

Abstract
Fanconi anemia, telomeropathies and ribosomopathies are members of the inherited bone marrow failure syndromes (IBMFS), rare genetic disorders that lead to failure of hematopoiesis, developmental abnormalities, and cancer predisposition. While each disorder is caused by different genetic defects in seemingly disparate processes of DNA repair, telomere maintenance, or ribosome biogenesis, they appear to lead to a common pathway characterized by premature senescence of hematopoietic stem cells. Here we review the experimental data on senescence and inflammation underlying marrow failure and malignant transformation. We conclude with a critical assessment of current and future therapies targeting these pathways in IBMFS patients.
Introduction
The classical inherited bone marrow failure syndromes (IBMFS) comprise a group of rare disorders caused by germline mutations, resulting in hematopoietic stem cell failure and a predisposition to cancer. Although different gene pathways are affected, these disorders have many strikingly similar clinical manifestations: progressive BMF due to altered cell senescence and strong cancer predisposition, particularly myeloid neoplasms. In this review, we will examine factors underlying cell senescence and malignant transformation in classical IBMFS patients, emphasizing the commonality between the different IBMFS.
Clinical Manifestations: BMF and cancer susceptibility
Fanconi anemia (FA) is the commonest IBMFS and is due to primarily biallelic (except for FANCB [X-linked] and FANCR [autosomal dominant]) germline defects in the DNA repair pathway;1 it may also present with limb abnormalities, café-au-lait spots, and short stature. Telomeropathies are due to monoallelic (TERT, TERC, TINF2), biallelic (RTEL1, CTC1), X-linked (DKC1), or even triallelic defects in telomere maintenance-related genes, and commonly involve other organs leading to liver cirrhosis and lung fibrosis.2,3 Dyskeratosis congenita (DC) is a severe form of telomere disease, usually presenting in the first decade of life, and marrow failure is accompanied by mucocutaneous abnormalities (oral leucoplakia, nail dystrophy, and skin changes). The ribosomopathies include Shwachman Diamond syndrome (SDS) and Diamond-Blackfan anemia (DBA).4 SDS is due to biallelic mutations in the SBDS gene and may present with isolated neutropenia, with a risk of progression to pancytopenia, and with pancreatic insufficiency and bony abnormalities. DBA is due to germline mutations in the ribosomal protein or RP genes and presents primarily as a pure red cell aplasia. However, some cases may eventually progress to pancytopenia, and DBA may associate with limb abnormalities and cardiac defects.
Marrow failure is the most typical clinical manifestation of all the IBMFS. In FA, the cumulative lifetime incidence of BMF is approximately 50%5. In DC, >94% of patients develop BMF by age 40 years, though this is less frequent in the milder phenotype adult-onset telomere diseases6. In both SDS and DBA, marrow failure occurs early in life. In SDS, neutropenia is the first and commonest cytopenia, though normocytic anemia is present in up to 80%, and patients may progress to pancytopenia and severe marrow failure. In DBA, most patients develop severe anemia prior to two years of age.7,8 The only curative therapy for severe marrow failure is hematopoietic stem cell transplant (HSCT), though steroid therapy is used in DBA and androgen therapy in FA and telomeropathies as a way to mitigate cytopenias.
All the classical IBMFS have an increased risk of solid and hematologic malignancies.9 Patients with FA incur the highest risk, followed by those with telomeropathies. In the National Cancer Institute long-term cohort, myelodysplastic syndrome (MDS) was the most common malignancy in both FA (50% CI by the age of 50) and telomeropathy (20% CI by the age of 50)10. In FA, 3% developed acute leukemia, and 12% developed solid cancers, with squamous cell carcinoma of the head, neck, or vulva and brain tumors predominating. Patients with telomeropathy had a similar risk of leukemia as compared to FA patients but a lower risk of solid malignancy at 7%. Increased cancer risk has also been described in the ribosomopathies, DBA and SDS. DBA patients have an increased risk of MDS/AML, colon cancer, and osteogenic sarcoma, whereas SDS patients appear to be at increased risk of MDS/AML but not solid malignancy.11
Cell senescence and bone marrow failure
Cell senescence underlies clinical bone marrow failure in these disorders. The classical IBMFS are caused by defects in gene products involved in seemingly disparate molecular housekeeping processes such as DNA repair, chromosome integrity, telomere maintenance, and ribosome biogenesis. Telomeropathy and FA are associated with molecular DNA damage and premature aging, whereas ribosomopathy disorders such as DBA and SDS primarily arise from defects in ribosome biogenesis. Despite these apparent differences, these syndromes bear biological and clinical similarities that suggest a common pathophysiologic mechanism involving cellular senescence. In line with this idea, cells from FA12, telomeropathy13, or ribosomopathy14 disorders may exhibit senescence features. These hallmarks15,16are generally recognized to include (1) enlarged and flattened cell morphology; (2) increased expression of cyclin-dependent kinase (CDK) inhibitors including p16 and p21; (3) increased senescence-associated β-galactosidase activity; (4) presence of DNA segments with chromatin alterations reinforcing senescence or DNA SCARS, including foci of the variant histone γ-H2AX; (5) persistent DNA damage with the arrest of proliferation; (6) senescence-associated secretory phenotype or SASP.
Experimental data from one of us are shown in Figure 1. By multicolor flow cytometry analysis, we found increased levels of senescence-associated β-galactosidase, γ-H2AX, and p16 in early-progenitor bone marrow cells isolated from SDS patients (JML, unpublished data).
Figure 1.
