

Abstract
Nanoparticles (NP) in biological fluids almost invariably become coated with proteins to form protein coronas. It is the NP–protein corona rather than the bare nanoparticle that determines the nanoparticle's bio-behavior. Here, ultrasmall gold nanoparticles (AuNPs) coated by a human serum albumin (HSA) corona were studied by Fourier transform infrared spectroscopy, denature experiments, fluorescence quenching. Moreover, the intracellular fate of AuNPs and the AuNP–HSA corona has also been investigated. The results show that HSA corona undergo a conformational transition (partial β-sheet changed to α-helicity) when they adsorb on AuNPs, which lead to an enhanced thermal stability. Importantly, we observed that the conformation-transited protein corona–AuNP complex could induce cell apoptosis. Meanwhile, for the first time, the conformation-transited HSA on the AuNPs surface are shown to disrupt living cell membranes. The results obtained here not only provide the detailed conformational behavior of HSA molecules on nanoparticles, but also reveal the structure–function relationship of protein corona, which is of utmost importance in the safe application of nanoscale objects in living organisms.
This study demonstrate that the AuNP–HSA corona could penetrate cell membranes and companied by substantial membrane disruption. However, the ultrasmall AuNPs can be internalized by cells without the destruction of cell membranes.
1. Introduction
Nanomaterials exposed to a biological milieu will strongly interact with proteins, forming a “protein corona” on the surface of nanomaterials.1–5 During the past decade, substantial progress has been made in understanding the physicochemical properties of the protein corona.2,5–17 Ultrasmall metal nanoparticles of core size <3 nm, sometimes presenting unique electrical and optical properties, have received enormous attention recently.18–21 The development of ultrasmall gold nanoparticles (AuNPs) provides an avenue for cancer therapy and in other nanomedicine fields. Better understanding of ultrasmall-AuNP–protein corona and its interact with cells is required for its safe and intended use.
The NP–corona-induced cell toxicity22,23 have been related to different individual parameters in vitro studies. On one hand, the physicochemical properties of NPs, such as size, shape, roughness, and surface properties, play a key role in determining the outcome of their interactions with cells.2,22,24,25 On the other hand, the proteomic composition, and aggregation effects of protein corona and its interaction with cancer cells or immune cells are also intensively studied.2,26–31 Proteomic analysis techniques, such as gel electrophoresis and mass analysis, have been proved that HSA, the most abundant protein in blood plasma, could be an important component of the protein corona.14,15,32
It is known that conformational change of proteins potentially associated with neurodegeneration, metabolic disorders, and other diseases.33,34 In previous work, we successfully applied Fourier transform infrared spectroscopy (FT-IR) to show that the protein corona is unfolding when it adsorbed on 40 nm Au nanoparticles.35,36 We also used fluorescence quenching and isothermal titration calorimetry to probe the binding affinity of Au nanoparticle–proteins.35,36 In the present research, ultrasmall AuNPs were synthesized and their functions on the structure and thermal stability of the absorbed HSA corona were analyzed. Our results revealed that the α-helical component of HSA was increased after HSA adsorbed on the ultrasmall AuNPs, which is much different from the large size AuNPs.36 Furthermore, by using laser scanning confocal microscopy (LSCM), dye diffusion assay, cytosolic enzyme leakage assays, we found that the conformation-transited HSA adsorbed on ultrasmall AuNPs could aggregate around cell membrane and then penetrate the cell membranes with membranes disruption, which lead to the increasement of cytotoxicity. These findings will advance our understanding of the structure–function relationship of protein corona, which will play a guide role for the application of ultrasmall AuNPs in biomedical fields.
2. Materials and methods
2.1. Materials
Bovine serum albumin was purchased from Sigma. Au(iii) chloride trihydrate, sodium citrate, NaCl, NaH2PO4·3H2O, and Na2HPO4·12H2O were all analytical grade and purchased from Chinese Sinopharm Chemical Reagent Co., Ltd. Double distilled water was used. 3-(4,5-Dimethyl-2-thiazolyl)-2-5-diphenyltetrazolium bromide (MTT) assay, Roswell Park Memorial Institute (RPMI) 1640 with 1% l-glutamine, phosphate buffered saline (0.01 M, pH 7.2) and fetal bovine serum were purchased form Gibco (Life Technologies, Thermo Fisher Scientific, Waltham, MA). Lactate dehydrogenase (LDH) cytotoxicity assay kit were purchased form Beyotime Institute of Biotechnology (Shanghai, China). FITC were purchased from Lumiprobe-Life Science.
2.2. Preparation of Au NPs
The procedure for the synthesis of ultrasmall Au NPs was adapted from a procedure published by Jana et al.37 with some modifications. A 10 mL aqueous solution containing 2 × 10−4 M HAuCl4 was prepared in a conical flask, and then added to 10 mL of 2 × 10−4 M sodium citrate solution, stirring for 10 min. Transfer the mixed solution above ice bath. Next, 0.6 mL of ice cold 0.1 M NaBH4 solution was added to the solution all at once while stirring. The solution turned pink immediately after adding NaBH4, indicating particle formation. The particles in this solution were used as seeds within 5 h after preparation.
2.3. Characterization of the Au NPs
Transmission electron microscopy (TEM) analysis were all performed on a JEM-2010 transmission electron microscope. Samples were prepared by dipping a carbon-coated copper grid into water containing the AuNPs, and the solvent evaporated at room temperature into the air. Dynamic light scattering (DLS) and zeta potential data were acquired from a MALVERN Zetasizer, Nano-ZS (Malvern Instruments, UK). 10 mg Au–HSA corona was dissociated in 3 mL of concentrated nitric acid, then heated to 120 °C for 3 h. The obtained solution were diluted to 100-fold, and then introduced into ICP-MS to detect the signal of Au ions. Thermogravimetry analysis (TGA) of the samples was conducted on a NNENETZNETZSCH STA 449 C instrument.
2.4. Establish the protein coronas
To date, most studies have used centrifugation and stringent washing to establish the protein coronas.1,38,39 In this article, 10 mg HSA lyophilized powder was dissolved in 20 mM phosphate buffer (pH 7.0, 1 mL). The obtained nanoparticle solution was incubated with HSA at 25 °C to form Au–HSA coronas. Centrifugation was used to separate the unbound protein as well as nanoparticle–corona complex. The amount of protein adsorbed by the particles after incubation and centrifugalization was quantified by SDS-PAGE experiments (4% stacking gel and a 12% separating gel).
