

Abstract
Mitochondrial dysfunction or loss of homeostasis is a central hallmark of many human diseases. Mitochondrial homeostasis is mediated by multiple quality control mechanisms including mitophagy, a form of selective autophagy that recycles terminally ill or dysfunctional mitochondria in order to preserve mitochondrial integrity. Our prior studies have shown that members of the insulin-like growth factor (IGF) family localize to the mitochondria and may play important roles in mediating mitochondrial health in the corneal epithelium, an integral tissue that is required for the maintenance of optical transparency and vision. Importantly, the IGF-binding protein-3, IGFBP-3, is secreted by corneal epithelial cells in response to stress and functions to mediate intracellular receptor trafficking in this cell type. In this study, we demonstrate a novel role for IGFBP-3 in mitochondrial homeostasis through regulation of the short isoform (s)BNIP3L/NIX mitophagy receptor in corneal epithelial cells and extend this finding to non-ocular epithelial cells. We further show that IGFBP-3-mediated control of mitochondrial homeostasis is associated with alterations in lamellar cristae morphology and mitochondrial dynamics. Interestingly, both loss and gain of function of IGFBP-3 drive an increase in mitochondrial respiration. This increase in respiration is associated with nuclear accumulation of IGFBP-3. Taken together, these findings support a novel role for IGFBP-3 as a key mediator of mitochondrial health in mucosal epithelia through the regulation of mitophagy and mitochondrial morphology.
Keywords: autophagy, insulin-like growth factor type 1 receptor, metabolism, mitochondria, mTOR
1 |. INTRODUCTION
Mitochondria are subcellular organelles that play a vital role in cellular homeostasis and energy production. It is well established that proper mitochondrial function is mediated by multiple quality control mechanisms including mitophagy, fission and fusion, the release of mitochondrial-derived vesicles, and mitochondrial biogenesis.1,2 Of these, mitophagy, a form of selective autophagy, is used to recycle terminally ill or dysfunctional mitochondria and preserve mitochondrial integrity.2–6 Like other types of selective autophagy, mitophagy uses the components of the macroautophagy pathway.5,7 When cells are exposed to stress or starvation, inhibition of the mechanistic target of rapamycin (mTOR) allows for phosphorylation of Unc-51-like kinase and phagophore formation.8–10 This is followed by elongation and closure of the autophagosome using a variety of pathways, including the mitophagy receptors BCL2/adenovirus E1B 19-kDa-interacting protein 3 (BNIP3), BNIP3-like (BNIP3L/NIX), and PTEN-induced putative kinase 1 (PINK1)/Parkin.11–13 The lysosome then fuses with the autophagosome to form an autophagolysosome, leading to degradation of damaged mitochondria. In addition to mitophagy, BNIP3 and BNIP3L/NIX have also been implicated in mitochondrial biogenesis and apoptotic cell death.14–17 Alternative splicing of the latter, results in the expression of a shorter isoform, sBNIP3L/NIX, that localizes to the nucleus to exert a cytoprotective effect.18 These findings suggest that together, BNIPL3/NIX and sBNIPL3/NIx may play an important role in mitochondrial homoestasis and quality control.
The type 1 IGF receptor (IGF-1R) is a receptor tyrosine kinase and member of the IGF family. Collectively, the IGF family is composed of multiple receptors, ligands, and binding proteins that regulate the phosphatidyl-inositol-3-kinase (PI3K)/Akt/mTOR signaling pathway and has essential roles in proliferation, differentiation, and cell survival.19–23 IGF-1R is a transmembrane glycoprotein composed of two intracellular beta subunits with tyrosine kinase activity and two extracellular alpha subunits that mediate ligand binding. IGF-1R is translated as a proreceptor and cleaved in the trans-Golgi complex to then be assembled and inserted into the plasma membrane.24 Upon ligand binding, receptor signaling is initiated and followed by internalization through clathrin and caveolin endocytic trafficking.25,26 IGF-1-mediated activation of IGF-1R has also been shown to induce nuclear translocation of the receptor and the expression of genes involved in cell cycle and proliferation.27,28 More recently, de novo nuclear trafficking has been demonstrated, in addition to mitochondrial localization in corneal epithelial cells, suggesting a potential role in mitochondrial homeostasis.29–31 Consistent with this, IGF-1R has been reported to regulate mitophagy.14 In breast cancer cells, treatment with IGF-1 induced BNIP3 expression and accumulation in mitochondria in a PI3K-dependent pathway. Similarly, suppression of IGF-1R activity led to a decrease in mitophagy and subsequent mitochondrial dysfunction.14
The IGF-binding protein-3 (IGFBP-3) is a pleiotropic N-linked glycosylated, phosphorylated secretory protein that antagonizes the effect of IGF-1. In circulation, IGFBP-3 is the predominant IGF-1 binding protein.32 It sequesters IGF-1 to prevent receptor activation and inhibit degradation, thereby extending the half-life of IGF-1.33,34 IGFBP-3 has also been shown to have IGF-independent functions. These include apoptosis, DNA repair, cell cycle, angiogenesis, hypoxia, and insulin resistance. 35–39 These effects, however, are cell and context dependent. Prior work in breast cancer cells has shown that IGFBP-3 induces autophagy to promote cell survival in adverse environments.37 In that study, environmental stress triggered an increase in non-glycosylated IGFBP-3 that strongly bound the glucose-regulated protein 78 (GRP78). IGFBP-3 has also been shown to activate autophagy in transformed lung epithelial cells.40 In this model, miRNA 34/449 downregulated IGFBP-3, leading to activation of Akt. In turn, the attenuation of miRNA 34/449 in the asthmatic airway resulted in increased IGFBP-3 expression.
The ocular surface is covered by two contiguous epithelia that are continuously bathed by tear fluid. Tear fluid is a composite of secreted proteins from corneal and conjunctival epithelial cells, glandular secretions, and derivatives from blood via capillaries found in the corneal limbus and conjunctiva.41 We have found that IGFBP-3 is present in human tear fluid and expression levels of IGFBP-3 are altered in diabetes. We have further shown that diabetes-induced damage of corneal nerves is directly related to tear levels of IGFBP-3, supporting an essential role for IGFBP-3 in the homeostasis of these epithelial tissues.42–45 Recent work has shown that IGFBP-3 has a diverse intracellular localization and in corneal epithelia, mediates intracellular trafficking of IGF-1R.30 Moreover, insulin, which regulates IGFBP-3 expression, was shown to block PINK1-mediated mitophagy induced by growth factor withdrawal in both corneal and bronchial epithelial cells.31 Like ocular surface epithelia, the bronchial epithelium is a mucosal surface subject to chronic stress.
In the present study, we investigated a role for IGFBP-3 in mediating macroautophagy and mitophagy. We furthered examined the effects of IGFBP-3 on mitochondrial morphology and respiration. To accomplish this, we used three different epithelial cell models: human corneal, conjunctival, and bronchial epithelial cells. Importantly, we demonstrate a novel role for IGFBP-3 in regulating mitophagic flux through the sBNIPL3/NIX pathway. Unlike prior work in tumor cell lines showing an induction of autophagy by IGFBP-3, here we show that IGFBP-3 blocks mitophagy through phosphorylation of mTOR.37,40 IGFBP-3-mediated inhibition of mitophagy is associated with remodeling of mitochondrial cristae to sustain mitochondrial function. Together, these data support a novel role for IGFBP-3 in the maintenance of mitochondrial structure and function in epithelial cells.
2 |. METHODS
2.1 |. Cell lines and primary cultures
For this study, we used three mucosal epithelial cell lines all immortalized with human telomerase reverse transcriptase (hTERT). These include human corneal epithelial (hTCEpi) cells (immortalized with hTERT alone), human bronchial epithelial cells (HBECs, immortalized with hTERT and overexpressed Cdk4), and human conjunctival cells (HCjECs) (immortalized with hTERT, mutant Cdk4, and a dominant negative p53).46–50 HBECs were a gift of Dr. Jerry Shay (Department of Cell Biology, UT Southwestern Medical Center). HCjECs were a gift of Drs. Pablo Argüeso (Schepens Eye Research Institute, Harvard Medical School) and Ajay Sharma (School of Pharmacy, Chapman University). All cell lines were cultured in serum-free keratinocyte basal media with growth supplements (KGM, Keratinocyte Growth Medium 2, PromoCell, VWR, Radnor, PA) at 37°C with 5% CO2. For optimal growth, calcium levels were increased to 0.15 mM using a commercially available calcium chloride solution (PromoCell, VWR, Radnor, PA). Since insulin is known to suppress IGFBP-3 expression, experiments were performed in basal conditions, defined as serum-free keratinocyte basal media containing 0.15 mM calcium without supplements (KBM).51 For experiments involving primary cultures, human donor corneas were obtained from Tissue Transplant Services, UT Southwestern Medical Center. Corneal epithelial cells were harvested as previously described.29 HCECs were initially cultured in CnT20 cell culture media enriched for progenitor cell culture (Zen Bio, Research Triangle Park, NC). After the first passage, primary cultures were transitioned to serum-free KGM media and cultured as described above.
2.2 |. Reagents
Recombinant human (rh)IGFBP-3 was purchased from Sino Biological (Wayne, PA) and used to treat cells at a concentration of 500 ng/ml. Lyophilized protein was resuspended in water and was free of preservatives. For select experiments, denatured rhIGFBP-3 was used as an additional control. Antibodies used for immunoblotting and immunofluorescence included: a rabbit polyclonal anti-IGF-1Rβ #3027, a rabbit monoclonal anti-S6 ribosomal protein #2217, a rabbit polyclonal anti-phospho-S6 ribosomal protein (Ser235/236) #2211, a rabbit monoclonal anti-mTOR #2983, a rabbit polyclonal anti-phospho-mTOR (Ser2448) #2971, a rabbit monoclonal anti-BNIP3L/NIX #12396, a rabbit monoclonal anti-mitofusion-2 #9482, a rabbit monoclonal anti-COX IV #4850, a rabbit polyclonal anti-VDAC1 #4661 (Cell Signaling, Danvers, MA); a mouse monoclonal anti-Akt1 #sc-5298, a rabbit polyclonal anti-phospho-serine 473 Akt1 #sc-7985-R; a mouse polyclonal anti-GAPDH #sc-66163, a mouse polyclonal anti-β-actin # sc-47778; a rabbit polyclonal anti-LC3B #L7543 (Sigma, St. Louis, MO); a mouse monoclonal anti-p62/ SQSTM1 #H00008878 (Novusbio, Littleton, CO); a rabbit polyclonal anti-PINK-1 #AB23707 (Abcam, Cambridge, MA), and a mouse monoclonal anti-LC3B #M152–3 (MLB, Woburn, MA). Secondary antibodies for immunofluorescence were anti-rabbit IgG conjugated to Alexa Fluor 488 and anti-mouse IgG conjugated to Alexa Fluor 555 (Cell Signaling, Danvers, MA). Secondary antibodies for immunoblotting were goat anti-rabbit IgG conjugated to horseradish peroxidase (HRP) #170–6515 and goat anti-mouse conjugated to HRP #170–6516 (Bio-rad, Hercules, CA).
2.3 |. Plasmid transfection
The GFP-mCherry-LC3 expression plasmid was transiently expressed in hTCEpi cells using our previously published protocol.31 Briefly, semi-confluent cells were transfected with a plasmid encoding GFP-mCherry-LC3 using Lipofectamine 3000 (Invitrogen, Carlsbad, CA) in antibiotic-free KGM. Five micrograms of plasmid DNA and 10 μl of P3000™ reagent were placed into 125 μl of Opti-MEM™ medium (Invitrogen, Carlsbad, CA). In a separate mixture, 5 μl of Lipofectamine 3000 (Invitrogen, Carlsbad, CA) was diluted in 125 μl of Opti-MEM™ medium. This was then added to the DNA mixture and incubated for 15 min before being added to hTCEpi cells containing 1 ml of KGM. Cells were incubated for 12 h. Following this, cells were subsequently transfected with double-stranded inhibitory RNA oligonucleotides targeting IGFBP-3 (described below). Cells were then cultured with or without 500 ng/ml of rhIGFBP-3 for 24 h. Cells were imaged on a Leica SP8 laser scanning confocal microscope (Leica Microsystems, Heidelberg, Germany) equipped with an environmental chamber to maintain cells at 37C°, 5% CO2. A 63×oil objective was used. In order to prevent spectral crosstalk between channels, images were sequentially scanned. Puncta were quantified using Fiji.52 A minimum of 20 cells were analyzed per condition per experiment.
2.4 |. siRNA knockdown of IGF-1R, IGFBP-3, and BNIP3L/NIX
For small interfering RNA (siRNA) experiments, hTCEpi cells were seeded at 50%–60% confluence into six-well plates and grown overnight. Cells were transfected with double-stranded inhibitory RNA oligonucleotides (FlexiTube GeneSolution, IGFBP-3 #GS3486, IGF-1R #GS3480, BNIP3L/NIX #SI02651544, Qiagen, Germantown, MD), using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA) in antibiotic-free KBM. Twelve pmol of siRNA oligonucleotides were added to 100 μl KBM and incubated for 5 min at room temperature. The siRNA mixture was then added to 2 μl Lipofectamine diluted in 100 μl KBM and allowed to incubate for an additional 15 min, after which the transfection mixture was added directly to hTCEpi cells containing 1 ml of KBM. Samples were then incubated for 24 h prior to treatment. Media was removed and cells were then cultured in KBM for another 24 h with or without IGFBP-3 as indicated. Allstars negative control siRNA was used as a non-targeting control (Qiagen, Germantown, MD) for all experiments.
2.5 |. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis and immunoblotting
For immunoblotting of whole cell lysate, adherent epithelial cells were directly lysed in six-well culture plates. The cell lysis buffer for isolation of autophagy proteins (ATG buffer) contained 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, and a protease and phosphatase inhibitor cocktail (Thermo Fisher, Rockford, IL). Samples were then placed on ice for 10 min, followed by centrifugation for 5 min at 12 000 rpm at 4°C (BioRad, Hercules, CA). Supernatants were removed and protein concentration was measured using a Qubit 3.0 Fluorometer (Thermo Fisher, Rockford, IL). Samples were then boiled for 5 min in 4×sample buffer pH 6.8 containing 65.8 mM Tris-HCl, 26.3% (w/v) glycerol, 2.1% SDS, 5.0% β-mercaptoethanol and 0.01% bromophenol blue (Bio-rad, Hercules, CA). Samples were electrophoresed through a 4%–15% precast linear gradient polyacrylamide gel (Bio-rad, Hercules, CA) and transferred to a polyvinyl difluoride membrane (Millipore, Temecula, CA). Transfer membranes were then blocked using 5% non-fat milk (Bio-rad, Hercules, CA) for 1 h at room temperature. Membranes were washed with phosphate-buffered saline (PBS) and incubated in primary antibody at 4°C. After overnight incubation, membranes were washed three times for 5 min and then incubated for 1 h with an anti-mouse or anti-rabbit secondary antibody (Santa Cruz, CA). An ECL Prime Detection Reagent (Amersham Biosciences, Piscataway, NJ) was used to visualize protein. Membranes were imaged on an Amersham Imager 600 (Amersham Biosciences, Piscataway, NJ). For immunoblots of whole cell lysates, β-actin or GAPDH were used as loading controls. Immunoblots were then analyzed using ImageQuant TL Toolbox v8.1 software (Amersham Bioscience, Piscataway, NJ). All protein bands were normalized using the respective control. Phosphorylated proteins were normalized to the respective total protein.
