

Abstract
The development of pulmonary hypertension (PH) is common and has adverse prognostic implications in patients with heart failure due to left heart disease (LHD), and thus far there are no known treatments specifically for PH-LHD, also known as Group 2 PH. Diagnostic thresholds for PH-LHD, and clinical classification of PH-LHD phenotypes, continue to evolve and therefore present a challenge for basic and translational scientists actively investigating PH-LHD in the preclinical setting. Furthermore, the pathobiology of PH-LHD is not well understood, although pulmonary vascular remodeling is thought to result from (1) increased wall stress due to increased left atrial pressures; (2) hemodynamic congestion-induced decreased shear stress in the pulmonary vascular bed; (3) comorbidity-induced endothelial dysfunction with direct injury to the pulmonary microvasculature; and (4) superimposed pulmonary arterial hypertension risk factors. To ultimately be able to modify disease, either by prevention or treatment, a better understanding of the various drivers of PH-LHD, including endothelial dysfunction, abnormalities in vascular tone, platelet aggregation, inflammation, adipocytokines, and systemic complications (including splanchnic congestion and lymphatic dysfunction) must be further investigated. Here we review the diagnostic criteria and various hemodynamic phenotypes of PH-LHD, the potential biological mechanisms underlying this disorder, and pressing questions yet to be answered about the pathobiology of PH-LHD.
Keywords: pulmonary hypertension, heart failure, pulmonary vascular disease, endothelial dysfunction, diagnosis, hemodynamics, Vascular Disease
INTRODUCTION
Left heart disease (LHD) affects nearly 10% of the population over the age of 20 years in the United States with coronary disease as the leading cause of death.1 Heart failure (HF), the end result of LHD, has a prevalence of 5.4% in the US with a growing burden of disease with the aging of the population.2, 3 HF as a clinical syndrome is divided into two primary classifications, HF with preserved ejection fraction (HFpEF) in which the left ventricular ejection fraction (LVEF) >40% and HF with reduced ejection fraction (HFrEF) in which LVEF is ≤ 40%.4 There is a growing move to subgroup patients with LVEF 41–49% as HF with mildly reduced (HFmrEF), although this population (particularly those with LVEF >45%) has been included in the epidemiologic, pathophysiologic, and clinical trial studies of HFpEF.. Of hospitalized patients with HF, HFpEF is at least 40%, and approximately half of the HF population in the community has HFpEF.5–10 HF is a highly morbid and mortal syndrome; both HFpEF and HF with reduced ejection fraction (HFrEF) are associated with a strikingly high 35% one-year mortality and HF readmission rates range from 18–30%.11–13 The development of concomitant pulmonary hypertension (PH) marks a significant event in the HF disease process with important survival implications.14 LHD, and specifically HF syndromes, make up the majority of patients with PH, comprising up to 80% of cases.15, 16 The most common etiologies of PH associated with LHD (World Health Organization Group 2 PH) in contemporary clinical practice are HFpEF and HFrEF, whereas mitral stenosis, in which Group 2 PH was initially described, is now relatively rare in developed countries. We therefore focus on Group 2 PH associated with HFpEF and HFrEF (PH-LHD). Despite the high prevalence of HF, the pathobiology of PH-LHD is not well understood, and preclinical models of PH-LHD typically do not investigate the full spectrum of hemodynamic PH-LHD phenotypes. Here we review the various hemodynamic phenotypes in patients with PH-LHD, the potential biological mechanisms underlying this disorder, and the highest priority unanswered questions.
DEFINING GROUP 2 PULMONARY HYPERTENSION
The 2018 World Symposium on PH updated the hemodynamic definitions of disease with a focus on the location of disease in the pulmonary vascular bed.17 The recommendations incorporated hemodynamic thresholds for defining PH based on normative data rather than expert opinion by reducing the mean pulmonary arterial (PA) pressure (mPAP) indicative of PH to ≥20mmHg.17, 18 In addition, the 2018 World Symposium highlighted the facts that pre-capillary pulmonary hypertension (defined as pulmonary vascular resistance [PVR] ≥ 3 Wood units) occurs in patients with PH-LHD and that combined pre- and post-capillary PH (Cpc-PH) represents a relatively common hemodynamic phenotype in clinical practice. Currently Group 2 PH is defined in two forms: (1) Cpc-PH (previously termed PH “out of proportion” to LHD) identified as a mPAP ≥20mmHg, PVR ≥3 Wood units, and PA wedge pressure (PAWP; a surrogate for left atrial [LA] pressure) >15 mmHg and (2) predominant pulmonary venous hypertension also known as isolated post-capillary PH (Ipc-PH), in which mPAP is ≥20mmHg, PAWP is >15 mmHg but PVR is less than 3 Wood units (Table 1).17
Table 1.
Hemodynamic Definitions of Pulmonary Hypertension Phenotypes in Patients with Left Heart Disease
| PH-LHD Phenotype | Hemodynamic Criteria |
|---|---|
| Isolated post-capillary PH | mPAP ≥20 mmHg PAWP >15 mmHg PVR <3 WU |
| Combined pre- and post-capillary PH | mPAP ≥20 mmHg PAWP >15 mmHg PVR ≥3 WU |
PH = pulmonary hypertension; LHD = left heart disease; mPAP = mean pulmonary artery pressure; PAWP = pulmonary artery wedge pressure; PVR = pulmonary vascular resistance.
The recent evolution of the definition of Group 2 PH is an improvement; however, several gaps in our understanding of its diagnosis remain. The 2018 diagnostic criteria use PAWP as the primary measure of left sided filling pressures and indicator of hemodynamic congestion. Clinically PAWP and LV end diastolic pressure (LVEDP) have historically been used interchangeably; however, multiple studies have shown that the two measurements frequently differ, and the 2 hemodynamic indices have different meanings.19–22 Variable agreement between LVEDP and PAWP is demonstrated by only a moderate correlation (R2=0.42) and impacted by the presence of atrial fibrillation, a comorbidity often seen in clinical practice.21 The reliability of the measures of hemodynamic congestion are essential to the diagnosis of pre- versus post-capillary PH, and the ultimate implementation of therapies.20 Rather than using these hemodynamic values interchangeably it is important to recognize that these two values measure different entities, even in the absence of mitral valvular disease or arrhythmia. Although LVEDP is often considered the gold standard for the hemodynamic diagnosis of LHD during the evaluation of patients with PH, PAWP reflects the health of the LA and is a more accurate representation of what pressures the pulmonary capillary bed sees over time. A true PAWP (as opposed to a partially damped PA pressure tracing) is easily confirmed during right heart catheterization if PAWP blood oxygen saturation is equal to systemic arterial oxygen saturation. When measured properly, PAWP is therefore a more inclusive assessment of chronic hemodynamic congestion reflecting the reaction of the LA to chronic pressure load, LA remodeling, and changes in LA compliance that develop over time.23
The LA response and adaptation to hemodynamic congestion is not universal between HF syndromes; HFpEF demonstrates a greater degree LA stiffness while HFrEF results in predominantly eccentric LA remodeling.24 LA dysfunction is associated with the development of PH and right ventricular dysfunction in both HFpEF and HFrEF.24 Impairment of LA function has prognostic value in the HFpEF population, marking an inability of the LA to buffer the pulmonary circulation against changes in LV pressure.19, 25 LA strain is impaired in both HFpEF and HFrEF.26 Clinically, LA strain, which is emerging as an easy-to-measure, reproducible measure of LA function that may have clinical and research utility, is divided into 3 phases: reservoir (LA filling during LV systole), conduit (LA passive emptying into the LV during early LV diastole), and booster (LA contraction at LV end-diastole).27, 28 Clinically LA dysfunction and congestion likely determines treatment responses to pulmonary vasodilators, as increased blood flow through the pulmonary bed may result in increased pulmonary congestion in the setting of a stiff, non-compliant LA. Decongestion with diuretics and systemic afterload reduction may help offset increased blood flow into the LA but ultimately degree of LA remodeling when pulmonary vasodilator therapies are implemented may prohibit significant symptomatic benefit especially given LA dysfunction is seen early in the left heart disease process.29 Other than treatment of atrial arrhythmias and anticoagulation, therapeutic options of LA dysfunction are non-existent at the present time.30 Atrial arrhythmias are clearly associated with worse outcomes and RV function in HFpEF.31, 32 Restoration of sinus rhythm by atrial fibrillation ablation has morbidity and mortality benefit in HF, but it is unclear what effect restoration of sinus rhythm has on development and severity of pulmonary hypertension, particularly when treatment of atrial arrhythmias only improves electrical function and not mechanical function of the LA.33–35 A more complete understanding of alterations LA structure and function in response to HFpEF and HFrEF is essential to determining risk for development of PH, differences between hemodynamic PH subtypes in HF, and potential therapeutics.
The diastolic pressure gradient (DPG), calculated as the difference between PA diastolic pressure and PAWP, and transpulmonary gradient (TPG), calculated as the difference between mPAP and PAWP, were previously key to PH diagnosis in patients with LHD and remain relevant despite their absence from the most recent diagnostic criteria in consensus guidelines. Elevated TPG and PVR in end-stage HF indicate worse prognosis and can prohibit cardiac transplantation.36 The differentiation between Ipc-PH and Cpc-PH, or “reactive” versus “fixed” PH in end stage HFrEF is a critical part of the cardiac transplant evaluation. Often a systemic vasodilator study is recommended for those patients with PA systolic pressure ≥50 mmHg, TPG ≥15mmHg, or PVR ≥3 WU if systemic systolic blood pressure is >85 mmHg to determine severity of PH and improvement in these indices with reduction in systemic afterload.37 Assessment of the degree of pulmonary vascular disease can be especially problematic in patients with severe, concomitant right-sided HF, where the right ventricle is unable to mount significant pulmonary pressures. Presumably the use of PVR partially overcomes this issue by incorporating the cardiac output into assessment of the pulmonary vascular disease; however, hemodynamic loading, congestion, and pulmonary vessel recruitment can all impact the TPG and DPG, although DPG less so.38 None of the currently used variables provide a complete understanding of the degree of pulmonary vascular disease present in PH-LHD, and these nuances in the diagnostic criteria of PH-LHD are not typically considered in pre-clinical models of PH-LHD.
HEMODYNAMIC PHENOTYPES OF PH-LHD
Theoretical pathophysiologic models of PH-LHD often show the LHD process (with both HFpEF and HFrEF combined) as a continuum of advancing severity of disease that starts with Ipc-PH and evolves into a pre-capillary component due to vascular remodeling and/or vasoconstrictive response to hemodynamic loading. This load occurs when hemodynamic congestion, an increase in LVEDP and LA pressure, progresses to pulmonary congestion with an increase in extravascular fluid in the lungs, and systemic congestion in series.39 The current progressive model of PH-LHD does not have clear longitudinal hemodynamic data to support it, and evidence has emerged that patients with Cpc-PH have overlapping features with PAH.40, 41 This overlap may indicate that some patients have an accelerated course that leads to Cpc-PH or have a predisposition to develop pre-capillary disease simultaneously with LHD. Proposed phenotypes of PH-LHD, and their relatedness to each other, are outlined in Figure 1.
Figure 1. Hemodynamic Phenotypes of Pulmonary Hypertension in Left Heart Disease.
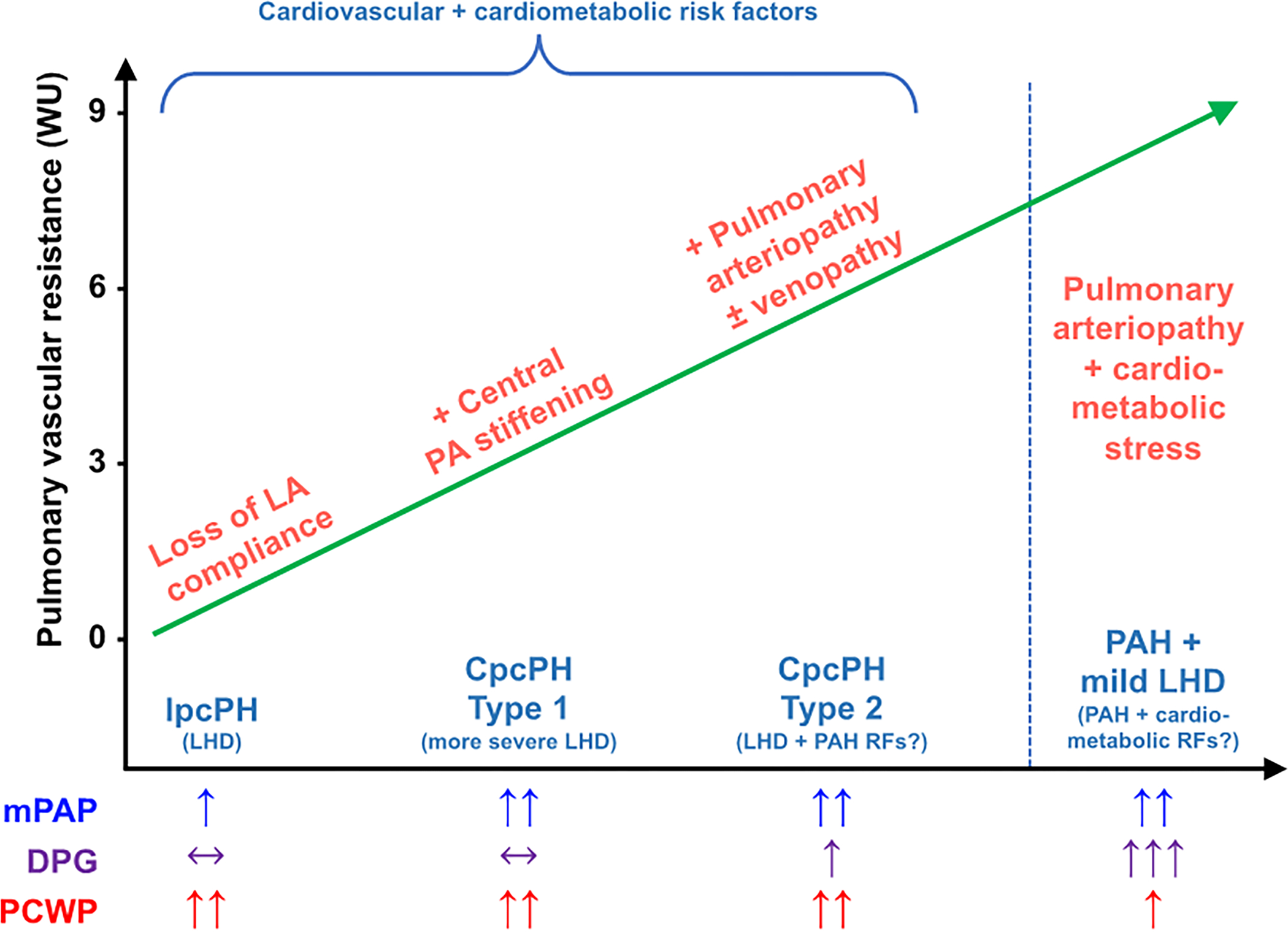
The current understanding of PH-LHD is a progressive worsening of PH starting with Ipc-PH with the ultimate development of Cpc-PH. However, the degree of elevation in PVR in a patient with LHD likely depends on both severity of LHD and the presence of risk factors for PAH. If risk factors for PAH are present, PVR will likely be higher, and Cpc-PH will be more severe. Alternatively, there are some patients who have very high PVR and DPG with only mildly elevated PAWP. These patients likely have predominant PAH but have cardiometabolic risk factors. Technically these patients would be classified as PH-LHD due to the elevated PAWP, but from a phenotypic standpoint are much more similar to Group 1 PAH.
Ipc-PH: isolated post-capillary pulmonary hypertension; Cpc-PH: combined pre and post capillary pulmonary hypertension; LHD: left heart disease; PAH: pulmonary arterial hypertension; RF: risk factors; mPAP: mean pulmonary artery pressure; DPG: diastolic pressure gradient; PAWP: pulmonary artery wedge pressure; LA: left atrial; PA: pulmonary artery
Since in the initial World Symposium on PH, much of the subgrouping, phenotyping, and implementation of therapy has been made under the assumption that patients have PH of one type or the other (pre-capillary vs. post-capillary) in isolation. In clinical practice rare is the patient who presents to the clinic or the hospital with isolated pre-capillary PH without other risk factors for LHD. The prevalence of obesity, sleep apnea, atrial fibrillation, hypertension, tobacco use, and diabetes, even in those younger than 50, means a significant proportion of the population inevitably has one or two risk factors for LHD.42–44 The pervasiveness of these risk factors in the general population indicate that PAH and LHD cannot be mutually exclusive, especially in patients with connective tissues diseases that are potent risk factors for both LHD and PAH. The reliance on one set of (variable and relatively poorly understood) hemodynamic data, taken almost in isolation without consideration of pre-test probability and overlapping disease processes has resulted in very strict definitions of those patients that are offered therapy for PAH. There is clear data of demographic, hemodynamic, and genetic similarities between populations of patients with PH-LHD (particularly the Cpc-PH phenotype) and PAH.40, 41 PH-LHD patients with evidence of pulmonary vascular disease (PVR≥3 Wood Units) have worse prognosis, and as such these changes in pulmonary vascular function and structure may be markers of divergent pathophysiology and may serve as potential therapeutic targets.16 Subgrouping PH-LHD into those with PAH risk factors may help differentiate a population of patients distinct from those with primarily congestion-driven PH. Figure 1 summarizes the 2 divergent theoretical models of PH-LHD (progressive continuum vs. risk factor overlap), which is relevant to the basic and translational scientific investigation of PH-LHD.
STRUCTURAL AND FUNCTIONAL CHANGES IN PULMONARY HYPERTENSION
Chronic hemodynamic congestion is believed to progressively drive structural and functional changes in the LA and pulmonary vasculature. In response to pressure and volume overload the LA dilates, and alterations in cardiomyocyte structure and mechanical function occur, which result in LA mechanical dysfunction. Ultimately, LA stiffness increases, and the function of the LA as a pressure and volume buffering reservoir between the LV and pulmonary circulation is impaired.24, 45 Loss of LA function (particularly loss of LA reservoir function—the ability of the LA to fill from the pulmonary veins during ventricular systole) correlates with lower PA compliance and higher PVR in the setting of HF46 (Figure 2), with loss of LA compliance precipitating increases in PA stiffness.24 PA stiffness directly correlates to the severity of right ventricular dysfunction, indicative that right ventricular dysfunction in this population is impacted by both afterload (PVR) and pulsatile load (PA stiffness), of which pulsatile load appears to have a larger impact.47, 48 These changes effectively switch the pulmonary vasculature from a low pressure, high compliance system to relatively high-pressure, low compliance system. However, some data departs from this theory as Cpc-PH had less severe cardiac structural remodeling on echocardiography compared to Ipc-PH in a large electronic health record-based cohort.40 Patients with Cpc-PH had smaller LA and ventricular dimensions and less LV hypertrophy (smaller LV mass and lower LV thickness) on echocardiography compared to Ipc-PH.16, 40 The lesser severity of these structural changes raise the question of a potential predisposition or increased sensitivity of the pulmonary vasculature to congestive stress, where small changes in diastolic function have a larger impact in Cpc-PH patients. Alternatively, in some Cpc-PH patients, elevated PVR may occur first, thereby resulting in reduced blood flow to the LA and LV, and PAWP increases later due to LHD risk factors. Finally, misdiagnosis is also a possibility in retrospective electronic health record-based cohorts, which may have explained the aforementioned findings.