2.5. FTIR spectroscopy
Protein spectra were obtained using our previously established protocol.40 FT-IR spectra were measured with a Bomem MB series Fourier transform infrared spectrometer (Quebec, Canada) equipped with a deuterated triglycine sulfate detector and purged constantly with dry air. Protein–nanoparticle samples were loaded in a CaF2 cell with a 7.5 μm spacer. For each spectrum, a 128-scan interferogram was collected in single-beam mode and at 4 cm−1 resolution. Reference spectra were recorded under identical conditions without protein in the cell. A straight baseline between 2000 and 1750 cm−1 was used as the standard to judge the success of water subtraction.40 Second-derivative spectra were obtained with a seven-point Savitsky–Golay derivative function, baseline-corrected, and area-normalized as described in our previous work.40 The secondary structure content of protein corona was calculated by curve-fitting analysis of the inverted second-derivative spectrum in the amide I band range of 1600–1700 cm−1.
2.6. Fluorescence quenching
Protein quenching experiments were performed on a Cary Eclipse fluorescence spectrometer with a temperature controller (Varian, Palo Alto, CA) using a 3 cm quartz cuvette at different temperatures. The protein concentration was set 0.2 mg mL−1, and the concentration range of AuNPs was 0 to 7.8 × 10−4 mM. The protein solution and NPs were equilibrated for 1 h at 290 K, 298 K, and 310 K. Excitation was performed at 295 nm with a slit width of 5 nm and the emission was performed at 340 nm with a slit width of 5 nm. For every point the fluorescence intensity was read at least 10 times to ensure that the result was not located at an extreme deviation region.
2.7. Equilibrium chemical unfolding
Chemical denaturation experiments were performed using high purity guanidine hydrochloride (GdmCl, Sigma) as denaturants. For experiments conducted, samples of 0.2 mg mL−1 HSA or AuNP–HSA corona (protein concentration 0.2 mg mL−1) were prepared in 10 mM phosphate buffer, respectively, and then diluted 10-fold in samples containing GdmCl concentrations ranging from 0 to 6 M urea. Samples were incubated for 24 h at room temperature to ensure equilibrium had been reached. Circular dichroism (CD) measurements were carried out with 0.1 cm pathlength cuvette, using Jasco J-810 polarimeter. Data were collected at a 0.5 nm interval with a response time of 4 s in the wavelength range of 200–260 nm. The corresponding buffer spectrum was subtracted from the protein spectrum.
2.8. Cell line and cell culture
The BRL 3A rat liver cell line was purchased from Shanghai institute of life sciences, Chinese academy of sciences. The cells were cultured in DMEM medium supplemented with 10% heat-inactivated FBS, penicillin (100 units per mL), streptomycin (100 μg mL−1), amphotericin B (0.25 μg mL−1) at 37 °C in a humidified incubator at fully humidified atmosphere at 37 °C, 5% CO2 and 95% room air.
2.9. Cytotoxicity assay
The cytotoxicity of Au NPs and Au–corona complexes to cell were determined by MTT assay. Cell (5 × 104 cells per mL per well) were seed in 96-well tissue culture plates for 24 h and then the culture media was discarded and washed by 1× PBS, and then incubated with 100 μL of fresh medium (0% FBS) containing different concentration of NPs and protein corona for another 6 h. Subsequently, 20 μL MTT solution was added to each well and incubated for 3 h. The AuNPs were used and contained concentrations of Au ion were 200, 100, 50, 25, 12.5, 6.25, 3.13, 1.56 μg mL −1. The concentrations of AuNP–HSA corona were established by ICP-MS and the same Au ion concentration were used. Then, the culture medium was removed and replaced with 150 μL per well DMSO. The color intensity of the medium was measured at 570 nm using a MK3 microplate reader (Thermo Fisher Scientific Inc.) to calculate the cell viability. All experiments were conducted at least twice to ensure the reproducibility of the results.
2.10. Cytotoxicity Assay-LDH
Cells were plated at a density of 1 × 10 4 cells per well in a 96-well plate and cultured for 24 h. Cells were then exposed to AuNPs and AuNP–HSA corona in basic culture medium (without FBS) with various concentrations and incubated for 6 h. LDH analysis was carried out according to the manufacturer's instructions of LDH Cytotoxicity Assay Kit (Beyotime). The absorbance of each well was read at 490 nm with Absorbance Microplate Reader (ELx800, BioTek Instruments, USA).
2.11. Conjugation of proteins and dyes
HSA (3 mL, 300 × 10−6 M−1) was conjugated with 100 μL 10−3 M FITC (excitation wavelength 495 nm and emission wavelength 519 nm), overnight at 4 °C with continuous stirring. The mixture was maintained at a pH of 8.5 to ensure monolabeling.41 The protein conjugates were separated from the mixture by passing through a Sephadex G-25 column and eluted with 10 × 10−3 M phosphate buffer solution (PBS, pH 7.2). The effluent was collected from 2.5 to 6 mL. The dye-labeled proteins were dialyzed against PBS buffer at 4 °C overnight to remove unreacted dye. The labeled HSA were incubated with AuNPs for 10 h and then centrifuged to separate the unbound HSA as well as AuNP–HSA* corona complex.
2.12. Dye diffusion
Live cells were first stained with calcein-AM (3 μM) for 30 min and then washed 3 times. Then AuNP–HSA corona were incubated with the stained cells for 0.5 h, 3 h and 24 h. For calcein stain, live cells were incubated with AuNP–HSA corona (3 μM) for 0.5 h, 3 h and 24 h and then washed 3 times for confocal microscopic imaging.
2.13. Confocal microscopic imaging
Olympus spectral confocal multiphoton microscope (FV1200) was used to image cell. These imaging results were obtained at the following parameters: internal PMTs at 16 bit and 1024 × 1024 pixels, excitation wavelength of 488 nm, and scan range of 500–540 nm (green channel).
2.14. Statistical analysis
Statistical significant was calculated using two-tailed Student's t-tests and defined as *P < 0.05, **P < 0.01, ***P < 0.001.