2.6 |. Immunoprecipitation and immunoblotting
To determine the presence of protein interactions between BNIP3L/NIX and LC3 in hTCEpi cells, whole cell lysates were immunoprecipitated with a rabbit monoclonal anti-BNIP3L/Nix #12396 antibody and a rabbit polyclonal anti-LC3B #L7543 antibody (Sigma, St. Louis, MO). A rabbit IgG was used as a control (sc2027, Santa Cruz, CA). Two micrograms of antibody were incubated with 500 μg of protein from whole cell lysates with continuous shaking overnight at 4°C in a refrigerated environmental chamber (Thermo Fisher, Rockville, IL). 25 μl of immobilized protein A/G plus magnetic agarose beads (Thermo Fisher, Rockville, IL) prewashed with 0.05% Tween-20 in TBS were added to the protein antibody complexes and incubated at room temperature for 1 h with shaking. A DynaMag-2 magnet (Thermo Fisher, Rockville, IL) was used to separate the beads from the lysis buffer and then beads were washed twice with TBS followed by one wash with deionized water. Following this, 60 μl of 4× sample buffer (pH 6.8) containing 0.25 M Tris, 8% lauryl sulfate, 40% glycerol, 20% mercaptoethanol, and 0.04% bromophenol blue was added for ten minutes at room temperature with shaking to elute the beads. The supernatant was electrophoresed through a 4%–5% precast linear gradient polyacrylamide gel (Bio-rad, Hercules, CA) and transferred to membranes as described above. Membranes were blotted for BNIP3L/NIX and LC3.
2.7 |. Nuclear and mitochondrial fractionation with immunoblotting
To determine the intracellular localization of IGF-1R, hTCEpi cells were subjected to mitochondrial and cytoplasmic fractionation. Trypsin-ethylenediaminetetraacetic acid (EDTA, Gibco, St. Louis, MO) was used to harvest cells after 24 h of treatment in KBM with or without rhIGFBP-3. Cells were washed with PBS prior to use. A mitochondrial fractionation kit (Thermo Fisher, Rockford, IL) was used to separate the cytosolic and mitochondrial fractions. A nuclear fractionation kit (Thermo Fisher, Rockford, IL) was used to separate the cytoplasmic, insoluble, and soluble nucleus. A Qubit 3.0 Fluorometer (Thermo Fisher, Rockford, IL) was used to determine protein concentrations. Both fractions were boiled with 4× sample buffer and electrophoresed through a 4%–15% polyacrylamide gel (BioRad, Hercules, CA). Immunoblotting was performed as described above.
2.8 |. Immunofluorescence
For immunofluorescent staining, hTCEpi cells were seeded onto 35-mm coverslip bottom dishes (MatTek Corporation, Ashland, MA). siRNA was used to knockdown IGFBP-3 as described and cells were incubated for 24 h. Cells were then cultured in KBM with or without rhIGFBP-3 for an additional 24 h. After, cells were washed twice with cold PBS and fixed in 1% paraformaldehyde (Electron Microscopy Sciences, Fort Washington, PA) in PBS for 10 min. Following this, cells were washed with PBS and then permeabilized in 0.1% Triton X-100 in PBS for another 10 min. Samples were then washed three times for 5 min each with PBS. Next, cells were blocked using 0.5% bovine serum albumin (Sigma, St. Louis, MO) in PBS for 30 min. Cells were incubated overnight at 4°C using one of the following primary antibodies diluted in 0.1% bovine serum albumin (Sigma, St. Louis, MO): PINK1, IGFBP-3, LC3B, BNIP3L/NIX, MFN2, and P62. Then, each sample was washed in PBS and stained with secondary antibodies, anti-rabbit IgG conjugated to Alexa Fluor 488 #4412 (Cell Signaling, Danvers, MA), and anti-mouse IgG conjugated to Alexa Fluor 555 #4409 (Cell Signaling, Danvers, MA) for 1 h at room temperature. Prolong gold anti-fade reagent containing 4′,6-diamidino-2-phenylindole (DAPI) was used for mounting and nuclear staining (Invitrogen, Carlsbad, CA). Cells were imaged on a Leica SP8 laser scanning confocal microscope (Leica Microsystems, Heidelberg, Germany) using a 63×oil objective. In order to prevent spectral crosstalk between channels, images were sequentially scanned. Fiji was used for quantification of immunofluorescence. 52 A minimum of 20 cells per condition per experiment were analyzed.
2.9 |. Mitochondrial polarization
To visualize mitochondrial changes, hTCEpi cells were seeded at 50% confluence and grown on coverslip bottom MatTek dishes (MatTek Corporation, Ashland, MA). Cells were transfected as described with siRNA oligonucleotides targeting IGFBP-3. After transfection, cells were treated for 24 h in KBM with and without 500 ng/ml rhIGFBP-3. Ten minutes prior to the end of the treatment period, cells were treated with either 10 μg/ml of tetraethylbenzimidazolylcarbocyanine iodide (JC-1) dye. For a depolarization control, hTCEpi cells were treated with 50 μM carbonyl cyanide m-chlorophenyl hydrazine (FCCP) in dimethyl sulfoxide (DMSO) for 10 min at 37°C. Cells were then washed three times in PBS and imaged with a Leica SP8 laser scanning confocal microscope (Leica Microsystems, Heidelberg, Germany) using a 63×oil objective. An environmental chamber was used during imaging to maintain cells at 5% CO2 and 37°C. JC-1 stained monomers were scanned using a 488 nm excitation laser and JC-1 aggregates were scanned using a 561 nm excitation laser. Sequential scanning was performed to prevent spectral crosstalk.
2.10 |. Fluorescent visualization of mitophagy
To visualize mitophagy, hTCEpi cells were seeded at 50% confluence on coverslip bottom MatTek dishes (MatTek Corporation, Ashland, MA). Cells were transfected as described above with siRNA oligonucleotides targeting IGFBP-3. Following transfection, cells were treated for 24 h in KBM with and without 500 ng/ml rhIGFBP-3. Ten minutes prior to the end of the 24-h treatment, cells were co-labeled with 100 nM of MitoTraker™ green (MTG, Invitrogen, Carlsbad, CA) to assess mitochondrial morphology and 100 nM of LysoTracker™ (Invitrogen, Carlsbad, CA) to visualize lysosomes. Co-localization of mitochondria with lysosomes indicates mitophagy.
2.11 |. Enzyme-linked immunoassay
Cells were transfected as described above and then placed in KBM media for 24 h with or without rhIGFBP-3. Media was collected and concentrated using protein concentrators containing a polyethersulfone membrane (3K MWCO; Millipore, Burlington, MA). Cell lysates were harvested using the ATG buffer as described. Whole cell lysates were incubated on ice with intermittent vortexing for 30 min. Protein concentration was measured using a Qubit 3.0 Fluorometer (Thermo Fisher, Rockford, IL). A human IGFBP-3 Quantikine ELISA (R&D systems, Minneapolis, MN) was used to determine levels of IGFBP-3 in cell supernatants and whole cell lysates. All samples were run in triplicate and repeated a minimum of two additional times.
2.12 |. Metabolic flux analysis
Using a Seahorse Metabolic Analyzer XFp (Agilent Technologies, Santa Clara, CA) simultaneous measurements of the cellular oxygen consumption rate (OCR) and intracellular acidification rate (ECAR) were made. hTCEpi cells were seeded into a six-well plate and grown overnight in KGM media. Media was removed and a transfection mixture to knockdown either IGFBP-3 or IGF-1R was added directly to hTCEpi cells and incubated for 24 h. Transfection media was then removed and the cells were trypsinized and centrifuged at 1500 rpm. They were then seeded onto Seahorse XFp miniplates and allowed to adhere overnight at 37°C, 5% CO2 for 24 h. The media was then removed and KBM with or without rhIGFBP-3 was added to the Seahorse XFp miniplates and cultured for another 24 h at 37°C, 5% CO2. Before measuring the OCR and ECAR, plates were incubated at 37°C with Seahorse XF base medium containing 1 mM pyruvate, 2 mM glutamine, and 10 mM glucose (pH 7.4) in a non-CO2 incubator for one hour. A Seahorse XFp Cell Mito Stress Test Kit (Agilent Technologies, Santa Clara, CA) was used to evaluate metabolic parameters. During the test, 10 μM oligomycin was added first to inhibit ATP synthase at 20 min followed by an injection of 10 μM carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) at 50 min to uncouple the proton gradient and disrupt the mitochondrial membrane potential, allowing for maximal oxygen consumption. Finally, an injection of rotenone and antimycin A at 80 min, which inhibits complexes 1 and 3 of the electron transport chain and shuts down mitochondrial respiration, was used to determine nonmitochondrial respiration. Measurements were taken every 6 min for a total of 94 min. Using the manufacturers Wave software, version 2.3.0, data was analyzed and the ratio for OCR/ECAR was calculated for each miniplate. Spare respiratory capacity and coupling efficiency were calculated using the following equations: spare respiratory capacity = (maximal respiration)/(basal respiration) × 100; coupling efficiency = (ATP-linked respiration rate)/(basal respiration rate) × 100. All six wells in the Seahorse plate were assessed per condition and repeated a minimum of two additional times. Data were normalized for cell number using a Celigo Image Cytometer (Nexelom Bioscience, Lawrence, MA).
2.13 |. Transmission electron microscopy
hTCEpi cells were seeded onto 35-mm coverslip bottom dishes (MatTek Corporation, Ashland, MA) and allowed to adhere overnight. Cells were transfected as described above and treated for 24 h in KBM with or without rhIGFBP-3. After treatment, cells were fixed with 2.5% glutaraldehyde/0.1 M cacodylate buffer pH 7.4 for 15 min at room temperature. Samples were then processed at the Electron Microscopy Core Facility at UT Southwestern. Briefly, cells were then rinsed three times for 5 min with 0.1 M sodium cacodylate buffer, and then post-fixed in 1% osmium tetroxide and 0.8% K3[Fe(CN6)] in 0.1 M sodium cacodylate buffer for 1 h at room temperature. Next, cells were again rinsed with water and stained overnight en bloc with 2% aqueous uranyl acetate. Cells were then rinsed in water and dehydrated with increasing concentrations of ethanol. Cells were then infiltrated with Embed-812 resin and polymerized at 60°C overnight. Embedded cell blocks were sectioned using a diamond knife (Diatome) on a Leica Ultracut UCT (7) ultramicrotome (Leica Microsystems) and placed onto copper grids. Samples were then post stained with 2% uranyl acetate in water and lead citrate. Images were acquired on a JEOL 1400 Plus (JEOL) equipped with a LaB6 source using a voltage of 120 kV. Mitochondrial length was measured using Fiji.52
2.14 |. Real time polymerase chain reaction
hTCEpi cells were seeded into a six-well plate and allowed to adhere overnight. Cells were transfected as described above and treated for 24 h in KBM with or without rhIGFBP-3. After treatment, RNA was extracted using an RNeasy kit according to the manufacturer protocol (Qiagen, Germantown, MD). Residual genomic DNA was removed using gDNA Wipeout Buffer (Qiagen, Germantown, MD). RNA concentration was then quantified using a Nanodrop One™ (Thermo Fisher, Rockford, IL) and 1 μg mRNA was reverse transcribed using a QuantiTect Reverse Transcription Kit (Qiagen, Germantown, MD). Real-time polymerase chain reaction (PCR) was performed using a QuantiFast SYBR Green PCR kit (Qiagen, Germantown, MD) to quantify levels of two nuclear encoded genes, Eukaryotic Initiation Factor 2 (EIF2) and NADH Dehydrogenase, and the mitochondrial encoded gene, Cyclooxygenase 2 (COX2). The experiments were run on a QuantStudio 6 Flex Real Time PCR machine (Applied Biosystems, Foster City, CA). During real time PCR, 100 ng of cDNA was amplified using 1 μM of each QuantiTect Primer Assay (Qiagen, Germantown, MD) for Hs_EIF2AK1 (QT01018920), Hs_PTGS2 (QT00040586) and Hs_ LOC100128596 (QT02432626). The total reaction volume was 10 μl and subject to 40 cycles of real-time PCR. Water was used as a no-template control and all controls were performed in parallel. Samples were plated in triplicate for all experiments. Experiments were repeated a minimum of two additional times. Data were analyzed using the 2-ΔΔCT method. Levels of COX2 were normalized to each of the nuclear-encoded genes.
2.15 |. Mitochondrial reactive oxygen species production
Mitochondrial produced H2O2 was measured using an Amplex Red Enzyme Assay (Thermo Fisher, Rockford, IL). hTCEpi cells were seeded onto a 96-well black plate and allowed to adhere overnight. A 96-well clear plate was plated in parallel to observe treatment effects. Cells were transfected as described above and treated for 24 h in KBM with or without rhIGFBP-3 for 24 h. Amplex Red was then added to cells to produce fluorescent resorufin when it reacts with H2O2. Florescence was measured at excitation/emission maxima of 530/590 nm on a Cellometer K2 Fluorescent Viability Cell Counter (Nexcelom, Lawrence, MA). All assays were done in triplicate and repeated a minimum of two additional times.
2.16 |. Mitochondrial mass
Mitochondrial mass was evaluated by seeding hTCEpi cells into a six-well plate. Cells were allowed to adhere overnight. Cells were then transfected as described above and placed in KBM for 24 h with or without rhIGFBP-3 prior to imaging. CellLight™ mitochondria-GFP (C10508, Thermo Fisher, Waltham, MA) was used to transiently express GFP in hTCEpi cells according to the manufacturer’s instructions. CellLight reagent was added to the cells 16 h before imaging. Cells were imaged on a Leica SP8 laser scanning confocal microscope (Leica Microsystems, Heidelberg, Germany) equipped with an environmental chamber. A 63×oil objective was used. Z-stacks were used to generate 3D images of mitochondria. Mitochondrial mass was then determined using Imaris software (Biplane, Concord, Massachusetts).
2.17 |. Statistical analysis
Data are expressed as mean ± standard deviation. For the comparison between two groups, Student’s t-test was utilized. For comparisons between more than two groups, a one-way ANOVA was used. A two-way ANOVA was used for the comparison of greater than two groups with multiple factors. For ANOVA analysis, an appropriate post hoc testing was performed. Statistical significance was set at p < .05.
