

Abstract
REV-ERBs are atypical nuclear receptors as they function as ligand-regulated transcriptional repressors. The natural ligand for the REV-ERBs (REV-ERBα and REV-ERBβ) is heme, and heme-binding results in recruitment of transcriptional corepressor proteins such as N-CoR that mediates repression of REV-ERB target genes. These two receptors regulate a large range of physiological processes including several important in the pathophysiology of non-alcoholic steatohepatitis (NASH). These include carbohydrate and lipid metabolism as well as inflammatory pathways. A number of synthetic REV-ERB agonists have been developed as chemical tools and they show efficacy in animal models of NASH. Here, we will review the functions of REV-ERB with regard to their relevance to NASH as well as the potential to target REV-ERB for treatment of this disease.
Keywords: NASH, REV-ERB, Heme, circadian clock, lipid and glucose metabolism, inflammation
Introduction
REV-ERB is a member of the nuclear receptor (NR) superfamily whose members play critical roles in regulation of the transcription of genes involved in myriad of aspects of physiology and disease. Two REV-ERB isotypes exist, REV-ERBα (NR1D1) and REV-ERBβ (NR1D2), which have overlapping expression patterns and activity as they have almost identical DNA binding domains (DBD) and share ~70% homology in their ligand-binding domains (LBD) (Fig. 1) [1–5]. While the reason for REV-ERB redundancy remains unknown, in REV-ERBα mouse knock out models, REV-ERBβ can compensate in certain tissues [6–11]. Like other NRs, REV-ERBs are ligand-gated transcription factors that bind specific DNA motifs via two zinc fingers within the DBD near the genes that they directly regulate. Unlike most other NRs, REV-ERBs are ligand-dependent transcriptional repressors. REV-ERBs lack helix 12 of the LBD that contains the carboxy-terminal activation function domain that aids in recognition of coactivator proteins enabling transcriptional activation of target genes (Fig. 1) [12, 13]. Instead, REV-ERBs act as a transcriptional repressor recruiting corepressors in the presence of their natural ligand, heme. Unliganded REV-ERB does not bind to the corepressor proteins. Additionally, REV-ERBs can repress transcription by competing for a “shared” class of DNA response element binding with another group of NRs that function as transcriptional activators (RORs; Retinoic Acid Receptor-Related Orphan Receptors) [14, 15]. This element, known as a ROR response element or RORE consists of a single core motif half site (5′-AGGTCA-3′) preceded by a short A/T rich sequence in which REV-ERB or ROR can bind as monomers to repress or activate transcription, respectively. Interestingly, while both REV-ERB and ROR compete for the identical “half-site” RORE, REV-ERB-mediated active repression depends on the presence of an intact dimeric REV-RE (REV-DR2) [16]. This dimeric site is required for the recruitment of NCoR to DNA, and recently it was shown that heme was necessary for this REV-ERB–NCOR interaction [17].
Fig. 1.
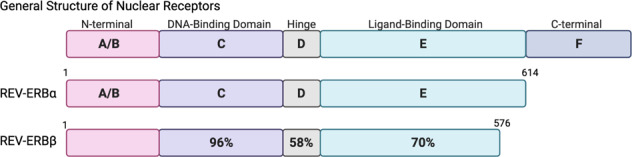
General organization of nuclear receptor structure and the REV-ERBs. The REV-ERB receptors lack the activation function-2 (AF-2) region within the C-terminal domain. Sequence identity (amino acid sequence was obtained from Uniprot) of each of the REV-ERB domains was compared and represented by percent identity as shown in the REV-ERBβ structure. The N-terminal domain was too variable for comparison between the isoforms. The A/B region, C region (DNA binding domain), D region (hinge), and E regions (ligand-binding domain) are indicated.
REV-ERB ligands—natural and synthetic
The REV-ERBs are transcriptional repressors whose natural ligand was identified as the iron-centered porphyrin heme [1, 18]. Cell-based and biochemical studies have been contradictory over the years in evaluating the role of heme in regulating REV-ERB activity. Cell-based experiments clearly showed heme-dependent recruitment of corepressor, but biochemical experiments displayed the opposite behavior. Recently, these conflicting data were resolved using a series of biophysical techniques demonstrating that heme binding does not displace NCOR but rather remodels the REV-ERB LBD, and changes thermodynamics to facilitate recruitment and binding of NCOR [16].
Classically, agonists are compounds that bind to the LBD of a NR and stimulate transcriptional activity by causing a conformational change that allows the recruitment of co-activator proteins and transcriptional apparatus to DNA. An antagonist blocks the effect of an agonist by competitive binding, and inverse agonists are ligands that can suppress activity below basal levels. Given that REV-ERB is a transcriptional repressor, agonists are ligands that enhance its repressive ability whereas antagonists cause increased transcription of REV-ERB target genes. Medicinal chemistry efforts over the last decade have identified a number of REV-ERB modulators that have proven to be useful as tool compounds in dissection the various physiological roles of this receptor in cell-based and animal studies (Fig. 2).
Fig. 2.

Synthetic REV-ERB ligands. Several unique chemical scaffolds that regulate REV-ERB activity are illustrated.
REV-ERB agonists
Initial efforts in ligand discovery for REV-ERB focused on the development of agonists. In 2008, Louden and colleagues described the first synthetic REV-ERB agonist, GSK4112 [19]. Using fluorescence resonance energy transfer (FRET) as the initial screening effort, they demonstrated that GSK4112 (EC50 = 0.4 μM) enhanced NCOR recruitment in a dose-dependent manner [19]. In a cell-based reporter assay using the Bmal1 promoter containing a REV-RE coupled to luciferase (BMAL1-Luc), GSK4112 displayed an EC50 of 2.3 μM. Aside from the robust suppression of Bmal1 expression, the group also demonstrated that administration of GSK4112 at various phases of Per2 oscillation induced bi-directional phase shifts or a weak re-setting of the circadian clock in cells. Unfortunately, GSK4112 is not very potent and it displays unfavorable pharmacokinetic (PK) properties making it sub-optimal for in vivo studies.
Building on the GSK4112 scaffold, our group expanded the structure-activity relationship (SAR) and identified two agonist compounds with improved potency and efficacy. In the same cell-based Bmal1-luc reporter assay, SR9009 and SR9011 displayed EC50 values of 0.7 and 0.62 μM, respectively [20]. Both compounds showed improvements in PK allowing our group and others to probe REV-ERB-mediated pharmacological effects in vivo for the first time. For example, twice per day dosing of SR9009 (100 mg/kg) reduced fat mass in diet-induced (DIO) and genetically obese (db/db) mice [20]. Single administration of SR9011 sufficiently altered locomotor activity and caused a delay in the initiation of diurnal activity in mice housed under dark:dark conditions. Recent work by the Lazar group suggested that SR9009 and SR9011 exhibited off-target or REV-ERB-independent effects in hepatocyte viability and metabolic regulation [21]. However, upon further examination of the deposited sequencing data, it appears that the mouse models were not complete REV-ERBα/β double knockout animals but rather hypomorphs with functional REV-ERB expression remaining. For example, in the REV-ERBα/β double KO hepatocytes described in Dierickx et al. [21] that were used to suggest that there were major off-target effects of SR9009, we found that when one compares the genes upregulated in the DKO hepatocytes (potential direct REV-ERB regulated genes) with the genes still downregulated by SR9009 in the DKO hepatocytes (potentially still regulated by residual REV-ERB expression) there was a highly significant (P < 10−152) overlap in genes identified as REV-ERB target genes (WT vs. DKO) that are still regulated by SR9009 in the DKO hepatocytes. In fact, of the 444 genes identified as still downregulated by SR9009 in the DKO hepatocytes 39% of them were identified as REV-ERB regulated in the WT vs. DKO comparison. These data clearly indicate that this DKO model cannot be used to probe the specificity of SR9009 or any other REV-ERB agonist for that matter. Nevertheless, like any pharmacological agent we do expect there to be off-target effects but as is the case in many of the studies mentioned below various methods to characterize the specificity of the pharmacological effects have been employed. Regardless, of the suggestion that these two agonists have wide off-target effects they been extensively utilized both in vitro and in vivo to demonstrate potential therapeutic activity of REV-ERB agonists in a variety of models ranging from NASH and hepatitis, muscle oxidative capacity, behavior and cognition, and neuroinflammation [1, 5, 6, 8–11, 20, 22–49]. As indicated above, a large number of these studies have employed various methods including genetic methods to confirm that the action examined is REV-ERB mediated.