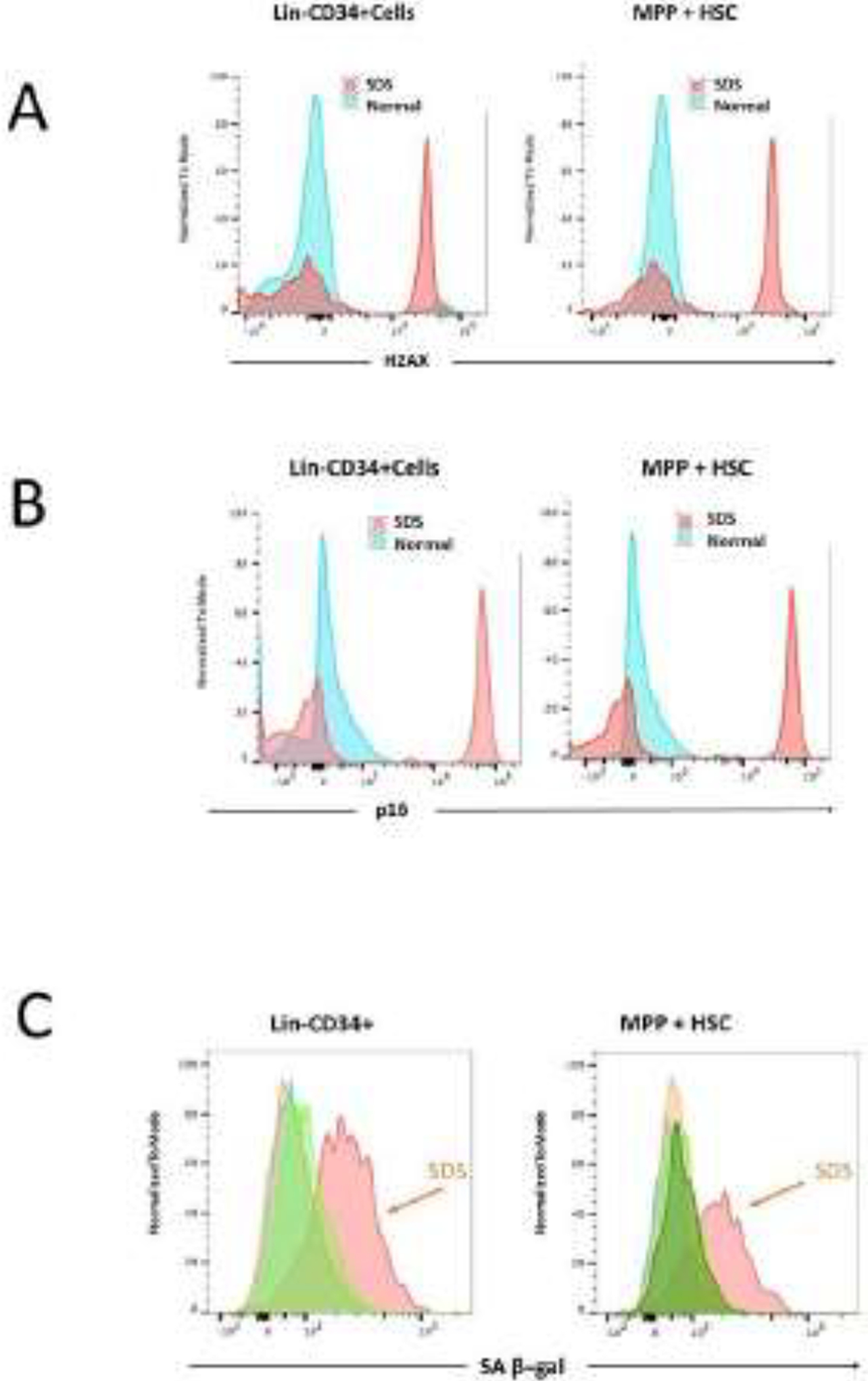
By multicolor flow cytometry analysis, we found increased levels of γ-H2AX (panel A), p16 (panel B), and senescence-associated β-galactosidase (panel C) in early-progenitor bone marrow cells isolated from an SDS patient. Progenitor cells were labeled as lineage-negative, CD34 positive (Lin-CD34+) or multipotent progenitors + hematopoietic stem cells (MPP + HSC).
Cell senescence: definition and association with the DNA Damage Response (DDR)
Telomeres are nucleoprotein structures protecting the ends of chromosomes by ensuring they are not treated as damaged DNA. Due to DNA polymerase’s inability to fully replicate the 3’ ends of chromosomes (the “end-replication problem”), telomeres shorten with each cell division. Replicative senescence was first defined as a state of total cell cycle arrest after non-transformed human fibroblasts undergoing successive cell divisions and telomere attrition reached the limit to their proliferative capacity17. In consequence, senescence has historically been confused with cell cycle arrest, growth inhibition, or aging, whereas we now understand that it encompasses a broad-ranging program triggered by multiple stressors. For the purposes of this critical review, we define senescence as a non-proliferative state in sub-lethally damaged cells, characterized by nearly irreversible arrest but metabolically active and paradoxically associated with the proliferation-like activity of mTORC1 (mechanistic target of rapamycin complex 1). The growth supporting and senescence-associated secretory phenotypes may promote inflammation and tumorigenesis.
Many types of senescence are now recognized: replicative senescence (RS), stress-induced senescence (SIS), and oncogene-induced senescence (OIS)14–16. All three major types may lead to persistent activation of the DNA damage response (DDR), which is a pathway that detects DNA lesions, signals their presence, and promotes their repair15,16. The DDR is characterized by increased deposition of the variant histone H2AX, γ-H2AX, phosphorylated at serine-139. Key DDR-signaling components include the protein kinases ATM (ataxia telangiectasia mutated) and ATR (ataxia telangiectasia and Rad3 related), which are recruited to and activated by DNA double-stranded breaks (DSBs) and single-stranded DNA, respectively. ATM/ATR along with DNA-dependent protein kinase (DNA-PK) lead to phosphorylation of ser139 of γ-H2AX, on chromatin bordering DSB sites. The end result of ATM/ATR signaling is to reduce CDK activity by various mechanisms and, together with the checkpoint kinases CHK1 and CHK2, activate the p53/p21 axis and halt cell proliferation.
Pathways to p53-mediated growth arrest and connection to nucleolar stress and ribosome biogenesis
As we have seen, senescent cell growth is arrested by activation of the p53/p21 and p16 axes, with both leading to hypophosphorylation of the retinoblastoma tumor suppressor Rb and exit from the cell cycle15. Concurrent with this growth arrest and genotoxic damage, senescent cells undergo global translation repression and inhibition of ribosomal RNA (rRNA) processing and ribosome biogenesis14. In the nucleolus, hundreds of trans-acting factors are normally involved in the complex and energy-intensive process of ribosome biogenesis14. The eukaryotic 80S ribosome is composed of the 40S small subunit (containing 18S rRNA and 33 RPS proteins) and 60S large subunit (containing the 28S, 5.8S, 5S rRNAs along with 47 RPL proteins).