3. Results
3.1. Physicochemical properties of AuNPs and binding HSA characteristics
AuNPs was prepared by the reduction of gold chloride in aqueous medium using sodium borohydride.37 This causes Au3+ ions be reduced into neutral gold atoms which after precipitation form nanoparticles. A representative transmission electron microscopy (TEM) image is shown in Fig. 1a. The primary particle size was measured to be ∼2.8 nm (Fig. 1b). Fig. 1c shows the molecular weight (MW) of HSA is 66KD and the saturated adsorption of AuNPs was ca. 0.08 mg-HSA/1 mg-AuNPs, which was evaluated by ImageJ software. Dynamic light scattering (DLS) measurement of the NPs with and without HSA were carried out. As shown in Fig. 1d, the hydrodynamic diameter of the prepared AuNPs in water is approximately ∼10 nm, while the size of HSA-coated AuNPs is ∼25 nm (Fig. 1d). Zeta-potential of AuNPs solution (0.1 mM in water) gave a slightly negative surface charge (−8.7 eV, Fig. 1e), while the HSA-coated AuNPs is −26.2 eV. The observation indicates that the HSA bind on the surface of AuNPs. The UV-vis absorption spectra and photograph of dispersions of AuNPs and AuNP–HSA corona showed in Fig. 1f. The AuNPs and AuNP–HSA corona exhibited UV absorbance band centered at 520 nm and both of them show a high colloidal stability in aqueous solution. The amount of the Au in Au–HSA corona has been confirmed by inductively-coupled-plasma-atomic-emission-spectroscopy (ICP-AES). The results show that the metal content is about 88.9% in protein corona (showed in Fig. S1(a)†). Thermogravimetric analysis (TGA) was conducted to determine the amount of HSA. Au–HSA corona show a weight loss at 200–300 °C as a result of HSA thermal decomposition. In this way, the content of HSA in Au–HSA corona was determined as 12% (showed in Fig. S1(b)†). The results are consisted with the SDS-PAGE results.
Fig. 1. Characterization of the synthesized ultra-small gold nanoparticles. (a) TEM images, (b) size histograms. (c) SDS-PAGE of HSA incubated with different concentration of AuNPs. (d) Dynamic light scattering spectra, (e) zeta-potential measurements. (f) UV-vis absorption spectra of AuNPs and AuNP–HSA corona (inset: photos of AuNPs and AuNP–HSA corona in PB).

3.2. Structure of protein corona on the surface of Au NPs
Albumin is the most abundant protein in human plasma.42 Data on the secondary structure of albumin is predominantly α-helical (∼65%) structure with the remaining being made up of β-sheet (16–18%).43,44 FT-IR spectroscopy is used for characterizing the structure of the protein corona since this technique is a useful tool in understanding the secondary structure of protein in various environments.45–51 The differential pattern in H-bonding, along with the geometric orientations of amide bonds in α-helices, β-sheets, and random coil structures, allow the different vibration frequencies to be associated with individual secondary structural folding.52 The IR spectra in Fig. 2a show a broad amide I band contour between 1610 and 1690 cm−1 that consists of overlapping component bands arising from different secondary structure elements. Amide I is the most sensitive region to the secondary structural components of proteins, from Fig. 2a we can see the amide I peak of AuNP–HSA corona shifts to lower wavenumbers compared with native HSA.
Fig. 2. FTIR spectra of native HSA and AuNP–HSA corona at 20 °C. (a) IR absorption spectra; (b) secondary-derivative amide I spectra; (c) and (d) curve-fitted inverted second-derivative amide I spectra of HSA and HSA coated on AuNPs at 20 °C. The second-derivative FTIR spectra using a seven-point Savitsky–Golay derivative function.

The amide I band (1700–1600 cm−1) is the most sensitive region to the secondary structural components, which has been widely used to quantify the individual elements of secondary structures.48,53 The second derivative analysis provides a mathematic solution to the overlapping components under the amide I band contour and reveals basic band components assignable to various secondary structural elements. From previous studies, the bands between 1656 ± 2 cm−1 are assigned to α-helical structures. The bands between 1642 and 1624 cm−1 are assigned to β-sheet components. The bands located at 1688, 1680, 1672, and 1666 cm−1 are assigned to β-turn structures. The characteristic band for random coil conformation is located at 1648 ± 2 cm−1.48,54Fig. 2b shows the representative secondary-derivative amide I spectra of native HSA and Au–HSA corona. The peak position and intensity show some changes. Compared with native BSA, α-helix absorbs at 1654 cm−1 was obviously become broader and the intensity was increased, indicating α-helical structure increased. Meanwhile, the bands between 1642 and 1624 cm−1 were decreased and the number of the resonance peaks also reduced, which demonstrate the transformation of β-sheet to α-helical structure (Fig. 2c and d).
The curve fitting of the inverted secondary derivative spectrum provides additional quantitative information about the secondary structures of HSA and HSA–AuNPs complex. As show in Table 1, HSA consists of 62% α-helices, 12% β-sheet, 8% β-turn, 10% random, HSA corona consists of 77% α-helices, 9% β-sheet, 11% β-turn at 20 °C. The α-helices component bands of HSA–AuNPs increased by 15%. These findings suggest that the ultrasmall AuNPs interaction with HSA, increased their helical secondary structure. This change in conformation may arise from a stronger interaction.
Curve-fitted inverted second-derivative amide I spectrum of ultrasmall AuNPs binding BSA at different temperature. The inversion of second-derivative spectrum was done by factoring by −1a.
| Assignment | Frequency (cm−1) | 20 °C, percent (%) | 37 °C, percent (%) | 50 °C, percent (%) | 70 °C, percent (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Corona | BSA | Corona | BSA | Corona | BSA | Corona | BSA | ||
| α-Helix | 1656 ± 2 | 77 | 62 | 68 | 59 | 65 | 56 | 46 | 36 |
| β-Sheet | 1643–1623 | 9 | 12 | 16 | 15 | 12 | 15 | 30 | 27 |
| 1698–1689 | |||||||||
| β-Turn | 1687–1666 | 11 | 8 | 8 | 6 | 10 | 10 | 7 | 6 |
| Random | 1648 ± 2 | 0 | 10 | 6 | 13 | 9 | 14 | 7 | 14 |
| Side chain | 1623–1600 | 3 | 3 | 2 | 2 | 2 | 4 | 16 | 17 |
3.3. Stability of protein corona
In order to study the effect of ultrasmall AuNPs on stability of protein coronas, HSA and AuNPs binding HSA at different temperature (37 °C, 50 °C and 70 °C) were also studied by FTIR in aqueous solution (Fig. S2,† and Table 1). From Table 1, we can find that when the HSA and AuNP–HSA complex were heated to 37 °C for 30 min, both HSA and HSA corona show a decrease in α-helical structural proportions as the temperature increasing, respectively. HSA consists of 59% α-helices, HSA corona consists of 68% α-helices. When the HSA and AuNPs–HSA complex incubated at 50 °C for 30 min, α-helices component of HSA and HSA corona were 56% and 65%. When the AuNPs–BSA complex incubated at 70 °C for 30 min, HSA consists of 36% α-helices, 27% β-sheet, 6% β-turn, 14% random, HSA corona consists of 46% α-helices, 30% β-sheet, 7% β-turn, 7% random. In contrast, the HSA–AuNPs are found to be keep higher component of α-helices than HSA. Therefore, it can be concluded that HSA–AuNPs possess a more thermal stability than pure HSA.
