. Annals of internal medicine. 1985;102:29–36. [DOI] [PubMed] [Google Scholar]
- 188.Weitzenblum E, Sautegeau A, Ehrhart M, Mammosser M and Pelletier A. Long-term oxygen therapy can reverse the progression of pulmonary hypertension in patients with chronic obstructive pulmonary disease. American Review of Respiratory Disease. 1985;131:493–498. [DOI] [PubMed] [Google Scholar]
- 189.Ozben B, Eryuksel E, Tanrikulu AM, Papila N, Ozyigit T, Celikel T and Basaran Y. Acute exacerbation impairs right ventricular function in COPD patients. Hellenic J Cardiol. 2015;56:324–31. [PubMed] [Google Scholar]
- 190.Olschewski H, Ardeschir Ghofrani H, Walmrath D, Schermuly R, Temmesfeld-Wollbruck B, GRIMMINGER F and Seeger W. Inhaled prostacyclin and iloprost in severe pulmonary hypertension secondary to lung fibrosis. American journal of respiratory and critical care medicine. 1999;160:600–607. [DOI] [PubMed] [Google Scholar]
- 191.Blanco I, Ribas J, Xaubet A, Gómez FP, Roca J, Rodriguez-Roisin R and Barberà JA. Effects of inhaled nitric oxide at rest and during exercise in idiopathic pulmonary fibrosis. Journal of applied physiology. 2011;110:638–645. [DOI] [PubMed] [Google Scholar]
- 192.Voswinckel R, Reichenberger F, Enke B, Kreckel A, Krick S, Gall H, Schermuly RT, Grimminger F, Rubin LJ and Olschewski H. Acute effects of the combination of sildenafil and inhaled treprostinil on haemodynamics and gas exchange in pulmonary hypertension. Pulmonary pharmacology & therapeutics. 2008;21:824–832. [DOI] [PubMed] [Google Scholar]
- 193.Chen X, Tang S, Liu K, Li Q, Kong H, Zeng X, Xie W and Wang H. Therapy in stable chronic obstructive pulmonary disease patients with pulmonary hypertension: a systematic review and meta-analysis. Journal of thoracic disease. 2015;7:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Corte TJ, Keir GJ, Dimopoulos K, Howard L, Corris PA, Parfitt L, Foley C, Yanez-Lopez M, Babalis D and Marino P. Bosentan in pulmonary hypertension associated with fibrotic idiopathic interstitial pneumonia. American journal of respiratory and critical care medicine. 2014;190:208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Raghu G, Behr J, Brown KK, Egan JJ, Kawut SM, Flaherty KR, Martinez FJ, Nathan SD, Wells AU and Collard HR. Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Annals of internal medicine. 2013;158:641–649. [DOI] [PubMed] [Google Scholar]
- 196.Goudie AR, Lipworth BJ, Hopkinson PJ, Wei L and Struthers AD. Tadalafil in patients with chronic obstructive pulmonary disease: a randomised, double-blind, parallel-group, placebo-controlled trial. The lancet Respiratory medicine. 2014;2:293–300. [DOI] [PubMed] [Google Scholar]
- 197.Vitulo P, Stanziola A, Confalonieri M, Libertucci D, Oggionni T, Rottoli P, Paciocco G, Tuzzolino F, Martino L and Beretta M. Sildenafil in severe pulmonary hypertension associated with chronic obstructive pulmonary disease: a randomized controlled multicenter clinical trial. The Journal of Heart and Lung Transplantation. 2017;36:166–174. [DOI] [PubMed] [Google Scholar]
- 198.Network IPFCR. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. New England Journal of Medicine. 2010;363:620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Han MK, Bach DS, Hagan PG, Yow E, Flaherty KR, Toews GB, Anstrom KJ, Martinez FJ and Investigators I. Sildenafil preserves exercise capacity in patients with idiopathic pulmonary fibrosis and right-sided ventricular dysfunction. Chest. 2013;143:1699–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Behr J, Nathan SD, Wuyts WA, Bishop NM, Bouros DE, Antoniou K, Guiot J, Kramer MR, Kirchgaessler K-U and Bengus M. Efficacy and safety of sildenafil added to pirfenidone in patients with advanced idiopathic pulmonary fibrosis and risk of pulmonary hypertension: a double-blind, randomised, placebo-controlled, phase 2b trial. The Lancet Respiratory Medicine. 2021;9:85–95. [DOI] [PubMed] [Google Scholar]
- 201.Nathan SD, Behr J, Collard HR, Cottin V, Hoeper MM, Martinez FJ, Corte TJ, Keogh AM, Leuchte H and Mogulkoc N. Riociguat for idiopathic interstitial pneumonia-associated pulmonary hypertension (RISE-IIP): a randomised, placebo-controlled phase 2b study. The Lancet Respiratory Medicine. 2019;7:780–790. [DOI] [PubMed] [Google Scholar]