Figure 2. Left Atrial Dysfunction Correlates with Pulmonary Vascular Resistance and Compliance in Heart Failure.
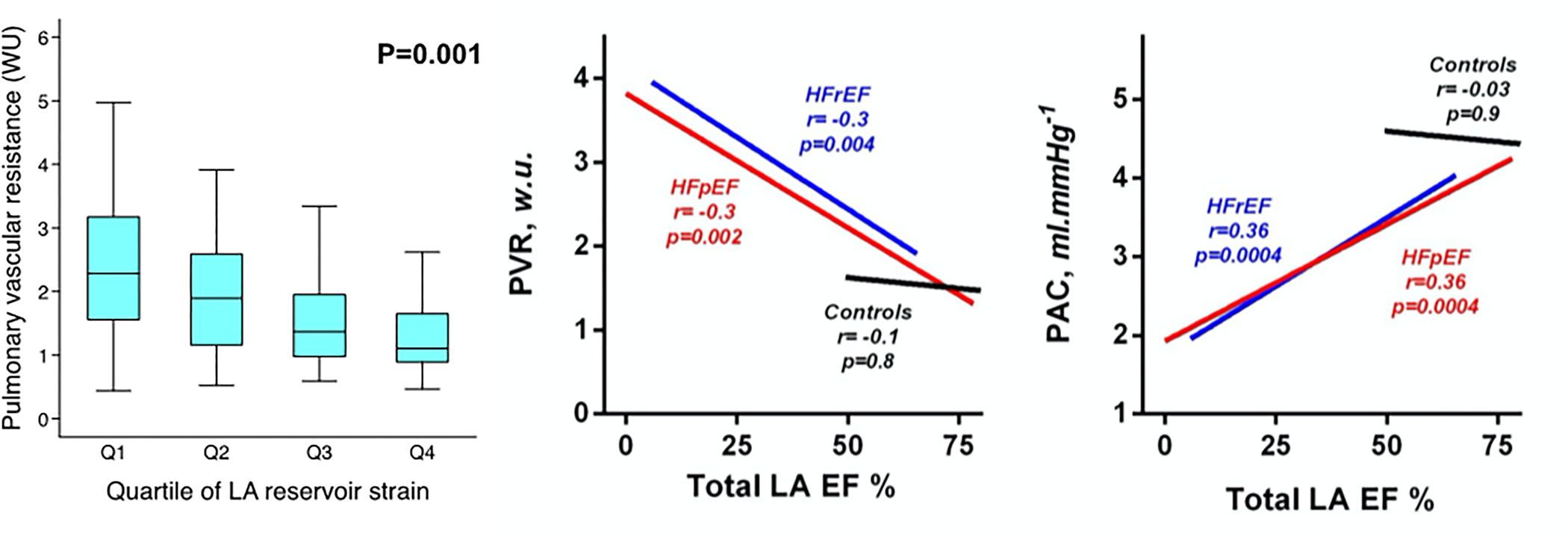
Left panel: In patients with HFpEF, worse LA reservoir strain (indicative of the inability of the LA to fill from the pulmonary veins during ventricular systole) is associated with increased PVR. Middle panel: In patients with HFpEF and HFrEF, lower LA emptying fraction (indicative of worse LA function) is also associated with elevated PVR. Right panel: Lower LA emptying fraction is also associated with reduced PA compliance in both HFpEF and HFrEF. Reproduced with permission from Freed, et al. Circulation: Cardiovascular Imaging 2016 and Melenovsky, et al. Circulation: Heart Failure 2015.
Molecular, functional, and structural changes in the pulmonary vasculature in response to reduced LA compliance and the resultant increase in hemodynamic stress is initially adaptive, but continued pulmonary vascular wall stress appears to drive maladaptive and pathologic changes. Changes in PA compliance coincide with loss of elastin and expansion of the extracellular matrix in both animal models of PH-LHD patients.49, 50 These structural changes are suspected to be related to “stress failure” or pressure injury of the capillary wall.51 Edema-driven injury to this alveolar-capillary interface in animal models activates matrix metalloproteinases, reducing the strength and increasing the permeability of the vascular endothelium.52, 53 Patients with acute cardiogenic pulmonary edema have elevated circulating levels of pulmonary surfactant proteins that leak across a stressed or disrupted alveolar-capillary interface.54, 55 Despite treatment and resolution of acute pulmonary edema, circulating pulmonary surfactant proteins remain elevated for up to 2 weeks indicating a persistent disruption of the alveolar-capillary interface.56 Along with pulmonary surfactant, circulating tumor necrosis factor-α (TNF-α) is elevated in acute pulmonary edema and similarly persist beyond clinical resolution. These findings suggest that injury to the capillary-alveolar interface persists past resolution of cardiogenic pulmonary edema due to LHD and may have the potential to trigger systemic inflammation.
Histologic structural modifications in the pulmonary vasculature occur frequently in PH-LHD, similar to vascular findings first described in severe mitral stenosis patients which manifested as pulmonary arteriopathy and venopathy.57, 58 Autopsy specimens of lung parenchyma in chronic HF patients show evidence of “congestive vasculopathy” with vascular remodeling across the spectrum of the pulmonary vascular bed.59, 60 Venous, capillary, and arterial remodeling are a result of LA hypertension and driven by adaptive and maladaptive pathways in the lung vasculature.61, 62 Wall stress is increased in pressure-loaded vasculature and drives structural changes in the vessels to accommodate stress, resulting in vasodilation and increased vessel wall thickness. Autopsy specimens from patients with PH-LHD demonstrate increased intimal and medial thickness in pulmonary arteries and veins compared to healthy controls but less structural changes than those seen in pulmonary veno-occlusive disease (Figure 3).60 More severe elevation of PA systolic pressure in PH-LHD correlated with greater degrees of arterial and venous remodeling with a greater percentage of arterialized veins in PH-LHD compared to pulmonary veno-occlusive disease (Figure 3). A separate examination of muscular arteries in patients with PH-LHD (defined as PA systolic pressure >30 mmHg or mPAP ≥19 mmHg) in patients with HFrEF who had undergone cardiac transplantation showed increased medial thickness in all patients without significant intimal fibrosis, although medial thickness did not correlate with post-transplant hemodynamics.59 A study of HFrEF patients with lung biopsy at time of LV assist device (LVAD) demonstrated increased volume of vascular media, intima, and adventitia in arteries and veins compared to controls.50 In some cases, the LVAD PH-LHD patients had media and intimal remodeling that nearly occluded the lumen indicative of the severe structural vessel changes that may occur in the setting of PH-LHD.
Figure 3. Histologic Pulmonary Vascular Remodeling in Pulmonary Hypertension due to Left Heart Disease.
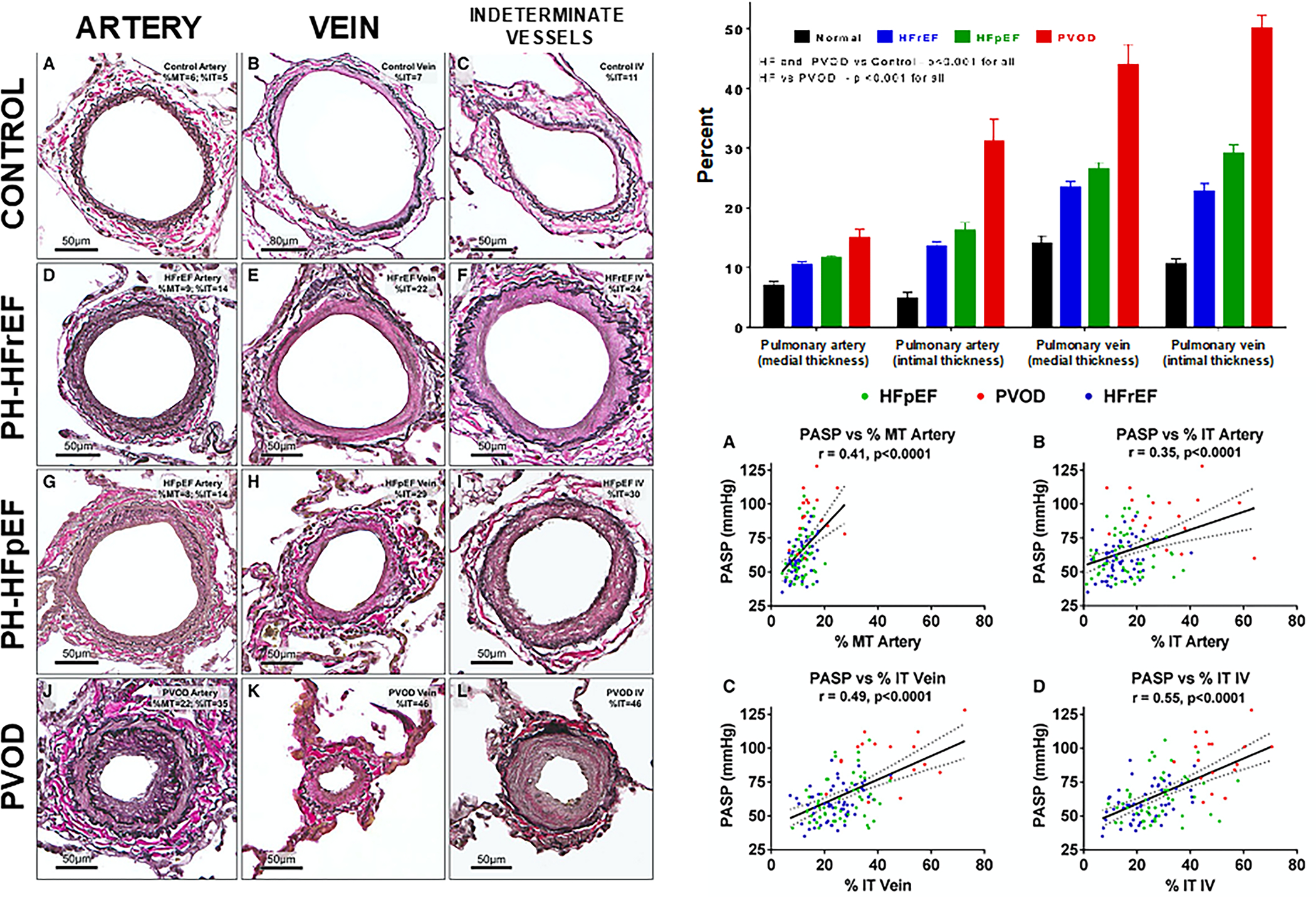
Left panel: Comparison of pulmonary arteries, veins, and indeterminate vessels among healthy controls, PH-HFrEF, PH-HFpEF, and PVOD patients. Representative vessels with remodeling approximating the median values for medial and intimal thickening of arteries, veins, and indeterminate vessels in each group are shown. Right, top panel: There is progressive severity of medial and intimal thickening in pulmonary arteries and veins from healthy controls (normal) to PH-HFrEF to PH-HFpEF to PVOD (most severely abnormal). Right, bottom panel: increases in medial and intimal thickening in the pulmonary arteries and veins correlates with increased severity of pulmonary hypertension (increased PASP). The solid line represents the estimated PASP via linear regression based on medial/intimal thickening, and the dotted lines represent the 95% confidence interval. HF, heart failure; HFpEF, HF with preserved ejection fraction; HFrEF, HF with reduced ejection fraction; %IT, percent intimal thickness; %MT, percent medial thickness; PH, pulmonary hypertension; PVOD, pulmonary veno-occlusive disease; IV, indeterminate vessels; PASP, pulmonary artery systolic pressure. Reproduced with permission from Fayyaz et al., Circulation 2018..; and PVOD, pulmonary veno-occlusive disease. Reproduced with permission from Fayyaz et al., Circulation 2018.
The differentiation between adaptive and maladaptive structural changes in PH-LHD is not well defined. The LA is sensitive to pressure and volume elevations and responds quickly to these hemodynamic derangements. A canine model of HFpEF demonstrated early adaptive response of the LA to pressure and volume stress with LA dilatation and augmented contractile function.63 However, with continued hemodynamic stress the pressure-volume relationship of the LA changes and LA compliance decreases, at which time these responses shift to maladaptive.63 These LA changes result in atrioventricular uncoupling and impaired cardiac output due to reduced LV stroke volume. Reduction of LA compliance impairs its reservoir function which buffers the pulmonary vascular bed from increased LV pressures; the resultant elevation in LA pressure results in a shift of the PVR/pulmonary arterial compliance (CPA) relationship to a state where compliance is lower at any given PVR. 48 Early mitigation of LA hypertension can modify these vascular changes, preventing the histologic pulmonary vascular remodeling and changes in pulmonary hemodynamics.64 The exposure of the pulmonary vascular bed exposure to hemodynamic stress drives pulmonary venous, capillary, and arterial remodeling, with impaired endothelial function, fibrosis, and smooth muscle proliferation. 60 These changes prove to be maladaptive, as even with relief of LA hypertension the abnormalities in PVR and CPA persist.64
There is evidence of genetic overlap between PAH and the Cpc-PH subset (but not Ipc-PH) that may contribute to the severity of pulmonary vascular disease. Single nucleotide polymorphisms (SNPs) resulting in increased lung gene expression of various proteins that have been identified in PAH have also been demonstrated in Cpc-PH.40 These SNPs encode modifications of extracellular matrix, basement membrane structure, as well as modifications in actin binding and structural molecular activity. These genetic variants were not found in Ipc-PH, suggesting that there may be a separate genetic subgroup predisposed to the Cpc-PH pattern of PH-LHD. These findings suggest the potential for a “two-hit hypothesis” where development of pre-capillary PH in the setting of LHD requires a second insult (e.g., genetic, metabolic, or hormonal) that predisposes to pulmonary arteriopathy. Below we outline the data surrounding potential influential contributors to pulmonary vascular disease in the setting of PH-LHD disease.
ANIMAL MODELS OF PH-LHD
Few animal models of PH-LHD exist, in HFpEF specifically the variability of human disease has made it difficult to create similar animal phenotypes (Table 2).
Table 2.
Animal Models of PH-LHD
| Model | Species | Strengths | Limitations |
|---|---|---|---|
| Metabolic Models | |||
| Metabolic Syndrome (ZSF1 model) + SU5416/hypoxia | Rat65 | Includes metabolic syndrome in phenotype | High Cost Limited molecular tools compared to mice Complicated phenotype |
| Metabolic Syndrome (ZSF1 model) + Aortic Banding | Rat74 | Combined pressure and metabolic injury | High cost Primarily mild PH Complicated phenotype |
| High fat diet/Obesity | Mouse66 Bovine75 |
Low cost Metabolic syndrome phenotype |
Longer timeline needed Variation in PH-HFpEF phenotype based on mouse strain used |
| High fat diet + L-NAME (or db/db model) + SU5416/hypoxia | Mouse | Low cost Metabolic syndrome phenotype with added pulmonary vascular disease |
Needs to be further studied and validated as a PH-HFpEF phenotype |
| Mechanical Obstructive/Pressure Overload Models | |||
| Aortic Banding | Mouse68 Rat76–78 Feline79 |
Simple Reproducible Low cost |
Does not induce severe PH and RV failure Most synonymous with Aortic stenosis induced-PH, less with non-valvular HFpEF |
| Transverse Aortic Constriction | Mouse 69, 80 | Simple Reproducible |
Associated pulmonary fibrosis Most synonymous with Aortic stenosis induced-PH, less with non-valvular HFpEF |
| Pulmonary vein banding | Porcine70 Bovine81 |
Represents pulmonary venous hypertension | Higher cost Multiple operators Large animal Not representative of underlying LV disease |
| Left atrial stenosis | Rat71, 72 | Simulates mitral stenosis | Not representative of underlying LV disease Difficult surgical intervention |
| Ischemic Models | |||
| Left coronary artery ligation | Rat73, 82, 84, 100 Mouse85, 86 Porcine83 |
Primary HFrEF model | Not representative of non-ischemic cardiomyopathy Minimal control of infarct size Low survival |
A Zucker (ZSF1) rat model of PH-HFpEF was created by inducing metabolic syndrome via a defect in leptin receptor combined with vascular endothelial growth factor receptor-2 antagonist Sugen 5416 (SU5416) which essentially layered features of the well-known SU5416-hypoxia model of PAH over the obese metabolic syndrome found in HFpEF. 65, 67 The SU5416-exposed obese ZSF1 rats had normal LVEF but developed elevated right and left sided pressures as well as higher PVR and pulmonary vascular remodeling compared to lean rats.65 Recently, a combined obesity and hypertensive stress (high fat diet plus L-NAME) has been utilized to produce a HFpEF phenotype in mice that mimics the human HFpEF phenotype. The db/db (leptin receptor-deficient) mouse is another model that mimics the HFpEF phenotype, albeit with less pulmonary congestion. SU5416 could theoretically be overlayed on top of these models to mimic the CpcPH phenotype of PH-HFpEF.
A similar mouse model was developed by observing the hemodynamics effects of a high fat diet in 36 strains of mice.66 Of the strains investigated the AKR/J, NON/shiLtJ, and WSB/EiJ developed hemodynamic changes consistent with PH-LHD. OF those strains the AKR/J mouse findings were the most reliably reproduced with the high fat diet inducing metabolic syndrome and the mice developing elevated right ventricular systolic pressures, LV end diastolic pressures, with biventricular hypertrophy compared to those fed a regular diet.66 This mouse model also showed higher PVR and pulmonary vascular remodeling, and these hemodynamic abnormalities progressed with continuation of the high fat diet. These metabolic models more closely approximate clinical disease than previous models of aortic banding in which increased LV afterload induces higher LV filling pressures, elevated mPAP, and PVR.68 While the aortic banding animal model is the most commonly used model in PH-LHD (specifically to model PH-HFpEF), this model does not mirror the metabolic syndrome component found in humans with HFpEF, and this model progresses to overt LV systolic dysfunction (HFrEF), which is not seen in the vast majority of patients with HFpEF who are followed longitudinally (i.e., in HFpEF, LVEF remains preserved over time and does not progress to HFrEF).
Similar models focusing on induction of HF without consideration of the metabolic features the co-exist in PH-LHD have been the basis of PH-LHD animal studies. Although aortic banding is the most widely used, several other models exist. Mouse models of transverse aortic constriction create phenotypes similar to aortic banding with development of HFpEF and pulmonary hypertension, although it also induced severe pulmonary fibrosis, which is not typically found in humans with HF.69 Pulmonary venous outflow obstruction by pulmonary vein banding or creation of left atrial stenosis have also been use to pressure load the pulmonary vascular bed in animal models, and induce PH and RV failure in rats and large animal models.70–72 The primary modality of inducing HFrEF in animal models has been coronary artery ligation to induce an ischemic cardiomyopathy, and this model can induce mild-to-moderate PH associated with low cardiac output and congestion, but infarct size is difficult to control, and the survival rates post-induced myocardial infarction can be low.73, 84–86 The mechanical, pressure overload models of PH-HFpEF do not mirror phenotypes seen in clinical practice, rather the metabolic-driven models are closer to human phenotypes.