Recently, proteomic studies have identified nucleolar proteins that function in both the DDR and in ribosome biogenesis18, suggesting that the nucleolus acts as a sensor of cellular stress and point of convergence between DNA damage and translational control/ribosome biogenesis. In normal cells, basal p53 activity is suppressed by the E3 ubiquitin ligase HDM2, which ubiquitinates and targets p53 for proteasomal degradation. When cells become senescent, they exhibit nucleolar stress14, with dysregulation of ribosome biogenesis leading to the release of free 5S ribonucleoprotein RNP particles that bind and inhibit HDM2, thus stabilizing and activating p53. This mechanism of p53 activation has been termed the impaired ribosome biogenesis checkpoint (IRBC).19
Cell senescence in FA, telomeropathies, and ribosomopathies can now be related on a molecular level to p53-mediated growth arrest by this convergence between the DDR and IRBC. Presumably, FA and telomeropathy defects activate the DDR, whereas ribosomopathies trigger the IRBC, but there may be significant overlaps in the pathophysiology (Figure 2).
Figure 2.
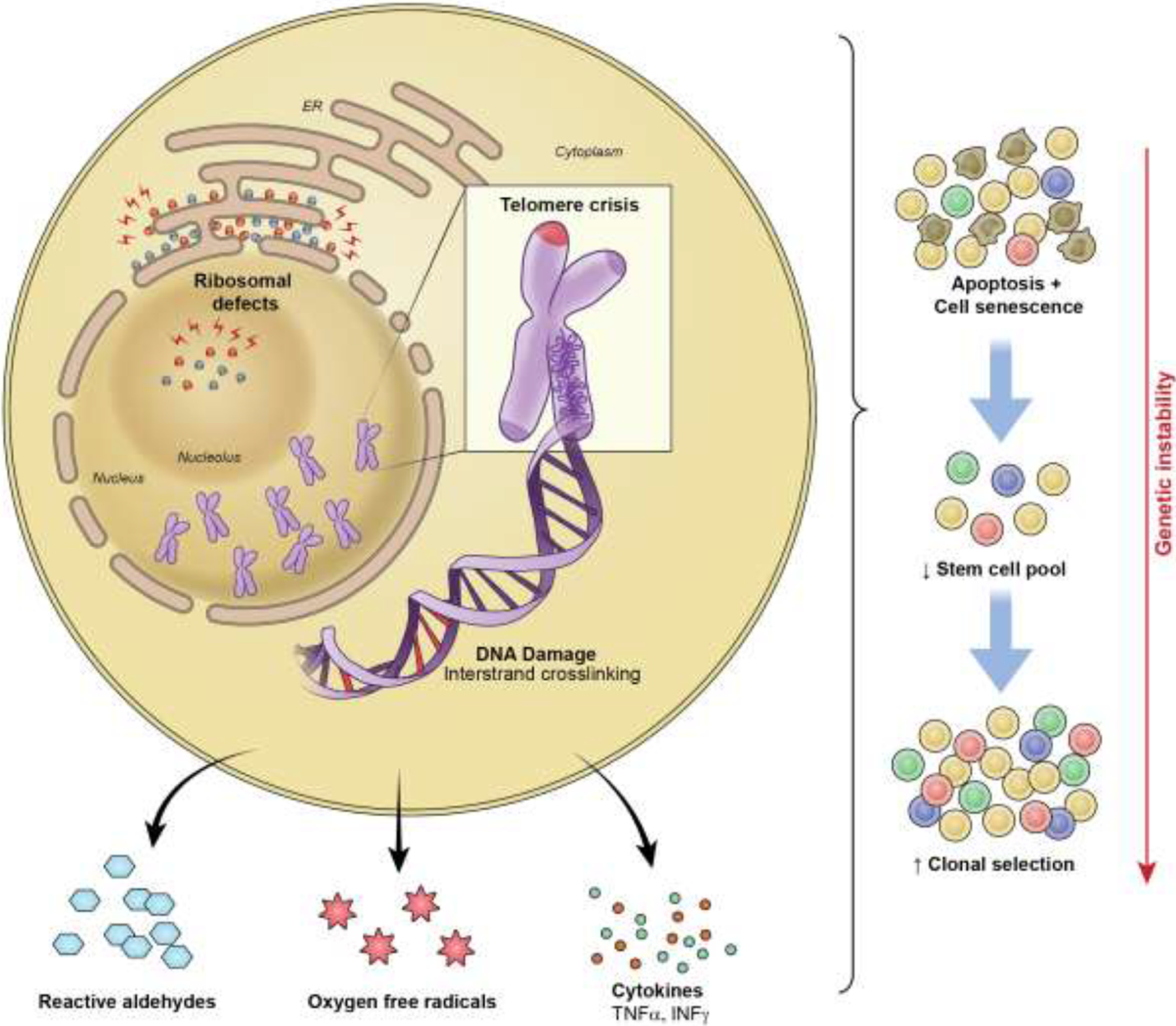
Cell senescence in FA, telomeropathies, and ribosomopathies are related on a molecular level to p53-mediated growth arrest with convergence between the DNA damage response and the impaired ribosome biogenesis checkpoint. Ongoing DNA damage chronically activates safety pathways to halt the proliferation of damaged cells and leads to an increase in cell apoptosis and senescence. Due to a depleted stem cell pool and consequent increased replicative stress, a selective advantage for mutant clones may occur.
The central paradox of senescence: growth arrest versus pro-survival pathways dependent upon mTORC1
Senescence is characterized by a highly stable state of cell cycle arrest, triggered by different stresses that converge on p53 and lead to global translation repression. Paradoxically, however, senescent cells also enlarge in size and maintain an active metabolic state with selective increases in the translation of specific proteins such as inflammatory proteins, as part of the so-called senescence-associated secretory phenotype or SASP20.
Driving cell metabolism and growth are the two mechanistic target of rapamycin (mTOR) complexes (mTOR complex-1 or mTORC1 and complex-2 or mTORC2)14. mTORC1 promotes protein synthesis by phosphorylation of p70S6 kinase 1 (S6K1) and the eIF4E-binding protein 4E-BP, both increasing translation initiation by promoting the formation of the pre-initiation complex and cap-dependent translation. A major consequence of this senescent translation program is the SASP20, which leads to secretion of cytokines (including IL-1a, IL-6, IL-8), chemokines (such as CCL2), and metalloproteinases (such as MMP-1 and MMP-3). Recent research has even suggested that activating the mTORC1 pathway may favor senescence over apoptosis in zebrafish undergoing telomere shortening21.