- 202.Saggar R, Khanna D, Vaidya A, Derhovanessian A, Maranian P, Duffy E, Belperio JA, Weigt SS, Dua S and Shapiro SS. Changes in right heart haemodynamics and echocardiographic function in an advanced phenotype of pulmonary hypertension and right heart dysfunction associated with pulmonary fibrosis. Thorax. 2014;69:123–129. [DOI] [PubMed] [Google Scholar]
- 203.Nathan SD, Waxman A, Rajagopal S, Case A, Johri S, DuBrock H, De La Zerda DJ, Sahay S, King C and Melendres-Groves L. Inhaled treprostinil and forced vital capacity in patients with interstitial lung disease and associated pulmonary hypertension: a post-hoc analysis of the INCREASE study. The Lancet Respiratory Medicine. 2021;9:1266–1274. [DOI] [PubMed] [Google Scholar]
- 204.Nikitopoulou I, Manitsopoulos N, Kotanidou A, Tian X, Petrovic A, Magkou C, Ninou I, Aidinis V, Schermuly RT and Kosanovic D. Orotracheal treprostinil administration attenuates bleomycin-induced lung injury, vascular remodeling, and fibrosis in mice. Pulmonary circulation. 2019;9:2045894019881954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lambers C, Roth M, Jaksch P, Muraközy G, Tamm M, Klepetko W, Ghanim B and Zhao F. Treprostinil inhibits proliferation and extracellular matrix deposition by fibroblasts through cAMP activation. Scientific reports. 2018;8:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Nathan SD, Tapson VF, Elwing J, Rischard F, Mehta J, Shapiro S, Shen E, Deng C, Smith P and Waxman A. Efficacy of Inhaled Treprostinil on Multiple Disease Progression Events in Patients with Pulmonary Hypertension Due to Parenchymal Lung Disease in the INCREASE Trial. American journal of respiratory and critical care medicine. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hoeper MM, Behr J, Held M, Grunig E, Vizza CD, Vonk-Noordegraaf A, Lange TJ, Claussen M, Grohe C and Klose H. Pulmonary hypertension in patients with chronic fibrosing idiopathic interstitial pneumonias. PloS one. 2015;10:e0141911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Vizza CD, Hoeper MM, Huscher D, Pittrow D, Benjamin N, Olsson KM, Ghofrani HA, Held M, Klose H and Lange T. Pulmonary hypertension in patients with chronic obstructive lung disease: results from COMPERA. Chest. 2021. [DOI] [PubMed] [Google Scholar]
- 209.Brewis MJ, Church AC, Johnson MK and Peacock AJ. Severe pulmonary hypertension in lung disease: phenotypes and response to treatment. European Respiratory Journal. 2015;46:1378–1389. [DOI] [PubMed] [Google Scholar]
- 210.Mathai SC, Puhan MA, Lam D and Wise RA. The minimal important difference in the 6-minute walk test for patients with pulmonary arterial hypertension. American journal of respiratory and critical care medicine. 2012;186:428–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Corboz MR, Zhang J, LaSala D, DiPetrillo K, Li Z, Malinin V, Brower J, Kuehl PJ, Barrett TE and Perkins WR. Therapeutic administration of inhaled INS1009, a treprostinil prodrug formulation, inhibits bleomycin-induced pulmonary fibrosis in rats. Pulmonary pharmacology & therapeutics. 2018;49:95–103. [DOI] [PubMed] [Google Scholar]
- 212.Leifer FG, Konicek DM, Chen K-J, Plaunt AJ, Salvail D, Laurent CE, Corboz MR, Li Z, Chapman RW and Perkins WR. Inhaled treprostinil-prodrug lipid nanoparticle formulations provide long-acting pulmonary vasodilation. Drug research. 2018;68:605–614. [DOI] [PubMed] [Google Scholar]
- 213.Corboz MR, Li Z, Malinin V, Plaunt AJ, Konicek DM, Leifer FG, Chen K-J, Laurent CE, Yin H and Biernat MC. Preclinical pharmacology and pharmacokinetics of inhaled hexadecyl-treprostinil (C16TR), a pulmonary vasodilator prodrug. Journal of Pharmacology and Experimental Therapeutics. 2017;363:348–357. [DOI] [PubMed] [Google Scholar]
- 214.Chapman RW, Li Z, Corboz MR, Gauani H, Plaunt AJ, Konicek DM, Leifer FG, Laurent CE, Yin H and Salvail D. Inhaled hexadecyl-treprostinil provides pulmonary vasodilator activity at significantly lower plasma concentrations than infused treprostinil. Pulmonary pharmacology & therapeutics. 2018;49:104–111. [DOI] [PubMed] [Google Scholar]