3 |. RESULTS
3.1 |. Nuclear IGFBP-3 accumulation parallels an increase mitochondrial respiration
Prior studies in our laboratory have indicated a potential regulatory role for IGFBP-3 in mitochondrial homeostasis.53 To investigate the potential effects of IGFBP-3 on metabolic activity, a Seahorse Mito-Stress assay was performed. To accomplish this, hTCEpi cells were cultured in KBM and transfected with siRNA oligonucleotides targeting IGFBP-3 or a non-targeting control. In parallel cultures, IGFBP-3 knockdown cells were co-treated with 500 ng/ml rhIGFBP-3. Knockdown efficiency and uptake of recombinant protein in whole cell lysates were measured using an ELISA (Figure 1A). As expected, there was a significant reduction in intracellular IGFBP-3 in siRNA-treated cells (p < .05). Treatment with rhIGFBP-3 increased intracellular levels 10-fold compared to the siRNA control (p < .05). Secreted or extracellular levels of rhIGFBP-3 were also measured using ELISA (Figure 1B). Similar to intracellular IGFBP-3, there was a significant decrease in IGFBP-3 in conditioned media following knockdown (p < .05). This was increased fourfold by treatment with rhIGFBP-3 (p < .05). We next tested the effect of increasing concentrations of rhIGFBP-3 on intracellular uptake. In whole cell lysates, the level of intracellular IGFBP-3 was increased in a concentration-dependent manner (Figure S1A,B) with no apparent difference in subcellular localization.
FIGURE 1.
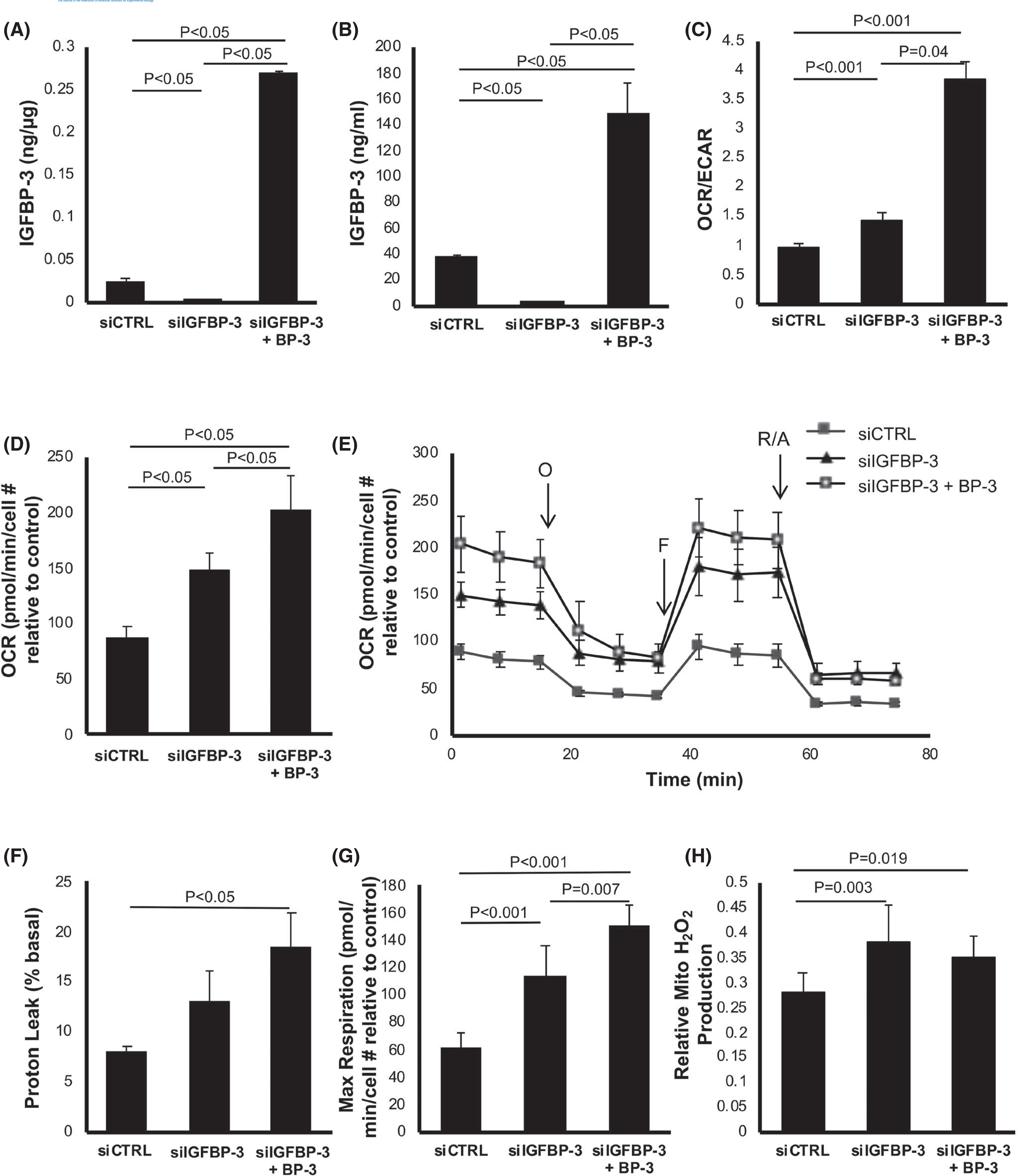
IGFBP-3 regulates mitochondrial respiration in corneal epithelial cells. hTCEpi cells were transfected with siRNA oligonucleotides targeting IGFBP-3. Non-targeting oligonucleotides were used as a control. Cells were cultured in KBM with or without 500 ng/ml rhIGFBP-3 for 24 h. Knockdown efficiency was confirmed using ELISA. (A) Intracellular IGFBP-3 expression was significantly decreased following knockdown (p < .05). Co-treatment with rhIGFBP-3 increased intracellular levels compared to the knockdown and siRNA control (p < .05). (B) Extracellular IGFBP-3 was similarly decreased after knockdown (p < .05) and increased after the addition of rhIGFBP-3 (p < .05). (C–H) Seahorse metabolic flux analysis. (C) The OCR/ECAR ratio was increased following knockdown of IGFBP-3 (p < .001). Co-treatment with rhIGFBP-3 further increased the OCR/ECAR ratio compared to the control (p < .001) and knockdown (p = .04). (D) OCR was increased in IGFBP-3 knockdown cells (p < .05) and further increased when treated with rhIGFBP-3 (p < .05). (E) OCR plotted as a function of time. Time points for the addition of oligomycin (O), FCCP (F), and rotenone/antimycin A (R/A) are indicated. (F) The proton leak was not increased following IGFBP-3 knockdown, but was increased by the addition of rhIGFBP-3 (p < .05). (G) In contrast, maximal respiration showed the same sequential increases as those seen with OCR in both knockdown and rhIGFBP-3-treated cells (p < .001 and p = .007, respectively). (H) Mitochondrial H2O2 concentrations were measured using an Amplex Red florescence assay. Knockdown of IGFBP-3 increased ROS levels compared to control (p = .003). Co-treatment with rhIGFBP-3 did not further increase ROS levels compared to the knockdown, but ROS levels were increased compared to control (p = .019). Data expressed as mean ± standard deviation from one representative experiment, N = 3. One-way ANOVA with Student–Newman–Keuls post hoc multiple comparison test. ECAR, extracellular acidification rate; KBM, keratinocyte basal media; OCR, oxygen consumption rate
We next tested the effects of IGFBP-3 on metabolic activity. The metabolic phenotype was determined by calculating the ratio of OCR, a measure of mitochondrial respiration, to the extracellular acidification rate (ECAR), a measure of glycolysis (Figure 1C). In agreement with our previously published data, the OCR/ECAR ratio was very low in control cells, indicating a glycolytic phenotype.31 IGFBP-3 knockdown resulted in a small but significant increase in mitochondrial respiration (p < .001). This effect was further enhanced by co-treatment with rhIGFBP-3 (p < .001 and p = .04, compared to the control and IGFBP-3 knockdown, respectively). Consistent with this, OCR was lowest in the control group and increased after IGFBP-3 knockdown (p < .05, Figure 1D). Interestingly, OCR was further increased in the knockdown group co-treated with rhIGFBP-3 (p < .05 compared to IGFBP-3 knockdown and the non-targeting control). The ability of exogenous IGFBP-3 to increase OCR was further tested at low (physiological) and high levels of rhIGFBP-3. Despite differential uptake of rhIGFBP-3, co-treatment with either 50 or 500 ng/ml both increased OCR (p < .05, Figure S2A). There were no differences in OCR between cells treated with low and high concentrations of rhIGFBP-3. Subcellular fractionation assays were used to further investigate the distribution of rhIGFBP-3. IGFBP-3 was not detected in the mitochondrial fraction (Figure S2B). Fractionation of lysates into nuclear and cytoplasmic components confirmed the presence of IGFBP-3 in the cytoplasm of control cells was greater than the knockdown condition (p = .024, Figure S2C). Cytoplasmic levels of IGFBP-3 were increased in both IGFBP-3-treated conditions compared to knockdown (p = .004 and p < .001 for 50 and 500 ng/ml rhIGFBP-3 treatment groups, respectively) and control (p = .006 for the 500 ng/ml rhIGFBP-3 treatment, Figure S2C). Interestingly, while IGFBP-3 knockdown showed a significant decrease in IGFBP-3 intracellular and extracellular expression, there was a corresponding increase in rhIGFBP-3 in the insoluble fraction compared to the control (p = .047, Figure S2C). In cells co-treated with rhIGFBP-3 following knockdown, the level of IGFBP-3 was further increased in the cytoplasm and insoluble nucleus compared to the control (p = .017 and p = .002 for 50 and 500 ng/ml rhIGFBP-3 treatment compared to the control, respectively, Figure S2C). These data suggest that decreasing levels of IGFBP-3 mRNA using siRNA oligonucleotides triggers an accumulation of IGFBP-3 in the insoluble nucleus. This is associated with the increase in mitochondrial respiration. The addition of exogenous IGFBP-3 further increased IGFBP-3 levels in the insoluble nucleus, along with an increase in mitochondrial and adenosine triphosphate (ATP)-linked respiration (Figure S3A).
To examine the various contributions of the electron transport chain to overall mitochondrial function, cells were treated with 1.0 μM oligomycin to inhibit ATP synthase and block ATP-linked respiration (Figure 1E). In IGFBP-3 knockdown cells, this triggered an increase in non-mitochondrial oxygen consumption, but not a significant increase in the proton leak (p < .001, Figures 1F and S3B). In contrast, co-treatment with rhIGFBP-3 did not alter non-mitochondrial respiration compared to the IGFBP-3 knockdown, but increased the proton leak (p < .05). Cells were then treated with the electron transport chain uncoupler, trifluoromethoxy carbonylcyanide phenylhydrazone (FCCP), to allow for uninhibited electron flow and determination of maximal and spare respiratory capacity. As shown in Figure 1G, maximal respiration was increased following IGFBP-3 knockdown (p < .001 compared to control) and was further increased by co-treatment with rhIGFBP-3 (p = .007 and p < .001 compared to IGFBP-3 knockdown and control, respectively). There was no change in spare respiratory capacity (Figure S3C). Similar to the total OCR, basal respiration was also increased after IGFBP-3 knockdown (p < .001) and further increased following co-treatment with rhIGFBP-3 (p < .001, Figure S3D). Although co-treatment with rhIGFBP-3 showed a distinct increase in respiration, there was not a corresponding increase in mitochondrial reactive oxygen species (ROS; Figure 1H). There was no measurable change in coupling efficiency between the test and control groups (Figure S3E). Collectively, these findings further support a role for IGFPB-3 in mediating mitochondrial respiration. To confirm that reduced expression of endogenous IGFPB-3 is required to trigger this increase in metabolic activity, hTCEpi cells were cultured in KGM, KBM, or KBM supplemented with rhIGFBP-3. As expected, (due to our prior work), culture in KBM increased the expression of endogenous IGFBP-3. The addition of exogenous protein without siRNA knockdown, had no further effects on OCR (Figure S4A–C).
3.2 |. IGFBP-3 negatively regulates autophagic flux
We next sought to examine the effects of IGFBP-3 on canonical autophagy and mitophagy, the latter a critical component of mitochondrial quality control. To determine the effect of IGFBP-3 on autophagic flux, hTCEpi cells were transfected with siRNA oligonucleotides targeting IGFBP-3 or non-targeting controls. After knockdown, cells were treated for 24 h with or without rhIGFBP-3. Cells were further treated with or without 10 nM bafilomycin to block lysosomal fusion. As shown in Figure 2A, knockdown of IGFBP-3 had no effect on the amount of the microtubule-associated protein 1A/1B-light chain (LC3-II) or sequestosome 1 (P62/SQSTM1). However, in cells treated with bafilomycin, exogenous rhIGFBP-3 decreased levels of LC3-II, indicating a reduction in autophagic flux. Similar patterns of expression were also seen for primary cultured HCECs, HBECs, and HCjECs, (Figure 2B for HCECs and Figure S5A,B for HBECs and HCjECs). Knockdown efficiency and re-uptake of IGFBP-3, measured by ELISA, are shown in Figure S5A–D.
FIGURE 2.