Mouse model of telomerase deficiency: the importance of mTOR for survival and implications
Genetically engineered mice deficient in telomerase (Terc−/−) may mimic aspects of the human telomeropathy disorders.
Abnormal metabolism is characteristic of telomeropathy. Although liver cirrhosis is commonly diagnosed in patients with telomere diseases, non-alcoholic steatohepatitis (NASH) also is observed in telomeropathy patients.22 In Terc and Tert “knockout” murine models fed with high-fat diet, hepatocytes display abnormalities in the tricarboxylic acid cycle and accumulate fat, indicating a dysfunctional metabolism upon dietary stress.23 These metabolic changes along with inflammation appear to contribute to liver dysfunction in telomeropathy.
Second-generation Terc−/− mice demonstrated accelerated telomere shortening and died young from premature aging pathologies such as intestinal atrophy.24 These mutant mice with short telomeres exhibited marked upregulation of the mTOR pathway and increased levels of phosphorylated ribosomal S6 protein in tissues. Inhibition of mTORC1 by rapamycin or deletion of the S6K1 downstream target of mTORC1 both decreased the survival of the mutant mice, demonstrating the importance of mTOR for survival in the context of critically short telomeres. mTORC1 hyperactivation, which appears to be critical for the survival of cells undergoing premature senescence (as in Terc−/− mice), may also explain cancer development later in the senescence program.
Clinically, patients with FA, telomeropathies, or ribosomopathies all present with bone marrow failure and developmental abnormalities, which mirror the consequences of p53-mediated growth arrest, nucleolar stress, and global translation repression. However, all these disorders also predispose to cancers, both solid and hematologic. Possibly, this cancer proneness reflects anabolic consequences of prolonged mTORC1 activity. In this context, mTORC1 has been reported to be constitutively active in certain AML types and may increase the synthesis of oncogenic proteins through phosphorylation of 4E-BP1, activation of eIF4E, and stimulation of cap-dependent translation.25
Cancer predisposition in the general population due to variants in DNA repair, telomere, and ribosomal protein genes
The FA pathway is thought to play a key role in carcinogenesis, though underlying pathophysiologic mechanisms remain uncertain. Along with the high cancer rates seen in FA patients, germline monoallelic variants in FANC genes, most commonly BRCA1 and BRCA2, have been shown to increase cancer susceptibility in the general population. Germline POT1 (gene in the shelterin complex) variants have also been described in familial CLL, myeloproliferative neoplasms, and other solid cancers.26,27
In addition to the described germline variants, the role of somatic variants and alterations in FANC, and telomere genes, as well as ribosomal proteins in cancer is also emerging via various mechanisms. Somatic mutations, as well as epigenetic silencing, upregulation, and overexpression of FANC genes have been described in several cancer types including AML, pancreatic cancer, head and neck squamous cell cancer, melanoma, and colorectal cancer28–32. When looking at cancer genome data from the Cancer Genome Atlas, FANC gene mutations and copy number variations can be seen in a number of cancer types in patients without a diagnosis of FA33. In addition, somatic mutations in the TERT promoter and in ribosomal proteins have been found in many solid and hematologic malignancies.34–36
Telomere length as a risk for cancer predisposition
Abnormal telomere length – either long or short – may also predispose to cancer. In large population studies, polymorphisms associated with long telomere length were correlated with increased cancer risk, in particular melanoma, lung cancer, and glioma37. In addition, long telomeres have also been seen in patients with hematologic malignancies such as chronic lymphocytic leukemia and B-cell lymphomas38,39. Short telomeres have been described in both MDS and AML, and in aplastic anemia as a risk factor for clonal evolution to myeloid malignancy.40
Impaired DNA damage response: pathophysiology underlying malignant transformation
As discussed, DDR pathways exist to repair DNA; dysregulation of DNA sensing and repair can lead to errors causing genomic instability and potentially drive malignant clones. The FA pathway plays an important role in DNA interstrand crosslink (ICL) repair. ICLs are toxic DNA lesions that inhibit DNA strand separation and thus prevent transcription and replication. FANC proteins, in combination with Fanconi anemia associated proteins (FAAP) act as a primary cell mechanism of recognizing and repairing ICLs41. This occurs in combination with other DNA repair pathways including the nucleotide excision repair pathway, translation synthesis, and homologous recombination.
Ongoing DNA damage chronically activates safety pathways to halt the proliferation of damaged cells and leads to an increase in cell apoptosis and senescence. Due to a depleted stem cell pool and consequent increased replicative stress, a selective advantage for mutant clones may occur. Failure of the G2 DNA damage checkpoint arrest during cell division, termed “attenuation” has been associated with lower expression of CHK1 and p53, resulting in protection from cell death, and a milder marrow failure phenotype but persistent risk of progression to MDS or AML42. As discussed, ribosome defects may result in increased ribosome proteins 5 and 11, which induce p53 activity by sequestering HDM2, a regulator of p5343.
Short telomeres lead to genomic instability
Telomeres are doubled-stranded repeat “caps” that ensure the ends of chromosomes do not activate the DDR due to being mistaken as DNA damage. The DDR is directly suppressed by shelterin, a complex of proteins that bind to and protect telomeres from DNA repair mechanisms. This occurs by preventing the activation of ATM and ATR, and the non-homologous end joining pathway (NHEJ), the primary repair mechanism for double-strand breaks in DNA. If cell senescence signaling induced by short telomeres is overridden and cell proliferation continues, telomeres may become excessively short and dysfunctional (termed “telomere crisis”) and may be recognized as DNA breaks and activate the DDR. Although most cells will undergo apoptosis in this scenario, end-to-end chromosome fusions may occur, resulting in aneuploidy, loss of heterozygosity, and chromosomal translocations;44 those that survive may then propagate and drive genomic instability and malignant transformation.45–47 Telomeropathy patients who develop MDS/AML have short telomeres and display hematopoietic clones bearing myeloid neoplasia-associated mutations.48 Even a single chromosomal dysfunctional telomere in an otherwise stable genome has been shown to fuse with multiple genomic loci, a risk that increases in the absence of adequate p53 checkpoint control49.
