

Abstract
The diverse pharmacological role of dihydropyrimidinone scaffold has made it to be an interesting drug target. Because of the high incidence and mortality rate of breast cancer, there is a dire need of discovering new pharmacotherapeutic agents in managing this disease. A series of twenty-two derivatives of 6-(chloromethyl)-4-(4-hydroxyphenyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (3a-3k) and ethyl 6-(chloromethyl)-4-(2-hydroxyphenyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (4a-4k) synthesized in a previous study were evaluated for their anticancer potential against breast cancer cell line. Molecular docking studies were performed to analyze the binding mode and interaction pattern of these compounds against nine breast cancer target proteins. The in vitro cell proliferation assay was performed against the breast cancer cell line MCF-7. The structure activity relationship of these compounds was further studied using QSARINS. Among nine proteins, the docking analysis revealed efficient binding of compounds 4f, 4e, 3e, 4g, and 4h against all target proteins. The in vitro cytotoxic assay revealed significant anticancer activity of compound 4f having IC50 of 2.15 μM. The compounds 4e, 3e, 4g, and 4h also showed anticancer activities with IC50 of 2.401, 2.41, 2.47 and 2.33 μM, respectively. The standard tamoxifen showed IC50 1.88 μM. The 2D qualitative structure-activity relationship (QSAR) analysis was also carried out to identify potential breast cancer targets through QSARINS. The final QSAR equation revealed good predictivity and statistical validation R2 and Q2 values for the model obtained from QSARINS was 0.98 and 0.97, respectively. The active compounds showed very good anticancer activities, and the binding analysis has revealed stable hydrogen bonding of these compounds with the target proteins. Moreover, the QSAR analysis has predicted useful information on the structural requirement of these compounds as anticancer agents with the importance of topological and autocorrelated descriptors in effecting the cancer activities.
1. Introduction
Many of the pharmacologically active natural and synthetic compounds are composed of the heterocyclic nucleus. The derivatives of these agents containing nitrogen, oxygen, and sulphur atoms act as an important scaffold in drug designing. They are also an integral part of nucleic acid base pairs DNA and RNA such as purines and pyrimidines [1].
Most of the alkaloids isolated from marine sources showed significant pharmacological properties which consisted of dihydropyrimidine nucleus. Batzelladine alkaloids A and B are one of these alkaloids isolated from marine sources and act as potent inhibitors of HIV gp-120-CD4. This extended their application in pharmaceutical industry after the identification of another novel cell permeable molecule, 4-(3-hydroxyphenyl)-2-thione derivative, also called manostrol, as anticancer agent. The anticancer activity of manostrol depends on a new mechanism of affecting cell division by specific and reversible inhibition of mitotic kinesis motility without targeting tubulin [2]. The inhibitory action has shown to be on human kinesin Eg5 which causes mitotic arrest followed by apoptosis. This motor protein causes mitotic spindle formation. Other possible targets of these moieties have also been studied including centrin, calcium channels, and topoisomerase I [3]. Analogs of manostrol such as oxomonastrol, thio, and 3,4-methylenedoxy derivatives were developed, and their activity against HT-29 cancer cell lines were tested. Various other synthetic analogs L-771,688 and SQ 32926 have also been developed [1].
Since pyrimidine derivatives shows significant pharmacological activities and are essential constituents of living nature. Biginelli compounds have gained interest since last two decades because of their structural similarity with the clinically active dihydropyrimidine. These compounds are called as the esters of 6-methyl-2-oxo-4- phenyl-1,2,3,4-tetrahydropyrimidine-5-carboxylic acid and were first synthesized by Pietro Biginelli by the condensation reaction of β-ketoesters, aryl aldehydes, and urea under acidic condition through one pot three component synthesis [4].
Manostrol is one of the most studied Biginelli adducts because of its promising anticancer activities providing an inspiration for the design of new compounds. Several manostrol analogs have shown potent anticancer activities against MCF-7 breast cancer cell lines. Globally, breast cancer has been diagnosed as the most commonly diagnosed malignancy having the highest incidence rate of mortality in women [5]. The progression of breast cancer is associated with several factors such as age, personal history of breast cancer, reproductive, environmental, and genetic factors. Prognostic factors can be used to predict the course and clinical outcome of breast cancer. These include ER, PR, Ki-67, and HER-2. Other factors that can be used to predict prognosis include cyclin E, cyclin D1, and cathepsin D but are not measured routinely. The status of progesterone receptor, estrogen receptor, and the human epidermal growth factor receptor 2 basically determines the scheme for the treatment of breast cancer along with the clinicopathological factors such as tumor grade, size, and status of lymph node [6]. The synthesis of derivatives of different scaffold having pharmacological importance has helped us in determining the biological activities of compounds that can further be screened for disease management [7–11].
In silico drug designing is a form of computer-based modeling and is a rapidly developing field. The development of in silico target identifications of drugs with the strategy of fast speed and low cost is receiving a huge attention worldwide because of the limitation of throughput, accuracy and cost, experimental techniques that cannot be applied widely [12]. Major roles of in silico approaches in drug discovery processes include virtual screening, in silico ADME/T prediction and advanced methods for determining protein-ligand binding and quantitative structure-based drug design.
The in silico quantitative structure activity relationship (QSAR) is another approach used to find out a statistical correlation between the structure and function with the help of chemometric technique. The structure represents the substituents, properties, or descriptors of the molecules and their interaction energy fields, while the function refers to a biological and experimental outcome [13]. The chemometric procedures in QSAR refer to MLR, PLS, PCR, PCA, GA, etc. Several tools are available for the prediction of QSAR models that perform specific QSAR steps such as modelling, validation of statistics, and the descriptor generation [14]. The Open3DQSAR or PyCoMFA generates the CoMFA-like models while CORAL, a freeware software, uses a specific set of descriptors (SMILE based) to generate the QSAR PLS model [15, 16]. Another standalone freeware QSAR tool is the QSAIRNS that can help in building the QSAR MLR having the ability of model validation, data partitioning, predicting a new activity of compound, and determination of applicability [17]. Ezqsar and camb are another R-package-based tools that are available openly. They are basically used for beginners that utilize a single function to do the entire job [18].
In view of finding new potential leads with effective chemotherapeutic activities, about twenty two derivatives of 6-(chloromethyl)-4-(4-hydroxyphenyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (3a-3k) and ethyl 6-(chloromethyl)-4-(2-hydroxyphenyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (4a-4k) were synthesized in a previous study [19] (Figure 1). A neat reaction of urea, 4-choloroethylacetoacetate, and substituted benzaldehyde were refluxed for 1 h to obtain 6-chloromethyl-DHPMs. The resulting compounds were further reacted with a series of benzyl amine derivatives in methanol. The crystals were recrystallized using ethanol. The compounds were then characterized using FT-IR, 1H NMR, and 13C NMR. The structures of the synthesized compounds are shown in Figure 2. The spectral analysis of these compounds is mentioned in supplementary Table 1. The compounds were screened for their anticancer activities. The anticancer activities of these synthesized compounds were evaluated against the breast cancer target proteins identified through system biology approach [20]. The system biology approach has helped in identifying several gene targets in better management of diseases [21]. The in silico molecular docking studies of these synthesized compounds were performed to screen for the best targets for these compounds. Furthermore, the in vitro efficacy of these compounds against breast cancer cell line MCF-7 was also performed to understand their antitumor effects. The in silico 2D-QSAR analysis was done to evaluate the structure activity relationship of synthesized compounds by QSARINS [22]. This was done to analyze the predicitivity and stability of models and the role of essential descriptors generated from both models.
Figure 1.
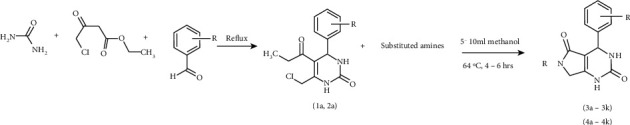
The general scheme used for the synthesis of dihydropyrimidinone derivatives.
Figure 2.
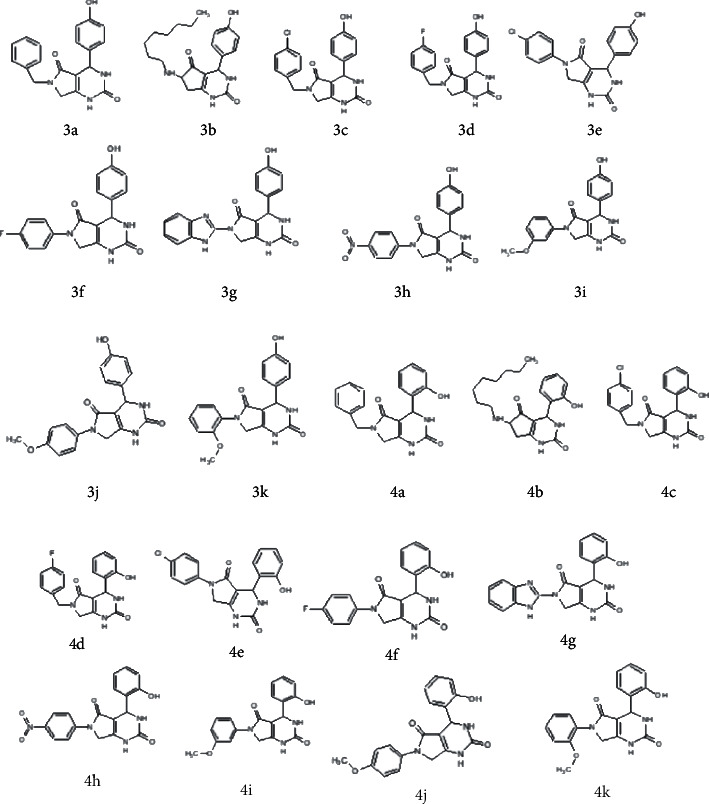
Structures of synthesized dihydropyrimidinones derivatives (3a-3k and 4a-4k).
2. Methodology
2.1. In Silico Chemoinformatics Analysis of Synthesized Ligands
The synthesized chemical structures were drawn in ChemBioDraw Ultra 14.0 and energy minimized using ChemBio3D Ultra 14.0. The Molsoft tool (http://www.molsoft.com/) was used to evaluate the basic chemical properties such as molecular weight (g/mol), hydrogen bond acceptor (HBA), hydrogen bond acceptor (HBA), LogP, molecular volume (A3), polarizibility, and drug likeness. Moreover, the ADMET properties were also evaluated using online pkCSM tool. The tool is used to predict the pharmacokinetics, drug likeness, and medicinal chemistry aspect of small molecules. The compounds having molecular weight < 500 g/mol, hydrogen bond donor < 5, hydrogen bond acceptor < 10, and number of rotatable bonds < 10 are drug likable compounds. The server also helps in identifying the absorption parameters such as the water solubility and intestinal absorption as well as skin permeation. The distribution properties such as blood brain barrier permeation and CNS permeation were also calculated. The total renal clearance and the toxicity profiling including Ames test, hepatotoxicity, and skin sensitivity was also evaluated. The ligand and lipophilic ligand efficiency (LE and LLE) as well as lipophilicity-corrected ligand efficiency (LELP) values were predicted using Data Warrior tool [23].