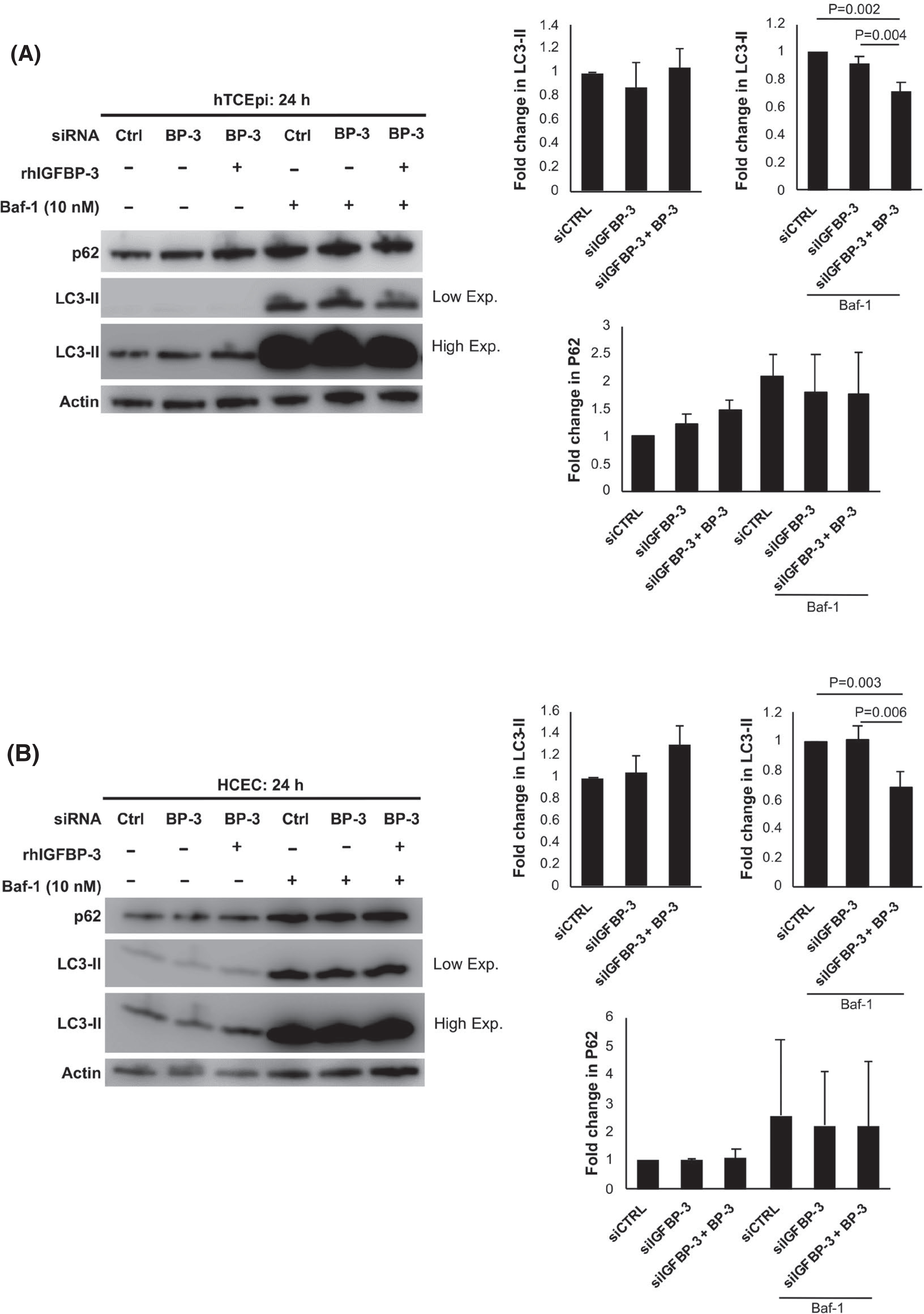
IGFBP-3 blocks autophagy in epithelial cells. Cells were transfected with siRNA oligonucleotides targeting IGFBP-3. Non-targeting oligonucleotides were used as a control. Cells were then cultured in KBM with or without 500 ng/ml rhIGFBP-3 for 24 h. Ten nM bafilomycin (Baf-1) was used to block lysosomal fusion. Lysates were immunoblotted for autophagy markers P62 and LC3-II. Low exposure (Low Exp) was used to visualize LC3-II in cells treated with Baf-1 due to robust protein accumulation. High exposure (High Exp) was used to visualize cells not treated with Baf-1 due to low accumulation of the LC3-II protein. (A) In hTCEpi cells, in the absence of Baf-1, no differences were observed in LC3-II. Treatment with rhIGFBP-3 did decrease LC3-II levels in Baf-1-treated cells (p = .004 and p = .002 compared to knockdown and control, respectively). (B) In HCECs, there was a similar decrease in LC3-II after treatment with rhIGFBP-3 in Baf-1-treated cells (p = .006 and p = .003 compared to knockdown and control, respectively). Data expressed as mean ± standard deviation from 3 repeated experiments. One-way ANOVA with Student–Newman–Keuls post hoc multiple comparison test. KBM, keratinocyte basal media
To further investigate changes in autophagic flux, hT-CEpi cells were transfected with an expression plasmid encoding green fluorescent protein (GFP)-mCherry-LC3. Cells were subsequently transfected with siRNA oligonucleotides targeting IGFBP-3 or non-targeting controls and treated with or without rhIGFBP-3. Consistent with our immunoblotting data, there was no accumulation of either autophagosomes (yellow) or autophagolysosomes (red) in cells cultured in growth media (Figure 3A). In KBM, there was an increase in both autophagosomes and autophagolysosomes, demonstrating an induction of autophagic flux. In IGFBP-3 knockdown cells, as shown by an increase in the ratio of red to green puncta (p < .05, Figure 3B), there was a further increase in autophagolysosomes, indicating an acceleration of the basal autophagic flux. In contrast, co-treatment with rhIGFBP-3 triggered an accumulation of autophagosomes that was confirmed by a decrease in the ratio of red to green puncta (p < .05). This decrease in autophagic flux is consistent with a block in autophagy. To further test the relationship between IGFBP-3 and mitophagy, cells were double-labeled with the lysosomal probe, LysoTracker, and the mitochondrial probe, MitoTracker Green, to assess for colocalization (Figure 3C). Cells were again transfected with siRNA oligonucleotides targeting IGFBP-3 or non-targeting controls and treated with or without rhIGFBP-3. MitoTracker Green staining showed elongated tubular mitochondria in KGM. In the KBM control, some shortening of the mitochondrial tubules was observed. In terms of mitophagy, there was an increase in colocalization of mitochondria and lysosomes in IGFBP-3 knockdown cells; whereas, there was no visible colocalization in cells treated with rhIGFBP-3. Taken together, these data indicate that uptake of exogenous IGFBP-3 functions to block mitophagy in corneal epithelial cells.
FIGURE 3.
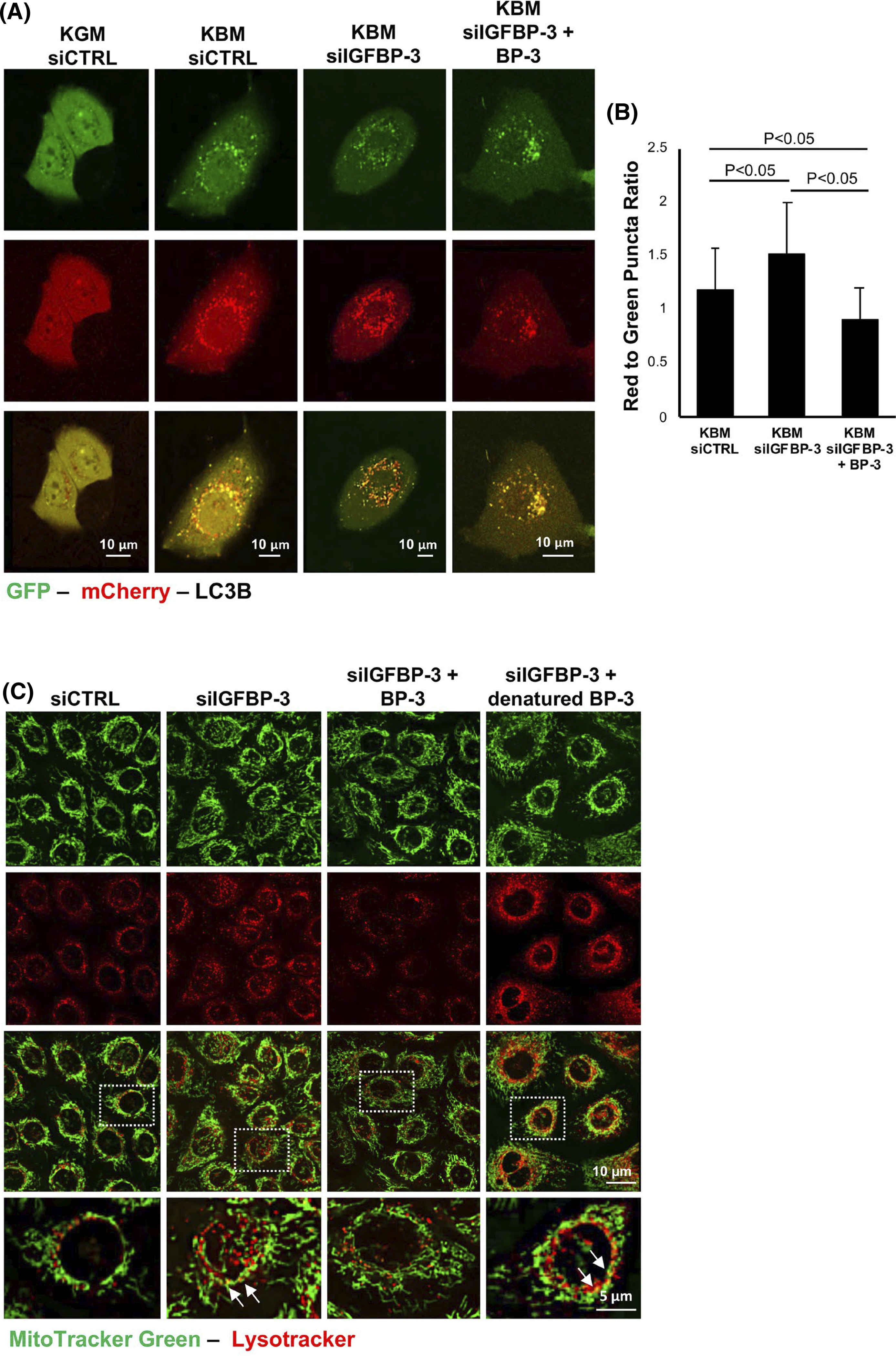
IGFBP-3 inhibits autophagic flux in corneal epithelial cells. (A and B) hTCEpi cells were sequentially transfected with siRNA oligonucleotides targeting IGFBP-3 or a non-targeting control, followed by transfection with a GFP-mCherry-LC3 expression plasmid. Cells were then cultured in either KGM or KBM with or without 500 ng/ml rhIGFBP-3 for 24 h. (A) In KBM, there was an increase in the accumulation of both autophagosomes (yellow puncta) and autophagolysosomes (red puncta). Knockdown of IGFBP-3 increased the number of autophagolysosomes, whereas the addition of rhIGFPB-3 led to an accumulation of autophagosomes. Scale bar: 10 μm. (B) In IGFBP-3 knockdown cells, there was an increase in the ratio of red to green puncta compared to the siRNA control (p < .05). In contrast, co-treatment with rhIGFBP-3 showed a decrease in the ratio of red to green puncta compared to knockdown with IGFBP-3 and the KBM control (p < .05). (C) MitoTracker Green (green) and LysoTracker (red) colocalization in hTCEpi cells. Colocalization was rarely observed in the growth condition transfected with a control siRNA. There was a slight increase in colocalization in KBM. Knockdown of IGFBP-3 increased colocalization of both probes, indicating an increase in mitophagy. Treatment with rhIGFBP-3 decreased colocalization, suggesting an inhibition of mitophagy. Zoomed images corresponding to the white box in the merged image are shown in the bottom row. Arrows indicate areas of colocalization. Scale bar: 10 μm. Data presented as mean ± standard deviation from one representative experiment, N = 3. Fifteen cells per group were analyzed. KBM, keratinocyte basal media; KGM, keratinocyte growth media
We next evaluated the localization of LC3 and P62. Using immunostaining, we found differences in intracellular localization with and without rhIGBP-3 (Figure S7A). In control cells, LC3 was detected in both the nucleus and the cytoplasm. Staining intensity for P62 was weak. Consistent with the GFP-mCherry-LC3 data, knockdown of IGFBP-3 induced nuclear translocation of LC3 and loss of detectable P62. In the nucleus, LC3 undergoes deacetylation. It then moves to the cytoplasm where it can interact with ATG7 to be utilized for autophagy.54 Together, the accumulation of LC3 in the nucleus following IGFBP-3 knockdown and the concurrent increase in LC3 in the cytoplasm following treatment with bafilomycin, suggests an increase in autophagic flux (p < .05, Figure S7B). In contrast, co-treatment with rhIGFBP-3 following knockdown prevented nuclear accumulation, with LC3 restricted to the cytoplasmic compartment (p < .05). This was associated with an increase in P62. The expression of LC3 and P62 after treatment with bafilomycin also showed an increasing trend.
3.3 |. Exogenous IGFBP-3 increases levels of mtDNA
Due to the observed shift in mitochondrial respiration and mitophagy, mtDNA levels were quantified for each test condition.Figure 4A shows SybrGreen staining of mtDNA. Quantification of SybrGreen fluorescence demonstrated a significant decrease in IGFBP-3 knockdown cells, while co-treatment with rhIGFBP-3 increased fluorescence compared to both the knockdown and KBM control (p < .05, Figure 4B). These changes were confirmed using qPCR (Figure 4C). We next quantified mitochondrial mass in live cells using a fluorescent mitochondrial probe. Unlike mtDNA, we were unable to detect a change in mitochondrial mass between the treatment and control groups (Figure S8A). To further investigate the effects on mitochondrial morphology, transmission electron microscopy (TEM) was used was used to quantify mitochondrial length (Figures 4D and S8B). In control cells cultured in KBM, mitochondria were sparse and fairly small. While occasional mitochondria were missing cristae, most cristae were present in a wavy, somewhat lamellar pattern. This is consistent with the mild stress induced by 24 h of culture in KBM. Knockdown of IGFBP-3, however, triggered a robust increase in small mitochondria with balloon-like mitochondrial cristae, suggestive of mitochondrial fission and/or decreased remodeling. These changes were associated with a decrease in mitochondrial length (p < .05, Figure S8B). Co-treatment with rhIGFBP-3 abrogated this effect, resulting in elongated mitochondria with lamellar-like cristae (p < .05, Figure S8B).
FIGURE 4.
