

Abstract
A novel orally bioavailable solid formulation to deliver a gaseous signaling molecule, carbon monoxide (CO), was developed by adsorbing oxalyl saccharin, a newly developed organic CO prodrug, in the activated charcoal (AC). The resulting solid dispersion formulation addresses key developability issues of this CO prodrug. By taking advantage of the large surface area of AC, the paradoxical problem of low water solubility of the prodrug and the requirement of hydrolysis to release CO is resolved, and the need for an organic cosolvent is completely circumvented. The AC formulation also mitigates the adverse effect of low pH on the CO release yield, allowing steady CO release in simulated gastric and intestine fluids. This formulation allows encapsulation in normal and enteric-coated gel capsules, which enables controllable CO delivery to the upper or lower GI system. It also features an advantage of trapping CO prodrug and release product in the AC, therefore lowering systemic absorption of these chemicals. Through in-vivo pharmacokinetic studies in mice, the AC formulation showed better CO delivery efficiency of delivering CO through oral administration compared to the prodrug dosed with an organic cosolvent. The AC formulation has also been applied to address similar developability issues of our cheletropic reaction-based CO prodrug. We envision the wide applicability of this formulation in facilitating the future development of CO-based therapeutics.
Keywords: Carbon monoxide, Activated charcoal, Formulation, Gasotransmitter, Gaseous signaling molecule, Solid dispersion, Drug delivery
Graphical Abstract
1. Introduction
In the past two decades, intensive studies have been conducted to understand carbon monoxide’s (CO) physiological and pharmacological activities, mechanism(s) of action, and therapeutic potentials (Motterlini and Otterbein, 2010; Tripathi et al., 2021; Wu and Wang, 2005; Yang et al., 2021a). The prospect of developing CO into a therapeutic agent for treating various diseases is on the horizon (Chu et al., 2021). The delivery of the gaseous molecule can be very different from conventional small-molecule drugs and poses unique challenges. CO gas inhalation has been the primary route of administration in animal studies and human clinical trials in the past. However, CO delivery efficiency is highly dependent on an individual respiratory function. Therefore, it requires special apparatus in a hospital setting to control the dosage, which could impede patient compliance to treat chronic diseases. Furthermore, there has been experimental evidence in dogs and rats showing non-airway routes such as gastrointestinal (GI) administration may have better safety profiles compared with inhaled CO (Goldbaum et al., 1976; McGrath and Leviseur, 1984; Yang et al., 2021b). There have been intensive efforts in developing non-airway approaches to deliver CO as potential therapeutic agents, including CO in solution (Yang et al., 2020; Yang et al., 2021a) and small-molecule CO donors (Abeyrathna et al., 2017a; Alghazwat et al., 2021; Cheng et al., 2020; Ji and Wang, 2018; Kueh et al., 2017; Motterlini and Otterbein, 2010; Poloukhtine and Popik, 2006; Sitnikov et al., 2015; Soboleva and Berreau, 2019; Steiger et al., 2014; Weinstain et al., 2020; Yang et al., 2020), which afford precise dosage of CO through oral or injection administration. In the field of chemical CO donors suitable for oral delivery (Yang et al., 2021b), earlier efforts were mostly focused on metal-based CO releasing molecules (CORMs) (Mann, 2012; Motterlini and Otterbein, 2010; Romao et al., 2012). Several of them have shown therapeutic effect in animal study via oral administration (Yang et al., 2021b). However, there has been debate about the clinical developability of the CORMs due to the toxicity concerns of the transition metal, CO release stochiometric incertitude (Southam et al., 2021), and CO independent reactivity and activities (Southam et al., 2021; Yuan et al., 2021a; Yuan et al., 2021b). To address such issues, a tablet formulation with a binary microreactor containing CORM-2 and a coated sodium dithionate as the CO release promotor was developed (Steiger et al., 2014). The controlled release of definitive amount of CO was achieved through this innovative formulation. However, the systemic exposure to metal byproduct was inevitable due to the disintegration of the tablet (Steiger et al., 2014).
Aimed at overcoming metal-related issues, recent years have seen much progress in developing organic CO donors (Abeyrathna et al., 2017; De La Cruz et al., 2018; Ji et al., 2019a; Ji et al., 2017a; Ji et al., 2017b; Ji et al., 2019b; Ji and Wang, 2018; Lazarus et al., 2020; Pan et al., 2018; Zheng et al., 2018). A key design is based on the cheletropic extrusion of carbon monoxide from a norbornadiene-7-one key intermediate generated from Diels-Alder reaction (Ji and Wang, 2018; Ji et al., 2016; Wang et al., 2014; Zheng et al., 2018), and elimination reaction (Ji et al., 2017a; Ji et al., 2019b; Kueh et al., 2017; Pan et al., 2018) under physiological conditions found in the GI lumen. Although the prodrug does not need to be absorbed into the blood circulation and has been shown to effectively deliver CO through oral administration (Wang et al., 2020), the systemic absorption of the “CO carrier” molecule and CO released byproduct is still concerning and warrant thorough evaluation (Chu et al., 2021). A way to work around is to use a benign molecule with known pharmacology/toxicology profiles as the carrier/byproduct of the CO prodrug. Along this line, we developed a CO prodrug based on the hydrolysis-initiated sequential decarboxylation-decarbonylation reactions of oxalic saccharine BW-CO-306 (Fig. 1).
Fig. 1.
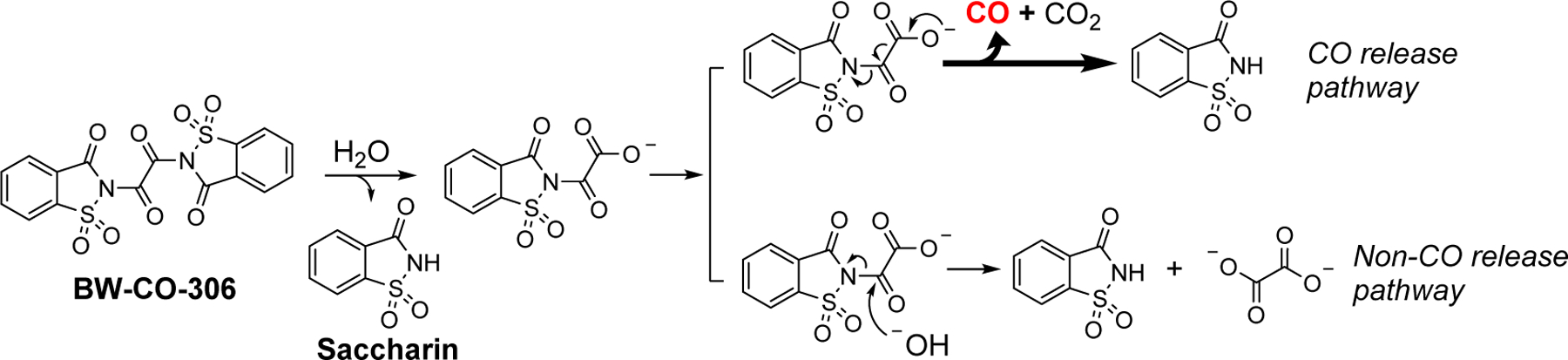
CO release pathways of BW-CO-306
BW-CO-306 reacts with water to release CO, leaving saccharine as the byproduct. As an artificial sweetener that has been used in the food industry for more than a century, the safety issue of the byproduct (LD50 of saccharin is about 17.5 g/kg in mice, according to MSDS) was largely resolved in this design. Unexpectedly, however, BW-CO-306 suffered from an unprecedented formulation challenge. It is virtually insoluble in water, though it relies on the hydrolysis reaction to generate CO. Directly incubating BW-CO-306 powder with phosphate buffer saline (PBS) did not lead to any CO production as determined by GC analysis. As a result, direct tablet formulation for oral administration would not be feasible. Therefore, in the initial proof-of-concept studies, a water-miscible aprotic organic solvent was used to prepare the stock solution of BW-CO-306. Mixing this stock solution with aqueous buffer solution leads to CO generation. Specifically, for CO release kinetic studies in solution, ACN was used to make the stock solution. In cell culture studies, DMA was used. However, for in-vivo studies and potential clinical applications, the choice of compatible organic solvents is very limited, and most of the common excipients, solubilizers, and formulations are not compatible due to the reactivity towards BW-CO-306. Even the available organic solvents may lead to undesirable bioactivity and/or injury to the administration site. In previous studies, DMA stock solution of BW-CO-306 was mixed with PEG400 before oral administration in mice for in-vivo pharmacokinetic studies (Martino et al., 2017). Though tolerated by the mice, DMA and PEG400 were known to have their own activity and toxicity issues (Kennedy and Sherman, 1986; Li et al., 2011), especially when given at a high dose. It is also not feasible for such vehicles to be used in human clinical studies. On the other hand, there are also chemical reasons that suggest the need for alternatives that do not rely on using organic cosolvents. Specifically, there is a competitive non-CO releasing hydrolysis reaction pathway between BW-CO-306 and water (Fig. 1). It decreases the CO release yield to about 60% by mixing ACN solution of BW-CO-306 with PBS (4:1, v:v). The lower pH in SGF further favors the hydrolysis pathway more than in PBS. For example, CO release yield decreased to 48% at pH 3 and to 28% at pH 1 (De La Cruz et al., 2021). The pH dependency of the CO release yield, if not further optimized, may render significant inconsistency and low efficiency of CO delivery in vivo. Especially for an oral formulation, the exposure to the acidic gastric environment and then neutral environments in the lower GI is expected. Lastly, a considerable amount of the saccharin byproduct after CO release was found to be absorbed into blood circulation after oral administration of the CO prodrug. Though less concerning due to the well-defined toxicology profile of saccharin, an increased systemic burden of such chemical is still undesirable. Interestingly, we found some of these developability issues also exist in other CO prodrugs we have developed. For example, the intramolecular Diels-Alder type CO prodrugs also suffered from low water solubility, and yet its CO release is driven by the aqueous environment (Ji and Wang, 2018). After CO release, the byproduct was also found to be absorbed systemically (Wang et al., 2020), which is undesirable due to its new chemical entity nature.
In this study, we aimed to address the above-mentioned hurdles by developing a novel formulation strategy that meets the following criteria. First, it should address the paradoxical water solubility issue by facilitating sufficient and consistent exposure of the CO prodrug to water to allow for the reactions needed for CO release. Second, it should mitigate the pH effect to allow for stable CO release under biologically relevant conditions with fluctuating pH, such as in the stomach or the lower GI. Third, preferably, it should allow for trapping of the side product after CO release in a form that mitigates systemic bioavailability. Fourth, the formulation should allow for stable storage. With these considerations, we envision a solid-state formulation, which is hydrophobic and yet allows for water access to generate CO; has high surface area and yet offers a chemical protective environment; and is highly absorbent, minimizing the systemic bioavailability of the prodrug or its byproduct after CO release. For all these reasons we propose to use activated charcoal (AC) as a carrier material to form a “solid dispersion” to tackle the above formulation problems and deliver CO through oral administration. It differs from the classical definition of the hydrophilic carrier material-based solid dispersion which is used to improve the solubility of the dispersed drug (Huang and Dai, 2014). On the contrary, solubilization of the CO prodrug dispersion matrix is not necessary as the gaseous CO is indeed the active “drug” to be delivered.