- 215.Hajian B, De Backer J, Vos W, Van Holsbeke C, Ferreira F, Quinn DA, Hufkens A, Claes R and De Backer W. Pulmonary vascular effects of pulsed inhaled nitric oxide in COPD patients with pulmonary hypertension. International journal of chronic obstructive pulmonary disease. 2016;11:1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Nathan SD, Flaherty KR, Glassberg MK, Raghu G, Swigris J, Alvarez R, Ettinger N, Loyd J, Fernandes P and Gillies H. A randomized, double-blind, placebo-controlled study of pulsed, inhaled nitric oxide in subjects at risk of pulmonary hypertension associated with pulmonary fibrosis. Chest. 2020;158:637–645. [DOI] [PubMed] [Google Scholar]
- 217.Rich S, Seidlitz M, Dodin E, Osimani D, Judd D, Genthner D, McLaughlin V and Francis G. The short-term effects of digoxin in patients with right ventricular dysfunction from pulmonary hypertension. Chest. 1998;114:787–792. [DOI] [PubMed] [Google Scholar]
- 218.Gerges C, Vollmers K, Pritzker MR, Gainor J, Scandurra J, Weir EK and Lang IM. Pulmonary Artery Endovascular Device Compensates for Loss of Vascular Compliance in Pulmonary Arterial Hypertension. Journal of the American College of Cardiology. 2020;76:2284–2286. [DOI] [PubMed] [Google Scholar]
- 219.Marsboom GR and Janssens SP. Models for pulmonary hypertension. Drug Discovery Today: Disease Models. 2004;1:289–296. [Google Scholar]
- 220.Janssens SP, Thompson BT, Spence CR and Hales CA. Polycythemia and vascular remodeling in chronic hypoxic pulmonary hypertension in guinea pigs. Journal of Applied Physiology. 1991;71:2218–2223. [DOI] [PubMed] [Google Scholar]
- 221.Steudel W, Scherrer-Crosbie M, Bloch KD, Weimann J, Huang PL, Jones RC, Picard MH and Zapol WM. Sustained pulmonary hypertension and right ventricular hypertrophy after chronic hypoxia in mice with congenital deficiency of nitric oxide synthase 3. The Journal of clinical investigation. 1998;101:2468–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Meyrick B and Reid L. Hypoxia-induced structural changes in the media and adventitia of the rat hilar pulmonary artery and their regression. The American journal of pathology. 1980;100:151. [PMC free article] [PubMed] [Google Scholar]
- 223.Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, Voelkel NF, McMurtry IF and Oka M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation. 2010;121:2747–2754. [DOI] [PubMed] [Google Scholar]
- 224.TARASEVICIENE-STEWART L, Kasahara Y, Alger L, Hirth P, MAHON GM, Waltenberger J, Voelkel NF and Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. The FASEB Journal. 2001;15:427–438. [DOI] [PubMed] [Google Scholar]
- 225.Ciuclan L, Bonneau O, Hussey M, Duggan N, Holmes AM, Good R, Stringer R, Jones P, Morrell NW and Jarai G. A novel murine model of severe pulmonary arterial hypertension. American journal of respiratory and critical care medicine. 2011;184:1171–1182. [DOI] [PubMed] [Google Scholar]
- 226.Maarman G, Lecour S, Butrous G, Thienemann F and Sliwa K. A comprehensive review: the evolution of animal models in pulmonary hypertension research; are we there yet? Pulmonary circulation. 2013;3:739–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.le Cras TD, Kim D-H, Gebb S, Markham NE, Shannon JM, Tuder RM and Abman SH. Abnormal lung growth and the development of pulmonary hypertension in the Fawn-Hooded rat. American Journal of Physiology-Lung Cellular and Molecular Physiology. 1999;277:L709–L718. [DOI] [PubMed] [Google Scholar]
- 228.Martinez-Lemus LA, Hester RK, Becker EJ, Jeffrey JS and Odom TW. Pulmonary artery endothelium-dependent vasodilation is impaired in a chicken model of pulmonary hypertension. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 1999;277:R190–R197. [DOI] [PubMed] [Google Scholar]
- 229.Xiang R, Sun W, Zhang K, Li J, Wang J and Wang X. Sodium chloride-induced acute and chronic pulmonary hypertension syndrome in broiler chickens. Poultry science. 2004;83:732–736. [DOI] [PubMed] [Google Scholar]
- 230.Cawthon D, Iqbal M, Brand J, McNew R and Bottje W. Investigation of proton conductance in liver mitochondria of broilers with pulmonary hypertension syndrome. Poultry science. 2004;83:259–265. [DOI] [PubMed] [Google Scholar]