The marrow environment contributes to carcinogenesis
Inflammation in the marrow environment may also play an important role in the development of cancer in IBMFS patients. In FA, the cytokines TNF-alpha and interferon-gamma have been shown to play a role in inducing apoptosis50,51. In a murine model of FA, while exposure of TNF-alpha initially inhibited stem cell growth, prolonged exposure led to abnormal cytogenetic clones and ultimately AML52. The clinical association of IBMFS with MDS, which in the absence of a genetic syndrome, would more commonly be seen in the elderly, has given rise to the idea that telomeropathies, FA, and ribosomopathies may be progeroid disorders. Currently, it is not clear how well these disorders mimic the process of normal aging.
Reactive oxygen species are the byproduct of normal cell metabolism and are known to damage DNA, RNA, intracellular proteins, and cellular structures. The FA pathway regulates oxidative response, and FA deficient cells demonstrate high levels of DNA oxidative damage and chromosome breakage53,54. Oxidative stress has been shown to cause telomere shortening and dysfunction in FA cells without provoking a p53 repair response, suggesting a potential mechanism for malignant transformation55. Ribosomal proteins are also involved in oxidative stress response, and mutations in these proteins may result in increased reactive oxygen species that have been demonstrated in ribosomopathies56. Reactive aldehydes are present both as byproducts of metabolism and in the external environment. Mouse model data and ex vivo experiments have shown that DNA damage due to aldehydes may contribute to cancer predisposition, especially in FA deficient cells57. Deficiency of aldehyde dehydrogenase 2 (ALDH2), an enzyme involved in aldehyde metabolism, has been associated with a more severe FA phenotype58.
Somatic genetic rescue may have differing effects on cancer risk
Somatic genetic rescue (SGR), the process whereby a somatic event occurs and offsets the pathogenic germline defect, is a well described phenomenon in IBMFS, particularly Fanconi anemia, where it occurs in up to 30% of patients.59 The prevalence of mosaicism in IBMFS suggests that development of clones that evade cell senescence and apoptosis (and thus evade early marrow failure) likely confers a selective advantage to the mutated cells. In general, SGR appears to be a positive phenomenon, and FA patients with SGR have reported superior long-term outcomes due to milder BMF phenotype and consequently a longer overall survival compared to non-mosaic FA patients60. In telomeropathies, SGR is less frequent among patients with marrow failure, but more common in older patients with pulmonary fibrosis, suggesting that SGR alleviates marrow failure.61 Somatic variants in telomere genes also were associated with reduced risk of MDS/AML62. However, not all SGR is positive. In SDS, somatic mutations in EIF6 and TP53 are commonly present; EIF6 is a clonal marker of SDS that compensates for the germline SBDS defect and ameliorates cell fitness whereas TP53 is linked to increased predisposition to clonal evolution. Interestingly monoallelic TP53 clones were stable for many years and cancer developed when the clones became biallelic, suggesting TP53 also acts as a compensation mechanism until a second hit develops that drives malignant evolution63.
How disease mechanisms may guide novel therapies
In general, cumulative DNA damage and critical telomere erosion drive hematopoietic stem cell (HSC) senescence and apoptosis, explaining marrow failure in FA and telomeropathies. However, as discussed above, the cellular and molecular mechanisms underlying these specific genetic aberrations are complex, involving inflammation, changes in the marrow microenvironment, and intrinsic HSC changes, preventing adequate cell cycle transit and proper hematopoietic differentiation, along with selection of HSC clones accumulating somatic mutations that may impact cell proliferation and malignant evolution. The dissection of these molecular pathways may help to develop novel therapeutic strategies to treat patients with marrow failure.
Three major approaches may be undertaken when developing novel strategies to treat inherited marrow failures: replacement with novel “healthy” hematopoiesis (allogeneic HSC transplant, HSCT), correction of the genetic defect (gene therapy, gene editing), and modulation of cellular pathways that compensate for the impaired cell proliferation and differentiation.
Androgens
That a minority of patients with aplastic anemia respond to treatment with androgens was observed in the early 1950’s64. Laboratory and clinical work demonstrated that these responders were likely to be patients with IBMFS. Approximately 90% of aplastic anemia patients with either FA or telomeropathy respond to androgens. At least two mechanisms may explain why androgens ameliorate hematopoiesis in inherited marrow failure syndromes65. First, steroids with androgenic effect increase the number of red blood cells by directly inducing erythroid progenitors either by increasing erythropoietin (EPO) production in the kidneys or by directly activating the EPO receptor on progenitor cells. Alternatively, The TERT gene promoter contains estrogen-response elements and high estrogen concentrations increase TERT transcription. Telomerase enzymatic activity of human hematopoietic cells is increased by treatment with androgens or estrogens but is abrogated by aromatase inhibitors (that blocks androgen to estrogen conversion) or tamoxifen (an estrogen antagonist)65. In agreement with these laboratory findings, murine models also display telomere elongation in leukocytes when animals are systemically treated with androgens, but elongation associated with androgen treatment does not occur in Tert “knockout” mice, indicating that the mechanism of elongation is directly mediated by telomerase.
These findings prompted a prospective clinical trial with telomere disease patients using danazol, an oral androgen. The use of danazol elongated telomeres of peripheral blood leukocytes, which correlated with hematologic improvement.66 These findings have been confirmed in independent studies.67 Of note, androgen therapy had an impact on hematopoietic clones bearing somatic mutations in the TERT promoter (TERTp). In one study, the TERTp clone diminished during androgen therapy, suggesting that the androgens have a direct impact on the HSC dynamics and mobilization.61 However, the androgen effect on clonal dynamics in FA is not known.
The effects of androgens on other organs, however, are less clear. In the pivotal study using danazol, lung function and liver fibrosis appeared to be stable during the study, although these were not study endpoints.66 In a clinical trial conducted by one of us, lung function was assessed by the diffusion capacity of carbon monoxide (DLCO) and also appeared to be stable in those patients with lung involvement (RTC, unpublished data).