2.2. Molecular Docking Studies
PyRx docking software, an open source software, was used to identify the best target proteins for the proposed compounds [24]. Several libraries of compounds can be screened for potential target identification using PyRx, starting from job preparation to submission and analysis of results. PyRx is an easy to use and a valuable tool for Computer-Aided Drug Design and has a docking wizard AutoDock Vina. The visual analysis of results in PyRx is based on embedded Python Molecular Viewer (ePMV), and the results are stored in a built-in SQLite database.
2.3. Selection of Breast Target Proteins
The target proteins identified through system biology approach were used in order to study the protein-ligand interaction of these proteins with the synthesized compounds [20]. The differentially expressed breast cancer genes were identified through extensive data mapping, and functional enrichment analysis was performed to screen the differentially expressed genes between breast tumor cells and treated tissues. Moreover, the interactions of these genes with several other proteins involved in breast cancer progression were studied. The shortlisted genes showed essential role in the progression of breast cancer. All the source proteins and the target proteins were shortlisted in order to identify the best target for these compounds. These proteins include ESR, PR, BRCA1, BRCA2, AKR1C2, HER2, CTNNB1, PLAUR, and RHEB.
2.4. Preparation of Proteins
Protein Data Bank was used to retrieve the atomic coordinates of proteins ESR (PDB ID =1L2I), PR (PDB ID = 1A28), BRCA1 (PDB ID = 4IGK), BRCA2 (PDB ID = 3EU7), AKR1C2 (PDB ID = 4JTR), HER2 (PDB ID = 1N8Z), CTNNB1 (PDB ID = 3SL9), PLAUR (PDB ID = 2FD6), and RHEB (PDB ID = 3T5G). The details of all proteins are mentioned in Table 1. All the proteins obtained from Protein Data Bank contained water molecules and the original ligands. For the preparation of protein structures, cocrystallized ligand and any water molecules that were present were removed using MGL Tools-1.5.6, nonpolar hydrogen bonds merged, AD4.2 type and Gasteiger charges were assigned, and proteins were saved in .pdbqt format.
Table 1.
List of target proteins used for docking purpose.
| Protein | PDB ID | Resolution (Å) | Structure title | Specie | Ref |
|---|---|---|---|---|---|
| ESR | 1L2I | 1.95 | Human estrogen receptor alpha ligand-binding domain in complex with (R,R)-5,11-cis-diethyl-5,6,11,12-tetrahydrochrysene-2,8-diol and a glucocorticoid receptor-interacting protein 1 NR box II peptide | Homo sapiens | [25] |
| PR | 1A28 | 1.80 | Hormone-bound human progesterone receptor ligand-binding domain | Homo sapiens | [26] |
| BRCA1 | 4IGK | 1.75 | Structure of human BRCA1 BRCT in complex with ATRIP peptide | Homo sapiens | [27] |
| BRCA2 | 3EU7 | 2.20 | Crystal structure of a PALB2/BRCA2 complex | Homo sapiens | [28] |
| AKR1C2 | 4JTR | 1.30 | AKR1C2 complex with ibuprofen | Homo sapiens | [29] |
| HER2 | 1N8Z | 2.52 | Crystal structure of extracellular domain of human HER2 complexed with Herceptin Fab | Homo sapiens | [30] |
| CTNNB1 | 3SL9 | 2.20 | X-ray structure of beta catenin in complex with Bcl9 | Homo sapiens | [31] |
| PLAUR | 2FD6 | 1.90 | Structure of human urokinase plasminogen activator in complex with urokinase receptor and an anti-upar antibody at 1.9 A | Homo sapiens | [32] |
| RHEB | 3T5G | 1.70 | Structure of fully modified farnesylated Rheb in complex with PDE6D | Homo sapiens | [33] |
2.5. Active Site Prediction
DOGSITESCORER was used to identify the active sites of the proteins from the 3D coordinates of the receptor. DOGSITESCORER is an automated tool for pocket prediction based on 3D structure of protein and calculates the druggability of protein cavities [34]. For the prediction of druggability of pockets, the supervised machine learning technique (SVM) is utilized that predicts the potential pocket and describes them through descriptors. The site provides a druggability score between 0 and 1 showing the higher the score, the more the pocket is druggable. PyMOL was used to visualize the active site of target proteins and the residues involved [35].
2.6. Preparation of Ligand
The structure of ligands was drawn using ChemBioDraw Ultra 14.0, and energy was minimized using MM2 with the help of ChemBio3D Ultra 14.0. The structures were saved in PDB format for AutoDock compatibility. The ligand.pdb files were converted to ligand.pdbqt format using MGL Tools-1.5.6 (The Scripps Research Institute).
2.7. AutoDock Run
The protein ligand binding was analyzed with the help of PyRx tool linked with AutoDock Vina in order to find the correct conformation and configuration of the ligands having the minimum energy structure. The grid centers were positioned on the active binding sites of both proteins, and the docked complexes were examined on the basis of their binding affinities (kcal/mol) and interaction patterns.
2.8. Analysis of Binding Affinity
The boxplot function in R-4.0.2 package was used to perform the scoring analysis of each protein with the synthesized compounds [36]. For interaction analysis, the Discovery Studio Visualizer Software, Version 4.0 (http://www.accelrys.com) was used to study the binding modes of synthesized compounds with the target proteins.
2.9. In Vitro Breast Cancer Activities of Synthesized Compounds
The anticancer activity of the synthesized compounds was determined against human breast (MCF-7) cancer cell line. The MCF-7 (ATCC® HTB-22™) cell lines were gifted by Dr Syed Shahzad ul Hussan from Lahore University of Management Sciences (LUMS). The cells were cryopreserved at -196°C. The cells were grown in RPMI (Roswell Park Memorial Institute Medium) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin purchased from Gibco, USA. The cultures were maintained in 5% CO2 atmosphere and a humidified incubator at 37°C. The different concentrations of synthesized compounds were used to assess the anticancer activity. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) (Sigma) assay was used as described by Mosmann with a slight modification of 72 h of incubation [37]. A spectrophotometer at 520 nm was used to read the assay plates. A dose-response curve was plotted from the data generated to evaluate the concentration of tested compounds required to kill 50% of cell population (IC50). The compounds having % inhibition less than 50% are considered inactive.
2.10. QSAR Studies
The QSARINS software was used to generate models according to OECD standards (Worth et al. 2007).
2.11. Molecular Descriptor Generations
The PaDEL descriptor software was used to generate the quantum molecular descriptors and to calculate the additional energy, where a total of 1875 descriptors were calculated. The use of all the available descriptors would be, however, difficult to calculate the models; hence, few descriptors per model were used to reduce the computation time and to explore all the combinations with the help of all subset technique. The model generation was run for up to 8 variables to see the effect of addition of new descriptor on the quality of model.
2.12. Data Division
The datasets were divided in a 4 : 1 ratio having both training sets and test sets. The training set constituted of 70% while the test set is 30% of the data according to the Kennard-Stone algorithm method.
2.13. Model Building and Validation
The genetic algorithm (GA) technique was employed in which the most appropriate descriptors were selected to develop models based on large number of descriptors. The MLR model was obtained by the ordinary least squares (OLS) algorithm [22]. Twenty models were generated using up to 8 different descriptors, and the best model was shortlisted according to the lowest lack of fit (LOF) value.
2.14. Internal Validation
The validation of model was done by OECD principle which states that the model should have a definite endpoint, a clear applicability domain, an ambiguous algorithm, appropriate measure of robustness and predictivity, and a systematic explanation [38].
2.15. Cross Validation
For cross validation (CV), the Q2LOO criteria were employed by iteratively removing from the dataset one compound while calculating the model with the rest of the compounds. The following parameters were considered to assess the quality of model:
R 2: highest value corresponds to the quality of the model, Q2LOO: highest values should be equal to R2, R2-Q2LOO: lower value indicates the stability of model, RMSE: value is low and close to training dataset, and other prediction methods.
Another method was used for cross validation, i.e., Leaving Many Out (LMO) allowing the study of compounds by excluding a large number of compounds. The stability of model was based on calculated values of R2 and Q2 (LMO), and their averages are close to R2 and Q2LOO values of the model.
2.16. y-Scrambling
The y-scrambling procedure was applied to validate that the generated model was not as a result of chance correlation. The responses were scuffled as to there be no correlation with the descriptors causing the performance of the models to decay drastically. For a good quality model, the R2 and Q2 values and their averages should be less than the values of the model.
2.17. External Validation
The generated model was then assessed for its performance by different measures such as RMSE external, Q2-F1, Q2-F2, Q2-F3, r2M, Δr2m, and CCC.
2.18. Applicability Domain
The domain of applicability was evaluated to confirm the consistency of the model within the chemical space it was developed [39]. The leverage approach was used, and the William's plot was generated between the standardized residuals and leverages.
3. Results
3.1. Pharmacokinetic Analysis
3.1.1. Drug-Likeness Properties
The drug-likeness properties were validated by evaluating the chemical properties of synthesized compounds and analyzing the Lipinski Rule. For drug absorption, the polar surface area (PSA) parameter is a significant tool and the molecular lipophilicity and molar refractivity values relate to protein binding and bioavailability. For compounds to be drug like the molar refractivity should be 40-130 cm, PSA < 89 Å, and molecular weight 160-480 g/mol. Table 2 shows the drug-likeness properties of synthesized compounds justifying a strong correlation with the standard values.
Table 2.