The conformational stabilities of protein corona and native protein were studied by chemical denaturation on CD instrument (Fig. 3). The AuNP–HSA corona shifts the conformational transition curve to a higher concentration of GdnHCl, yielding a midpoint of 2.3 M vs.2.0 M for pure protein. This data reveal that the GdnHCl-induced unfolding of protein corona is less cooperative than that of native protein, suggests that the protein absorbed on the ultrasmall AuNPs is more stable than the pure protein. This result confirms that the ultrasmall AuNPs binding HSA shows an improvement in stability.
Fig. 3. Chemical denaturation of native HSA and AuNP–HSA corona in the presence of increasing concentration of GdnHCl by CD instrument. Data were fitted to a standard two-state model.

3.4. Interaction of HSA with ultrasmall AuNPs
The interaction of HSA with ultrasmall AuNPs was studied by quenching of the tryptophan fluorescence of protein by AuNPs. A notable decrease in fluorescence intensity (Fig. 4a) of Trp fluorophore was observed by successive addition of AuNPs. An intense tryptophan fluorescence at ∼344 nm indicates that the tryptophans are present in nonpolar environment. Upon adding of AuNPs, a progressive reduction in tryptophan fluorescence intensity was observed. The fluorescence quenching can be described by the well-known Stern–Volmer (SV) equation,55,56
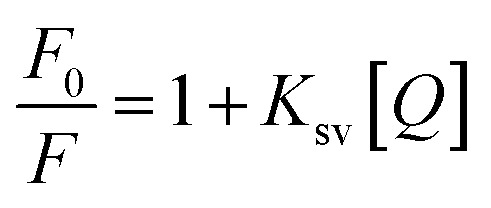 |
1 |
where F0/F is the fractional decrease in fluorescence due to the quencher (Q), and Ksv is the quenching constants, respectively. F0/F against Q have been plotted and the results are shown in Fig. 4b and c. The Stern–Volmer quenching constant (Ksv) values were 8.25 × 105 M−1, 2.23 × 105 M−1, 2.04 × 105 M−1 at 290 K, 298 K and 310 K. The decreased Ksv with the elevated temperature demonstrate that there is a specific interaction between the proteins and AuNPs and the probable quenching mechanism was initiated by static quenching. From these results, we can deduced that the AuNPs bind with HSA and formed a complex. The binding constants of AuNPs and HSA can be calculated using eqn (2).57–59
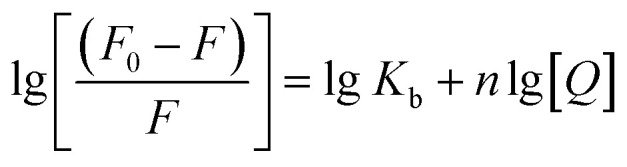 |
2 |
In which Kb is the binding constant and n is the number of binding sites. Under the assumption of no significant variation of the enthalpy change (ΔH) within the range of temperature studied, the enthalpy change (ΔH) can be evaluated from the van't Hoff equation:
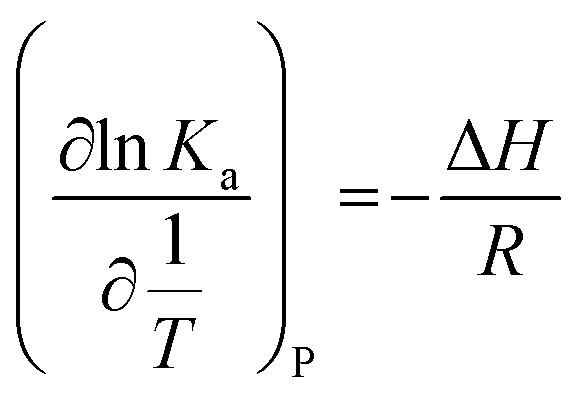 |
3 |
where Ka is analogous to the associative binding constants at the corresponding temperature, and R is the gas constant.
Fig. 4. AuNPs quenching of tryptophan fluorescence of HSA at 290 K, 298 K and 310 K. (a) Effect of AuNPs on HSA fluorescence emission spectra. (b) Stern–Volmer plots and (c) modified Stern–Volmer plots for the HSA binding on AuNPs.

The free energy change (ΔG) and entropy change (ΔS) of the process are estimated from the following relationship:
| ΔG = RT ln Ka | 4 |
| ΔG = ΔH − TΔS | 5 |
According to Ross' report60 the main binding forces can be judged by thermodynamic parameters. ΔH > 0 and ΔS > 0 imply a hydrophobic interaction; ΔH < 0 and ΔS < 0 reflect van der Waals forces or hydrogen bonding; ΔH < 0 and ΔS > 0 suggest electrostatic forces. The positive value of ΔS and the negative value of ΔH reveal the predominance of electrostatic interactions in the binding of Au NPs with HSA (Table 2).