PROPOSED PATHOBIOLOGIC CONTRIBUTORS TO PH-LHD
The pathophysiology of Group 2 PH and contributing factors to disease severity and development of PH-LHD are not well understood, especially in HFpEF. There is emerging evidence of underlying inflammatory, hormonal, and metabolic derangements that appear to contribute to pulmonary vascular disease in LHD. Figure 4 summarizes our current understanding of the pathobiology of PH-LHD, as explained in detail below.
Figure 4. Pathobiologic Contributors to Pulmonary Hypertension in Left Heart Disease.
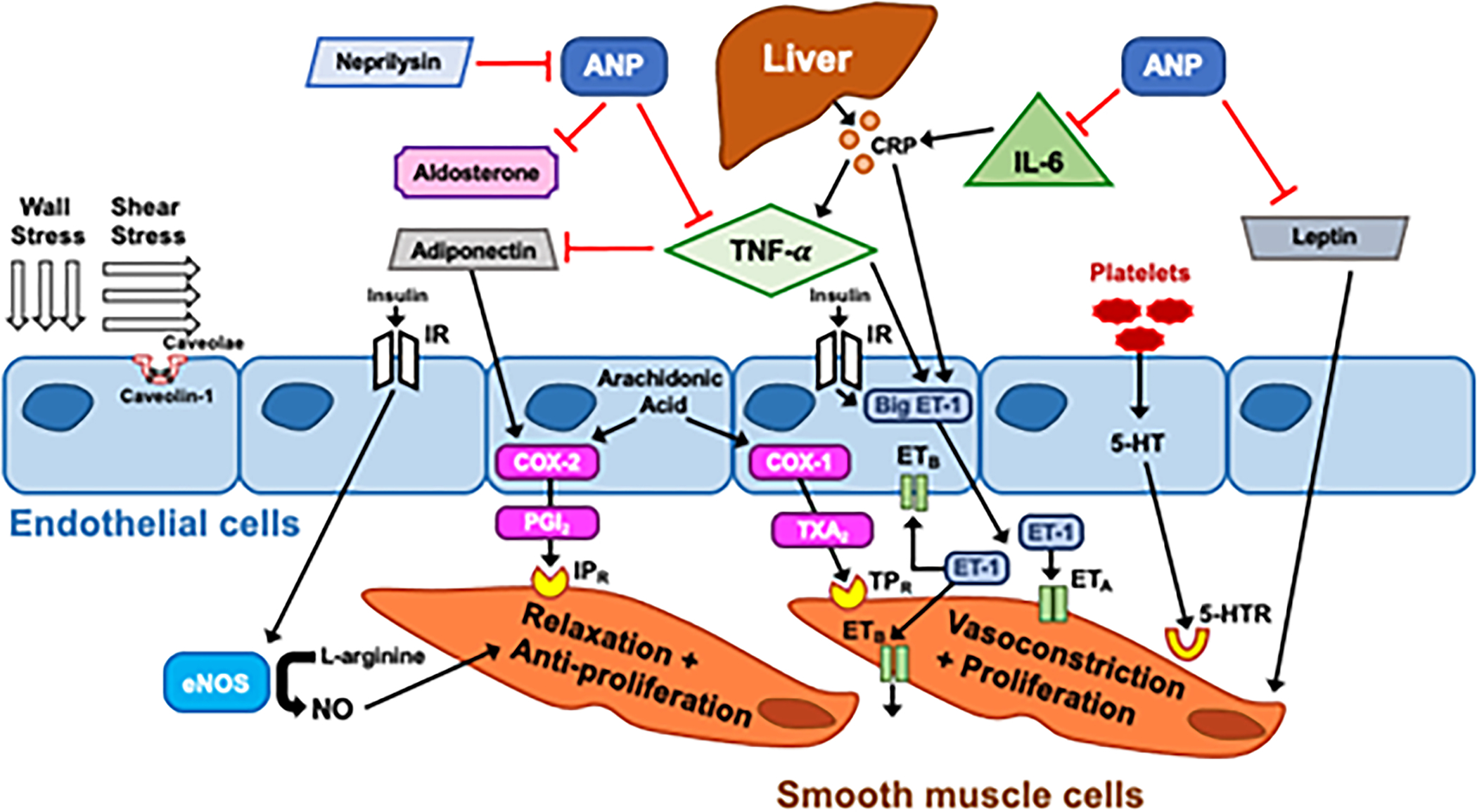
Wall stress and shear stress are drivers of adaptive and maladaptive processes resulting in endothelial dysfunction, smooth muscle hypertrophy, and inflammation. Many of these mediators of endothelial function in PH-LHD that have effects both systemically and locally. eNOS: endothelial nitric oxide synthase, IR:insulin receptor, NO: nitric oxide, ANP: atrial natriuretic peptide, TNF-α:Tumor necrosis factor alpha, COX: cyclooxygenase, PGI2:Prostaglandin I2, IPR: Prostacyclin I2 receptor, TXA2: thromboxane A2, TPR: Thromboxane A2 receptor, ET-1: endothelin-1, ETA: Endothelin receptor A, 5-HT: serotonin, 5-HTR: serotonin receptor, IL-6: Interleukin-6, CRP: C-reactive protein
Endothelial Injury and Dysfunction
Both HF and PH are independently associated with systemic endothelial dysfunction in venous and arterial beds.87, 88 Animals models of HF have demonstrated evidence of pulmonary endothelial dysfunction.89–91 Multiple endothelial vasoactive mediators have been implicated in this dysfunction with overlap in both PH and LHD; however, the initial insult has been difficult to elucidate. Shear stress is the mechanical force on the endothelium caused by blood flow and is instrumental in both vascular homeostasis as well as development of disease.92 Reduced shear stress is a known driver of endothelial dysfunction in the systemic vasculature and plays a role in atherogenesis.93 Reductions in shear stress also correlate with higher PA pressures in patients with PAH 94, 95 Changes in shear stress stimulate endothelial responses to adapt to alterations in pressure and flow; in endothelial dysfunction this response in muted or maladaptive. Here we describe a variety of mediators of endothelial function that have potential involvement in PH-LHD.
Caveolae and Caveolin-1
Changes in shear stress are sensed by endothelial cells, resulting in alterations in cellular signaling, in which caveolae and caveolin-1 expression and activity are key drivers.96 Caveolae are plasma membrane invaginations that sense and convert hemodynamic changes in the vasculature into intracellular signals. Caveolin-1 is the main structural membrane protein within caveolae that enacts messaging across the cell membrane, with roles in intracellular calcium signaling, regulation of nitric oxide (NO) production, and activation of PA smooth muscle cell (PASMC) proliferative pathways.97 Changes in caveolin-1 signaling are present in HF and PAH. Chronic beta-adrenergic stimulation, which occurs in HF as well as hypoxia, reduces caveolin gene expression.98, 99 Caveolin-1 knock-out mice develop dilated cardiomyopathy with associated PH and exhibit pulmonary vascular pathologic changes similar to PH-LHD.100, 101 In the vasculature, NO availability is partly controlled by the interaction between caveolin-1 and endothelial NO synthase (eNOS).102 Caveolin-1 binds to eNOS rendering it inactive and unable to produce NO, a potent regulator of vascular tone.103, 104 Caveolin-1 expression is lost in structurally normal arteries in PAH but upregulated in PASMCs surrounding remodeled arteries.105 While caveolin-1 has been implicated in PH, the role of caveolin-3 is less clear, with inconsistent changes in HF. Murine models of HF show selective reduction in expression of caveolin-3 expression rather than caveolin-1 or 2.106 Overexpression of caveolin-3 in rodent models reveals reduction in hemodynamically driven cardiomyocyte hypertrophy.107 Conversely, increased expression of caveolin-3 is present in a canine pacing-induced HF model.108
At the present time, it is not clear how caveolin expression and function contribute to PH-LHD; decreased expression in some instances appears to modify cardiomyocyte adaptive responses, whereas caveolin gene expression is variable in pulmonary vascular disease and it is unknown whether these changes are reactive to or independent of LA hypertension. Nevertheless, caveolae and the caveolin proteins are essential to signal transduction responses to shear stress (and therefore the endothelial cellular response to hemodynamic alterations); therefore, further investigation of the role of caveolae PH-LHD is worth pursuing.
Nitric Oxide
NO is well known to be deranged in the setting of HF. Peripheral venous dysfunction is associated with imbalance in NO synthesis and degradation (resulting in reduced NO bioavailability), and increased NADPH oxidase superoxide generation, which are both also regulators of pulmonary vascular homeostasis.109, 110 Peripheral endothelial dysfunction occurs in HFpEF, where response to flow mediated vasodilation correlates with higher PVR on invasive hemodynamics.111 NO-mediated endothelium-dependent vasodilation in chronic HFrEF is also impaired and has also been attributed to reduction in NO synthesis, increased degradation, and reduced vasodilatory responsiveness.112–115 The beneficial pulmonary vascular effects of NO also include reduction in smooth muscle hypertrophy and proliferation.116 A rodent model of HF showed impairment of alveolar-capillary endothelial function and absence of NO synthesis in response to mechanical and chemical stress driven by impairment of endothelial calcium handling.91 Notably, the expression of eNOS was not different from controls; rather, activation of eNOS by calcium was reduced. NO production and NO-dependent pulmonary vasodilation are impaired in PH-LHD, although it is unclear to what degree the vascular response is mediated through hemodynamic-driven changes in biochemical signaling and NO synthesis or scavenging mechanisms are causal or consequential.117 Furthermore, restoring NO-cyclic guanosine monophosphate signaling via phosphodiesterase-5 inhibition and soluble guanylate cyclase stimulation has not proven beneficial in PH-LHD (especially in PH-LHD due to HFpEF, which has been studied extensively in clinical trials).118–124
Endothelin-1
Endothelial cells secrete endothelin-1 (ET-1) with the primary receptors located on vascular smooth muscle cells through which ET-1 acts as a potent vasoconstrictor and pro-proliferative agent.125, 126 The ET-1 peptide is important for maintenance of vascular tone and has mitogenic effects on endothelial and vascular smooth muscle cells.127 Balance between vasoconstrictive and vasodilatory effects are modulated by concentrations of ETA and ETB receptors, respectively, on vascular smooth muscle and endothelial cells.128, 129 Endothelin-1 levels are elevated in chronic HF and correlate with severity of PH and 1 year mortality.130–133 The downregulation of ETB receptors and upregulation of ETA receptors seen in chronic HF tips the balance into predominantly vasoconstrictive and smooth muscle cell proliferative ET-1 effects 117, 130, 134 Local administration of ETA receptor antagonist causes a dose-dependent reduction in PVR in patients with chronic HF owing to the integral role of ET-1 in pulmonary vasoconstriction.135 Additionally, ET-1 mediates pulmonary vascular remodeling in PH-LHD by inducing smooth muscle proliferation and hypertrophy as well as collagen production.136 The pulmonary vasculature is particularly rich in ET-1 production and sensitive to its effects.137
Despite the critical role of ET-1 in the pathogenesis of HF and PH-LHD, multiple clinical trials of endothelin receptor antagonists (ERAs) have failed to demonstrate benefit in the setting of PH-LHD. Vascular tone and smooth muscle tone is in part driven by the balance between ET-1 and NO activity as well as prostacyclin-, cyclooxygenase-, and thromboxane-mediated vascular effects, which may explain the lack of benefit of ERAs in PH-LHD (Figure 4).138 ET-1 also reduces cardiomyocyte gene expression of natriuretic peptides,139 which may counteract their beneficial vasodilatory and natriuretic effects. Indeed ERA-induced sodium and fluid retention have been the Achilles heel of ERA therapy in patients with HF. For these reasons, strategies to selectively reduce ET-1 or block endothelin receptors in the pulmonary vasculature while preventing the natriuretic peptide reduction and avoiding adverse renal effects may be a fruitful endeavor in the future.
Prostacyclin and Thromboxane A2
In the normal pulmonary circulation, prostacyclin (also known as prostaglandin I2 [PGI2]) is a key to contributor to vascular tone via stimulation of cyclic adenosine monophosphate production-induced smooth muscle relaxation.140 Thromboxane-A2 (TXA2), a countermeasure to the vasodilatory and anti-proliferative functions of PGI2, drives vasoconstriction and promotes platelet aggregation.141 Both PGI2 and TXA2 are products of arachidonic acid metabolism by cyclo-oxygenase (COX) activity, and the balance of PGI2/TXA2 activity regulates vascular tone. This PGI2/TXA2 ratio depends on many variables including the amount of COX isoform present with COX-1 favoring TXA2 and COX-2 favoring PGI2.142 In rat models of cardiovascular disease, angiotensin-II induces production and release of PGI2 from cardiac fibroblasts which in turn prevents TGF-β-induced upregulation of extracellular matrix genes and therefore functions as an anti-fibrotic.143, 144 In a canine model of HF, expression of arterial eNOS and COX-1 gene expression was reduced as was resultant NO production in response to vasodilators.145 Circulating PGI2 is increased in chronic HF and correlates with plasma renin and angiotensin-II concentrations.146 Conflicting data exist about the ability of TXA2 to stimulate smooth muscle proliferation, although it appears that the presence of serotonin (5-HT) is synergistic with TXA2 producing a mitogen effect on smooth muscle cells.147
Platelet Bioenergetics
Platelet aggregation occurs at the site of vascular injury and platelet-derived factors influence vascular repair and remodeling at these sites utilizing peptide growth factors, two of which are serotonin (5-HT), and TXA2.148 Platelet aggregation and activation at sites of “stress failure” or shear injury in the pulmonary vascular bed specifically have not been well characterized. The pulmonary vasculature exhibits metabolic abnormalities in PH that favor glycolysis and alter electron transport chain function149, 150 Platelet metabolism and function in PH-LHD patients is similarly altered; mitochondrial maximal oxygen consumption rate and reserve respiratory capacity are both increased as is seen in Group 1 PAH patients.151 Reserve respiratory capacity the amount of ATP that can be produced via oxidative phosphorylation in the setting of an increased demand and is a marker of mitochondrial function. The increased reserve capacity in PH-LHD platelets appears to be in part due to increased fatty acid oxidation, as has been shown in PAH.152 Increased respiratory reserve capacity is associated with resistance to apoptosis and improved survival with oxidative stress.153 Platelet glycolytic rate, however, is not increased in PH-LHD patients in comparison with Group 1 PAH.151, 152 These metabolic alterations do not correlate with PVR in PH-LHD, as in Group 1 PAH, but are associated with worse right ventricular function.151 NO-induced platelet activation did not differ from healthy age-matched controls. Platelet function is altered in PH-LHD but it is unclear to what degree these differences are reactive versus pathologic. Furthermore, while prostacyclin analogues are one of the mainstays of therapy in patients with Group 1 PAH, their use in PH-LHD has not been well studied, though in a trial of an intravenous prostacyclin (epoprostenol) with severe HFrEF was associated with worse outcomes compared to placebo.154
Serotonin
Serotonin hormone is released by activated platelets, can induce pulmonary vasoconstriction and smooth muscle proliferation, and is metabolized in the lung, and has been studied extensively in PAH.155, 156 Local serotonin release is instrumental in platelet-induced vasoconstriction, as shown in acute coronary syndromes.157 HF is associated with in enhanced platelet aggregation and activation in which the local serotonin release may drive pulmonary vascular remodeling.158, 159 Notably, serotonin appears to have a substantial contribution to the stimulation of platelet induced-smooth muscle cell proliferation.160 Circulating serotonin levels are elevated in patients with HF and the degree of elevation correlates with decompensation and worse functional class.161 Currently, it is unknown if or how much the serotonin pathway is involved in the development of PH-LHD.
Tumor Necrosis Factor-α and Interleukin-6
Tumor necrosis factor-α (TNF-α) is an inflammatory cytokine produced by macrophages and monocytes and upregulated in HF.162–164 Rodent models of pulmonary congestion in HF demonstrate increased circulating and pulmonary venous concentrations of Interleukin-6 (IL-6) and TNF-α which were exacerbated with increased pulmonary venous mechanical stretch used to simulate the distention that occurs in response to elevated LA pressure.165 In other animal models, higher levels of circulating TNF-α are associated with increased circulating ET-1 levels while IL-6 induces C-reactive protein (CRP) production in the liver.166–168 Endothelin-1 and TNF-α are ultimately stimulated by CRP-induced complement activation, thereby creating and deleterious positive feedback loop.168, 169 Known as drivers of chronic inflammation, TNF- α and IL-6 have increased expression in patients with HF.170, 171 Notably, IL-6 and TNF-α levels correlate with elevated PA pressures in HFrEF and HFpEF.172 If increased levels of inflammatory cytokines are drivers of pulmonary vascular disease, targeting inflammation would be a worthwhile therapeutic target. Indeed, autoimmune disease, which is associated with increased inflammation, is known to be major risk factor for PAH and likely plays a key role in the development of pulmonary vascular disease in LHD and supports the role of a dysregulated immune system in driving PH-LHD, particularly Cpc-PH. Alternatively, increased severity of HF (which is present in patients with elevated PA pressures and pulmonary vascular disease) is known to result in increased inflammation, which would mean that therapies targeting the immune system are unlikely to be successful in PH-LHD. Both scenarios are likely true, and targeted studies of both innate and adaptive immunity aimed at deciphering the role of the immune system in the development and progression of PH in LHD remains a worthwhile pursuit, with the hope of ultimately identifying therapeutic targets in specific phenotypes of PH-LHD.