Small molecules
There is a large ongoing effort to identify molecules that can modulate DNA repair and telomere elongation and thus be applied in the clinic. The non-canonical poly(A) polymerase PAPD5 recognizes and targets nascent TERC, the RNA component of telomerase. PAPD5 inhibition increases TERC expression and elongates telomere length in vitro.68 High-throughput screening identified the BCH001 small molecule as a PAPD5 inhibitor. Using an induced pluripotent stem cell model for human telomeropathies, BCH001 was able to restore telomerase activity and elongate telomeres in patient-specific derived cells, suggesting this molecule as a potential application in the clinic21.
Eltrombopag has been successful in the treatment of acquired immune aplastic anemia, but information on its effects on IBMFS is scarce. In a clinical trial setting, one older patient with DBA and pancytopenia was treated with eltrombopag with significant improvement.69 For FA or telomeropathies, there are anedoctal cases only on the use of eltrombopag and in combination with other drugs. Thus, these results are too preliminary to support its use for IBMFS outside of a clinical trial.
Hematopoietic stem cell transplant strategies
Early reports on HSCT for dyskeratosis congenita identified a prohibitive number of lung and liver complications post-transplant70. We now know that liver and lungs are affected in telomeropathies.2 In addition, murine models also suggested that excessive telomere attrition modify the marrow microenvironment and may limit hematopoietic engraftment,71 but larger transplant reports did not observe obvious increased graft failure in human patients.72 However, the high incidence of non-hematopoietic complications as causes of post-transplant mortality limits the use of conventional allogenic HSCT for telomeropathy patients to patients with severe marrow failure and no evidence of other organ involvement. These observations have also prompted novel reduced intensity conditioning regimen strategies aimed at mitigating transplant-related toxicity.
Gene therapy and gene editing
Various somatic mutations occur in patients with IBMFS partially or completely rescuing hematopoiesis, suggesting that gene correction in the HSC may restore hematopoietic function. Gene therapy has been successfully applied in some monogenic hematologic disorders, especially in hemoglobinopathies, rescuing erythropoiesis.73 These are genetic monogenic disorders in which a single gene is involved and, in sickle cell disease, a specific gene variant. For IBMFS, although monogenic for each patient, a myriad of genes are affected in FA, telomeropathies, or ribosomopathies, and multiple gene lesions are observed (point mutations, frameshifts, deletions, splicing variants), complicating gene therapy strategies. Additionally, the expression of genes affected in IBMFS is tightly regulated and overexpression may be associated with cancer development, as mentioned above. Thus, potential gene therapy strategies should be tailored to each case, requiring intricate strategies.
In FA, mutations in the FANCA gene correspond to >60% of cases, facilitating gene therapy design. A pioneer clinical trial used lentiviral-based gene therapy to correct the FANCA gene in non-conditioned patients with FA and marrow failure, demonstrated feasibility.74 Corrected cells had a repopulation advantage, and marrow failure was ameliorated in patients with high gene marking. These results show that gene therapy as a potential therapy for IBMFS patients in the future.
Future directions
Understanding molecular and cellular mechanisms causing HSC failure is essential for the development of novel therapeutic strategies for IBMFS patients. Specific targeting of senescence and inflammation pathways may improve hematopoiesis, prevent cancer development, or modulate involvement of other organs. Gene editing is a powerful and specific new therapeutic strategy for patients with IBMFS that has great potential but also comes with unknown long-term risks. Future strategies will focus on the early recognition of undesired clones with potential malignant transformation and methods for its suppression. Gene therapy also may be a tool to modulate malignant clonal evolution, although current knowledge from gene therapy for hemoglobinopathies has shown that unbalanced clonal dynamics may arise.
Acknowledgements:
Figure 2 credit to Erina He, medical illustrator, NIH. JML is supported by the Conley Family Endowed Chair of Hematology.
Funding and conflicts of interest:
EMG receives funding from the intramural research program of the National Heart, Lung and Blood Institute. No authors have conflicts of interest to declare.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Fiesco-Roa MO, Giri N, McReynolds LJ, Best AF, Alter BP. Genotype-phenotype associations in Fanconi anemia: A literature review. Blood Rev 2019;37:100589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calado RT, Young NS. Mechanisms of Disease: Telomere Diseases. New England Journal of Medicine 2009;361:2353–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Collopy LC, Walne AJ, Cardoso S, et al. Triallelic and epigenetic-like inheritance in human disorders of telomerase. Blood 2015;126:176–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruggero D, Shimamura A. Marrow failure: a window into ribosome biology. Blood 2014;124:2784–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenberg PS, Huang Y, Alter BP. Individualized risks of first adverse events in patients with Fanconi anemia. Blood 2004;104:350–5. [DOI] [PubMed] [Google Scholar]
- 6.Dokal I Dyskeratosis congenita in all its forms. British Journal of Haematology 2000;110:768–79. [DOI] [PubMed] [Google Scholar]
- 7.Jean D, Odile F, Blandine B, et al. Classification of and risk factors for hematologic complications in a French national cohort of 102 patients with Shwachman-Diamond syndrome. Haematologica 2012;97:1312–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Willig T-N, Niemeyer CM, Leblanc T, et al. Identification of New Prognosis Factors from the Clinical and Epidemiologic Analysis of a Registry of 229 Diamond-Blackfan Anemia Patients. Pediatric Research 1999;46:553–. [DOI] [PubMed] [Google Scholar]
