

Recently in Cell Research, Zhang et al. demonstrate that hydrogen voltage-gated channel 1 (Hv1) is expressed in sensory neurons and regulates the production of reactive oxygen species. Furthermore, inhibition of Hv1 with YHV98-4 reduces inflammation and inflammatory pain.
Hydrogen voltage-gated channel 1 (Hv1) was identified as a voltage-gated proton channel in rodents and humans.1,2 It is activated by membrane depolarization. The channel’s voltage-sensitivity regulation depends on the pH difference between the intracellular and extracellular fluids: the greater the pH difference, the stronger the depolarization required to activate Hv1. This pH-dependent threshold shift results in a unique property of Hv1, i.e., the channel only opens when the membrane potential exceeds the H+ equilibrium potential. Furthermore, protons are only transported outward across the channel, leading to acid extrusion from cells.2 In immune cells, Hv1 regulates the production of reactive oxygen species (ROS) through functional coupling with NADPH oxidase (NOX).3 In mammalian nervous systems, Hv1 is expressed in microglia and contributes to ischemic brain damage.4 Hv1 has also been implicated in the pathogenesis of neuropathic pain by regulating microglial production of ROS and microglia-astrocyte communication.5 In a recent paper of Cell Research, Zhang et al.6 revealed the expression and function of Hv1 in primary sensory neurons. Importantly, this study also identified a novel Hv1 inhibitor that has analgesic and anti-inflammatory actions in mice.6
Using RNAscope in situ hybridization, Zhang et al. showed wide expression of Hvcn1 mRNA in 60% of sensory neurons in dorsal root ganglia (DRG), including small-sized neurons (presumably nociceptors) and medium-large sized neurons. Interestingly, Hv1 expression was upregulated by acute and persistent inflammation, inflammatory cytokines, NOX activators, and nerve injury. Thus, Hv1 expression in sensory neurons may serve as a marker for inflammatory insult and nociceptor depolarization.
To investigate the role of Hv1 in pain regulation, Zhang et al. employed multiple approaches, such as genetic approaches including generation of Hvcn1-knockout mice and virus-mediated knockdown of Hvcn1 in DRG neurons, behavioral testing of pain in different animal models, and electrophysiological and biochemical approaches to investigate Hv1-mediated currents and intracellular signaling. Genetic deletion of Hcvn1 did not affect baseline pain sensitivity, as mechanical and thermal sensitivities are comparable between wild-type and Hcvn1-knockout mice. However, complete Freund’s adjuvant (CFA)-induced inflammatory pain, which causes mechanical and thermal hypersensitivity, was diminished in knockout mice.
Notably, the authors identified a novel Hv1 inhibitor, YHV98-4, using computer simulations and virtual screening of a chemical library. YHV98-4 specifically inhibited Hv1 with a half-maximal inhibitory concentration of 1 µM without inhibiting other ion channels. YHV98-4 at 20 µM could inhibit Hv1-mediated currents in DRG neurons from both naïve mice and CFA-treated mice. As expected, the magnitude of current inhibition was dependent on the intracellular pH (pHi). Hv1 plays an active role in modifying pHi in nociceptive neurons following inflammation, and accordingly, intracellular alkalinization in DRG is associated with pain. Akin to immune cells and microglia, inhibition of Hv1 reduced ROS production in sensory neurons following inflammation or neuronal hyperactivation with depolarization.
To demonstrate the translational clinical potential of Hv1 inhibition, Zhang et al. assessed the pharmacokinetic profiles of YHV98-4. After a single intraperitoneal injection of YHV98-4 (10 mg/kg), the plasma concentration of this compound reached 4 µg/mL with a half-life of 1.75 h. Systemic injection of YHV98-4 caused rapid analgesia in mouse models of inflammatory and neuropathic pain within 30–60 min following administration. The analgesic potency of YHV98-4 for inflammatory pain inhibition is similar to that of diclofenac sodium, a nonsteroidal anti-inflammatory drug (NSAID) used to treat mild-to-moderate clinical pain, swelling (inflammation), and joint stiffness caused by arthritis. It is noteworthy that YHV98-4 does not change baseline pain thresholds. The authors also found that this inhibitor had no analgesic effect in Hvcn1- knockout mice or in mice with Hvcn1 knockdown in sensory neurons. Furthermore, they found that ROS scavenger alleviated CFA-induced mechanical and thermal allodynia in wild-type mice but not in mice with Hv1 knockdown in sensory neurons. Together, these results suggest that YHV98-4 attenuates inflammatory pain via inhibition of Hv1 and ROS production.
Mechanistically, Zhang et al. revealed a signaling pathway by which Hv1 promotes pain via inhibition of the phosphatase Src homology 2 domain-containing tyrosine phosphatase 1 (SHP-1) in sensory neurons (Fig. 1). Recent studies have highlighted an important role of SHP-1 in sensory neurons for the control of pain.7,8 The Hv1/SHP-1 signaling pathway was initially discovered in immune cells, in which proton channels in B cells regulate the generation of ROS and oxidation of SHP-1, leading to altered AKT signaling.9 In this study, the authors provided several lines of evidence to support the involvement of the neuronal SHP-1/AKT pathway in Hv1-mediated pain control. Firstly, CFA induced the downregulation of SHP-1 phosphorylation in DRG neurons, which could be reversed by pharmacological and genetic inhibition of Hv1. Secondly, treatment with SHP-1 inhibitor abolished the analgesia caused by Hv1 inhibition, and the expression of SHP-1 was increased in Hvcn1-knockout mice. Finally, inhibition of neuronal Hv1 also reversed inflammation-induced upregulations of the PI3K/AKT pathway and pronociceptive inflammatory cytokines (e.g., CXCL1). Therefore, inhibition of neuronal Hv1 can restore homeostatic function of the SHP-1-PI3K/AKT-CXCL1 signaling pathway disrupted by tissue injury and inflammation (Fig. 1).