Vascular Endothelial Growth Factor
Angiogenesis is dependent on vascular endothelial growth factor (VEGF), which, in turn is often stimulated by both mechanical and metabolic stress. 173 The VEGF-D subtype appears to be most altered in HF and is instrumental in lymphangiogenesis.174 Systemic VEGF-D levels are elevated in patients with pulmonary congestion on chest imaging, and circulating levels of VEGF-D are higher in PH-LHD.175 In HFrEF, higher PAWP and lower cardiac output are associated with higher VEGF-D levels.176 VEGF-D appears to be elevated in hemodynamic and pulmonary congestion, the presumptive precursors of PH-LHD, but the role of VEGF in development of PH in HF is not well characterized. Preclinical models of PAH and PH-HFpEF have used the VEGF receptor antagonist SU5416 in combination with hypoxia stimulus to recapitulate features of PH.177 However, clinical studies of PAH and PH-LHD demonstrate significant elevation of VEGF-D, with PH-LHD demonstrating the highest concentrations. 178, 179 This presents a paradox given VEGF antagonists in combination with hypoxia induce PH in murine models, yet in clinical studies elevations in VEGF levels are associated with more severe PH. One study hypothesized that VEGF activity through the un-blocked VEGF3 receptor (SU5416 antagonizes receptors VEGF-1 and −2) and change in expression of VEGF isoforms. In the lungs of SU5416/hypoxia rat PAH model there was increased expression of VEGF-C and VEGF-D as well as VEGF-3 receptor.180 Interestingly the addition of a VEGF-3 antagonist prevented but did not ameliorate structural changes and development of PH in the SU5416/hypoxia model. The SU5416 model blocks VEGF-1 and VEGF-2, ultimately shifting activity to VEGF- C and VEGF-D, of which VEGF-D predominates on PH-LHD and PAH, isoforms and activation through available VEGF-3 receptor. This VEGF-3 effect produces vascular changes and lumen obliteration driving pulmonary hypertension.180 However, VEGF-3 knockout mouse models are also associated with development of severe PH with hypoxia and VEGF-3 receptor expression is reduced in PAH and PH-LHD.181, 182 It has been hypothesized that knock-out of VEGF-3 receptor leads to increased VEGF-C signaling through VEGF-2 receptor. 183 VEGF-2 and VEGF-3 receptors both play a role in the vascular endothelial mechanosensitive response to shear stress. 184 VEGF-C and VEGF-3 also play a primary role in lymphangiogenesis and there is increasing recognition of the importance of the lymphatic system in HF.185 A preclinical model of HFpEF demonstrated early increase in VEGF-C and VEGF-3 protein expression which then dropped with worsening HF and decompensation.185 VEGF antagonism results in PH and increased expression appears to have a protective effect, yet, in the presence of cardiovascular or pulmonary disease VEGF may promote cellular proliferation and vascular remodeling.87, 186 Further preclinical studies may take advantage of combining metabolic stress (e.g., high fat diet), hypertensive stress (e.g., L-NAME), and SU5416/hypoxia to create a model of Cpc-PH in HFpEF to better elucidate the role of VEGF and VEGF receptors in the disease process. This type of strategy has been employed successfully in rat models as well, by combining the Zucker diabetic rat (ZSF1) model of HFpEF with SU5416/hypoxia, as detailed above.187
Estrogen
There is an association between post-menopausal reduction in estrogen production and endothelial dysfunction, partly explained by estrogen modulation of NO, endothelin, and prostacyclin pathways.188–190 Female predominance in PAH had led to suspicion of sex hormone influence on pulmonary disease, but this association is incompletely characterized. In addition, estrogen appears to have some protective effects on RV function.191 Visceral adipose is a primary site of estrogen production, and estrogen modulates leptin production.192, 193 The influence of estrogen and sex hormones on development of PH-LHD requires further investigation, but seems worthwhile given the postulated role of loss of estrogen on development of HFpEF, pulmonary vascular disease, and right ventricular dysfunction.
Obesity and Metabolic Syndrome
Obesity is associated with the development of invasively diagnosed pulmonary hypertension, with risk of PH increasing in parallel with increasing BMI.194 Hemodynamic markers of PH such as mPAP, PAWP, and TPG increase with advancing classes of obesity. There is a higher prevalence of CPC-PH and IPC-PH with more advanced obesity classes. Unsupervised machine learning (phenomapping) of HFpEF patients has shown that the pheno-group of patients who had higher prevalence of obesity, diabetes mellitus, and obstructive sleep apnea had higher PVR.195 Metabolic syndrome (MetS) represents a milieu of physiologic and biochemical abnormalities including central obesity, glucose intolerance, insulin resistance, and dyslipidemia; the presence of which increases risk of development of cardiovascular disease and overt diabetes mellitus. Metabolic syndrome is highly prevalent in HFpEF but also occurs in up to 39% of patients with PAH.196
While inflammation has been quoted as the driver of obesity related PH-LHD, the mechanisms are complex and variable, and likely go beyond inflammation alone. Adiponectin is a fat-derived hormone that in animal models reduces intimal thickening in injured arteries and suppresses proliferation of smooth muscle cells.197, 198 Vascular effects of adiponectin are mediated by AMPK activation of eNOS which increases NO synthesis while anti-inflammatory effects stem from reduction of TNF-α (and subsequently interleukin-8) production by endothelial cells in response to injury.199–201 COX2 expression increases with higher levels of adiponectin which favors PGI2 vasodilatory and anti-proliferative effects and improving endothelial function in mouse models.202 Adiponectin levels are negatively correlated with BMI, more specifically visceral adipose, as opposed to leptin which increases with increasing BMI.203, 204 Adiponectin levels are lower in diabetic patients compared to non-diabetes, and among diabetics, those with coronary disease have less circulating adiponectin.205 Tumor necrosis factor-α (elevated in HF) inhibits adiponectin promoter activity and may therefore modifies plasma levels of adiponectin in systemic inflammatory states. Hypoadiponectinemia appears to be integral in the development of metabolic syndrome phenotype.206 Adiponectin and leptin have been implicated in the development of PH in MetS and obesity.
Leptin, a proinflammatory cytokine that is key in glycemic and lipid metabolism, plays a role on endothelial function and is a the product of a hypoxia-inducible factor-dependent gene.207, 208 Leptin normally has a vasodilatory effect; however, in vitro pulmonary endothelial cells in PAH have been shown to overproduce leptin, and PASMCs overexpress leptin’s primary receptor.209 The subsequent activity of leptin on PASMCs augments hypoxic driven proliferation and appears to be involvement in PAH development.209 Higher circulating leptin levels are also associated with increased risk of incident HF and disease progression.210–213 Patients with advanced HFrEF have elevated circulating leptin levels and soluble leptin receptor even when adjusted for BMI, although there is no data in humans describing differences between those with and without PH.214 Rodent models of PH-HFpEF have shown that elevated circulating leptin levels cause impairment of myocardial relaxation.74, 215 Natriuretic peptides appear to decrease the secretion of leptin in adipose tissue and suppress secretion of other inflammatory cytokines, including IL-6 and TNF-α.216
Atrial natriuretic peptide exerts anti-inflammatory effects by suppression of circulating markers of chronic inflammation, IL-6 and TNF-α.216 Circulating levels of these natriuretic peptides are known to be reduced in the obese HFpEF phenotype which also demonstrated more RV dysfunction and dilation, higher PAWP, and higher exercise pulmonary pressures.217 Patients with HFpEF have lower natriuretic peptide levels compared to those with HFrEF.218 Animal and human models of HFpEF-PH demonstrate upregulated activation of IL-6/signal transducer and activator of transcription 3 (STAT3) pathways in MetS-LHD, which exacerbates PH, with pulmonary vascular remodeling, macrophage infiltration, and IL-6/STAT3 upregulation in human lung tissue.74 Increased levels of IL-6 are also known to alter endothelial function and arterial stiffness.219 Treatment with metformin and an anti-IL-6 antibody in a rodent model of MetS-LHD induced regression of pulmonary hypertension implicating a role for multimodal suppression of the leptin/IL-6/STAT3 pathway as a promising target in PH-HFpEF.74
Metabolic syndrome and each of its independent components are associated with increased activity of neprilysin, which degrades natriuretic peptides, which reduces normal physiologic inhibition of aldosterone secretion by circulating ANP.220, 221 Obesity also results in increased aldosterone production and renin-angiotensin activation outside of ANP-mediated effects.222–224 The ultimate result of these factors is hyperaldosteronism, with sodium retention, insulin resistance, increased leptin expression, and an increase in inflammatory markers. 225–227 The renin-angiotensin-aldosterone system has a role in pulmonary vascular remodeling in PAH with elevated levels of angiotensin and aldosterone correlating with hemodynamics and pulmonary vascular remodeling through similar mechanisms implicated in heart disease.228 Pulmonary angiotensin-II levels and angiotensin-II type 1 receptor expression on myofibroblasts have been shown to be increased in post-MI rat models of PH-LHD.229 Aldosterone antagonism may have clinical benefit in patients with PAH and prevent pulmonary vascular remodeling in pre-clinical models.230, 231 Currently while pathologic changes in aldosterone in HF and PAH exist, the role of aldosterone in PH-LHD is less clear.
Splanchnic Circulation
Right-sided HF plays a significant role in prognosis in patients with LHD, regardless of underlying ejection fraction, and is significantly impacted by pulmonary vascular function.232–234 The systemic venous congestion in RV failure affects multiple other organ systems, including the kidneys, gastrointestinal tract, and liver. Splanchnic venous congestion caused by RV failure has multiple potential effects on the gastrointestinal tract with increases in systemic inflammation and alteration of sodium and phosphate hemostasis.235, 236 Venous congestion contributes to the development of cardiorenal syndrome, particularly driven by central venous pressure, and not PAWP or cardiac output.237 Renal dysfunction results in altered calcium and phosphate metabolism which causes increased stiffness of the pulmonary artery thereby increasing RV afterload.238 Renal dysfunction associated with splanchnic congestion causes dysregulation of mineral metabolism, specially calcium and phosphate homeostasis regulated by parathyroid hormone. A preclinical canine model of CKD showed and association between secondary hyperparathyroidism elevated pulmonary pressures and pulmonary vascular calcification.239 Interestingly, in this model prophylactic parathyroidectomy resulted in significantly lower mPAP and RV pressure despite the same degree of renal dysfunction. Central venous congestion-induced splanchnic congestion may result in upregulation of NHE3 in the gut (by creating an acidotic environment in enterocytes due to poor blood flow and increased anerobic metabolism), thereby enhancing sodium resorption and resulting in lower pH in the gut lumen. Reduced pH in the gut lumen, in turn, can (1) reduce short-chain fatty acids (reducing integrity of gut gap junctions and increasing gastrointestinal permeability, a risk factor for systemic infections) and (2) increase the population of bacteria that produce TMAO, which is known to increase inflammation.235, 238 Increased sodium resorption in the gut facilitates additional fluid retention and congestion, loading the central vasculature, raising intracardial filling pressures, and driving elevation in pulmonary pressures. These abnormalities may be reasons why patients with PH-LHD, particularly those with right-sided HF, are predisposed to adverse outcomes.
Systemic inflammation is a suspected feature of splanchnic congestion that contributes to HF and development of PH-LHD. Altered gut permeability due to edema allows bacterial translocation and systemic exposure to endotoxin.240 Decompensated HF patients have higher circulating levels of endotoxin than compensated, as well as higher concentrations of serum cytokines (CRP, TNF-α, and IL-6). Diuresis and relief of peripheral edema resulted in a significant reduction in circulating serum endotoxin levels, but cytokine levels remained elevated.240 Gut edema contributes to the systemic inflammatory milieu in decompensated HF. Many of the circulating inflammatory markers we have described have important effects on vascular biology and endothelial function that contribute to development of PH in HF.
The splanchnic circulation holds a large proportion of the total blood volume, acting as a reservoir of volume able to be mobilized by alterations in venous tone. Venous capacitance, the ability of the vessel to accommodate changes in volume with minimal changes in pressure, is reduced in HF, obesity, diabetes, and inflammatory states, leading to increased central blood volume when stressed.241–243 The volume loading of the central vascular compartment results in pulmonary and hemodynamic congestion, driving factors in development of PH and pulmonary vascular remodeling. Reduced PA capacitance in PH-LHD, which signals a lack of ability of the pulmonary vasculature to accommodate changes in volume loading, is associated with adverse outcomes.244 Coupled with sodium-driven plasma volume expansion due to upregulation of gastrointestinal NHE3, the splanchnic circulation appears to play an integral role in the pathogenesis of PH-LHD. In patients with PH-LHD due to HFpEF, levosimendan (a calcium sensitizer and KATP channel opener) was associated with improved hemodynamics and exercise capacity compared with placebo.245 Although levosimendan is known to increase contractility, it was reduction of the stressed blood volume via splanchnic vasodilation that appeared to be the underlying cause for improvement.246 Given these encouraging results, further investigation of the effects splanchnic vasoconstriction on the development and progression of PH-LHD in animal models and humans is therefore warranted.
Lymphatic Dysfunction
The lymphatic system controls extracellular fluid volume, holding and transporting up to 8 liters per day, of which approximately 1.5L/day travels through the thoracic duct to the left subclavian vein.247 This function is possible through both intrinsic factors, the pumping function of the lymphangion via smooth muscle, and extrinsic factors such as the respiratory cycle, skeletal muscle contraction, and surrounding blood vessel pulsations.247 In HF, elevated central venous pressure can impede outflow from the thoracic duct, thereby increasing hydrostatic pressure and ultimately resulting in increased extracellular fluid.248 Recent data indicates reduced lymphatic reserve as a byproduct of microvascular disease and capillary rarefaction accompanied by dilated, dysfunctional lymphatics in HFpEF.249
Lymphatics may play a role in the acuity of HF and the development of pulmonary edema, as patients with longstanding HF tolerate higher filling pressures without development of pulmonary edema even as PH progresses. Hemodynamic congestion results in more equally distributed blood flow through the lung fields through capillary recruitment.250 The thoracic lymphatics act to buffer the lungs from elevated intracardiac filling pressures by accommodating extracellular fluid influx.251 Lymphatic vessels in the pulmonary connective tissues and pleural space can accommodate fluid removal as a high capacitance reservoir with up to ten-fold increase in volume chronically.252 In PH-LHD, where left atrial pressure is one of the primary drivers of development of pulmonary hypertension, the capacity of the lymphatic system to act as a reservoir for excess volume serves to mitigate to some degree the hydrostatic pressure that drives PH.253 When maximal lymphatic efflux rate is overwhelmed, both fluid and protein stalls in the interstitial compartment leading to ongoing edema development.254, 255 Patients with HFpEF have lower numbers of lymphatic vessels with larger diameters although despite this drainage was impaired.249 The lymphatic system contributes to pulmonary vascular hemodynamics as a reservoir for volume and pressure. In response to chronic congestion lymphatics muscularize and hypertrophy similar to the pulmonary vascular bed.251, 252 VEGF-D, which is elevated in chronic HF, has strong lymphangiogenic and angiogenic functions. 176 The potential implications of this is important as increased circulating VEGF-D in chronic HF may, in part, be adaptive to drive the development of a larger lymphatic reservoir.256 These changes indicate that the lymphatic system may play a critical role in the development and progression of PH-LHD given the role of lymphatic system in buffering congestion, and therefore should be studied more extensively in the pre-clinical and clinical settings.
FUTURE DIRECTIONS
Large gaps knowledge gaps exist in our understanding of PH-LHD, starting with accurate diagnostic criteria and rational pathophysiological and pathobiological disease classification. The most basic requirement for improved understanding PH-LHD is to establish a widely accepted, reproducible definition of disease, especially with provocative maneuvers. Determination of the impact of chronicity and severity of LHD as well as genetic, epigenetic, and inherited factors on risk for development of Cpc-PH versus Ipc-PH is also critical. Understanding the interplay between chronic inflammation in association with metabolic syndrome, insulin resistance, and the imbalance of vasoconstrictors and vasodilators in the pulmonary vascular bed is essential to finding intervenable pathways in these disease processes. Given the high prevalence, morbidity, and mortality of PH-LHD, it is essential that we continue to push the field forward in basic, translational, clinical, and epidemiological research so that we can identify patients at risk for PH-LHD, diagnose it early and accurately, treat it, and prevent progression of disease. Table 3 outlines important unanswered questions for improved understanding of PH-LHD and the underlying pathobiology which drives disease progression, which we hope will serve as a stimulus to the scientific community.
Table 3.
Pulmonary Hypertension due to Left Heart Disease: Unanswered Questions
| 1 | Which provocative maneuver is most specific for the diagnosis of PH-LHD and what are the optimal diagnostic thresholds indicative of pulmonary vascular disease? |
| 2 | How does left atrial dysfunction lead to development of PH-LHD? |
| 3 | Is Cpc-PH a further progressed and more severe form of Ipc-PH or a different disease entity with specific risk factors? |
| 4 | At which point in the adaptive response to shear stress does intracellular signaling and pulmonary vascular remodeling become pathologic? What are the biologic drivers that differ between adaptive and maladaptive remodeling? |
| 5 | How does platelet aggregation and activation impact pulmonary vascular function, and which drivers of platelet activation are dominant in hemodynamic congestion? |
| 6 | In metabolic syndrome, what features are most influential in the development of PH in the setting of LHD? How does the presence of insulin resistance, chronic inflammation, and changes in natriuretic peptides influence congestion? |
| 7 | Does increased circulating adiponectin modify endothelial function in PH-LHD? How does adiponectin effect the pulmonary vasculature? Are there other adipokines that are relevant to the development and progression of PH-LHD. |
| 8 | What are the roles of venous capacitance, peripheral venous dysfunction, and stress blood volume in development and progression of PH-LHD? What are the molecular drivers of splanchnic vasoconstriction, and can those pathways be targeted with specific treatments? |
| 9 | How does lymphatic dysfunction impact different degrees of congestion and the ability of the pulmonary vasculature and RV to accommodate congestive stress? |
| 10 | What are the ideal small and large animal models for studying PH-LHD and its various phenotypes? |
CONCLUSIONS
There are multiple phenotypes of Group 2 PH, which likely have significant variation in metabolic, hemodynamic, and inflammatory derangements. Hemodynamically, patients with Ipc-PH and Cpc-PH vary by severity of pulmonary vascular disease potentially driven by underlying wall stress and injury related to both the chronicity of exposure to LA hypertension over time and superimposed PAH risk factors. The current understanding of the pathobiology of PH-LHD is very limited. While endothelial dysfunction, obesity/metabolic syndrome, splanchnic vasoconstriction, and lymphatic dysfunction may all play a role, phenotypes vary, and the primary drivers of disease have not been fully identified. Furthermore, there are no direct treatments of PH-LHD, with a track record of multiple failed trials of pulmonary vasodilators. Improved understanding of the drivers of PASMC homeostasis and the biologic drivers of disease is essential to intervening in this disease process, whether via prevention or for amelioration.
FUNDING SOURCES
SJS is supported by research grants from the National Institutes of Health (U54 HL160273, R01 HL107577, R01 HL140731, and R01 HL149423).
Nonstandard Abbreviations and Acronyms:
- PH
pulmonary hypertension
- LHD
left heart disease
- HF
heart failure
- HFpEF
heart failure with preserved ejection fraction
- HFrEF
heart failure with reduced ejection fraction
- LVEF
left ventricular ejection fraction
- HFmrEF
Heart failure with mid-range ejection fraction
- PA
pulmonary artery
- mPAP
mean pulmonary artery pressure
- PVR
pulmonary vascular resistance
- Cpc-PH
combined pre and post capillary pulmonary hypertension
- Ipc-PH
isolated post capillary pulmonary hypertension
- PAWP
pulmonary artery wedge pressure
- LVEDP
left ventricular end diastolic pressure
- LA
left atrium
- LV
left ventricle
- RV
right ventricle
- DPG
diastolic pressure gradient
- TPG
transpulmonary gradient
- PAH
pulmonary arterial hypertension
- TNF-α
tumor necrosis factor alpha
- LVAD
left ventricular assist device
- CPA
pulmonary arterial compliance
- SNP
single nucleotide polymorphisms
- ZSF1
Zucker
- SU5416
Sugen 5416
- NO
nitric oxide
- PASMC
pulmonary artery smooth muscle cells
- eNOS
Endothelial nitric oxide synthase
- NADPH
nicotinamide adenine dinucleotide phosphate
- ET-1
endothelin-1
- ETA
endothelin receptor A
- ETB
endothelin receptor B
- ERAs
endothelin receptor antagonists
- PGI2
prostaglandin I2
- TXA2
thromboxane-A2
- COX
cyclo-oxygenase
- 5-HT
serotonin
- ATP
adenosine triphosphate
- IL-6
interleukin-6
- CRP
C-reactive protein
- VEGF
vascular endothelial growth factor
- MetS
Metabolic syndrome
- AMPK
adenosine monophosphate protein kinase
- BMI
body mass index
- STAT-3
signal transducer and activator of transcription-3
- CKD
chronic kidney disease
- NHE3
sodium-hydrogen antiporter 3
- TMAO
trimethylamine N-oxide
- KATP
adenosine triphosphate sensitive potassium channel
- RF
risk factors
- PVOD
pulmonary veno-occlusive disease
- PASP
pulmonary artery systolic pressure
- IR
insulin receptor
- TPR
thromboxane A2 receptor
- 5-HTR
serotonin receptor
Footnotes
DISCLOSURES
SJS has received research grants from Actelion, AstraZeneca, Corvia, Novartis, and Pfizer; and consulting fees from Abbott, Actelion, AstraZeneca, Amgen, Aria CV, Axon Therapies, Bayer, Boehringer-Ingelheim, Boston Scientific, Bristol-Myers Squibb, Cardiora, Coridea, CVRx, Cyclerion, Cytokinetics, Edwards Lifesciences, Eidos, Eisai, Imara, Impulse Dynamics, Intellia, Ionis, Ironwood, Lilly, Merck, MyoKardia, Novartis, Novo Nordisk, Pfizer, Prothena, Regeneron, Rivus, Sanofi, Shifamed, Tenax, Tenaya, and United Therapeutics.