2. Materials and methods
All chemicals, including oxalyl chloride, saccharin, activated charcoal (denoted as AC, purists. p.a., powder, 31616, Lot#BCBX9192), acetonitrile, carboxymethylcellulose sodium, and phosphate-buffered saline (pH 7.4) were purchased from Sigma-Aldrich (St Louis, USA). Activated charcoal was dried in an oven overnight at 180 °C before use. Acetonitrile (ACN) was dried by a solvent purification system (Pure Process Technology, Nashua, USA). Simulated gastric fluid (SGF, pH 1.2) and simulated intestinal fluid (SIF, pH 6.8) were prepared according to USP methods (Klein, 2010). Size-3 cellulose and gelatin gel capsules were purchased from XPRS NUTRA (South Jordan, Utah, USA). The enteric coating material Eudragit® L 30 D-55 was a gift from Evonik (New Jersey, USA). HPLC analyses were conducted on an Agilent (Santa Clara, USA) 1100 HPLC system equipped with a Kromasil C18 5 μm 4.6×150 mm column. Mobile phase: A: 0.1% TFA in water; B: 0.1% TFA in acetonitrile. Elution gradient:0–15 min (5% B to 95% B). UV spectra were recorded on a Varian Cary 100 UV/Vis spectrometer (Santa Clara, USA). Gas chromatography was analyzed on an Agilent 7820A GC system equipped with a thermal conductivity detector (TCD) and a Supelco Carboxen 1000 carbon molecular sieve column (15 ft × 2.1 mm, O.D.: 1/8 in) using helium as the carrier gas. GC conditions: total flow: 30 ml/min; reference flow: 30 ml/min, TCD detector: 125 °C; oven temperature program: 35 °C (0–5 min), 35–225 °C (ramp: 20 °C/min), 225 °C (hold 5min), 225–35 °C (ramp: 50 °C/min). Scanning electron microscope (SEM) studies were conducted using a TESCAN VEGA-3 (Brno-Kohoutovice, Czech Republic) at voltage setting of 6 kV. Thermogravimetric analyses (TGA) were conducted on an SDT-Q600 instrument (Delaware, USA) at Georgia Institute of Technology, ramp: 30 °C/min. Cross-Polarization Magic-Angle-Spinning solid-state NMR data were acquired on a Bruker Avance III HD 500 NMR spectrometer at Georgia Institute of Technology. BW-CO-306 (De La Cruz et al., 2021) and BW-CO-103 (Ji et al., 2016) were synthesized and characterized according to previously published procedures.
2.1. Adsorption of BW-CO-306 in activated charcoal
To determine the maximal loading capacity, a saturation adsorption assay was performed. BW-CO-306 was dissolved in anhydrous ACN in preparing calibration samples with concentrations ranging from 0.019 to 0.3 mg/ml. The UV absorption was measured with a UV-Vis spectrometer and peak intensity at 280 nm was plotted against the concentration to give the calibration curve of BW-CO-306. At 25 °C, 1–6 mg BW-CO-306 was mixed with 10 mg of AC followed by the addition of 0.5 ml anhydrous ACN in a headspace vial sealed with a crimped cap with a silicon-PTFE septum. The mixture was shaken for 1 h followed by centrifuging at 12000 × g for 5 min. The supernatant was drawn out and diluted by 50 folds with ACN, followed by UV absorption measurement at 280 nm. The concentration of BW-CO-306 in the supernatant was determined via calculation using the standard curve. The adsorbed mass was calculated by Eq. 1.
| Eq.1 |
The adsorbed mass was plotted against the concentration in the solution. Nonlinear regression (Eq. 2) was applied by assuming the adsorption follows a one-phase association process in an exponential manner.
| Eq.2 |
The calculated plateau indicates the theoretical maximal mass that can be adsorbed in 10 mg of AC at 25 °C.
2.2. Preparation of BW-AC-306 formulation and AC-saccharin control formulation
BW-CO-306 (50 mg) was dissolved in 20 mL dry ACN followed by mixing with 334 mg dry activated charcoal at room temperature for 30 mins on an orbital shaker. The mixture was concentrated slowly by using a rotavapor under controlled vacuum (80 mmHg) to a stage of a free-flowing charcoal powder. Then the black powder (BW-AC-306) was further dried by a lyophilizer (vacuum 35~50 Pa, condensation trap −70 °C) overnight at room temperature. Saccharin was also separately loaded into AC by the same method. A technical point to note is the need to ensure anhydrous conditions. Besides using anhydrous ACN to dissolve BW-CO-306, the evaporation system needs to be completely dry. Dry argon or nitrogen was used to balance the pressure after evaporation. BW-CO-103 was adsorbed in AC by a soaking method (see supporting information for detail). The resulting products were named as Sac-AC and BW-AC-103, respectively.
2.3. Determination of CO release profile in physiological medium
BW-AC-306 or BW-AC-103 (about 10 mg) was weighed into a headspace vial, followed by the addition of 3 ml of the appropriate medium (PBS, SGF, SIF). Then the vial was sealed immediately with a crimped cap with a silicon-PTFE septum. The vial was incubated at 37 °C; and the headspace gas was injected into GC to determine CO release yield.
2.4. Determination of saccharin desorption from the charcoal matrix in various media
BW-AC-306 or BW-AC-103 (about 10 mg) was weighed into a headspace vial followed by addition of 3 ml of the appropriate medium (PBS, SGF, SIF). Then the vial was sealed immediately with a crimped cap with a silicon-PTFE septum. The vial was incubated at 37 °C for 24 h. The supernatant was injected into HPLC to determine the concentration of the CO release byproduct saccharin or BW-CP-103 (Wang et al., 2020).
2.5. Shelf stability of BW-AC-306
BW-AC-306 was stored at −80 °C, −15 °C, and room temperature respectively with or without silica gel desiccant. At the designated time (7 days, 6 months, and 12 months), 20 mg BW-AC-306 was weighed into a 6-ml headspace vial. Then 3 ml of PBS was added to each vial, which was sealed immediately with a crimped cap with a silicon-PTFE septum. After incubation at 37 °C for 2 h, the headspace gas was injected into and analyzed by GC. The results are presented as percentage of the CO release yield from freshly made BW-AC-306.
2.6. Preparation of dosing formulation for oral administration
BW-AC-306 (100 mg) was weighed into a 3-ml syringe. Vehicle (0.6 ml) was filled into another syringe. Then these two syringes were connected by a one-way female Luer-to-Luer adapter. Timer was started as soon as the vehicle was mixed with BW-AC-306 by injecting the vehicle to the other syringe containing the BW-AC-306. Mixing was achieved by moving both plungers back-and-forth until a uniform black slurry was formed. A known amount of slurry was injected into several headspace vials at an interval of 1 to 5 minutes. After measuring the precise weight of the slurry, 3 ml of PBS was added to each vial, which was sealed immediately. After incubation at 37 °C for 2 h, the headspace gas (250 μl) was injected into GC for analysis.
2.7. Packing BW-AC-306 in cellulose and enteric gelatin capsule and CO release in SGF and SIF
About 20 mg of BW-AC-306 was precisely weighed into size-3 cellulose capsules. The capsule was then transferred to a 6 ml headspace vial (8.8 ml actual volume) containing 3 ml of SGF (pH 1.2). The headspace vial was immediately sealed with a PTFE-silicone rubber crimp septum and incubated at 37 °C on an orbital shaker. At designated time points, 250 μl of headspace gas was injected into GC followed by injecting 250 μl air back to the vial to balance the pressure. CO in the headspace was quantified by calculation using a standard curve as described earlier. For studies using enteric capsules, size-3 gelatin capsules were firstly coated by spraying the capsule with Eudragit® L 30 D-55-based coating formula (L30D-55:triethyl citrate:talc:water = 41.67:1.25:6.25:50.83, w/w) according to the manufacture’s protocol and dried overnight at 40 °C. Spray coating was done with a lab-built oscillation spray coating apparatus (see supplemental information for detail). To prevent prodrug from contacting with moisture during the coating process, BW-AC-306 was loaded into the dried capsules, followed by sealing the seam with UV curable epoxy resin (Fig S14, SI).
2.8. In-vivo evaluation of CO delivery efficiency of BW-AC-306 formulation
CD-1 mice (25–30 g) were purchased from Envigo (Indianapolis, USA), and were fed with food and drinking water ad libitum. Animals were maintained in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institute of Health. All the animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Mississippi (IACUC protocol: 19–012).
BW-AC-306 (CO loading degree: 0.31 mmol/g) or AC-saccharin (loading degree: 0.21 mmol/g) was suspended in 3% carboxymethylcellulose (CMC) solution; free BW-CO-306 was dissolved in anhydrous dimethylacetamide (DMA) and diluted to 20% (v/v) with PEG400 as the free drug control. The concentration and stability of the free BW-CO-306 control was confirmed by HPLC analysis before the experiment. Immediately after preparation, BW-CO-306 in DMA/PEG400, BW-AC-306 in 3% CMC or the blank vehicle was administered to fed mice (n ≥ 3) via oral gavage (p.o.). At pre-determined time points post-administration, blood samples were collected by retro-orbital bleeding. Fecal samples were also collected. Circulating CO was monitored by measuring the COHb level in whole blood using a CO-oximeter (AVOXimeter 4000, Avox Systems, New York, NY, USA). The CO-oximetry measurements were made following the manufacturer’s protocol and were validated by performing the specified quality control protocol. The baseline COHb level of each mouse was also measured before administration. Due to the fast conversion of BW-CO-306 to saccharin in the aqueous solution, only the concentration of saccharin in plasma was quantified by HPLC. Non-compartmental analysis was performed by using WinNonlin® version 6 (Pharsight, MountainView, CA, USA) to obtain the pharmacokinetic parameters of saccharin, including the terminal half-life (t1/2), and area under the curve (AUC0−∞). The area under the COHb level curve (COHb AUC) was estimated using GraphPad Prism (San Diego, CA, USA).
2.9. Plasma Sample extraction
The plasma samples were extracted by a salting-out assisted liquid/liquid extraction method reported previously (Wang et al., 2020). Briefly, for a 5 μl plasma sample, 2 μl acetaminophen (200 μM) was spiked in as an internal standard. Then 23 μl of sodium chloride (4 M) and 300 μl of acetonitrile were added and mixed. After 10 min of vigorous vortex and centrifugation at 10,000×g for 5 min, the acetonitrile supernate was aspirated, evaporated to dryness in vacuum, and reconstituted in HPLC mobile phase solution prior to HPLC analysis.
2.10. HPLC Analytical methods of PK studies
An Agilent 1260 Infinity II HPLC system (Agilent Technologies, Palo Alto, CA) equipped with an autosampler, a PDA detector, and an OpenLAB CDS computer system was used for the quantitative analysis of both CO-306 and saccharin. An Agilent Poroshell 120 EC-C18 column (2.7 μm, 3 × 150 mm) with a guard column (3.5 μm, 3 × 3 mm) was used. Acetaminophen (200 μM) was used as the internal standard. The detection wavelengths for acetaminophen, BW-CO-306, and saccharin were 244 nm, 253 nm, and 266 nm, respectively. An isocratic mobile phase of 94% (v/v) phosphate buffer (20 mM, pH 3.0) and 6% (v/v) acetonitrile was used at a flow rate of 0.5 mL/min. The calibration curves for both BW-CO-306 and saccharin were established in plasma and fecal samples and validated with respect to selectivity, linearity, sensitivity, extraction recovery, stability, precision, and accuracy.