Additional efforts to optimize REV-ERB agonists in terms of PK properties and potency yielded GSK2945, GSK0999, GSK5072, and GSK2667 that displayed higher potency in a FRET assay (EC50s ranged from 50–200 nM) [46]. While these compounds showed significant improvement in the cell-based assays as compared to GSK4112, only GSK2945 showed an improved PK profile with ~23% oral bioavailability in rodents. An additional REV-ERB agonist, SR12418, was recently reported by the Solt group to have an EC50 of 68 nM in cell-based assays and improved PK properties as compared to SR9011 [50]. SR12418 was used in vivo to demonstrate the effectiveness of REV-ERB agonism on the suppression of TH17 cell driven autoimmunity diseases in mouse models [50].
In addition to classical medicinal chemistry SAR strategies, computational approaches have been taken in recent years to discover several structurally diverse REV-ERB agonists. Of note is M35, which activates REV-ERB in a chimeric Gal4-REV-ERB-LBD fusion luciferase assay with a reported EC50 = 0.52 μM [46, 51]. It was suggested that M35 binds at the LBD interface with the NCOR binding site to promote co-repressor recruitment and complex stability; however, no crystal structures have been published to validate this interaction.
REV-ERB antagonists
While characterizing the chemical scaffold that led to the discovery of REV-ERB agonists SR9009 and SR9011, our group also identified a ligand that increased expression of REV-ERB target genes in cell-based assays. SR8278 was identified as a REV-ERB antagonist with an EC50 = 0.47 μM in the BMAL-Luc reporter assays, and was able to inhibit the activity of the REV-ERB agonist GSK4112 in competition assays [12, 46]. While the PK properties of SR8278 are suboptimal, this ligand has been successfully used in vivo for a number of studies demonstrating the role of REV-ERB in muscle function and osteoblast differentiation [12, 52]. Recently two additional antagonist compounds have been described, ARN5187 and GSK1362, unfortunately both have poor PK properties and SR8278 remains the most potent REV-ERB antagonist that can be used for in vivo studies to date.
Despite recent efforts, there is still a need to develop REV-ERB ligands with improved efficacy and PK profiles. Until then, these reported tool compounds are extremely useful in not only validating REV-ERB as a therapeutic target for various diseases, but also in elucidating the regulatory network the REV-ERB drives in physiology and disease.
Pathophysiology of fatty liver diseases
REV-ERB regulates a variety of physiological processes from central and peripheral circadian clocks, lipid and glucose metabolism, and inflammation [5, 24, 35, 37, 52, 53]. Based on its role in these processes, it has been identified as a potential therapeutic target for several diseases including non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) [5, 24, 45, 54, 55]. Over the last few decades, the prevalence of metabolic diseases including NAFLD and NASH, have increased dramatically with a worldwide incidence around 25%. Moreover, NAFLD has emerged as the most common chronic liver disease and is closely associated with other metabolic complications including obesity, type 2 diabetes, and cardiovascular diseases [56–58]. More concerning is the fact that ~25% of NAFLD patients progress and develop NASH, the leading factor in developing cirrhosis and/or requiring liver transplantation [58].
NAFLD incorporates a spectrum of disease pathologies that typically begins with excessive triglyceride storage in hepatocytes (simple steatosis) and considered commonly as a benign or protective state [59]. Chronic steatosis drives the disease progression towards NASH, which is characterized by inflammation and ballooning or hepatocyte damage [60, 61]. NASH itself is a highly complex disease state as many patients will develop fibrosis, a condition that may lead to cirrhosis or even hepatocellular carcinoma. Genetic, nutritional, and environmental factors often contribute to the disease progression, however NAFLD is a highly complex, heterogenic disease, causing drug development to lag other metabolic-related diseases [57, 62–67]. Many compounds that are in later stages of clinical evaluation are frequently found to have limited efficacy and no approved therapies are available for metabolic liver diseases [68–71].
The development of NAFLD has been classically described as a “two-hit” model with the initial “hit” considered the chronic hepatic accumulation of triglycerides, followed by a secondary insult which often include oxidative stress or prolonged inflammatory response [72]. Recent studies have clarified that the pathogenesis of NAFLD is an intricate metabolic disorder where functionality of several tissues including liver, intestine, pancreas, adipose, and the immune system are dysregulated and lead to NAFLD development. Perturbations of bioenergetics, imbalances in free fatty acid supply and utilization, lipid accumulation, and the inability of cells to effectively metabolize carbohydrates and free fatty acids are at the forefront of NAFLD development and progression [58, 72–74].
Hepatic fibrosis occurs as a result of chronic insult that causes a progressive accumulation and remodeling of the extracellular matrix (ECM) [75]. Activation of hepatic stellate cells (HSCs) is recognized as the primary event in the development of fibrosis in NASH. HSCs are activated by a variety of pro-inflammatory cytokines and this activation results in collagen and ECM deposition [75]. Based on experiments utilizing several different animal models as well as analysis of clinical studies, fructose is recognized as a major mediator of the development of NAFLD [76]. Its consumption has increased significantly over the last few decades correlating with increased obesity and a continuous rise in metabolic complications. Ingestion of high amounts of fructose can lead to the activation of both ChREBP and SREBP which can induce fructolytic and lipogenic enzymes [76]. In humans, ~70% of fructose is metabolized by the liver—the process of fructose catabolism is faster and more lipogenic than that of glucose [76]. Chronic activation of these transcription factors increases lipogenesis, eventually leading to increased cardiometabolic risk and NASH.
Fructose-induced liver damage promotes hepatic toll-like receptor (TLR-4) activation, which plays a pivotal role during the development of NASH. In mice that were chronically fed a high fructose diet, increased levels of endotoxins in portal blood and unregulated inflammatory mediators of Kupffer cells were reported [77]. These endotoxins activate the proinflammatory signaling cascade by binding TLR-4 on Kupffer cells, inducing a pro-inflammatory cascade by upregulating TNFα, NF-κB, IL-6, and IL-1β [76]. While this signaling cascade promotes inflammation within the liver, it also stimulates fructose-driven steatosis by upregulating lipogenic enzymes SREBP1, ACC, and FASN. Rodent studies have provided significant evidence that fructose triggers the infiltration and activation of Kupffer cells (and macrophages), driving necrosis of hepatocytes and inflammation which may ultimately lead to the progression of NAFLD toward NASH [76]. While the development of NASH is multifactorial, significant evidence supports that inflammasome activation, specifically NLRP3 (NOD-like receptor family pyrin-containing 3; inflammasome), is critically important.