REFERENCES
- 1.Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Cheng S, Delling FN, et al. American Heart Association Council on E, Prevention Statistics C and Stroke Statistics S. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation. 2021;143:e254–e743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heidenreich PA, Albert NM, Allen LA, Bluemke DA, Butler J, Fonarow GC, Ikonomidis JS, Khavjou O, Konstam MA, Maddox TM, et al. Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart Fail. 2013;6:606–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bragazzi NL, Zhong W, Shu J, Abu Much A, Lotan D, Grupper A, Younis A and Dai H. Burden of heart failure and underlying causes in 195 countries and territories from 1990 to 2017. Eur J Prev Cardiol 2021. [DOI] [PubMed] [Google Scholar]
- 4.Bozkurt B, Hershberger RE, Butler J, Grady KL, Heidenreich PA, Isler ML, Kirklin JK and Weintraub WS. 2021 ACC/AHA Key Data Elements and Definitions for Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Data Standards (Writing Committee to Develop Clinical Data Standards for Heart Failure). Circ Cardiovasc Qual Outcomes. 2021;14:e000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steinberg BA, Zhao X, Heidenreich PA, Peterson ED, Bhatt DL, Cannon CP, Hernandez AF, Fonarow GC, Get With the Guidelines Scientific Advisory C and Investigators. Trends in patients hospitalized with heart failure and preserved left ventricular ejection fraction: prevalence, therapies, and outcomes. Circulation. 2012;126:65–75. [DOI] [PubMed] [Google Scholar]
- 6.Vasan RS, Larson MG, Benjamin EJ, Evans JC, Reiss CK and Levy D. Congestive heart failure in subjects with normal versus reduced left ventricular ejection fraction: prevalence and mortality in a population-based cohort. J Am Coll Cardiol. 1999;33:1948–55. [DOI] [PubMed] [Google Scholar]
- 7.Kitzman DW, Gardin JM, Gottdiener JS, Arnold A, Boineau R, Aurigemma G, Marino EK, Lyles M, Cushman M, Enright PL, et al. Importance of heart failure with preserved systolic function in patients > or = 65 years of age. CHS Research Group. Cardiovascular Health Study. Am J Cardiol 2001;87:413–9. [DOI] [PubMed] [Google Scholar]
- 8.Paulus WJ, Tschope C, Sanderson JE, Rusconi C, Flachskampf FA, Rademakers FE, Marino P, Smiseth OA, De Keulenaer G, Leite-Moreira AF, et al. How to diagnose diastolic heart failure: a consensus statement on the diagnosis of heart failure with normal left ventricular ejection fraction by the Heart Failure and Echocardiography Associations of the European Society of Cardiology. Eur Heart J. 2007;28:2539–50. [DOI] [PubMed] [Google Scholar]
- 9.Pfeffer MA, Shah AM and Borlaug BA. Heart Failure With Preserved Ejection Fraction In Perspective. Circ Res. 2019;124:1598–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shah SJ, Borlaug BA, Kitzman DW, McCulloch AD, Blaxall BC, Agarwal R, Chirinos JA, Collins S, Deo RC, Gladwin MT, et al. Research Priorities for Heart Failure With Preserved Ejection Fraction: National Heart, Lung, and Blood Institute Working Group Summary. Circulation. 2020;141:1001–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khan MS, Sreenivasan J, Lateef N, Abougergi MS, Greene SJ, Ahmad T, Anker SD, Fonarow GC and Butler J. Trends in 30- and 90-Day Readmission Rates for Heart Failure. Circ Heart Fail. 2021;14:e008335. [DOI] [PubMed] [Google Scholar]
- 12.van Walraven C, Jennings A and Forster AJ. A meta-analysis of hospital 30-day avoidable readmission rates. J Eval Clin Pract. 2012;18:1211–8. [DOI] [PubMed] [Google Scholar]
- 13.Cheng RK, Cox M, Neely ML, Heidenreich PA, Bhatt DL, Eapen ZJ, Hernandez AF, Butler J, Yancy CW and Fonarow GC. Outcomes in patients with heart failure with preserved, borderline, and reduced ejection fraction in the Medicare population. Am Heart J. 2014;168:721–30. [DOI] [PubMed] [Google Scholar]
- 14.Dzudie A, Kengne AP, Thienemann F and Sliwa K. Predictors of hospitalisations for heart failure and mortality in patients with pulmonary hypertension associated with left heart disease: a systematic review. BMJ Open. 2014;4:e004843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iorio A, Senni M, Barbati G, Greene SJ, Poli S, Zambon E, Di Nora C, Cioffi G, Tarantini L, Gavazzi A, et al. Prevalence and prognostic impact of non-cardiac co-morbidities in heart failure outpatients with preserved and reduced ejection fraction: a community-based study. Eur J Heart Fail. 2018;20:1257–1266. [DOI] [PubMed] [Google Scholar]
- 16.Miller WL, Grill DE and Borlaug BA. Clinical features, hemodynamics, and outcomes of pulmonary hypertension due to chronic heart failure with reduced ejection fraction: pulmonary hypertension and heart failure. JACC Heart Fail. 2013;1:290–299. [DOI] [PubMed] [Google Scholar]
- 17.Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG and Souza R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kovacs G, Berghold A, Scheidl S and Olschewski H. Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J. 2009;34:888–94. [DOI] [PubMed] [Google Scholar]
- 19.Mascherbauer J, Zotter-Tufaro C, Duca F, Binder C, Koschutnik M, Kammerlander AA, Aschauer S and Bonderman D. Wedge Pressure Rather Than Left Ventricular End-Diastolic Pressure Predicts Outcome in Heart Failure With Preserved Ejection Fraction. JACC Heart Fail. 2017;5:795–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halpern SD and Taichman DB. Misclassification of pulmonary hypertension due to reliance on pulmonary capillary wedge pressure rather than left ventricular end-diastolic pressure. Chest. 2009;136:37–43. [DOI] [PubMed] [Google Scholar]
- 21.Dickinson MG, Lam CS, Rienstra M, Vonck TE, Hummel YM, Voors AA and Hoendermis ES. Atrial fibrillation modifies the association between pulmonary artery wedge pressure and left ventricular end-diastolic pressure. Eur J Heart Fail. 2017;19:1483–1490. [DOI] [PubMed] [Google Scholar]
- 22.Bitar A, Selej M, Bolad I and Lahm T. Poor agreement between pulmonary capillary wedge pressure and left ventricular end-diastolic pressure in a veteran population. PLoS One. 2014;9:e87304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reddy YNV, El-Sabbagh A and Nishimura RA. Comparing Pulmonary Arterial Wedge Pressure and Left Ventricular End Diastolic Pressure for Assessment of Left-Sided Filling Pressures. JAMA Cardiol. 2018;3:453–454. [DOI] [PubMed] [Google Scholar]
- 24.Melenovsky V, Hwang SJ, Redfield MM, Zakeri R, Lin G and Borlaug BA. Left atrial remodeling and function in advanced heart failure with preserved or reduced ejection fraction. Circ Heart Fail. 2015;8:295–303. [DOI] [PubMed] [Google Scholar]
- 25.Okura H, Kataoka T and Yoshida K. Comparison of Left Ventricular Relaxation and Left Atrial Function in Patients With Heart Failure and Preserved Ejection Fraction Versus Patients With Systemic Hypertension and Healthy Subjects. Am J Cardiol. 2016;118:1019–23. [DOI] [PubMed] [Google Scholar]
- 26.Jin X, Nauta JF, Hung CL, Ouwerkerk W, Teng TK, Voors AA, Lam CS and van Melle JP. Left atrial structure and function in heart failure with reduced (HFrEF) versus preserved ejection fraction (HFpEF): systematic review and meta-analysis. Heart Fail Rev 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Santos AB, Roca GQ, Claggett B, Sweitzer NK, Shah SJ, Anand IS, Fang JC, Zile MR, Pitt B, Solomon SD, et al. Prognostic Relevance of Left Atrial Dysfunction in Heart Failure With Preserved Ejection Fraction. Circ Heart Fail. 2016;9:e002763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas L, Muraru D, Popescu BA, Sitges M, Rosca M, Pedrizzetti G, Henein MY, Donal E and Badano LP. Evaluation of Left Atrial Size and Function: Relevance for Clinical Practice. J Am Soc Echocardiogr. 2020;33:934–952. [DOI] [PubMed] [Google Scholar]
- 29.Sanchis L, Gabrielli L, Andrea R, Falces C, Duchateau N, Perez-Villa F, Bijnens B and Sitges M. Left atrial dysfunction relates to symptom onset in patients with heart failure and preserved left ventricular ejection fraction. Eur Heart J Cardiovasc Imaging. 2015;16:62–7. [DOI] [PubMed] [Google Scholar]
- 30.Wang YC, Lin JL, Hwang JJ, Lin MS, Tseng CD, Huang SK and Lai LP. Left atrial dysfunction in patients with atrial fibrillation after successful rhythm control for > 3 months. Chest. 2005;128:2551–6. [DOI] [PubMed] [Google Scholar]
- 31.Goyal P, Almarzooq ZI, Cheung J, Kamel H, Krishnan U, Feldman DN, Horn EM and Kim LK. Atrial fibrillation and heart failure with preserved ejection fraction: Insights on a unique clinical phenotype from a nationally-representative United States cohort. Int J Cardiol. 2018;266:112–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melenovsky V, Kotrc M, Borlaug BA, Marek T, Kovar J, Malek I and Kautzner J. Relationships between right ventricular function, body composition, and prognosis in advanced heart failure. J Am Coll Cardiol. 2013;62:1660–1670. [DOI] [PubMed] [Google Scholar]
- 33.Marrouche NF, Brachmann J, Andresen D, Siebels J, Boersma L, Jordaens L, Merkely B, Pokushalov E, Sanders P, Proff J, et al. Catheter Ablation for Atrial Fibrillation with Heart Failure. N Engl J Med. 2018;378:417–427. [DOI] [PubMed] [Google Scholar]
- 34.Black-Maier E, Ren X, Steinberg BA, Green CL, Barnett AS, Rosa NS, Al-Khatib SM, Atwater BD, Daubert JP, Frazier-Mills C, et al. Catheter ablation of atrial fibrillation in patients with heart failure and preserved ejection fraction. Heart Rhythm. 2018;15:651–657. [DOI] [PubMed] [Google Scholar]
- 35.Packer DL, Piccini JP, Monahan KH, Al-Khalidi HR, Silverstein AP, Noseworthy PA, Poole JE, Bahnson TD, Lee KL, Mark DB, et al. Ablation Versus Drug Therapy for Atrial Fibrillation in Heart Failure: Results From the CABANA Trial. Circulation. 2021;143:1377–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Costard-Jackle A and Fowler MB. Influence of preoperative pulmonary artery pressure on mortality after heart transplantation: testing of potential reversibility of pulmonary hypertension with nitroprusside is useful in defining a high risk group. J Am Coll Cardiol. 1992;19:48–54. [DOI] [PubMed] [Google Scholar]
- 37.Mehra MR, Canter CE, Hannan MM, Semigran MJ, Uber PA, Baran DA, Danziger-Isakov L, Kirklin JK, Kirk R, Kushwaha SS, et al. The 2016 International Society for Heart Lung Transplantation listing criteria for heart transplantation: A 10-year update. J Heart Lung Transplant. 2016;35:1–23. [DOI] [PubMed] [Google Scholar]
- 38.Naeije R, Vachiery JL, Yerly P and Vanderpool R. The transpulmonary pressure gradient for the diagnosis of pulmonary vascular disease. Eur Respir J. 2013;41:217–23. [DOI] [PubMed] [Google Scholar]
- 39.Girerd N, Seronde MF, Coiro S, Chouihed T, Bilbault P, Braun F, Kenizou D, Maillier B, Nazeyrollas P, Roul G, et al. Integrative Assessment of Congestion in Heart Failure Throughout the Patient Journey. JACC Heart Fail. 2018;6:273–285. [DOI] [PubMed] [Google Scholar]
- 40.Assad TR, Hemnes AR, Larkin EK, Glazer AM, Xu M, Wells QS, Farber-Eger EH, Sheng Q, Shyr Y, Harrell FE, et al. Clinical and Biological Insights Into Combined Post- and Pre-Capillary Pulmonary Hypertension. J Am Coll Cardiol. 2016;68:2525–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Assad TR, Brittain EL, Wells QS, Farber-Eger EH, Halliday SJ, Doss LN, Xu M, Wang L, Harrell FE, Yu C, et al. Hemodynamic evidence of vascular remodeling in combined post- and precapillary pulmonary hypertension. Pulm Circ. 2016;6:313–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mohammed SF, Borlaug BA, Roger VL, Mirzoyev SA, Rodeheffer RJ, Chirinos JA and Redfield MM. Comorbidity and ventricular and vascular structure and function in heart failure with preserved ejection fraction: a community-based study. Circ Heart Fail 2012;5:710–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gami AS, Hodge DO, Herges RM, Olson EJ, Nykodym J, Kara T and Somers VK. Obstructive sleep apnea, obesity, and the risk of incident atrial fibrillation. J Am Coll Cardiol. 2007;49:565–71. [DOI] [PubMed] [Google Scholar]
- 44.Fuchs FD and Whelton PK. High Blood Pressure and Cardiovascular Disease. Hypertension. 2020;75:285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gottdiener JS, Kitzman DW, Aurigemma GP, Arnold AM and Manolio TA. Left atrial volume, geometry, and function in systolic and diastolic heart failure of persons > or =65 years of age (the cardiovascular health study). Am J Cardiol 2006;97:83–9. [DOI] [PubMed] [Google Scholar]
- 46.Freed BH, Daruwalla V, Cheng JY, Aguilar FG, Beussink L, Choi A, Klein DA, Dixon D, Baldridge A, Rasmussen-Torvik LJ, et al. Prognostic Utility and Clinical Significance of Cardiac Mechanics in Heart Failure With Preserved Ejection Fraction: Importance of Left Atrial Strain. Circ Cardiovasc Imaging. 2016;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stevens GR, Garcia-Alvarez A, Sahni S, Garcia MJ, Fuster V and Sanz J. RV dysfunction in pulmonary hypertension is independently related to pulmonary artery stiffness. JACC Cardiovasc Imaging. 2012;5:378–87. [DOI] [PubMed] [Google Scholar]
- 48.Tedford RJ, Hassoun PM, Mathai SC, Girgis RE, Russell SD, Thiemann DR, Cingolani OH, Mudd JO, Borlaug BA, Redfield MM, et al. Pulmonary capillary wedge pressure augments right ventricular pulsatile loading. Circulation. 2012;125:289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ooi CY, Wang Z, Tabima DM, Eickhoff JC and Chesler NC. The role of collagen in extralobar pulmonary artery stiffening in response to hypoxia-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2010;299:H1823–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hunt JM, Bethea B, Liu X, Gandjeva A, Mammen PP, Stacher E, Gandjeva MR, Parish E, Perez M, Smith L, et al. Pulmonary veins in the normal lung and pulmonary hypertension due to left heart disease. Am J Physiol Lung Cell Mol Physiol. 2013;305:L725–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dayeh NR, Ledoux J and Dupuis J. Lung Capillary Stress Failure and Arteriolar Remodelling in Pulmonary Hypertension Associated with Left Heart Disease (Group 2 PH). Prog Cardiovasc Dis. 2016;59:11–21. [DOI] [PubMed] [Google Scholar]
- 52.Conforti E, Fenoglio C, Bernocchi G, Bruschi O and Miserocchi GA. Morpho-functional analysis of lung tissue in mild interstitial edema. Am J Physiol Lung Cell Mol Physiol. 2002;282:L766–74. [DOI] [PubMed] [Google Scholar]
- 53.Guazzi M and Naeije R. Pulmonary Hypertension in Heart Failure: Pathophysiology, Pathobiology, and Emerging Clinical Perspectives. J Am Coll Cardiol. 2017;69:1718–1734. [DOI] [PubMed] [Google Scholar]
- 54.De Pasquale CG, Arnolda LF, Doyle IR, Aylward PE, Chew DP and Bersten AD. Plasma surfactant protein-B: a novel biomarker in chronic heart failure. Circulation. 2004;110:1091–6. [DOI] [PubMed] [Google Scholar]
- 55.Hermans C and Bernard A. Lung epithelium-specific proteins: characteristics and potential applications as markers. Am J Respir Crit Care Med. 1999;159:646–78. [DOI] [PubMed] [Google Scholar]
- 56.De Pasquale CG, Arnolda LF, Doyle IR, Grant RL, Aylward PE and Bersten AD. Prolonged alveolocapillary barrier damage after acute cardiogenic pulmonary edema. Crit Care Med. 2003;31:1060–7. [DOI] [PubMed] [Google Scholar]
- 57.Parker F and Weiss S. The Nature and Significance of the Structural Changes in the Lungs in Mitral Stenosis. Am J Pathol. 1936;12:573–598 15. [PMC free article] [PubMed] [Google Scholar]
- 58.Naeije R, Gerges M, Vachiery JL, Caravita S, Gerges C and Lang IM. Hemodynamic Phenotyping of Pulmonary Hypertension in Left Heart Failure. Circ Heart Fail. 2017;10. [DOI] [PubMed] [Google Scholar]
- 59.Delgado JF, Conde E, Sanchez V, Lopez-Rios F, Gomez-Sanchez MA, Escribano P, Sotelo T, Gomez de la Camara A, Cortina J and de la Calzada CS. Pulmonary vascular remodeling in pulmonary hypertension due to chronic heart failure. Eur J Heart Fail. 2005;7:1011–6. [DOI] [PubMed] [Google Scholar]
- 60.Fayyaz AU, Edwards WD, Maleszewski JJ, Konik EA, DuBrock HM, Borlaug BA, Frantz RP, Jenkins SM and Redfield MM. Global Pulmonary Vascular Remodeling in Pulmonary Hypertension Associated With Heart Failure and Preserved or Reduced Ejection Fraction. Circulation. 2018;137:1796–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsukimoto K, Mathieu-Costello O, Prediletto R, Elliott AR and West JB. Ultrastructural appearances of pulmonary capillaries at high transmural pressures. J Appl Physiol (1985). 1991;71:573–82. [DOI] [PubMed] [Google Scholar]
- 62.West JB and Mathieu-Costello O. Vulnerability of pulmonary capillaries in heart disease. Circulation. 1995;92:622–31. [DOI] [PubMed] [Google Scholar]