2.11. Fecal analysis
Following oral administration of BW-AC-306 or BW-CO-306 in mice, feces were collected at the 24-h and 48-h time points. To extract both the detached and activated charcoal adsorbed saccharin from feces, the fecal samples were dried, weighed, and ground thoroughly using a mortar and pestle. The fecal samples (50 mg per aliquot) were spiked with acetaminophen as an internal standard, soaked in 300 μl of a sodium chloride solution (4 M), and then homogenized using a vortex mixer and sonication bath. The homogenized fecal samples were extracted by 600 μl acetonitrile with vigorous vortex for 60 min. The samples were then centrifuged at 10,000 g for 5 min. 5 μl supernatants were mixed with 15 μl of the phosphate buffer for HPLC analysis.
3. Result and Discussion
3.1. Solid dispersion formulation of CO prodrug BW-CO-306
AC has large surface areas and is a safe and benign excipient and detoxicant widely used in the drug and food industry. Several studies have demonstrated the effectiveness of AC in trapping organic molecules of various types (Guillossou et al., 2020; Lin et al., 2019). We hypothesize that the large surface area of AC would allow the adsorbed BW-CO-306 to be readily exposed to water when immersed in an aqueous solution, leading to the desired hydrolysis reaction for CO release. Further, as an insoluble matrix, AC is expected to adsorb both the CO prodrug and the resulting byproduct in the GI, minimizing their systemic exposure.
To prepare the solid dispersion, a solvent evaporation method was chosen for loading the prodrug into the AC matrix for two reasons. First, the evaporation of the solvent gradually increases the concentration of the adsorbate, thus increases the adsorption rate. Second, the process is done in a vacuum system, thus minimizing the risk of moisture exposure. ACN was chosen as the solvent because it has been used in our previous CO release kinetic study (De La Cruz et al., 2021), and the moderate boiling point of ACN is neither too low to cause rapid precipitation of the prodrug nor too high to generate stability problem of the prodrug during evaporation process (Bhujbal et al., 2021). Additionally, adsorption of ACN in AC is known to be relatively low comparing to other aprotic solvents (Tsai et al., 2008). Due to the saturable nature of AC to adsorb organic compound, the saturation point at which BW-CO-306 forms the solid dispersion with the highest CO release was examined. We first established a saturation curve of BW-CO-306 in AC by an external calibration method (Fig. S1, supplemental information). Specifically, a calibration curve was established by measuring UV absorption of a series solutions of BW-CO-306 at different concentrations in ACN. After equilibrating AC with increasing concentrations of BW-CO-306 in ACN, the residual BW-CO-306 in the supernatant was measured by using the calibration curve. Thus, the adsorbed amount was calculated to generate the saturation curve of BW-CO-306 in AC (Fig. 2a). A maximal loading capacity of BW-CO-306 in AC was calculated to be 17.3% by weight, which gave a prodrug loading degree of 0.35 mmol/g. Further, we studied the effect of prodrug loading on CO release yield. Specifically, various quantities of BW-CO-306 was loaded into AC under the same conditions to form a series of AC formulations with different loading degrees. The actual CO release yield and the bona fide CO loading degree of these AC formulations were determined by mixing them in PBS in a headspace vial. The subsequent CO release was measured by GC analysis. It was found that the bona-fide CO loading plateaued at about 0.32 mmol/g when the prodrug loading degree was 0.36 mmol/g. Further increasing the prodrug loading to 0.4 mmol/g did not lead to a meaningful increase in CO production (Fig. 2b). CO release yield peaked at a prodrug loading degree of 0.32 mmol/g and decreased at higher loading degrees, presumably due to insufficient dispersion and thus inefficient water exposure when loading was beyond the adsorption saturation point. Therefore, we chose a prodrug loading degree of 0.32 mmol/g as the standard formulation for subsequent studies. At this loading degree, the CO release yield was 96%, giving about 0.31 mmol/g of CO. The resulting formulation, BW-AC-306, is a free-flowing black powder without macroscopic inhomogeneity (Fig. S2, supplemental information). For all subsequent studies, the batch with a loading degree of 0.32 mmol/g was used.
Fig. 2.
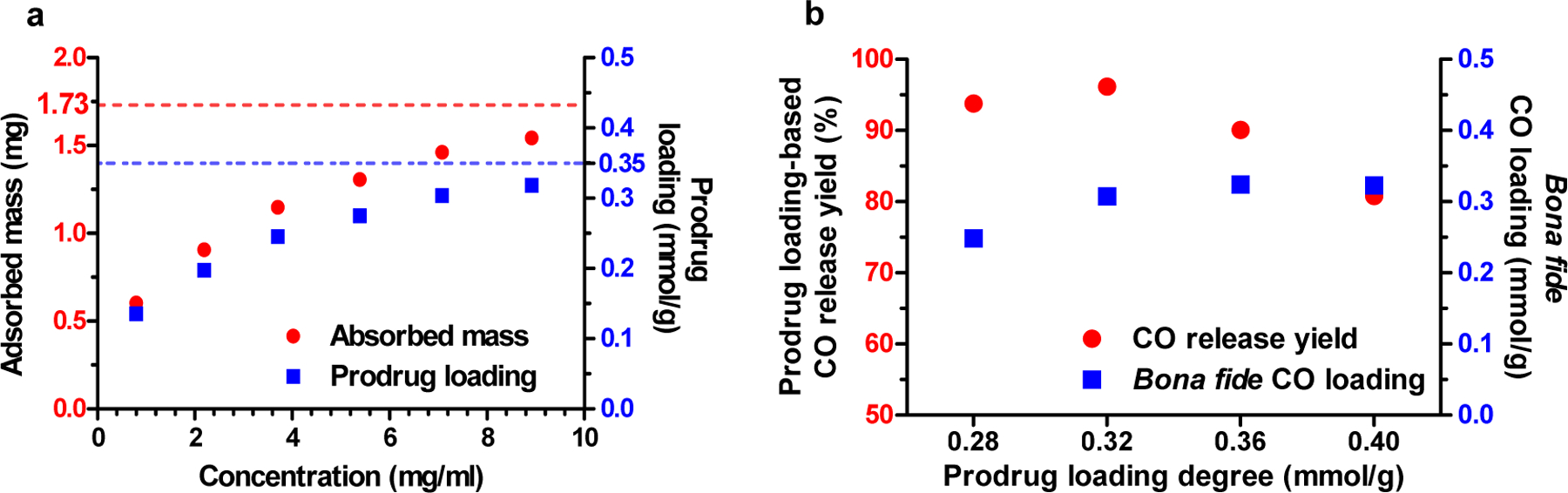
Adsorption of BW-CO-306 in activated charcoal. a. Adsorption profile of BW-CO-306 in 10 mg AC; b. Relationship between prodrug loading degree and CO release yield and bona fide CO loading degree.
To characterize the formed solid dispersion, we first verified chemical loading degree (LD, mmol/g) based on sulfur contents (2.03%) by elemental analyses and calculation by Eq.3. The calculated loading degree was 0.32 mmol/g if assuming the sulfur is from the loaded BW-CO-306. As a control, the AC sample before loading did not show any sulfur content.
| Eq.3 |
Then we assessed the thermal stability characteristics of the formulation to gain an understanding of the nature of the prodrug loading into AC. In DSC analysis (Fig. 3a), BW-AC-306 did not show a melting peak. In contrast, BW-CO-306 showed a distinct endothermic peak at 269 °C (melting followed by decomposition). Such results indicate a lack of crystallinity of BW-AC-306, supporting the fact that a molecular dispersion complex was formed instead of a physical mixture (Miriyala et al., 2017). TGA of BW-CO-306 (Fig. S4, supplemental information) showed a total weight loss about 69% (260–430 °C), presumably due to decomposition of the oxalyl group and the saccharin’s sulfonamide ring. As for BW-AC-306, the matrix started to lose weight at 182 °C, and the overall weight lost was about 5.1% at 520 °C. The weight loss for BW-AC-306 was calculated to account for 54% of the loaded prodrug if assuming the same 69% loss applies to the prodrug in the AC matrix (BW-AC-306). It is possible that the decomposition pattern is different between the ones trapped in the AC matrix and in the pure crystalline form, thus resulting in the observed difference in decomposition behaviors.
Fig. 3.
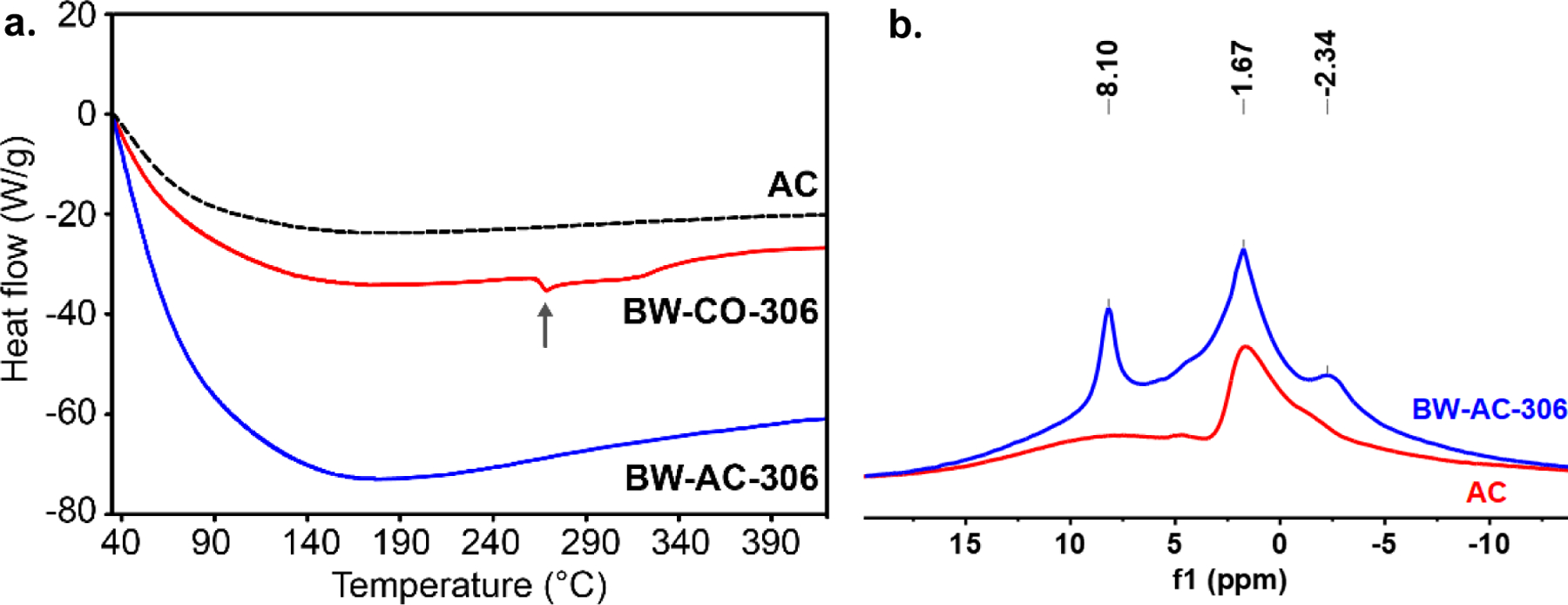
Characterization of BW-AC-306. a. DSC analysis of BW-AC-306, BW-CO-306, and AC (arrow marked the melting point at 269°C); b. MAS-NMR of BW-AC-306 and AC.