The innate immune system is the first line of defense to clear bacteria and virus via recognition of various molecules within these pathogens (pathogen-associated molecular patterns; PAMPs), however it also plays an important role in the removal of abnormal accumulation of cellular debris, cholesterol crystals, and protein tangles (damage-associated molecular patterns; DAMPs). DAMPs are released by damaged or dying cells at sites of tissue damage, which in the liver, can be caused by excess of fructose and fats in the diet, as well as a variety of other factors. Pattern Recognition Receptors that recognize various PAMP and DAMP signals are found on a variety of immune cells (monocytes, neutrophils, and dendritic cells) and trigger immune responses by signaling via TLRs for the activation of immune mediators that secrete pro-inflammatory cytokines to signal adaptive immunity response [78]. The NLRP3 inflammasome plays an important role in the recognition of a large variety of PAMPs and DAMPs and has been identified as an important mediator of NAFLD progression toward NASH. Activation of the inflammasome is dependent on TLR signaling and leads to the cleavage of caspase-1, causing the secretion of pro-inflammatory cytokines IL-1β and IL-18 [73]. In the liver, inflammasomes can facilitate responses to gut-derived microbiota (from the intestinal lumen) as well as viral responses. Recent research has uncovered significant evidence implicating the inflammasome in the pathogenesis of NAFLD and NASH. For example, mice deficient in Caspase-1 or Nlrp3 did not develop a NASH-like phenotype when challenged with high fat/fructose diet while in the wildtype littermates, NASH was apparent. Additional studies showed that the NLRP3 inhibitor, MCC950, was sufficient in preventing the development of fibrosis in mice challenged with a methionine/choline deficient diet for six weeks [79, 80], suggesting that inhibition of the inflammasome may be a therapeutic avenue for the treatment of NASH.
The REV-ERBs and circadian control of liver disease
The complexities of development and progression of the NAFLD spectrum of diseases remans poorly understood, however the role of circadian regulation in maintaining metabolic homeostasis has been identified as a major factor in the development of liver diseases. Following the discovery of the REV-ERBs, it was demonstrated that its expression fluctuated rhythmically in a 24-h period [81–84]. The role of REV-ERB in the regulation of central and peripheral circadian clocks is now well-established but recently many in the field have been identifying how the REV-ERB—circadian clock axis is involved in disease pathogenesis [2, 3, 6, 20, 22, 54, 84, 85]. The circadian system relies on the presence of the molecular clockwork in every cell of the body, which generates multiple autonomous clocks that are both self-sustaining and synchronized by the central circadian clock located within the suprachiasmatic nucleus (SCN). This central circadian clock receives input via the retina as well as non-photon-based input from tissues and organs, integrates the signaling, and generates output signal cascades to the peripheral clocks on a 24-h basis [86]. While the peripheral clocks can act with some autonomy, the central clock within the SCN is a necessary component to coordinate a regulatory network among peripheral clocks.
Mammalian clock and liver disease
Circadian rhythms, regardless of peripheral or central location, are orchestrated by a tightly regulated transcription/translation feedback loop containing both activator and repressor components. As mentioned above, REV-ERB is a well-established component of this feedback system, with other clock regulators being rhythmically regulated by the REV-ERBs as well [3, 15, 86, 87]. The activator arm of the circadian clock consists of a dimer of CLOCK or NPAS2 with BMAL1 (ARNTL) which then activates the transcription of Cry, Per, REV-ERB, and ROR. Once threshold levels of CRY and PER expression is reached, the CRY/PER dimer then inhibits the activity of the activator arm, leading to a reduction of expression of these transcription factors. A secondary loop also maintains the activity of the clock where REV-ERB and ROR compete for RORE binding to either repress (REV-ERB) or induce (ROR) Bmal1, Clock, and NPAS2 expression [3, 15]. Disruptions in this tightly controlled circadian system, either within the central or peripheral clocks, can lead to the increased incidence of metabolic related diseases [86].
How does circadian dysfunction lead to increased incidence of NAFLD and NASH? The most widely studied peripheral clock is that of hepatocytes, as disruptions in liver circadian rhythms negatively impact metabolism and overall health [6, 54, 55, 88, 89]. Circadian functions play a major role in the regulation of both whole body and liver homeostasis, and lipid and glucose metabolism. It has been reported that ~50% of liver metabolites have a circadian rhythm coupled to REV-ERB expression [90]. Disruptions to circadian regulation of liver homeostasis induce a variety of epigenetic and microcellular changes that aid in the development of liver diseases by disrupting lipid, glucose, cholesterol, and bile acid metabolism [91–94]. Other factors including nutrient changes (high fat, high sugar diets) and restricted feeding schedules also profoundly affect the regulation of hepatic circadian function and lead to activation of pathways involved in NAFLD and NASH [91, 95].
While work is continuously being done to understand circadian control of liver diseases, recent evidence points to the REV-ERBα cistrome in the (mouse) liver which consists of thousands of binding sites that follow the oscillating expression of REV-ERB and are highly coordinated with NCOR and HDAC cistromes [96]. More specifically, these cistromes are enriched for genes involved in lipid metabolism, and hepatic loss of REV-ERBα leads to moderate hepatosteatosis in mice. REV-ERBβ is less understood, however its cistrome is highly similar to that of REV-ERBα in the liver, suggesting that the moderate levels of hepatosteatosis observed in REV-ERBα KO mice was due to a compensatory mechanism of REV-ERBβ [96]. Interestingly, depletion of REV-ERBβ in REV-ERBα-null mice increases hepatosteatosis and elevates hepatic triglyceride levels, suggesting that both isotypes regulate the metabolic factors often associated with NAFLD [96]. Likely, this redundancy in REV-ERB expression and function is an evolutionary mechanism to protect both circadian and metabolic function. With the development of more sophisticated sequencing technology, the role of the two REV-ERBs will likely be solved in the near future.
Ablation of both Rev-Erbα and Rev-Erbβ specifically in mouse hepatocytes leads to the loss of circadian expression of genes resulting in the disruption of de novo lipogenesis [14]. REV-ERB also controls the accumulation of nuclear SREBP in hepatocytes, thereby controlling the synthesis of cholesterols within those cells. In DIO mouse studies, treatment with REV-ERB pan-agonists modifies both clock and metabolic gene expression, reduces hepatic triglyceride storage, and suppressed hepatic cholesterol synthesis [20]. Another study using a genetically obese mouse model fed a NASH-inducing western diet showed that REV-ERB pan-agonists inhibited the development of fibrosis, signaling that REV-ERB may be a valid target for the treatment of an array of fatty liver diseases [24].
Clearly a number of studies suggest a strong link between circadian regulation and metabolic dysfunction, however defining the actions of REV-ERB is critical to understanding this complex interaction and discovering novel therapeutic approaches to combating NAFLD. A recent study by Hunter et al. [27] described the development of transgenic models to study REV-ERB function and chromatin binding specifically in the liver by employing endogenous expression of HaloTag epitope-tagged REV-ERBα. Using this system, they defined a robust liver cistrome without the dependency on antibodies, that have been associated with specificity issues and lead to inaccurate profiling of REV-ERB within the genome over the last few years. This study performed Halo-REV-ERBα ChIP-seq and identified expected signal enrichment at clock gene loci in the liver. They also found evidence of co-repressor recruitment to REV-ERBα occupied sites and sequence motif analysis showing a high enrichment of classic RORE and REV-RE (REV-DR2) sequences. Unlike previous studies, Hunter et al. found no evidence for REV-ERBα tethering to other transcription factors (HNF6, C/EBP, HNF4α, etc.) in the liver. They validated this finding by generating a DBD-mutant animal (ReverbαDBDm) and reported that hepatic lipid accumulation was not observed in these animals as was reported in the global REV-ERBα-null mouse [27].