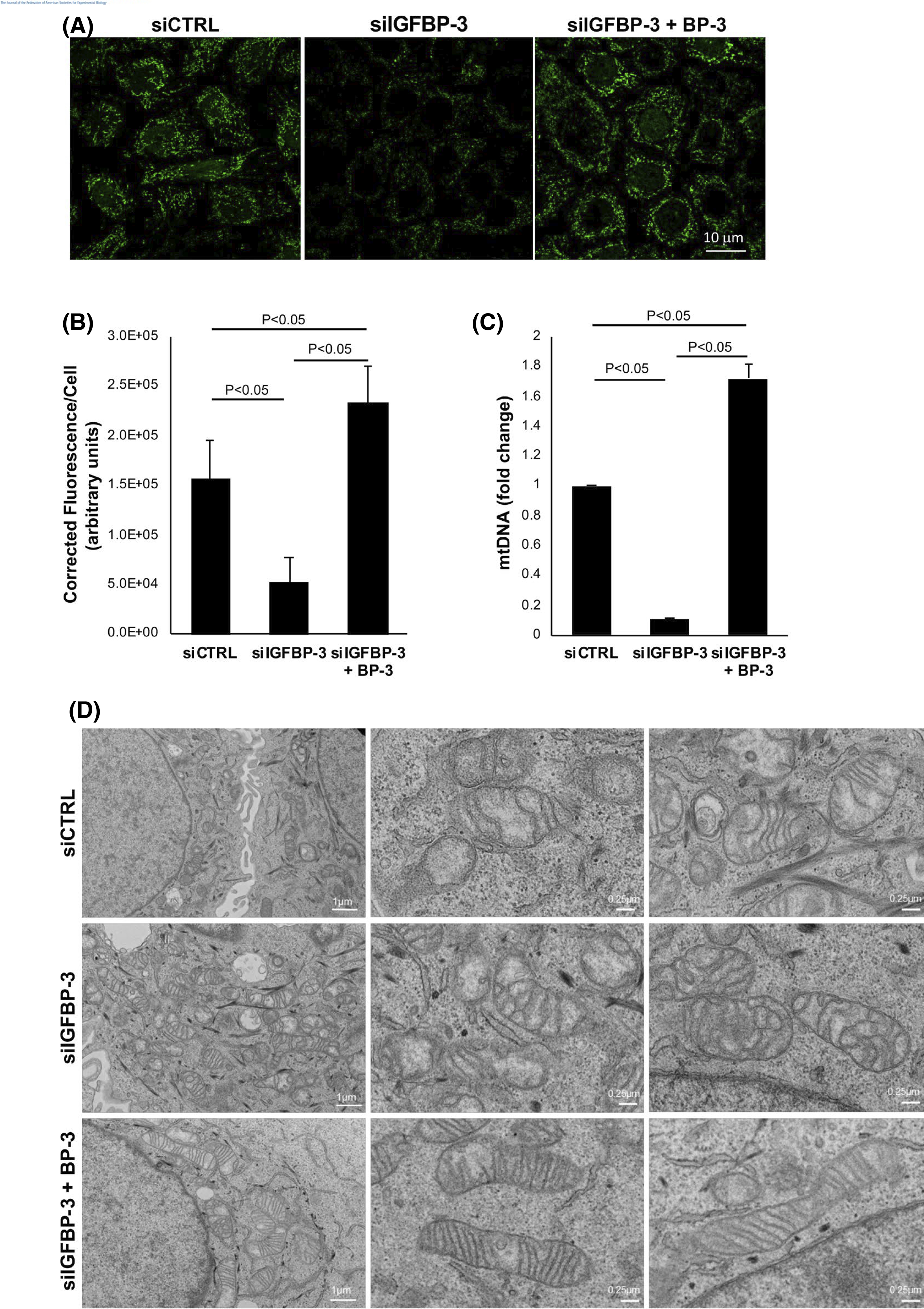
IGFBP-3 regulates mitochondrial morphology and mtDNA levels. hTCEpi cells were transfected with siRNA oligonucleotides targeting IGFBP-3. Non-targeting oligonucleotides were used as a control. Cells were then cultured in KBM with or without 500 ng/ml rhIGFBP-3 for 24 h. (A) mtDNA measured by SYBR green (shown in green) was decreased after IGFBP-3 knockdown. This decrease was blocked by co-treatment with rhIGFBP-3. Scale bar: 10 μm. (B) Quantification of total corrected florescence per cell showed identical changes (p < .05). (C) mtDNA measured by qPCR. IGFBP-3 knockdown decreased mtDNA (p < .05), while co-treatment with rhIGFBP-3 increased mtDNA compared to the knockdown and control conditions (p < .05). (C) TEM showing distinct changes in mitochondrial morphology and lamellar structure across treatment groups. Scale bar: 1 μm (first column), 0.25 μm (second and third columns). All data are expressed as mean ± standard deviation of 3 repeated experiments. One-way ANOVA with Student–Newman–Keuls post hoc multiple comparison test. KBM, keratinocyte basal media
3.4 |. Loss of IGF-1R drives mitochondrial respiration
We have previously reported that a reciprocal relationship exists between IGF-1R and IGFBP-3.51 While IGFBP-3 is critical to regulating nuclear translocation of IGF-1R, IGF-1R is essential in the regulation of IGFBP-3. This was supported by our prior studies showing that IGFBP-3 expression was abrogated by knockdown of IGF-1R. To analyze the effect of IGF-1R knockdown on mitochondrial function, a Seahorse Mito-Stress assay was performed. hT-CEpi cells were again transfected with siRNA oligonucleotides targeting IGF-1R or non-targeting controls. Cells were cultured in KBM with or without rhIGFBP-3. Knockdown efficiency of IGF-1R was confirmed using western blot (Figure 5A). Compared to control, knockdown of IGF-1R showed an increase in OCR (p < .001, Figure 5B,C), basal respiration (p < .001, Figure 5D), ATP-linked respiration (p = .001, Figure S9A), and non-mitochondrial oxygen consumption (p = .009, Figure 5E). Co-treatment with rhIGFBP-3 increased OCR compared to the control (p < .001, Figure 5C), but was unchanged compared to the IGF-1R knockdown. Basal respiration and ATP-linked respiration were further increased by the addition of rhIGFBP-3 (p = .004 and p = .01, respectively, Figures 5D and S9A). Non-mitochondrial respiration was unchanged (Figure 5E).
FIGURE 5.
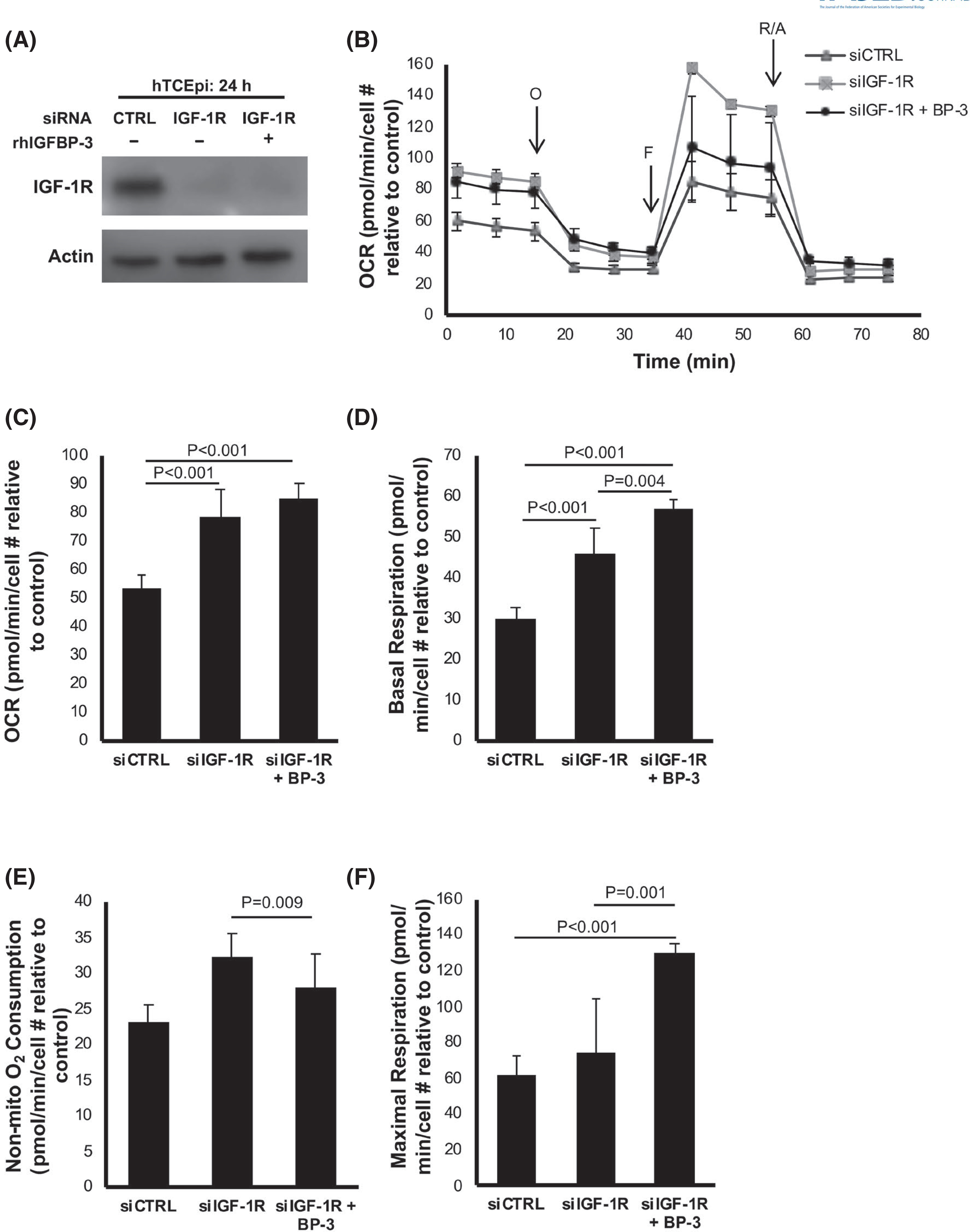
IGF-1R regulates mitochondrial and non-mitochondrial oxygen consumption. hTCEpi cells were transfected with siRNA oligonucleotides targeting IGF-1R. Non-targeting oligonucleotides were used as a control. Cells were then cultured in KBM with or without 500 ng/ml rhIGFBP-3 for 24 h. (A) Immunoblotting was used to confirm knockdown of IGF-1R. ß-actin was used as a loading control. (B) OCR plotted as a function of time. The time points for addition of oligomycin (O), FCCP (F), and rotenone/antimycin A (R/A) are indicated. (C) OCR increased after IGF-1R knockdown (p < .001). OCR was unchanged in cells treated with rhIGFBP-3 compared to IGF1R knockdown. (D) IGF-1R knockdown increased basal OCR (p < .001), which was further increased by treatment with rhIGFBP-3 (p = .004). (E) Non-mitochondrial oxygen consumption was increased after IGF-1R knockdown (p = .009) but was unchanged with the addition of rhIGFBP-3 (F) Maximal respiration was not affected by IGF-1R knockdown, but was increased in cells co-treated with rhIGFBP-3 (p < .001 compared to control, p = .001 compared to IGF-1R knockdown). Data expressed as mean ± standard deviation from one representative experiment, N = 3. One-way ANOVA with Student–Newman–Keulspost hoc multiple comparison test. N = 3 repeated experiments. KBM, keratinocyte basal media; OCR, oxygen consumption rate
In contrast to knockdown of IGFBP-3, maximal respiration was unaffected by IGF-1R knockdown but was increased by co-treatment with rhIGFBP-3 (p = .001 compared to IGF-1R knockdown, p < .001 compared to control, Figure 5F). rhIGFBP-3 also increased the spare respiratory capacity compared to the IGF-1R knockdown (p = .009, Figure S9B) and increased the proton leak (p < .001, Figure S9C). There were no differences in coupling efficiency in any of the test groups (Figure S9D). Taken together, these data show that loss of IGF-1R increases mitochondrial respiration. This parallels the changes observed with loss of IGFBP-3. These data further show that supplementation with exogenous IGFBP-3 under these conditions is associated with a substantial increase in maximal respiration.
3.5 |. IGFBP-3 inhibits sBNIP3L/NIX-mediated mitophagy through mTOR
To investigate whether loss of IGFBP-3 also modulates mitophagy, we first examined expression levels of BNIP3L/NIX and PINK1 in hTCEpi cells after IGFBP-3 knockdown with and without rhIGFBP-3. As shown in Figure 6A, only the short isoform (s) of BNIP3L/NIX was detected in hTCEpi cells. This isoform was increased following knockdown of IGFBP-3 in hTCEpi cells (p < .05). Co-treatment with rhIGFBP-3 maintained sBNIP3L/NIX expression at basal levels (p < .05). Unlike sBNIP3L/NIX, there were no differences in PINK1 expression between test group and the basal control. Opposite sBNIP3L/NIX, expression of Mitofusin-2 (MFN2) was decreased in IGFBP-3 knockdown cells (p = .013), but was unchanged in rhIGFBP-3-treated cells compared to control (p = .024). Immunofluorescent staining for sBNIP3L/NIX also showed an increase in fluorescence intensity after IGFBP-3 knockdown (Figures 6B and S10A). In addition to cytosolic staining, sBNIP3L/NIX also localized to the nucleus in certain cells. Both the increase in staining intensity and the observed shift to the nucleus were abrogated by treatment with rhIGFBP-3 (p < .05). In Figures 6C and S10B, immunofluorescent staining for MFN2 again showed a decrease in IGFBP-3 knockdown cells, which was blocked by co-treatment with rhIGFBP-3. Finally, no significant changes were noted for PINK1 (Figures 6D and S10C).
FIGURE 6.