In solid-state NMR (MAS-NMR) analysis, BW-AC-306 showed characteristic peaks of aromatic protons of the saccharin moiety (Fig. 3b). In contrast, AC had none. SEM shows similarity in the surface structure between BW-AC-306 and AC, further supporting the notion of BW-CO-306 adsorption on AC, not a physical mixture (Fig 4, Fig.S5).
Fig. 4.

SEM image of (A) AC, and (B) BW-AC-306. (Scale bar: 20 μm)
3.2. In-vitro evaluation of CO release profiles
Excessive hydrolysis leads to the non-CO release pathway (Fig 1), thus lowers the overall CO release yield. In previous study, mixing an ACN solution of BW-CO-306 with PBS gave a 76 ± 6% CO release yield (De La Cruz et al., 2021). However, to our delight, the CO release yield of BW-AC-306 in PBS was tested to be 96.3 ± 0.5% calculated from the percentage of the overall quantity of the released CO (analyzed by TCD-GC) and the loaded BW-CO-306 in the test sample (Fig. 5a). It indicates that AC mitigated the excessive hydrolysis of the adsorbed BW-CO-306, potentially due to the hydrophobic environment in the AC matrix. This protective effect can also be seen under lower pH. At lower pH, the non-CO release pathway is dominant in the aqueous solution of BW-CO-306 due to the protonation of the carboxyl group of the oxalic intermediate. It was found that CO release yield was decreased to about 30% by mixing ACN solution of BW-CO-306 with pH 1.2 SGF (De La Cruz et al., 2021). BW-AC-306 formulation showed a much higher CO yield (79.1 ± 1.7%, Fig. 5a) than the non-formulated BW-CO-306. In SIF, the CO release yield of BW-AC-306 was 95.0 ± 0.3%, similar to that in PBS (Fig. 5a). Therefore, it shows that the charcoal formulation can attenuate the pH-dependency of the CO release yield and should improve the CO delivery consistency through oral delivery.
Fig. 5.
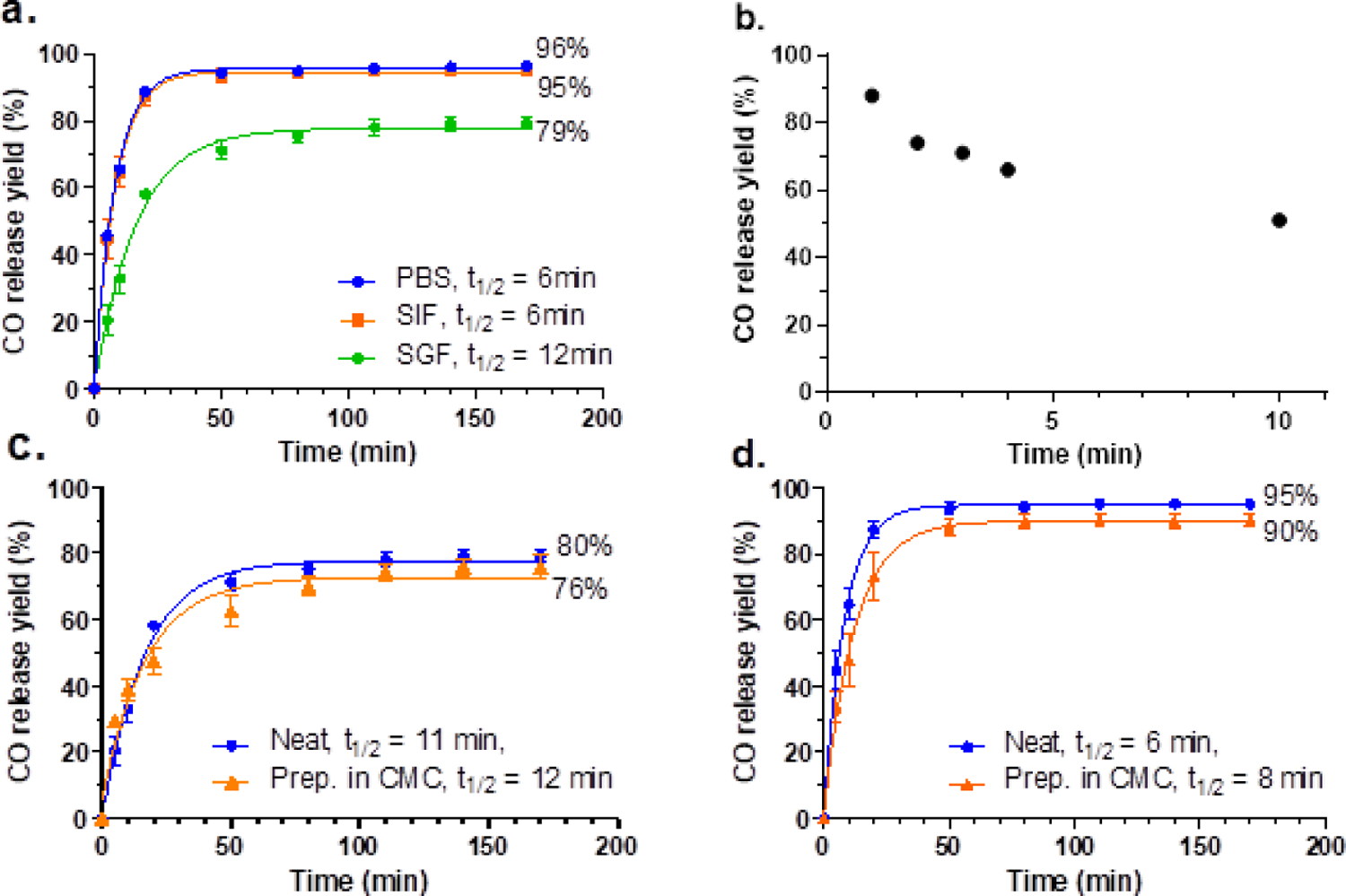
CO release profiles of BW-AC-306. a. CO release yield and kinetics of BW-AC-306 in different media. b. Complete CO release yield after pre-mixing BW-AC-306 with 3% CMC for a designated time. CO release yield of BW-AC-306 dosing formulation in SGF (c) and in SIF (d); the dosing formulation was directly (neat) or premixed with CMC for 1 min followed by adding SGF or SIF to release CO (n = 3).
Release kinetic studies in these simulated physiological media showed the release half-life being longer (t1/2 = 6–12 min) than that of BW-CO-306 in the corresponding medium (t1/2 = 1.28 min) (De La Cruz et al., 2021). possibly due to the slower reaction kinetic between water and the adsorbed prodrug in BW-AC-306 because of the hydrophobic environment of the charcoal surface, the heterogeneous nature of the BW-AC-306 formulation, and possibly other forms of physical hindrance to water diffusion. Thus, compared to BW-CO-306, the BW-AC-306 formulation led to a higher CO release yield, slower CO release kinetics, and reduced pH dependence in gastric-intestinal fluids. The overall increased CO production yield of BW-AC-306 also suggests that the factors that led to a slower reaction rate in the charcoal formulation were favorable for the CO release process, possibly by attenuating the non-CO releasing hydrolysis pathway as shown in Fig.1.
3.3. Shelf stability of BW-AC-306 formulation
With the demonstration of the aforementioned beneficial effects of the charcoal formulation to the CO release profile, we were interested in examining the effect on the stability of the adsorbed prodrug. Therefore, we assessed the shelf-life of the prodrug formulation under various conditions, including variations of storage temperature and the addition of silica gel desiccant. Specifically, we determined the CO release yield after a defined period of storage and found that in the presence of silica gel desiccant, BW-AC-306 was stable for at least 12 months (the duration of the study) at −78 °C and 6 months at −18 °C (Table 1). Room-temperature storage led to significant decomposition with or without desiccant. Such results demonstrate the possibility for long-term storage and suggest the need for further optimization of the formulation process to increase its stability at room temperature.
Table 1.
Shelf stability of BW-AC-306
| Desiccant | Storage temperature |
||
|---|---|---|---|
| −80 °C | −18 °C | 25 °C | |
| Without desiccant | 55% (12 month) | 86% (14 days) | 26% (7 days) |
| With desiccant | 96% (12 month) | 91% (6 month) | 72% (7 days) |
Percentage of the overall CO release (%) after storage.
3.4. Dosing formulations for oral administration
BW-AC-306 showed robust CO release without the need for organic solvents in buffer solutions. To study its in-vivo CO delivery, we needed a proper vehicle to facilitate oral gavage in mice. As an insoluble powder, it has to be prepared in a suspension with a viscous vehicle so that it can be dosed through a gavage needle. It is critical that the CO release yield should not be significantly compromised by the vehicle between the time of preparation of the dosage formulation and oral gavage. We at first tried to use PEG400 to make the dosage formulation of BW-AC-306. However, it led to a significantly lower yield than direct incubation (Fig S6a and S6b, see supplemental information for discussion). On the other hand, due to the impact of PEG400 in COHb level (Fig S6c), we explored if BW-AC-306 can be prepared in a more benign vehicle without sacrificing CO release yield. CMC is a widely used thickener for food and drug excipient due to its viscosity and low toxicity. In the food industry, there are no quantitative restrictions on its use, nor does its addition to food require to be declared (Martino et al., 2017). Therefore, we tested if the CMC solution could be used as a vehicle to form a suspension without significant loss of CO delivery capability. It was found that the suspension made with 3% CMC solution in water was able to pass through an 18-gauge gavage needle, and the minimum ratio of CMC solution to the weight of BW-AC-306 was 6 μL (3% CMC) per 1 mg of BW-AC-306 (supplemental information, Movie 1). Because the aqueous medium of the CMC solution may trigger CO release, we studied the CO release yield after preparing BW-AC-306 in a CMC suspension and found that within a 10-min period, CO release yield gradually decreased to 51% (Fig.5b). Nevertheless, 51% is similar to the yield we obtained from the BW-CO-306 in a DMA/PEG400 formulation (De La Cruz et al., 2021) and BW-AC-306 prepared in a PEG400 formulation in 1 minute (Figure 3c, 3d, S6a, and S6b). Because 10 min is within the time required for oral gavage in mice, we found it was feasible to deliver the required amounts of CO to mice using this method. Regarding the release kinetics, BW-AC-306 CMC suspension gave a similar CO release rate in both SGF and SIF when compared to the ones without CMC (neat), which were all faster than the one prepared in PEG400 (Fig. 5c, 5d). Such results indicate that, unlike PEG400, 3% CMC did not affect the reaction between the adsorbed BW-CO-306 and water. In general, 3% CMC was shown to be a good and practical vehicle that had minor influence on CO releasing kinetic and CO release yield compared to PEG400. It has to be mentioned that a vehicle is not obligatory for studies in larger animals in which a gel capsule administration is feasible.