Hunter et al. [27] further investigated REV-ERB action in the liver by generating a hepatocyte-specific Reverbα-null mouse model that allowed for deletion of REV-ERB in adulthood. The results from this model were not only surprising but may explain a new paradigm for REV-ERB action in NAFLD. When REV-ERBα is deleted in the hepatocytes during adulthood, there was limited impact in lipid metabolism or lipid accumulation, which is in direct contrast to previously published work demonstrating significant phenotypic changes and transcriptional dysregulation in Reverbα-null livers [27]. However, this group then showed that up-regulated genes identified in previous systemic deletions of REV-ERB and targeted deletions contribute to the regulation of metabolic genes in a metabolic or nutritional-dependent context. While many studies have described the role that nutritional signals play in hepatic chromatin landscape, this study also demonstrated that daytime recruitment of REV-ERB to the genome serves to repress activity at enhancers that are sensitive to the feeding state via HDAC3 recruitment and regulating enhancer-promoter loop formation suggesting that REV-ERB serves a metabolic state-dependent role [27]. This was further investigated by challenging the hepatocyte-specific knockout mice with mistimed feeding, which revealed a separate cluster of REV-ERB-directed metabolic genes. This exciting work proposes that the disordered feeding behavior of systemic Reverbα-/- mice described in previous studies is due to the loss of REV-ERB in the brain (central clock) [27]. This clock dysfunction is then responsible for the upregulation of lipogenic processes observed in these animals, suggesting that targeting the core clock may be therapeutic for the treatment of metabolic diseases including NAFLD and NASH.
Circadian regulation of the immune system in NAFLD
REV-ERB is a well-established regulator of inflammation. Recently, REV-ERBα was identified as a direct regulator of the NLRP3 inflammasome, a component of the innate immune system that has been implicated in the development and progression of NAFLD and NASH [5, 97]. NLRP3 expression and activation displays a circadian rhythmicity, driven by REV-ERB. Deletion or silencing of REV-ERBα in macrophages exacerbate both IL-1β and IL-18 production and secretion, while the pharmacological activation of REV-ERBα attenuates NLRP3-driven inflammation in several mouse models including fulminant hepatitis, suggesting that activating REV-ERB may be a promising strategy to treat NLRP3-mediated diseases including NASH [5].
Like other cell types, molecular clocks are present in immune cells including macrophages, lymphocytes, and neutrophils, as well as lymphatic tissues including the spleen and lymph nodes. Not only do circulating immune cell numbers oscillate during the 24-h period (highest during rest phase), immune cell functions including cytokine production, phagocytosis, and pathogenic response also display daily oscillations. The intricate timing of immune regulation ensures an optimized immune response to maintain or restore homeostasis after infection or tissue damage/injury [78]. Disruptions in the molecular clock are often associated with inflammatory-mediated diseases.
As mentioned earlier, REV-ERBα directly regulates both Nlrp3 and IL-1β by binding specific response elements within the promoter of these genes, silencing the expression and controlling activation of the inflammasome assembly. Directly inhibiting NLRP3 with the small molecule MCC950 has been successful in inhibiting fibrosis in rodent models of NASH; however clinical trials with this and similar analogs have been suspended due to increased risk of hepatotoxicity [98]. While more research is necessary to understand the intricate regulation of NASH progression, the innate immune system, and therapeutics, it is likely that complete inhibition of the inflammasome is detrimental as a long-term solution. This lends the idea that suppressing NLRP3 activity via REV-ERB activation may be a preferential model for treatment of NAFLD and NASH.
Our group recently demonstrated that the REV-ERB agonist SR9009 inhibited fibrosis in a mouse model of NASH by actively suppressing profibrogenic factors including TGFβ1 and Mmp13 in addition to pro-inflammatory mediators IL-1β, NF-κB, and TNFα [24]. While effects on lipid accumulation and steatosis were not observed in this study, it is possible that activating REV-ERB pharmacologically initiates active suppression of inflammatory components via both direct regulation of this pathway and circadian modulation, which will need to be probed further to fully elucidate REV-ERB’s role in NASH.
While it plays a significant role in the pathogenesis of NASH, the inflammasome is not the only inflammatory mediator in this disease. Over the past few years, studies have demonstrated that IL-17A contributes significantly to NAFLD pathogenesis [99–101]. In fact, in mouse models fed high fats or high sugar diets, DNA damage and hepatic infiltration of Th17 cells were described [100–108]. This infiltration of Th17 cells in the liver subsequently correlate to the increased IL-17A concentration and promotion of hepatic inflammation, fibrosis, and NASH progression. Inhibition of IL-17A signaling improves the condition and suggests an alternative anti-inflammatory route for NASH treatment [99, 101]. There has been a significant amount of work demonstrating key factors that drive Th17 cell development and pathogenicity, yet mechanisms that regulate this pathway are still less understood. RORγt, another nuclear receptor encoded by Rorc that works in concert with REV-ERB as the activating arm of the circadian clock, is often thought of as a master regulator of Th17 cell differentiation and IL-17A activation. Since both ROR and REV-ERB bind to the same RORE within the promoter of target genes, REV-ERB suppresses Th17 cell activity and subsequent signaling [50]. Previous work by Yu et al. demonstrated that REV-ERBα diurnally regulated Th17 cell number and function and is necessary for the dampening of IL-17A and other Th17 cell-mediated cytokine expression [109]. The same study also demonstrated that genetic deletion of REV-ERBα resulted in enhanced Th17 cell development and exacerbated autoimmune responses in mouse models [109]. Interestingly, REV-ERBα directly represses the expression of Rorc by binding its promoter, thereby suggesting potential autoregulation in controlling Th17 expression and activity [50, 109]. While it has been suggested that the cell autonomous clock is not required for lymphocyte development and function [110], it is clear in many cases that the ROR and REV-ERB components of the clock are very important for the functioning of at least a subset of lymphocytes based on the work described above. While RORγt inhibitors are currently being explored in preclinical and clinical trials to suppress Th17 mediated inflammatory pathways, it is likely that developing small molecule activators for REV-ERB will provide similar beneficial effects as REV-ERB modulators do not appear to affect the thymus unlike modulators of RORγ. The efficacy of targeting REV-ERB to suppress Th17 cell activation and IL-17A release in NASH has yet to be explored.
Conclusions and perspectives
NAFLD is a spectrum of liver pathologies ranging from simple steatosis to lobular inflammation to pericellular inflammation. The accumulation of hepatic lipids and the reduced capacity to metabolize them lead to lipotoxicity, increased oxidative stress, and fibrogenesis that ultimately promotes NASH progression. Despite significant progress in this field to characterize molecular etiologies occurring during this transition toward NASH, there have been significant challenges in the discovery of novel therapeutics with no drug yet approved for NASH. Current therapies are focused on bile acid signaling (FXR agonists; obeticholic acid) and pan-PPAR agonists (lanifibranor). Moreover, the focus on various signaling pathways involved in NASH progression including inflammatory processes have shed light on the potential for targeting circadian clock components for the treatment of various metabolic disorders. The nuclear receptor REV-ERB is not only a master regulator of inflammatory processes and lipid metabolism but is also a circadian regulator that may provide a novel route for NASH therapies. REV-ERB is a repressor of transcription and therefore ligands that activate its function suppress pro-inflammatory molecules (i.e., IL-1β, IL-6, TNFα, and NLRP3), lipogenic enzymes (i.e., SREBP1c, FASN), and can promote re-synchronization of hepatic clocks which are often disrupted in NAFLD, NASH, and metabolic syndrome. While more research is warranted and compounds are needed to further understand the role of REV-ERB in NASH, it is likely that these efforts will catalyze the discovery of new therapeutic strategies.
Competing interests
The authors declare no competing interests.