- 63.Zakeri R, Moulay G, Chai Q, Ogut O, Hussain S, Takahama H, Lu T, Wang XL, Linke WA, Lee HC, et al. Left Atrial Remodeling and Atrioventricular Coupling in a Canine Model of Early Heart Failure With Preserved Ejection Fraction. Circ Heart Fail. 2016;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guazzi M and Borlaug BA. Pulmonary hypertension due to left heart disease. Circulation. 2012;126:975–90. [DOI] [PubMed] [Google Scholar]
- 65.Lai YC, Tabima DM, Dube JJ, Hughan KS, Vanderpool RR, Goncharov DA, St Croix CM, Garcia-Ocana A, Goncharova EA, Tofovic SP, et al. SIRT3-AMP-Activated Protein Kinase Activation by Nitrite and Metformin Improves Hyperglycemia and Normalizes Pulmonary Hypertension Associated With Heart Failure With Preserved Ejection Fraction. Circulation. 2016;133:717–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meng Q, Lai YC, Kelly NJ, Bueno M, Baust JJ, Bachman TN, Goncharov D, Vanderpool RR, Radder JE, Hu J, Goncharova E, et al. Development of a Mouse Model of Metabolic Syndrome, Pulmonary Hypertension, and Heart Failure with Preserved Ejection Fraction. Am J Respir Cell Mol Biol. 2017;56:497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ciuclan L, Bonneau O, Hussey M, Duggan N, Holmes AM, Good R, Stringer R, Jones P, Morrell NW, Jarai G, Walker C, Westwick J, et al. A novel murine model of severe pulmonary arterial hypertension. Am J Respir Crit Care Med. 2011;184:1171–82. [DOI] [PubMed] [Google Scholar]
- 68.Hentschel T, Yin N, Riad A, Habbazettl H, Weimann J, Koster A, Tschope C, Kuppe H and Kuebler WM. Inhalation of the phosphodiesterase-3 inhibitor milrinone attenuates pulmonary hypertension in a rat model of congestive heart failure. Anesthesiology. 2007;106:124–31. [DOI] [PubMed] [Google Scholar]
- 69.Chen Y, Guo H, Xu D, Xu X, Wang H, Hu X, Lu Z, Kwak D, Xu Y, Gunther R, et al. Left ventricular failure produces profound lung remodeling and pulmonary hypertension in mice: heart failure causes severe lung disease. Hypertension. 2012;59:1170–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pereda D, Garcia-Alvarez A, Sanchez-Quintana D, Nuno M, Fernandez-Friera L, Fernandez-Jimenez R, Garcia-Ruiz JM, Sandoval E, Aguero J, Castella M, et al. Swine model of chronic postcapillary pulmonary hypertension with right ventricular remodeling: long-term characterization by cardiac catheterization, magnetic resonance, and pathology. J Cardiovasc Transl Res. 2014;7:494–506. [DOI] [PubMed] [Google Scholar]
- 71.Fujimoto Y, Urashima T, Kawachi F, Akaike T, Kusakari Y, Ida H and Minamisawa S. Pulmonary hypertension due to left heart disease causes intrapulmonary venous arterialization in rats. J Thorac Cardiovasc Surg. 2017;154:1742–1753 e8. [DOI] [PubMed] [Google Scholar]
- 72.Xiong PY, Baba S, Nishioka N, Fujimoto Y, Archer SL and Minamisawa S. Left Atrial Stenosis Induced Pulmonary Venous Arterialization and Group 2 Pulmonary Hypertension in Rat. J Vis Exp. 2018. [DOI] [PubMed] [Google Scholar]
- 73.Nguyen QT, Nsaibia MJ, Sirois MG, Calderone A, Tardif JC, Fen Shi Y, Ruiz M, Daneault C, Gagnon L, Grouix B, et al. PBI-4050 reduces pulmonary hypertension, lung fibrosis, and right ventricular dysfunction in heart failure. Cardiovasc Res. 2020;116:171–182. [DOI] [PubMed] [Google Scholar]
- 74.Ranchoux B, Nadeau V, Bourgeois A, Provencher S, Tremblay E, Omura J, Cote N, Abu-Alhayja’a R, Dumais V, Nachbar RT, et al. Metabolic Syndrome Exacerbates Pulmonary Hypertension due to Left Heart Disease. Circ Res. 2019;125:449–466. [DOI] [PubMed] [Google Scholar]
- 75.Krafsur GM, Neary JM, Garry F, Holt T, Gould DH, Mason GL, Thomas MG, Enns RM, Tuder RM, Heaton MP, et al. Cardiopulmonary remodeling in fattened beef cattle: a naturally occurring large animal model of obesity-associated pulmonary hypertension with left heart disease. Pulm Circ. 2019;9:2045894018796804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dai ZK, Tan MS, Chai CY, Yeh JL, Chou SH, Chiu CC, Jeng AY, Chen IJ and Wu JR. Upregulation of endothelial nitric oxide synthase and endothelin-1 in pulmonary hypertension secondary to heart failure in aorta-banded rats. Pediatr Pulmonol. 2004;37:249–56. [DOI] [PubMed] [Google Scholar]
- 77.Yin J, Kukucka M, Hoffmann J, Sterner-Kock A, Burhenne J, Haefeli WE, Kuppe H and Kuebler WM. Sildenafil preserves lung endothelial function and prevents pulmonary vascular remodeling in a rat model of diastolic heart failure. Circ Heart Fail. 2011;4:198–206. [DOI] [PubMed] [Google Scholar]
- 78.Yin N, Kaestle S, Yin J, Hentschel T, Pries AR, Kuppe H and Kuebler WM. Inhaled nitric oxide versus aerosolized iloprost for the treatment of pulmonary hypertension with left heart disease. Crit Care Med. 2009;37:980–6. [DOI] [PubMed] [Google Scholar]
- 79.Wallner M, Eaton DM, Berretta RM, Borghetti G, Wu J, Baker ST, Feldsott EA, Sharp TE 3rd, Mohsin S, Oyama MA, et al. A Feline HFpEF Model with Pulmonary Hypertension and Compromised Pulmonary Function. Sci Rep. 2017;7:16587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang H, Kwak D, Fassett J, Hou L, Xu X, Burbach BJ, Thenappan T, Xu Y, Ge JB, Shimizu Y, et al. CD28/B7 Deficiency Attenuates Systolic Overload-Induced Congestive Heart Failure, Myocardial and Pulmonary Inflammation, and Activated T Cell Accumulation in the Heart and Lungs. Hypertension. 2016;68:688–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.LaBourene JI, Coles JG, Johnson DJ, Mehra A, Keeley FW and Rabinovitch M. Alterations in elastin and collagen related to the mechanism of progressive pulmonary venous obstruction in a piglet model. A hemodynamic, ultrastructural, and biochemical study. Circ Res. 1990;66:438–56. [DOI] [PubMed] [Google Scholar]
- 82.Mulder P, Richard V, Derumeaux G, Hogie M, Henry JP, Lallemand F, Compagnon P, Mace B, Comoy E, Letac B, et al. Role of endogenous endothelin in chronic heart failure: effect of long-term treatment with an endothelin antagonist on survival, hemodynamics, and cardiac remodeling. Circulation. 1997;96:1976–82. [DOI] [PubMed] [Google Scholar]
- 83.Houweling B, Merkus D, Sorop O, Boomsma F and Duncker DJ. Role of endothelin receptor activation in secondary pulmonary hypertension in awake swine after myocardial infarction. J Physiol. 2006;574:615–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ravi Y, Selvendiran K, Naidu SK, Meduru S, Citro LA, Bognar B, Khan M, Kalai T, Hideg K, Kuppusamy P, et al.Pulmonary hypertension secondary to left-heart failure involves peroxynitrite-induced downregulation of PTEN in the lung. Hypertension. 2013;61:593–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dayeh NR, Tardif JC, Shi Y, Tanguay M, Ledoux J and Dupuis J. Echocardiographic validation of pulmonary hypertension due to heart failure with reduced ejection fraction in mice. Sci Rep. 2018;8:1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Philip JL, Murphy TM, Schreier DA, Stevens S, Tabima DM, Albrecht M, Frump AL, Hacker TA, Lahm T and Chesler NC. Pulmonary vascular mechanical consequences of ischemic heart failure and implications for right ventricular function. Am J Physiol Heart Circ Physiol. 2019;316:H1167–H1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Budhiraja R, Tuder RM and Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004;109:159–65. [DOI] [PubMed] [Google Scholar]
- 88.Katz SD, Hryniewicz K, Hriljac I, Balidemaj K, Dimayuga C, Hudaihed A and Yasskiy A. Vascular endothelial dysfunction and mortality risk in patients with chronic heart failure. Circulation. 2005;111:310–4. [DOI] [PubMed] [Google Scholar]
- 89.Ontkean M, Gay R and Greenberg B. Diminished endothelium-derived relaxing factor activity in an experimental model of chronic heart failure. Circ Res. 1991;69:1088–96. [DOI] [PubMed] [Google Scholar]
- 90.Ben Driss A, Devaux C, Henrion D, Duriez M, Thuillez C, Levy BI and Michel JB. Hemodynamic stresses induce endothelial dysfunction and remodeling of pulmonary artery in experimental compensated heart failure. Circulation. 2000;101:2764–70. [DOI] [PubMed] [Google Scholar]
- 91.Kerem A, Yin J, Kaestle SM, Hoffmann J, Schoene AM, Singh B, Kuppe H, Borst MM and Kuebler WM. Lung endothelial dysfunction in congestive heart failure: role of impaired Ca2+ signaling and cytoskeletal reorganization. Circ Res. 2010;106:1103–16. [DOI] [PubMed] [Google Scholar]
- 92.Traub O and Berk BC. Laminar shear stress: mechanisms by which endothelial cells transduce an atheroprotective force. Arterioscler Thromb Vasc Biol. 1998;18:677–85. [DOI] [PubMed] [Google Scholar]
- 93.Fisher AB, Chien S, Barakat AI and Nerem RM. Endothelial cellular response to altered shear stress. Am J Physiol Lung Cell Mol Physiol. 2001;281:L529–33. [DOI] [PubMed] [Google Scholar]
- 94.Schafer M, Kheyfets VO, Schroeder JD, Dunning J, Shandas R, Buckner JK, Browning J, Hertzberg J, Hunter KS and Fenster BE. Main pulmonary arterial wall shear stress correlates with invasive hemodynamics and stiffness in pulmonary hypertension. Pulm Circ. 2016;6:37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tang BT, Pickard SS, Chan FP, Tsao PS, Taylor CA and Feinstein JA. Wall shear stress is decreased in the pulmonary arteries of patients with pulmonary arterial hypertension: An image-based, computational fluid dynamics study. Pulm Circ. 2012;2:470–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Frank PG and Lisanti MP. Role of caveolin-1 in the regulation of the vascular shear stress response. J Clin Invest. 2006;116:1222–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chettimada S, Yang J, Moon HG and Jin Y. Caveolae, caveolin-1 and cavin-1: Emerging roles in pulmonary hypertension. World J Respirol. 2015;5:126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shi Y, Pritchard KA, Jr., Holman P, Rafiee P, Griffith OW, Kalyanaraman B and Baker JE. Chronic myocardial hypoxia increases nitric oxide synthase and decreases caveolin-3. Free Radic Biol Med. 2000;29:695–703. [DOI] [PubMed] [Google Scholar]
- 99.Oka N, Asai K, Kudej RK, Edwards JG, Toya Y, Schwencke C, Vatner DE, Vatner SF and Ishikawa Y. Downregulation of caveolin by chronic beta-adrenergic receptor stimulation in mice. Am J Physiol. 1997;273:C1957–62. [DOI] [PubMed] [Google Scholar]
- 100.Jasmin JF, Mercier I, Hnasko R, Cheung MW, Tanowitz HB, Dupuis J and Lisanti MP. Lung remodeling and pulmonary hypertension after myocardial infarction: pathogenic role of reduced caveolin expression. Cardiovasc Res. 2004;63:747–55. [DOI] [PubMed] [Google Scholar]
- 101.Zhao YY, Liu Y, Stan RV, Fan L, Gu Y, Dalton N, Chu PH, Peterson K, Ross J, Jr. and Chien KR. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci U S A. 2002;99:11375–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Levine AB, Punihaole D and Levine TB. Characterization of the role of nitric oxide and its clinical applications. Cardiology. 2012;122:55–68. [DOI] [PubMed] [Google Scholar]
- 103.Grayson TH, Chadha PS, Bertrand PP, Chen H, Morris MJ, Senadheera S, Murphy TV and Sandow SL. Increased caveolae density and caveolin-1 expression accompany impaired NO-mediated vasorelaxation in diet-induced obesity. Histochem Cell Biol. 2013;139:309–21. [DOI] [PubMed] [Google Scholar]
- 104.Garcia-Cardena G, Martasek P, Masters BS, Skidd PM, Couet J, Li S, Lisanti MP and Sessa WC. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J Biol Chem. 1997;272:25437–40. [DOI] [PubMed] [Google Scholar]
- 105.Patel HH, Zhang S, Murray F, Suda RY, Head BP, Yokoyama U, Swaney JS, Niesman IR, Schermuly RT, Pullamsetti SS, et al. Increased smooth muscle cell expression of caveolin-1 and caveolae contribute to the pathophysiology of idiopathic pulmonary arterial hypertension. FASEB J. 2007;21:2970–9. [DOI] [PubMed] [Google Scholar]
- 106.Feiner EC, Chung P, Jasmin JF, Zhang J, Whitaker-Menezes D, Myers V, Song J, Feldman EW, Funakoshi H, Degeorge BR Jr., et al. Left ventricular dysfunction in murine models of heart failure and in failing human heart is associated with a selective decrease in the expression of caveolin-3. J Card Fail 2011;17:253–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Markandeya YS, Phelan LJ, Woon MT, Keefe AM, Reynolds CR, August BK, Hacker TA, Roth DM, Patel HH and Balijepalli RC. Caveolin-3 Overexpression Attenuates Cardiac Hypertrophy via Inhibition of T-type Ca2+ Current Modulated by Protein Kinase Calpha in Cardiomyocytes. J Biol Chem. 2015;290:22085–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hare JM, Lofthouse RA, Juang GJ, Colman L, Ricker KM, Kim B, Senzaki H, Cao S, Tunin RS and Kass DA. Contribution of caveolin protein abundance to augmented nitric oxide signaling in conscious dogs with pacing-induced heart failure. Circ Res. 2000;86:1085–92. [DOI] [PubMed] [Google Scholar]
- 109.Dworakowski R, Walker S, Momin A, Desai J, El-Gamel A, Wendler O, Kearney MT and Shah AM. Reduced nicotinamide adenine dinucleotide phosphate oxidase-derived superoxide and vascular endothelial dysfunction in human heart failure. J Am Coll Cardiol. 2008;51:1349–56. [DOI] [PubMed] [Google Scholar]
- 110.Frazziano G, Champion HC and Pagano PJ. NADPH oxidase-derived ROS and the regulation of pulmonary vessel tone. Am J Physiol Heart Circ Physiol. 2012;302:H2166–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Farrero M, Blanco I, Batlle M, Santiago E, Cardona M, Vidal B, Castel MA, Sitges M, Barbera JA and Perez-Villa F. Pulmonary hypertension is related to peripheral endothelial dysfunction in heart failure with preserved ejection fraction. Circ Heart Fail. 2014;7:791–8. [DOI] [PubMed] [Google Scholar]
- 112.Katz SD, Biasucci L, Sabba C, Strom JA, Jondeau G, Galvao M, Solomon S, Nikolic SD, Forman R and LeJemtel TH. Impaired endothelium-mediated vasodilation in the peripheral vasculature of patients with congestive heart failure. J Am Coll Cardiol. 1992;19:918–25. [DOI] [PubMed] [Google Scholar]
- 113.Treasure CB, Vita JA, Cox DA, Fish RD, Gordon JB, Mudge GH, Colucci WS, Sutton MG, Selwyn AP, Alexander RW and et al. Endothelium-dependent dilation of the coronary microvasculature is impaired in dilated cardiomyopathy. Circulation. 1990;81:772–9. [DOI] [PubMed] [Google Scholar]
- 114.Maguire SM, Nugent AG, McGurk C, Johnston GD and Nicholls DP. Abnormal vascular responses in human chronic cardiac failure are both endothelium dependent and endothelium independent. Heart. 1998;80:141–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Katz SD, Khan T, Zeballos GA, Mathew L, Potharlanka P, Knecht M and Whelan J. Decreased activity of the L-arginine-nitric oxide metabolic pathway in patients with congestive heart failure. Circulation. 1999;99:2113–7. [DOI] [PubMed] [Google Scholar]
- 116.Zuckerbraun BS, Shiva S, Ifedigbo E, Mathier MA, Mollen KP, Rao J, Bauer PM, Choi JJ, Curtis E, Choi AM and Gladwin MT. Nitrite potently inhibits hypoxic and inflammatory pulmonary arterial hypertension and smooth muscle proliferation via xanthine oxidoreductase-dependent nitric oxide generation. Circulation. 2010;121:98–109. [DOI] [PubMed] [Google Scholar]
- 117.Moraes DL, Colucci WS and Givertz MM. Secondary pulmonary hypertension in chronic heart failure: the role of the endothelium in pathophysiology and management. Circulation. 2000;102:1718–23. [DOI] [PubMed] [Google Scholar]
- 118.Hoendermis ES, Liu LC, Hummel YM, van der Meer P, de Boer RA, Berger RM, van Veldhuisen DJ and Voors AA. Effects of sildenafil on invasive haemodynamics and exercise capacity in heart failure patients with preserved ejection fraction and pulmonary hypertension: a randomized controlled trial. Eur Heart J. 2015;36:2565–73. [DOI] [PubMed] [Google Scholar]
- 119.Liu LC, Hummel YM, van der Meer P, Berger RM, Damman K, van Veldhuisen DJ, Voors AA and Hoendermis ES. Effects of sildenafil on cardiac structure and function, cardiopulmonary exercise testing and health-related quality of life measures in heart failure patients with preserved ejection fraction and pulmonary hypertension. Eur J Heart Fail. 2017;19:116–125. [DOI] [PubMed] [Google Scholar]
- 120.Lewis GD, Shah R, Shahzad K, Camuso JM, Pappagianopoulos PP, Hung J, Tawakol A, Gerszten RE, Systrom DM, Bloch KD and Semigran MJ. Sildenafil improves exercise capacity and quality of life in patients with systolic heart failure and secondary pulmonary hypertension. Circulation. 2007;116:1555–62. [DOI] [PubMed] [Google Scholar]
- 121.Guazzi M, Vicenzi M, Arena R and Guazzi MD. Pulmonary hypertension in heart failure with preserved ejection fraction: a target of phosphodiesterase-5 inhibition in a 1-year study. Circulation. 2011;124:164–74. [DOI] [PubMed] [Google Scholar]