The thermodynamic parameters (ΔH, ΔS and ΔG) for HSA–AuNPs interaction.
| T (K) | K sv (105 L mol −1) | K b (104 M) | n | ΔG (kJ mol−1 K−1) | ΔH (105 kJ mol−1) | ΔS (J mol−1 K−1) |
|---|---|---|---|---|---|---|
| 290 | 8.25 | 8.51 | 0.99 | −5162 | −1.86 | 659 |
| 298 | 2.23 | 6.92 | 1.01 | |||
| 310 | 2.04 | 1.51 | 1.28 |
3.5. Interaction of the AuNP–HSA corona with cells
3-(4,5-Dimethyl-2-thiazolyl)-2-5-diphenyltetrazolium bromide (MTT) assay was employed to determine the cytotoxicity of AuNPs and AuNP–HSA corona at different concentration (contain Au ion were 200, 100, 50, 25, 12.5, 6.25, 3.13, 1.56 μg mL −1). The normal liver cell (3A) line was used as the research model. From Fig. 5a, we can find that the toxicity of AuNP–HSA corona was much higher than the Au NPs. The cell viabilities were all above 80% after being exposed to AuNPs even at the high concentration of 200 μg mL−1 for 6 h. However, when cells were exposed to AuNP–HSA corona, the cytotoxicity showed a concentration-dependent, and the cell viabilities decreased dramatically to 38% at the concentration of 200 μg mL−1. The AuNP–HSA corona regulate 293T cell cytotoxicity and the integrity of cell membranes have been also investigated. The result shows that the toxicity of AuNP–HSA corona was also much higher than the Au NPs (Fig. S3†). This result indicates that ultrasmall AuNPs used in our current study present almost no cytotoxicity, but the AuNP–HSA corona showed significant toxicity to normal cells. MTT assay was also employed to determine the cytotoxicity of AuNPs and AuNP–HSA corona in complete medium (with 10% fetal bovine serum). The result also show that the cell toxicity of AuNP–HSA corona was higher than the AuNPs (Fig. S4†).
Fig. 5. AuNP–HSA corona internalization in cells. (a) Cell viability of 3A cells incubated with AuNPs and Au–HSA corona for 6 h at different concentrations (200, 100, 50, 25, 12.5, 6.25, 3.13, 1.56 μg mL−1). (b) LDH release from cells exposed to AuNPs and Au–HSA corona or 6 h. (c) Confocal laser scanning microscopy (CLSM) upper line: fluorescent detection of 3A cells by AuNP–HSA corona was stained by FITC (green); middle line: 3A cells were pre-loaded with calcein-AM, and then co-incubated with AuNP–HSA corona for 0.5 h, 3 h and 24 h; bottom line: 3A cells incubated with calcein and AuNP–HSA corona. (d) and (e) Mean fluorescence intensities from confocal images at (c). All data are presented as the mean ± SD of three replicates. *P < 0.05 and **P < 0.01.

The integrity of cell membranes treated with different concentration of AuNPs and AuNP–HSA corona were studied by LDH release assay (Fig. 5b). If NPs permeabilize cell membranes and introduced nanoscale holes into the living cell membranes, cytosolic enzymes such as LDH may be released into the cell media.61 The results show that the ultrasmall AuNPs have only subtle influence on the cell membrane, however, AuNP–HSA corona cause the leakage of LDH. When the integrity of plasma membrane is compromised, LDH is rapidly released into the cell culture supernatant. The membrane damage caused by AuNP–HSA corona was also found to be concentration dependent. We deduced that AuNP–HSA corona penetrate into the cell and its concomitant membrane disruptions. The integrity of 293T cell membranes also treated with different concentration of AuNPs and AuNP–HSA corona were studied by LDH release assay (Fig. S5†). The results show that the AuNP–HSA corona cause more leakage of LDH than ultrasmall AuNPs.
To determine how the AuNP–HSA corona interact with cells, we labelled the corona with FITC dye, and confocal laser scanning microscopy was employed to monitor the process that the labelled HSA–AuNP interact with living cells (Fig. 5c). At 0.5 h, we cannot see significant labelled HSA–Au NP attach to cells. At 3 h, the labelled HSA corona attached to the cell membrane, and it exhibit punctate fluorescence patterns. It is in agreement with previous findings.61,62 At 6 h, the AuNP–HSA could be internalized by cells. Therefore, we can conclude that AuNP–HSA corona interaction with cells is a two-step process, the nanoparticles initially adhere to the cell membrane and then internalized by cells.63
To further explore the interaction of AuNP–HSA corona and cells, a cytosol-localized experiment was designed and carried out. Calcein-AM is an acetomethoxy form of the dye that is trapped in the cytosol by conversion from a membrane-permeable to membrane-impermeable form by intracellular esterases. Firstly, 3A cells were pre-loaded with calcein-AM. Then, AuNP–HSA corona incubated with the calcein-AM-dyed cells. Finally, CLSM was used to detect measurable leakage of calcein-AM from cells at 0.5 h, 3 h, and 24 h (Fig. 5c). As shown in Fig. 5c, the fluorescence signals at 0.5 h have no significant difference with the untreated cell. When cells and AuNP–HSA corona were incubated for 3 h, the fluorescence intensity has decreased 20%. At 24 h, it exhibited a punctate extracellular distribution of dye, and the fluorescent intensity of calcein-AM in cell decreased dramatically, which may cause calcein-AM escape to extracellular. To confirm the Au–HSA corona penetrate cell membranes and can create transient holes, we also tested whether the internalization of Au–HSA corona was accompanied by entry of an initially extracellular tracer dye.64 First, cells were co-incubated with Au–HSA corona and calcein; calcein can only stay in endolysosomal compartments in cells unless it is co-internalized with a membrane-disrupting agent. Cells were incubated for 0 h, 3 h, 24 h with calcein and Au–HSA corona, washed and imaged by confocal microscopy. As shown in Fig. 5c, Au–HSA corona caused vast calcein to escape into the cytosol of the cells, whereas AuNPs incubated with calcein alone exhibited slightly distribution of dye. Combined the data described above showing that Au–HSA corona clearly access the cytosolic compartment, and induce calcein AM escape cell and promote calcein entrance into cytosol, we can deduce that the presence of AuNP–HSA corona disrupt integrity of the cell membrane, which may cause the leakage of ions and molecules out of the cell, and lead to cell death.61,65
4. Discussion
In this work, ultrasmall AuNPs with the average size about 2.8 nm were prepared using ice-cold sodium borohydride. The obtained AuNPs have a high affinity binding to HSA. SDS-PAGE, DLS and zeta potential confirm the formation of AuNP–HSA coronas. FT-IR spectroscopy was employed for characterizing the secondary structure of the protein corona. We found the α-helices component bands of AuNP–HSA corona increased by 15% when compared with native HSA. Fluorescence quenching results shows the changes in corona conformation may arise from a stronger electrostatic interaction between AuNPs and HSA. Furthermore, the corona was found to have a special ability to keep higher component of α-helices than native HSA at elevated temperatures. Chemical denaturation of HSA and AuNP-bound HSA in the presence of GdnHCl also showed the improvement in stability of protein corona. Taken together with these results, it can be deduced that HSA corona may undergo conformational transition at the interface, and it might form an intermolecular H-bond containing α-helices conformation. The evaluation of cellular cytotoxicity shows that ultrasmall AuNPs used in our current study present almost no cytotoxicity in basic medium for 6 h, but the AuNP–HSA corona showed significant toxicity to 3A cells at the same condition. This is in stark contrast with large-sized Au nanorod–BSA corona, which showed the unfolding protein can protective effect of protein coronas against the toxicity of nanomaterial.65 A possible explanation is the “rigid” conformation of serum protein and NP size influence the interaction of AuNPs and cells.