References
- 1.Burris TP. Nuclear hormone receptors for heme: REV-ERBalpha and REV-ERBbeta are ligand-regulated components of the mammalian clock. Mol Endocrinol. 2008;22:1509–20. doi: 10.1210/me.2007-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duez H, Staels B. Rev-erb alpha gives a time cue to metabolism. FEBS Lett. 2008;582:19–25. doi: 10.1016/j.febslet.2007.08.032. [DOI] [PubMed] [Google Scholar]
- 3.Duez H, Staels B. Rev-erb-alpha: an integrator of circadian rhythms and metabolism. J Appl Physiol. 2009;107:1972–80. doi: 10.1152/japplphysiol.00570.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ikeda R, Tsuchiya Y, Koike N, Umemura Y, Inokawa H, Ono R, et al. REV-ERBα and REV-ERBβ function as key factors regulating mammalian circadian output. Sci Rep. 2019;9:10171. doi: 10.1038/s41598-019-46656-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pourcet B, Zecchin M, Ferri L, Beauchamp J, Sitaula S, Billon C, et al. Nuclear receptor subfamily 1 group D member 1 regulates circadian activity of NLRP3 inflammasome to reduce the severity of fulminant hepatitis in mice. Gastroenterology. 2018;154:1449–64. doi: 10.1053/j.gastro.2017.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cho H, Zhao X, Hatori M, Yu RT, Barish GD, Lam MT, et al. Regulation of circadian behaviour and metabolism by REV-ERB-α and REV-ERB-β. Nature. 2012;485:123–7. doi: 10.1038/nature11048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alibhai FJ, LaMarre J, Reitz CJ, Tsimakouridze EV, Kroetsch JT, Bolz SS, et al. Disrupting the key circadian regulator CLOCK leads to age-dependent cardiovascular disease. J Mol Cell Cardiol. 2017;105:24–37. doi: 10.1016/j.yjmcc.2017.01.008. [DOI] [PubMed] [Google Scholar]
- 8.Amador A, Wang Y, Banerjee S, Kameneka TM, Solt LA, Burris TP. Pharmacological and genetic modulation of REV-ERB activity and expression affects orexigenic gene expression. PLoS One. 2016;11:e0151014. doi: 10.1371/journal.pone.0151014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amador A, Huitron-Resendiz S, Roberts AJ, Kamenecka TM, Solt LA, Burris TP. Pharmacological targeting the REV-ERBs in sleep/wake regulation. PLoS One. 2016;11:e0162452. doi: 10.1371/journal.pone.0162452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amador A, Kamenecka TM, Solt LA, Burris TP. REV-ERBβ is required to maintain normal wakefulness and the wake-inducing effect of dual REV-ERB agonist SR9009. Biochem Pharmacol. 2018;150:1–8. doi: 10.1016/j.bcp.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Griffin P, Dimitry JM, Sheehan PW, Lananna BV, Guo C, Robinette ML, et al. Circadian clock protein Rev-erbα regulates neuroinflammation. Proc Natl Acad Sci USA. 2019;116:5102–7. doi: 10.1073/pnas.1812405116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kojetin D, Wang Y, Kamenecka TM, Burris TP. Identification of SR8278, a synthetic antagonist of the nuclear heme receptor REV-ERB. ACS Chem Biol. 2011;6:131–4. doi: 10.1021/cb1002575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kojetin DJ, Burris TP. REV-ERB and ROR nuclear receptors as drug targets. Nat Rev Drug Discov. 2014;13:197–216. doi: 10.1038/nrd4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim YH, Marhon SA, Zhang Y, Steger DJ, Won KJ, Lazar MA. Rev-erbα dynamically modulates chromatin looping to control circadian gene transcription. Science. 2018;359:1274–7. doi: 10.1126/science.aao6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Solt LA, Kojetin DJ, Burris TP. The REV-ERBs and RORs: molecular links between circadian rhythms and lipid homeostasis. Future Med Chem. 2011;5:623–38. doi: 10.4155/fmc.11.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mosure SA, Strutzenberg TS, Shang J, Munoz-Tello P, Solt LA, Griffin PR, et al. Structural basis for heme-dependent NCoR binding to the transcriptional repressor REV-ERBβ. Sci Adv. 2021;5:eabc6479. doi: 10.1126/sciadv.abc6479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Varadinova MG, Valcheva-Traykova ML, Boyadjieva NI. Effect of circadian rhythm disruption and alcohol on the oxidative stress level in rat brain. Am J Ther. 2016;6:e1801–e1805. doi: 10.1097/MJT.0000000000000363. [DOI] [PubMed] [Google Scholar]
- 18.Raghuram S, Stayrook KR, Huang P, Rogers PM, Nosie AK, McClure DB, et al. Identification of heme as the ligand for the orphan nuclear receptors REV-ERBalpha and REV-ERBbeta. Nat Struct Mol Biol. 2007;14:1207–13. doi: 10.1038/nsmb1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meng QJ, McMaster A, Beesley S, Lu WQ, Gibbs J, Parks D, et al. Ligand modulation of REV-ERBalpha function resets the peripheral circadian clock in a phasic manner. J Cell Sci. 2008;121:3629–35. doi: 10.1242/jcs.035048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Solt LA, Wang Y, Banerjee S, Hughes T, Kojetin DJ, Lundasen T, et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature. 2012;485:62–68. doi: 10.1038/nature11030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dierickx P, Emmett MJ, Jiang C, Uehara K, Liu M, Adlanmerini M, et al. SR9009 has REV-ERB-independent effects on cell proliferation and metabolism. Proc Natl Acad Sci USA. 2019;116:12147–52. doi: 10.1073/pnas.1904226116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duez H, Staels B. The nuclear receptors Rev-erbs and RORs integrate circadian rhythms and metabolism. Diab Vasc Dis Res. 2008;2:82–8. doi: 10.3132/dvdr.2008.0014. [DOI] [PubMed] [Google Scholar]
- 23.Feillet CA, Bainier C, Mateo M, Blancas-Velázquez A, Salaberry NL, Ripperger JA, et al. Rev-erbα modulates the hypothalamic orexinergic system to influence pleasurable feeding behaviour in mice. Addict Biol. 2017;2:411–22. doi: 10.1111/adb.12339. [DOI] [PubMed] [Google Scholar]
- 24.Griffett K, Bedia-Diaz G, Elgendy B, Burris TP. REV-ERB agonism improves liver pathology in a mouse model of NASH. PLoS One. 2020;15:e0236000. doi: 10.1371/journal.pone.0236000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Griffin P, Sheehan PW, Dimitry JM, Guo C, Kanan MF, Lee J, et al. REV-ERBα mediates complement expression and diurnal regulation of microglial synaptic phagocytosis. Elife. 2020;9:e58765. doi: 10.7554/eLife.58765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo DK, Zhu Y, Sun HY, Xu XY, Zhang S, Hao ZB, et al. Pharmacological activation of REV-ERBα represses LPS-induced microglial activation through the NF-κB pathway. Acta Pharmacol Sin. 2019;40:26–34. doi: 10.1038/s41401-018-0064-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hunter AL, Pelekanou CE, Adamson A, Downton P, Barron NJ, Cornfield T, et al. Nuclear receptor REVERBα is a state-dependent regulator of liver energy metabolism. Proc Natl Acad Sci USA. 2020;117:25869–79. doi: 10.1073/pnas.2005330117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishimaru K, Nakajima S, Yu G, Nakamura Y, Nakao A. The putatively specific synthetic REV-ERB agonist SR9009 inhibits IgE- and IL-33-mediated mast cell activationindependently of the circadian clock. Int J Mol Sci. 2019;20:E6320. doi: 10.3390/ijms20246320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim K, Kim JH, Kim I, Seong S, Kim N. Rev-erbα negatively Regulates osteoclast and osteoblast differentiation through p38 MAPK signaling pathway. Mol Cells. 2020;43:34–47. doi: 10.14348/molcells.2019.0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin Y, Lin L, Gao L, Wang S, Wu B. Rev-erbα regulates hepatic ischemia-reperfusion injury in mice. Biochem Biophys Res Commun. 2020;529:916–21. doi: 10.1016/j.bbrc.2020.06.152. [DOI] [PubMed] [Google Scholar]