Fig. 1. Hv1 regulates inflammation and pain via modulation of ROS production and inhibition of SHP-1 signaling in DRG neurons.
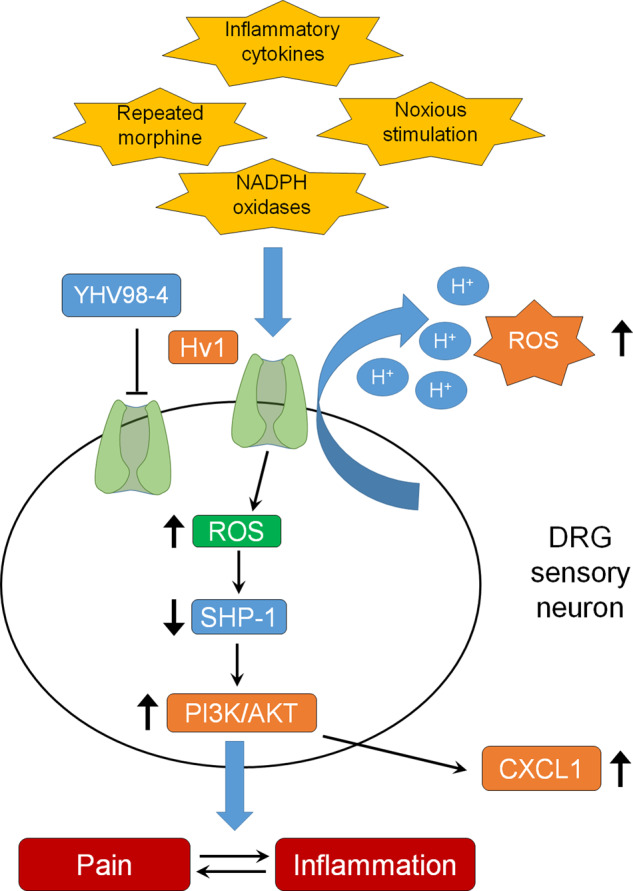
Hv1 can be upregulated by inflammatory cytokines, activation of NADPH oxidases, noxious stimulation, and repeated morphine treatment.
It is well established that repeated administration of opioids such as morphine induces antinociceptive tolerance and hyperalgesia. Interestingly, Hv1 inhibition prevented the development of morphine-induced hyperalgesia and tolerance. The authors also observed that production of ROS in DRG increased following chronic morphine exposure, and this increase was blocked by pharmacological inhibition of Hv1 or knockdown of Hv1 in sensory neurons.
In summary, this study revealed a novel role of Hv1 in sensory neurons in regulating ROS production. The research team identified a novel Hv1 inhibitor YHV98-4, which showed analgesic effects in several animal models of pain, including inflammatory pain, neuropathic pain, and opioid-induced hyperalgesia. It will be of great interest to test YHV98-4 in cancer pain conditions, as cancer pain is typically treated with opioids and Hv1 is itself implicated in cancer progression.10 The inhibitor may exert dual effects of tumor suppression and pain relief. Future studies are helpful to study possible Hv1 interaction with opioid receptors in sensory neurons. Hv1 is a notable novel target for pain control, but the analgesic efficacy of YHV98-4 can be further improved. The benefits of the current Hv1 inhibitor vs NSAIDs and antioxidants will need to be investigated. Furthermore, immune cells and microglia express Hv1, and the half-life of YHV98-4 in the brain is longer than in plasma, suggesting that the brain may be an important target of this compound. It would be also interesting to explore whether targeting central Hv1 proton channels may alleviate CNS neurological disorders such as stroke4 in the future.
References
- 1.Ramsey IS, Moran MM, Chong JA, Clapham DE. Nature. 2006;440:1213–1216. doi: 10.1038/nature04700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sasaki M, Takagi M, Okamura Y. Science. 2006;312:589–592. doi: 10.1126/science.1122352. [DOI] [PubMed] [Google Scholar]
- 3.Demaurex N, El Chemaly A. J. Physiol. 2010;588:4659–4665. doi: 10.1113/jphysiol.2010.194225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu LJ, et al. Nat. Neurosci. 2012;15:565–573. doi: 10.1038/nn.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peng J, et al. Mol. Brain. 2021;14:99. doi: 10.1186/s13041-021-00812-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Q, et al. Cell Res. 2022 doi: 10.1038/s41422-022-00616-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiao X, et al. Pain. 2015;156:597–608. doi: 10.1097/01.j.pain.0000460351.30707.c4. [DOI] [PubMed] [Google Scholar]
- 8.Chen G, et al. Nat. Neurosci. 2017;20:917–926. doi: 10.1038/nn.4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Capasso M, et al. Nat. Immunol. 2010;11:265–272. doi: 10.1038/ni.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Li SJ, Wu X, Che Y, Li Q. J. Biol. Chem. 2012;287:13877–13888. doi: 10.1074/jbc.M112.345280. [DOI] [PMC free article] [PubMed] [Google Scholar]