Cheminformatic properties of compounds (3a-4k).
| Properties | Mol. weight (g/mol) | No. HBA | No. HBD | Mol. LogP | Mol. PSA (A2) | Stereo centers | Mol. Vol (A3) | Molar refractivity (cm3) | Surface tension (dyne/cm) | Density (g/cm3) | Polarizability (cm3) | Lipinski Rule |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3a | 335.13 | 3 | 3 | 1.41 | 70.35 | 1 | 234.3 | 93.56 | 77.4 | 1.43 | 37.09 | Yes |
| 3b | 357.21 | 3 | 3 | 3.365 | 70.60 | 1 | 399.41 | 99.87 | 57.4 | 1.24 | 39.59 | Yes |
| 3c | 369.09 | 6 | 3 | 2.00 | 81.67 | 1 | 363.31 | 98.39 | 79.5 | 1.51 | 39.0 | Yes |
| 3d | 353.12 | 3 | 3 | 1.47 | 70.35 | 1 | 352.04 | 93.67 | 74.8 | 1.48 | 37.13 | Yes |
| 3e | 415.05 | 3 | 4 | 3.01 | 78.73 | 1 | 363.19 | 95.26 | 84.2 | 1.74 | 37.76 | Yes |
| 3f | 355.13 | 3 | 4 | 2.24 | 78.73 | 1 | 347.25 | 87.67 | 75.2 | 1.53 | 34.74 | Yes |
| 3g | 363.13 | 3 | 4 | 0.75 | 89.55 | 3 | 397.93 | 96.82 | 101.3 | 1.62 | 38.38 | Yes |
| 3h | 366.10 | 5 | 3 | 1.58 | 103.2 | 1 | 361.92 | 93.57 | 89.6 | 1.60 | 37.06 | Yes |
| 3i | 351.12 | 9 | 3 | 1.49 | 77.24 | 1 | 364.27 | 93.90 | 75.3 | 1.47 | 37.22 | Yes |
| 3j | 351.12 | 4 | 3 | 1.60 | 77.45 | 1 | 366.49 | 93.90 | 75.3 | 1.47 | 37.22 | Yes |
| 3k | 351.12 | 4 | 3 | 1.74 | 77.45 | 1 | 366.56 | 93.90 | 75.3 | 1.47 | 37.22 | Yes |
| 4a | 335.13 | 3 | 3 | 1.86 | 69.28 | 1 | 348.41 | 93.56 | 77.4 | 1.43 | 37.09 | Yes |
| 4b | 357.21 | 3 | 3 | 3.97 | 69.53 | 1 | 401.7 | 99.87 | 57.4 | 1.24 | 39.59 | Yes |
| 4c | 369.09 | 3 | 3 | 2.45 | 69.28 | 1 | 365.60 | 98.39 | 79.5 | 1.51 | 39.0 | Yes |
| 4d | 353.12 | 3 | 3 | 1.92 | 69.28 | 1 | 354.32 | 93.67 | 74.8 | 1.48 | 37.13 | Yes |
| 4e | 399.02 | 3 | 3 | 2.93 | 68.84 | 1 | 358.78 | 95.26 | 84.2 | 1.74 | 37.76 | Yes |
| 4f | 339.10 | 3 | 3 | 2.16 | 68.84 | 1 | 342.84 | 87.67 | 75.2 | 1.53 | 34.74 | Yes |
| 4g | 363.13 | 5 | 4 | 1.20 | 88.48 | 3 | 400.23 | 96.82 | 101.3 | 1.62 | 38.38 | Yes |
| 4h | 366.10 | 5 | 3 | 2.02 | 102.2 | 1 | 361.92 | 93.57 | 89.6 | 1.60 | 37.06 | Yes |
| 4i | 351.12 | 4 | 3 | 1.93 | 76.17 | 1 | 366.55 | 93.90 | 75.3 | 1.47 | 37.22 | Yes |
| 4j | 351.12 | 4 | 3 | 2.18 | 76.38 | 1 | 368.85 | 93.90 | 75.3 | 1.47 | 37.22 | Yes |
| 4k | 351.12 | 4 | 3 | 2.05 | 76.38 | 1 | 368.77 | 93.90 | 75.3 | 1.47 | 37.22 | Yes |
3.2. ADMET Studies of Synthesized Compounds
The pharmacokinetic ADMET properties were evaluated to assess the effectiveness of the synthesized compounds. The compounds having good pharmacokinetic properties and better activities are considered in the drug discovery and development. To evaluate the pharmacokinetic properties, the pkCSM tool was used. The water and intestinal solubility (log mol/L, % absorbed) and the skin permeability (LogKp) predicted values revealed efficient absorption of these compounds as well as efficient skin permeability as compared to standard value (>30% abs and -2.5 LogKp). Effective absorption of compound leads to effective potency due to passive penetration to reach the target molecule. All the compounds showed poor permeability to the blood brain barrier when compared to standard value (>0.3 to <-1), and the compounds having <-1 are considered poorly distributed in the brain. However, all the compounds showed good penetration to the CNS having LogPS > −2 when compared to standard value (>-2 to <-3 LogPS). The compounds having LogPS < −3 are impossible to cross in the CNS. The toxicity profiling revealed all the compounds are nonmutagenic and nontoxic except for compounds 3a, 3g, 3h, and 4h. All the compounds also showed hepatotoxicity but no skin sensitization was revealed (Table 3).
Table 3.
ADMET properties of synthesized compounds.
| Absorption | Distribution | Excretion | Toxicity | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| WS | IS | SP | BBBP | CNSP | CYP3A4 | TC | AMES toxicity | Max tolerated dose | HT | SS | |
| 3a | -3.57 | 94.724 | -2.738 | -0.854 | -2.369 | No | 0.181 | Yes | -0.238 | Yes | No |
| 3b | -3.395 | 93.057 | -2.739 | -1.09 | -2.689 | No | 1.147 | No | -0.781 | Yes | No |
| 3c | -3.639 | 93.342 | -2.739 | -1.038 | -2.258 | No | 0.055 | No | -0.293 | Yes | No |
| 3d | -3.571 | 94.41 | -2.738 | -1.076 | -2.418 | No | 0.067 | No | -0.35 | Yes | No |
| 3e | -3.738 | 93.138 | -2.769 | -0.904 | -2.176 | No | -0.102 | No | -0.492 | Yes | No |
| 3f | -3.533 | 94.3 | -2.797 | -0.922 | -2.363 | No | -0.136 | No | -0.542 | Yes | No |
| 3g | -2.987 | 82.281 | -2.735 | -1.014 | -2.54 | No | 0.998 | Yes | 0.221 | Yes | No |
| 3h | -3.935 | 82.358 | -2.802 | -0.82 | -2.455 | No | -0.112 | Yes | -0.781 | Yes | No |
| 3i | -3.77 | 93.634 | -2.996 | -.8360 | -2.468 | No | 0.061 | No | -0.428 | Yes | No |
| 3j | -3.757 | 93.437 | -3.007 | -0.833 | -2.457 | No | -0.009 | No | -0.44 | Yes | No |
| 3k | -3.748 | 93.182 | -3.01 | -0.823 | -2.466 | No | -0.042 | No | -0.463 | Yes | No |
| 4a | -3.79 | 91.409 | -3.044 | -0.688 | -2.391 | No | 0.211 | No | -0.092 | Yes | No |
| 4b | -4.184 | 93.036 | -2.912 | -0.971 | -2.711 | No | 1.071 | No | -0.781 | Yes | No |
| 4c | -4.213 | 90 | -3.02 | -0.875 | -2.282 | No | 0.172 | No | -0.079 | Yes | No |
| 4d | -3.963 | 91.069 | -3.101 | -0.914 | -2.445 | No | 0.097 | No | -0.014 | Yes | No |
| 4e | -4.271 | 89.694 | -3.344 | -0.824 | -2.149 | No | 0.02 | No | -0.104 | Yes | No |
| 4f | -3.843 | 90.829 | -3.386 | -0.842 | -2.335 | No | -0.072 | No | -0.03 | Yes | No |
| 4g | -2.785 | 73.392 | -2.735 | -0.675 | -2.774 | No | 0.656 | No | 0.409 | Yes | No |
| 4h | -4.066 | 82.551 | -2.807 | -0.828 | -2.454 | No | -0.044 | Yes | -0.579 | Yes | No |
| 4i | -3.76 | 94.307 | -3.176 | -0.961 | -2.512 | No | 0.105 | No | -0.472 | Yes | No |
| 4j | -3.754 | 94.11 | -3.213 | -0.958 | -2.501 | No | 0.035 | No | -0.488 | Yes | No |
| 4k | -3.723 | 93.819 | -3.243 | -0.946 | -2.513 | No | 0.002 | No | -0.53 | Yes | No |
Abbreviations: WS: water solubility (Log mol/L); ISA: intestinal solubility (%abs); SP: skin permeability (LogKp); BBBP: blood brain barrier permeability (LogBB); CNSP: CNS permeability (LogPS); TC: total clearance (log ml/min/kg); ORAT: Oral Rat Acute Toxicity; HT: hepatotoxicity; SS: skin sensitization.
3.3. Lead Optimization
Further drug-likeness properties of all compounds such as ligand efficiency (LE), lipophilic ligand efficiency (LLE), and lipophilic-corrected ligand efficiency (LELP) values were predicted. The lipophilicity is considered to be a basic parameter to enhance structure efficiency making it from lead to drug candidate. ThecLogP, LE, LLE, and LELP of all compounds showed comparable results with that of standard values forLE > 0.30 kcal/mol/HA,LLE > 0.5 kcal/mol,LELP − 10<to <10, andcLogP < 3. All the synthetic compounds showed to have none mutagenic and irritant behavior (Table 4).
Table 4.
Ligand efficacy prediction value.
| Ligands | cLogP | cLogS | LE | LLE | LELP | Mutagenic | Tumorigenic | Irritant |
|---|---|---|---|---|---|---|---|---|
| 3a | 1.65 | -2.722 | 0.4819 | 7.1314 | 3.4256 | None | None | None |
| 3b | 3.3655 | -3.319 | 0.4707 | 5.1075 | 7.5279 | None | None | None |
| 3c | 2.2568 | -3.458 | 0.4562 | 6.389 | 4.9469 | None | None | None |
| 3d | 1.7517 | -3.036 | 0.46204 | 7.0048 | 3.7913 | None | None | None |
| 3e | 2.456 | -4.078 | 0.4724 | 6.1571 | 5.1921 | None | None | None |
| 3f | 1.828 | -3.558 | 0.47949 | 6.909 | 3.814 | None | None | None |
| 3g | 1.6971 | -3.726 | 0.44562 | 7.0732 | 3.8084 | None | None | None |
| 3h | 0.8064 | -3.704 | 0.46204 | 8.287 | 1.7453 | None | None | None |
| 3i | 1.658 | -3.262 | 0.4633 | 7.1224 | 3.5787 | None | None | None |
| 3j | 1.658 | -3.262 | 0.4633 | 7.1224 | 3.5787 | None | None | None |
| 3k | 1.658 | -3.262 | 0.4633 | 7.1224 | 3.5787 | None | None | None |
| 4a | 1.6509 | -2.722 | 0.4819 | 7.1314 | 3.4256 | None | None | None |
| 4b | 3.3655 | -3.319 | 0.44707 | 5.1075 | 7.5279 | None | None | None |
| 4c | 2.2569 | -3.458 | 0.4562 | 6.3896 | 4.9469 | None | None | None |
| 4d | 1.7517 | -3.036 | 0.46204 | 7.0048 | 3.7913 | None | None | None |
| 4e | 2.4532 | -4.078 | 0.47249 | 6.1571 | 5.1921 | None | None | None |
| 4f | 1.8288 | -3.558 | 0.47949 | 6.909 | 5.1921 | None | None | None |
| 4g | 1.6971 | -3.726 | 0.47949 | 6.909 | 3.814 | None | None | None |
| 4h | 0.8064 | -3.704 | 0.44562 | 7.0732 | 3.8084 | None | None | None |
| 4i | 1.658 | -3.262 | 0.46204 | 8.287 | 1.7453 | None | None | None |
| 4j | 1.658 | -3.262 | 0.4633 | 70122 | 3.5787 | None | None | None |
| 4k | 1.658 | -3.262 | 0.4633 | 70122 | 3.5787 | None | None | None |
3.4. Molecular Docking
The molecular docking studies of synthesized compounds against nine target proteins were performed to analyze the best target for these compounds based on docking scores. The boxplot was generated to present the docking scores of all target proteins. Figure 3 shows the boxplot of all synthesized compounds on the basis of their interactions with all target proteins.