- 31.Liu H, Zhu Y, Gao Y, Qi D, Zhao L, Zhao L, et al. NR1D1 modulates synovial inflammation and bone destruction in rheumatoid arthritis. Cell Death Dis. 2020;11:129. doi: 10.1038/s41419-020-2314-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mayeuf-Louchart A, Thorel Q, Delhaye S, Beauchamp J, Duhem C, Danckaert A, et al. Rev-erb-α regulates atrophy-related genes to control skeletal muscle mass. Sci Rep. 2017;7:14383. doi: 10.1038/s41598-017-14596-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morioka N, Tomori M, Zhang FF, Saeki M, Hisaoka-Nakashima K, Nakata Y. Stimulation of nuclear receptor REV-ERBs regulates tumor necrosis factor-induced expression of proinflammatory molecules in C6 astroglial cells. Biochem Biophys Res Commun. 2016;469:151–7. doi: 10.1016/j.bbrc.2015.11.086. [DOI] [PubMed] [Google Scholar]
- 34.Morioka N, Kodama K, Tomori M, Yoshikawa K, Saeki M, Nakamura Y, et al. Stimulation of nuclear receptor REV-ERBs suppresses production of pronociceptive molecules in cultured spinal astrocytes and ameliorates mechanical hypersensitivity of inflammatory and neuropathic pain of mice. Brain Behav Immun. 2019;78:116–30. doi: 10.1016/j.bbi.2019.01.014. [DOI] [PubMed] [Google Scholar]
- 35.Alibhai FJ, LaMarre J, Reitz CJ, Tsimakouridze EV, Kroetsch JT, Bolz SS, et al. Disrupting the key circadian regulator CLOCK leads to age-dependent cardiovascular disease. Mol Cell Cardiol. 2017;105:24–37. [DOI] [PubMed]
- 36.Reitz CJ, Alibhai FJ, Khatua TN, Rasouli M, Bridle BW, Burris TP, et al. SR9009 administered for one day after myocardial ischemia-reperfusion prevents heart failure in mice by targeting the cardiac inflammasome. Commun Biol. 2019;2:353. doi: 10.1038/s42003-019-0595-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roby DA, Ruiz F, Kermath BA, Voorhees JR, Niehoff M, Zhang J, et al. Pharmacological activation of the nuclear receptor REV-ERB reverses cognitive deficits and reduces amyloid-β burden in a mouse model of Alzheimer’s disease. PLoS One. 2019;14:e0215004. doi: 10.1371/journal.pone.0215004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shen W, Zhang W, Ye W, Wang H, Zhang Q, Shen J, et al. SR9009 induces a REV-ERB dependent anti-small-cell lung cancer effect through inhibition of autophagy. Theranostics. 2020;10:4466–80. doi: 10.7150/thno.42478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Burris TP. Anti-proliferative actions of a synthetic REV-ERBα/β agonist in breast cancer cells. Biochem Pharm. 2015;96:315–22. doi: 10.1016/j.bcp.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sitaula S, Billon C, Kamenecka TM, Solt LA, Burris TP. Suppression of atherosclerosis by synthetic REV-ERB agonist. Biochem Biophys Res Commun. 2015;460:566–71. doi: 10.1016/j.bbrc.2015.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Q, Robinette ML, Billon C, Collins PL, Bando JK, Fachi JL, et al. Circadian rhythm-dependent and circadian rhythm-independent impacts of the molecular clock on type 3 innate lymphoid cells. Sci Immunol. 2019;4:eaay7501. doi: 10.1126/sciimmunol.aay7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sitaula S, Zhang J, Ruiz F, Burris TP. Rev-erb regulation of cholesterologenesis. Biochem Pharmacol. 2017;13:168–77. doi: 10.1016/j.bcp.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song C, Tan P, Zhang Z, Wu W, Dong Y, Zhao L, Liu H, Guan H, Li F. REV-ERB agonism suppresses osteoclastogenesis and prevents ovariectomy-induced bone loss partially via FABP4 upregulation. FASEB J. 2018;32:3215–28. doi: 10.1096/fj.201600825RRR. [DOI] [PubMed] [Google Scholar]
- 44.Stujanna EN, Murakoshi N, Tajiri K, Xu D, Kimura T, Qin R, Feng D, Yonebayashi S, Ogura Y, Yamagami F, Sato A, Nogami A, Aonuma K. Rev-erb agonist improves adverse cardiac remodeling and survival in myocardial infarction through an anti-inflammatory mechanism. PLoS ONE. 2017;12:e0189330. doi: 10.1371/journal.pone.0189330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thomes PG, Brandon-Warner E, Li T, Donohue TM, Schrum LW. Rev-erb agonist and TGF-β similarly affect autophagy but differentially regulate hepatic stellate cell fibrogenic phenotype. Int J Biochem Cell Biol. 2016;81:137–47. doi: 10.1016/j.biocel.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 46.Uriz-Huarte A, Date A, Ang H, Ali S, Brady HJM, Fuchter MJ. The transcriptional repressor REV-ERB as a novel target for disease. Bioorg Med Chem Lett. 2020;30:127395. doi: 10.1016/j.bmcl.2020.127395. [DOI] [PubMed] [Google Scholar]
- 47.Yuan X, Dong D, Li Z, Wu B. Rev-erbα activation down-regulates hepatic Pck1 enzyme to lower plasma glucose in mice. Pharmacol Res. 2019;141:310–8. doi: 10.1016/j.phrs.2019.01.010. [DOI] [PubMed] [Google Scholar]
- 48.Zhang L, Zhang R, Tien CL, Chan RE, Sugi K, Fu C, Griffin AC, Shen Y, Burris TP, Liao X, Jain MK. REV-ERBα ameliorates heart failure through transcription repression. JCI Insight. 2017;2:e95177. doi: 10.1172/jci.insight.95177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao W, Cui L, Huang X, Wang S, Li D, Li L, Sun Y, Du M. Activation of Rev-erbα attenuates lipopolysaccharide-induced inflammatory reactions in human endometrial stroma cells via suppressing TLR4-regulated NF-κB activation. Acta Biochim Biophys Sin. 2019;51:908–14. doi: 10.1093/abbs/gmz078. [DOI] [PubMed] [Google Scholar]
- 50.Chang C, Loo CS, Zhao X, Solt LA, Liang Y, Bapat SP, Cho H, Kamenecka TM, Leblanc M, Atkins AR, Yu RT, Downes M, Burris TP, Evans RM, Zheng Y. The nuclear receptor REV-ERBα modulates Th17 cell-mediated autoimmune disease. Proc Natl Acad Sci USA. 2019;116:18528–36. doi: 10.1073/pnas.1907563116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Westermaier Y, Ruiz-Carmona S, Theret I, Perron-Sierra F, Poissonnet G, Dacquet C, Boutin JA, Ducrot P, Barril X. Binding mode prediction and MD/MMPBSA-based free energy ranking for agonists of REV-ERBα/NCoR. J Comput Aided Mol Des. 2017;31:755–75. doi: 10.1007/s10822-017-0040-7. [DOI] [PubMed] [Google Scholar]