3.5. In-vivo evaluation of CO delivery efficiency of the BW-AC-306
After successful demonstration of CO release from BW-AC-306 in-vitro, we tested its CO delivery profiles in animal models. To do so, the COHb level in whole blood measured by a CO-oximeter is commonly used as the key exposure parameter. Even though COHb may not always reflect the systemic CO exposure levels, especially after inhalation exposure (Yang et al., 2021b), this is the most practical and reliable way of assessing systemically available CO in the current state of the art. Following oral administration, the COHb level is expected to parallel systemic exposure much more so than after inhalation exposure (Yang et al., 2021b). It was observed that, in healthy CD-1 mice, the baseline level of COHb was typically between 0.5–1.8%. As shown in Fig. 6, the COHb level increased quickly following p.o. administration of BW-AC-306 (50 mg/kg and 100 mg/kg of the equivalent doses of BW-CO-306) prepared in 3% CMC solution with a tmax of around 20 min and remained elevated above the baseline for 1–1.5 h (p < 0.05). In comparison, 100 mg/kg of BW-CO-306 dosed with DMA/PEG400 vehicle gave a peak COHb level of 5.1% with a shorter tmax (10 min), indicating a burst of CO release. For BW-AC-306 dosed with CMC, the peak COHb level was 6.0% for the 100 mg/kg dose and 3.1% for the 50 mg/kg dose, showing a clear dose-response relationship. As a reference, the COHb level was unaffected (data not shown) following p.o. administration of AC-saccharin or blank AC-CMC vehicle, indicating the origin of the elevated COHb level being from BW-AC-306. The COHb level gradually returned to the baseline after the peak was reached at 20 min. The results are expected based on the release t1/2 of 12 min in CMC (Fig. 5) and elimination half-life of about 22 min for CO in mouse (Tyuma et al., 1981). BW-AC-306 dosed with CMC vehicle also gave a higher peak COHb level than using PEG400 as the vehicle in the previous study (De La Cruz et al., 2021), consistent with the in-vitro study result that PEG400 resulted in a lower CO yield. The overall CO delivery efficiency and availability could be assessed by calculating the area under the COHb curve (COHb AUC), which is defined as the area under the blood COHb curve subtracted by the pre-administration baseline from time zero to the last sampling time point. Compared to BW-CO-306, BW-AC-306 yielded a higher COHb AUC (Table 2). Therefore, with a higher COHb Cmax level (6.0 vs. 5.1% at 100 mg/kg), a more sustained elevation of COHb, and a higher COHb AUC (4.6 vs. 2.9 at 100 mg/kg), systemic CO exposure after oral administration of BW-AC-306 is more efficient than that of BW-CO-306. As a reference, these COHb levels (3–7.5%) were previously shown to be therapeutically relevant in kidney protection (De La Cruz et al., 2021), liver injury protection (Zheng et al., 2018), postoperative ileus (Nakao et al., 2006), sickle cell disease (Belcher et al., 2018), among others (Yang et al., 2020; Yang et al., 2021a). We also verified the distribution of the ingested AC formulation in the GI system. Thirty minutes after oral administration of BW-AC-306, isolation of the GI tract of the mice showed black charcoal residues in the stomach and ileum. Considering the CO release half-life of about 10 min, the distribution of the charcoal indicates stomach and ileum being the primary sites of CO release.
Fig. 6.
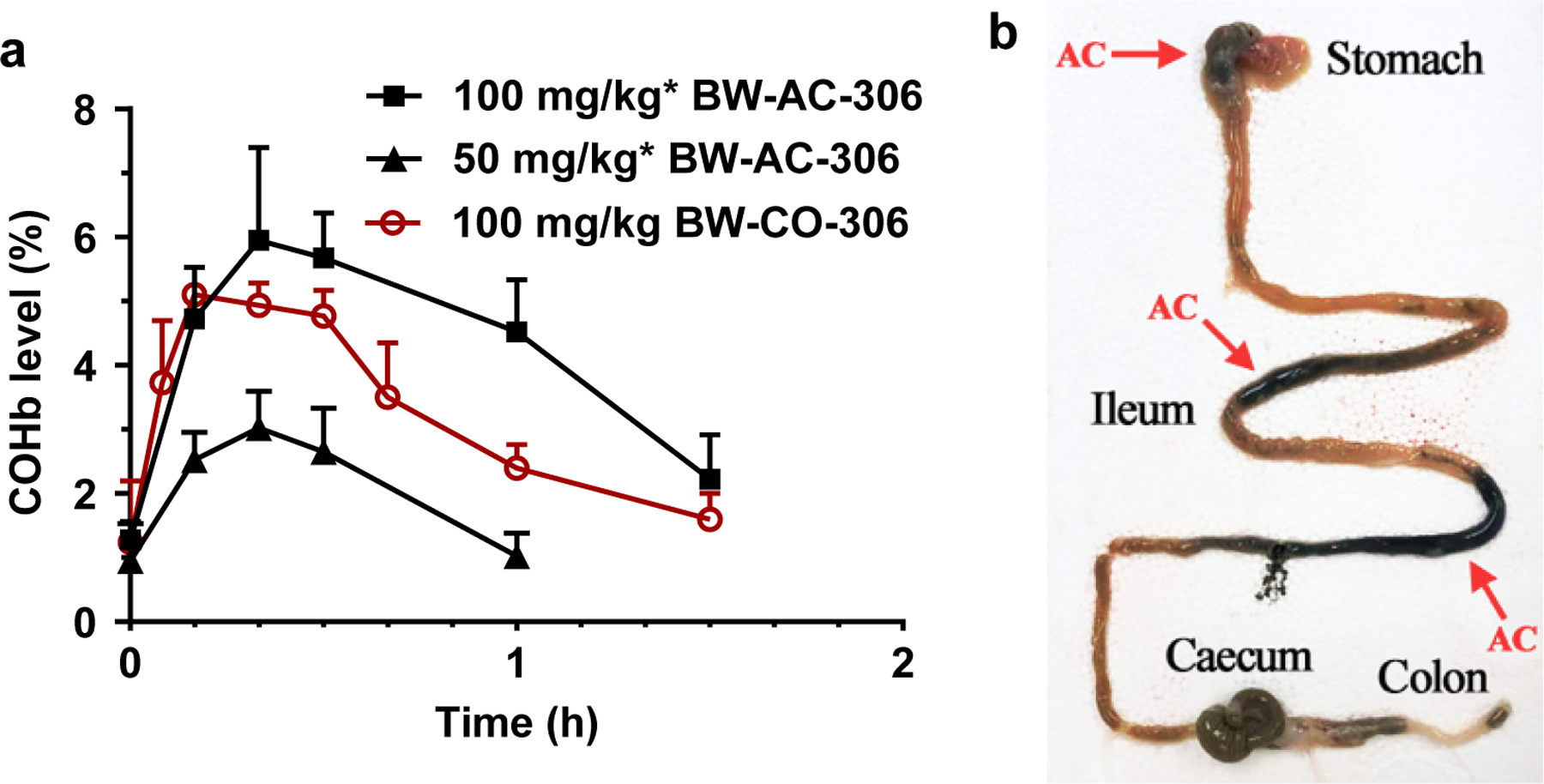
In-vivo CO delivery by oral administration of BW-AC-306. a. COHb level in blood following p.o. administration of BW-AC-306 (50 and 100 mg/kg). Results are presented as the mean ± SD (n ≥ 3). (*BW-AC-306 was dosed at same equivalent dosage of BW-CO-306 according to the loading degree); b. Distribution of AC in GI tract at 30 min after oral gavage BW-AC-306 (arrows marked the location of AC).
Table 2.
Pharmacokinetic parameters of blood COHb levels following p.o. administration of AC-CO-306 and free CO-306.
| Compound | Vehicle | Dose (mg/kg) | COHb AUC (%·h) | COHb Cmax | COHb Tmax |
|---|---|---|---|---|---|
| BW-AC-306 | 3% CMC | 100 | 4.6 | 6.0% | 20 min |
| 50 | 1.3 | 3.1% | 20 min | ||
| BW-CO-306 | DMA/PEG400 | 100 | 2.9 | 5.1% | 10 min |
COHb AUC: area under the curve for blood COHb level subtracting the pre-administration baseline from time zero to the last sampling time point; COHb Cmax: peak carboxyhemoglobin level; COHb Tmax: time at which peak COHb level was reached; Results are presented as the mean ± SD (n ≥ 3).
3.6. BW-AC-306 lowers systemic exposure to the CO-release byproduct
Saccharin as the CO-release byproduct is known to be adsorbed on AC (Bernardo et al., 2006), which is expected to lower its systemic exposure. By analyzing the saccharine concentration in the supernatant of the in-vitro CO release assays with HPLC, we were unable to detect saccharin in SGF after incubating BW-AC-306 (neat or prepared in 3% CMC) for 24 h (Table 3, Fig. S7). On the other hand, we were able to detect free saccharine in the supernatant after CO release in SIF at a level that corresponds to 11% or 13% of the total loaded saccharin for either neat BW-AC-306 or the one prepared in 3% CMC, respectively. Such results are consistent with the higher percentage of ionization of saccharin under the pH in SIF (pH 6.8) than in SGF (pH 1.2), which likely leads to a lower level of saccharine adsorption on AC. However, when BW-AC-306 was prepared in PEG400, much more dissociated saccharin was detected in the supernatant, indicating the desorption ability of an organic solvent such as PEG400.
Table 3.
In-vitro evaluation of the dissociated saccharin after CO release from BW-AC-306.
| Media | Preparation | Saccharin in supernatant (% of the loaded amount) |
|---|---|---|
| SGF | Neat | N.D. |
| 3% CMC | N.D. | |
| PEG400 | 7% | |
|
| ||
| SIF | Neat | 11% |
| 3% CMC | 13% | |
| PEG400 | 71% | |
N.D.: not detected.
In animal model studies, both the plasma Cmax and AUC0–2h of saccharin following p.o. administration of BW-AC-306 was lower than that of BW-CO-306, indicating a lower exposure level of saccharin (Fig. 7 and Table 4). This contrasts with the higher COHb AUC resulting from BW-AC-306 administration. Tmax of plasma saccharin after dosing BW-AC-306 was also about 10 min longer than that of BW-CO-306, consistent with the slower CO release kinetics of BW-AC-306 than BW-CO-306. The plasma saccharin level then declined gradually after the peak level was reached. The time course of plasma saccharin level is the net result of saccharin generation following CO release from BW-AC-306 and the absorption and disposition of saccharin itself. The pharmacokinetics of saccharin has been extensively studied in humans as well as in animals (Colburn et al., 1981; Renwick, 1985). As a low-pKa small organic molecule, saccharin is almost completely ionized at physiological pH, and yet has high oral bioavailability. The majority of the orally given saccharin is known to be eliminated in the urine unchanged at a fast rate due to the hydrophilic nature of its ionized form and metabolic inertness. When orally given a formulation with sustained drug release, the terminal phase of plasma drug concentration can sometimes be rate-limited by the drug absorption rather than the elimination kinetics, a phenomenon known as flip-flop pharmacokinetics (Colburn et al., 1981; Yanez et al., 2011). The elimination half-life of saccharin following p.o. administration of BW-AC-306 was longer than that of BW-CO-306, suggesting that saccharin was generated and absorbed in a more sustained manner from BW-AC-306 and the elimination rate of saccharin was slower due to flip-flop kinetics.