Figure 3.
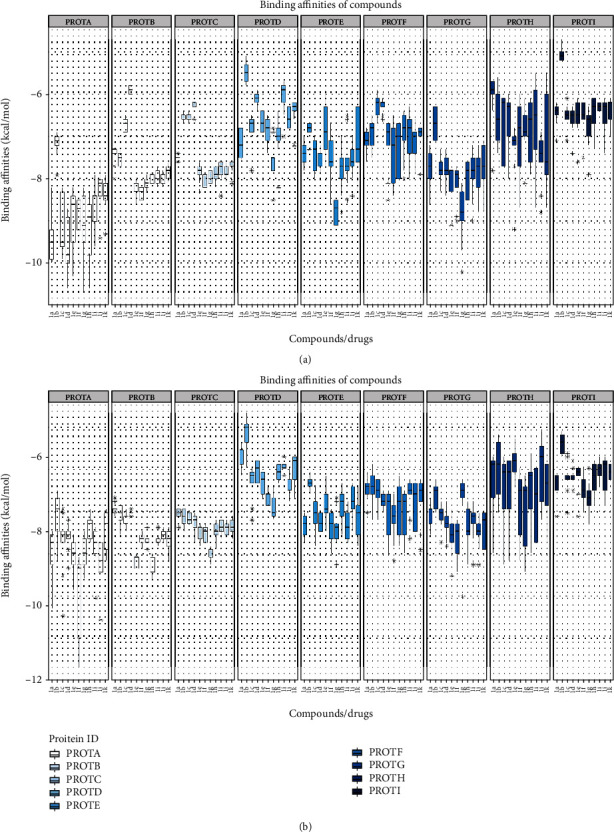
Boxplot for docking scores generated by RStudio 4.0. (a) 6-(Chloromethyl)-4-(4-hydroxyphenyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate derivatives (3a-3k). (b) Ethyl 6-(chloromethyl)-4-(2-hydroxyphenyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate derivatives (4a-4k). The y-axis represents the docking scores while the x-axis shows the synthesized compounds. ProtA: CTNNB1; ProtB: BRCA1; ProtC: BRCA2; ProtD: AKR1C2; ProtE: ESR; ProtF: HER2; ProtG: PLAUR; ProtH: PR; ProtI: RHEB.
In case of protein A (CTNNB1) according to the median value, the compound 4f is having the lowest median score of -11.7 with 80% of data in lower quartile and 20% in upper quartile. The compounds 4h and 4e showed the lowest median score of -10.4 and -10.3, respectively, with equal distribution of data. The compound 4k showed the median score of -10.3 with 75% of data in lower quartile and 25% in upper quartile. The compounds 3e and 3f showed the median score of -10.1 and -9.8 with 60% of data in lower quartile and 40% in upper quartile. The compounds 4g, 4i, 3g, 4j, and 4h showed median score in the range of -9.8 to -9 kcal/mol. In protein B (BRCA1), the compound 4h showed the median score of -9.3 with 90% of data in upper quartile, 4e showed -8.5 median score with equal distribution, and 4f showed median score of -9.2 with 90% of data in upper quartile; 4k and 3e showed -8.8 with equal distribution and -8.6 with 90% in upper quartile, 3f had median score of -8.5 with 80% in lower quartile and 20% in upper quartile, 4g showed -9.8, and 4i had a score of -8.4 with 90% in upper quartile. The protein C (BRCA2) also showed a similar pattern but with the median score in the range of -8.9 to -6.6. The compound 4f showed the lowest median score of -8.3 with 80% of data in lower quartile, 4e showed -8.7, and 4h showed -8.9. Similarly, the compounds 3f and 3g had the lowest median score of -8.4 and 3e -8.3 with equal distribution.
The protein D (AKR1C2) showed the median score in the range of -8.5 to -5.8 with low range and varying distribution. The binding affinities for AKR1C2 were less when compared to proteins A, B, and C. The protein E (IGFR1) had the median score ranging between -9.2 and 7 with binding affinities better than protein D and high range. In protein G (RHEB), all the compounds had median score in the range of -7.8 to -6 kcal/mol with varying distribution and high range. Similarly, the protein F (HER2) showed the lowest median score in the range of -8.8 to -6.6 with varying distribution. The protein H (PLAUR) showed the median score of -9.2 to -6.8 kcal/mol. A highest median range was observed in protein H with varying distribution. Moreover, the protein I (PR) showed the median score of -7.8 to -5.1 kcal/mol with low median range and varying distribution of data. In all the proteins, the compounds 4f and 4h showed the lowest binding scores. The docking of ligands into the active binding site of CTNNB1 showed the lowest binding scores.
3.5. Interaction Analysis with Target Proteins
The protein ligand interaction analysis was performed to study the interaction patterns of ligands with different proteins in order to find the common binding sites in proteins subjecting to new functional roles. Figure 4 shows the binding mode of active compounds 4e, 4f, 4g, and 4h and standard against target proteins CTNNB1 (Figures 4(a)–4(e)), BRCA1 (Figures 4(f)–4(j)), BRCA2 (Figures 4(k)–4(o)), and AKR1C2 (Figures 4(p)–4(t)). Similarly, Figure 5 shows the binding mode of active compounds against target proteins ESR (Figures 5(a)–5(e)), HER-2 (Figures 5(f)–5(j)), RHEB (Figures 5(k)–5(o)), PLAUR (Figures 5(p)–5(t)), and PR (Figures 5(u)–5(y)).
Figure 4.
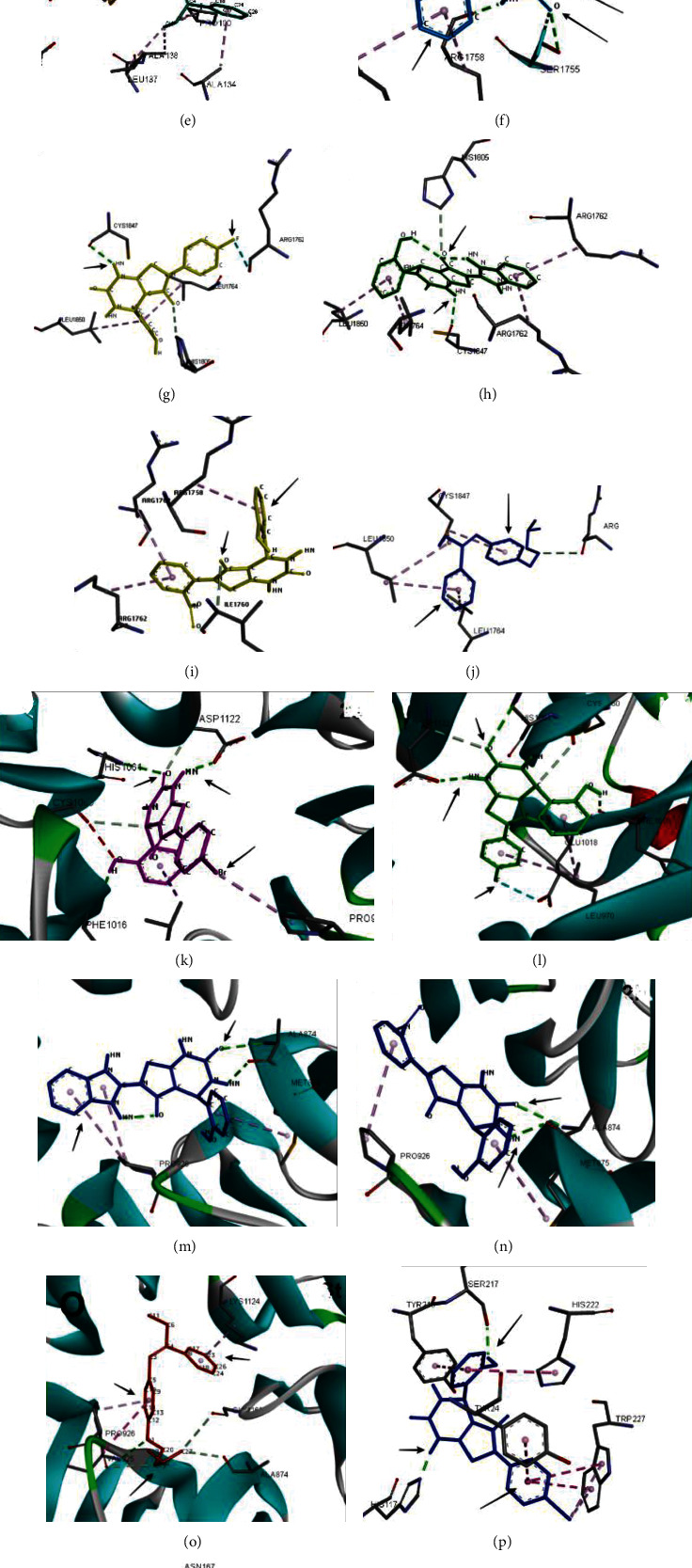
Protein-ligand interactions of target proteins with active compounds 4e, 4f, 4g, and 4h. (a–e) CTNNB1, (f–j) BRCA1, (k–o) BRCA2, and (p–t) AKR1C2.
Figure 5.