The cell membrane, as the primary defensive barrier of the cell, plays a key role in the uptake of NPs.66,67 Previous studies have shown some polymer nanoparticles penetrate the plasma membranes of cells by generating transient holes which can lead to cell death.61 NPs can adhere to the amphiphilic lipid bilayer surface or become inserted into the bilayer alkane chain region, where they can disturb phospholipid bilayers and block membrane proteins.68 Verma et al. found that cationic nano-objects can pass through cell membranes by generating transient holes, a process associated with cytotoxicity.64 Leroueil et al. showed 5 nm poly(amidoamine) NPs could cause LDH leakage, but the poly(amidoamine)-folic acid enters the cell with no LDH leakage.61 Our results show that the ultrasmall AuNPs have subtle influences on the cell membrane. However, the AuNP–HSA corona cause LDH release. This is different from previous reports, where the surface-adsorbed serum provides some degree of protection to the cells and reduces damage.69 We deduced that AuNP–HSA corona penetration the cell and its concomitant membrane disruptions. This result was confirmed through fluorescent confocal studies, which shows the AuNP–HSA corona interaction with cells is a two-step process whereby the nanoparticles initially adhere to the cell membrane and then internalized by cells. Furthermore, from studies with tracer dyes (with noticeable leakage), we determined the AuNP–HSA corona penetrate cell membranes and cause substantial membrane disruption.
The mechanism of AuNP–HSA corona interaction with cell is very diverse, complex and hard to clarify. Wang reports that the secondary structure of the proteins can be altered by adsorption on the surface.65 Schnitzer et al. have demonstrated that albumin conjugated to colloidal gold particles can bind with cell membrane-associated glycoproteins.62 Our further work toward determining the molecular mechanism of the conformational transition take effect on the cell membrane and subcellular components was undergoing in our laboratory.
5. Conclusion
In conclusion, a typical protein corona binding on ultrasmall AuNPs was systemically studied here. FT-IR was applied to address the key question of the secondary structure of protein corona on nanoparticles, and found that ultrasmall AuNPs can induce the transformation of the conformation of protein. The in vitro experiments showed that AuNP–HSA corona were toxic to cells but the bare AuNPs were nontoxic. Further studies show that the AuNP–HSA corona interaction with cells is a two-step process whereby the nanoparticles initially adhere to the cell membrane and then internalized by cells. Further exploration demonstrate that the AuNP–HSA corona could penetrate cell membranes and companied by substantial membrane disruption, which lead to death of the cell. These findings and the combined strategy will provide guidance between NPs and biological interfaces, which is of utmost importance in the safe application of nanoscale objects in living organisms.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (21505033 and 21571053), and the 111 project (D17007), China Postdoctoral Science Foundation (2018M642755), key scientific and technological project of Henan Province (172102310618), the Program for Innovative Research Team in Science and Technology in University of Henan Province (18IRTSTHN002), the PhD Start-up Fund of Henan Normal University, China (qd15112 and qd16104), key scientific research projects in colleges and universities of Henan province (19A350004).
Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10049g
References
- Cedervall T. Lynch I. Lindman S. Berggard T. Thulin E. Nilsson H. Dawson K. A. Linse S. Proc. Natl. Acad. Sci. U. S. A. 2007;104:2050–2055. doi: 10.1073/pnas.0608582104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monopoli M. P. Aberg C. Salvati A. Dawson K. A. Nat. Nanotechnol. 2012;7:779–786. doi: 10.1038/nnano.2012.207. [DOI] [PubMed] [Google Scholar]
- Lindman S. Lynch I. Thulin E. Nilsson H. Dawson K. A. Linse S. Nano Lett. 2007;7:914–920. doi: 10.1021/nl062743+. [DOI] [PubMed] [Google Scholar]
- Lundqvist M. Nat. Nanotechnol. 2013;8:701–702. doi: 10.1038/nnano.2013.196. [DOI] [PubMed] [Google Scholar]
- Nel A. E. Madler L. Velegol D. Xia T. Hoek E. M. Somasundaran P. Klaessig F. Castranova V. Thompson M. Nat. Mater. 2009;8:543–557. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- Harper S. L. Carriere J. L. Miller J. M. Hutchison J. E. Maddux B. L. Tanguay R. L. ACS Nano. 2011;5:4688–4697. doi: 10.1021/nn200546k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenzer S. Docter D. Rosfa S. Wlodarski A. Kuharev J. Rekik A. Knauer S. K. Bantz C. Nawroth T. Bier C. Sirirattanapan J. Mann W. Treuel L. Zellner R. Maskos M. Schild H. Stauber R. H. ACS Nano. 2011;5:7155–7167. doi: 10.1021/nn201950e. [DOI] [PubMed] [Google Scholar]