Fig. 7.
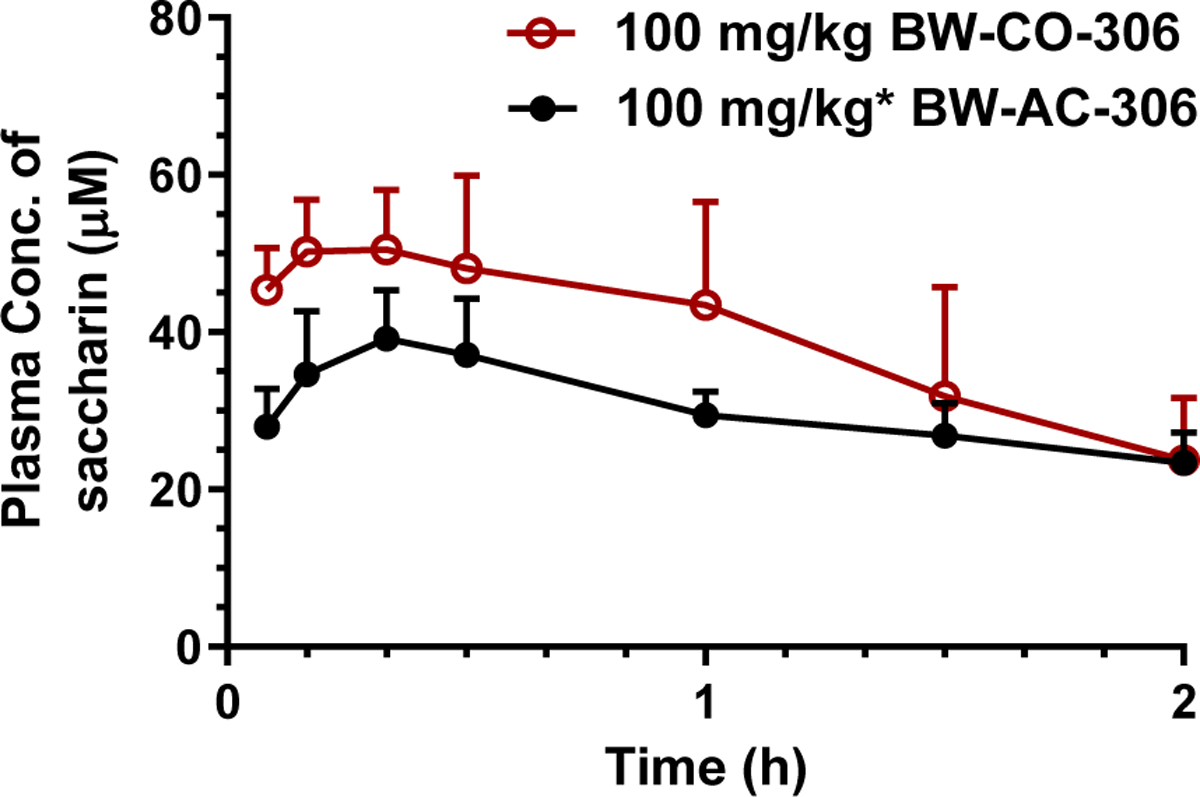
Comparison of plasma saccharin concentration between BW-CO-306 dosed with DMA/PEG400 and BW-AC-306 dosed with 3% CMC. (mean ± SD, n ≥ 3. *BW-AC-306 dosed as same equivalent of BW-CO-306 at 100 mg/kg according to the loading degree.)
Table 4.
Pharmacokinetic parameters of saccharin following p.o. administration of BW-AC-306 and BW-CO-306.
| Saccharin origin | Dose (mg/kg) | Tmax (min) | t1/2 (h) | AUC0–2h (μM·h) | Cmax (μM) |
|---|---|---|---|---|---|
| BW-AC-306 | 100 | 20 | 2.98 | 58.4 | 39.2 |
| BW-CO-306 | 100 | 10 | 1.14 | 76.2 | 50.5 |
t1/2: terminal half-life; AUC0–2h: area under the plasma drug concentration curve from time zero to 2 h; Results are presented as the mean ± SD (n ≥ 3).
Mouse fecal analysis after BW-AC-306 administration showed that 68 ± 5% of saccharin was recovered within 24 h after oral administration. Beyond 24 h, saccharine was below the detectable level (Table S1, supplemental information). As orally administered saccharin is known to mainly undergo renal excretion in its original form (Magnuson et al., 2016; Renwick, 1985), the high recovery rate of saccharin from the fecal sample indicates that most of the saccharin is still adsorbed in the AC and eliminate in feces without systemic absorption. Together with the facts of the pharmacokinetic study, BW-AC-306 offers a lower systemic exposure to the saccharine byproduct than BW-CO-306 in-vivo.
3.7. Encapsulation of BW-AC-306 for controlled CO release
Considering the future studies in larger animals and human, in which oral administration in a capsule form is feasible, and premixing BW-AC-306 with CMC is not needed owning to its inherent ability to release CO upon contacting with water. We were interested in encapsulating BW-AC-306 to achieve the concept of “CO in a pill.” For this, BW-AC-306 was encapsulated in either cellulose or gelatin capsules (Fig. 8a) with or without enteric coating to allow for targeted CO release in the upper or lower GI system. In SGF, the overall CO release yield from BW-AC-306 packed in normal cellulose capsules (Fig. 8a) was similar to that of incubating neat BW-AC-306 with SGF (Fig. 5c). However, the CO release rate (t1/2 = 25 min) was slower than the latter case, presumably due to delays caused by the gradual disintegration of the capsule. The results indicate that the encapsulation does not change the overall CO release yield and the release kinetics can be tuned by controlling the dissolution profile of the capsule. Because of the ability for an enteric coating to protect the capsule from dissolution in gastric fluid and to allow for dissolution at a higher pH (>5.5) in SIF, we also studied BW-AC-306 packed in the enteric gelatin capsule (Fig. 8a) for controlled CO release in SIF (Fig. 8c) by first exposing the capsules to SGF and then SIF. There was no CO release by incubating the loaded enteric capsule in SGF within 60 min. After changing the medium to SIF, complete CO release was achieved within 2 h with the overall CO yield (Fig. 8c) being similar to that from incubating neat BW-AC-306 (Fig. 5d). Although variation in release kinetic was seen among different capsules, probably due to coating variance, the results clearly showed that BW-AC-306 was able to release defined amounts of CO in a controllable manner in a simulated intestinal environment, but not a gastric environment. It should be noted that there have been newly developed ready-to-fill enteric capsule (such as Eudracap™ by Evonik) that allows for direct encapsulation of BW-AC-306. The availability of such ready-to-fill capsules will significantly facilitate the manufacturing process in the future and improve the uniformity of the “CO in a capsule.”
Fig. 8.
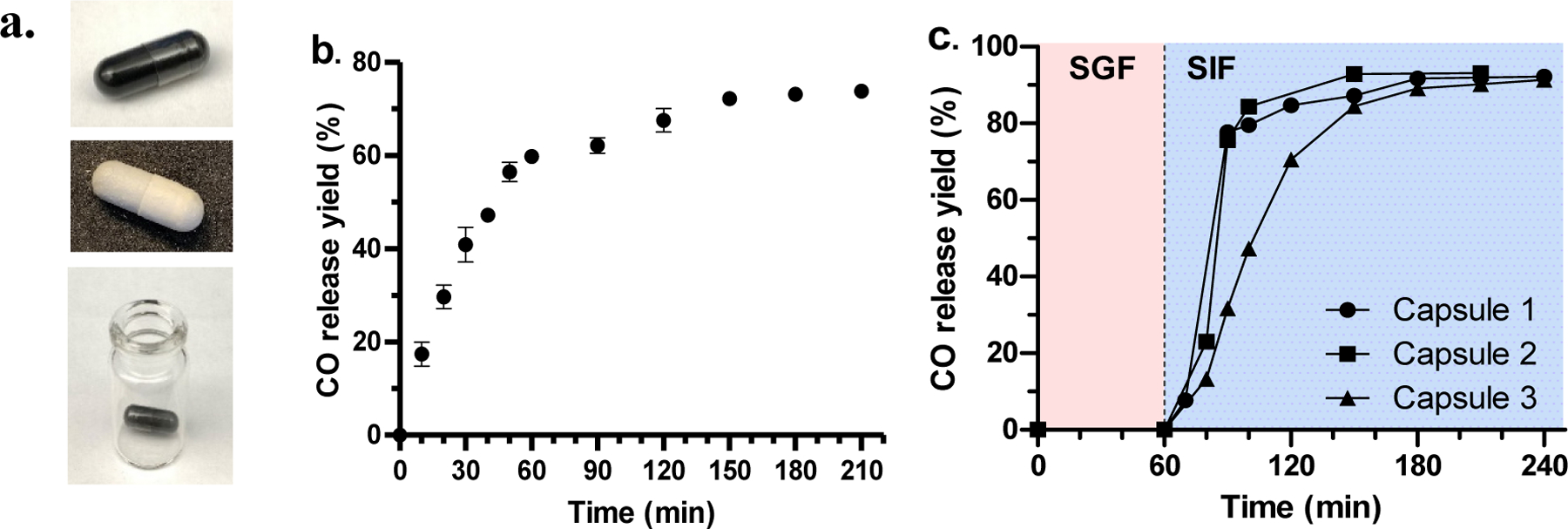
Encapsulated CO prodrug formulation. a. BW-AC-306 packed in cellulose capsule (top), enteric gelatin capsule (middle), and kept in headspace vial for CO release study (bottom); CO release profile of BW-AC-306: b. packed in cellulose capsule and incubated in SGF; c. packed in enteric coated gelatin capsule and incubated in SGF for 1 h followed by changing the medium to SIF.
3.8. Application of charcoal formulation beyond BW-AC-306
With the success of applying activated charcoal to resolve the paradoxical problems in formulating the decarboxylation-decarbonylation type of CO prodrug BW-CO-306, we further tested the same strategy for other organic CO prodrugs to study its general applicability (Ji and Wang, 2018). As an example, we chose BW-CO-103, which utilizes an intramolecular Diels-Alder reaction to initiate the formation the norbornadienone moiety for CO generation (Ji et al., 2016). Water exposure plays a critical role in promoting Diels-Alder reaction partially through hydrophobic forces. BW-CO-103 has been studied for efficacy in multiple animal models as well as pharmacokinetics through different administration routes (Wang et al., 2020).