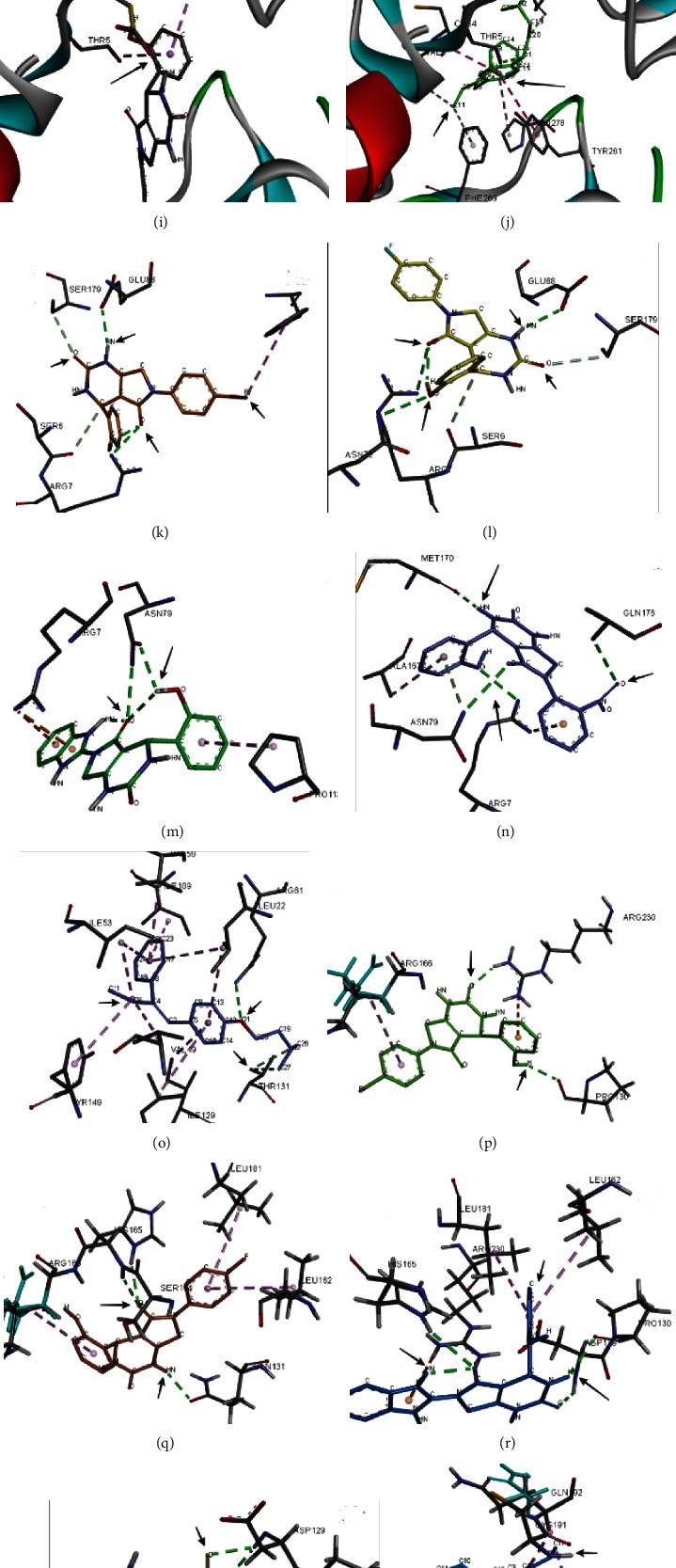
Protein-ligand interactions of target proteins with active compounds 4e, 4f, 4g, and 4h. (a–e) ESR, (f–j) HER-2, (k–o) RHEB, (p–t) PLAUR, and (u–y) PR.
Figure 4 shows the residue interactions of active compounds 4e, 4f, 4g, and 4h with the protein CTNNB1 (ProtA). These compounds showed the lowest binding scores of -10.3, -11.7, -9.8, and -10.4 kcal/mol, respectively. The interaction analysis revealed stable hydrogen bond interactions of compound 4i with ASP199, while compound 4j showed two stable hydrogen bond interactions with LEU177 and GLU176. The standard tamoxifen showed pi-alkyl with PRO100, ALA138, LEU137, LYS199, and ALA134 and amide-pi stacked interactions with VAL197 (Figures 4(a)–4(e)).
In protein BRCA1 (ProtB), stable conventional hydrogen bonding was observed in compounds 4f and 4g with CYS1847 and 4e with amino acid TYR1845, SER 1755, ARG 1758, and ILE 1760. In compound 4h, no hydrogen bonds were observed; however, pi-alkyl interactions were seen with ARG1762. The standard tamoxifen showed no hydrogen bonds, and pi-alkyl interactions were observed with LEU1764, LEU1850, and CYS1847 (Figures 4(f)–4(j)). In BRCA2 (ProtC), the compounds 4e and 4f revealed hydrogen bond interaction with ASP1122, HIS1061, and PHE1016 and 4g and 4h with ALA874. The standard tamoxifen showed hydrogen bonding with VAL925 (Figures 4(k)–4(o)). The interaction analysis of protein D (AKR1C2) showed that the compound 4e showed hydrogen bonding with SER217 and HIS117. 4f showed hydrogen bonding with TYR24 and ASN167. About four hydrogen bonding were observed in compound 4h with amino acid TYR272, ARG 276, LEU219, and SER221. The compound 4i showed hydrogen bonding with GLN190 and 2k with GLN224 (Figures 4(p)–4(t)).
In protein E (ESR), compound 4e showed 3 hydrogen bonding with SER329, TYR328, and ARG352. Similarly, compound 4f showed hydrogen bonding with GLY521 while 4g showed four hydrogen bonding with THR347, TYR537, GLY344, and GLU330, and compound 4h showed stable interactions with ASP538 and LEU539 (Figures 5(a)–5(e)). The interaction patterns of protein F (HER2) showed stable interactions of compounds 4e, 4f, 4g, and 4h with ASP8, GLY270, and THR7 along with some van der Waal interactions (Figures 5(f)–5(j)). In protein G (RHEB), the compounds 4e, 4f, and 4g showed three hydrogen bond interactions with ARG7, SER179, ASN79, and GLU88. The compound 4h showed ARG7, ASN79, and MET 170 (Figures 5(k)–5(o)). In protein H (PLAUR), the compound 4e showed hydrogen bonding with ASP697 and LYS769 and in 4f with LYS769, SER728, and GLU695. 4h showed hydrogen bonding with ILE699 and ARG766 (Figures 5(p)–5(t)). The interaction analysis of protein I (PR) is shown in Figure 5. The compound 4e showed conventional hydrogen bonding with ASP697, LYS769, and LEU755. In compound 4f, the interaction between fluorine and nitrogen group of dihydropyrimidinone was observed with amino acid SER726, GLU695, and LYS769. 4g showed hydrogen bonding with ILE699 and ARG766 (Figures 5(u)–5(y)). Table 5 highlights the important common residues involved in interactions with the active compounds.
Table 5.
Some common amino acid residues involved in interaction with target proteins.
| Protein | Common residues involved |
|---|---|
| CTNNB1 | Arg360, ASP361, PRO368, PHE367, MET365 |
| BRCA1 | THR1802, CYS1847, HIS1805, ARG1762, ARG1758, ILE1760, LEU1764, ARG1762 |
| BRCA2 | PRO924, ASP1122, HIS1061, CYS1060, LEU970, CYS1060, VAL969, ALA1017, GLU1018, ALA874, CYS1060, PHE1016 |
| AKR1C2 | LYS270, HIS222, SER217, TRP227, VAL54 |
| ESR | ALA340, VAL534, PRO535, TYR537, SER329, LEU327, GLU330, ALA350, PHE404, LEU391 |
| HER-2 | PRO278, ASP8, THR7, THR281, ARG81, LEU414, THR5, SER288 |
| RHEB | ARG7, GLU88, SER179, PRO2, ASN79, MET179 |
| PLAUR | ARG166, LEU181, PRO130, CYS182, ARG230, LEU162, HIS165, SER214 |
| PR | ILE699, ARG766, VAL698, PRO696, MET759, PHE718 |
3.6. Anticancer Activity
In this study, the in vitro anticancer activity of 22 derivatives of synthesized compounds was determined against the human breast (MCF-7) cancer cell lines with the help of MTT assay (Table 6). The results revealed that the compounds having p-hydroxyl group of benzaldehyde (2) showed excellent anticancer activities when compared to standard against the breast cancer cell line. The compounds that showed more than 50% of inhibition were considered active. The compound 4f showed 85% inhibition of cells with an IC50 of 2.19 at 200 μM concentration. The standard tamoxifen showed IC50 of 1.88 μM. The compounds 4e and 4 g showed 82% inhibition with an IC50 of 2.401 and 2.47, respectively. The compound 4h also showed 80% inhibition of cells with IC50 of 2.33. The % inhibition of compounds 3e and 3f was 79.4 and 77.2% with IC50 of 2.41 μM. The compounds 4k, 4i, and 4j showed up to 75% inhibition with IC50 of 2.40, 2.699, and 2.88, respectively. The compounds 3h, 3i, 3j, and 3k showed approximately 55% inhibition at the same concentration, while the compounds 3a, 3b, 3c, 3d, 4a, 4b, 4c, and 4d showed less than 50% of inhibition (Figure 6).
Table 6.
The percentage inhibition of breast cancer cells using MTT assay by the tested compounds at 72 h.
| Compounds | % inhibition (μM) | IC50 | |||
|---|---|---|---|---|---|
| 25 | 50 | 100 | 200 | ||
| 3a | 0.98 | 15 | 23.2 | 48 | 4.38 |
| 3b | 5.2 | 12.6 | 19.5 | 39 | 5.35 |
| 3c | 6.5 | 13.6 | 21.2 | 32.2 | 6.23 |
| 3d | 2.1 | 9.8 | 18.5 | 29.3 | 6.38 |
| 3e | 25.6 | 36.8 | 54.8 | 79.4 | 2.41 |
| 3f | 28.9 | 39.6 | 59.6 | 77.2 | 2.41 |
| 3g | 22.5 | 33.6 | 49.9 | 71.1 | 2.88 |
| 3h | 19.9 | 28.5 | 46.6 | 69.3 | 3.03 |
| 3i | 11.5 | 25.3 | 33.6 | 58.3 | 3.69 |
| 3j | 8.6 | 19.9 | 29.6 | 54.4 | 3.98 |
| 3k | 9.7 | 15.6 | 27.5 | 55.9 | 4.01 |
| 4a | 0.85 | 16.5 | 19.8 | 32.2 | 5.85 |
| 4b | 11.2 | 21.3 | 31.5 | 51.1 | 4.13 |
| 4c | 5.5 | 16.6 | 28.9 | 49.2 | 4.23 |
| 4d | 6.7 | 14.9 | 23.2 | 35 | 5.72 |
| 4e | 21 | 38.7 | 66.5 | 82.2 | 2.401 |
| 4f | 28.9 | 41 | 69.5 | 85 | 2.19 |
| 4g | 22.6 | 37.4 | 59.8 | 82.3 | 2.47 |
| 4h | 24 | 47.1 | 61.2 | 80.01 | 2.33 |
| 4i | 19.8 | 32.5 | 56.8 | 75.6 | 2.699 |
| 4j | 15.5 | 28.9 | 53.6 | 72.3 | 2.88 |
| 4k | 21.3 | 41.1 | 66.5 | 78.9 | 2.401 |
| Tamoxifen | 29.1 | 55.2 | 75.4 | 91.2 | 1.88 |
Figure 6.