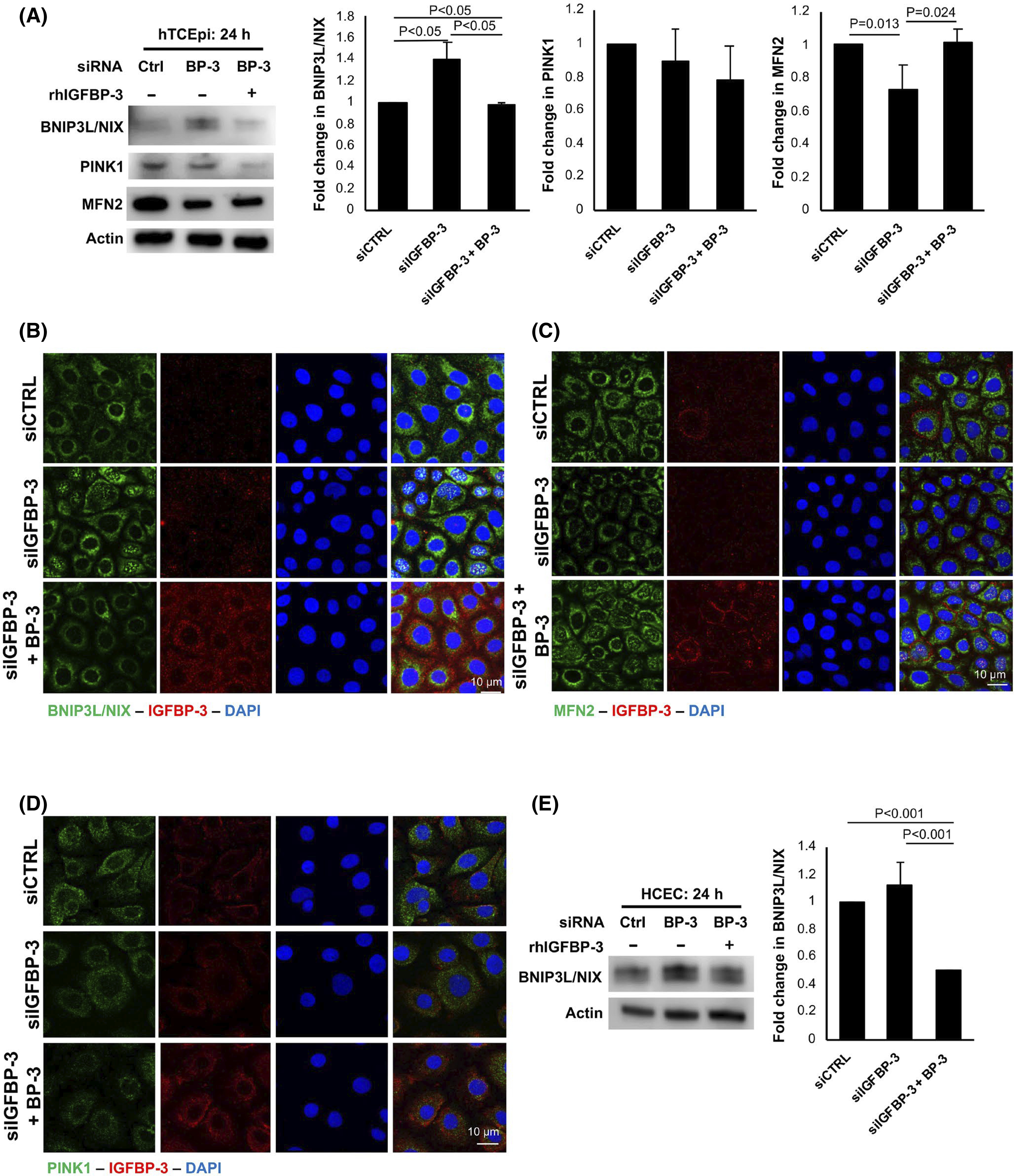
IGFBP-3 blocks mitophagy through sBNIP3L/NIX. Cells were transfected with siRNA oligonucleotides targeting IGFBP-3. Non-targeting oligonucleotides were used as a control. Cells were then cultured in KBM with or without 500 ng/ml rhIGFBP-3 for 24 h. Knockdowns were confirmed with ELISA or immunofluorescent staining. (A) Immunoblotting for mitophagy markers BNIP3L/NIX and PINK1, and the mitochondrial fusion protein MFN2 in hTCEpi cells. There was an increase in sBNIP3L/NIX in IGFBP-3 knockdown treated cells (p < .05), whereas co-treatment with rhIGFBP-3 decreased sBNIP3L/NIX compared to the knockdown (p < .05). In contrast, MFN2 was decreased in IGFBP-3 knockdown cells (p = .013). The addition of rhIGFBP-3 prevented this decrease (p = .024 compared to knockdown), maintaining MFN2 expression at control levels. ß-actin was used as a loading control. There were no significant changes in PINK1. (B) Immunofluorescent labeling of hTCEpi cells for sBNIP3L/NIX (green) and IGFBP-3 (red). Nuclei were labelled with DAPI. Scale bar: 10 μm. (C) Immunofluorescent labeling of hTCEpi cells for MFN2 (green) and IGFBP-3 (red). Nuclei were labeled with DAPI (blue). Scale bar: 10 μm. (D) Immunofluorescent labeling of PINK1 (green) and IGFBP-3 (red) in hTCEpi cells. Nuclei were labeled with DAPI (blue). Scale bar: 10 μm. (E) Immunoblotting of BNIP3L/NIX and PINK1 in primary cultured HCECs. Similar to cell line findings, the presence or absence of IGFBP-3 modulated sBNIP3L/NIX expression. Data presented as mean ± standard deviation from three repeated experiments. One-way ANOVA with Student–Newman–Keuls post hoc multiple comparison test. KBM, keratinocyte basal media
Further interrogation of BNIP3L/NIX in primary cultured HCECs showed the same increase in expression of the short isoform after IGFBP-3 knockdown that was abrogated by the addition of rhIGFBP-3 (p < .001, Figure 6E). The full-length isoform was undetectable. Since PINK1-mediated mitophagy requires mitochondrial depolarization for protein stability, polarization was evaluated using J-aggregate-forming cationic dye (JC-1). As shown in Figure S11, there was no mitochondrial depolarization following knockdown of either IGFBP-3 (Figure S11A) or IGF-1R (Figure S11B). Thus, the absence of mitochondrial depolarization prevented the accumulation of PINK1 at the outer mitochondrial membrane, shifting cells toward a receptor-mediated mitophagy pathway involving sBNIP3L/NIX.
mTOR is known to play an important role in the initiation of canonical autophagy.10 To examine the effects of IGFBP-3 on components of the mTOR pathway, whole cell lysates from hTCEpi cells were immunoblotted with antibodies for phosphorylated Akt, mTOR, and S6 (Figure 7A). Knockdown of IGFBP-3 resulted in a decrease in Thr(308)-p-Akt (p = .004) and Ser(2448)-p-mTOR (p < .05). This decrease in mTOR activity was consistent with the increase in mitophagy in IGFBP-3 knockdown cells. In contrast to this, there was an increase in phosphorylated proteins in cells co-treated with rhIGFBP-3 (p-Akt p = .011, p-S6 p = .001, and p-mTOR p < .05). This increase in phosphorylation indicates activation of the mTOR pathway, which is known to inhibit autophagy. To further interrogate a potential relationship between sBNIP3L/NIX and mTOR, cells were transfected with siRNA oligonucleotides targeting BNIP3L/NIX. Compared to the non-targeting control, knockdown of sBNIP3L/NIX increased the level of Ser(2448)-p-mTOR (p = .001, Figure 7B). Collectively, these data support a role for sBNIP3L/NIX in the regulation of mTOR activity.
FIGURE 7.
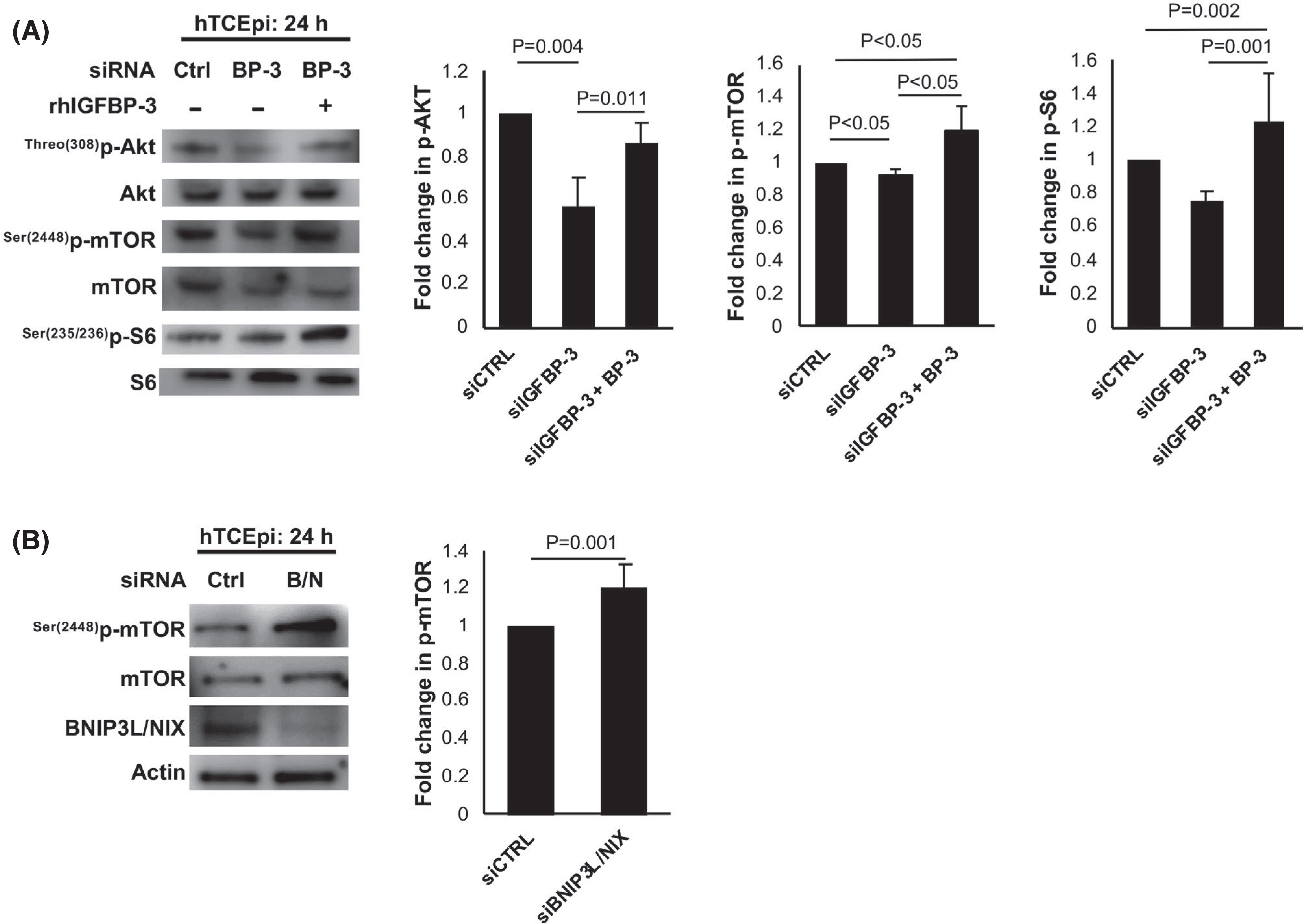
IGFBP-3 blocks mitophagy through activation of mTOR. (A) hTCEpi cells were transfected with siRNA oligonucleotides targeting IGFBP-3. Non-targeting oligonucleotides were used as a control. Cells were then cultured in KBM with or without 500 ng/ml rhIGFBP-3 for 24 h. Immunoblotting for phosphorylation of Akt, mTOR, and S6 proteins in hTCEpi cells. There was a decrease in phosphorylation of AKT (p = .004) and mTOR (p < .05) in knockdown treated cells. Treatment with rhIGFBP-3 increased phosphorylation of mTOR (p < .05 compared to knockdown and control), p-Akt (p = .011 compared to knockdown), and S6 (p = .001 and p = .002 compared to knockdown and control). All phosphorylated proteins were normalized to the respective total protein expression. (B) Cells were transfected with siRNA oligonucleotides targeting BNIP3L/NIX. Non-targeting oligonucleotides were used as a control. Cells were then cultured in KBM for 24 h. Knockdown was confirmed using immunoblotting. Immunoblotting for phosphorylation of mTOR was completed by normalizing phosphorylated protein to total protein expression. Knockdown of BNIP3L/NIX increased mTOR phosphorylation (p = .001). Data representative of mean ± standard deviation from three repeated experiments. One-way ANOVA with Student–Newman–Keuls post hoc multiple comparison test. KBM, keratinocyte basal media
To confirm that this pathway was not specific to corneal epithelial cells, we further investigated the expression of BNIP3L/NIX, PINK1, and MFN2 in bronchial and conjunctival epithelial cells. In agreement with our findings in corneal epithelial cells, only the short isoform of BNIP3L/NIX was detectable by immunoblotting. Knockdown of IGFBP-3 in HBECs also increased sBNIP3L/NIX expression in whole cell lysates (p = .035, Figure S12A) but was subsequently decreased after co-treatment with rhIGFBP-3 (p = .031). Immunostaining for sBNIP3L/NIX also showed an increase in staining intensity with occasional nuclear localization in IGFBP-3 knockdown cells, with loss of expression in rhIGFBP-3-treated cells (Figure S12B). In contrast, MFN2 was decreased in knockdown cells and maintained at basal levels when treated with rhIGFBP-3 (p = .006 compared to IGFBP-3 knockdown and p = .026 compared to the siRNA control, Figure S12A,C). Similar findings in terms of expression and localization were also seen in conjunctival epithelial cells with knockdown of IGFBP-3 (p = .013, Figure S13A) and treatment with rhIGFBP-3 decreasing sBNIP3L/NIX (p = .012 compared to the knockdown and p = .014 compared to control), while increasing MFN2 (p < .001 and p = .007, compared to the knockdown and control, respectively). Immunostaining showed similar findings as both HBEC and HTCEpi cells (Figure S13B,C). The expression of PINK1 was unchanged in both cell lines (Figures S12A and S13A).
To further investigate the role of sBNIP3L/NIX in the autophagic changes observed with IGFBP-3, we tested for the interaction of sBNIP3L/NIX and LC3-II by immunoprecipitation (Figure 8). An IgG was used as a control. Consistent with our other findings, reciprocal co-immunoprecipitations demonstrated an interaction between the sBNIP3L/NIX and LC3-II in epithelial cells with IGFBP-3 knockdown (Figure 8A,B). This interaction was decreased by the addition of rhIGFBP-3. In Figure 8C, immunofluorescence showed an increase in co-localization and overall protein expression for sBNIP3L/NIX and LC3-II in cells without IGFBP-3. Nuclear sBNIP3L/NIX coincided with strong nuclear expression of LC3-II. As expected, treatment with rhIGFBP-3 blunted this effect. Together, these findings support a role for IGFBP-3 in the regulation of mitophagy and mitochondrial dynamics through downregulation of sBNIP3L/NIX and activation of mTOR. A summary of these findings is detailed in Figure 9.
FIGURE 8.