- 52.Welch RD, Billon C, Valfort AC, Burris TP, Flaveny CA. Pharmacological inhibition of REV-ERB stimulates differentiation, inhibits turnover and reduces fibrosis in dystrophic muscle. Sci Rep. 2017;7:17142. doi: 10.1038/s41598-017-17496-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ni Y, Zhao Y, Ma L, Wang Z, Ni L, Hu L, Fu Z. Pharmacological activation of REV-ERBα improves nonalcoholic steatohepatitis by regulating intestinal permeability. Metabolism. 2021;114:154409. doi: 10.1016/j.metabol.2020.154409. [DOI] [PubMed] [Google Scholar]
- 54.Bugge A, Feng D, Everett LJ, Briggs ER, Mullican SE, Wang F, Jager J, Lazar MA. Rev-erbα and Rev-erbβ coordinately protect the circadian clock and normal metabolic function. Genes Dev. 2012;26:657–67. doi: 10.1101/gad.186858.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Delezie J, Dumont S, Dardente H, Oudart H, Gréchez-Cassiau A, Klosen P, Teboul M, Delaunay F, Pévet P, Challet E. The nuclear receptor REV-ERBα is required for the daily balance of carbohydrate and lipid metabolism. FASEB J. 2012;26:3321–35. doi: 10.1096/fj.12-208751. [DOI] [PubMed] [Google Scholar]
- 56.Dwivedi DK, Jena GB. NLRP3 inhibitor glibenclamide attenuates high-fat diet and streptozotocin-induced non-alcoholic fatty liver disease in rat: studies on oxidative stress, inflammation, DNA damage and insulin signalling pathway. Naunyn Schmiedebergs Arch Pharm. 2020;393:705–16. doi: 10.1007/s00210-019-01773-5. [DOI] [PubMed] [Google Scholar]
- 57.Michelotti GA, Machado MV, Diehl AMNAFLD. NASH and liver cancer. Nat Rev Gastroenterol Hepatol. 2013;10:656–65. doi: 10.1038/nrgastro.2013.183. [DOI] [PubMed] [Google Scholar]
- 58.Zhang C, Yang M. Current options and future directions for NAFLD and NASH treatment. Int J Mol Sci. 2021;22:7571. doi: 10.3390/ijms22147571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rinella ME, Loomba R, Caldwell SH, Kowdley K, Charlton M, Tetri B, Harrison SA. Controversies in the diagnosis and management of NAFLD and NASH. Gastroenterol Hepatol. 2014;10:219–27. [PMC free article] [PubMed] [Google Scholar]
- 60.Dongiovanni P, Anstee QM, Valenti L. Genetic predisposition in NAFLD and NASH: impact on severity of liver disease and response to treatment. Curr Pharm Des. 2013;19:5219–38. doi: 10.2174/13816128113199990381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Farrell GC, Haczeyni F, Chitturi S. Pathogenesis of NASH: how metabolic complications of overnutrition favour lipotoxicity and pro-inflammatory fatty liver disease. Adv Exp Med Biol. 2018;106:119–44. doi: 10.1007/978-981-10-8684-7_3. [DOI] [PubMed] [Google Scholar]
- 62.Friedman SL. Liver fibrosis in 2012: convergent pathways that cause hepatic fibrosis in NASH. Nat Rev Gastroenterol Hepatol. 2013;10:71–2. doi: 10.1038/nrgastro.2012.256. [DOI] [PubMed] [Google Scholar]
- 63.Handa P, Morgan-Stevenson V, Maliken BD, Nelson JE, Washington S, Westerman M, Yeh MM, Kowdley KV. Iron overload results in hepatic oxidative stress, immune cell activation, and hepatocellular ballooning injury, leading to nonalcoholic steatohepatitis in genetically obese mice. Am J Physiol Gastrointest Liver Physiol. 2016;310:G117–27. doi: 10.1152/ajpgi.00246.2015. [DOI] [PubMed] [Google Scholar]
- 64.Kashi MR, Torres DM, Harrison SA. Current and emerging therapies in nonalcoholic fatty liver disease. Semin Liver Dis. 2008;28:396–406. doi: 10.1055/s-0028-1091984. [DOI] [PubMed] [Google Scholar]
- 65.Suguro R, Pang XC, Yuan ZW, Chen SY, Zhu YZ, Xie Y. Combinational applicaton of silybin and tangeretin attenuates the progression of non-alcoholic steatohepatitis (NASH) in mice via modulating lipid metabolism. Pharmacol Res. 2019;151:104519. doi: 10.1016/j.phrs.2019.104519. [DOI] [PubMed] [Google Scholar]
- 66.Szabo G, Iracheta-Vellve A. Inflammasome activation in the liver: focus on alcoholic and non-alcoholic steatohepatitis. Clin Res Hepatol Gastroenterol. 2015;39:S18–23. doi: 10.1016/j.clinre.2015.06.012. [DOI] [PubMed] [Google Scholar]
- 67.Wan X, Xu C, Yu C, Li Y. Role of NLRP3 inflammasome in the progression of NAFLD to NASH. Can J Gastroenterol Hepatol. 2016;2016:6489012. doi: 10.1155/2016/6489012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chalasani N, Abdelmalek MF, Garcia-Tsao G, Vuppalanchi R, Alkhouri N, Rinella M, Noureddin M, Pyko M, Shiffman M, Sanyal A, Allgood A, Shlevin H, Horton R, Zomer E, Irish W, Goodman Z, Harrison SA, Traber PG. Effects of belapectin, an inhibitor of galectin-3, in patients With nonalcoholic steatohepatitis with cirrhosis and portal hypertension. Gastroenterology. 2020;158:1334–45. doi: 10.1053/j.gastro.2019.11.296. [DOI] [PubMed] [Google Scholar]
- 69.Neuschwander-Tetri BA. Pharmacologic management of nonalcoholic wteatohepatitis. Gastroenterol Hepatol. 2018;14:582–9. [PMC free article] [PubMed] [Google Scholar]
- 70.Neuschwander-Tetri BA. Therapeutic landscape for NAFLD in 2020. Gastroenterology. 2020;158:1984–98. doi: 10.1053/j.gastro.2020.01.051. [DOI] [PubMed] [Google Scholar]
- 71.Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2019;394:2184–96. doi: 10.1016/S0140-6736(19)33041-7. [DOI] [PubMed] [Google Scholar]
- 72.Wree A, Broderick L, Canbay A, Hoffman HM, Feldstein AE. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat Rev Gastroenterol Hepatol. 2013;10:627–36. doi: 10.1038/nrgastro.2013.149. [DOI] [PubMed] [Google Scholar]
- 73.Wree A, McGeough MD, Peña CA, Schlattjan M, Li H, Inzaugarat ME, et al. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J Mol Med. 2001; 92:1069–82. [DOI] [PMC free article] [PubMed]
- 74.Xia MF, Bian H, Gao X. NAFLD and diabetes: two sides of the same coin? Rationale for gene-based personalized NAFLD treatment. Front Pharmacol. 2019;10:877. doi: 10.3389/fphar.2019.00877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dibba P, Li AA, Cholankeril G, Iqbal U, Gadiparthi C, Khan MA, Kim D, et al. The role of cannabinoids in thesetting of cirrhosis. Medicines. 2018;5:E52. doi: 10.3390/medicines5020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Muriel P, López-Sánchez P, Ramos-Tovar E. Fructose and theliver. Int J Mol Sci. 2021;22:6969. doi: 10.3390/ijms22136969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Boland ML, Oró D, Tølbøl KS, Thrane ST, Nielsen JC, Cohen TS, et al. Towards a standard diet-induced and biopsy-confirmed mouse model of non-alcoholic steatohepatitis: Impact of dietary fat source. World J Gastroenterol. 2019;25:4904–20. doi: 10.3748/wjg.v25.i33.4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pourcet B, Duez H. Circadian control of inflammasome pathways: implications for circadian medicine. Front Immunol. 2020;11:1630. doi: 10.3389/fimmu.2020.01630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol. 2017;66:1037–46. doi: 10.1016/j.jhep.2017.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thomas H. NAFLD: a critical role for the NLRP3 inflammasome in NASH. Nat Rev Gastroenterol Hepatol. 2017;14:197. doi: 10.1038/nrgastro.2017.21. [DOI] [PubMed] [Google Scholar]