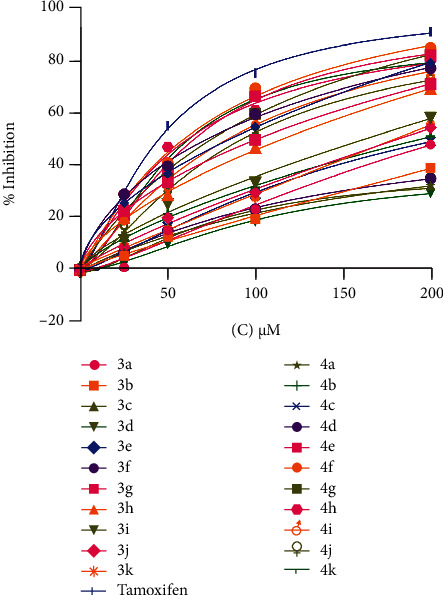
The antiproliferative effects of synthesized compounds in breast cancer cell lines (MCF-7) after 72 h treatment. The cell viability was measured by MTT assay.
3.7. QSAR Studies
The dataset consisting of 22 compounds was divided into training set of 15 compounds and test set of 6 compounds, where training set was used to develop the model while test set to evaluate the predictive ability of the model. Using the PaDEL software, 1872 descriptors were calculated which were then filtered using the QSARINS software. The descriptors having 80% constant values and 90% correlation were eliminated. About 1058 variables were excluded from the study based on all subset method. Several models were developed having good correlation with the response and a low multicollinearity between descriptors. The genetic algorithm–multiple linear regression (GA-MLR) method provided 4 descriptors which were then used for calculating the anticancer activities of the compounds. The average values of R2 and Q2LOO (with their standard deviation) were plotted to evaluate the model performances versus the size of the developed models. It also revealed whether any overfitting in the models exists (Figure 7). The plot showed that by adding a new descriptor, the values of R2 and Q2LOO increased. The model with four variables was selected based on the lowest LOF value to predict the anticancer activities.
Figure 7.
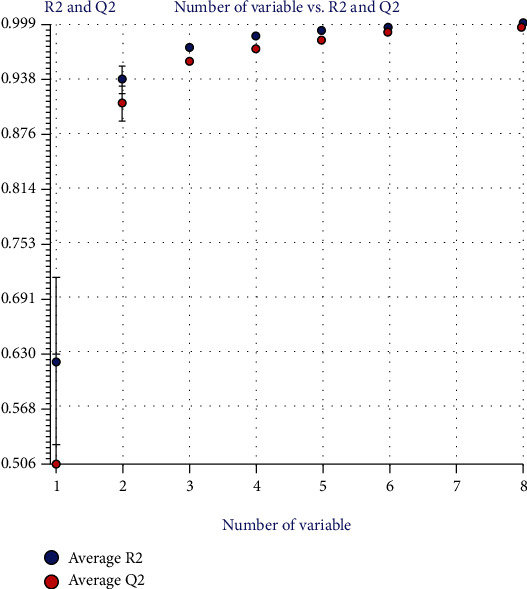
Performance of models according to different variable obtained from QSARINS.
The best MLR model equation obtained is shown below.
| (1) |
Table 7 shows the experimental IC50 and the results predicted by MLR model for training set. Table 8 shows the Pearson correlation matrix which describes that a low value in coefficient (<0.7) between each pair of descriptor shows no significant multicollinearity among descriptors in the developed model. The internal validation of the model that is the scatter plot, scatter plot by LOO, scatter plot by LMO, and y-scrambling predicted the reliability of the model as shown in Figure 8. The applicability domain also defined the reliability of the model (Figure 9).
Table 7.
Chemical structure and corresponding observed and predicted activities obtained from QSARINS.
| Compounds | R | R′ | Experimental endpoint | Predicted by model equation | Predicted model equation residual |
|---|---|---|---|---|---|
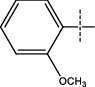
| 3a | 4-OH |
|
4.38 | 5.22 | 2.39 |
| 3b | 4-OH |

|
5.35 | 5.22 | -0.12 |
| 3c | 4-OH |
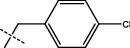
|
6.23 | 7.16 | 0.93 |
| 3d | 4-OH |
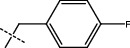
|
6.38 | 6.57 | 0.192 |
| 3e | 4-OH |
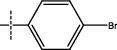
|
2.41 | 2.39 | -0.015 |
| 3f | 4-OH |
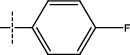
|
2.41 | 2.69 | -0.035 |
| 3g | 4-OH |
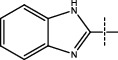
|
2.88 | 1.56 | -1.31 |
| 3h | 4-OH |
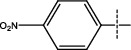
|
3.03 | 2.94 | -0.08 |
| 3i | 4-OH |
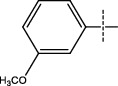
|
3.69 | 3.54 | -0.14 |
| 3j | 4-OH |
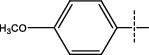
|
3.98 | 3.36 | -0.6 |
| 3k | 4-OH |
|
4.01 | 2.82 | -1.18 |
| 4a | 2-OH |
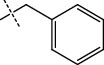
|
5.85 | 5.92 | 0.07 |
| 4b | 2-OH |

|
4.13 | 4.25 | 0.12 |
| 4c | 2-OH |
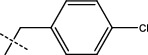
|
4.23 | 6.04 | 1.81 |
| 4d | 2-OH |
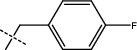
|
5.72 | 5.47 | -0.24 |
| 4e | 2-OH |
|
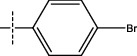
2.40 | 2.55 | 0.155 |
| 4f | 2-OH |
|
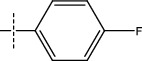
2.19 | 2.188 | -0.0015 |
| 4g | 2-OH |

|
2.47 | 2.46 | -0.003 |
| 4h | 2-OH |
|
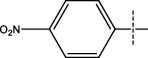
2.33 | 2.59 | 0.26 |
| 4i | 2-OH |
|
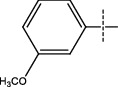
2.69 | 2.68 | -0.013 |
| 4j | 2-OH |
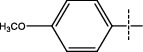
|
2.88 | 2.62 | -0.25 |
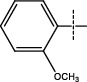
| 4k | 2-OH |
|
2.40 | 2.47 | 0.074 |
Table 8.
Pearson correlation matrix.
| MATS3i | VR2_Dzi | ASP-5 | GGI10 | |
|---|---|---|---|---|
| MATS3i | 1.0000 | |||
| VR2_Dzi | 0.5057 | 1.0000 | ||
| ASP-5 | -0.0805 | 0.2648 | 1.0000 | |
| GGI10 | 0.1194 | -0.0831 | 0.0844 | 1.0000 |
Figure 8.

The internal validation of models through different methods. (a) The scatter plot of experimental IC50 versus predicted by model equation. (b) The scatter plot obtained by LOO method. (c) Plot comparing the original model with the LMO validations. (d) Plot comparing the original model with the y-scrambling model.
Figure 9.
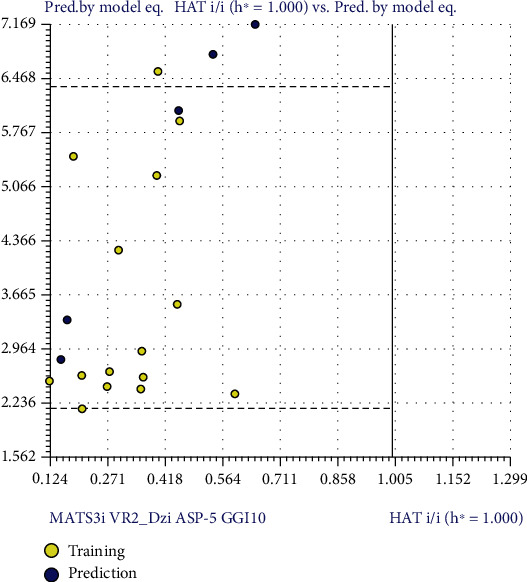
William's plot of the dataset of IC50 standardized against its descriptor space.
4. Discussion
Breast cancer pathogenesis and progression has been studied extensively with the discovery of several agents that have proved potential in the management of this disease. However, till date, the incidence rate of breast cancer is still significant and requires further strategies to combat the mortality and morbidity rate. This study uses the computational technology to identify the breast cancer targets for the synthesized compounds that can have potential role as breast cancer activities.
The in silico ADMET and lead optimization studies revealed all the compounds to be nonmutagenic and noncarcinogenic having drug-like properties. The results depicted compounds may act as therapeutically active against target proteins. All the synthesized compounds also followed the Lipinski Rule of 5 having HBA < 10 and HBD < 5, LogP < 500 g/mol. The increase number of HBA and HBD results in poor permeation. The molecular docking analysis was performed to analyze the binding of synthesized compounds with the identified target proteins. In the protein-ligand docking analysis, when comparing the binding energies and the interaction pattern, all the compounds showed the lowest binding affinity towards the target protein A (CTNNB1). The interaction analysis revealed stable hydrogen bond interactions of compound 4i with ASP199, while compound 4j showed two stable hydrogen bond interactions with LEU177 and GLU176. The standard tamoxifen showed pi-alkyl with PRO100, ALA138, LEU137, LYS199, and ALA134 and amide-pi stacked interactions with VAL197. The energy scores revealed efficient binding of these compounds with the target proteins. All the other proteins also showed efficient binding and interaction pattern, and the common amino acid residues involved in interaction are mentioned in Table 5.
The breast cancer activities of all the synthesized compounds were performed against the cell line MCF-7. The MCF-7 cell line is considered estrogen receptor- (ER-) positive and progesterone receptor- (PR-) positive expressing high level of Erα transcripts [40, 41]. The epidermal growth factor receptor (EGFR) and the human epidermal growth factor receptor-2 (HER2) are also present in MCF-7 cells [40]. The MCF-7 cells are also positive for β-catenin [42]. Due to the expression of these proteins by MCF-7 cell line, it was used to analyze the role of synthesized compounds as cytotoxic agents. It was observed that the activities of compounds 4f, 4h, and 4e were greater than all the compounds and were due to the –F, NO2, and –Br aniline groups with fluorine having the most potent activity due to its high electronegative nature. By replacing the groups with benzylamine (3a and 4a), –Br benzylamine (3c and 4c), and –F benzylamine (3d and 4d), the activity dropped significantly suggesting the more cytotoxic activities of aniline derivatives when compared to benzylamine derivatives. The compounds 3e, 3f, and 3g also showed better activities due to the aniline nature of compounds with –NO2 group of 3h showing the least activity. The benzimidazole moiety of compounds 3g and 4 g also showed effective nature of this molecule. The compounds 4k, 4i, and 4j also showed good activities having the anisidine moieties. The ortho anisidine showed more % inhibition than para and meta. This study was carried due to the existence of several evidences on the antiproliferative activities of dihydropyrimidinones by scientists. In a similar study, about 22 manostrol analogs were synthesized by Matias and coworkers and studied for their antiproliferative activities against five different cancer cell lines. Their result also showed stronger antiproliferative activities of their compounds against MCF-7 cancer cell line with compounds having chlorine moiety displaying significant effects on the proliferation of hepatic (HepaRG), colon (Caco-2), and breast (MCF-7) cancer cell lines [43]. Another series of 32 novel Biginelli dihydropyrimidinones were synthesized by Kumar and colleagues and were studied for their in vitro antioxidant and anticancer activities. The compounds exhibited significant anticancer activities against breast cancer cell line MCF-7 at 10 μg concentration [44]. The cytotoxic activities of another synthesized library of dihydropyrimidinone benzopyran hybrids were evaluated for their cytotoxic activities against four human cancer cell lines A549 (lung carcinoma), MCF-7 (mammary gland adenocarcinoma), HCT-116 (colorectal carcinoma), and PANC-1 (pancreatic duct carcinoma) and showed consistent cytotoxic activities against these cell lines [45]. The antiproliferative activities of dihydropyrimidinones were also studied in another study depicting potent cytotoxic activities of dihydropyrimidinone analogs against melanoma (UACC.62), kidney (786-0), breast (MCF-7), ovarian (OVCAR03), and, particularly, colon (HT-29) cancer cell lines [46]. All the evidences support the significant role of dihydropyrimidone in breast cancer cell line. Moreover, the significant activities of these compounds against breast cancer cell line and optimum binding energies of these compounds against identified target proteins support the effectiveness of these compounds as anticancer agents.