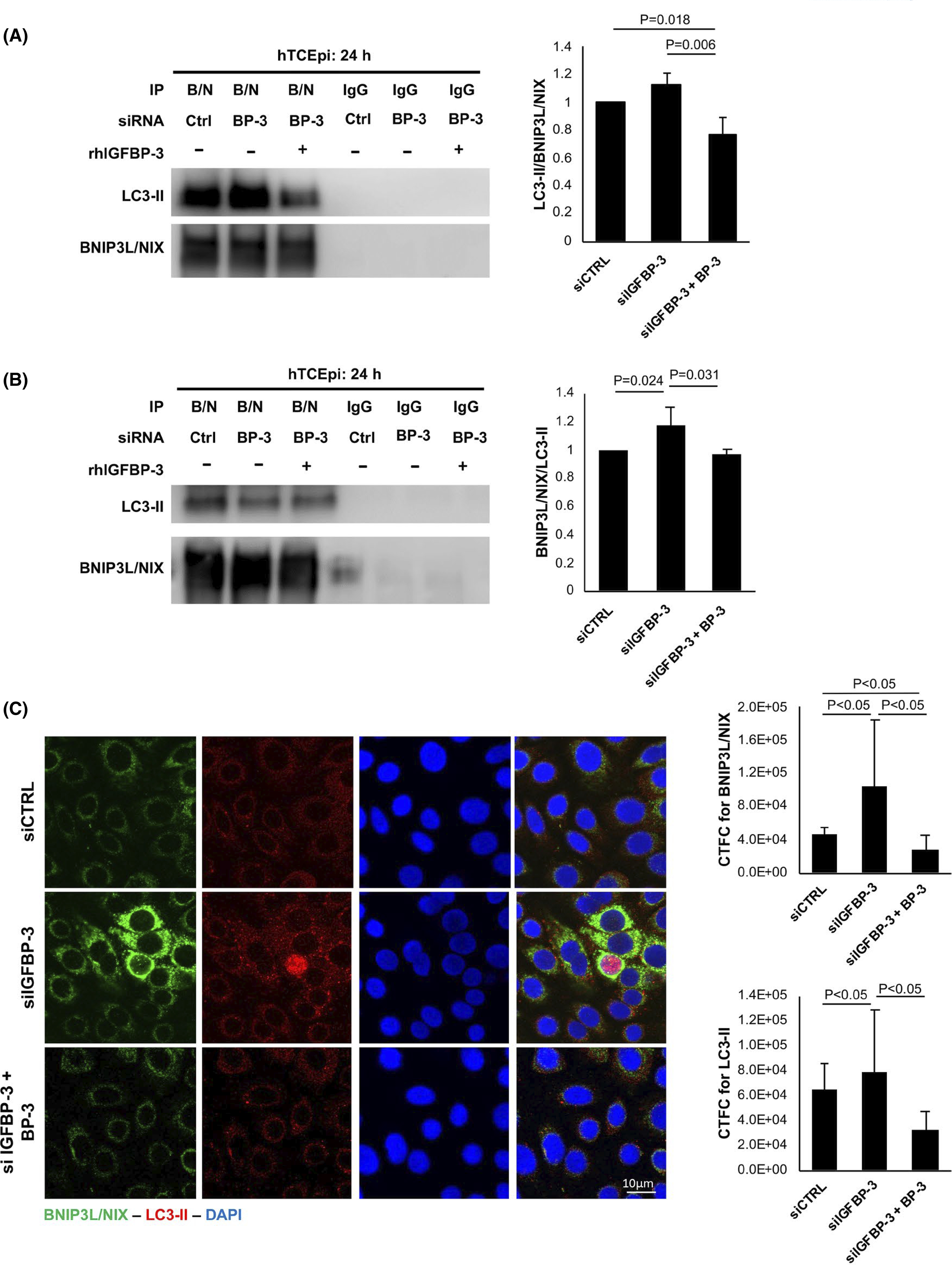
IGFBP-3 increases mitophagy through increased interaction of sBNIP3L/NIX and LC3-II. Cells were transfected with siRNA oligonucleotides targeting IGFBP-3. Non-targeting oligonucleotides were used as a control. Cells were then cultured in KBM with or without 500 ng/ml rhIGFBP-3 for 24 h. (A) Immunoprecipitation of sBNIP3L/NIX and immunoblotting for LC3-II confirmed a decrease in the interaction between LC3-II and sBNIP3L/NIX in cells co-treated with rhIGFBP-3 (p = .006 and p = .018 compared to knockdown and control, respectively). (B) Immunoprecipitation for LC3-II and immunoblotting sBNIP3L/NIX showed an increase in the interaction between LC3-II and sBNIP3L/NIX in knockdown cells (p = .024 compared to control). This was decreased by co-treatment with rhIGFBP-3 (p = .031 compared to knockdown). (C) Immunofluorescent labeling and quantification for sBNIP3L/NIX (green) and LC3-II (red) in hTCEpi cells. Nuclei were labeled with DAPI (blue). Findings paralleled the immunoprecipitation data shown in panels A and B. Scale bar: 10 μm. All data are expressed as mean ± standard deviation from three repeated experiments. One-way ANOVA with Student–Newman–Keuls post hoc multiple comparison test. KBM, keratinocyte basal media
FIGURE 9.
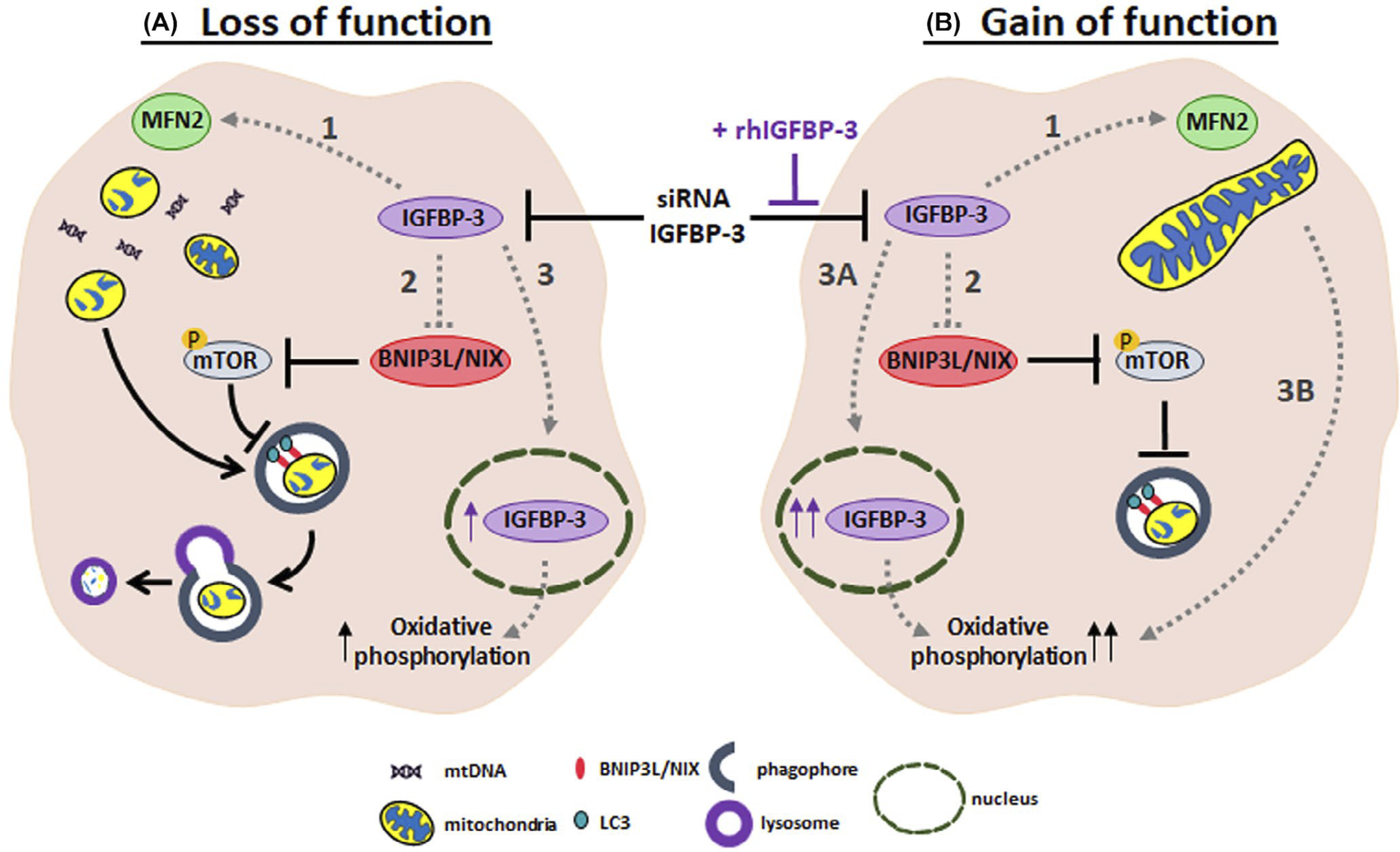
IGFBP-3 functions as a molecular switch that mediates mitochondrial and metabolic homeostasis. (A) Loss of function of IGFBP-3: knockdown of IGFBP-3 using siRNA oligonucleotides abrogated cytoplasmic expression. This was associated with (1) a decrease in MFN2 that coincided with the appearance of numerous small mitochondria and potential release of mtDNA; (2) an increase in sBNIP3L/NIX that activated mitophagy through mTOR; and (3) residual IGFBP-3 protein accumulation in the nucleus and increased oxidative phosphorylation. (B) Gain of function of IGFBP-3: Co-treatment with exogenous IGFBP-3 in cells subject to IGFBP-3 knockdown resulted in opposite effects with (1) an increase in MFN2 that coincided with an enhancement in lamellar cristae morphology; (2) a decrease in sBNIP3L/NIX that inhibited mitophagy through activation of mTOR; and (3) robust accumulation of IGFBP-3 in the nucleus and further increase in oxidative phosphorylation. Dotted lines highlight the pathways identified in this study. Further experiments are required to define the molecular mechanisms responsible for the observed phenotypes
4 |. DISCUSSION
Here we report the novel identification of IGFBP-3 as a critical regulator of mitochondrial structure and function in epithelial cells. Our first key finding was the observation that loss of IGFBP-3 increased mitochondrial respiration, while co-treatment with the recombinant protein further increased it. This finding was unexpected and appeared to be associated with the subcellular localization of IGFBP-3. IGFBP-3 is a pleiotropic secretory protein that functions in both an IGF-dependent and -independent manner. In our cell model, exogenous IGFBP-3 triggers cell cycle arrest in actively proliferating cells in growth media.51 In contrast to this, culture in basal media upregulates IGFBP-3, the extent of which, is influenced by cell confluence.51 Thus, treatment of cells in basal media with exogenous IGFBP-3 had little effect, due to the increased presence of the endogenous protein. To examine the effects of exogenous IGFBP-3 while controlling for confluence, siRNA knockdown was employed. Surprisingly, while knockdown of IGFBP-3 decreased IGFBP-3 expression in whole cell lysates, there was a corresponding increase in IGFBP-3 in the insoluble nucleus. This observed increase in nuclear IGFBP-3 was either due to the presence of residual intracellular protein accumulating in the nucleus after knockdown or the uptake of extracellular IGFBP-3 from conditioned media. Supplementation with exogenous IGFBP-3 after knockdown further increased nuclear levels, indicating that at least a portion of the nuclear localized protein originated from the extracellular environment.
IGFBP-3 is known to localize to the nucleus and contains a nuclear localization signal.55 Nuclear activities of IGFBP-3 include nonhomologous endjoining to repair DNA double stranded breaks, apoptosis, and transcriptional regulation.56 While we previously reported on the translocation of IGFBP-3 to the insoluble nucleus, we now show that this response is dose-dependent and increases with an increase in exogenous IGFBP-3.51 Moreover, increasing levels of IGFBP-3 in the nucleus are associated with increasing levels of OCR. These data suggest a new role for IGFBP-3 in mediating mitochondrial respiration. Potential metabolic functions for IGFBP-3 have been proposed that include glucose regulation and insulin intolerance.57,58 The mechanisms that underlie these essential metabolic functions however, remain undefined and conflicting results argue for and against IGFBP-3 as a protector or initiator of cell death. Within the context of epithelial cells, our data support a protective role for IGFBP-3 in mitochondrial structure and function. This is due in part to the enhanced lamellar cristae architecture, evident by treatment with exogenous IGFPG-3, and mitophagy regulation. Further studies are needed to determine the factors that are upregulated upon IGFBP-3 accumulation in the nucleus. These findings will shed new light on the metabolic function of this protein.
We initially postulated that IGFBP-3 functions as a stress response protein in the corneal epithelium. This was due to our prior observations that growth factor withdrawal, hyperglycemia, and hypoxia (unpublished data) all increase IGFBP-3 expression and secretion.43,44,51 The ability of IGFBP-3 to modulate mitochondrial respiration and mitophagy supports a role for IGFBP-3 in mediating epithelial stress responses. Indeed, we have recently reported on the ability of IGFBP-3 to block a hyperosmolar-mediated drop in respiration and glycolysis.53 In agreement with this, we now report that exogenous IGFBP-3 functions to block an increase in ROS production, despite increased respiration. Exogenous IGFBP-3 also increased the proton leak. The increased protein leak may illustrate a novel protective mechanism against uncoupling to prevent excessive ROS produced by increased oxidative phosphorylation in our epithelial cell models.59 These findings contrast with recent work by Wang and colleagues who reported on an IGFBP-3-mediated increase in ROS in cancer cells. This occurred through the induction of apoptosis and mitochondrial compromise.
IGFBP-3 has been previously reported to induce autophagy in SV40-transformed human lung epithelial cells and breast cancer cells.37,40 In the first study, IGFBP-3 stimulated autophagy through binding interactions with Nurr77 and BCL2 and the subsequent release of Beclin.40 In the second, IGFBP-3 was found to bind directly to the glucose-regulated protein 78 (GRP78), an endoplasmic reticulum protein involved in the regulation of cellular stress.37 In contrast to these prior studies, we found that IGFBP-3 blocked autophagy in three different telomerase-immortalized epithelial cell lines and primary cultured corneal epithelial cells. IGFBP-3 exerts these effects through the short isoform of the mitophagy receptor, BNIP3L/NIX. In IGFBP-3 knockdown cells, not only were changes in cristae ultrastructure observed, but the mitochondria themselves were small and numerous, suggestive of a potential increase in mitochondrial fission or biogenesis. Mitochondria are known to continuously undergo fission and fusion in effort to remove damaged portions of the organelle via mitophagy and maintain homeostasis. Disruption of fusion triggers mitochondrial fragmentation.60 While loss of MFN2 does not confirm a shift in mitochondrial dynamics, it has been established that activation of mitophagy leads to a decline in MFN2.61,62 In concert with this, there was an increase in the overall autophagic flux in IGFBP-3 knockdown cells. This was supported by the increase in autophagolysosomes using live cell imaging and a corresponding increase in nuclear accumulation of LC3, an important step for deacetylation and activation.54 Our subsequent investigation into mTOR activity also showed changes in the phosphorylation of several key proteins up and downstream of mTOR including Akt and S6. In knockdown cells, loss of IGFBP-3 increased expression levels of sBNIP3L/NIX. This in turn blocked phosphorylation of mTOR, allowing for the induction of autophagy. In contrast to loss of function, co-treatment with exogenous IGFBP-3 again yielded the opposite effect. The increase in cytosolic IGFBP-3 was associated with an increase in MFN2. This led to the appearance of longer, fused mitochondria. Increased cytosolic IGFBP-3 also triggered a block in sBNIP3L/NIX and the subsequent phosphorylation mTOR, a known inhibitor of autophagy. Taken together, these data show that changes in the level of cytosolic IGFBP-3 play a key role in mediating mitochondrial morphology and mitophagy through sBNIP3L/NIX and MFN2. The ability of IGFBP-3 to block autophagy in our epithelial cell models, as opposed to other prior reports, may be reconciled by the tissue and context specific functions of IGFBP-3. The one variation in these findings was that knockdown of IGFBP-3 in conjunctival epithelial cells did not increase sBNIP3L/NIX to the same extent as seen in the other two cell types. While the conjunctival epithelial cell line was developed using hTERT, similar to the other cell lines used in this study, it also expresses a dominant negative p53.50 In tumor cells, BNIP3L/NIX has been to shown to be a p53-regulated gene in response to hypoxia.63 Thus, the absence of a functional p53 may have abrogated the upregulation of sBNIP3L/NIX in conjunctival epithelial cells after siRNA knockdown of IGFBP-3.
Despite the stark changes in mitochondrial morphology observed using TEM, we were unable to detect a change in mitochondrial mass. This is most likely due to the shift from the rounder, swollen mitochondria in IGFBP-3 knockdown cells to more thin, elongated structures in the presence of exogenous IGFBP-3. This is consistent with the measurable difference in mitochondrial length across test groups. Since the electron transport chain localizes to the mitochondrial cristae, the enhanced cristae ultrastructure would increase surface area allowing for an increase in mitochondrial respiration. Interestingly, the pattern of reduced mitochondrial length in knockdown cells and elongation in IGFBP-3-treated cells paralleled changes in levels of mtDNA. Mitochondria are known to release mtDNA into the cytosol and extracellular milieu in response to stress. Moreover, fusion defects have also been shown to result in a decrease in mtDNA replication, loss of mitochondrial nucleoids and mtDNA.60 The ability of exogenous IGFBP-3 to restore mitochondrial fusion through the increase in MFN2 and maintenance of mitochondrial ultrastructure is essential to sustain healthy levels of mtDNA for optimal oxidative phosphorylation.