At first, the saturation point was determined by the adsorption curve method using a similar method for BW-AC-306 (see supporting information for detail). From the saturation curve (Fig. S8b), the maximal adsorption capacity in 10 mg AC at 25 °C was calculated to be about 2.7 mg. However, the attempt to load BW-CO-103 through evaporation led to the precipitation during the evaporation process due to the lower solubility of BW-CO-103. As BW-CO-103 is not as sensitive to moisture as BW-AC-306 and to avoid precipitation problems, we chose to use an adsorption-centrifugation process to prepare the activated charcoal formulation (see supporting information for detail). In such a process, AC was soaked in ACN solution of BW-CO-103 followed by centrifugation and removal of the supernatant and drying to get BW-CO-103 loaded AC: BW-AC-103. Based on the saturation studies of BW-CO-103, a concentration at 9 mg/ml reached the plateauing point for efficient loading (Fig. S8B). The ratio between AC and BW-CO-103 was 3.5:1 (w/w), which was slightly lower than the calculated ratio (3.7:1) to assure sufficient loading. Therefore, in a loading experiment of soaking 98.5 mg of AC in a solution of BW-CO-103 (9 mg/ml, 3.1 ml), the weight increase of AC after loading was found to be 26 mg. This agrees with the precalculated weight increase (26.6 mg) according to the saturation curve (see supplemental information for detail), which gave a prodrug loading degree of 0.5 mmol/g.
The AC-loaded formulation BW-AC-103 was further characterized with DSC, TGA and solid-state NMR (Fig. S9 and S10). The thermal decomposition patterns of BW-CO-103 with or without AC adsorption were found to be very different, indicating an essential difference in the molecular arrangement patterns. In solid-state NMR studies, new peaks were found in the aromatic region compared to AC itself, presumably can be attributed to the aromatic proton of BW-CO-103. CO release kinetics and yields were determined in PBS, SGF, and SIF (Fig 9). The half-lives were about 1.3 h, similar to that of free BW-CO-103 in DMSO:PBS (1:4) solution (Ji et al., 2016), and pH was found to have no influence on CO release kinetics. The bona fide CO loading degree was determined to be 0.18 mmol/g, which gave a CO release yield of 59.2 ± 0.3%. The lower CO release yield might be attributed to the spontaneous intramolecular D-A reaction leading to CO release during the loading process. BW-AC-103 benefits from the AC formulation that it circumvents the need for an organic cosolvent such as DMSO or a solubilizer such as Kolliphor HS 15 for drug administration; and CO release can be implemented in a completely aqueous medium. HPLC analysis of the PBS supernatant showed neither BW-CO-103 nor the byproduct BW-CP-103 after CO release (Wang et al., 2020) (Fig S11). Such results indicate that AC may also lower systemic exposure to the CO prodrug and CO release byproduct by trapping them in the AC matrix. Therefore, the AC formulation could be a potential platform to address developability issues such as low water solubility, systemic exposure to the carrier molecule, and targeted GI delivery of organic CO prodrugs.
Figure 9.
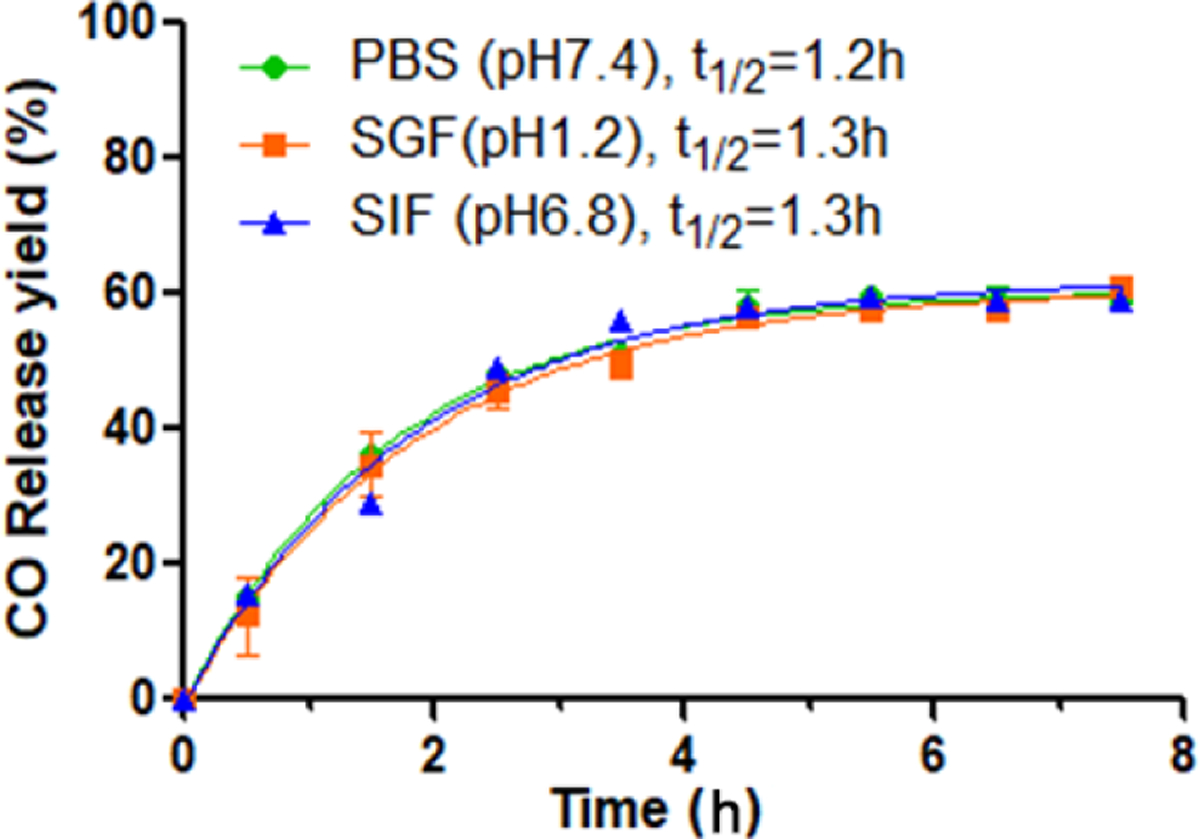
CO release profile of BW-AC-103 in physiological mediums (n=3).
4. Conclusion
We have developed a solid formulation BW-AC-306 by adsorption of organic CO prodrug BW-CO-306 in activated charcoal. The resulting non-classic solid dispersion addressed the paradoxical issue of the CO prodrug being insoluble in water and requiring water for activation. Activated charcoal facilitated the reaction between the adsorbed BW-CO-306 and water for CO release, increased CO release yield, minimized pH dependency, trapped BW-CO-306 and byproduct saccharine in the charcoal matrix, further minimizing the systemic exposure to these chemical entities. Through in-vitro and in-vivo CO release studies, we have demonstrated that BW-AC-306 can deliver defined amounts of CO in a controllable manner. Along the same line, activated charcoal was also applied to one of our CO prodrugs based on the cheletropic release of CO from norbornadienone generated from a Diels-Alder reaction. The resulting formulation BW-AC-103 also features CO release in aqueous medium and retention of the byproduct after CO release in the AC matrix. By simply choosing a readily available encapsulation method, controlled CO delivery to the stomach or the lower GI can be achieved. With the in-vivo verification of its therapeutic activity in our previous study of kidney injury protection model, we envision this CO delivery approach being used in further preclinical and clinical assessment in the near future.
Supplementary Material
Acknowledgment
The authors are grateful for financial support from National Institutes of Health (R01DK119202), the Georgia Research Alliance for an Eminent Scholar fund, and internal resources at Georgia State University. The DSC/TGA analyses were performed at the Georgia Institute of Technology for Electronics and Nanotechnology, a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (Grant ECCS-2025462). We thank Mr. Philip Anschutz and Dr. Paul J Joseph for the DSC/TGA analyses. We also thank Evonik for gift samples of the enteric coating material and technical support in enteric formulation preparation.
Declaration of Competing Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Binghe Wang reports financial support was provided by National Institutes of Health. The co-authors Xiaoxiao Yang, Ladie Kimberly De La Cruz, and Binghe Wang, have patent (US20210238155A1) pending to Georgia State University Research Foundation Inc.
Abbreviations:
- CO
carbon monoxide
- AC
activated charcoal
- ACN
acetonitrile
- DMA
dimethylacetamide
- PEG
polyethylene glycol
- SGF
simulated gastric fluid
- SIF
simulated intestine fluid
- PBS
phosphate buffered saline
- DSC
differential scanning calorimetry
- TGA
thermogravimetry analysis
- MAS-NMR
magic angel spinning-NMR
- SEM
surface scanning electron microscopy
- GC
CMC carboxyl methylcellulose
- COHb
carboxyhemoglobin
- AUC
area under the curve.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT authorship contribution statement
X.Y.: Conceptualization, Methodology, Investigation, Writing-Original Draft. W.L.: Conceptualization, Methodology, Investigation, Writing - Review & Editing. M.W.: Methodology, Investigation, Writing - Review & Editing. L.K.D.L.C.: Conceptualization, Investigation. C.T., and B.W.: Conceptualization, Writing - Review & Editing, Supervision, Funding acquisition. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Abeyrathna N, Washington K, Bashur C, Liao Y, 2017. Nonmetallic carbon monoxide releasing molecules (CORMs). Org Biomol Chem 15, 8692–8699. [DOI] [PubMed] [Google Scholar]
- Alghazwat O, Talebzadeh S, Oyer J, Copik A, Liao Y, 2021. Ultrasound responsive carbon monoxide releasing micelle. Ultrason Sonochem 72, 105427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belcher JD, Gomperts E, Nguyen J, Chen C, Abdulla F, Kiser ZM, Gallo D, Levy H, Otterbein LE, Vercellotti GM, 2018. Oral carbon monoxide therapy in murine sickle cell disease: Beneficial effects on vaso-occlusion, inflammation and anemia. PLoS One 13, e0205194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardo EC, Fukuta T, Fujita T, Ona EP, Kojima Y, Matsuda H, 2006. Enhancement of saccharin removal from aqueous solution by activated carbon adsorption with ultrasonic treatment. Ultrason Sonochem 13, 13–18. [DOI] [PubMed] [Google Scholar]
- Bhujbal SV, Mitra B, Jain U, Gong Y, Agrawal A, Karki S, Taylor LS, Kumar S, Tony Zhou Q, 2021. Pharmaceutical amorphous solid dispersion: A review of manufacturing strategies. Acta Pharm Sin B 11, 2505–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Zheng B, Cheng S, Zhang G, Hu J, 2020. Metal-free carbon monoxide-releasing micelles undergo tandem photochemical reactions for cutaneous wound healing. Chem Sci 11, 4499–4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu LM, Shaefi S, Byrne JD, Alves de Souza RW, Otterbein LE, 2021. Carbon monoxide and a change of heart. Redox Biol 48, 102183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colburn WA, Bekersky I, Blumenthal HP, 1981. A preliminary report on the pharmacokinetics of saccharin in man: single oral dose administration. J Clin Pharmacol 21, 147–151. [DOI] [PubMed] [Google Scholar]
- De La Cruz LK, Yang X, Menshikh A, Brewer M, Lu W, Wang M, Wang S, Ji X, Cachuela A, Yang H, Gallo D, Tan C, Otterbein L, de Caestecker M, Wang B, 2021. Adapting decarbonylation chemistry for the development of prodrugs capable of in vivo delivery of carbon monoxide utilizing sweeteners as carrier molecules. Chem Sci 12, 10649–10654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Cruz LKC, Benoit SL, Pan Z, Yu B, Maier RJ, Ji X, Wang B, 2018. Click, Release, and Fluoresce: A Chemical Strategy for a Cascade Prodrug System for Codelivery of Carbon Monoxide, a Drug Payload, and a Fluorescent Reporter. Org Lett 20, 897–900. [DOI] [PubMed] [Google Scholar]
- Goldbaum LR, Orellano T, Dergal E, 1976. Mechanism of the toxic action of carbon monoxide. Ann Clin Lab Sci 6, 372–376. [PubMed] [Google Scholar]