- 9.Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in dyskeratosis congenita. Blood 2009;113:6549–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica 2018;103:30–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vlachos A, Rosenberg PS, Atsidaftos E, et al. Increased risk of colon cancer and osteogenic sarcoma in Diamond-Blackfan anemia. Blood 2018;132:2205–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Helbling-Leclerc A, Garcin C, Rosselli F. Beyond DNA repair and chromosome instability-Fanconi anaemia as a cellular senescence-associated syndrome. Cell Death Differ 2021;28:1159–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jose SS, Tidu F, Burilova P, Kepak T, Bendickova K, Fric J. The Telomerase Complex Directly Controls Hematopoietic Stem Cell Differentiation and Senescence in an Induced Pluripotent Stem Cell Model of Telomeropathy. Front Genet 2018;9:345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Payea MJ, Anerillas C, Tharakan R, Gorospe M. Translational Control during Cellular Senescence. Mol Cell Biol 2021;41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herranz N, Gil J. Mechanisms and functions of cellular senescence. J Clin Invest 2018;128:1238–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of Cellular Senescence. Trends Cell Biol 2018;28:436–53. [DOI] [PubMed] [Google Scholar]
- 17.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res 1961;25:585–621. [DOI] [PubMed] [Google Scholar]
- 18.Gueiderikh A, Maczkowiak-Chartois F, Rosselli F. A new frontier in Fanconi anemia: From DNA repair to ribosome biogenesis. Blood Rev 2021:100904. [DOI] [PubMed] [Google Scholar]
- 19.Pelletier J, Riano-Canalias F, Almacellas E, et al. Nucleotide depletion reveals the impaired ribosome biogenesis checkpoint as a barrier against DNA damage. EMBO J 2020;39:e103838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Birch J, Gil J. Senescence and the SASP: many therapeutic avenues. Genes Dev 2020;34:1565–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.El Mai M, Marzullo M, de Castro IP, Ferreira MG. Opposing p53 and mTOR/AKT promote an in vivo switch from apoptosis to senescence upon telomere shortening in zebrafish. Elife 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calado RT, Regal JA, Kleiner DE, et al. A Spectrum of Severe Familial Liver Disorders Associate with Telomerase Mutations. Plos One 2009;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alves-Paiva RM, Kajigaya S, Feng X, et al. Telomerase enzyme deficiency promotes metabolic dysfunction in murine hepatocytes upon dietary stress. Liver International 2018;38:144–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferrara-Romeo I, Martinez P, Saraswati S, et al. The mTOR pathway is necessary for survival of mice with short telomeres. Nat Commun 2020;11:1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tamburini J, Green AS, Bardet V, et al. Protein synthesis is resistant to rapamycin and constitutes a promising therapeutic target in acute myeloid leukemia. Blood 2009;114:1618–27. [DOI] [PubMed] [Google Scholar]
- 26.Speedy HE, Kinnersley B, Chubb D, et al. Germ line mutations in shelterin complex genes are associated with familial chronic lymphocytic leukemia. Blood 2016;128:2319–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lim TL, Lieberman DB, Davis AR, et al. Germline POT1 variants can predispose to myeloid and lymphoid neoplasms. Leukemia 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tischkowitz MD, Morgan NV, Grimwade D, et al. Deletion and reduced expression of the Fanconi anemia FANCA gene in sporadic acute myeloid leukemia. Leukemia 2004;18:420–5. [DOI] [PubMed] [Google Scholar]
- 29.van der Heijden MS, Yeo CJ, Hruban RH, Kern SE. Fanconi Anemia Gene Mutations in Young-onset Pancreatic Cancer. Cancer Research 2003;63:2585–8. [PubMed] [Google Scholar]
- 30.Wreesmann VB, Estilo C, Eisele DW, Singh B, Wang SJ. Downregulation of Fanconi anemia genes in sporadic head and neck squamous cell carcinoma. ORL J Otorhinolaryngol Relat Spec 2007;69:218–25. [DOI] [PubMed] [Google Scholar]
- 31.Kauffmann A, Rosselli F, Lazar V, et al. High expression of DNA repair pathways is associated with metastasis in melanoma patients. Oncogene 2008;27:565–73. [DOI] [PubMed] [Google Scholar]
- 32.Ozawa H, Iwatsuki M, Mimori K, et al. FANCD2 mRNA overexpression is a bona fide indicator of lymph node metastasis in human colorectal cancer. Ann Surg Oncol 2010;17:2341–8. [DOI] [PubMed] [Google Scholar]
- 33.Nalepa G, Clapp DW. Fanconi anaemia and cancer: an intricate relationship. Nature Reviews Cancer 2018;18:168–85. [DOI] [PubMed] [Google Scholar]
- 34.Colebatch AJ, Dobrovic A, Cooper WA. TERT gene: its function and dysregulation in cancer. J Clin Pathol 2019;72:281–4. [DOI] [PubMed] [Google Scholar]
- 35.Sulima SO, Kampen KR, De Keersmaecker K. Cancer Biogenesis in Ribosomopathies. Cells 2019;8:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Panero J, Alves-Paiva RM, Roisman A, et al. Acquired TERT promoter mutations stimulate TERT transcription in mantle cell lymphoma. American Journal of Hematology 2016;91:481–5. [DOI] [PubMed] [Google Scholar]
- 37.Rode L, Nordestgaard BG, Bojesen SE. Long telomeres and cancer risk among 95 568 individuals from the general population. International Journal of Epidemiology 2016;45:1634–43. [DOI] [PubMed] [Google Scholar]
- 38.Machiela MJ, Lan Q, Slager SL, et al. Genetically predicted longer telomere length is associated with increased risk of B-cell lymphoma subtypes. Hum Mol Genet 2016;25:1663–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ojha J, Codd V, Nelson CP, et al. Genetic Variation Associated with Longer Telomere Length Increases Risk of Chronic Lymphocytic Leukemia. Cancer Epidemiol Biomarkers Prev 2016;25:1043–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scheinberg P, Cooper JN, Sloand EM, Wu CO, Calado RT, Young NS. Association of Telomere Length of Peripheral Blood Leukocytes With Hematopoietic Relapse, Malignant Transformation, and Survival in Severe Aplastic Anemia. Jama-Journal of the American Medical Association 2010;304:1358–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu W, Palovcak A, Li F, Zafar A, Yuan F, Zhang Y. Fanconi anemia pathway as a prospective target for cancer intervention. Cell & Bioscience 2020;10:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ceccaldi R, Briot D, Larghero J, et al. Spontaneous abrogation of the G2 DNA damage checkpoint has clinical benefits but promotes leukemogenesis in Fanconi anemia patients. The Journal of Clinical Investigation 2011;121:184–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bursać S, Brdovčak MC, Pfannkuchen M, et al. Mutual protection of ribosomal proteins L5 and L11 from degradation is essential for p53 activation upon ribosomal biogenesis stress. Proc Natl Acad Sci U S A 2012;109:20467–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Calado RT, Cooper JN, Padilla-Nash HM, et al. Short telomeres result in chromosomal instability in hematopoietic cells and precede malignant evolution in human aplastic anemia. Leukemia 2012;26:700–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Artandi SE, Chang S, Lee SL, et al. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 2000;406:641–5. [DOI] [PubMed] [Google Scholar]