In summary, these findings demonstrate a novel regulatory role for IGFBP-3 as a molecular switch that mediates mitochondrial and metabolic homeostasis. This occurs through (1) control of sBNIP3L/NIX-mediated mitophagy, alterations in mitochondrial dynamics, cristae remodeling, and mTOR signaling; and (2) nuclear accumulation of IGFBP-3. As shown in Figure 9, it remains unclear whether changes in MFN2 expression are a result of direct or indirect changes in cytosolic IGFBP-3. Along these lines, further characterization of fusion and fission proteins are required to fully understand these changes. Finally, while we speculate that the decrease in IGFBP-3 mRNA following siRNA knockdown is the stimulus for IGFBP-3 nuclear accumulation, additional experimentation to test this hypothesis is needed. Thus, future studies will be focused on deciphering the underlying mechanisms that mediate these pathways and their impact on mitochondrial homeostasis in the context of disease.
Supplementary Material
Funding information
This study was funded by NEI grants R01 EY029258 (DMR), R01 EY024546 (DMR), F30 EY031559 (WLS), P30 EY030413, NIH grant 1S10OD021685, Lions Foundation for Sight (WLS/DMR), Eye Bank Association of America (WLS/DMR), and an unrestricted grant from Research to Prevent Blindness, New York, NY
Abbreviations:
- Akt
protein kinase B
- ATG
autophagy
- ATP
adenosine triphosphate
- Baf
Bafilomycin
- BNIP3
BCL2 and adenovirus E1B 19-kDa-interacting
- BNIP3L/NIX
BCL2 and adenovirus E1B 19-kDa-interacting like
- COXIV
cytochrome c oxidase 4
- ECAR
intracellular acidification rate
- ELISA
enzyme-linked immunosorbent assay
- FCCP
Trifluoromethoxy carbonylcyanide phenylhydrazone
- GFP
green fluorescent protein
- HBECs
human bronchial epithelial cells
- HCjECs
human conjunctival epithelial cells
- hTCEpi
human corneal epithelial cells
- IGF
insulin-like growth factor
- IGF-1R
type 1 insulin-like growth factor receptor
- IGFBP-3
insulin-like growth factor binding protein-3
- JC-1
J-aggregate-forming cationic dye
- KBM
keratinocyte basal media
- KGM
keratinocyte growth media
- LC3
microtubule-associated protein 1A/1B-light chain 3
- MFN2
mitofusion 2
- MTG
MitoTracker Green
- mTOR
mammalian target of rapamycin
- OCR
oxygen consumption rate
- p62/SQSTM1
Sequestosome 1
- PBS
phosphate-buffered saline
- PI3K
phosphatidylinositol-3-kinase
- PINK1
PTEN-induced putative kinase 1
- siRNA
small interfering RNA
- TEM
transmission electron microscopy
- ULK
Unc-51-like kinase
- VDAC
voltage dependent anion channel
Footnotes
DISCLOSURES
None of the authors of this manuscript have any conflicts of interest to disclose.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of the article at the publisher’s website.
REFERENCES
- 1.Ott M, Gogvadze V, Orrenius S, et al. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12(5):913–922. [DOI] [PubMed] [Google Scholar]
- 2.Pickles S, Vigié P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 2018;28(4):R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem. 2012;393(7):547–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013;20(1):31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wei H, Liu L, Chen Q. Selective removal of mitochondria via mitophagy: distinct pathways for different mitochondrial stresses. Biochim Biophys Acta. 2015;1853(10):2784–2790. [DOI] [PubMed] [Google Scholar]
- 6.Levine B Cell biology: autophagy and cancer. Nature. 2007;446(7137):745–747. [DOI] [PubMed] [Google Scholar]
- 7.Jin M, Liu X, Klionsky DJ. SnapShot: selective autophagy. Cell. 2013;152(1):368–368.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Papinski D, Kraft C. Regulation of autophagy by signaling through the Atg1/ULK1 complex. J Mol Biol. 2016;428(9): 1725–1741. [DOI] [PubMed] [Google Scholar]
- 9.Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Munson MJ, Ganley IG. MTOR, PIK3C3, and autophagy: signaling the beginning from the end. Autophagy. 2015;11(12): 2375–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoo SM, Jung YK. A molecular approach to mitophagy and mitochondrial dynamics. Mol Cells. 2018;41(1):18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu L, Sakakibara K, Chen Q, et al. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014;24(7): 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lyons A, Coleman M, Riis S, et al. Insulin-like growth factor 1 signaling is essential for mitochondrial biogenesis and mitophagy in cancer cells. J Biol Chem. 2017;292(41):16983–16998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi JW, Jo A, Kim M, et al. BNIP3 is essential for mitochondrial bioenergetics during adipocyte remodelling in mice. Diabetologia. 2016;59(3):571–581. [DOI] [PubMed] [Google Scholar]
- 16.Palikaras K, Lionaki E, Tavernarakis N. Coupling mitogenesis and mitophagy for longevity. Autophagy. 2015;11(8):1428–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16(7):939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y, Decker KF, Zheng D, et al. A nucleus-targeted alternately spliced Nix/Bnip3L protein isoform modifies nuclear factor κB (NFκB)-mediated cardiac transcription. J Biol Chem. 2013;288(22):15455–15465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.King ER, Wong KK. Insulin-like growth factor: current concepts and new developments in cancer therapy. Recent Pat Anticancer Drug Discov. 2012;7(1):14–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Connor R, Fennelly C, Krause D. Regulation of survival signals from the insulin-like growth factor-I receptor. Biochem Soc Trans. 2000;28(2):47–51. [DOI] [PubMed] [Google Scholar]
- 21.Valentinis B, Baserga R. IGF-I receptor signalling in transformation and differentiation. Mol Pathol. 2001;54(3):133–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vincent AM, Feldman EL. Control of cell survival by IGF signaling pathways. Growth Horm IGF Res. 2002;12(4):193–197. [DOI] [PubMed] [Google Scholar]
- 23.Werner H, LeRoith D. The role of the insulin-like growth factor system in human cancer. Adv Cancer Res. 1996;68:183–223. [DOI] [PubMed] [Google Scholar]
- 24.Leroith D, Werner H, Beitner-johnson D, et al. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr Rev. 1995;16(2):143–163. [DOI] [PubMed] [Google Scholar]
- 25.Huo H, Guo X, Hong S, et al. Lipid rafts/caveolae are essential for insulin-like growth factor-1 receptor signaling during 3T3-L1 preadipocyte differentiation induction. J Biol Chem. 2003;278(13):11561–11569. [DOI] [PubMed] [Google Scholar]
- 26.Salani B, Passalacqua M, Maffioli S, et al. IGF-IR internalizes with Caveolin-1 and PTRF/Cavin in HaCat cells. PLoS One. 2010;5(11):e14157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aleksic T, Chitnis MM, Perestenko OV, et al. Type 1 insulin-like growth factor receptor translocates to the nucleus of human tumor cells. Cancer Res. 2010;70(16):6412–6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warsito D, Sjöström S, Andersson S, et al. Nuclear IGF1R is a transcriptional co-activator of LEF1/TCF. EMBO Rep. 2012;13(3):244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu YC, Zhu M, Robertson DM. Novel nuclear localization and potential function of insulin-like growth factor-1 receptor/insulin receptor hybrid in corneal epithelial cells. PLoS One. 2012;7(8):e42483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Titone R, Zhu M, Robertson DM. Insulin mediates de novo nuclear accumulation of the IGF-1/insulin hybrid receptor in corneal epithelial cells. Sci Rep. 2018;8(1):4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Titone R, Robertson DM. Insulin receptor preserves mitochondrial function by binding VDAC1 in insulin insensitive mucosal epithelial cells. FASEB J. 2020;34(1):754–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ranke MB. growth factor binding-protein-3 (IGFBP-3). Best Pract Res Clin Endocrinol Metab. 2015;29(5): 701–711. [DOI] [PubMed] [Google Scholar]
- 33.Furstenberger G, Senn HJ. Insulin-like growthfactors and cancer. Lancet Oncol. 2002;3(5):298–302. [DOI] [PubMed] [Google Scholar]
- 34.Kim J-H, Choi DS, Lee O-H, et al. Antiangiogenic antitumor activities of IGFBP-3 are mediated by IGF-independent suppression of Erk1/2 activation and Egr-1-mediated transcriptional events. Blood. 2011;118(9):2622–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chua MWY, Lin MZ, Martin JL, et al. Involvement of the insulin-like growth factor binding proteins in the cancer cell response to DNA damage. J Cell Commun Signal. 2015;9(2):167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin MZ, Marzec KA, Martin JL, et al. The role of insulin-like growth factor binding protein-3 in the breast cancer cell response to DNA-damaging agents. Oncogene. 2014;33(1):85–96. [DOI] [PubMed] [Google Scholar]
- 37.Grkovic S, O’Reilly VC, Han S, et al. IGFBP-3 binds GRP78, stimulates autophagy and promotes the survival of breast cancer cells exposed to adverse microenvironments. Oncogene. 2013;32(19):2412–2420. [DOI] [PubMed] [Google Scholar]
- 38.Granata R, Trovato L, Lupia E, et al. Insulin-like growth factor binding protein-3 induces angiogenesis through IGF-I- and SphK1-dependent mechanisms. J Thromb Haemost. 2007;5(4): 835–845. [DOI] [PubMed] [Google Scholar]
- 39.Rajah R, Valentinis B, Cohen P. Insulin-like growth factor (IGF)-binding protein-3 induces apoptosis and mediates the effects of transforming growth factor-beta1 on programmed cell death through a p53- and IGF-independent mechanism. J Biol Chem. 1997;272(18):12181–12188. [DOI] [PubMed] [Google Scholar]
- 40.Yin H, Zhang S, Sun Y, et al. MicroRNA-34/449 targets IGFBP-3 and attenuates airway remodeling by suppressing Nur77-mediated autophagy. Cell Death Dis. 2017;8(8):e2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Janssen PT, van Bijsterveld OP. Origin and biosynthesis of human tear fluid proteins. Invest Ophthalmol Vis Sci. 1983;24(5): 623–630. [PubMed] [Google Scholar]
- 42.Robertson DM, Ho S-I, Hansen B, et al. Insulin-like growth factor binding protein-3 expression in the human corneal epithelium. Exp Eye Res. 2007;85(4):492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stuard WL, Titone R, Robertson DM. Tear levels of insulin-like growthfactor binding protein 3 correlate with subbasal nerve plexus changes in patients with type 2 diabetes mellitus. Invest Ophthalmol Vis Sci. 2017;58(14):6105–6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu Y-C, Buckner BR, Zhu M, et al. Elevated IGFBP3 levels in diabetic tears: a negative regulator of IGF-1 signaling in the corneal epithelium. Ocul Surf. 2012;10(2):100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stuard WL, Titone R, Robertson DM. The IGF/Insulin-IGFBP axis in corneal development, wound healing, and disease. Front Endocrinol. 2020;11:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robertson DM, Li LI, Fisher S, et al. Characterization of growth and differentiation in a telomerase-immortalized human corneal epithelial cell line. Invest Ophthalmol Vis Sci. 2005;46(2):470–478. [DOI] [PubMed] [Google Scholar]
- 47.Hoi Y, Spurr-Michaud S, Russo CL, et al. Differential regulation of membrane-associated mucins in the human ocular surface epithelium. Invest Ophthalmol Vis Sci. 2004;45(1):114–122. [DOI] [PubMed] [Google Scholar]
- 48.Delgado O, Kaisani AA, Spinola M, et al. Multipotent capacity of immortalized human bronchial epithelial cells. PLoS One. 2011;6(7):e22023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vaughan MB, Ramirez RD, Wright WE, et al. A three-dimensional model of differentiation of immortalized human bronchial epithelial cells. Differentiation. 2006;74(4):141–148. [DOI] [PubMed] [Google Scholar]
- 50.Gipson IK, Spurr-Michaud S, Argüeso P, et al. Mucin gene expression in immortalized human corneal-limbal and conjunctival epithelial cell lines. Invest Ophthalmol Vis Sci. 2003;44(6):2496–2506. [DOI] [PubMed] [Google Scholar]
- 51.Titone R, Zhu M, Robertson DM. Mutual regulation between IGF-1R and IGFBP-3 in human corneal epithelial cells. J Cell Physiol. 2019;234(2):1426–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bogdan ED, Stuard WL, Titone R, et al. IGFBP-3 mediates metabolic homeostasis during hyperosmolar stress in the corneal epithelium. Invest Ophthalmol Vis Sci. 2021;62(7):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang R, Xu Y, Wan W, et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell. 2015;57(3):456–466. [DOI] [PubMed] [Google Scholar]
- 55.Redfern RL, Barabino S, Baxter J, et al. Dry eye modulates the expression of toll-like receptors on the ocular surface. Exp Eye Res. 2015;134:80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Varma Shrivastav S, Bhardwaj A, Pathak KA, et al. Insulin-like growth factor binding protein-3 (IGFBP-3): unraveling the role in mediating IGF-independent effects within the cell. Front Cell Dev Biol. 2020;8:286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yamada PM, Mehta HH, Hwang D, et al. Evidence of a role for insulin-like growth factor binding protein (IGFBP)-3 in metabolic regulation. Endocrinology. 2010;151(12):5741–5750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ning Y, Schuller AGP, Bradshaw S, et al. Diminished growth and enhanced glucose metabolism in triple knockout mice containing mutations of insulin-like growth factor binding protein-3, -4, and -5. Mol Endocrinol. 2006;20(9):2173–2186. [DOI] [PubMed] [Google Scholar]
- 59.Jastroch M, Divakaruni AS, Mookerjee S, et al. Mitochondrial proton and electron leaks. Essays Biochem. 2010;47:53–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kondadi AK, Anand R, Reichert AS. Functional interplay between cristae biogenesis, mitochondrial dynamics and mitochondrial DNA integrity. Int J Mol Sci. 2019;20(17):4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Benischke A-S, Vasanth S, Miyai T, et al. Activation of mitophagy leads to decline in Mfn2 and loss of mitochondrial mass in Fuchs endothelial corneal dystrophy. Sci Rep. 2017;7(1):6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xiong W, Ma Z, An D, et al. Mitofusin 2 participates in mitophagy and mitochondrial fusion against angiotensin II-induced cardiomyocyte injury. Front Physiol. 2019;10:411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fei P, Wang W, Kim S-H, et al. Bnip3L is induced by p53 under hypoxia, and its knockdown promotes tumor growth. Cancer Cell. 2004;6(6):597–609. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.