- 122.Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, LeWinter MM, Rouleau JL, Bull DA, Mann DL, et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2013;309:1268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bonderman D, Pretsch I, Steringer-Mascherbauer R, Jansa P, Rosenkranz S, Tufaro C, Bojic A, Lam CSP, Frey R, Ochan Kilama M, Unger S, et al. Acute hemodynamic effects of riociguat in patients with pulmonary hypertension associated with diastolic heart failure (DILATE-1): a randomized, double-blind, placebo-controlled, single-dose study. Chest. 2014;146:1274–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bonderman D, Ghio S, Felix SB, Ghofrani HA, Michelakis E, Mitrovic V, Oudiz RJ, Boateng F, Scalise AV, Roessig L, Semigran MJ and Left Ventricular Systolic Dysfunction Associated With Pulmonary Hypertension Riociguat Trial Study G. Riociguat for patients with pulmonary hypertension caused by systolic left ventricular dysfunction: a phase IIb double-blind, randomized, placebo-controlled, dose-ranging hemodynamic study. Circulation. 2013;128:502–11. [DOI] [PubMed] [Google Scholar]
- 125.Salani D, Taraboletti G, Rosano L, Di Castro V, Borsotti P, Giavazzi R and Bagnato A. Endothelin-1 induces an angiogenic phenotype in cultured endothelial cells and stimulates neovascularization in vivo. Am J Pathol. 2000;157:1703–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Marasciulo FL, Montagnani M and Potenza MA. Endothelin-1: the yin and yang on vascular function. Curr Med Chem. 2006;13:1655–65. [DOI] [PubMed] [Google Scholar]
- 127.Vignon-Zellweger N, Heiden S, Miyauchi T and Emoto N. Endothelin and endothelin receptors in the renal and cardiovascular systems. Life Sci. 2012;91:490–500. [DOI] [PubMed] [Google Scholar]
- 128.Tsukahara H, Ende H, Magazine HI, Bahou WF and Goligorsky MS. Molecular and functional characterization of the non-isopeptide-selective ETB receptor in endothelial cells. Receptor coupling to nitric oxide synthase. J Biol Chem. 1994;269:21778–85. [PubMed] [Google Scholar]
- 129.Tirapelli CR, Casolari DA, Yogi A, Montezano AC, Tostes RC, Legros E, D’Orleans-Juste P and de Oliveira AM. Functional characterization and expression of endothelin receptors in rat carotid artery: involvement of nitric oxide, a vasodilator prostanoid and the opening of K+ channels in ETB-induced relaxation. Br J Pharmacol. 2005;146:903–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zamora MA, Dempsey EC, Walchak SJ and Stelzner TJ. BQ123, an ETA receptor antagonist, inhibits endothelin-1-mediated proliferation of human pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol. 1993;9:429–33. [DOI] [PubMed] [Google Scholar]
- 131.McCulloch KM, Docherty CC, Morecroft I and MacLean MR. EndothelinB receptor-mediated contraction in human pulmonary resistance arteries. Br J Pharmacol. 1996;119:1125–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cody RJ, Haas GJ, Binkley PF, Capers Q and Kelley R. Plasma endothelin correlates with the extent of pulmonary hypertension in patients with chronic congestive heart failure. Circulation. 1992;85:504–9. [DOI] [PubMed] [Google Scholar]
- 133.Pacher R, Stanek B, Hulsmann M, Koller-Strametz J, Berger R, Schuller M, Hartter E, Ogris E, Frey B, Heinz G, et al. Prognostic impact of big endothelin-1 plasma concentrations compared with invasive hemodynamic evaluation in severe heart failure. J Am Coll Cardiol. 1996;27:633–41. [DOI] [PubMed] [Google Scholar]
- 134.Bauer M, Wilkens H, Langer F, Schneider SO, Lausberg H and Schafers HJ. Selective upregulation of endothelin B receptor gene expression in severe pulmonary hypertension. Circulation. 2002;105:1034–6. [DOI] [PubMed] [Google Scholar]
- 135.Ooi H, Colucci WS and Givertz MM. Endothelin mediates increased pulmonary vascular tone in patients with heart failure: demonstration by direct intrapulmonary infusion of sitaxsentan. Circulation. 2002;106:1618–21. [DOI] [PubMed] [Google Scholar]
- 136.Kim H, Yung GL, Marsh JJ, Konopka RG, Pedersen CA, Chiles PG, Morris TA and Channick RN. Endothelin mediates pulmonary vascular remodelling in a canine model of chronic embolic pulmonary hypertension. Eur Respir J. 2000;15:640–8. [DOI] [PubMed] [Google Scholar]
- 137.Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, Kimura S, Masaki T, Duguid WP and Stewart DJ. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328:1732–9. [DOI] [PubMed] [Google Scholar]
- 138.Rich S and McLaughlin VV. Endothelin receptor blockers in cardiovascular disease. Circulation. 2003;108:2184–90. [DOI] [PubMed] [Google Scholar]
- 139.Valero-Munoz M, Li S, Wilson RM, Boldbaatar B, Iglarz M and Sam F. Dual Endothelin-A/Endothelin-B Receptor Blockade and Cardiac Remodeling in Heart Failure With Preserved Ejection Fraction. Circ Heart Fail. 2016;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jones RL, Qian Y, Wong HN, Chan H and Yim AP. Prostanoid action on the human pulmonary vascular system. Clin Exp Pharmacol Physiol. 1997;24:969–72. [DOI] [PubMed] [Google Scholar]
- 141.Cheng Y, Austin SC, Rocca B, Koller BH, Coffman TM, Grosser T, Lawson JA and FitzGerald GA. Role of prostacyclin in the cardiovascular response to thromboxane A2. Science. 2002;296:539–41. [DOI] [PubMed] [Google Scholar]
- 142.Caughey GE, Cleland LG, Penglis PS, Gamble JR and James MJ. Roles of cyclooxygenase (COX)-1 and COX-2 in prostanoid production by human endothelial cells: selective up-regulation of prostacyclin synthesis by COX-2. J Immunol. 2001;167:2831–8. [DOI] [PubMed] [Google Scholar]
- 143.Chan EC, Dusting GJ, Guo N, Peshavariya HM, Taylor CJ, Dilley R, Narumiya S and Jiang F. Prostacyclin receptor suppresses cardiac fibrosis: role of CREB phosphorylation. J Mol Cell Cardiol. 2010;49:176–85. [DOI] [PubMed] [Google Scholar]
- 144.Yu H, Gallagher AM, Garfin PM and Printz MP. Prostacyclin release by rat cardiac fibroblasts: inhibition of collagen expression. Hypertension. 1997;30:1047–53. [DOI] [PubMed] [Google Scholar]
- 145.Smith CJ, Sun D, Hoegler C, Roth BS, Zhang X, Zhao G, Xu XB, Kobari Y, Pritchard K Jr., Sessa WC and Hintze TH. Reduced gene expression of vascular endothelial NO synthase and cyclooxygenase-1 in heart failure. Circ Res. 1996;78:58–64. [DOI] [PubMed] [Google Scholar]
- 146.Dzau VJ, Packer M, Lilly LS, Swartz SL, Hollenberg NK and Williams GH. Prostaglandins in severe congestive heart failure. Relation to activation of the renin--angiotensin system and hyponatremia. N Engl J Med. 1984;310:347–52. [DOI] [PubMed] [Google Scholar]
- 147.Pakala R, Willerson JT and Benedict CR. Effect of serotonin, thromboxane A2, and specific receptor antagonists on vascular smooth muscle cell proliferation. Circulation. 1997;96:2280–6. [DOI] [PubMed] [Google Scholar]
- 148.Rana A, Westein E, Niego B and Hagemeyer CE. Shear-Dependent Platelet Aggregation: Mechanisms and Therapeutic Opportunities. Front Cardiovasc Med 2019;6:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dromparis P, Sutendra G and Michelakis ED. The role of mitochondria in pulmonary vascular remodeling. J Mol Med (Berl). 2010;88:1003–10. [DOI] [PubMed] [Google Scholar]
- 150.Freund-Michel V, Khoyrattee N, Savineau JP, Muller B and Guibert C. Mitochondria: roles in pulmonary hypertension. Int J Biochem Cell Biol. 2014;55:93–7. [DOI] [PubMed] [Google Scholar]
- 151.Nguyen QL, Wang Y, Helbling N, Simon MA and Shiva S. Alterations in platelet bioenergetics in Group 2 PH-HFpEF patients. PLoS One. 2019;14:e0220490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Nguyen QL, Corey C, White P, Watson A, Gladwin MT, Simon MA and Shiva S. Platelets from pulmonary hypertension patients show increased mitochondrial reserve capacity. JCI Insight. 2017;2:e91415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Pfleger J, He M and Abdellatif M. Mitochondrial complex II is a source of the reserve respiratory capacity that is regulated by metabolic sensors and promotes cell survival. Cell Death Dis. 2015;6:e1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Califf RM, Adams KF, McKenna WJ, Gheorghiade M, Uretsky BF, McNulty SE, Darius H, Schulman K, Zannad F, Handberg-Thurmond E, et a.. A randomized controlled trial of epoprostenol therapy for severe congestive heart failure: The Flolan International Randomized Survival Trial (FIRST). Am Heart J. 1997;134:44–54. [DOI] [PubMed] [Google Scholar]
- 155.Ulrich S, Huber LC, Fischler M, Treder U, Maggiorini M, Eberli FR and Speich R. Platelet serotonin content and transpulmonary platelet serotonin gradient in patients with pulmonary hypertension. Respiration. 2011;81:211–6. [DOI] [PubMed] [Google Scholar]
- 156.MacLean MMR. The serotonin hypothesis in pulmonary hypertension revisited: targets for novel therapies (2017 Grover Conference Series). Pulm Circ 2018;8:2045894018759125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Golino P, Piscione F, Benedict CR, Anderson HV, Cappelli-Bigazzi M, Indolfi C, Condorelli M, Chiariello M and Willerson JT. Local effect of serotonin released during coronary angioplasty. N Engl J Med. 1994;330:523–8. [DOI] [PubMed] [Google Scholar]
- 158.Mehta J and Mehta P. Platelet function studies in heart disease. VI. Enhanced platelet aggregate formation activity in congestive heart failure: inhibition by sodium nitroprusside. Circulation. 1979;60:497–503. [DOI] [PubMed] [Google Scholar]
- 159.Chung I and Lip GY. Platelets and heart failure. Eur Heart J. 2006;27:2623–31. [DOI] [PubMed] [Google Scholar]
- 160.Crowley ST, Dempsey EC, Horwitz KB and Horwitz LD. Platelet-induced vascular smooth muscle cell proliferation is modulated by the growth amplification factors serotonin and adenosine diphosphate. Circulation. 1994;90:1908–18. [DOI] [PubMed] [Google Scholar]
- 161.Selim AM, Sarswat N, Kelesidis I, Iqbal M, Chandra R and Zolty R. Plasma Serotonin in Heart Failure: Possible Marker and Potential Treatment Target. Heart Lung Circ. 2017;26:442–449. [DOI] [PubMed] [Google Scholar]
- 162.Idriss HT and Naismith JH. TNF alpha and the TNF receptor superfamily: structure-function relationship(s). Microsc Res Tech. 2000;50:184–95. [DOI] [PubMed] [Google Scholar]
- 163.Yoshioka K, Azukari K, Ashida K, Kasamatsu Y, Yokoo S, Yoshida T and Kondo M. The efficacy of voglibose on daily glycemic excursions assessed by the “J”-index in non-insulin dependent diabetes mellitus. Horm Metab Res. 1997;29:407–8. [DOI] [PubMed] [Google Scholar]
- 164.Sanders-van Wijk S, Tromp J, Beussink-Nelson L, Hage C, Svedlund S, Saraste A, Swat SA, Sanchez C, Njoroge J, Tan RS, et al. Proteomic Evaluation of the Comorbidity-Inflammation Paradigm in Heart Failure With Preserved Ejection Fraction: Results From the PROMIS-HFpEF Study. Circulation. 2020;142:2029–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Huang W, Liu H, Pan Y, Yang H, Lin J and Zhang H. Mechanical stretching of the pulmonary vein mediates pulmonary hypertension due to left heart disease by regulating SAC/MAPK pathway and the expression of IL-6 and TNF-alpha. J Cardiothorac Surg 2021;16:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Marsden PA and Brenner BM. Transcriptional regulation of the endothelin-1 gene by TNF-alpha. Am J Physiol. 1992;262:C854–61. [DOI] [PubMed] [Google Scholar]
- 167.Klemm P, Warner TD, Hohlfeld T, Corder R and Vane JR. Endothelin 1 mediates ex vivo coronary vasoconstriction caused by exogenous and endogenous cytokines. Proc Natl Acad Sci U S A. 1995;92:2691–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Yin WH, Chen JW, Jen HL, Chiang MC, Huang WP, Feng AN, Young MS and Lin SJ. Independent prognostic value of elevated high-sensitivity C-reactive protein in chronic heart failure. Am Heart J. 2004;147:931–8. [DOI] [PubMed] [Google Scholar]
- 169.Reina-Couto M, Pereira-Terra P, Quelhas-Santos J, Silva-Pereira C, Albino-Teixeira A and Sousa T. Inflammation in Human Heart Failure: Major Mediators and Therapeutic Targets. Front Physiol. 2021;12:746494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Seta Y, Shan K, Bozkurt B, Oral H and Mann DL. Basic mechanisms in heart failure: the cytokine hypothesis. J Card Fail. 1996;2:243–9. [DOI] [PubMed] [Google Scholar]
- 171.Collier P, Watson CJ, Voon V, Phelan D, Jan A, Mak G, Martos R, Baugh JA, Ledwidge MT and McDonald KM. Can emerging biomarkers of myocardial remodelling identify asymptomatic hypertensive patients at risk for diastolic dysfunction and diastolic heart failure? Eur J Heart Fail 2011;13:1087–95. [DOI] [PubMed] [Google Scholar]
- 172.Dolenc J, Sebestjen M, Vrtovec B, Kozelj M and Haddad F. Pulmonary hypertension in patients with advanced heart failure is associated with increased levels of interleukin-6. Biomarkers. 2014;19:385–90. [DOI] [PubMed] [Google Scholar]
- 173.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–60. [DOI] [PubMed] [Google Scholar]
- 174.Lohela M, Bry M, Tammela T and Alitalo K. VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. Curr Opin Cell Biol. 2009;21:154–65. [DOI] [PubMed] [Google Scholar]
- 175.Borne Y, Gransbo K, Nilsson J, Melander O, Orho-Melander M, Smith JG and Engstrom G. Vascular Endothelial Growth Factor D, Pulmonary Congestion, and Incidence of Heart Failure. J Am Coll Cardiol. 2018;71:580–582. [DOI] [PubMed] [Google Scholar]
- 176.Houston BA, Tedford RJ, Baxley RL, Sykes B, Powers ER, Nielsen CD, Steinberg DH, Maran A, Fernandes VLC, Todoran T, et al. Relation of Lymphangiogenic Factor Vascular Endothelial Growth Factor-D to Elevated Pulmonary Artery Wedge Pressure. Am J Cardiol. 2019;124:756–762. [DOI] [PubMed] [Google Scholar]
- 177.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF and Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001;15:427–38. [DOI] [PubMed] [Google Scholar]
- 178.Saleby J, Bouzina H, Ahmed S, Lundgren J and Radegran G. Plasma receptor tyrosine kinase RET in pulmonary arterial hypertension diagnosis and differentiation. ERJ Open Res. 2019;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Saleby J, Bouzina H, Lundgren J and Radegran G. Angiogenic and inflammatory biomarkers in the differentiation of pulmonary hypertension. Scand Cardiovasc J. 2017;51:261–270. [DOI] [PubMed] [Google Scholar]
- 180.Al-Husseini A, Kraskauskas D, Mezzaroma E, Nordio A, Farkas D, Drake JI, Abbate A, Felty Q and Voelkel NF. Vascular endothelial growth factor receptor 3 signaling contributes to angioobliterative pulmonary hypertension. Pulm Circ 2015;5:101–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ma Z, Yu YR, Badea CT, Kovacs JJ, Xiong X, Comhair S, Piantadosi CA and Rajagopal S. Vascular Endothelial Growth Factor Receptor 3 Regulates Endothelial Function Through beta-Arrestin 1. Circulation. 2019;139:1629–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hwangbo C, Lee HW, Kang H, Ju H, Wiley DS, Papangeli I, Han J, Kim JD, Dunworth WP, Hu X, et al. Modulation of Endothelial Bone Morphogenetic Protein Receptor Type 2 Activity by Vascular Endothelial Growth Factor Receptor 3 in Pulmonary Arterial Hypertension. Circulation. 2017;135:2288–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hamada K, Oike Y, Takakura N, Ito Y, Jussila L, Dumont DJ, Alitalo K and Suda T. VEGF-C signaling pathways through VEGFR-2 and VEGFR-3 in vasculoangiogenesis and hematopoiesis. Blood. 2000;96:3793–800. [PubMed] [Google Scholar]
- 184.Park YG, Choi J, Jung HK, Song IK, Shin Y, Park SY and Seol JW. Fluid shear stress regulates vascular remodeling via VEGFR-3 activation, although independently of its ligand, VEGF-C, in the uterus during pregnancy. Int J Mol Med. 2017;40:1210–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lin QY, Zhang YL, Bai J, Liu JQ and Li HH. VEGF-C/VEGFR-3 axis protects against pressure-overload induced cardiac dysfunction through regulation of lymphangiogenesis. Clin Transl Med. 2021;11:e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Partovian C, Adnot S, Raffestin B, Louzier V, Levame M, Mavier IM, Lemarchand P and Eddahibi S. Adenovirus-mediated lung vascular endothelial growth factor overexpression protects against hypoxic pulmonary hypertension in rats. Am J Respir Cell Mol Biol. 2000;23:762–71. [DOI] [PubMed] [Google Scholar]
- 187.Schauer A, Draskowski R, Jannasch A, Kirchhoff V, Goto K, Mannel A, Barthel P, Augstein A, Winzer E, Tugtekin M, et al. ZSF1 rat as animal model for HFpEF: Development of reduced diastolic function and skeletal muscle dysfunction. ESC Heart Fail. 2020;7:2123–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Taddei S, Virdis A, Ghiadoni L, Mattei P, Sudano I, Bernini G, Pinto S and Salvetti A. Menopause is associated with endothelial dysfunction in women. Hypertension. 1996;28:576–82. [DOI] [PubMed] [Google Scholar]
- 189.Hisamoto K and Bender JR. Vascular cell signaling by membrane estrogen receptors. Steroids. 2005;70:382–7. [DOI] [PubMed] [Google Scholar]