- 81.Balsalobre A, Damiola F, Schibler U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell. 1998;93:29–37. doi: 10.1016/S0092-8674(00)81199-X. [DOI] [PubMed] [Google Scholar]
- 82.Ramakrishnan SN, Muscat GE. The orphan Rev-erb nuclear receptors: a link between metabolism, circadian rhythm and inflammation. Nucl Recept Signal. 2006;4:e009. doi: 10.1621/nrs.04009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yin L, Wu N, Curtin JC, Qatanani M, Szwergold NR, Reid RA, et al. Rev-erbalpha, a heme sensor that coordinates metabolic and circadian pathways. Science. 2007;318:1786–9. doi: 10.1126/science.1150179. [DOI] [PubMed] [Google Scholar]
- 84.Fontaine C, Staels B. The orphan nuclear receptor Rev-erbalpha: a transcriptional link between circadian rhythmicity and cardiometabolic disease. Curr Opin Lipidol. 2007;18:141–6. doi: 10.1097/MOL.0b013e3280464ef6. [DOI] [PubMed] [Google Scholar]
- 85.Teboul M, Guillaumond F, Gréchez-Cassiau A, Delaunay F. The nuclear hormone receptor family round the clock. Mol Endocrinol. 2008;22:2573–82. doi: 10.1210/me.2007-0521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Griffett K, Burris TP. The mammalian clock and chronopharmacology. Bioorg Med Chem Lett. 2013;23:1929–34. doi: 10.1016/j.bmcl.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Adan A, Archer SN, Hidalgo MP, Di Milia L, Natale V, Randler C. Circadian typology: a comprehensive review. Chronobiol Int. 2012;29:1153–75. doi: 10.3109/07420528.2012.719971. [DOI] [PubMed] [Google Scholar]
- 88.Feng D, Liu T, Sun Z, Bugge A, Mullican SE, Alenghat T, et al. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science. 2011;331:1315–9. doi: 10.1126/science.1198125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kovač U, Skubic C, Bohinc L, Rozman D, Režen T. Oxysterols and gastrointestinal cancers around the clock. Front Endocrinol. 2019;10:483. doi: 10.3389/fendo.2019.00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Crespo M, Leiva M, Sabio G. Circadian clock and liver cancer. Cancers. 2021;13:3631. doi: 10.3390/cancers13143631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li D, Ikaga R, Ogawa H, Yamazaki T. Different expressions of clock genes in fatty liver induced by high-sucrose and high-fat diets. Chronobiol Int. 2021;38:762–78. doi: 10.1080/07420528.2021.1889579. [DOI] [PubMed] [Google Scholar]
- 92.Marjot T, Ray DW, Williams FR, Tomlinson JW, Armstrong MJ. Sleep and liver disease: a bidirectional relationship. Lancet Gastroenterol Hepatol. 2021;6:850–63. doi: 10.1016/S2468-1253(21)00169-2. [DOI] [PubMed] [Google Scholar]
- 93.Vernia F, Di Ruscio M, Ciccone A, Viscido A, Frieri G, Stefanelli G, et al. Sleep disorders related to nutrition and digestive diseases: a neglected clinical condition. Int J Med Sci. 2021;18:593–603. doi: 10.7150/ijms.45512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.McCommis KS, Butler AA. The importance of keeping time in the liver. Endocrinology. 2021;162:bqaa230. doi: 10.1210/endocr/bqaa230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jialal I, Patel A, Devaraj S, Adams-Huet B. Metabolites that activate the inflammasome in nascent metabolic syndrome. J Diabetes Complicat. 2021;5:107836. doi: 10.1016/j.jdiacomp.2020.107836. [DOI] [PubMed] [Google Scholar]
- 96.Zhang Y, Fang B, Emmett MJ, Damle M, Sun Z, Feng D, et al. Discrete functions of nuclear receptor Rev-erbα couple metabolism to the clock. Science. 2015;348:1488–92. doi: 10.1126/science.aab3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pourcet B, Duez H. Nuclear receptors in the control of the NLRP3 Inflammasome pathway. Front Endocrinol. 2021;12:630536. doi: 10.3389/fendo.2021.630536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lee YA, Friedman SL. Inflammatory and fibrotic mechanisms in NAFLD-Implications for new treatment strategies. J Intern Med. 2022;291:11–31. doi: 10.1111/joim.13380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chackelevicius CM, Gambaro SE, Tiribelli C, Rosso N. Th17 involvement in nonalcoholic fatty liver disease progression to non-alcoholic steatohepatitis. World J Gastroenterol. 2016;22:9096–103. doi: 10.3748/wjg.v22.i41.9096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li N, Yamamoto G, Fuji H, Kisseleva T. Interleukin-17 in liver disease pathogenesis. Semin Liver Dis. 2021;41:507–15. doi: 10.1055/s-0041-1730926. [DOI] [PubMed] [Google Scholar]
- 101.Van Herck MA, Weyler J, Kwanten WJ, Dirinck EL, De Winter BY, Francque SM, et al. The differential roles of T cells innon-alcoholic fatty liver disease andobesity. Front Immunol. 2019;10:82. doi: 10.3389/fimmu.2019.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24:908–22. doi: 10.1038/s41591-018-0104-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Neuschwander-Tetri BA, Ford DA, Acharya S, Gilkey G, Basaranoglu M, Tetri LH, et al. Dietary trans-fatty acid induced NASH is normalized following loss of trans-fatty acids from hepatic lipid pools. Lipids. 2012;47:941–50. doi: 10.1007/s11745-012-3709-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Neuschwander-Tetri BA. Carbohydrate intake and nonalcoholic fatty liver disease. Curr Opin Clin Nutr Metab Care. 2013;16:446–52. doi: 10.1097/MCO.0b013e328361c4d1. [DOI] [PubMed] [Google Scholar]
- 105.Neuschwander-Tetri BA, Wang DQ. Excess cholesterol and fat in the diet: a dangerous liaison for energy expenditure and the liver. Hepatology. 2013;57:7–9. doi: 10.1002/hep.25953. [DOI] [PubMed] [Google Scholar]
- 106.Neuschwander-Tetri BA. Too much sugar-the not-so-sweet reality of its impact on our health. Hepatology. 2020;71:377–9. doi: 10.1002/hep.30910. [DOI] [PubMed] [Google Scholar]
- 107.Trevaskis JL, Griffin PS, Wittmer C, Neuschwander-Tetri BA, Brunt EM, Dolman CS, et al. Glucagon-like peptide-1 receptor agonism improves metabolic, biochemical, and histopathological indices of nonalcoholic steatohepatitis in mice. Am J Physiol Gastrointest Liver Physiol. 2012;302:G762–72. doi: 10.1152/ajpgi.00476.2011. [DOI] [PubMed] [Google Scholar]
- 108.Amir M, Chaudhari S, Wang R, Campbell S, Mosure SA, Chopp LB, et al. REV-ERBα regulates TH17 cell development and autoimmunity. Cell Rep. 2018;25:3733–49. doi: 10.1016/j.celrep.2018.11.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yu X, Rollins D, Ruhn KA, Stubblefield JJ, Green CB, Kashiwada M, et al. TH17 cell differentiation is regulated by the circadian clock. Science. 2013;342:727–30. doi: 10.1126/science.1243884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hemmers S, Rudensky AY. The cell-intrinsic circadian clock is dispensable for lymphocyte differentiation and function. Cell Rep. 2015;11:1339–49. doi: 10.1016/j.celrep.2015.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]