- Guillossou R, Le Roux J, Brosillon S, Mailler R, Vulliet E, Morlay C, Nauleau F, Rocher V, Gaspéri J, 2020. Benefits of ozonation before activated carbon adsorption for the removal of organic micropollutants from wastewater effluents. Chemosphere 245, 125530. [DOI] [PubMed] [Google Scholar]
- Huang Y, Dai WG, 2014. Fundamental aspects of solid dispersion technology for poorly soluble drugs. Acta Pharm Sin B 4, 18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X, Aghoghovbia RE, De La Cruz LKC, Pan Z, Yang X, Yu B, Wang B, 2019a. Click and Release: A High-Content Bioorthogonal Prodrug with Multiple Outputs. Org Lett 21, 3649–3652. [DOI] [PubMed] [Google Scholar]
- Ji X, de La Cruz LK, Pan Z, Chittavong V, Wang B, 2017a. pH-sensitive Metal-free Carbon Monoxide Prodrugs with Tunable and Predictable Release Rates. Chem Commun 53, 9628–9631. [DOI] [PubMed] [Google Scholar]
- Ji X, Ji K, Chittavong V, Yu B, Pan Z, Wang B, 2017b. An Esterase-activated Click and Release Approach to Metal-free CO-prodrugs. Chem Commun 53, 8296–8299. [DOI] [PubMed] [Google Scholar]
- Ji X, Pan Z, Li C, Kang T, De La Cruz LKC, Yang L, Yuan Z, Ke B, Wang B, 2019b. Esterase-Sensitive and pH-Controlled Carbon Monoxide Prodrugs for Treating Systemic Inflammation. J Med Chem 62, 3163–3168. [DOI] [PubMed] [Google Scholar]
- Ji X, Wang B, 2018. Strategies toward Organic Carbon Monoxide Prodrugs. Accounts of Chemical Research 51, 1377–1385. [DOI] [PubMed] [Google Scholar]
- Ji X, Zhou C, Ji K, Aghoghovbia RE, Pan Z, Chittavong V, Ke B, Wang B, 2016. Click and Release: A Chemical Strategy toward Developing Gasotransmitter Prodrugs by Using an Intramolecular Diels–Alder Reaction. Angew Chem Int Ed Engl 55, 15846–15851. [DOI] [PubMed] [Google Scholar]
- Kennedy GL Jr., Sherman H, 1986. Acute and subchronic toxicity of dimethylformamide and dimethylacetamide following various routes of administration. Drug Chem Toxicol 9, 147–170. [DOI] [PubMed] [Google Scholar]
- Klein S, 2010. The use of biorelevant dissolution media to forecast the in vivo performance of a drug. AAPS J 12, 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kueh JTB, Stanley NJ, Hewitt RJ, Woods LM, Larsen L, Harrison JC, Rennison D, Brimble MA, Sammut IA, Larsen DS, 2017. Norborn-2-en-7-ones as physiologically-triggered carbon monoxide-releasing prodrugs. Chem Sci 8, 5454–5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus LS, Benninghoff AD, Berreau LM, 2020. Development of Triggerable, Trackable, and Targetable Carbon Monoxide Releasing Molecules. Acc Chem Res 53, 2273–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B. q., Dong X, Fang S. h., Gao J. y., Yang G. q., Zhao H, 2011. Systemic toxicity and toxicokinetics of a high dose of polyethylene glycol 400 in dogs following intravenous injection. Drug Chem Toxicol 34, 208–212. [DOI] [PubMed] [Google Scholar]
- Lin Y, Ma J, Liu W, Li Z, He K, 2019. Efficient removal of dyes from dyeing wastewater by powder activated charcoal/titanate nanotube nanocomposites: adsorption and photoregeneration. Environ Sci Pollut Res Int 26, 10263–10273. [DOI] [PubMed] [Google Scholar]
- Magnuson BA, Carakostas MC, Moore NH, Poulos SP, Renwick AG, 2016. Biological fate of low-calorie sweeteners. Nutr Rev 74, 670–689. [DOI] [PubMed] [Google Scholar]
- Mann BE, 2012. CO-Releasing Molecules: A Personal View. Organometallics 31, 5728–5735. [Google Scholar]
- Martino JV, Van Limbergen J, Cahill LE, 2017. The Role of Carrageenan and Carboxymethylcellulose in the Development of Intestinal Inflammation. Front Pediatr 5, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JJ, Leviseur C, 1984. Cardiorespiratory responses to intestinal injection of carbon monoxide. Pharmacol Biochem Behav 21 Suppl 1, 103–107. [DOI] [PubMed] [Google Scholar]
- Miriyala N, Ouyang D, Perrie Y, Lowry D, Kirby DJ, 2017. Activated carbon as a carrier for amorphous drug delivery: Effect of drug characteristics and carrier wettability. Eur J Pharm Biopharm 115, 197–205. [DOI] [PubMed] [Google Scholar]
- Motterlini R, Otterbein LE, 2010. The therapeutic potential of carbon monoxide. Nat Rev Drug Discov 9, 728–743. [DOI] [PubMed] [Google Scholar]
- Nakao A, Toyokawa H, Tsung A, Nalesnik MA, Stolz DB, Kohmoto J, Ikeda A, Tomiyama K, Harada T, Takahashi T, Yang R, Fink MP, Morita K, Choi AM, Murase N, 2006. Ex vivo application of carbon monoxide in University of Wisconsin solution to prevent intestinal cold ischemia/reperfusion injury. Am J Transplant 6, 2243–2255. [DOI] [PubMed] [Google Scholar]
- Pan Z, Zhang J, Chittavong V, Ji X, Wang B, 2018. Organic CO Prodrugs Activated by Endogenous ROS. Org Lett 20, 8–11. [DOI] [PubMed] [Google Scholar]
- Poloukhtine A, Popik VV, 2006. Mechanism of the cyclopropenone decarbonylation reaction. A density functional theory and transient spectroscopy study. J Phys Chem A 110, 1749–1757. [DOI] [PubMed] [Google Scholar]
- Renwick AG, 1985. The disposition of saccharin in animals and man--a review. Food Chem Toxicol 23, 429–435. [DOI] [PubMed] [Google Scholar]
- Romao CC, Blattler WA, Seixas JD, Bernardes GJ, 2012. Developing drug molecules for therapy with carbon monoxide. Chem Soc Rev 41, 3571–3583. [DOI] [PubMed] [Google Scholar]
- Sitnikov NS, Li Y, Zhang D, Yard B, Schmalz HG, 2015. Design, synthesis, and functional evaluation of CO-releasing molecules triggered by Penicillin G amidase as a model protease. Angew Chem Int Ed Engl 54, 12314–12318. [DOI] [PubMed] [Google Scholar]
- Soboleva T, Berreau LM, 2019. 3-Hydroxyflavones and 3-Hydroxy-4-oxoquinolines as Carbon Monoxide-Releasing Molecules. Molecules 24, 1252–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southam HM, Williamson MP, Chapman JA, Lyon RL, Trevitt CR, Henderson PJF, Poole RK, 2021. ‘Carbon-monoxide-releasing molecule-2 (CORM-2)’ is a misnomer: ruthenium toxicity, not CO release, accounts for Its antimicrobial effects. Antioxidants (Basel) 10, 915–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiger C, Luhmann T, Meinel L, 2014. Oral drug delivery of therapeutic gases - carbon monoxide release for gastrointestinal diseases. J Control Release 189, 46–53. [DOI] [PubMed] [Google Scholar]
- Tripathi R, Yang X, Ryter WS, Wang B, 2021. Carbon Monoxide as a Therapeutic for Airway Diseases: Contrast and Comparison of Various CO Delivery Modalities. Curr Top Med Chem 21, 2890–2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai J-H, Chiang H-M, Huang G-Y, Chiang H-L, 2008. Adsorption characteristics of acetone, chloroform and acetonitrile on sludge-derived adsorbent, commercial granular activated carbon and activated carbon fibers. J Hazard Mater 154, 1183–1191. [DOI] [PubMed] [Google Scholar]
- Tyuma I, Ueda Y, Imaizumi K, Kosaka H, 1981. Prediction of the carbonmonoxyhemoglobin levels during and after carbon monoxide exposures in various animal species. Jpn J Physiol 31, 131–143. [DOI] [PubMed] [Google Scholar]
- Wang D, Viennois E, Ji K, Damera K, Draganov A, Zheng Y, Dai C, Merlin D, Wang B, 2014. A click-and-release approach to CO prodrugs. Chem Commun 50, 15890–15893. [DOI] [PubMed] [Google Scholar]
- Wang M, Yang X, Pan Z, Wang Y, De La Cruz LK, Wang B, Tan C, 2020. Towards “CO in a pill”: Pharmacokinetic studies of carbon monoxide prodrugs in mice. J Control Release 327, 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstain R, Slanina T, Kand D, Klán P, 2020. Visible-to-NIR-Light Activated Release: From Small Molecules to Nanomaterials. Chem Rev 120, 13135–13272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Wang R, 2005. Carbon monoxide: endogenous production, physiological functions, and pharmacological applications. Pharmacol Rev 57, 585–630. [DOI] [PubMed] [Google Scholar]
- Yanez JA, Remsberg CM, Sayre CL, Forrest ML, Davies NM, 2011. Flip-flop pharmacokinetics--delivering a reversal of disposition: challenges and opportunities during drug development. Ther Deliv 2, 643–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Ke B, Lu W, Wang B, 2020. CO as a therapeutic agent: discovery and delivery forms. Chin J Nat Med 18, 284–295. [DOI] [PubMed] [Google Scholar]
- Yang X, Lu W, Hopper CP, Ke B, Wang B, 2021a. Nature’s marvels endowed in gaseous molecules I: carbon monoxide and its physiological and therapeutic roles. Acta Pharm Sin B 11, 1434–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Lu W, Wang M, Tan C, Wang B, 2021b. “CO in a pill”: Towards oral delivery of carbon monoxide for therapeutic applications. Journal of Controlled Release 338, 593–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z, Yang X, Wang B, 2021a. Redox and catalase-like activities of four widely used carbon monoxide releasing molecules (CO-RMs). Chem Sci 12, 13013–13020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z, Yang X, Ye Y, Tripathi R, Wang B, 2021b. Chemical Reactivities of Two Widely Used Ruthenium-Based CO-Releasing Molecules with a Range of Biologically Important Reagents and Molecules. Anal Chem 93, 5317–5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Ji X, Yu B, Ji K, Gallo D, Csizmadia E, Zhu M, Choudhury MR, De La Cruz LKC, Chittavong V, Pan Z, Yuan Z, Otterbein LE, Wang B, 2018. Enrichment-triggered prodrug activation demonstrated through mitochondria-targeted delivery of doxorubicin and carbon monoxide. Nat Chem 10, 787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.