The QSAR studies were performed by two different software to analyze the model quality and their reliability by both methods.
The model was generated by the QSARINS having the following fitting criteria:
N (number of compounds in the training set) = 15, R2 (coefficient of determination) = 0.989, R2adj (adjusted R2) = 0.985, s (standard error of estimate) = 0.182, F (variance ratio) = 234.487, and RMSEtr (Root Mean Square Error in fitting of training set) = 0.148.
According to the fitting criteria, the R2 value is 0.989 that is closer to 1 that shows a good quality model for anticancer inhibition. Moreover, the lower value of LOF and the R2adj of 0.985 depicting the convenience to add a new descriptor to the model suggest no overfitting in the model. The model showed to be a good model having least amount of descriptors. The higher value of F (234.487) and the low value of kxx (0.324) show minimum correlation between the descriptors. Similarly, the Delta k (0.084) and the small error on training sets (RMSEtr = 0.148) showed appropriate correlation between the descriptors. The scatter plot obtained by the model equation versus the experimental IC50 for training set determines the availability of potential outliers (Figure 8(a)). The scatter plot detects the grouping of the data and the possibility of any outlier present.
4.1. Internal and External Validation of the Model
The internal validation of the model was done to check the fitting and stability of the models. The cross validation by Leave-One-Out (LOO) method showed good internal prediction as the Q2LOO = 0.977 (variance explained by LOO) has a comparable value with R2 = 0.989. Moreover, the small error in prediction of RMSEcv = 0.217 shows a robust and stable model. A plot was generated between the predicted values by LOO versus the experimental values of IC50 (Figure 8(b)). Another method was employed for internal validation that is Leaving-Many-Out (LMO) that leaves out 30% of the dataset to study the model behavior. The values of Q2LMO = 0.9721 and the calculation in each iteration of LMO and their averages are comparable to the values of R2 and Q2LOO of the model revealing the stability of the model. Figure 8(c) displays the plot between the Q2LMO and the correlation between descriptors and IC50 (kxy) showing that the model is a good fit having robustness and stability. The y-scrambling method was employed to determine whether the model is the result of chance correlation. For a good model with low chance of correlation, the values of R2 and Q2 and their averages R2y−scr and Q2y−scr should be lower than the values obtained previously. Here, R2y−scr = 0.28 and Q2y−scr = −0.66 that are far from the values obtained for R2 and Q2 indicating the model has not been obtained by random correlation. Figure 8(d) shows the plot between the R2y−scr and Q2y−scr values against the R2 and Q2 of the model.
The external validation of the model was also performed to test the predictive ability of the model. The model showed R2ext (external determination coefficient [47]): 0.97, R2ext: 0.6479, Q2-F1: 0.7320, Q2-F2: 0.8682, and Q2-F3 (variances explained in external prediction [48]): 0.702. The parameters were equivalent to the value of R2 model. The predictions of compound in external set are shown in Figure 8(a).
The reliability of the model is based on the compounds falling in the applicability domain (AD). The leverage (h) and standardized residuals were used as described by [49]. William's graph was generated to observe the compounds lying in the applicability domain of the model (Figure 9) by plotting the standardized residuals for each compound against the leverage values. In the applicability domain, a defined domain is set up constituting all the data points within the boundary for residuals having a leverage threshold of HAT i/i h∗ = 1.000 [50]. Most of the compounds fall in the applicability domain except for the compound 3f having value greater than critical leverage (h = 1.29) that can be considered as an outlier.
4.2. Interpretation of Descriptors
In model generated by QSARINS, 70% of the anticancer activity can be described using four descriptors. All the variables belong to 2-dimensional family (MATS3i, ASP-5, VR2, and GGI10). The descriptor GGI10 belongs to the GALVEZ family and is a topological charge index that has its origin in first ten eigenvalues. There are two categories for the GALVEZ class, that is, the topological charge index of order n (GGIn) and the mean topological charge index of order n (JGGIn). The “n” is the order of eigenvalue. The GGI10 is the topological charge index of order 10 and has shown positive correlations to the activity, suggesting an increase in value of GGI10 would augment the anticancer activities of synthesized compounds. The descriptor VR2_Dzi also belongs to topological distance matrix and is defined as the normalized Randic-like eigenvalue-based index from Barysz matrix weighted by ionization potential. The negative correlation suggests lower value is associated with the activity of compounds. The 2D-AUTO descriptor (MATS3i) is the topological structure of Moran autocorrelation of lag 3 weighted by ionization potential. It is the summation of different autocorrelation functions giving different vectors based on lengths of structural fragment. The weighted component in the descriptor is linked to the physicochemical property suggesting the association of topology of the structure with the selected property. The autocorrelation vector of lag k is indicative of the number of edges in the fragment, while the last character of the descriptor “i” shows the physicochemical property that is the ionization potential. The negative correlation of MATS3i in the model suggests unfavorable conditions associated with lag 3 weighted by ionization potential. All the descriptors were not correlated with each other.
5. Conclusion
In this study, 22 derivatives of ethyl 6-(chloromethyl)-4-(4-hydroxyphenyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate were evaluated for their potential for anticancer activities. The compounds 4e, 4f, 4g, and 4h showed good anticancer activities against the breast cancer cell line MCF-7 when compared to standard tamoxifen. The in silico data also revealed best binding affinity and interaction pattern of these compounds against target proteins; moreover, the lead optimization revealed that the compounds have drug-like properties and may act as a lead. The QSAR analysis was carried out to investigate the role of molecular descriptors in attributing anticancer activities of synthesized compounds. The models developed to predict the structural features of these compounds as anticancer revealed useful information about the structural requirement of these compounds suggesting the importance of topological and autocorrelated descriptors. Further, in vitro assays will be carried out to confirm the role of these compounds in targeting these proteins.
Contributor Information
Reem Altaf, Email: reem.altaf@iqraisb.edu.pk.
Faisal Raza, Email: faisalraza@sjtu.edu.cn.
Data Availability
All the data has been included in the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Authors' Contributions
All authors contributed to data analysis, drafting, or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agreed to be accountable for all aspects of the work.
Supplementary Materials
Supplementary Table 1: the spectral analysis of twenty two derivatives of dihydropyrimidinones.
References
- 1.Kaur R., Chaudhary S., Kumar K., Gupta M. K., Rawal R. K. Recent synthetic and medicinal perspectives of dihydropyrimidinones: a review. European Journal of Medicinal Chemistry . 2017;132:108–134. doi: 10.1016/j.ejmech.2017.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Phucho I., Nongpiur A., Tumtin S., Nongrum R., Nongkhlaw R. Recent progress in the chemistry of dihydropyrimidinones. ChemInform . 2010;41(31) doi: 10.1002/chin.201031260. [DOI] [Google Scholar]
- 3.Matos L. H. S., Masson F. T., Simeoni L. A., Homem-de-Mello M. Biological activity of dihydropyrimidinone (DHPM) derivatives: a systematic review. European Journal of Medicinal Chemistry . 2018;143:1779–1789. doi: 10.1016/j.ejmech.2017.10.073. [DOI] [PubMed] [Google Scholar]
- 4.Rathwa S. K., Vasava M. S., Bhoi M. N., Borad M. A., Patel H. D. Recent advances in the synthesis of C-5-substituted analogs of 3, 4-dihydropyrimidin-2-ones: a review. Synthetic Communications . 2018;48(9):963–994. doi: 10.1080/00397911.2017.1423503. [DOI] [Google Scholar]
- 5.Friedenreich C. M. Physical activity and breast cancer: review of the epidemiologic evidence and biologic mechanisms. Recent Results in Cancer Research . 2010;188:125–139. doi: 10.1007/978-3-642-10858-7_11. [DOI] [PubMed] [Google Scholar]
- 6.Guarino M., Rubino B., Ballabio G. The role of epithelial-mesenchymal transition in cancer pathology. Pathology . 2007;39(3):305–318. doi: 10.1080/00313020701329914. [DOI] [PubMed] [Google Scholar]
- 7.Ilyas U., Altaf R., Aun Muhammad S., Qadir M. I., Nadeem H., Ahmed S. Computational drug designing of newly synthesized triazoles against potential targets of methicillin resistant Staphylococcus aureus. Pakistan Journal of Pharmaceutical Sciences . 2017;30(6):2271–2279. [PubMed] [Google Scholar]
- 8.Ilyas U., Naz S., Altaf R., et al. Design, synthesis and biological evaluations of 2-aminothiazole scaffold containing amino acid moieties as anti-cancer agents. Pakistan Journal of Pharmaceutical Sciences . 2021;34(4(Supplementary)):1509–1517. [PubMed] [Google Scholar]
- 9.Muhammad S. A., Ali A., Ismail T., Zafar R., Ilyas U., Ahmad J. Insilico study of anti-carcinogenic lysyl oxidase-like 2 inhibitors. Computational Biology and Chemistry . 2014;51:71–82. doi: 10.1016/j.compbiolchem.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Faheem M., Ali S. H., Khan A. W., et al. 1, 3, 4-Oxadiazole derivative attenuates chronic constriction injury induced neuropathic pain: a computational, behavioral, and molecular approach. Brain Sciences . 2020;10(10):p. 731. doi: 10.3390/brainsci10100731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faheem M., Althobaiti Y. S., Khan A. W., Ullah A., Ali S. H., Ilyas U. Investigation of 1, 3, 4 oxadiazole derivative in PTZ-induced neurodegeneration: a simulation and molecular approach. Journal of Inflammation Research . 2021;14:5659–5679. doi: 10.2147/JIR.S328609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rao V. S., Srinivas K. Modern drug discovery process: an in silico approach. Journal of Bioinformatics and Sequence Analysis . 2011;3(5):89–94. [Google Scholar]