- 231.Iqbal M, Cawthon D, Wideman R Jr and Bottje W. Lung mitochondrial dysfunction in pulmonary hypertension syndrome. I. Site-specific defects in the electron transport chain. Poultry Science. 2001;80:485–495. [DOI] [PubMed] [Google Scholar]
- 232.Wideman R Pathophysiology of heart/lung disorders: pulmonary hypertension syndrome in broiler chickens. World's Poultry Science Journal. 2001;57:289–307. [Google Scholar]
- 233.Janwari AQ, Mir MS, Mariam A, Altaf R, Amin U, Shafi M, Shah SA, Khan HM and Kamil SA. Pathomorphological alterations due to pulmonary hypertension syndrome in broiler chicken reared under temperate climatic conditions of Northern Himalayas. J Entomol Zool Stud. 2018;6:1347–1353. [Google Scholar]
- 234.Harrison J and Lazo J. High dose continuous infusion of bleomycin in mice: a new model for drug-induced pulmonary fibrosis. Journal of Pharmacology and Experimental Therapeutics. 1987;243:1185–1194. [PubMed] [Google Scholar]
- 235.Lynch DA, Hirose N, Cherniack RM and Doherty DE. Bleomycin-induced lung disease in an animal model: correlation between computed tomography-determined abnormalities and lung function. Academic Radiology. 1997;4:102–107. [DOI] [PubMed] [Google Scholar]
- 236.Bowden D Unraveling pulmonary fibrosis: the bleomycin model. Laboratory investigation; a journal of technical methods and pathology. 1984;50:487–488. [PubMed] [Google Scholar]
- 237.Albertine KH. Utility of large-animal models of BPD: chronically ventilated preterm lambs. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2015;308:L983–L1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Berger J and Bhandari V. Animal models of bronchopulmonary dysplasia. The term mouse models. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2014;307:L936–L947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.D'Angio CT and Ryan RM. Animal models of bronchopulmonary dysplasia. The preterm and term rabbit models. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2014;307:L959–L969. [DOI] [PubMed] [Google Scholar]
- 240.O'Reilly M and Thébaud B. Animal models of bronchopulmonary dysplasia. The term rat models. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2014;307:L948–L958. [DOI] [PubMed] [Google Scholar]
- 241.Silva DM, Nardiello C, Pozarska A and Morty RE. Recent advances in the mechanisms of lung alveolarization and the pathogenesis of bronchopulmonary dysplasia. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2015;309:L1239–L1272. [DOI] [PubMed] [Google Scholar]
- 242.Yoder BA and Coalson JJ. Animal models of bronchopulmonary dysplasia. The preterm baboon models. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2014;307:L970–L977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Ambalavanan N and Morty RE. Searching for better animal models of BPD: a perspective. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2016;311:L924–L927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Nogueira-Ferreira R, Faria-Costa G, Ferreira R and Henriques-Coelho T. Animal models for the study of pulmonary hypertension: potential and limitations. Cardiol Cardiovasc Med. 2016;1:01–22. [Google Scholar]
- 245.King CS and Shlobin OA. The trouble with group 3 pulmonary hypertension in interstitial lung disease: dilemmas in diagnosis and the conundrum of treatment. Chest. 2020;158:1651–1664. [DOI] [PubMed] [Google Scholar]
- 246.Stolz D, Rasch H, Linka A, Di Valentino M, Meyer A, Brutsche M and Tamm M. A randomised, controlled trial of bosentan in severe COPD. European Respiratory Journal. 2008;32:619–628. [DOI] [PubMed] [Google Scholar]
- 247.Valerio G, Bracciale P and Grazia D'Agostino A. Effect of bosentan upon pulmonary hypertension in chronic obstructive pulmonary disease. Therapeutic advances in respiratory disease. 2009;3:15–21. [DOI] [PubMed] [Google Scholar]
- 248.Collard HR, Anstrom KJ, Schwarz MI and Zisman DA. Sildenafil improves walk distance in idiopathic pulmonary fibrosis. Chest. 2007;131:897–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Rao RS, Singh S, Sharma BB, Agarwal V and Singh V. Sildenafil improves six-minute walk distance in chronic obstructive pulmonary disease: a randomised, double-blind, placebo-controlled trial. Indian Journal of Chest Diseases and Allied Sciences. 2011;53:81. [PubMed] [Google Scholar]