- 190.Moriarty K, Kim KH and Bender JR. Minireview: estrogen receptor-mediated rapid signaling. Endocrinology. 2006;147:5557–63. [DOI] [PubMed] [Google Scholar]
- 191.Ventetuolo CE, Praestgaard A, Palevsky HI, Klinger JR, Halpern SD and Kawut SM. Sex and haemodynamics in pulmonary arterial hypertension. Eur Respir J. 2014;43:523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Hardie L, Trayhurn P, Abramovich D and Fowler P. Circulating leptin in women: a longitudinal study in the menstrual cycle and during pregnancy. Clin Endocrinol (Oxf). 1997;47:101–6. [DOI] [PubMed] [Google Scholar]
- 193.Gao Q and Horvath TL. Cross-talk between estrogen and leptin signaling in the hypothalamus. Am J Physiol Endocrinol Metab. 2008;294:E817–26. [DOI] [PubMed] [Google Scholar]
- 194.Frank RC, Min J, Abdelghany M, Paniagua S, Bhattacharya R, Bhambhani V, Pomerantsev E and Ho JE. Obesity Is Associated With Pulmonary Hypertension and Modifies Outcomes. J Am Heart Assoc. 2020;9:e014195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Shah SJ, Katz DH, Selvaraj S, Burke MA, Yancy CW, Gheorghiade M, Bonow RO, Huang CC and Deo RC. Phenomapping for novel classification of heart failure with preserved ejection fraction. Circulation. 2015;131:269–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Ussavarungsi K, Thomas CS and Burger CD. Prevalence of metabolic syndrome in patients with pulmonary hypertension. Clin Respir J. 2017;11:721–726. [DOI] [PubMed] [Google Scholar]
- 197.Achari AE and Jain SK. Adiponectin, a Therapeutic Target for Obesity, Diabetes, and Endothelial Dysfunction. Int J Mol Sci. 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Matsuda M, Shimomura I, Sata M, Arita Y, Nishida M, Maeda N, Kumada M, Okamoto Y, Nagaretani H, Nishizawa H, et al. Role of adiponectin in preventing vascular stenosis. The missing link of adipo-vascular axis. J Biol Chem. 2002;277:37487–91. [DOI] [PubMed] [Google Scholar]
- 199.Kobashi C, Urakaze M, Kishida M, Kibayashi E, Kobayashi H, Kihara S, Funahashi T, Takata M, Temaru R, Sato A, et al. Adiponectin inhibits endothelial synthesis of interleukin-8. Circ Res. 2005;97:1245–52. [DOI] [PubMed] [Google Scholar]
- 200.Ouchi N, Kobayashi H, Kihara S, Kumada M, Sato K, Inoue T, Funahashi T and Walsh K. Adiponectin stimulates angiogenesis by promoting cross-talk between AMP-activated protein kinase and Akt signaling in endothelial cells. J Biol Chem. 2004;279:1304–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Ouchi N, Kihara S, Arita Y, Okamoto Y, Maeda K, Kuriyama H, Hotta K, Nishida M, Takahashi M, Muraguchi M, et al. Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-kappaB signaling through a cAMP-dependent pathway. Circulation. 2000;102:1296–301. [DOI] [PubMed] [Google Scholar]
- 202.Ohashi K, Ouchi N, Sato K, Higuchi A, Ishikawa TO, Herschman HR, Kihara S and Walsh K. Adiponectin promotes revascularization of ischemic muscle through a cyclooxygenase 2-dependent mechanism. Mol Cell Biol. 2009;29:3487–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Matsuzawa Y, Funahashi T, Kihara S and Shimomura I. Adiponectin and metabolic syndrome. Arterioscler Thromb Vasc Biol. 2004;24:29–33. [DOI] [PubMed] [Google Scholar]
- 204.Takahashi M, Funahashi T, Shimomura I, Miyaoka K and Matsuzawa Y. Plasma leptin levels and body fat distribution. Horm Metab Res. 1996;28:751–2. [DOI] [PubMed] [Google Scholar]
- 205.Hotta K, Funahashi T, Arita Y, Takahashi M, Matsuda M, Okamoto Y, Iwahashi H, Kuriyama H, Ouchi N, Maeda K, et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol. 2000;20:1595–9. [DOI] [PubMed] [Google Scholar]
- 206.Lara-Castro C, Luo N, Wallace P, Klein RL and Garvey WT. Adiponectin multimeric complexes and the metabolic syndrome trait cluster. Diabetes. 2006;55:249–59. [PubMed] [Google Scholar]
- 207.Huertas A, Tu L, Gambaryan N, Girerd B, Perros F, Montani D, Fabre D, Fadel E, Eddahibi S, Cohen-Kaminsky S, et al. Leptin and regulatory T-lymphocytes in idiopathic pulmonary arterial hypertension. Eur Respir J. 2012;40:895–904. [DOI] [PubMed] [Google Scholar]
- 208.Chai S, Wang W, Liu J, Guo H, Zhang Z, Wang C and Wang J. Leptin knockout attenuates hypoxia-induced pulmonary arterial hypertension by inhibiting proliferation of pulmonary arterial smooth muscle cells. Transl Res. 2015;166:772–82. [DOI] [PubMed] [Google Scholar]
- 209.Huertas A, Tu L, Thuillet R, Le Hiress M, Phan C, Ricard N, Nadaud S, Fadel E, Humbert M and Guignabert C. Leptin signalling system as a target for pulmonary arterial hypertension therapy. Eur Respir J. 2015;45:1066–80. [DOI] [PubMed] [Google Scholar]
- 210.Martin SS, Blaha MJ, Muse ED, Qasim AN, Reilly MP, Blumenthal RS, Nasir K, Criqui MH, McClelland RL, Hughes-Austin JM, et al. Leptin and incident cardiovascular disease: the Multi-ethnic Study of Atherosclerosis (MESA). Atherosclerosis. 2015;239:67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Lieb W, Sullivan LM, Harris TB, Roubenoff R, Benjamin EJ, Levy D, Fox CS, Wang TJ, Wilson PW, Kannel WB, et al. Plasma leptin levels and incidence of heart failure, cardiovascular disease, and total mortality in elderly individuals. Diabetes Care. 2009;32:612–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Wannamethee SG, Shaper AG, Whincup PH, Lennon L and Sattar N. Obesity and risk of incident heart failure in older men with and without pre-existing coronary heart disease: does leptin have a role? J Am Coll Cardiol. 2011;58:1870–7. [DOI] [PubMed] [Google Scholar]
- 213.Bobbert P, Jenke A, Bobbert T, Kuhl U, Rauch U, Lassner D, Scheibenbogen C, Poller W, Schultheiss HP and Skurk C. High leptin and resistin expression in chronic heart failure: adverse outcome in patients with dilated and inflammatory cardiomyopathy. Eur J Heart Fail. 2012;14:1265–75. [DOI] [PubMed] [Google Scholar]
- 214.Schulze PC, Kratzsch J, Linke A, Schoene N, Adams V, Gielen S, Erbs S, Moebius-Winkler S and Schuler G. Elevated serum levels of leptin and soluble leptin receptor in patients with advanced chronic heart failure. Eur J Heart Fail. 2003;5:33–40. [DOI] [PubMed] [Google Scholar]
- 215.Van den Bergh A, Vanderper A, Vangheluwe P, Desjardins F, Nevelsteen I, Verreth W, Wuytack F, Holvoet P, Flameng W, Balligand JL, et al. Dyslipidaemia in type II diabetic mice does not aggravate contractile impairment but increases ventricular stiffness. Cardiovasc Res. 2008;77:371–9. [DOI] [PubMed] [Google Scholar]
- 216.Moro C, Klimcakova E, Lolmede K, Berlan M, Lafontan M, Stich V, Bouloumie A, Galitzky J, Arner P and Langin D. Atrial natriuretic peptide inhibits the production of adipokines and cytokines linked to inflammation and insulin resistance in human subcutaneous adipose tissue. Diabetologia. 2007;50:1038–47. [DOI] [PubMed] [Google Scholar]
- 217.Obokata M, Reddy YNV, Pislaru SV, Melenovsky V and Borlaug BA. Evidence Supporting the Existence of a Distinct Obese Phenotype of Heart Failure With Preserved Ejection Fraction. Circulation. 2017;136:6–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Khalid U, Wruck LM, Quibrera PM, Bozkurt B, Nambi V, Virani SS, Jneid H, Agarwal S, Chang PP, Loehr L, Basra SS, Rosamond W, Ballantyne CM and Deswal A. BNP and obesity in acute decompensated heart failure with preserved vs. reduced ejection fraction: The Atherosclerosis Risk in Communities Surveillance Study. Int J Cardiol. 2017;233:61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Weiss TW, Arnesen H and Seljeflot I. Components of the interleukin-6 transsignalling system are associated with the metabolic syndrome, endothelial dysfunction and arterial stiffness. Metabolism. 2013;62:1008–13. [DOI] [PubMed] [Google Scholar]
- 220.Standeven KF, Hess K, Carter AM, Rice GI, Cordell PA, Balmforth AJ, Lu B, Scott DJ, Turner AJ, Hooper NM and Grant PJ. Neprilysin, obesity and the metabolic syndrome. Int J Obes (Lond). 2011;35:1031–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Ganguly A. Atrial natriuretic peptide-induced inhibition of aldosterone secretion: a quest for mediator(s). Am J Physiol. 1992;263:E181–94. [DOI] [PubMed] [Google Scholar]
- 222.Bentley-Lewis R, Adler GK, Perlstein T, Seely EW, Hopkins PN, Williams GH and Garg R. Body mass index predicts aldosterone production in normotensive adults on a high-salt diet. J Clin Endocrinol Metab. 2007;92:4472–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Huby AC, Antonova G, Groenendyk J, Gomez-Sanchez CE, Bollag WB, Filosa JA and Belin de Chantemele EJ. Adipocyte-Derived Hormone Leptin Is a Direct Regulator of Aldosterone Secretion, Which Promotes Endothelial Dysfunction and Cardiac Fibrosis. Circulation. 2015;132:2134–45. [DOI] [PubMed] [Google Scholar]
- 224.Briones AM, Nguyen Dinh Cat A, Callera GE, Yogi A, Burger D, He Y, Correa JW, Gagnon AM, Gomez-Sanchez CE, Gomez-Sanchez EP, et al. Adipocytes produce aldosterone through calcineurin-dependent signaling pathways: implications in diabetes mellitus-associated obesity and vascular dysfunction. Hypertension. 2012;59:1069–78. [DOI] [PubMed] [Google Scholar]
- 225.Rozansky DJ. The role of aldosterone in renal sodium transport. Semin Nephrol. 2006;26:173–81. [DOI] [PubMed] [Google Scholar]
- 226.Kraus D, Jager J, Meier B, Fasshauer M and Klein J. Aldosterone inhibits uncoupling protein-1, induces insulin resistance, and stimulates proinflammatory adipokines in adipocytes. Horm Metab Res. 2005;37:455–9. [DOI] [PubMed] [Google Scholar]
- 227.Gerling IC, Sun Y, Ahokas RA, Wodi LA, Bhattacharya SK, Warrington KJ, Postlethwaite AE and Weber KT. Aldosteronism: an immunostimulatory state precedes proinflammatory/fibrogenic cardiac phenotype. Am J Physiol Heart Circ Physiol. 2003;285:H813–21. [DOI] [PubMed] [Google Scholar]
- 228.Maron BA and Leopold JA. The role of the renin-angiotensin-aldosterone system in the pathobiology of pulmonary arterial hypertension (2013 Grover Conference series). Pulm Circ. 2014;4:200–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Lefebvre F, Prefontaine A, Calderone A, Caron A, Jasmin JF, Villeneuve L and Dupuis J. Modification of the pulmonary renin-angiotensin system and lung structural remodelling in congestive heart failure. Clin Sci (Lond). 2006;111:217–24. [DOI] [PubMed] [Google Scholar]
- 230.Maron BA, Waxman AB, Opotowsky AR, Gillies H, Blair C, Aghamohammadzadeh R, Loscalzo J and Leopold JA. Effectiveness of spironolactone plus ambrisentan for treatment of pulmonary arterial hypertension (from the [ARIES] study 1 and 2 trials). Am J Cardiol. 2013;112:720–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Boehm M, Arnold N, Braithwaite A, Pickworth J, Lu C, Novoyatleva T, Kiely DG, Grimminger F, Ghofrani HA, Weissmann N, et al. Eplerenone attenuates pathological pulmonary vascular rather than right ventricular remodeling in pulmonary arterial hypertension. BMC Pulm Med. 2018;18:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Puwanant S, Priester TC, Mookadam F, Bruce CJ, Redfield MM and Chandrasekaran K. Right ventricular function in patients with preserved and reduced ejection fraction heart failure. Eur J Echocardiogr. 2009;10:733–7. [DOI] [PubMed] [Google Scholar]
- 233.Lundorff IJ, Sengelov M, Pedersen S, Modin D, Bruun NE, Fritz-Hansen T, Biering-Sorensen T and Godsk Jorgensen P. Prognostic value of right ventricular echocardiographic measures in patients with heart failure with reduced ejection fraction. J Clin Ultrasound. 2021;49:903–913. [DOI] [PubMed] [Google Scholar]
- 234.Gorter TM, Hoendermis ES, van Veldhuisen DJ, Voors AA, Lam CS, Geelhoed B, Willems TP and van Melle JP. Right ventricular dysfunction in heart failure with preserved ejection fraction: a systematic review and meta-analysis. Eur J Heart Fail. 2016;18:1472–1487. [DOI] [PubMed] [Google Scholar]
- 235.Polsinelli VB, Marteau L and Shah SJ. The role of splanchnic congestion and the intestinal microenvironment in the pathogenesis of advanced heart failure. Curr Opin Support Palliat Care. 2019;13:24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Sandek A, Rauchhaus M, Anker SD and von Haehling S. The emerging role of the gut in chronic heart failure. Curr Opin Clin Nutr Metab Care. 2008;11:632–9. [DOI] [PubMed] [Google Scholar]
- 237.Mullens W, Abrahams Z, Francis GS, Sokos G, Taylor DO, Starling RC, Young JB and Tang WHW. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol. 2009;53:589–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Polsinelli VB, Sinha A and Shah SJ. Visceral Congestion in Heart Failure: Right Ventricular Dysfunction, Splanchnic Hemodynamics, and the Intestinal Microenvironment. Curr Heart Fail Rep 2017;14:519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Akmal M, Barndt RR, Ansari AN, Mohler JG and Massry SG. Excess PTH in CRF induces pulmonary calcification, pulmonary hypertension and right ventricular hypertrophy. Kidney Int. 1995;47:158–63. [DOI] [PubMed] [Google Scholar]
- 240.Niebauer J, Volk HD, Kemp M, Dominguez M, Schumann RR, Rauchhaus M, Poole-Wilson PA, Coats AJ and Anker SD. Endotoxin and immune activation in chronic heart failure: a prospective cohort study. Lancet. 1999;353:1838–42. [DOI] [PubMed] [Google Scholar]
- 241.Fallick C, Sobotka PA and Dunlap ME. Sympathetically mediated changes in capacitance: redistribution of the venous reservoir as a cause of decompensation. Circ Heart Fail. 2011;4:669–75. [DOI] [PubMed] [Google Scholar]
- 242.Stepniakowski K and Egan BM. Additive effects of obesity and hypertension to limit venous volume. Am J Physiol. 1995;268:R562–8. [DOI] [PubMed] [Google Scholar]
- 243.Lindenberger M Reduced Venous Compliance in Young Women with Type 1 Diabetes - Further Aggravated by Prolonged Elevated Levels of HbA1c. Front Endocrinol (Lausanne). 2016;7:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Al-Naamani N, Preston IR, Paulus JK, Hill NS and Roberts KE. Pulmonary Arterial Capacitance Is an Important Predictor of Mortality in Heart Failure With a Preserved Ejection Fraction. JACC Heart Fail. 2015;3:467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Burkhoff D, Borlaug BA, Shah SJ, Zolty R, Tedford RJ, Thenappan T, Zamanian RT, Mazurek JA, Rich JD, Simon MA, Chung ES, Raza F, Majure DT, Lewis GD, Preston IR and Rich S. Levosimendan Improves Hemodynamics and Exercise Tolerance in PH-HFpEF: Results of the Randomized Placebo-Controlled HELP Trial. JACC Heart Fail. 2021;9:360–370. [DOI] [PubMed] [Google Scholar]
- 246.Brener MI, Hamid NB, Sunagawa K, Borlaug BA, Shah SJ, Rich S and Burkhoff D. Changes in Stressed Blood Volume with Levosimendan in Pulmonary Hypertension from Heart Failure with Preserved Ejection Fraction: Insights Regarding Mechanism of Action From the HELP Trial. J Card Fail. 2021;27:1023–1026. [DOI] [PubMed] [Google Scholar]
- 247.Itkin M, Rockson SG and Burkhoff D. Pathophysiology of the Lymphatic System in Patients With Heart Failure: JACC State-of-the-Art Review. J Am Coll Cardiol. 2021;78:278–290. [DOI] [PubMed] [Google Scholar]
- 248.Herpertz U. [Differential diagnosis of edemas]. Z Lymphol. 1988;12:42–7. [PubMed] [Google Scholar]
- 249.Rossitto G, Mary S, McAllister C, Neves KB, Haddow L, Rocchiccioli JP, Lang NN, Murphy CL, Touyz RM, Petrie MC and Delles C. Reduced Lymphatic Reserve in Heart Failure With Preserved Ejection Fraction. J Am Coll Cardiol. 2020;76:2817–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.West JB, Dollery CT and Naimark A. Distribution of Blood Flow in Isolated Lung; Relation to Vascular and Alveolar Pressures. J Appl Physiol. 1964;19:713–24. [DOI] [PubMed] [Google Scholar]
- 251.Uhley HN, Leeds SE, Sampson JJ and Friedman M. Role of pulmonary lymphatics in chronic pulmonary edema. Circ Res. 1962;11:966–70. [DOI] [PubMed] [Google Scholar]
- 252.Gehlbach BK and Geppert E. The pulmonary manifestations of left heart failure. Chest. 2004;125:669–82. [DOI] [PubMed] [Google Scholar]
- 253.Verbrugge FH, Dupont M, Steels P, Grieten L, Malbrain M, Tang WH and Mullens W. Abdominal contributions to cardiorenal dysfunction in congestive heart failure. J Am Coll Cardiol. 2013;62:485–95. [DOI] [PubMed] [Google Scholar]
- 254.Aukland K and Reed RK. Interstitial-lymphatic mechanisms in the control of extracellular fluid volume. Physiol Rev. 1993;73:1–78. [DOI] [PubMed] [Google Scholar]
- 255.Moore JE Jr. and Bertram CD. Lymphatic System Flows. Annu Rev Fluid Mech. 2018;50:459–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Rissanen TT, Markkanen JE, Gruchala M, Heikura T, Puranen A, Kettunen MI, Kholova I, Kauppinen RA, Achen MG, Stacker SA, Alitalo K and Yla-Herttuala S. VEGF-D is the strongest angiogenic and lymphangiogenic effector among VEGFs delivered into skeletal muscle via adenoviruses. Circ Res. 2003;92:1098–106. [DOI] [PubMed] [Google Scholar]