- Monopoli M. P. Walczyk D. Campbell A. Elia G. Lynch I. Baldelli Bombelli F. Dawson K. A. J. Am. Chem. Soc. 2011;133:2525–2534. doi: 10.1021/ja107583h. [DOI] [PubMed] [Google Scholar]
- Martel J. Young D. Young A. Wu C. Y. Chen C. D. Yu J. S. Young J. D. Anal. Biochem. 2011;418:111–125. doi: 10.1016/j.ab.2011.06.018. [DOI] [PubMed] [Google Scholar]
- Monopoli M. P. Pitek A. S. Lynch I. Dawson K. A. Methods Mol. Biol. 2013;1025:137–155. doi: 10.1007/978-1-62703-462-3_11. [DOI] [PubMed] [Google Scholar]
- Landry M. P. Kruss S. Nelson J. T. Bisker G. Iverson N. M. Reuel N. F. Strano M. S. Sensors. 2014;14:16196–16211. doi: 10.3390/s140916196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke P. C. Lin S. Parak W. J. Davis T. P. Caruso F. ACS Nano. 2017;11:11773–11776. doi: 10.1021/acsnano.7b08008. [DOI] [PubMed] [Google Scholar]
- Roach P. Farrar D. Perry C. C. J. Am. Chem. Soc. 1994;84:8168–8173. doi: 10.1021/ja042898o. [DOI] [PubMed] [Google Scholar]
- Schottler S. Becker G. Winzen S. Steinbach T. Mohr K. Landfester K. Mailander V. Wurm F. R. Nat. Nanotechnol. 2016;11:372–377. doi: 10.1038/nnano.2015.330. [DOI] [PubMed] [Google Scholar]
- Hadjidemetriou M. Kostarelos K. Nat. Nanotechnol. 2017;12:288–290. doi: 10.1038/nnano.2017.61. [DOI] [PubMed] [Google Scholar]
- Rocker C. Potzl M. Zhang F. Parak W. J. Nienhaus G. U. Nat. Nanotechnol. 2009;4:577–580. doi: 10.1038/nnano.2009.195. [DOI] [PubMed] [Google Scholar]
- Bekdemir A. Stellacci F. Nat. Commun. 2016;7:13121. doi: 10.1038/ncomms13121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips E. Penate-Medina O. Zanzonico P. B. Carvajal R. D. Mohan P. Ye Y. Humm J. Gonen M. Kalaigian H. Schoder H. Strauss H. W. Larson S. M. Wiesner U. Bradbury M. S. Sci. Transl. Med. 2014;6:260ra149. doi: 10.1126/scitranslmed.3009524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boselli L. Polo E. Castagnola V. Dawson K. A. Angew. Chem., Int. Ed. Engl. 2017;56:4215–4218. doi: 10.1002/anie.201700343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo S. Jin S. Ma X. Xue X. Yang K. Kumar A. Wang P. C. Zhang J. Hu Z. Liang X. J. ACS Nano. 2014;8:5852–5862. doi: 10.1021/nn5008572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B. H. Hackett M. J. Park J. Hyeon T. Chem. Mater. 2014;26:59–71. doi: 10.1021/cm402225z. [DOI] [Google Scholar]
- Saha K. Kim S. T. Yan B. Miranda O. R. Alfonso F. S. Shlosman D. Rotello V. M. Small. 2013;9:300–305. doi: 10.1002/smll.201201129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albanese A. Tang P. S. Chan W. C. W. Annu. Rev. Biomed. Eng. 2012;14:1–16. doi: 10.1146/annurev-bioeng-071811-150124. [DOI] [PubMed] [Google Scholar]
- Pelaz B. del Pino P. Maffre P. Hartmann R. Gallego M. Rivera-Fernandez S. de la Fuente J. M. Nienhaus G. U. Parak W. J. ACS Nano. 2015;9:6996–7008. doi: 10.1021/acsnano.5b01326. [DOI] [PubMed] [Google Scholar]
- Walkey C. D. Olsen J. B. Guo H. Emili A. Chan W. C. J. Am. Chem. Soc. 2012;134:2139. doi: 10.1021/ja2084338. [DOI] [PubMed] [Google Scholar]
- Moyano D. F. Rotello V. M. Langmuir. 2011;27:10376–10385. doi: 10.1021/la2004535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monopoli M. P. Aberg C. Salvati A. Dawson K. A. Nat. Nanotechnol. 2012;7:779–786. doi: 10.1038/nnano.2012.207. [DOI] [PubMed] [Google Scholar]
- Calzolai L. Franchini F. Gilliland D. Rossi F. Nano Lett. 2010;10:3101–3105. doi: 10.1021/nl101746v. [DOI] [PubMed] [Google Scholar]
- Chen D. Ganesh S. Wang W. Amiji M. Nanomedicine. 2017;12:2113. doi: 10.2217/nnm-2017-0178. [DOI] [PubMed] [Google Scholar]
- Ahsan S. M., Rao C. M. and Ahmad M. F., Oxygen Transport to Tissue XXXIII, 2018, vol. 1048, pp. 175–198 [Google Scholar]
- Chinen A. B. Guan C. M. Ko C. H. Mirkin C. A. Small. 2017;13:1603847. doi: 10.1002/smll.201603847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. Wang Z. Dong L. Trends Biotechnol. 2018;36:661–672. doi: 10.1016/j.tibtech.2018.02.016. [DOI] [PubMed] [Google Scholar]
- Liu Y. Fares M. Dunham N. P. Gao Z. Miao K. Jiang X. Bollinger S. S. Boal A. K. Zhang X. Angew. Chem., Int. Ed. 2017;56:8672. doi: 10.1002/anie.201702417. [DOI] [PubMed] [Google Scholar]
- Liu Y. Wolstenholme C. H. Carter G. C. Liu H. Hu H. Grainger L. S. Miao K. Fares M. Hoelzel C. A. Yennawar H. P. Ning G. Du M. Bai L. Li X. Zhang X. J. Am. Chem. Soc. 2018;140:7381–7384. doi: 10.1021/jacs.8b02176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu C. Yang H. Wang M. Xiong H. Yu S. Chem. Commun. 2015;51:3634–3636. doi: 10.1039/C4CC08372E. [DOI] [PubMed] [Google Scholar]
- Zhang X. Zhang J. Zhang F. Yu S. Nanoscale. 2017;9:4787–4792. doi: 10.1039/C7NR01523B. [DOI] [PubMed] [Google Scholar]