- 13.Entezari Heravi Y., Sereshti H., Saboury A. A., Ghasemi J., Amirmostofian M., Supuran C. T. 3D QSAR studies, pharmacophore modeling, and virtual screening of diarylpyrazole-benzenesulfonamide derivatives as a template to obtain new inhibitors, using human carbonic anhydrase II as a model protein. Journal of Enzyme Inhibition and Medicinal Chemistry . 2017;32(1):688–700. doi: 10.1080/14756366.2016.1241781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verma J., Khedkar V. M., Coutinho E. C. 3D-QSAR in drug design-a review. Current Topics in Medicinal Chemistry . 2010;10(1):95–115. doi: 10.2174/156802610790232260. [DOI] [PubMed] [Google Scholar]
- 15.Benfenati E., Toropov A. A., Toropova A. P., Manganaro A., Gonella D. R. Coral software: QSAR for anticancer agents. Chemical Biology and Drug Design . 2011;77(6):471–476. doi: 10.1111/j.1747-0285.2011.01117.x. [DOI] [PubMed] [Google Scholar]
- 16.Tosco P., Balle T. Open 3DQSAR: a new open-source software aimed at high-throughput chemometric analysis of molecular interaction fields. Journal of Molecular Modeling . 2011;17(1):201–208. doi: 10.1007/s00894-010-0684-x. [DOI] [PubMed] [Google Scholar]
- 17.Gramatica P., Sangion A. A historical excursus on the statistical validation parameters for QSAR models: a clarification concerning metrics and terminology. Journal of Chemical Information and Modeling . 2016;56(6):1127–1131. doi: 10.1021/acs.jcim.6b00088. [DOI] [PubMed] [Google Scholar]
- 18.Shamsara J. Ezqsar: an R package for developing QSAR models directly from structures. The Open Medicinal Chemistry Journal . 2017;11(1):212–221. doi: 10.2174/1874104501711010212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Altaf R., Nadeem H., Iqbal M. N., et al. Synthesis, biological evaluation, 2D-QSAR, and molecular simulation studies of dihydropyrimidinone derivatives as alkaline phosphatase Inhibitors. Omega . 2022;7(8):7139–7154. doi: 10.1021/acsomega.1c06833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Altaf R., Nadeem H., Babar M. M., Ilyas U., Muhammad S. A. Genome-scale meta-analysis of breast cancer datasets identifies promising targets for drug development. Journal of Biological Research . 2021;28(1) doi: 10.1186/s40709-021-00136-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ilyas U., Altaf R., Nadeem H., Muhammad S. A. Genome wide meta-analysis of cDNA datasets reveals new target gene signatures of colorectal cancer based on systems biology approach. Journal of Biological Research-Thessaloniki . 2020;27(1):1–13. doi: 10.1186/s40709-020-00118-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gramatica P., Chirico N., Papa E., Cassani S., Kovarich S. QSARINS: A new software for the development, analysis, and validation of QSAR MLR models. Journal of Computational Chemistry . 2013;34(24):2121–2132. doi: 10.1002/jcc.23361. [DOI] [Google Scholar]
- 23.Sander T., Freyss J., von Korff M., Rufener C. Data Warrior: an open-source program for chemistry aware data visualization and analysis. Journal of Chemical Information and Modeling . 2015;55(2):460–473. doi: 10.1021/ci500588j. [DOI] [PubMed] [Google Scholar]
- 24.Dallakyan S., Olson A. J. Methods in Molecular Biology . Springer; 2015. Small-Molecule Library Screening by Docking with PyRx; pp. 243–250. [DOI] [PubMed] [Google Scholar]
- 25.Shiau A. K., Barstad D., Radek J. T., et al. Structural characterization of a subtype-selective ligand reveals a novel mode of estrogen receptor antagonism. Nature Structural Biology . 2002;9(5):359–364. doi: 10.1038/nsb787. [DOI] [PubMed] [Google Scholar]
- 26.Williams S. P., Sigler P. B. Atomic structure of progesterone complexed with its receptor. Nature . 1998;393(6683):392–396. doi: 10.1038/30775. [DOI] [PubMed] [Google Scholar]
- 27.Liu X., Ladias J. A. Structural basis for the BRCA1 BRCT interaction with the proteins ATRIP and BAAT1. Biochemistry . 2013;52(43):7618–7627. doi: 10.1021/bi400714v. [DOI] [PubMed] [Google Scholar]
- 28.Oliver A. W., Swift S., Lord C. J., Ashworth A., Pearl L. H. Structural basis for recruitment of BRCA2 by PALB2. EMBO Reports. . 2009;10(9):990–996. doi: 10.1038/embor.2009.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bauman D. R., Rudnick S. I., Szewczuk L. M., Jin Y., Gopishetty S., Penning T. M. Development of nonsteroidal anti-inflammatory drug analogs and steroid carboxylates selective for human aldo-keto reductase isoforms: potential antineoplastic agents that work independently of cyclooxygenase isozymes. Molecular Pharmacology . 2005;67(1):60–68. doi: 10.1124/mol.104.006569. [DOI] [PubMed] [Google Scholar]
- 30.Cho H.-S., Mason K., Ramyar K. X., et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature . 2003;421(6924):756–760. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- 31.de La Roche M., Rutherford T. J., Gupta D., et al. An intrinsically labile α-helix abutting the BCL9-binding site of β-catenin is required for its inhibition by carnosic acid. Nature Communications . 2012;3(1) doi: 10.1038/ncomms1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huai Q., Mazar A. P., Kuo A., et al. Structure of human urokinase plasminogen activator in complex with its receptor. Science . 2006;311(5761):656–659. doi: 10.1126/science.1121143. [DOI] [PubMed] [Google Scholar]
- 33.Ismail S. A., Chen Y.-X., Rusinova A., et al. Arl 2-GTP and Arl 3-GTP regulate a GDI-like transport system for farnesylated cargo. Nature Chemical Biology . 2011;7(12):942–949. doi: 10.1038/nchembio.686. [DOI] [PubMed] [Google Scholar]
- 34.Volkamer A., Kuhn D., Rippmann F., Rarey M. DoGSiteScorer: a web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics . 2012;28(15):2074–2075. doi: 10.1093/bioinformatics/bts310. [DOI] [PubMed] [Google Scholar]
- 35.Yuan S., Chan H. S., Hu Z. Using PyMOL as a platform for computational drug design. WIREs Computational Molecular Science . 2017;7(2, article e1298) doi: 10.1002/wcms.1298. [DOI] [Google Scholar]
- 36.Horton N. J., Kleinman K. Using R and RStudio for Data Management, Statistical Analysis, and Graphics . CRC Press; 2015. [DOI] [Google Scholar]
- 37.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Journal of Immunological Methods . 1983;65(1-2):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 38.Worth A. P., Bassan A., De Bruijn J., et al. The role of the European Chemicals Bureau in promoting the regulatory use of (Q) SAR methods. SAR and QSAR in Environmental Research . 2007;18(1-2):111–125. doi: 10.1080/10629360601054255. [DOI] [PubMed] [Google Scholar]
- 39.Tropsha A., Gramatica P., Gombar V. K. The Importance of Being Earnest: Validation is the Absolute Essential for Successful Application and Interpretation of QSPR Models. QSAR & Combinatorial Science . 2003;22(1):69–77. doi: 10.1002/qsar.200390007. [DOI] [Google Scholar]
- 40.Baguley B. C., Leung E. Heterogeneity of Phenotype in Breast Cancer Cell Lines. Breast Cancer - Carcinogenesis, Cell Growth and Signalling Pathways . 2011. https://www.intechopen.com/chapters/23376 .
- 41.Buteau-Lozano H., Ancelin M., Lardeux B., Milanini J., Perrot-Applanat M. Transcriptional regulation of vascular endothelial growth factor by estradiol and tamoxifen in breast cancer cells: a complex interplay between estrogen receptors α and β. Cancer Research . 2002;62(17):4977–4984. [PubMed] [Google Scholar]
- 42.D’Anselmi F., Masiello M. G., Cucina A., et al. Microenvironment promotes tumor cell reprogramming in human breast cancer cell lines. PloS One . 2013;8(12, article e83770) doi: 10.1371/journal.pone.0083770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matias M., Campos G., Santos A. O., Falcão A., Silvestre S., Alves G. Synthesis, in vitro evaluation and QSAR modelling of potential antitumoral 3, 4-dihydropyrimidin-2-(1H)-thiones. Arabian Journal of Chemistry . 2019;12(8):5086–5102. doi: 10.1016/j.arabjc.2016.12.007. [DOI] [Google Scholar]
- 44.Kumar B. P., Sankar G., Baig R. N., Chandrashekaran S. Novel Biginelli dihydropyrimidines with potential anticancer activity: a parallel synthesis and CoMSIA study. European Journal of Medicinal Chemistry . 2009;44(10):4192–4198. doi: 10.1016/j.ejmech.2009.05.014. [DOI] [PubMed] [Google Scholar]
- 45.Dash A. K., Nayak D., Hussain N., et al. Synthesis and investigation of the role of benzopyran dihydropyrimidinone hybrids in cell proliferation, migration and tumor growth. Anti-Cancer Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry-Anti-Cancer Agents). . 2019;19(2):276–288. doi: 10.2174/1871520618666180903101422. [DOI] [PubMed] [Google Scholar]
- 46.Russowsky D., Canto R. F., Sanches S. A., et al. Synthesis and differential antiproliferative activity of Biginelli compounds against cancer cell lines: monastrol, oxo-monastrol and oxygenated analogues. Bioorganic Chemistry . 2006;34(4):173–182. doi: 10.1016/j.bioorg.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 47.Golbraikh A., Tropsha A. QSAR modeling using chirality descriptors derived from molecular topology. Journal of Chemical Information and Computer Sciences . 2003;43(1):144–154. doi: 10.1021/ci025516b. [DOI] [PubMed] [Google Scholar]
- 48.Consonni V., Ballabio D., Todeschini R. Comments on the definition of the Q 2 parameter for QSAR validation. Journal of Chemical Information and Modeling . 2009;49(7):1669–1678. doi: 10.1021/ci900115y. [DOI] [PubMed] [Google Scholar]
- 49.Atkinson A. C. JSTOR; 1985. Plots, Transformations and Regression; An Introduction to Graphical Methods of Diagnostic Regression Analysis. [Google Scholar]
- 50.Roy K., Kar S., Ambure P. On a simple approach for determining applicability domain of QSAR models. Chemometrics and Intelligent Laboratory Systems . 2015;145:22–29. doi: 10.1016/j.chemolab.2015.04.013. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: the spectral analysis of twenty two derivatives of dihydropyrimidinones.
Data Availability Statement
All the data has been included in the manuscript.