- 46.Riboni R, Casati A, Nardo T, et al. Telomeric fusions in cultured human fibroblasts as a source of genomic instability. Cancer Genet Cytogenet 1997;95:130–6. [DOI] [PubMed] [Google Scholar]
- 47.Maciejowski J, de Lange T. Telomeres in cancer: tumour suppression and genome instability. Nature Reviews Molecular Cell Biology 2017;18:175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schratz KE, Haley L, Danoff SK, et al. Cancer spectrum and outcomes in the Mendelian short telomere syndromes. Blood 2020;135:1946–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liddiard K, Ruis B, Takasugi T, et al. Sister chromatid telomere fusions, but not NHEJ-mediated inter-chromosomal telomere fusions, occur independently of DNA ligases 3 and 4. Genome Res 2016;26:588–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rathbun RK, Faulkner GR, Ostroski MH, et al. Inactivation of the Fanconi anemia group C gene augments interferon-gamma-induced apoptotic responses in hematopoietic cells. Blood 1997;90:974–85. [PubMed] [Google Scholar]
- 51.Haneline LS, Broxmeyer HE, Cooper S, et al. Multiple inhibitory cytokines induce deregulated progenitor growth and apoptosis in hematopoietic cells from Fac−/− mice. Blood 1998;91:4092–8. [PubMed] [Google Scholar]
- 52.Li J, Sejas DP, Zhang X, et al. TNF-alpha induces leukemic clonal evolution ex vivo in Fanconi anemia group C murine stem cells. J Clin Invest 2007;117:3283–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Degan P, Bonassi S, Caterina MD, et al. In vivo accumulation of 8-hydroxy-2’-deoxyguanosine in DNA correlates with release of reactive oxygen species in Fanconi’s anaemia families. Carcinogenesis 1995;16:735–42. [DOI] [PubMed] [Google Scholar]
- 54.Pagano G, Korkina LG, Degan P, et al. In Vitro Hypersensitivity to Oxygen of Fanconi Anemia (FA) Cells Is Linked to Ex Vivo Evidence for Oxidative Stress in FA Homozygotes and Heterozygotes. Blood 1997;89:1111–2. [PubMed] [Google Scholar]
- 55.Uziel O, Reshef H, Ravid A, et al. Oxidative stress causes telomere damage in Fanconi anaemia cells – a possible predisposition for malignant transformation. British Journal of Haematology 2008;142:82–93. [DOI] [PubMed] [Google Scholar]
- 56.Zambetti NA, Ping Z, Chen S, et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-leukemia. Cell Stem Cell 2016;19:613–27. [DOI] [PubMed] [Google Scholar]
- 57.Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 2011;475:53–8. [DOI] [PubMed] [Google Scholar]
- 58.Van Wassenhove LD, Mochly-Rosen D, Weinberg KI. Aldehyde dehydrogenase 2 in aplastic anemia, Fanconi anemia and hematopoietic stem cells. Molecular Genetics and Metabolism 2016;119:28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Revy P, Kannengiesser C, Fischer A. Somatic genetic rescue in Mendelian haematopoietic diseases. Nat Rev Genet 2019;20:582–98. [DOI] [PubMed] [Google Scholar]
- 60.Ramírez MJ, Pujol R, Trujillo-Quintero JP, et al. Natural gene therapy by reverse mosaicism leads to improved hematology in Fanconi anemia patients. Am J Hematol 2021;96:989–99. [DOI] [PubMed] [Google Scholar]
- 61.Gutierrez-Rodrigues F, Donaires FS, Pinto A, et al. Pathogenic TERT promoter variants in telomere diseases. Genet Med 2019;21:1594–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schratz KE, Gaysinskaya V, Cosner ZL, et al. Somatic reversion impacts myelodysplastic syndromes and acute myeloid leukemia evolution in the short telomere disorders. J Clin Invest 2021;131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kennedy AL, Myers KC, Bowman J, et al. Distinct genetic pathways define pre-malignant versus compensatory clonal hematopoiesis in Shwachman-Diamond syndrome. Nature Communications 2021;12:1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sanchez-Medal L, Gomez-Leal A, Duarte L, Guadalupe Rico M. Anabolic androgenic steroids in the treatment of acquired aplastic anemia. Blood 1969;34:283–300. [PubMed] [Google Scholar]
- 65.Calado RT, Yewdell WT, Wilkerson KL, et al. Sex hormones, acting on the TERT gene, increase telomerase activity in human primary hematopoietic cells. Blood 2009;114:2236–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Townsley DM, Dumitriu B, Liu D, et al. Danazol Treatment for Telomere Diseases. New England Journal of Medicine 2016;374:1921–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kirschner M, Vieri M, Kricheldorf K, et al. Androgen derivatives improve blood counts and elongate telomere length in adult cryptic dyskeratosis congenita. Br J Haematol 2021;193:669–73. [DOI] [PubMed] [Google Scholar]
- 68.Boyraz B, Moon DH, Segal M, et al. Posttranscriptional manipulation of TERC reverses molecular hallmarks of telomere disease. J Clin Invest 2016;126:3377–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fan X, Desmond R, Winkler T, et al. Eltrombopag for patients with moderate aplastic anemia or uni-lineage cytopenias. Blood Adv 2020;4:1700–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rocha V, Devergie A, Socie G, et al. Unusual complications after bone marrow transplantation for dyskeratosis congenita. Br J Haematol 1998;103:243–8. [DOI] [PubMed] [Google Scholar]
- 71.Ju Z, Jiang H, Jaworski M, et al. Telomere dysfunction induces environmental alterations limiting hematopoietic stem cell function and engraftment. Nat Med 2007;13:742–7. [DOI] [PubMed] [Google Scholar]
- 72.Bonfim C Special pre- and posttransplant considerations in inherited bone marrow failure and hematopoietic malignancy predisposition syndromes. Hematology Am Soc Hematol Educ Program 2020;2020:107–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thompson AA, Walters MC, Kwiatkowski J, et al. Gene Therapy in Patients with Transfusion-Dependent beta-Thalassemia. N Engl J Med 2018;378:1479–93. [DOI] [PubMed] [Google Scholar]
- 74.Rio P, Navarro S, Wang W, et al. Successful engraftment of gene-corrected hematopoietic stem cells in non-conditioned patients with Fanconi anemia. Nat Med 2019;25:1396–401. [DOI] [PubMed] [Google Scholar]