- Jana N. R. Gearheart L. Murphy C. J. J. Phys. Chem. B. 2001;105:4065–4067. doi: 10.1021/jp0107964. [DOI] [Google Scholar]
- Capriotti A. L. Caracciolo G. Caruso G. Cavaliere C. Pozzi D. Samperi R. Lagana A. Anal. Bioanal. Chem. 2010;398:2895–2903. doi: 10.1007/s00216-010-4104-y. [DOI] [PubMed] [Google Scholar]
- Cedervall T. Lynch I. Foy M. Berggård T. Donnelly S. C. Cagney G. Linse S. Dawson K. A. Angew. Chem., Int. Ed. 2007;46:5754–5756. doi: 10.1002/anie.200700465. [DOI] [PubMed] [Google Scholar]
- Dong A. Malecki J. M. Lee L. Carpenter J. F. Lee J. C. Biochemistry. 2002;41:6660–6667. doi: 10.1021/bi020036z. [DOI] [PubMed] [Google Scholar]
- Zhang X. Shi H. Zhang R. Zhang J. Xu F. Qiao L. Yu S. Part. Part. Syst. Charact. 2019;36:1800257. doi: 10.1002/ppsc.201800257. [DOI] [Google Scholar]
- Farrugia A. Transfus. Med. Rev. 2010;24:53–63. doi: 10.1016/j.tmrv.2009.09.005. [DOI] [PubMed] [Google Scholar]
- Takeda K. Wada A. Yamamoto K. Moriyama Y. Aoki K. J. Protein Chem. 1989;8:653–659. doi: 10.1007/BF01025605. [DOI] [PubMed] [Google Scholar]
- Carter D. C. Ho J. X. Adv. Protein Chem. 1994;45:153–203. doi: 10.1016/S0065-3233(08)60640-3. [DOI] [PubMed] [Google Scholar]
- Griebenow K. Klibanov A. M. J. Am. Chem. Soc. 1996;118:11695–11700. doi: 10.1021/ja961869d. [DOI] [Google Scholar]
- Arrondo J. L. R. Goñi F. M. Prog. Biophys. Mol. Biol. 1999;72:367–405. doi: 10.1016/S0079-6107(99)00007-3. [DOI] [PubMed] [Google Scholar]
- Goormaghtigh E. Raussens V. Ruysschaert J. M. Biochim. Biophys. Acta. 1999;1422:105–185. doi: 10.1016/S0304-4157(99)00004-0. [DOI] [PubMed] [Google Scholar]
- Kong J. Yu S. Acta Biochim. Biophys. Sin. 2007;39:549–559. doi: 10.1111/j.1745-7270.2007.00320.x. [DOI] [PubMed] [Google Scholar]
- Barth A. Biochim. Biophys. Acta, Bioenerg. 2007;1767:1073–1101. doi: 10.1016/j.bbabio.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Liu K. Z. Shaw R. A. Man A. Dembinski T. C. Mantsch H. H. Clin. Chem. 2002;48:499–506. [PubMed] [Google Scholar]
- Lenk T. J. Horbett T. A. Ratner B. D. Chittur K. K. Langmuir. 1991;7:1755–1764. doi: 10.1021/la00056a030. [DOI] [Google Scholar]
- Singh Bal R., in Infrared Analysis of Peptides and Proteins, American Chemical Society, 1999, vol. 750, ch. 1, pp. 2–37 [Google Scholar]
- Arrondo J. L. Muga A. Castresana J. Goni F. M. Prog. Biophys. Mol. Biol. 1993;59:23–56. doi: 10.1016/0079-6107(93)90006-6. [DOI] [PubMed] [Google Scholar]
- Bawendi M. G. Steigerwald M. L. Brus L. E. Annu. Rev. Phys. Chem. 1990;41:477–496. doi: 10.1146/annurev.pc.41.100190.002401. [DOI] [Google Scholar]
- Chen R. Lee J. C. J. Biol. Chem. 2003;278:13235–13243. doi: 10.1074/jbc.M211551200. [DOI] [PubMed] [Google Scholar]
- Shen X. Li H. Ou Y. Tao W. Dong A. Kong J. Ji C. Yu S. J. Biol. Chem. 2008;283:11407–11413. doi: 10.1074/jbc.M708513200. [DOI] [PubMed] [Google Scholar]
- Xiao C. Q. Jiang F. L. Zhou B. Li R. Liu Y. Photochem. Photobiol. Sci. 2011;10:1110–1117. doi: 10.1039/C1PP05008G. [DOI] [PubMed] [Google Scholar]
- Abou-Zied O. K. Al-Shihi O. I. J. Am. Chem. Soc. 2008;130:10793–10801. doi: 10.1021/ja8031289. [DOI] [PubMed] [Google Scholar]
- Paul B. K. Bhattacharjee K. Bose S. Guchhait N. Phys. Chem. Chem. Phys. 2012;14:15482–15493. doi: 10.1039/C2CP42415K. [DOI] [PubMed] [Google Scholar]
- Ross P. D. Subramanian S. Biochemistry. 1981;20:3096–3102. doi: 10.1021/bi00514a017. [DOI] [PubMed] [Google Scholar]
- Leroueil P. R. Seungpyo H. Almut M. Baker J. R. Orr B. G. Banaszak Holl M. M. ChemInform. 2010;38:335–342. [Google Scholar]
- Schnitzer J. E. Bravo J. J. Biol. Chem. 1993;268:7562–7570. [PubMed] [Google Scholar]
- Wilhelm C. Billotey C. Roger J. Pons J. N. Bacri J. C. Gazeau F. Biomaterials. 2003;24:1001–1011. doi: 10.1016/S0142-9612(02)00440-4. [DOI] [PubMed] [Google Scholar]
- Verma A. Uzun O. Hu Y. Hu Y. Han H. S. Watson N. Chen S. Irvine D. J. Stellacci F. Nat. Mater. 2008;7:588–595. doi: 10.1038/nmat2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liming W. Jingyuan L. Jun P. Xiumei J. Yinglu J. Yufeng L. Ying Q. Yuliang Z. Xiaochun W. Chunying C. J. Am. Chem. Soc. 2013;135:17359–17368. doi: 10.1021/ja406924v. [DOI] [PubMed] [Google Scholar]
- Lin J. Zhang H. Chen Z. Zheng Y. ACS Nano. 2010;4:5421–5429. doi: 10.1021/nn1010792. [DOI] [PubMed] [Google Scholar]
- Wang X. Wang X. Bai X. Yan L. Liu T. Wang M. Song Y. Hu G. Gu Z. Miao Q. Chen C. Nano Lett. 2019;19:8–18. doi: 10.1021/acs.nanolett.8b02638. [DOI] [PubMed] [Google Scholar]
- Anna L. Anna S. Santos-Martinez M. J. Radomski M. W. Dawson K. A. Christoffer B. J. Am. Chem. Soc. 2013;135:1438. doi: 10.1021/ja309812z. [DOI] [PubMed] [Google Scholar]
- Tedja R. Lim M. Amal R. Marquis C. ACS Nano. 2012;6:4083–4093. doi: 10.1021/nn3004845. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.